

Abstract
Background:
The TP53 tumor suppressor gene is the most commonly mutated gene in human cancers. Humans who inherit mutant TP53 alleles develop a wide range of early onset cancers, a disorder called Li-Fraumeni Syndrome (LFS). Trp53-deficient mice recapitulate most but not all of the cancer phenotypes observed in TP53-deficient human cancers, indicating that new animal models may complement current mouse models and better inform on human disease development.
Materials and Methods:
The recent application of CRISPR/Cas9 genetic engineering technology has permitted the emergence of golden Syrian hamsters as genetic models for wide range of diseases, including cancer. Here, the first cancer phenotype of TP53 knockout golden Syrian hamsters is described.
Results:
Hamsters that are homozygous for TP53 mutations become moribund on average ~ 139 days of age, while hamsters that are heterozygous become moribund at ~ 286 days. TP53 homozygous knockout hamsters develop a wide range of cancers, often synchronous and metastatic to multiple tissues, including lymphomas, several sarcomas, especially hemangiosarcomas, myeloid leukemias and several carcinomas. TP53 heterozygous mutants develop a more restricted tumor spectrum, primarily lymphomas.
Conclusions:
Overall, hamsters may provide insights into how TP53 deficiency leads to cancer in humans and can become a new model to test novel therapies.
Keywords: cancer, hamsters, TP53
Introduction
The TP53 gene, which encodes for the p53 protein, is the most commonly mutated gene in human cancers, estimated to occur in more than 50% of all human cancers, including cancers in all major tissue types, while its activity is almost abrogated in the rest of cancers by other mechanisms.[1,2,3] For this reason, p53 has been referred to as the “guardian of the genome.”[4] A majority of TP53 mutations are loss of function (LOF) or dominant-negative gain of function (GOF) missense mutations that are primarily clustered in exons 4–9 that include the DNA-binding domain of the p53 protein. These missense mutations have been associated with cancer cell motility, invasion, and metastasis.[5] Overall, TP53 mutations are important prognostic predictors of cancer cell aggressiveness and are associated with poor clinical outcomes.[5,6] Humans who inherit germline mutations in TP53 develop Li-Fraumeni Syndrome (LFS) that is characterized by a range of early onset cancers including premenopausal breast cancer, soft-tissue and bone sarcomas, carcinomas of the lung, pancreas, skin, and adrenal cortex, several types of brain tumors, and several leukemias.[7,8
A wide range of mouse models of p53 (its gene is referred to as Trp53 in the mouse) deficiency has been generated that include both LOF and GOF models that are used to approximate the effect of TP53 point mutations in human cancers.[9] To originally model LFS, two homozygous knockout (KO) models (Trp53−/−) were generated in the early 1990s.[10,11] Notably, these early LFS mouse models (both heterozygous and homozygous KO mice) did not recapitulate the spectrum, frequency, or latency of tumors observed in human LFS patients. The Trp53−/− LFS mice rapidly developed and succumbed to thymic T-cell lymphomas by ~ 4 months of age. At a much lower frequency, they also developed B-cell lymphomas, and sarcomas, especially hemangiosarcomas. Carcinomas are very rare in the Trp53−/− LFS mouse models. Approximately, a decade later, new missense heterozygous GOF and LOF point mutant mouse models of LFS were developed, such as the structural mutant Trp53R172H and the DNA contact mutant Trp53R270H.[9] These models better approximated the cancers found in human LFS patients, as they developed a variety of carcinomas and endothelial cancers. However, to date, no Trp53 mutant mouse model fully recapitulates the full spectrum of cancers observed in human LFS patients. These phenotypic differences may be due to both species and mouse strain differences.[12] Accordingly, new nonmurine animal models of LFS may help inform on how TP53 dysregulation leads to cancer in humans while providing valuable mammalian interspecies comparisons of TP53-mediated cancer susceptibility. Here, we describe the cancer spectrum in a TP53 mutant golden Syrian hamster model of LFS generated by CRISPR/Cas9 technology.
Materials and Methods
Animals
Golden Syrian hamsters used for pronuclear (PN) embryo production were bred in-house using founder animals purchased from Charles River (LVG Golden Syrian Hamster, Strain Code: 049). All hamsters were raised and maintained in an air-conditioned room with a 14 L: 10D light cycle (light from 0600). Hamsters were fed standard rodent chow, and food and water were available ad libitum. Hamsters were bred at three institutions: Utah State University, the University of Minnesota, and Zhengzhou University (ZZU). At all locations, TP53+/− crosses were the predominant mating strategy employed. We did determine that crosses employing TP53−/− females produced smaller litter sizes, regardless of the genotype of the offspring. Two TP53−/− dams also failed to deliver full-term pups and did not survive the pregnancy.
Generation of TP53 knockout golden Syrian hamsters by CRISPR/Cas9
sgRNA design, construct, embryo manipulation, and embryo transfer were performed for the establishment of TP53 KO golden Syrian hamster as described previously.[13] In brief, sgRNA was designed to target a site-specific sequence within the fifth exon of the hamster TP53 gene[14] (NW_004801614.1). Microinjection was performed under red light and HECM-9 medium covered by mineral oil was used as the injection medium. For PN injection, the DNA injection solution was diluted to 10 ng/μl with TE buffer. The injected embryos were cultured in HECM-9 medium covered by mineral oil at 37.5°C under 10% CO2, 5% O2, and 85% N2 for 0.5 h before embryo transfer. Viable embryos after injection were transferred to each oviduct (15 embryos per oviduct) of pseudopregnant females. Genotyping of pups produced from microinjected embryos was performed by Cleavage Detection assay followed by Sanger sequencing with genomic DNA isolated from toes collected from 2-week-old pups. Genomic regions flanking the CRISPR targeted sites were amplified by the polymerase chain reaction (PCR) primers p53-F 5'-CTA AAC CAA CTG GTG TGT AGA ACC CC-3', P53-R 5'-GCT CAT AGG GCA CCA CCA CA-3'. All pups were genotyped using a T7 endo I assay that detects heteroduplex DNA that results from annealing DNA stands that have been modified after a sgRNA/Cas9 mediated cut to DNA strands without modifications. Potential off-target effects were analyzed based on the rule that sequences matching the final 12 nt of the target sequence and protospacer adjacent motif sequence might cause the off-target effect. The off-target fragments were amplified from the founder's genomic DNA and identified by Sanger sequence. PCR primers for analysis of off-target effects are listed in Table 1.
