

Abstract
Cerebrotendinous xanthomatosis (CTX) is an autosomal recessive lipid storage disorder caused by mutations in the CYP27A1 gene, which encodes the mitochondrial enzyme sterol 27-hydroxylase. Decreased sterol 27-hydroxylase activity results in impaired bile acid synthesis, leading to reduced production of bile acids, especially chenodeoxycholic acid (CDCA), as well as elevated serum cholestanol and urine bile alcohols. The accumulation of cholestanol and cholesterol mainly in the brain, lenses, and tendons results in the characteristic clinical manifestations of CTX. Clinical presentation is characterized by systemic symptoms including neonatal jaundice or cholestasis, refractory diarrhea, juvenile cataracts, tendon xanthomas, osteoporosis, coronary heart disease, and a broad range of neuropsychiatric manifestations. The combinations of symptoms vary from patient to patient and the presenting symptoms, especially in the early disease phase, may be nonspecific, which leads to a substantial diagnostic delay or underdiagnosis. Replacement of CDCA has been approved as a first-line treatment for CTX, and can lead to biochemical and clinical improvements. However, the effect of CDCA treatment is limited once significant neuropsychiatric manifestations are established. The age at diagnosis and initiation of CDCA treatment correlate with the prognosis of patients with CTX. Therefore, early diagnosis and subsequent treatment initiation are essential.
Keywords: Cerebrotendinous xanthomatosis, CTX, CYP27A1, Cholestanol, Chenodeoxycholic acid
Introduction
Cerebrotendinous xanthomatosis (CTX: OMIM#213700), first described by van Bogaert et al. in 1937, is a rare autosomal-recessive lipid storage disease caused by deficiency of the mitochondrial cytochrome P 450 enzyme, sterol 27-hydroxylase (CYP27A1, EC 1.14.15.15) due to mutations in the CYP27A1 gene 1) . Clinical presentation is characterized by neonatal jaundice or cholestasis, refractory diarrhea, juvenile cataracts, tendon xanthomas, osteoporosis, coronary heart disease, and progressive neuropsychiatric disturbances including mental retardation or dementia, psychiatric symptoms, pyramidal and cerebellar signs, progressive myelopathy, peripheral neuropathy, extrapyramidal manifestations, and seizures 2 - 9) . CTX is associated with considerable variability in clinical manifestations among patients and even within the same family 2) . The broad and diverse clinical symptoms cause a substantial diagnostic delay 2 - 4 , 9) . Replacement treatment with chenodeoxycholic acid (CDCA) in the early stage of the disease has been reported to improve or even prevent clinical symptoms of CTX 10 , 11) ; however, after significant neurological pathology is established, the effect of the treatment is limited and deterioration of clinical manifestations may continue 3 , 8 , 12 , 13) . Therefore, it is crucial to treat CTX patients at the initial stage of the disease. In this article, we provide the current understanding of the underlying pathomechanisms, clinical manifestations, diagnosis, and treatment of CTX.
Pathophysiology
CTX is caused by mutations in the CYP27A1 gene encoding sterol 27-hydroxylase, a key enzyme in the bile acid synthesis pathway. A schematic representation of the bile acid synthesis pathway is shown in Fig.1 . The classical pathway is initiated by 7α-hydroxylation of cholesterol, catalyzed by the rate-limiting enzyme cholesterol 7α-hydroxylase. The alternative pathway is initiated by 27-hydroxylation of cholesterol, which is catalyzed by sterol 27-hydroxylase. Decreased activity of sterol 27-hydroxylase leads to impaired bile acid synthesis in both the classical and alternative pathways 14) , resulting in reduced production of bile acids, especially CDCA, and to a lesser extent cholic acid 15) . The absence of a negative feedback effect of CDCA on cholesterol 7α-hydroxylase accelerates these metabolic abnormalities, leading to increased levels of the bile acid intermediate 7α-hydroxy-4-cholesten-3-one as a precursor for cholestanol and bile alcohols 16) . Elevated serum cholestanol and urine bile alcohols are the biochemical diagnostic hallmarks in CTX. Consequently, increased cholesterol metabolites, such as cholestanol, accumulate mainly in the brain, lenses, and tendons, leading to the characteristic clinical manifestations of CTX. Elevated levels of cholestanol have been found in the serum and tissues, including those of the central nervous system, tendon xanthomas, and atheromatous lesions, in CTX patients. Although the cholestanol-to-cholesterol ratios of various tissues were higher than that of serum, cholesterol was more abundant than cholestanol in both serum and tissues 17) . Cholestanol is widely used as a diagnostic marker but the usefulness of 7α-hydroxy-4-cholesten-3-one quantification in both the diagnosis and monitoring of CTX has also been reported 18) . It has also been shown that quantification of a panel of plasma ketosterol bile acid precursors (7α-hydroxy-4-cholesten-3-one, 7α,12α-dihydroxy-4-cholesten-3-one, and 7α,12α-dihydroxy-5β-cholestan-3-one) provides a more sensitive biochemical approach when compared with measurement of cholestanol 19) .
