

Abstract
Maintenance of the proteome (proteostasis) is essential for cellular homeostasis and prevents cytotoxic stress responses that arise from protein misfolding. However, little is known about how different types of misfolded proteins impact homeostasis, especially when protein degradation pathways are compromised. We examined the effects of misfolded protein expression on yeast growth by characterizing a suite of substrates possessing the same aggregation-prone domain but engaging different quality control pathways. We discovered that treatment with a proteasome inhibitor was more toxic in yeast expressing misfolded membrane proteins, and this growth defect was mirrored in yeast lacking a proteasome-specific transcription factor, Rpn4p. These results highlight weaknesses in the proteostasis network’s ability to handle the stress arising from an accumulation of misfolded membrane proteins.
Keywords: quality control, ubiquitin proteasome system, Rpn4, yeast growth, proteasome stress response, chaperone, protein misfolding, Hsp104, endoplasmic reticulum associated degradation, cytoplasmic quality control
Introduction
Proteins, including those destined for the cytoplasm or secretory pathway, must fold into their native conformations to support cellular health and homeostasis. Maintaining the proper balance of folded proteins within the cell is referred to as proteostasis [1]. Molecular chaperones facilitate protein folding and refolding and maintain the solubility of non-native proteins [2–4].
Nevertheless, protein misfolding is a common occurrence [5], and can result from environmental stressors, errors in gene expression, or genetic mutations [6]. Misfolding can result in a loss-of-function phenotype, as is the case with the cystic fibrosis transmembrane conductance regulator (CFTR) [7], or a toxic gain-of-function phenotype, as seen when misfolded proteins accumulate in neurodegenerative diseases, including Alzheimer’s, Parkinson’s, and Huntington’s [8, 9] or antitrypsin-associated liver disease [10]. Therefore, eukaryotic cells evolved stress response pathways to preserve homeostasis, including the unfolded protein response (UPR) in the endoplasmic reticulum (ER), the heat shock response (HSR) in the cytosol, and the recently described proteasome stress response (PSR) [11–15].
One mechanism by which the proteostasis network (PN) maintains the proteome is by regulated protein degradation. Elimination of misfolded proteins is accomplished by three main degradation pathways, the ubiquitin proteasome system (UPS), protein sorting to the vacuole/lysosome, and autophagy [16–18]. For secretory proteins, such as select mutant forms of CFTR, elimination occurs via ER-associated degradation (ERAD) [7, 19, 20]. Other misfolded secretory proteins can be delivered through the Golgi for degradation in the vacuole/lysosome [21–23]. Misfolded cytoplasmic proteins can also be degraded by the UPS via a process termed cytoplasmic quality control (CytoQC) [24–28]. Alternatively, misfolded cytoplasmic proteins can be degraded by autophagy, as seen for select neurodegenerative disease-associated protein aggregates [29], and ER-phagy is a specialized form of autophagy which can degrade fragments of the ER and disease-associated misfolded proteins that accumulate in the ER [30–33]. Unfortunately, the PN—and especially the UPS—decline with age, which increases the toxicity from misfolded proteins that escape or overwhelm these degradative pathways [34, 35].
To define the relative contributions of select degradation pathways on homeostasis, we asked whether topologically distinct misfolded proteins differentially impact yeast cell growth when proteasome activity is reduced. We developed and employed a panel of misfolded proteins, all of which contain the identical misfolded domain, but that are targeted to the vacuole, CytoQC, or ERAD. We demonstrate that the greatest toxicity arises from integral membrane ERAD substrates that accumulate when the proteasome is inhibited. Similar effects were seen in yeast lacking a transcription factor, Rpn4, which responds to an increase in misfolded protein and activates proteasome expression. Our data reveal a hierarchy of quality control pathways that overcome proteotoxicity arising from compromised PN function.
Materials and Methods
Yeast strains, plasmids, and plasmid construction –
Yeast were treated as described previously [36]. A complete list of the Saccharomyces cerevisiae strains used in this study is shown in Table S1. Strains expressing proteins induced by β-estradiol under the control of the GAL1 promoter were constructed as described [37].
Oligonucleotides and plasmids used in this study are listed in Table S2. To drive expression using β-estradiol, the GAL1 promoter from pCG163 was amplified using primers oCG336 and oCG337 (for NBD2*, Chimera A*, Chimera N*, and Ste6p*), and oCG338 and oCG339 (for SZ*). To generate the SZ* GAL1-regulated expression plasmid, the PCR product was subcloned into pRS416 TEF SZ* [38] after first removing the TEF promoter using SacI and XbaI, generating pCG217. For all other plasmids, the PCR product was subcloned into the HindIII and XmaI sites following removal of the PGK promoter, which generated pCG213, pCG214, pCG215, pCG216. DNA sequencing (Genewiz) was performed to confirm the desired construct. Where indicated, protein expression was induced 300 nM β-estradiol.
Cycloheximide chase assays –
Protein degradation was monitored using a cycloheximide chase assay essentially as described [39]. In brief, after yeast cells expressing the indicated protein were grown overnight in selective media to log phase, and a 1 mL aliquot of culture was mixed with NaN3 to provide the 0 min time point. Cycloheximide was then added to the remaining culture to a final concentration of 200 μg/mL, the cultures were incubated at 26° or 37°C, as indicated, and an aliquot was removed at each time point before cells were lysed [40]. Pelleted protein samples were incubated in sample buffer plus fresh β-mercaptoethanol (final concentration of 5%), the samples were incubated at 37°C for 30 min, and an aliquot was analyzed by SDS-PAGE and immunoblotting. Antibodies used in this study were rat monoclonal anti-HA-HRP high affinity (3F10, Roche Applied Science) and rabbit anti-glucose-6-phosphate dehydrogenase (A9521, Sigma-Aldrich), and immunoblots were probed with anti-rabbit secondary antibody (Jackson ImmunoResearch) as a qualitative measure of equal protein loading. Proteins were visualized with SuperSignal Chemiluminescence (ThermoFisher Scientific), images were taken using a Bio-Rad ChemiDoc XRS+ imager (Bio-Rad Laboratories), and the results were quantified using ImageJ version 1.51 software (National Institutes of Health). All images captured for quantification were unsaturated to assure accurate band intensity measurements. Western blots with low protein signal were acquired using binning which decreases the final resolution of the images and sometimes produces a grainy appearance. Statistical analysis was performed using Student’s T-test Prism 9 for Windows (GraphPad, San Diego, CA). Differences were considered statistically significant at p < 0.05 and were indicated by an asterisk. In several cases, the standard error bars are smaller than the symbols displayed on the graph and were thus invisible.
Growth assays –
The indicated strains were transformed with plasmids harboring the cDNAs to express the indicated proteins from either constitutive or GAL1-regulated promoters. Overnight cultures (in selective medium containing glucose) were diluted to an OD600 of 0.2 into 96-well plates and were treated with DMSO or 50 μM MG132. Where noted, protein expression was induced by the addition 300 nM β-estradiol. Growth was monitored for 21–23 hrs at 37°C in a Cytation 5 Multi-Mode Reader (BioTek, Winooski, VT), using double orbital shaking, and the OD600 was measured every 30 min. Relative growth rates were determined after subtracting a media background and plotting the OD600 over time. For growth curve analysis one-way ANOVA (GraphPad Prism 9.02) followed by Dunnett’s post hoc test was performed using the recorded OD600 values to determine differences in growth compared to an empty vector control. Differences were considered statistically significant at p<0.05.