Table 1.
Polymerase chain reaction primers for polymerase chain reaction off-target assay
| Primers - off targets | |
|---|---|
|
| |
| Name | Sequence (5`-3`) |
| OT1F | TGGTTGCTTTACATCTTCTG |
| OT1R | GAGTCATTGAGCTAGGTCTG |
| OT2F | GTGTCAAGAAGGAGAGGCGC |
| OT2R | GCAAAGCTGAGGTGATGGGG |
| OT3F | GCTTTCTGATCACACCCAAG |
| OT3R | TAGCATTATCTGGCCTGCCC |
| OT4F | ATTGCGCTCTCTTCATTCGG |
| OT4R | AGAGAGGGGAACTGACAAGC |
| OT5F | CACTTGTGTGGATGTAAAAG |
| OT5R | ATCTCTTATGTGCAGGCATC |
| OT6F | AGGGAGAATTTTGTTGCTTG |
| OT6R | TCACTGTGAACACCTGAGAG |
| OT7F | TTGGTGTGGGGCAGTGACTG |
| OT7R | TTCTGCGCCAAGGGAAATGG |
| OT8F | ATTGAACTCACAGAGATCTG |
| OT8R | GGACTAGTATATTGTCTTCCTC |
Histopathology
Tissues for histological analyses were formalin fixed for a minimum of 2 days, followed by rinsing and storage in 70% ethanol. Histological analysis was conducted at the Utah Veterinary Diagnostic Laboratory at Utah State University, the Department of Pathology in the First Affiliated Hospital of ZZU, and at the University of Minnesota Masonic Cancer Center Comparative Pathology Shared Resource (CPSR). Gross necropsy was performed at USU by a graduate student assistant. Gross necropsy at ZZU was performed by animal care staff. Collection and formalin-fixation of tissues at the University of Minnesota was performed by Dr. Robert Cormier with follow-up full body necropsies carried out at the CPSR by Dr. Alexandru-Flaviu Tabaran or Dr. Timothy Carlson. Analyses were performed at USU by A. C. V. P.-certified veterinary pathologist Arnaud J. Van Wettere (D. V. M, Ph. D.), at ZZU by Kuisheng Chen (MD, Ph. D.) and Huixiang Li (MD, Ph. D.), trained and Chinese Society of Pathologists-Certified pathologists, and at the UMN by three A. C. V. P.-certified veterinary pathologists, M. Gerald O'Sullivan.(M. V. B., Ph. D.), Director of the CPSR, Alexandru-Flaviu Tabaran (D. V. M., Ph. D.), and Timothy Carlson (D. V. M, Ph. D.). All gross lesions observed in the hamster carcasses were analyzed, with representative slides of tissue sections stained with hematoxylin and eosin.
Immunohistochemistry
Anti-myeloperoxidase (MPO). Whole body was formalin fixed for 3 days, followed by storage in 70% ethanol. Tissues were processed for paraffin embedding, and 10 μm sections were mounted on gelatin-coated histological slides followed by standard deparaffinization steps. Target Retrieval: Citrate low pH 6.0, 30 min; Abcam antibody: cat # ab9535, dilution 1:50, 30 min; Detection: Dako Rabbit Envision-30 min; Chromagen: DAB 5 min. Anti-CD3. Anti-CD3 antibody, DAKO/Agilent, Santa Clara, CA, USA; Cat# A0452. Tissues were processed identically as for MPO as was target retrieval and detection.
Western blotting
Ear biopsies were collected from the wild-type (WT) and homozygous one-nucleotide insertion mutants. Tissues were ground in liquid nitrogen and then lysed in lysis buffer (150 mM NaCl, 1.0% Triton X-100 and 50 mM Tris, pH 8.0, with a protease inhibitor cocktails (Sigma)). Gross protein mass was quantified by a Pierce BCA protein assay kit (Thermo Scientific) according to the manufacturer's protocol. Tissue lysates were separated on NuPAGE 4%–12% Bis-Tris gel (Life Technologies) and then electrotransferred to a polyvinylidene fluoride membrane (Life Technologies). The blocking and washing steps were performed using the WesternBreeze chemiluminescent kit (Life Technologies) and strictly followed the manufacturer's protocol. The membrane was incubated at RT with the primary antibody mouse monoclonal anti-P53 antibody (Abcam, PAb 240, ab26), 1:500 dilution in 1% bovine serum albumin/phosphate-buffered saline for 1 h. After washing, the membrane was incubated with the alkaline phosphatase-conjugated secondary antibody (anti-rabbit) for 30 min and was exposed.
Kaplan–Meier analysis
Overall survival was defined as the time from birth to sacrifice. TP531+/+ animals were sacrificed at 365 days while still healthy and so were censored. TP53+/− and TP53−/− animals were immediately sacrificed when moribund or otherwise in distress such as demonstrating presumptive cancers (e.g., lumps and skin lesions), swelling or weight loss, lethargy, and difficulty in moving. Ages of animals found dead in cage were also included. No animals in these groups were censored. Kaplan–Meier analysis was used to visualize differences in survival between genotypes. The significance of differences between genotypes was determined using a log-rank test with Bonferroni correction for multiple comparisons. P ≤0.05 was considered significant.