Fig.1. Impaired bile acid synthesis in cerebrotendinous xanthomatosis (CTX).

In CTX, mutations in the CYP27A1 gene lead to sterol 27-hydroxylase deficiency, resulting in reduced production of chenodeoxycholic acid and upregulation of the rate-limiting enzyme in the bile acid synthesis pathway, cholesterol 7α-hydroxylase. Increased levels of serum cholestanol and urinary bile alcohols are biological markers in CTX. HMG-CoA: 3-hydroxy-3-methylglutaryl-CoA.
In 1968, Menkes et al. discovered accumulation of cholestanol and cholesterol in the cerebrum and cerebellum of patients with CTX 20) . Although the mechanism by which cholestanol accumulates in the brain remains unclear, one possible explanation is that the bile acid precursor 7α-hydroxy-4-cholesten-3-one, which passes through the blood-brain barrier (BBB) more efficiently than cholestanol, can be converted to cholestanol by neurons, astrocytes, microglia, and human monocyte-derived macrophages 21 , 22) . Another possible explanation is impairment of the BBB. Increased levels of cholestanol and apolipoprotein B were observed in the cerebrospinal fluid of patients with CTX, indicating disrupted function of the BBB 23) . It has also been proposed that large plasma bile alcohol glucuronides play a role in the abnormal BBB permeability in CTX, leading to increased transport of cholestanol and cholesterol in the brain 24) .
Although the major pathway for production of cholestanol in CTX has been clarified, little is known about its metabolism. Under normal conditions, the 7α-hydroxy-4-cholesten-3-one-dependent pathway accounts for only about 30% of cholestanol biosynthesis in the brain, and cerebral cholestanol is mainly formed from cholesterol 25) . Using Cyp27a1 and Cyp46a1 knockout mice, Mast et al. demonstrated that CYP46A1 plays an important role in cholestanol removal from the brain and that CYP27A1 deficiency results in a preferential increase in cholestanol in the cerebellum 25) .
CTX patients develop premature atherosclerosis and xanthomas despite normal serum cholesterol concentrations. However, abundant deposits of cholesterol are detected in addition to cholestanol in the respective lesions in CTX 17) . Although the mechanism leading to premature arteriosclerosis and tendon xanthomas in CTX remains unclear, reduced capacity for reverse cholesterol transport has been proposed as a possible cause 26 - 31) . Sterol 27-hydroxylase, which is expressed in macrophages, endothelial cells, and tenocytes as well as in the liver, seems to contribute to the transport of peripheral cholesterol to the liver by transforming intracellular cholesterol into 27-hydroxycholesterol, which has a higher capacity for passing through lipophilic membranes compared with cholesterol 26 - 28) . In addition, 27-hydroxycholesterol is an endogenous ligand for liver X receptor (LXR). LXR activation induces upregulation of ATP-binding cassette transporter A1 (ABCA1) expression, leading to increased cholesterol efflux 29 - 31) . Fu et al. demonstrated that upregulation of ABCA1 in response to cholesterol loading was impaired in primary fibroblasts derived from a CTX patient 29) . In addition, since 27-hydroxycholesterol was found to be the major oxysterol in human atherosclerotic lesions 28) , extrahepatic sterol 27-hydroxylase is thought to be an anti-atherosclerotic enzyme. Absence of the two above defense mechanisms may contribute to premature atherosclerosis and xanthoma formation in CTX.