Results
Characterization of misfolded protein substrates that utilize different protein degradation pathways
We previously reported on several model misfolded proteins that were based on the yeast ATP-binding cassette (ABC) transporter, Sterile 6 (Ste6p) (Fig. 1A) [41]. A Ste6p mutant containing a 42 amino acid truncation in the second nucleotide binding domain (NBD2*) (Ste6p*; Fig. 1B), results in ER retention and degradation by the UPS/ERAD [42–45]. Thus, to minimize secondary effects from the expression of distinct misfolded protein domains, all of the model substrates used in the current study contained this truncated NBD from Ste6p* (Fig. 1B). They included a dual-pass transmembrane protein fused to NBD2* oriented toward the cytoplasm (Chimera A*), a single-pass transmembrane protein depositing NBD2* in the ER lumen (Chimera N*), a single-pass transmembrane protein fused to NBD2* that localized in the cytoplasm (SZ*), and a soluble, i.e., transmembrane-free form of the domain (NBD2*) (Fig. 1C–F) [38, 39, 46, 47].
Figure 1. Misfolded proteins that contain NBD2*, a truncated second nucleotide binding domain (NBD2) derived from Ste6p.
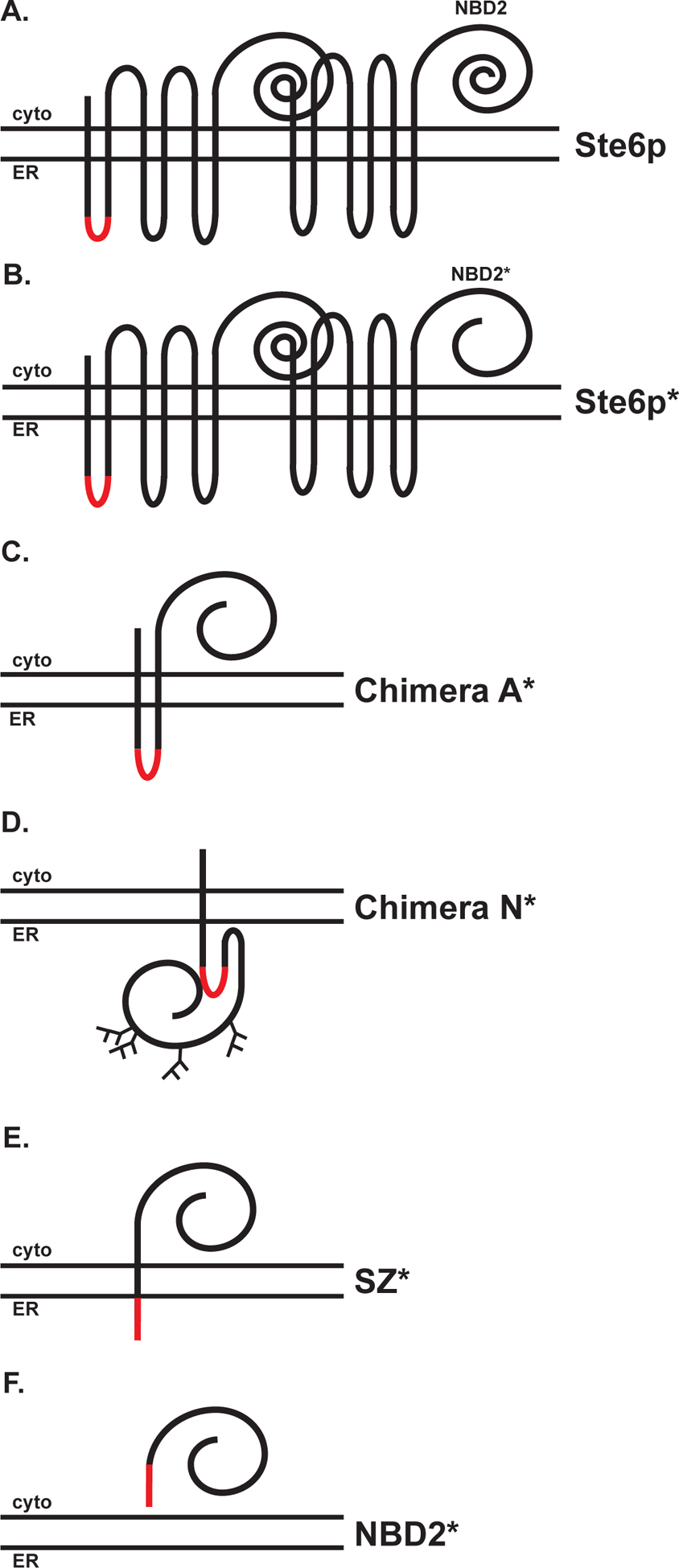
The topologies of (A) Ste6p and Ste6p*, (B) Chimera A*, (C) Chimera N*, (D) SZ*, and (E) NBD2* are shown. Each substrate contains the same truncated NBD2 domain. The red segment represents a 3X HA-tag.
We reported previously that Chimera A*, SZ*, and NBD2* rely on the cytoplasmic Hsp70, Ssa1p, for maximal degradation [38, 39, 47], but the requirements for the degradation of Chimera N* were not defined. Chimera N* is unique among the substrates tested, as inefficient insertion of TMH2 results in deposition of NBD2* into the ER lumen [46]. Because the NBD2* moiety in Chimera N* resides in the ER lumen, we predicted that degradation would instead require the ER lumenal Hsp70, Kar2p. As expected, Chimera N* degradation was Ssa1p-independent (Fig. S1A), but surprisingly the substrate was modestly stabilized in yeast harboring a KAR2 mutant (kar2-1), which we previously showed is required for the degradation of lumenal substrates (Fig. S1B) [48–50]. Because Hsp40s typically enhance Hsp70 activity but can also recognize substrates directly [51–53], we next examined the requirement for the ER luminal Hsp40s, Scj1p and Jem1p, on Chimera N* turnover. As shown in Fig. S1C, Chimera N* was stabilized to the same degree in an scj1Δjem1Δ strain as in the kar2-1 strain, consistent with the notion that these chaperones work together to select Chimera N* for degradation. As the NBD2* motif is deposited into the ER lumen in Chimera N*, the protein also presents several additional consensus sites for N-glycan modification to the cellular glycosylation machinery. Indeed, we showed previously that there appear to be four glycosylation sites utilized, adding an additional 12 kDa to the molecular mass [46]. For other glycosylated substrates, degradation requires ER-lumenal mannosidases [54–57]. However, Chimera N* showed minimal stabilization in a yos9Δ strain (Fig. S1D), suggesting that glycans do not constitute the primary determinant in Chimera N* recognition.
We next confirmed that Chimera N* is an ERAD substrate and thus investigated the contributions of the ERAD-associated E3 ligases, Hrd1p and Doa10p, on degradation [58, 59]. ERAD substrates with lesions in the ER lumen, such as Chimera N*, are classified as ERAD-L substrates and are typically degraded in a Hrd1p-dependent manner [60, 61]. Chimera N* was stabilized in yeast lacking Hrd1p (Fig. S2A) but was considerably stabilized in a strain containing a temperature-sensitive mutant form of Cdc48p (cdc48–2) (Fig. S2B). This result is consistent with the role of this AAA-ATPase on ERAD substrate extraction from the ER membrane [62, 63].