Ethics statement
The experiments were conducted in strict accordance with guidelines of the AAALAC-accredited Laboratory Animal Research Center at the Utah State University and approved by the Institutional Animal Care and Use Committee of Utah State University (IACUC Protocol: 2091). The animal experiments conformed to the Provision and General Recommendation of Chinese Experimental Animals Administration Legislation accepted principles for the care and use of laboratory animals by the Ethics Committee on Animal Experiment of ZZU (IACUC-ZZU-2016/Wang). All procedures on golden Syrian hamsters followed a protocol approved by the University of Minnesota Institutional Animal Care and Use Committee (IACUC protocol: 1807-36189A). Hamsters were housed in a separate room at the University of Minnesota Medical School Duluth Animal Services Facility. They were tested for a panel of common rodent laboratory pathogens. They were fed Teklad 501 Lab Chow and tap water ad libitum. All cages included enrichment such as plastic tubes.
Results
Generation of TP53 knockout golden Syrian Hamsters using CRISPR/Cas9
The CRISPR/Cas9 system[15] has been demonstrated to be a highly efficient system for generating targeted mutations in the golden Syrian hamster genome.[13] We designed gRNA/Cas9 expressing vectors targeting hamster TP53 exon 5 [Figure 1a]. Employing PN injection 10 ng/μL of vector was injected into the male nucleus of zygotes of the golden Syrian hamster. A total of 76 injected zygotes were transferred to 3 pseudopregnant female golden Syrian hamsters, and 30 pups were born. Genotyping results by the Cleavage Detection assay are shown for three golden Syrian hamster pups [Figure 1b]. We determined that pups F0#1 and F0#3 were WT; and pup F0#2 was genetically modified in the TP53 locus. Furthermore, subcloning sequencing confirmed that one founder hamster (F0#2) was genetically mosaic, carrying a frame-shift mutation (1 bp insertion) and 21-nucleotide deletions, as well as the WT allele [Figure 1c]. To detect germline transmission of the mutant alleles to the F1 generation founder, F0#2 was chosen to establish a breeding colony (designated as TP53−/−), which carried a 1 bp insertion, and resulted in a termination codon thioglycolic acid at amino acid 311, that produces a truncation mutation that disrupts the DNA binding domain of TP53. [Figure 1d] shows the genotyping results with the Cleavage Detection assay for the eighteen F1 pups produced by breeding founder animal F0#2 with a WT littermate. Sub-cloning sequencing shows an individual clone had either WT or TP53 KO alleles. Sanger sequencing results comparing between WT sequence and the sequencing result of F1#15 at the TP53 genomic locus further demonstrated the success in making a germline transmission TP53 mutant golden Syrian hamster. The red box shows the insertion of 1 bp in TP53 genomic loci [Figure 1e]. Western blotting using the cell lysates from the F5 homozygous fibroblasts cells and WT fibroblasts cells showed that P53 protein expression was totally abolished in tissues containing the 1 bp insertion [Figure 1f].
Figure 1.

Generation of p53 knockout golden Syrian hamsters using the CRISPR/Cas9 system. (a) Schematic of the golden Syrian hamster p53 genomic locus showing the exons and gene targeting site. The sequence of sgRNA in green letters, with the protospacer adjacent motif in red letters to target a region of exon 5 in the p53 golden Syrian hamster gene. The blue two arrows indicate the locations of polymerase chain reaction primers for the sequence. (b). Genotyping results by the Cleavage Detection assay on three golden Syrian hamster pups produced by pronuclear injections of sgRNA/Cas9-p53. Pups F0#1 and F0#3 are wild type; pup F0#2 is genetically modified in the p53 locus. (c) The frequencies of DNA sub-clones with F0#2 are indicated by flanking regions surrounding the sgRNA targeting site identified F0#2 with frame shift mutations. −21 bp: 21-nucleotide deletions; +1 bp: one-nucleotide insertion. (d) Genotyping results with the Cleavage Detection assay on the eighteen F1 pups produced by breeding founder animal F0#2 with a wild type littermate. (e) Subcloning sequencing shows an individual clone has either wild type or p53 knockout alleles. Comparison between wild type sequence and the sequencing result of F1#15 at the p53 genomic locus. The red box shows the insertion of 1 nucleotide in p53 genomic loci. (f) Western blotting assay to compare the expression of p53 protein in wild type and homozygous one-nucleotide insertion hamsters
Analysis of off-site targeting in TP53 KO golden Syrian hamsters
Studies have reported that CRISPR/Cas9 can introduce off-target cleavage.[16] We investigated off-target cleavage in the CRISPR/Cas9 generated TP53 mutant hamsters using a blast search of the golden Syrian hamster sequence database (http://www.ncbi.nlm.nih.gov/genome/11998) with the targeting sequence by sgRNA/Cas9-p53 as the query to find the genomic sequences with the highest homology. We then tested the top eight potential off-target sites which share the highest sequence homology to the seed sequence of the target-site [Figure 2a]. A region flanking each off-target site was amplified by using the genomic DNA isolated from WT and TP53-targeted founders [Figure 2b]. All samples resulted in the same molecular size band indicating that there were no large deletions flanking the sites. We confirmed the sequence with primers flanking each off-target site by Sanger sequencing [Figure 2c]. The sequencing data confirmed no off-target cleavage, indicating that the TP53 gene has been specifically targeted in the golden Syrian hamster genome.
Figure 2.