Epidemiology
CTX patients have been reported worldwide but prevalence of the disease is considered to be underestimated 32) . Based on the carrier frequency of the pathogenic CYP27A1 c.1183C>T (p.R395C) mutation in 115 control subjects, the prevalence of CTX in the USA among Caucasians of European ancestry was estimated to be 3-5:100,000 individuals 32) . Pilo-de-la-Fuente et al. estimated a minimum prevalence of 1/1,8000,000 individuals in Spain 3) . Estimates of the incidence of CTX vary among locations. A recent genetic epidemiological study based on the Exome Aggregation Consortium (ExAC) cohort, a large cohort of over 60,000 unrelated subjects, evaluated the allele frequency of 57 known and 29 predicted CTX-causing variants and estimated the incidence of CTX to be 1:134,970-1:461,358 in Europeans, 1:263,222-1:468,624 in Africans, 1:71,677-1:148,914 in Americans, 1:64,247-1:64,712 in East Asians, and 1:36,072-1:75,601 in South Asians 33) . Prevalence among Jews of Moroccan origin and the Druze sect in Israel has been reported to be particularly high 34 , 35) .
Molecular Genetics
In 1991, human sterol 27-hydroxylase cDNA was isolated from a liver cDNA library. The CYP27A1 gene consists of nine exons and eight introns and spans 18.6 kb of DNA on chromosome 2q33-qter 36 , 37) . Sterol 27-hydroxylase consists of a 33-residue mitochondrial signal sequence followed by a mature protein of 498 amino acids containing putative binding sites for heme and adrenodoxin 1) . Cali et al. first identified two CYP27A1 missense mutations, p.R395C and p.R479C, in patients with CTX and demonstrated that a loss-of-function mechanism is responsible for CTX 1) .
CYP27A1 is the only gene known to be associated with CTX. Therefore, the diagnostic gold standard is genetic analysis of the CYP27A1 gene 4 , 38) . The diagnosis is confirmed by the presence of biallelic pathogenic CYP27A1 mutations 4 , 38) . To date, over 99 pathogenic mutations, including missense mutations, nonsense mutations, splice-site mutations, and insertion/deletion mutations, have been reported worldwide 6) . A relatively high frequency of CYP27A1 mutations in particular ethnic groups has been reported: c.1016C>T (p.T339M), c.1183C>T (p.R395C), and c.1263+1G>A in the Netherlands 13) , c.646G>C (p.A216P), c.1183C>T (p.R395C), c.1184+1G>A, c.1263+1G>A, and a 1.9 kb deletion including exons 7-9 in Italy 38) , c.1183C >T (p.R395C) in northwestern Spain and c.1213C>T (p.R405W) in southern Spain 3) , c.1214G>A (p.R405Q), c.1421G>A (p.R474Q), c.435G>T (p.G145=), and c.1420C>T (p.R474W) in Japan 9) , which seems to be reasonable considering the allele frequency reported in the ExAC 33) . The allele frequencies of these variants in six global populations according to the ExAC database (version 0.3) are shown in Supplementary Table 1 . Although no genotype-phenotype correlation has been reported 2 , 3 , 13) , our nationwide survey revealed possible associations between c.1421G>A (p.R474Q) and classical form CTX, c.1241G>A (p.R405Q) and spinal form CTX, and c.435G>T (p.G145=) and non-neurological form CTX despite considerable phenotypic variation among patients with the same genotype 9) .