In contrast to Chimera N*, Chimera A*, SZ*, and NBD2* deposit misfolded domains in the cytoplasm (Fig. 1). Chimera A* and NBD2* are handled by the UPS, but SZ* is targeted to both ERAD and the vacuole (see below) [38, 39, 47]. Moreover, Chimera A* is targeted for ERAD but NBD2* requires the CytoQC machinery for its disposal [39, 47]. Therefore, it was unclear whether the recognition of the truncated and misfolded NBD2 in Chimera A* and NBD2* occurred similarly. Of note, we previously reported that a 42 amino acid truncation in Chimera A* destabilizes this ERAD substrate, but the half-lives of different C-terminal truncations varied significantly [64]. For example, a truncation that removed 51 amino acids was “hyperstable”, whereas the removal of 47 amino acids from the C-terminus led to hyper-instability. Consequently, we asked whether these alternate truncations in Chimera A* would similarly affect NBD2* stability.
As previously reported, the full-length NBD2 species was stable over a 60 min time course, and NBD2* (“Q247X”) was unstable (Fig. 2) [39]. In addition and in accordance with the effects on Chimera A*, the 47 (I242X) and 51 (L238X) amino acid truncated NBD2 species were degraded either more quickly (t1/2 ~ 13 min) or more slowly (t1/2 > 60 min) than NBD2* (t1/2 ~ 30 min), respectively (Fig. 2B). The degradation of each substrate was also proteasome-dependent, as anticipated (Fig. S3A–C) [39]. Moreover, each of the truncations also shared similar Ssa1p-dependent degradation profiles (Fig. S3D–F). These data suggest that the same mode of chaperone-based recognition is used for the NBD2 truncations, regardless of whether the substrate is targeted for ERAD or CytoQC.
Figure 2. Relative stabilities of NBD2 truncations.
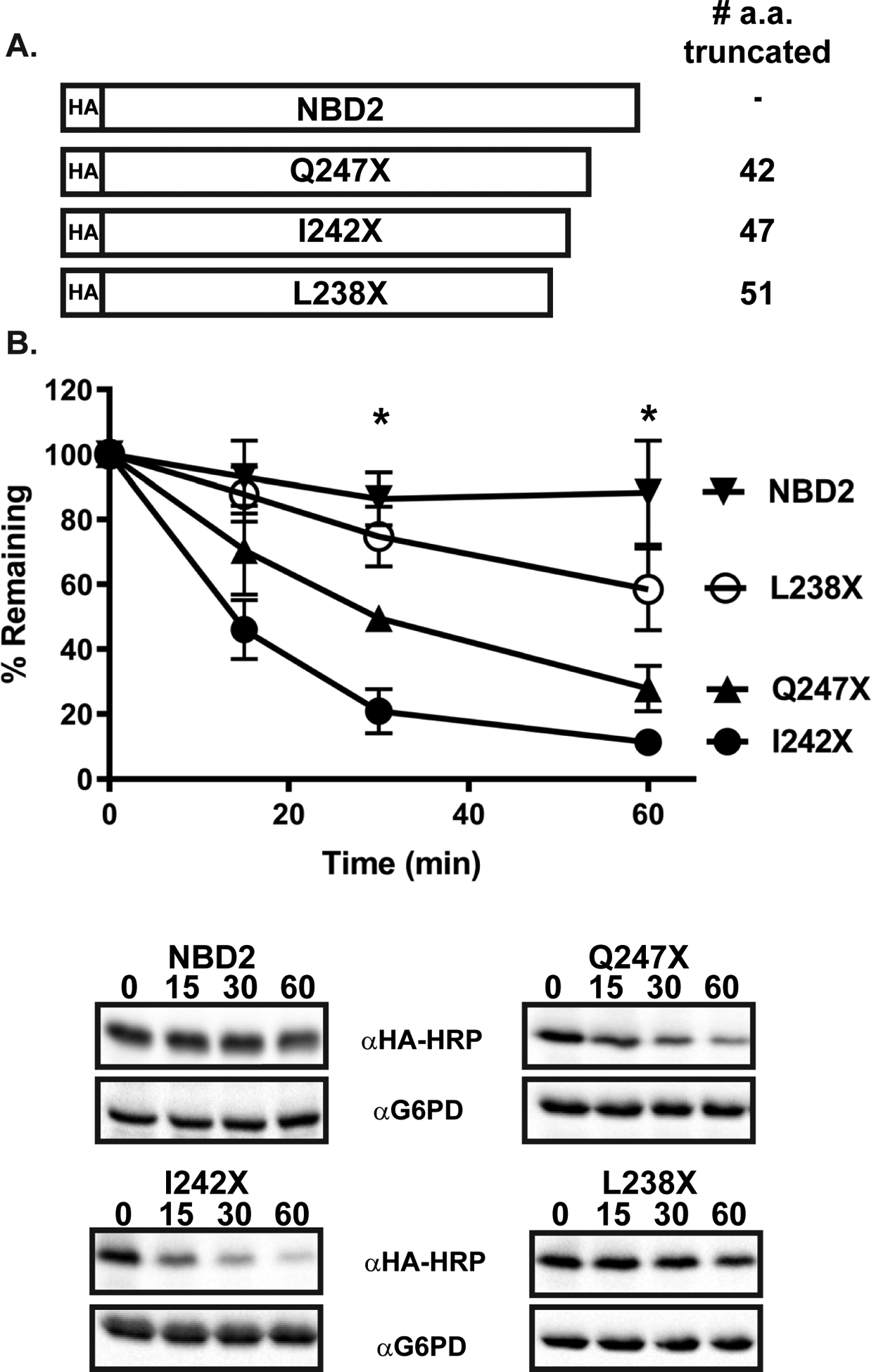
(A) NBD2 truncations used in this study are truncated by 51, 42, and 47 amino acids, generating L238X, Q247X (also referred to herein as NBD2*), and I242X respectively. (B) Protein stability was assessed by a cycloheximide chase analysis as described in Materials and methods in pdr5Δ yeast expressing L238X, Q247X, and I242X treated with DMSO. Data represent the means ± S.E., n = 6–12. All p values were determined using Student’s t-test by comparing each truncation to full length NBD2, * denotes p<0.05 for the indicated time points. The degradation of all three truncations were found to be statistically different from NBD2 at both the 30 and 60 min time points, as indicated by an asterisk above the time points.
Hsp104 is required only for the degradation of a truncated cytosolic NBD in the cytosol
To facilitate its targeting to the ERAD pathway, the aggregation-prone Chimera A* substrate requires the cytoplasmic AAA+ ATPase/disaggregase, Hsp104 [64]. Because SZ* and NBD2* contain the same truncated second nucleotide-binding domain, we asked if these substrates were also Hsp104-dependent. As shown in Fig. 3, Hsp104 facilitated the degradation of both substrates. In contrast and as might be expected based on its topology, Chimera N* degradation was Hsp104-independent. In addition, Ste6p* was also Hsp104-independent, most likely due to the maintenance of proper intramolecular interactions, thereby allowing Ste6p* to avoid aggregation. These data demonstrate that for the artificial substrates—regardless of whether a substrate is selected for ERAD (Chimera A*), both ERAD and vacuolar degradation (SZ*), or CytoQC (NBD2*)—the turnover of a protein containing an aggregation-prone domain in the cytoplasm requires Hsp104.
Figure 3. Differential effects of Hsp104 on substrates containing the NBD2* moiety residing in the cytosol versus ER lumen.