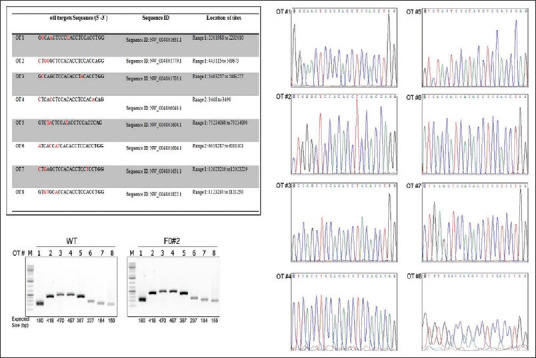
Analysis of off-site target candidates at predicted sites in the golden Syrian hamster genome. (a) The top eight predicted off-site targets for mutagenesis. The mismatch sequence is labeled in red and protospacer adjacent motif is indicated in bold letters. (b) Polymerase chain reaction products flanking each predicted off-target site (#1-8) in wild type and mutant founder (F0#2) hamster. (c) Polymerase chain reaction product sequencing confirmed no off-target cleavage. A representative sequencing analysis of F0#2 is shown
Survival rate of TP53−/− and TP53+/− golden Syrian hamsters
We determined the survival rate of 105 TP53−/− and 105 TP53+/− hamsters, along with a cohort of seven TP53+/+ control hamsters. None of the WT hamsters developed cancers by 1 year of age, nor did we commonly see cancers in aged WT hamsters that have a normal lifespan of up to 30 months. All of the TP53 mutant hamsters (both homozygous and heterozygous KOs) became ill or in some cases were found dead in their cage starting at 52 days of age. Animals that became ill were immediately sacrificed and analyzed. Figure 3 shows a Kaplan–Meier survival plot for the three TP53 genotypes. On average, TP53−/− hamsters survived to 139 days, and TP53+/− hamsters survived to 286 days. The great majority (>95%) of the hamsters studied by experienced pathologists showed evidence of various types of neoplasia, often synchronous.
Figure 3.
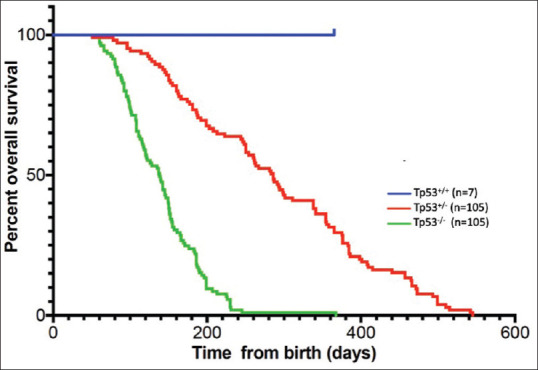
Kaplan–Meier plot showing the association of TP53 genotypes with overall survival. P values were determined by log-rank test using a Bonferroni adjustment for multiple comparisons. All comparisons show statistically significant survival differences. Tp53+/+ versus Tp53+/−, P ≤ 0.05; Tp53+/+ versus Tp53−/−, P ≤ 0.001; Tp53+/− versus Tp53−/−, P ≤ 0.001
Cancer types and frequency
Table 2 lists the various types of cancers observed and their frequency. In all, 82 mutant hamsters received full pathological analysis. These consisted of 52 TP53−/− and 30 TP53+/− animals.
Table 2.
Cancer incidence and frequency
| Incidence | Frequency (%) | |
|---|---|---|
| P53−/− (n=52) | ||
| Lymphoma | 15 | 29 |
| Hemangiosarcoma | 14 | 27 |
| Myeloid leukemia | 11 | 21 |
| Anaplastic sarcoma | 9 | 17 |
| Myeloid proliferation | 5 | 10 |
| Adrenocortical carcinoma | 4 | 8 |
| Osteosarcoma | 3 | 6 |
| Pancreatic adenocarcinoma | 2 | 4 |
| Plasma cell tumor | 1 | 2 |
| Squamous cell carcinoma | 1 | 2 |
| Renal adenocarcinoma | 0 | 0 |
| P53+/− (n=30) | ||
| Lymphoma | 20 | 67 |
| Hemangiosarcoma | 5 | 17 |
| Myeloid leukemia | 2 | 6 |
| Anaplastic sarcoma | 0 | 0 |
| Myeloid proliferation | 1 | 3 |
| Adrenocortical carcinoma | 1 | 3 |
| Osteosarcoma | 1 | 3 |
| Pancreatic adenocarcinoma | 0 | 0 |
| Plasma cell tumor | 0 | 0 |
| Squamous cell carcinoma | 0 | 0 |
| Renal adenocarcinoma | 1 | 3 |
Loss of heterozygosity analysis
We tested cancers that were primarily diagnosed as lymphomas, from 17 TP53+/− hamsters. Using Sanger sequencing flanking the TP53 mutation site, we found that all 17 cancers demonstrated loss of heterozygosity (LOH) [Figure 4]. It is noted that while lymphomas were predominant in the TP53+/− cohort, and we tested 17 of the 25 lymphomas for LOH, several sarcomas were also found in this cohort that was not tested for LOH.
Figure 4.