Supplementary Table 1. Allele frequencies of CYP27A1 variants according to Exome Aggregation Consortium database (version 0.3) .
| variant | AFR | AMR | EAS | FIN | NFE | SAS |
|---|---|---|---|---|---|---|
| p.G145 = | 0.00000 | 0.00000 | 0.00040 | 0.00000 | 0.00000 | 0.00000 |
| p.A216P | 0.00000 | 0.00000 | 0.00000 | 0.00000 | 0.00004 | 0.00000 |
| p.T339M | 0.00000 | 0.00000 | 0.00010 | 0.00000 | 0.00002 | 0.00007 |
| p.R395C | 0.00010 | 0.00020 | 0.00000 | 0.00030 | 0.00020 | 0.00006 |
| p.R405W | 0.00000 | 0.00009 | 0.00000 | 0.00000 | 0.00000 | 0.00006 |
| p.R405Q | 0.00010 | 0.00009 | 0.00050 | 0.00000 | 0.00004 | 0.00000 |
| p.R474W | 0.00000 | 0.00000 | 0.00000 | 0.00000 | 0.00002 | 0.00000 |
| p.R474Q | 0.00000 | 0.00000 | 0.00010 | 0.00000 | 0.00002 | 0.00000 |
| c.1184+1G> A | 0.00000 | 0.00000 | 0.00000 | 0.00000 | 0.00006 | 0.00070 |
| c.1263+1G> A | 0.00000 | 0.00009 | 0.00010 | 0.00000 | 0.00007 | 0.00000 |
AFR: African; AMR: Admixed American; EAS: East Asian; FIN: Finnish; NFE: Non-Finnish European; SAS: South Asian.
Clinical Features
Clinical Phenotypes
Clinical presentation of CTX is characterized by diverse systemic and neuropsychiatric manifestations and combinations of symptoms vary from patient to patient. Systemic symptoms include neonatal jaundice or cholestasis, chronic diarrhea, juvenile cataracts, xanthomas, osteoporosis, and coronary heart disease. The neurological and psychiatric manifestations of CTX vary widely. Intellectual disability as well as pyramidal and cerebellar signs are the most frequent and are cardinal clinical features 2 - 4 , 7 , 9) . In addition, CTX patients can present with extrapyramidal manifestations, peripheral neuropathy, epilepsy, and psychiatric disturbances. Autonomic involvement has also been reported 39) .
A representative clinical course of classical form CTX, the most common form of this condition, is shown in Fig.2 . Patients with classical form CTX develop neuropsychiatric symptoms attributed to the cerebrum, cerebellum, and/or brainstem, in combination with various systemic manifestations. The concept of spinal form CTX, also called spinal xanthomatosis, was proposed by Virrips et al. in 1999 40) . Patients exhibit clinical symptoms and signs related to involvement of the corticospinal tracts and dorsal columns of the spinal cord, without intellectual impairment, cerebellar signs, or peripheral neuropathy, at the time of presentation of the spinal cord syndrome 40) . Although most patients with spinal form CTX also exhibit various systemic and neurological symptoms, spinal form CTX without other neurological manifestations has been reported 41 - 46) . Spinal form CTX has a relatively mild clinical course compared with classical form CTX 40) .
Fig.2. Representative clinical course of classical form CTX.

Figure shows typical ages of onset of CTX-related symptoms.
We have proposed non-neurological form CTX 9) as another clinical phenotype. Although patients with the non-neurological form may develop neurological symptoms later in life, two genetically confirmed CTX patients in their fifties showed no evidence of neurological manifestations ≥ 20 years after disease onset. Therefore, we regarded the non-neurological form as a distinct clinical phenotype of CTX 9) .
All CTX patients exhibit increased serum cholestanol levels at the time of diagnosis 2 - 4 , 9) . While a significant relationship between serum cholestanol and clinical phenotype or disability was not detected 3) , Sekijima et al. showed that classical form patients had significantly higher levels of cholestanol than spinal form patients 9) .
Systemic Manifestations/Neonatal Jaundice or Cholestasis
Prolonged neonatal jaundice or cholestasis could be the earliest clinical presentation of CTX 47) . Laboratory findings have revealed conjugated hyperbilirubinemia with raised transaminases and alkaline phosphatase, whereas levels of γ-glutamyl transferase were normal or minimally elevated 47 - 50) , which is the characteristic feature of inborn errors of bile acid synthesis 51) . In one study, hepatomegaly or hepatosplenomegaly was evident 50) . Liver biopsy specimens have revealed nonspecific chronic active hepatitis with giant cell transformation, piecemeal or focal bridging necrosis, and fibrosis, in addition to intralobular cholestasis 47 - 50) . Cirrhosis was detected in an explanted liver 50) . In addition, retrospective cohort studies have demonstrated that about 8–16% of patients had a past medical history of neonatal cholestatic jaundice 4 , 13 , 47) . Furthermore, family histories have revealed fetal deaths or jaundice-related infantile deaths among siblings of affected individuals 47) .