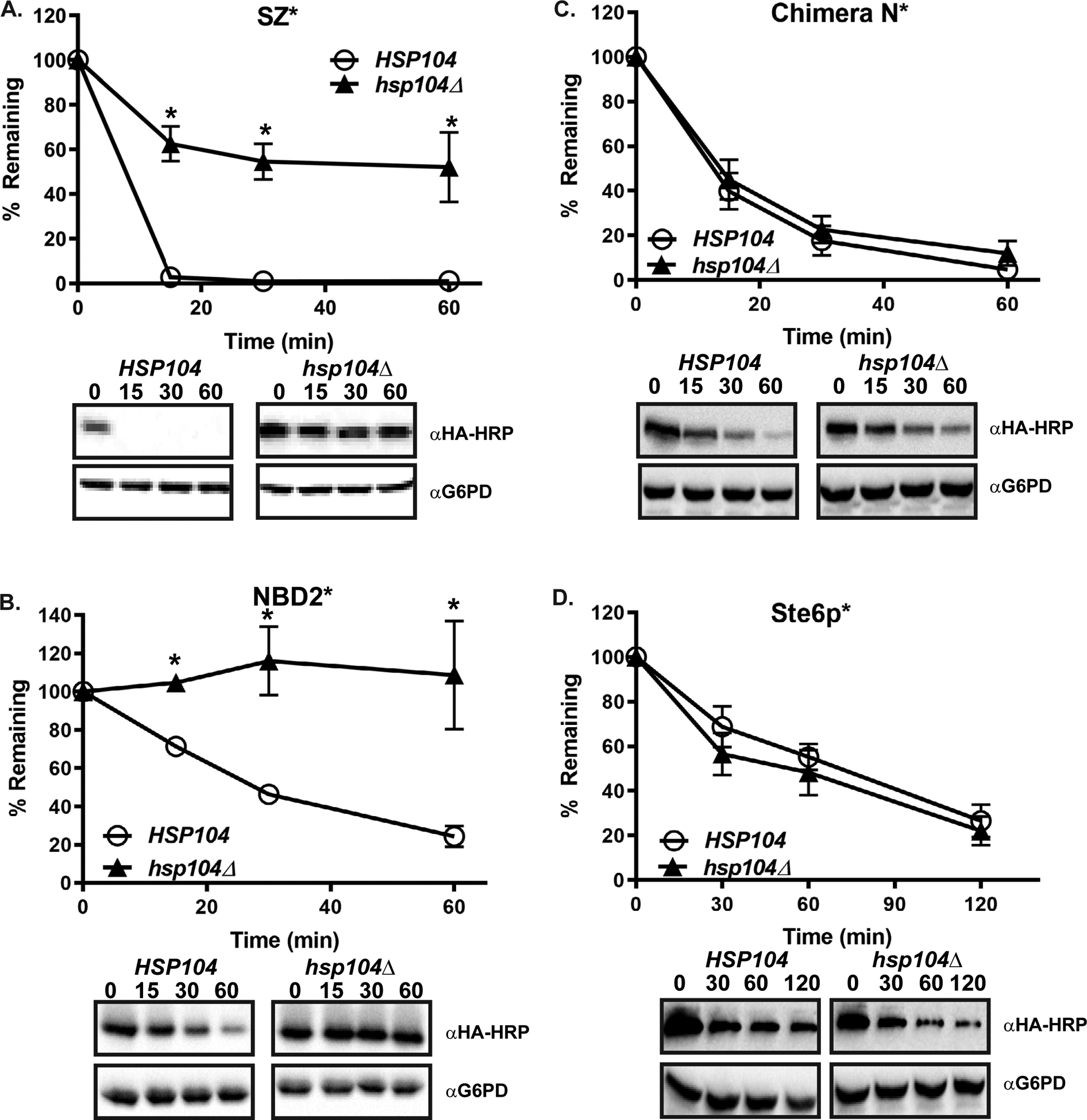
Wild-type (HSP104) and hsp104Δ yeast were transformed with plasmids engineered for the expression of (A) SZ*, (B) NBD2* (C) Chimera N*, and (D) Ste6p*. A cycloheximide chase analysis was performed following a 30 min temperature shift to 37°C. Data represent the means ± S.E., n = 3–7. All p values determined using Student’s t-test vs the wild-type control, * denotes p<0.05.
Misfolded membrane proteins compromise cell growth when proteasome function is suppressed
Even though each substrate examined in this study, as well as Ste6p*, contains the same misfolded domain, they exhibit a set of non-overlapping requirements for their degradation (see Table 1 for a summary of data from the current study and past work). This afforded us the unique opportunity to examine how the demand on different nodes of the PN might affect cell growth.
Table 1.
Factors required for model substrate degradation.
| Ste6p* | Chimera A* | Chimera N* | SZ* | NBD2* | |
|---|---|---|---|---|---|
| Proteasome | Yes | Yes | Partial | Partial | Yes |
| Vacuole | No | No | No | Yes | No |
| Hsp70 | Ssa1 | Ssa1 | Kar2 | Ssa1 | Ssa1 |
| Hsp40 | Ydj1 HIJ1 | Ydj1 Hlj1 | Scj1 Jem1 | Ydj1 | Ydj1 Hlj1 |
| E3 Ubiquitin Ligase | Doa10 | Doa10 | Hrd1 | Doa10 | San1 Ubr1 |
| Hsp104 | No | Yes | No | Yes | Yes |
| Cdc48 | Yes | Yes | Yes | Yes | Yes |
| References | [42, 45] | [47] | [46] | [38] | [39] |
The requirements are listed for the indicated substrates and QC machinery. Ste6p* and Chimera A* can be classified as ERAD-C substrates, Chimera N* is an ERAD-L substrate, SZ* is a substrate for degradation by ERAD and the vacuole, and NBD2* is a CytoQC substrate.
To this end, we transformed each substrate listed in Table 1 into yeast lacking PDR5, which allows for the effective administration of a proteasome inhibitor (e.g., MG-132) into the strain. We first monitored yeast growth at 37°C. Consistent with a robust PN in wild-type yeast, no significant effects on cell growth were evident (Fig. 4A).
Figure 4. Limiting proteasome function sensitizes cells to the expression of integral membrane ERAD substrates.