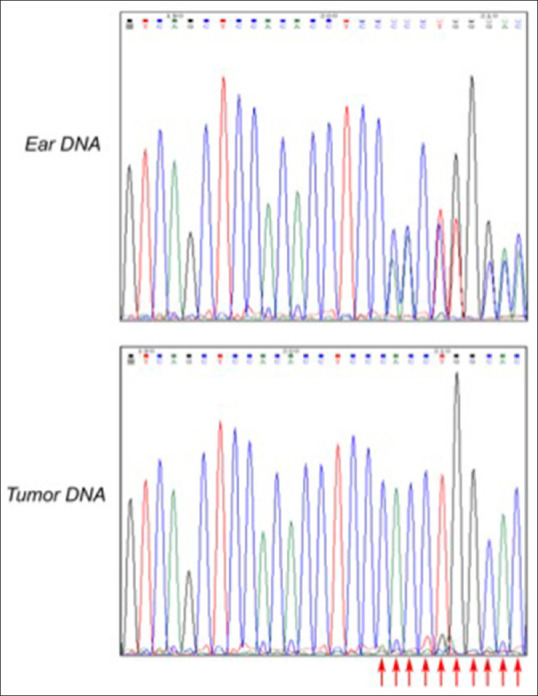
Loss of heterozygosity analysis. Sanger sequencing was employed to detect the upstream and downstream sequence of 300 bp above and below the knockout site of the targeted TP53 mutation in tumor tissue. Ear DNA was isolated at genotyping. Tumor tissue clearly shows the loss of the wild type allele. Cancers were predominantly T cell lymphomas and were isolated from the following 17 animal IDs: F2M2, F2M3, F2M6, F3F25, F3F16, F3M21, F3M51, F4F41, F3M27, F3F15, F3F4, F4F65, F2M1, F3F30, F1F15, F10M25, and F5F8. All 17 animals tested showed loss of heterozygosity. Table S1 for a full description of the animals and pathology. Polymerase chain reaction primers: F: CTAAACCAACTGGTGTGTAGACCCC, R: GCTCATAGGGCACCACCACA
Cancer phenotypes
The great majority of hamsters analyzed by histopathology developed cancers, with some developing multiple synchronous cancers. Cancers were often metastatic to multiple organs. Figure 5 depicts H and E images of several cancers, notably invasive lymphoma cells in skeletal muscle, heart, spleen, and kidney [Figure 5a-c, f, and g], hemangiosarcoma in liver and dermis [Figure 5d and h], and squamous cell carcinoma in dermis [Figure 5e]. Overall, combing both genotypes, 43% of mutant hamsters developed lymphomas, with T-cell lymphomas positive for CD3 likely representing the majority of lymphomas. We note that some of the lymphomas could be B-cell lymphomas as not all lymphomas were tested for CD3 and CD20 immunohistochemistry (IHC) for B cells was inconclusive. Thirty-nine percent of hamsters developed a range of sarcomas, with hemangiosarcomas the most common type, followed by anaplastic sarcomas and osteosarcomas. Several hamsters developed vertebral sarcomas that caused paraparesis and paraplegia. A depiction of gross pathology images of various sarcomas, including several that are unusual in anatomic location, are shown in Figure 6. Several hamsters developed sarcomas in the mouth and facial area [Figure 6a and b]. Osteosarcomas were found in four hamsters [Figure 6c and d]. Figure 6f depicts a sarcoma at T7 suggestive of hemangiosarcoma and osteosarcoma in a hamster with paraplegia. Figure 6g and h depicts a very large abdominal anaplastic sarcoma. Thirteen hamsters (16%) were diagnosed with neoplasia consistent with myeloid leukemias and an additional 5 hamsters (7%) exhibited myeloid proliferation in multiple tissues including bone marrow that could represent a precancerous stage. A biomarker of myeloid-derived cells, MPO, was detected in these tumors by IHC [Figure 7]. It is noted that eleven cases also had extensive myeloid infiltration of bone marrow consistent primarily with a diagnosis of myeloid leukemia and secondarily of myeloid hyperplasia. Six of the 13 lesions diagnosed as myeloid leukemia based on morphological analysis were stained for MPO and all showed varying degrees of focal positive MPO staining. A subset (but not all) of the suspect myeloid leukemias tested negative for CD3 IHC (data not shown). Similarly, some of the lymphomas untested for MPO could have been MPO positive. Nevertheless, it is probable that the relative frequency of lymphomas and myeloid leukemias is roughly accurate, with lymphomas, most likely T-cell lymphomas, predominant. Five hamsters developed adrenocortical carcinomas, cancer commonly observed in human LFS patients[17] [Figure 6e]. The distribution of cancers in the two mutant cohorts showed some similarities but also distinct differences. The frequency of lymphomas was higher in TP53+/− hamsters (67% vs. 29% in TP53−/− hamsters), and the frequency of myeloid leukemias (and myeloid proliferation) was significantly higher in TP53−/− hamsters versus TP53+/− animals. TP53+/− hamsters developed only two epithelial cancers, a renal cell carcinoma and an adrenocortical carcinoma while TP53−/− hamsters developed a total of 6 epithelial cancers, all at low frequency, including adrenocortical carcinomas, renal cell carcinoma, and pancreatic adenocarcinoma, plus one oral squamous cell carcinoma. Sarcomas in general and anaplastic sarcomas in particular were found more frequently in the TP53−/− cohort. It is noted that no mammary cancers were observed in mutant hamsters, an absence also seen in all Trp53 mutant mice. Breast cancers are common in human LFS patients.
Figure 5.
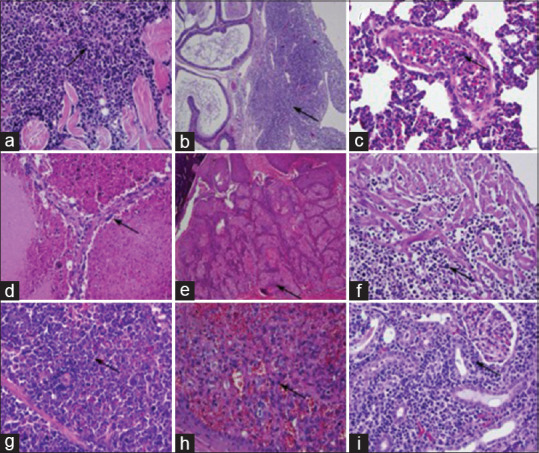
Histopathology of representative tumors from TP53−/− and TP53+/− hamsters. Neoplastic lymphocytes (lymphoma) infiltrate among skeletal muscle fibers (a), peri-epididymal collagen fibers (b), and cardiac myocytes (f), and expand the splenic red pulp (g) and the renal interstitium (i). Neoplastic lymphocytes within a pulmonary blood vessel (leukemia; c). Neoplastic endothelial cells (hemangiosarcoma) form vascular channels within the liver (d) and dermis (h). Neoplastic squamous epithelial cells (squamous cell carcinoma) form an expansive mass within the dermis (e). (a-i) Hematoxylin and eosin stains. Scale bars: 150 μm
Figure 6.