Von Bahr et al. described a patient with genetically confirmed CTX who had fatal cholestatic liver damage 48) . Recently, Gong et al. reported on eight patients who presented with neonatal cholestasis. Among their cohort, this was fatal in four and one underwent liver transplantation. Although neonatal cholestasis associated with CTX has been generally assessed as transient and self-limiting with patient survival, a substantial proportion of patients could experience a more severe clinical course than previously recognized 50) . The mechanism by which mutations in the CYP27A1 gene lead to cholestasis may involve nuclear receptors such as farnesoid X receptor (FXR). CDCA is a potent stimulator of FXR 52) . Marked reduction of CDCA in CTX leads to decreased activation of FXR, which results in reduced expression of the bile salt export pump, causing a decrease in canalicular bile salt transportation 48 , 52) .
Systemic Manifestations/Chronic Diarrhea
Chronic unexplained diarrhea begins in infancy and continues into adulthood 2) . It may be the earliest symptom of CTX and could start within the first year of life 53) . Gastrointestinal tract investigations in patients with diarrhea did not produce any abnormal findings 54) . Also, rectal biopsy did not demonstrate any accumulation of cholestenol or cholesterol and fatty acids could not be detected in the feces 54) . Usually, diarrhea ceases immediately after starting treatment with CDCA 11) . Although the pathogenesis of diarrhea is still unclear, presence of bile alcohol in the lumen of the gut and/or intraluminal deficiency of CDCA are the most likely causes 54) .
Systemic Manifestations/Ocular Manifestations
Juvenile cataracts are one of the earliest clinical signs and often precede tendon xanthomas and neurological symptoms, and are usually noted in the second decade of life. Lens nuclei from CTX patients had a greater cholestanol content compared with the senile lens nuclei used as a control 55) . Although stabilization of cataracts with CDCA treatment has been reported 11) , complete resolution is unlikely 56) and operations should be considered. Early onset of cataracts is uncommon and therefore, juvenile cataracts are arguably an important cue for early diagnosis of CTX. A screening for CTX among 170 patients with idiopathic bilateral cataracts diagnosed between the ages of 2 and 21 years identified 3 cases 57) .
In addition to cataracts, ophthalmological manifestations include optic neuropathy with optic disc paleness, premature retinal vessel sclerosis, and cholesterol-like deposits 58) . Optic neuropathy with features suggestive of optic neuritis has also been reported 59) .
Systemic Manifestations/Xanthomas
Xanthomas usually appear during the second or third decade of life. They typically occur on the Achilles tendon, but may be found on the elbow, neck, knee, and the bottom of the foot ( Fig.3 ) . The patellar and finger extensor tendons are also common sites for development of tendon xanthomas 60 - 64) . Xanthomas in the lung 65) and choroid plexus have also been reported 60 , 66 , 67) . It is noteworthy that presence of xanthomas is a characteristic feature of the disease, but it is not mandatory for CTX diagnosis 68) . Biopsy specimens of xanthomas show lipid crystal clefts with infiltration of foamy macrophages 64 , 69) . In gallium-67 scintigraphy, there can be abnormal uptake in Achilles tendons 70) , even if Achilles tendon xanthomas were not evident in a physical examination or on MRI 44) . In addition, positron emission tomography (PET) using 18 F-2-deoxy-2-fluoro-glucose (FDG) showed abnormally high radioactivity in the Achilles tendons and adjacent regions 5) . Although CDCA treatment does not significantly reduce tendon xanthomas 10) , a decrease in size has been reported in some subjects 71 , 72) .
Fig.3. Xanthomas in a patient with CTX.

Figure shows xanthoma on the knee (A) and one on the Achilles tendon (B).