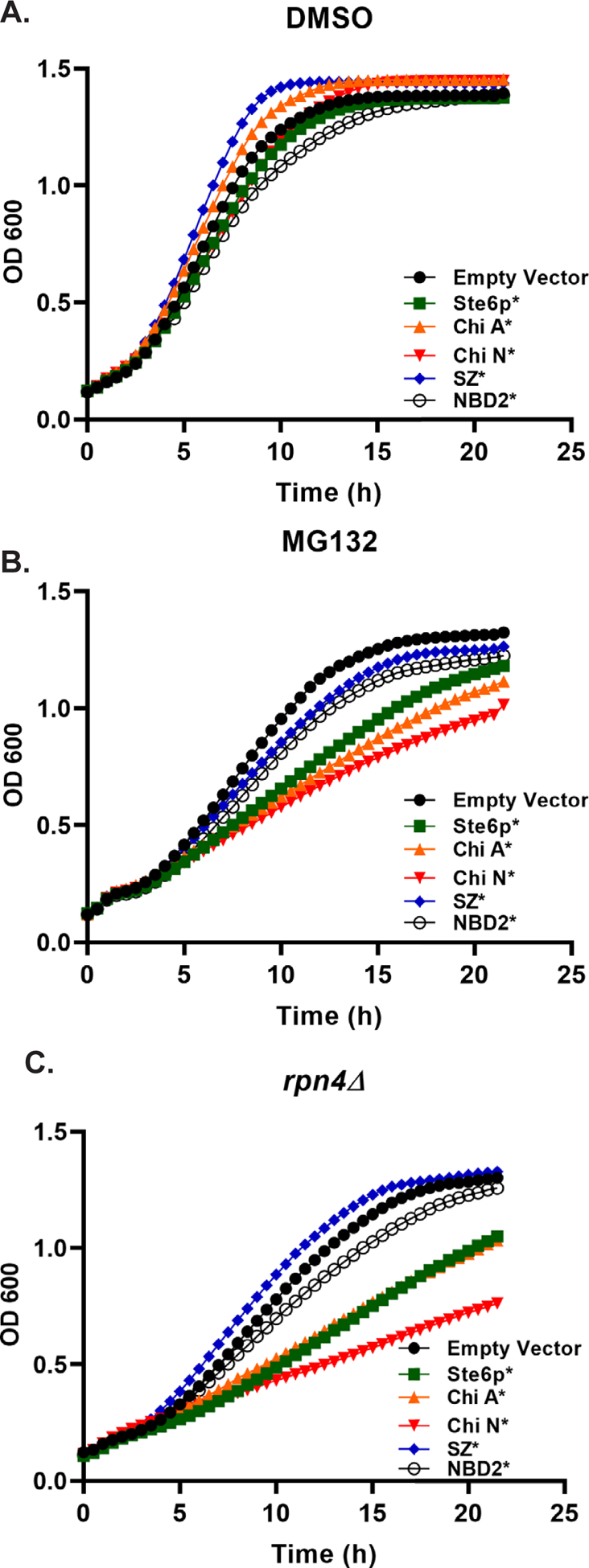
(A and B) The pdr5Δ strain or (C) rpn4Δ yeast were transformed with the indicated expression plasmids and grown to stationary phase in selective media. The next day, they were diluted back to a starting OD600 = 0.2, and growth was monitored over time at 37°C. Yeast were treated with (A) DMSO (B) 50 μM MG132, or (C) 300 nM β-estradiol (to induce protein expression). Plots represent the average data from n = 8–16. A one-way ANOVA (GraphPad Prism 9.02) followed by a Dunnett’s test was performed using the OD600 measurements recorded for growth curve replicates in order to assess reduced growth resulting from (B) MG132 treatment compared to an empty vector control and (C) the rpn4Δ strain expressing each substrate compared to an empty vector control. Data were statistically significant (p<0.05) from the 7.5 h time point onward (B) or starting at 6 h time point onward (C).
Because the efficiency of the UPS declines with age or under stress conditions, thus leading to severe effects on several nodes of the PN [34, 35], we next asked if a particular class of misfolded proteins is more toxic when proteasome function is altered. In this case, when MG132 was included in the growth media, maximal growth was achieved when cells contained a vector control, and the expression of SZ* and NBD2* had only minor effects on growth (Fig. 4B). In contrast, pronounced effects on growth were evident in yeast expressing Ste6p*, Chimera A*, or Chimera N*. In order to assure that the observed toxicity was not the result of elevated levels of expression, we also compared steady state levels by immunoblot (Fig. S4). Surprisingly, despite use of the same expression system for Ste6p*, Chimera A*, Chimera N*, and NBD2*, expression levels differed. Nevertheless, the three substrates with the lowest expression levels were the most toxic, excluding the possibility that toxicity arises by high levels of substrate expression.
Earlier work indicated that Ste6p* expression induced a unique stress response pathway that relied on the Rpn4 transcription factor that responds to reduced proteasome activity and thereby activates the expression of proteasome subunits [15, 65]. To test if the toxicity of expressing Ste6p*, Chimera A*, and Chimera N* was related to a failure of this stress response to mitigate proteotoxic stress, we put the substrates under control of an inducible promoter in order to test growth in rpn4Δ yeast. The use of an inducible promoter was necessary as expression of Ste6p* in an rpn4Δ strain is toxic [65]. As observed in the MG132-treated yeast and consistent with those data, expression of Ste6p*, Chimera A*, and Chimera N* all resulted in severely delayed growth in rpn4Δ yeast (Fig. 4C). These data indicate that cells require optimal proteasome activity to avoid the proteotoxicity associated with integral membrane, aggregation-prone ERAD substrates.
Discussion
In this study, we report first on the completed characterization of a set of misfolded proteins, all of which contain the same misfolded region but which engage different QC pathways. Through our previous work and this study, we have now categorized these substrates (Table 1), an undertaking that underscores the diversity of substrate selection and participating degradation pathways during PQC. To arrive at this conclusion, no prior study employed such a diverse array of substrates, particularly those containing the same misfolded degradation-targeting region (i.e., “degron”) which limits secondary effects. Armed with this set of reagents, we were then able to examine the growth of yeast expressing each substrate in the presence or absence of a proteasome inhibitor, or in the absence of a transcription factor, Rpn4, that regulates the expression of proteasome subunits. Based on this second aspect of our work, three substrates (Ste6p*, Chimera A*, and Chimera N*) negatively impacted growth, indicating that integral membrane ERAD substrates present a toxic challenge to cells with compromised proteasome capacity. In contrast, SZ* which is degraded primarily in the vacuole after ER exit [38], does not induce the same level of toxicity to proteasome-challenged cells (Fig. 4B and C). We propose that the ability of this protein to largely escape the ER decreases its toxicity.
The levels of Rpn4p in the cell are regulated by degradation, so when degradation is slowed due to reduced proteasome efficiency, Rpn4p is available to trigger proteasome subunit expression [66]. Alternatively, if misfolded proteins induce a HSR, then heat shock transcription factor 1 can induce RPN4 transcription [67]. Interestingly, in our growth assays, the presumed stabilization of Rpn4 by MG132 was unable to adapt cells to the proteotoxic stress accompanying the expression of misfolded integral membrane proteins. In contrast, yeast treated with another proteasome inhibitor fully adapted to the expression of misfolded proteins and degraded proteins with similar efficiency to untreated cells [14]. However, the shorter treatments used in by Work and Brandman may have missed the long-term effects of proteasome inhibition. Indeed, one interpretation of our data is that integral membrane proteins become increasingly toxic over time because they can accumulate in the cytoplasm and aggregate into Lewy Body-like structures [68, 69]. For example, when proteasome activity is reduced, retrotranslocated CFTR amasses in perinuclear aggresomes [70]. Alternatively, Neal and colleagues recently demonstrated that expression of an integral membrane ERAD substrate could induce toxicity in a yeast strain with impaired retrotranslocation [71]. Therefore, further work will be essential to establish whether the accumulation of misfolded integral membrane proteins in the membrane or in the cytoplasm presents a unique threat to proteasome-challenged cells, and whether this effect applies to a wide group of membrane proteins.
Unlike membrane proteins, misfolded cytoplasmic proteins are instead targeted to and degraded by the UPS via CytoQC [24, 28, 72–74]. Although some of the mechanistic details of CytoQC have been explored [24–26, 72, 75], the machinery required for the degradation of truncated species has not been fully elucidated. We demonstrate here that changing the position of the truncation in a cytosolic protein influences stability (Fig. 2), an observation that correlates with our previous findings on altering the truncations in Ste6p* and Chimera A* [64]. One scenario to explain these findings is that chaperone recognition mediates degradation rate, however all three truncations are equally dependent on Ssa1p (Fig. S3). Alternatively, selection by ubiquitin ligases might dictate stability. Interestingly, L238X and Q247X exhibit a strong dependence on the canonical CytoQC E3 ubiquitin ligases, San1p and Ubr1p for turnover, whereas I242X is incompletely stabilized in strains lacking San1p and Ubr1p or even in a strain that also lacks the ER membrane-associated E3 Doa10p (Fig. S5), which can also participate in CytoQC [28, 39, 76].