Gross pathology images of sarcomas. (a) Hamster# 22880, anaplastic sarcoma in mouth. (b) Hamster# 22880, arrow-same anaplastic sarcoma, interface with tongue. (c) Hamster# 22832, osteosarcoma in body wall. (d) Hamster# 22832, same osteosarcoma, rib. (e) Hamster# 22857, arrow-adrenal carcinoma. (f) Hamster# 22861, sarcoma at T7 suggestive of hemangiosarcoma and osteosarcoma. (g) Hamster# 22873, abdominal tumor– bizarre anaplastic sarcoma. (h) Hamster# 22873, same abdominal tumor– bizarre anaplastic sarcoma
Figure 7.
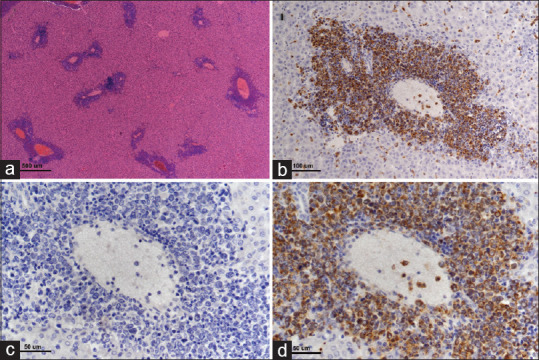
Myeloid leukemia involvement of liver. There are multifocal infiltrates of neoplastic cells predominantly involving the portal triads (a, hematoxylin and eosin, ×4 objective lens). Infiltrating cells surrounding hepatic portal veins are strongly myeloperoxidase positive (b and d, ×20 and × 40 objective lens, respectively). (c) Corresponding negative control (myeloperoxidase-specific primary antibody is substituted with normal rabbit immunoglobulin on a serial step section, ×40 objective lens) for image in d
Other lesions in TP53 mutant hamsters
In addition to cancers, both TP53+/− and TP53−/− mutant hamsters showed a wide range of tissue abnormalities that were putatively either secondary to cancers, possible preneoplastic tissue events, or incidental findings. These abnormalities included facial dermatitis and epidermal hyperplasia; bone marrow myeloid hyperplasia; hemothorax and hemoabdomen; interstitial pneumonia and pulmonary hemorrhage; liver necrosis, hepatitis, fibrosis, telangiectasia, and biliary hyperplasia; chronic nephropathy and hydronephrosis; myocardial necrosis and inflammation, myonecrosis of skeletal muscle; intestinal abscesses, protozoa and bacterial hyperproliferation in the large intestine and stomach, cecum, and colon inflammation; and splenic lymphoid hyperplasia and extramedullary hematopoiesis.
Discussion
With the successful application of CRISPR/Cas9 technology to genetic engineering in the golden Syrian hamster,[13,18] the hamster has become an increasingly powerful emerging genetic model for human diseases[19] including cancer.[20] Here, we describe the cancer phenotype of the first genetic KO of the TP53 tumor suppressor gene in hamsters. TP53 homozygous KO hamsters developed cancers early, becoming moribund as early as 52 days with a mean age of 139 days. They developed a wide range of cancers that, with two notable exceptions are similar to cancers observed in human LFS patients and in patients whose cancers harbor sporadic mutations in TP53. Similar to human cancers with TP53 mutations, a variety of sarcomas were found in TP53 homozygous KO hamsters, especially hemangiosarcomas but also anaplastic sarcomas and osteosarcomas, along with low-frequency carcinomas of the kidney, pancreas, and adrenal glands. The two exceptions were the absence of mammary cancers, which are common in LFS patients and in sporadic TP53-mutant human cancers and the high prevalence in hamsters of lymphomas, which are rare in LFS patients and sporadic TP53 mutant human cancers. The cancer spectrum in TP53+/− hamster cancers, who became moribund at a mean age of 286 days, is much more restricted to lymphomas and a low incidence of hemangiosarcomas.
Notably, one other TP53-dependent cancer occurring in hamsters and humans is myeloid leukemias. TP53 homozygous KO hamsters demonstrated invasive neoplasia consistent with myeloid leukemia with an incidence of 16%. While TP53 mutant myeloid leukemias are not common in LFS patients (leukemias overall have an incidence of only 4%), they are a very serious problem in therapy-relatedacute myeloid leukemia (AML s) and myelodysplastic syndromes (MDS) in LFS patients.[21,22] In sporadic human cancers, ~10% of AMLs harbor TP53 mutations, representing a subset of AMLs that are very aggressive and that respond poorly to chemotherapy.[23,24,25] In non-LFS patients, TP53 mutations are also observed in ~ 20% of MDS.[26] Moreover, p53 dysfunction, measured by large-scale sequencing, proteomics, and clinical studies, is highly prevalent in human AML, independent of TP53 mutational status, owing to dysregulation of p53 inhibitors such as MDM2 and MDM4.[27,28,29] As discussed by Barbosa et al.,[30] dysregulation of TP53 in AML, either by mutation or by inhibition of WT p53, presents both therapeutic challenges and opportunities for the development of novel therapies for AML. Accordingly, hamster models of TP53-dependent myeloid diseases can provide important insights into how these disorders develop in humans and provide models for the testing of novel therapies.
Conclusion
It is important to compare the cancer phenotypes of TP53 mutant hamsters with Trp53 mutant mouse models and in turn with human TP53-deficient cancers. As mentioned in the introduction, mouse models fail to fully recapitulate the spectrum and phenotypes of TP53-deficient cancers in humans, thus arguing for the need for new animal models that can either complement mouse models or better inform on human cancer etiology. TP53 mutant hamsters, as a fully new model of TP53-deficient cancers in a new species, provide unique opportunities for comparisons with human TP53-deficient cancers and valuable mammalian interspecies comparisons with other animal models such as Trp53-deficient mice. As discussed in the paragraph above, an example of where TP53-deficient hamsters are superior to mouse models is the development in hamsters of an aggressive form of acute myelogenous leukemia (AML). TP53-deficient AML in humans is one of the most lethal types of AML with poor prognosis and median survival of ~ 6 months, and TP53-deficient human AML is refractory to all current AML therapies. TP53-deficient mice are poor models of AML and generally do not develop AML, MDS, or other myeloid diseases,[31] unlike hamsters, and this fact has hindered studies to understand stages of AML progression in human AML. Thus, hamsters provide a much needed rodent model of TP53-deficient AML, and TP53-deficient hamsters thereby also offer a valuable model for testing of novel therapeutics that may have failed or be unfeasible in mouse models.