Systemic Manifestations/Skeletal System Involvement
Osteoporosis and increased bone fractures are CTX-associated systemic manifestations. However, the underlying pathogenesis of osteoporosis in this condition is still unknown. Decreased levels of serum 25-hydroxyvitamin D were detected in CTX patients 73 - 75) . In contrast, however, Federico et al. reported that levels of 25-hydroxyvitamin D were substantially within the normal range 76) , indicating that a deficiency in vitamin D metabolites may not be the only factor responsible for the development of osteoporosis in CTX 74) . An alternative hypothesis for explaining the cause of osteoporosis is impairment of intestinal calcium absorption due to changes in the quantity and composition of bile acids 76) . In general, osteoporosis has been considered to occur in the later stages of the disease 4 , 9) . However, teenage CTX patients could have early osteoporosis and a history of bone fracture 77) . CDCA treatment has been shown to improve bone mineral density (BMD) 74 , 76) .
Skeletal deformities including kyphosis, pectus excavates, pes equinovarus, and pes cavus were found in CTX patients 77) and Ginanneschi et al. reported that pes cavus occurrence was not significantly different in groups with and without peripheral nerve abnormalities 78) .
Systemic Manifestations/Cardiovascular System Involvement
Premature atherosclerosis and cardiovascular disease have been reported as systemic manifestations in CTX patients even in their thirties 79 - 82) . Myocardial infarction is a cause of premature death in this condition 82) . Kuriyama et al. reviewed 144 cases of CTX and reported that 15 patients (10.4%) had cardiovascular disease, consisting of coronary artery disease in ten patients, ischemic changes on electrocardiogram in four, and mitral valve insufficiency in one patient 79) . Coronary artery disease was evident in 8 of 40 CTX patients (20%) in a nationwide survey on CTX in Japan 9) . In this survey, the mean age at onset of coronary artery disease was 52.5.±5.8 years (mean±standard deviation (SD)) 9) . Duell et al. reported that 3 of 43 CTX patients (7%) had premature cardiovascular disease, consisting of myocardial infarction in two patients, and angina pectoris in one patient in the USA 8) . Abdominal aortic aneurysm, coronary artery dissection, aneurysmal coronary artery disease, advanced carotid atherosclerotic lesions, and thickening of the interatrial septum compatible with lipomatous hypertrophy have also been reported in CTX 81 , 83 - 86) .
Systemic Manifestations/Pulmonary Involvement
Elevated levels of cholestanol in bronchoalveolar lavage fluid as well as in serum have been reported in CTX patients without pulmonary symptoms, or radiological and pulmonary function abnormalities. Transbronchial lung biopsy specimens have revealed foamy macrophages and small granulomas in alveolar septa 87) .
Neuropsychiatric Manifestations/Intellectual disability
Among CTX patients, 48–74% present with intellectual disability 2 - 4 , 9) , which is one of the most frequent neurological symptoms. It is particularly important to take developmental delays, mental retardation, and learning difficulties beginning in childhood into consideration for early diagnosis of CTX 2 - 4 , 7 , 9 , 53) . Cognitive decline, presenting in adolescence or early adulthood, is also frequently observed 2 , 3 , 9) . Although a neuropsychological profile of patients with CTX remains undetermined, a fronto-temporal dementia phenotype exhibiting behavioral and personality changes 88) , extensive cerebral cortex symptoms including left-right disorientation, constructional apraxia, and temporal and spatial disorientation in addition to frontal lobe dysfunction 89) , and a corticobasal syndrome phenotype 90) have been reported.
Neuropsychiatric Manifestations/Pyramidal and Cerebellar Signs
Pyramidal and/or cerebellar signs typically emerge in the third or fourth decade and lead to gait disturbance in CTX patients 2 , 4 , 9) . Pyramidal and cerebellar signs have been detected in 64–92% and 36–83% of patients with CTX, respectively 2 - 4 , 9) . Pyramidal signs such as spasticity, hyperreflexia, and extensor plantar response can be cardinal clinical signs especially in patients with spinal form CTX 40 - 46) . Owing to dorsal column involvement, simultaneous occurrence of impaired position and vibration sensation in the lower extremities can lead to spastic-ataxic gait in this form 41 , 45) . Mignarri et al. have reported the usefulness of transcranial magnetic stimulation in detecting corticospinal tract damage 91) . Cerebellar signs include nystagmus, ataxic dysarthria, as well as limb and truncal ataxia 69 , 92 - 94) . Pyramidal signs frequently coexist with cerebellar signs 2 , 3) .