Hsp104 is a key modulator of CytoQC and ERAD [37, 64, 77] and exhibits disaggregase activity, especially when cells are incubated at elevated temperatures [78]. Hsp104 works in concert with the Hsp70 and Hsp40 machinery to rescue heat-damaged proteins [79], and previous work from our lab and as shown in this study demonstrate that Hsp104 supports the degradation of Chimera A*, SZ*, and NBD2* (Table 1). Like Chimera A*, NBD2* is also aggregation-prone [39, 64], which likely accounts for the Hsp104 requirement. More specifically, Hsp104 retains Chimera A* in a retrotranslocation-competent state [64]. Therefore, we propose that Hsp104 disaggregates NBD2*, allowing for the maintenance of a species that can enter the proteasome aperture. Future work in which the effect of Hsp104 on substrate solubility is reconstituted will allow us to test this hypothesis.
One unexpected outcome from our work is that Chimera N* degradation showed a relatively subtle dependence on the ER lumenal Hsp70 and Hsp40s (Fig. S1A). However, this is not without precedence, as degradation of the alpha subunit of the epithelial sodium channel was minimally dependent on Kar2p despite the presence of a large ER lumenal domain [80]. Our work also suggests that the Yos9 lectin, which recognizes glycosylated ERAD substrates [81], like Chimera N*, plays a minor role in Chimera N* turnover (Fig. S1D). Therefore, we suggest that a cadre of chaperones and chaperone-like lectins contribute to the selection of this ERAD substrate.
Finally, in the future, it will be critical to relate our work to studies in higher cells. As noted elsewhere, the reduction in PN and especially UPS activity during aging and implies that the altered homeostasis and cell death that accompanies age might arise from the accumulation of misfolded membrane proteins. Future proteomic studies in aged mammalian cells will allow us to test this hypothesis.
Supplementary Material
Acknowledgements
We are grateful to all the members of the Brodsky lab for insightful comments during the course of this work. We also thank Mikala Aleksandruk for her early contributions. J.L.B. was supported by National Institute of Health grants R35 GM131731 and P30 DK079307, and C.J.G. was supported by DK101584 and DK124659. G.B. acknowledges a grant from the Duquesne University Undergraduate Research Program and a Howard Hughes Medical Institute Summer Undergraduate Research Fellowship.
Abbreviations
- CFTR
cystic fibrosis transmembrane conductance regulator
- UPR
unfolded protein response
- ER
endoplasmic reticulum
- HSR
heat shock response
- PSR
proteasome stress response
- PN
proteostasis network
- UPS
ubiquitin proteasome system
- ERAD
endoplasmic reticulum-associated degradation
- QC
quality control
- CytoQC
cytoplasmic quality control
- PQC
protein quality control
- ABC
ATP binding cassette
- Ste6
Sterile 6
- NBD
nucleotide binding domain
Data Availability Statement
The data that supports the findings of this study are available in Figures 1–4 and the supplementary material of this article.
References
- 1.Balch WE, Morimoto RI, Dillin A & Kelly JW (2008) Adapting proteostasis for disease intervention, Science. 319, 916–9. [DOI] [PubMed] [Google Scholar]
- 2.Lindquist S (1986) THE HEAT-SHOCK RESPONSE, Annual Review of Biochemistry. 55, 1151–1191. [DOI] [PubMed] [Google Scholar]
- 3.Pearse BR & Hebert DN (2010) Lectin chaperones help direct the maturation of glycoproteins in the endoplasmic reticulum, Biochimica et Biophysica Acta, Molecular Cell Research. 1803, 684–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Feige MJ & Hendershot LM (2011) Disulfide bonds in ER protein folding and homeostasis, Current Opinion in Cell Biology. 23, 167–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chaudhuri TK & Paul S (2006) Protein-misfolding diseases and chaperone-based therapeutic approaches, Febs J. 273, 1331–49. [DOI] [PubMed] [Google Scholar]
- 6.Chen B, Retzlaff M, Roos T & Frydman J (2011) Cellular Strategies of Protein Quality Control, Cold Spring Harbor Perspectives in Biology. 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cheng SH, Gregory RJ, Marshall J, Paul S, Souza DW, White GA, O’Riordan CR & Smith AE (1990) Defective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosis, Cell. 63, 827–34. [DOI] [PubMed] [Google Scholar]
- 8.Winklhofer KF, Tatzelt J & Haass C (2008) The two faces of protein misfolding: gain- and loss-of-function in neurodegenerative diseases, EMBO J. 27, 336–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ross CA & Poirier MA (2004) Protein aggregation and neurodegenerative disease, Nature Medicine. 10, S10–S17. [DOI] [PubMed] [Google Scholar]
- 10.Guerriero C & Brodsky J (2012) The Delicate Balance Between Secreted Protein Folding and Endoplasmic Reticulum-Associated Degradation in Human Physiology, Physiological reviews. 92, 537–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Karagöz GE, Acosta-Alvear D & Walter P (2019) The Unfolded Protein Response: Detecting and Responding to Fluctuations in the Protein-Folding Capacity of the Endoplasmic Reticulum, Cold Spring Harb Perspect Biol. 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sinnige T, Yu A & Morimoto RI (2020) Challenging Proteostasis: Role of the Chaperone Network to Control Aggregation-Prone Proteins in Human Disease, Adv Exp Med Biol. 1243, 53–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gomez-Pastor R, Burchfiel ET & Thiele DJ (2018) Regulation of heat shock transcription factors and their roles in physiology and disease, Nature Reviews Molecular Cell Biology. 19, 4–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Work JJ & Brandman O (2021) Adaptability of the ubiquitin-proteasome system to proteolytic and folding stressors, J Cell Biol. 220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schmidt RM, Schessner JP, Borner GH & Schuck S (2019) The proteasome biogenesis regulator Rpn4 cooperates with the unfolded protein response to promote ER stress resistance, Elife. 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ciechanover A (2017) Intracellular protein degradation: From a vague idea thru the lysosome and the ubiquitin-proteasome system and onto human diseases and drug targeting, Best Pract Res Clin Haematol. 30, 341–355. [DOI] [PubMed] [Google Scholar]
- 17.Varshavsky A (2017) The Ubiquitin System, Autophagy, and Regulated Protein Degradation, Annu Rev Biochem. 86, 123–128. [DOI] [PubMed] [Google Scholar]
- 18.Dikic I (2017) Proteasomal and Autophagic Degradation Systems, Annu Rev Biochem. 86, 193–224. [DOI] [PubMed] [Google Scholar]
- 19.Jensen TJ, Loo MA, Pind S, Williams DB, Goldberg AL & Riordan JR (1995) Multiple proteolytic systems, including the proteasome, contribute to CFTR processing, Cell. 83, 129–35. [DOI] [PubMed] [Google Scholar]
- 20.Ward CL, Omura S & Kopito RR (1995) Degradation of CFTR by the ubiquitin-proteasome pathway, Cell. 83, 121–7. [DOI] [PubMed] [Google Scholar]
- 21.Holkeri H & Makarow M (1998) Different degradation pathways for heterologous glycoproteins in yeast, FEBS Letters. 429, 162–166. [DOI] [PubMed] [Google Scholar]