Regarding other cancer comparisons between TP53-deficient mice and hamsters, lymphomas, likely predominantly T-cell lymphomas (although some B-cell lymphomas cannot be excluded), are common in both hamsters and mice, but as noted above, are not very common in human patients, and both hamster and mouse models fail to develop TP53-deficient mammary cancers. TP53+/− hamsters and Trp53+/− mice both develop cancers at a latency of about double the age of homozygous mutants, with a more restricted range of cancers, and in both hamsters and mice, LOH is observed in the cancers, although the reported rate of LOH in Trp53 point mutant mice[9] was ~50% compared with 100% in the 17 hamster cancers that were analyzed in our study. Trp53 homozygous mutant mice rapidly develop and succumb to T-cell lymphomas,[10,11] with an occasional sarcoma and no carcinomas. Targeted Trp53+/− GOF point mutants such as Trp53R172H and Trp53R270H[11] develop a much wider range of cancers that are similar, with one exception, to cancers found in TP53 homozygous KO hamsters, and Trp53R270H/+ mice were reported to also develop B-cell lymphomas.
Finally, it should be noted that golden Syrian hamsters are susceptible to lymphomagenesis caused by hamster polyomavirus (HaPV).[32,33] Lymphomas caused by HaPV was first identified in a colony of HASH: Sal hamsters in Spain that were susceptible to audiogenic seizures, and HaPV has since been considered endemic in hamster colonies worldwide including commercial sources. Therefore, it is possible that some of the lymphomas observed in the TP53 mutant hamsters were in fact caused by HaPV. We did not routinely test TP53 mutant hamsters for HaPV infection. However, we did test hamsters of differing genotypes at the hamster colonies of all three institutions collaborating on this project: Utah State University, University of Minnesota Medical School, and ZZU in China. We employed a sensitive PCR test that is considered highly accurate for HaPV detection.[34] The USU hamster colony where the TP53 KO hamster was created initially tested positive for HaPV but since become HaPV-free following cleanup. The ZZU colony tested 12 current hamster samples and all tested negative. At the UMN colony, the first pups from the initial breeding pair acquired from USU in 2017 were tested, along with samples from 2018, 2019, 2020, and 2021. All samples tested negative. While more conclusive ELISA tests would be necessary, the PCR data indicates that the colony in Minnesota has been HaPV free that the colony at ZZU is currently HaPV free but could have been HaPV positive in the past, while the colony at USU was definitely HaPV positive in the past, when the studies described here were underway.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Acknowledgments
This work was supported by the National Key R and D Program of China, No. 2016YFE0200800; the Nature Sciences Foundation of China, No. 81771776 and No. U1704282 and the MRC MR/M015696/1. At the University of Minnesota, this research project received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors; however, Essentia Health Systems and the University of Minnesota Medical School provided general research support to RC.
We wish to thank Dr. Kati Kovacs and Ms. Paula Overn at the University of Minnesota Masonic Cancer Center Comparative Pathology Shared Resource for excellent histological and IHC preparations.
References
- 1.Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502:333–9. doi: 10.1038/nature12634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hollstein M, Sidransky D, Vogelstein B, Harris CC. p53 mutations in human cancers. Science. 1991;253:49–53. doi: 10.1126/science.1905840. [DOI] [PubMed] [Google Scholar]
- 3.Zhou X, Hao Q, Lu H. Mutant p53 in cancer therapy-the barrier or the path. J Mol Cell Biol. 2019;11:293–305. doi: 10.1093/jmcb/mjy072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lane DP. Cancer.p53, guardian of the genome. Nature. 1992;358:15–6. doi: 10.1038/358015a0. [DOI] [PubMed] [Google Scholar]
- 5.Tang Q, Su Z, Gu W, Rustgi AK. Mutant p53 on the path to metastasis. Trends Cancer. 2020;6:62–73. doi: 10.1016/j.trecan.2019.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hall C, Muller P. The diverse functions of mutant 53, its family members and isoforms in cancer. Int J Mol Sci. 2019;20:6188. doi: 10.3390/ijms20246188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kleihues P, Schäuble B, Zur Hausen A, Estève J, Ohgaki H. Tumors associated with p53 germline mutations: A synopsis of 91 families. Am J Pathol. 1997;150:1–13. [PMC free article] [PubMed] [Google Scholar]
- 8.Malkin D. Li-Fraumeni syndrome. Genes Cancer. 2011;2:475–84. doi: 10.1177/1947601911413466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Olive KP, Tuveson DA, Ruhe ZC, Yin B, Willis NA, Bronson RT, et al. Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell. 2004;119:847–60. doi: 10.1016/j.cell.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 10.Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA, Jr, Butel JS, et al. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992;356:215–21. doi: 10.1038/356215a0. [DOI] [PubMed] [Google Scholar]
- 11.Jacks T, Remington L, Williams BO, Schmitt EM, Halachmi S, Bronson RT, et al. Tumor spectrum analysis in p53-mutant mice. Curr Biol. 1994;4:1–7. doi: 10.1016/s0960-9822(00)00002-6. [DOI] [PubMed] [Google Scholar]
- 12.Harvey M, McArthur MJ, Montgomery CA, Jr, Bradley A, Donehower LA. Genetic background alters the spectrum of tumors that develop in p53-deficient mice. FASEB J. 1993;7:938–43. doi: 10.1096/fasebj.7.10.8344491. [DOI] [PubMed] [Google Scholar]
- 13.Fan Z, Li W, Lee SR, Meng Q, Shi B, Bunch TD, et al. Efficient gene targeting in golden Syrian hamsters by the CRISPR/Cas9 system. PLoS One. 2014;9:e109755. doi: 10.1371/journal.pone.0109755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Legros Y, McIntyre P, Soussi T. The cDNA cloning and immunological characterization of hamster p53. Gene. 1992;112:247–50. doi: 10.1016/0378-1119(92)90384-2. [DOI] [PubMed] [Google Scholar]
- 15.Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–23. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ran FA, Hsu PD, Lin CY, Gootenberg JS, Konermann S, Trevino AE, et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell. 2013;154:1380–9. doi: 10.1016/j.cell.2013.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Crona J, Beuschlein F. Adrenocortical carcinoma – Towards genomics guided clinical care. Nat Rev Endocrinol. 2019;15:548–60. doi: 10.1038/s41574-019-0221-7. [DOI] [PubMed] [Google Scholar]
- 18.Li R, Miao J, Fan Z, Song S, Kong IK, Wang Y, et al. Production of genetically engineered golden Syrian hamsters by pronuclear injection of the CRISPR/Cas9 complex. J Vis Exp. 2018;131:562. doi: 10.3791/56263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sanchez-Felipe L, Vercruysse T, Sharma S, Ma J, Lemmens V, Van Looveren D, et al. A single-dose live-attenuated YF17D-vectored SARS-CoV-2 vaccine candidate. Nature. 2021;590:320–5. doi: 10.1038/s41586-020-3035-9. [DOI] [PubMed] [Google Scholar]
- 20.Li R, Miao J, Tabaran AF, O'Sullivan MG, Anderson KJ, Scott PM, et al. A novel cancer syndrome caused by KCNQ1-deficiency in the golden Syrian hamster. J Carcinog. 2018;17:6. doi: 10.4103/jcar.JCar_5_18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Swaminathan M, Bannon SA, Routbort M, Naqvi K, Kadia TM, Takahashi K, et al. Hematologic malignancies and Li-Fraumeni syndrome. Cold Spring Harb Mol Case Stud. 2019;5:a003210. doi: 10.1101/mcs.a003210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Anensen N, Skavland J, Stapnes C, Ryningen A, Børresen-Dale AL, Gjertsen BT, et al. Acute myelogenous leukemia in a patient with Li-Fraumeni syndrome treated with valproic acid, theophyllamine and all-trans retinoic acid: A case report. Leukemia. 2006;20:734–6. doi: 10.1038/sj.leu.2404117. [DOI] [PubMed] [Google Scholar]
- 23.Chung J, Sallman DA, Padron E. TP53 and therapy-related myeloid neoplasms. Best Pract Res Clin Haematol. 2019;32:98–103. doi: 10.1016/j.beha.2019.02.009. [DOI] [PubMed] [Google Scholar]
- 24.Hunter AM, Sallman DA. Current status and new treatment approaches in TP53 mutated AML. Best Pract Res Clin Haematol. 2019;32:134–44. doi: 10.1016/j.beha.2019.05.004. [DOI] [PubMed] [Google Scholar]
- 25.Welch JS. Patterns of mutations in TP53 mutated AML. Best Pract Res Clin Haematol. 2018;31:379–83. doi: 10.1016/j.beha.2018.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hunter AM, Sallman DA. Targeting TP53 mutations in Myelodysplastic syndromes. Hematol Oncol Clin North Am. 2020;34:421–40. doi: 10.1016/j.hoc.2019.11.004. [DOI] [PubMed] [Google Scholar]
- 27.Papaemmanuil E, Gerstung M, Bullinger L, Gaidzik VI, Paschka P, Roberts ND, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med. 2016;374:2209–21. doi: 10.1056/NEJMoa1516192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Post SM, Kornblau SM, Quintás-Cardama A. p53 pathway dysfunction in AML: Beyond TP53 mutations. Oncotarget. 2017;8:108288–9. doi: 10.18632/oncotarget.22713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Quintás-Cardama A, Hu C, Qutub A, Qiu YH, Zhang X, Post SM, et al. p53 pathway dysfunction is highly prevalent in acute myeloid leukemia independent of TP53 mutational status. Leukemia. 2017;31:1296–305. doi: 10.1038/leu.2016.350. [DOI] [PubMed] [Google Scholar]
- 30.Barbosa K, Li S, Adams PD, Deshpande AJ. The role of TP53 in acute myeloid leukemia: Challenges and opportunities. Genes Chromosomes Cancer. 2019;58:875–88. doi: 10.1002/gcc.22796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pant V, Quintás-Cardama A, Lozano G. The p53 pathway in hematopoiesis: Lessons from mouse models, implications for humans. Blood. 2012;120:5118–27. doi: 10.1182/blood-2012-05-356014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McNees AL, Vilchez RA, Heard TC, Sroller V, Wong C, Herron AJ, et al. SV40 lymphogenesis in Syrian golden hamsters. Virology. 2009;384:114–24. doi: 10.1016/j.virol.2008.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Munoz LJ, Ludena D, Gedvilaite A, Zvirbliene A, Jandrig B, Voronkova T, et al. Lymphoma outbreak in a HASH: Sal hamster colony. Arch Virol. 2013;158:2255–65. doi: 10.1007/s00705-013-1737-0. [DOI] [PubMed] [Google Scholar]
- 34.Simmons JH, Riley LK, Franklin CL, Besch-Williford CL. Hamster polyomavirus infection in a pet Syrian hamster (Mesocricetus auratus) Vet Pathol. 2001;38:441–6. doi: 10.1354/vp.38-4-441. [DOI] [PubMed] [Google Scholar]