- 22.Hong E, Davidson AR & Kaiser CA (1996) A pathway for targeting soluble misfolded proteins to the yeast vacuole, Journal of Cell Biology. 135, 623–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang S & Ng DTW (2010) Evasion of Endoplasmic Reticulum Surveillance Makes Wsc1p an Obligate Substrate of Golgi Quality Control, Molecular Biology of the Cell. 21, 1153–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Heck JW, Cheung SK & Hampton RY (2010) Cytoplasmic protein quality control degradation mediated by parallel actions of the E3 ubiquitin ligases Ubr1 and San1, Proceedings of the National Academy of Sciences of the United States of America. 107, 1106–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nillegoda NB, Theodoraki MA, Mandal AK, Mayo KJ, Ren HY, Sultana R, Wu K, Johnson J, Cyr DM & Caplan AJ (2010) Ubr1 and Ubr2 Function in a Quality Control Pathway for Degradation of Unfolded Cytosolic Proteins, Molecular Biology of the Cell. 21, 2102–2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McClellan AJ, Scott MD & Frydman J (2005) Folding and Quality Control of the VHL Tumor Suppressor Proceed through Distinct Chaperone Pathways, Cell. 121, 739–748. [DOI] [PubMed] [Google Scholar]
- 27.Prasad R, Kawaguchi S & Ng DTW (2010) A Nucleus-based Quality Control Mechanism for Cytosolic Proteins, Molecular Biology of the Cell. 21, 2117–2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maurer MJ, Spear ED, Yu AT, Lee EJ, Shahzad S & Michaelis S (2016) Degradation Signals for Ubiquitin-Proteasome Dependent Cytosolic Protein Quality Control (CytoQC) in Yeast, G3 (Bethesda). 6, 1853–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Martin DDO, Ladha S, Ehrnhoefer DE & Hayden MR (2015) Autophagy in Huntington disease and huntingtin in autophagy, Trends in Neurosciences. 38, 26–35. [DOI] [PubMed] [Google Scholar]
- 30.Lipatova Z & Segev N (2015) A Role for Macro-ER-Phagy in ER Quality Control, PLOS Genetics. 11, e1005390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bernales S, McDonald KL & Walter P (2006) Autophagy counterbalances endoplasmic reticulum expansion during the unfolded protein response, PLoS Biol. 4, e423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fregno I & Molinari M (2019) Proteasomal and lysosomal clearance of faulty secretory proteins: ER-associated degradation (ERAD) and ER-to-lysosome-associated degradation (ERLAD) pathways, Critical Reviews in Biochemistry and Molecular Biology. 54, 153–163. [DOI] [PubMed] [Google Scholar]
- 33.Ferro-Novick S, Reggiori F & Brodsky JL (2021) ER-Phagy, ER Homeostasis, and ER Quality Control: Implications for Disease, Trends Biochem Sci. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hipp MS, Kasturi P & Hartl FU (2019) The proteostasis network and its decline in ageing, Nature Reviews Molecular Cell Biology. 20, 421–435. [DOI] [PubMed] [Google Scholar]
- 35.Vilchez D, Saez I & Dillin A (2014) The role of protein clearance mechanisms in organismal ageing and age-related diseases, Nature Communications. 5, 5659. [DOI] [PubMed] [Google Scholar]
- 36.Kaiser C, Michaelis S & Mitchell A (1994) Methods in yeast genetics.
- 37.Doonan LM, Guerriero CJ, Preston GM, Buck TM, Khazanov N, Fisher EA, Senderowitz H & Brodsky JL (2019) Hsp104 facilitates the endoplasmic-reticulum-associated degradation of disease-associated and aggregation-prone substrates, Protein Sci. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sun Z & Brodsky JL (2018) The degradation pathway of a model misfolded protein is determined by aggregation propensity, Mol Biol Cell. 29, 1422–1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Guerriero CJ, Weiberth KF & Brodsky JL (2013) Hsp70 targets a cytoplasmic quality control substrate to the San1p ubiquitin ligase, J Biol Chem. 288, 18506–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang Y, Michaelis S & Brodsky JL (2002) CFTR expression and ER-associated degradation in yeast, Methods in Molecular Medicine. 70, 257–65. [DOI] [PubMed] [Google Scholar]
- 41.Berkower C & Michaelis S (1991) Mutational analysis of the yeast a-factor transporter STE6, a member of the ATP binding cassette (ABC) protein superfamily, EMBO Journal. 10, 3777–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huyer G, Piluek WF, Fansler Z, Kreft SG, Hochstrasser M, Brodsky JL & Michaelis S (2004) Distinct Machinery Is Required in Saccharomyces cerevisiae for the Endoplasmic Reticulum-associated Degradation of a Multispanning Membrane Protein and a Soluble Luminal Protein, Journal of Biological Chemistry. 279, 38369–78. [DOI] [PubMed] [Google Scholar]
- 43.Loayza D, Tam A, Schmidt WK & Michaelis S (1998) Ste6p mutants defective in exit from the endoplasmic reticulum (ER) reveal aspects of an ER quality control pathway in Saccharomyces cerevisiae, Molecular Biology of the Cell. 9, 2767–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stolz A, Besser S, Hottmann H & Wolf DH (2013) Previously unknown role for the ubiquitin ligase Ubr1 in endoplasmic reticulum-associated protein degradation, Proceedings of the National Academy of Sciences. 110, 15271–15276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nakatsukasa K, Huyer G, Michaelis S & Brodsky JL (2008) Dissecting the ER-associated degradation of a misfolded polytopic membrane protein, Cell. 132, 101–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Guerriero CJ, Gomez YK, Daskivich GJ, Reutter KR, Augustine AA, Weiberth KF, Nakatsukasa K, Grabe M & Brodsky JL (2019) Harmonizing Experimental Data with Modeling to Predict Membrane Protein Insertion in Yeast, Biophys J. 117, 668–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Guerriero CJ, Reutter KR, Augustine AA, Preston GM, Weiberth KF, Mackie TD, Cleveland-Rubeor HC, Bethel NP, Callenberg KM, Nakatsukasa K, Grabe M & Brodsky JL (2017) Transmembrane helix hydrophobicity is an energetic barrier during the retrotranslocation of integral membrane ERAD substrates, Mol Biol Cell. 28, 2076–2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zacchi LF, Wu HC, Bell SL, Millen L, Paton AW, Paton JC, Thomas PJ, Zolkiewski M & Brodsky JL (2014) The BiP molecular chaperone plays multiple roles during the biogenesis of torsinA, an AAA+ ATPase associated with the neurological disease early-onset torsion dystonia, J Biol Chem. 289, 12727–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Brodsky JL, Werner ED, Dubas ME, Goeckeler JL, Kruse KB & McCracken AA (1999) The requirement for molecular chaperones during endoplasmic reticulum-associated protein degradation demonstrates that protein export and import are mechanistically distinct, Journal of Biological Chemistry. 274, 3453–60. [DOI] [PubMed] [Google Scholar]
- 50.Kabani M, Kelley SS, Morrow MW, Montgomery DL, Sivendran R, Rose MD, Gierasch LM & Brodsky JL (2003) Dependence of endoplasmic reticulum-associated degradation on the peptide binding domain and concentration of BiP, Mol Biol Cell. 14, 3437–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nillegoda NB, Wentink AS & Bukau B (2018) Protein Disaggregation in Multicellular Organisms, Trends Biochem Sci. 43, 285–300. [DOI] [PubMed] [Google Scholar]
- 52.Kityk R, Kopp J & Mayer MP (2018) Molecular Mechanism of J-Domain-Triggered ATP Hydrolysis by Hsp70 Chaperones, Mol Cell. 69, 227–237.e4. [DOI] [PubMed] [Google Scholar]
- 53.Kampinga HH & Craig EA (2010) The HSP70 chaperone machinery: J proteins as drivers of functional specificity, Nat Rev Mol Cell Biol. 11, 579–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Izawa T, Nagai H, Endo T & Nishikawa S. i. (2012) Yos9p and Hrd1p mediate ER retention of misfolded proteins for ER-associated degradation, Molecular biology of the cell. 23, 1283–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bhamidipati A, Denic V, Quan EM & Weissman JS (2005) Exploration of the topological requirements of ERAD identifies Yos9p as a lectin sensor of misfolded glycoproteins in the ER lumen, Mol Cell. 19, 741–51. [DOI] [PubMed] [Google Scholar]
- 56.Kim W, Spear ED & Ng DT (2005) Yos9p detects and targets misfolded glycoproteins for ER-associated degradation, Mol Cell. 19, 753–64. [DOI] [PubMed] [Google Scholar]
- 57.Szathmary R, Bielmann R, Nita-Lazar M, Burda P & Jakob CA (2005) Yos9 protein is essential for degradation of misfolded glycoproteins and may function as lectin in ERAD, Mol Cell. 19, 765–75. [DOI] [PubMed] [Google Scholar]
- 58.Ravid T, Kreft SG & Hochstrasser M (2006) Membrane and soluble substrates of the Doa10 ubiquitin ligase are degraded by distinct pathways, EMBO Journal. 25, 533–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bays NW, Gardner RG, Seelig LP, Joazeiro CA & Hampton RY (2001) Hrd1p/Der3p is a membrane-anchored ubiquitin ligase required for ER-associated degradation, Nature Cell Biology. 3, 24–9. [DOI] [PubMed] [Google Scholar]
- 60.Vashist S & Ng DT (2004) Misfolded proteins are sorted by a sequential checkpoint mechanism of ER quality control, Journal of Cell Biology. 165, 41–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ruggiano A, Foresti O & Carvalho P (2014) ER-associated degradation: Protein quality control and beyond, Journal of Cell Biology. 204, 869–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wolf DH & Stolz A (2012) The Cdc48 machine in endoplasmic reticulum associated protein degradation, Biochimica et Biophysica Acta, Molecular Cell Research. 1823, 117–124. [DOI] [PubMed] [Google Scholar]
- 63.Barthelme D & Sauer RT (2016) Origin and Functional Evolution of the Cdc48/p97/VCP AAA+ Protein Unfolding and Remodeling Machine, J Mol Biol. 428, 1861–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Preston GM, Guerriero CJ, Metzger MB, Michaelis S & Brodsky JL (2018) Substrate Insolubility Dictates Hsp104-Dependent Endoplasmic-Reticulum-Associated Degradation, Molecular Cell. 70, 242–253.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Metzger MB & Michaelis S (2008) Analysis of Quality Control Substrates in Distinct Cellular Compartments Reveals a Unique Role for Rpn4p in Tolerating Misfolded Membrane Proteins, Molecular Biology of the Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Xie Y & Varshavsky A (2001) RPN4 is a ligand, substrate, and transcriptional regulator of the 26S proteasome: A negative feedback circuit, Proceedings of the National Academy of Sciences. 98, 3056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hahn J-S, Neef DW & Thiele DJ (2006) A stress regulatory network for co-ordinated activation of proteasome expression mediated by yeast heat shock transcription factor, Molecular Microbiology. 60, 240–251. [DOI] [PubMed] [Google Scholar]
- 68.Mahul-Mellier A-L, Burtscher J, Maharjan N, Weerens L, Croisier M, Kuttler F, Leleu M, Knott GW & Lashuel HA (2020) The process of Lewy body formation, rather than simply α-synuclein fibrillization, is one of the major drivers of neurodegeneration, Proceedings of the National Academy of Sciences. 117, 4971–4982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.McNaught KS, Shashidharan P, Perl DP, Jenner P & Olanow CW (2002) Aggresome-related biogenesis of Lewy bodies, Eur J Neurosci. 16, 2136–48. [DOI] [PubMed] [Google Scholar]
- 70.Johnston JA, Ward CL & Kopito RR (1998) Aggresomes: a cellular response to misfolded proteins, Journal of Cell Biology. 143, 1883–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Neal S, Syau D, Nejatfard A, Nadeau S & Hampton RY (2020) HRD Complex Self-Remodeling Enables a Novel Route of Membrane Protein Retrotranslocation, iScience. 23, 101493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Metzger MB, Maurer MJ, Dancy BM & Michaelis S (2008) Degradation of a cytosolic protein requires endoplasmic reticulum-associated degradation machinery, Journal of Biological Chemistry. 283, 32302–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chan SN, Prasad R & Matsudaira P (2020) Genetic Selection Based on a Ste6(*)C-HA-Ura3 Substrate Identifies New Cytosolic Quality Control Alleles in Saccharomyces cerevisiae, G3 (Bethesda). 10, 1879–1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gardner RG, Nelson ZW & Gottschling DE (2005) Degradation-Mediated Protein Quality Control in the Nucleus, Cell. 120, 803–815. [DOI] [PubMed] [Google Scholar]
- 75.Park SH, Bolender N, Eisele F, Kostova Z, Takeuchi J, Coffino P & Wolf DH (2007) The Cytoplasmic Hsp70 Chaperone Machinery Subjects Misfolded and Endoplasmic Reticulum Import-incompetent Proteins to Degradation via the Ubiquitin-Proteasome System, Molecular Biology of the Cell. 18, 153–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Swanson R, Locher M & Hochstrasser M (2001) A conserved ubiquitin ligase of the nuclear envelope/endoplasmic reticulum that functions in both ER-associated and Matalpha2 repressor degradation, Genes & Development. 15, 2660–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lee DH & Goldberg AL (2010) Hsp104 is essential for the selective degradation in yeast of polyglutamine expanded ataxin-1 but not most misfolded proteins generally, Biochemical and Biophysical Research Communications. 391, 1056–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Shorter J & Southworth DR (2019) Spiraling in Control: Structures and Mechanisms of the Hsp104 Disaggregase, Cold Spring Harb Perspect Biol. 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kummer E, Szlachcic A, Franke KB, Ungelenk S, Bukau B & Mogk A (2016) Bacterial and Yeast AAA+ Disaggregases ClpB and Hsp104 Operate through Conserved Mechanism Involving Cooperation with Hsp70, Journal of Molecular Biology. 428, 4378–4391. [DOI] [PubMed] [Google Scholar]
- 80.Buck TM, Kolb AR, Boyd CR, Kleyman TR & Brodsky JL (2010) The endoplasmic reticulum-associated degradation of the epithelial sodium channel requires a unique complement of molecular chaperones, Mol Biol Cell. 21, 1047–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kanehara K, Kawaguchi S & Ng DT (2007) The EDEM and Yos9p families of lectin-like ERAD factors, Semin Cell Dev Biol. 18, 743–50. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that supports the findings of this study are available in Figures 1–4 and the supplementary material of this article.