Neuropsychiatric Manifestations/Extrapyramidal Manifestations
CTX patients can present with a wide range of movement disorders including parkinsonism 90 , 95 , 96) , dystonia 97 - 99) , myoclonus 98 , 100 , 101) , and postural tremor 100 , 102) . When movement disorders are diagnosed, patients have a tendency to present with other CTX-associated systemic and neuropsychiatric manifestations 103) . Parkinsonism usually occurs later in life 7 , 95 , 103) and is the most frequently reported type of movement disorder in CTX, followed by dystonia, myoclonus, and postural tremor 103) . Parkinsonism seems to be a treatment-resistant feature in CTX 13) , with CDCA treatment seemingly having no effect. In addition, CTX patients may develop parkinsonism during treatment with CDCA 103) . The effect of L-dopa is controversial 90 , 95 , 103 - 105) . In addition to the characteristic brain MRI findings of CTX, signal hyperintensities on T2-weighted images in the substantia nigra, globus pallidus, and striatum 90 , 95 , 96 , 103) have been described and functional dopaminergic imaging has demonstrated a pre-synaptic dopaminergic deficit in CTX patients presenting with parkinsonism 90 , 95 , 96 , 104 , 105) . Although movement disorders are considered a late disease manifestation, Zubarioglu et al. reported that all six patients who were diagnosed before 18 years of age had intention tremor 77) .
Neuropsychiatric Manifestations/Peripheral Nervous System Involvement
Peripheral neuropathy is an established clinical feature of CTX; however, it is still being debated whether the underlying pathogenesis of CTX-related polyneuropathy is demyelinating or axonal in origin. Based on the presence of onion bulbs, which are generally considered a hallmark of chronic demyelination, the pathological process has been interpreted as demyelinating 106 , 107) . On the other hand, Verrips et al. reported that axonal degeneration was the predominant process on the basis of nerve conduction velocity (NCV) studies and sural nerve biopsy specimens showing features of axonal degeneration 108) . In addition to axonal polyneuropathy and demyelination polyneuropathy, a mixed type of neuropathy has been reported, indicating that CTX could exhibit any type of neuropathy 109) . CTX-related polyneuropathy seems to be predominantly motor neuropathy 78 , 109) . Although neurophysiologically confirmed neuropathy frequently occurs in CTX, signs and symptoms related to polyneuropathy are often absent or difficult to appreciate because central nervous system involvement may dominate the clinical picture 4 , 78 , 109) . The disease severity of polyneuropathy varies greatly among patients, ranging from asymptomatic presentations to severe polyneuropathy 78 , 109 , 110) . Thickening of the nerve roots and trunks of the lumbosacral plexus or cauda equina has been reported 90 , 111) .
Neuropsychiatric Manifestations/Muscle Involvement
Controversy exists regarding whether muscle involvement is a characteristic feature in CTX. Federico et al. noted mild myopathic changes 112) , while Verrips et al. reported that muscle biopsies demonstrated neurogenic changes without any definite myopathic characteristics 108) . The results for mitochondrial respiratory chain enzymatic activity are also controversial 108 , 113) . Abnormal findings from ultrastructural studies of muscles include changes in the mitochondria and membranous system, and an increased amount of lipid droplets, lipofuscin, and glycogen; however, the significance of these findings remains to be determined 39 , 108 , 112) .
Neuropsychiatric Manifestations/Epilepsy
In CTX, 10–33% of patients have epileptic seizures 2 - 4 , 7 , 9) . Epilepsy can develop at any stage in life and is often seen in the early phase of the disease 7) . Epilepsy could be a diagnostic cue in some cases 114 - 116) . A CTX patient presenting with infantile spasms has also been reported, but this is a rare case 117) . Electroencephalographic abnormalities are frequently observed in cases of CTX even without clinical signs of seizures 11 , 118) . In addition to slow background activity composed of theta and delta waves, bursts of high voltage slow activity are frequently demonstrated. Spike and sharp wave complexes can also be detected 10 , 11 , 118) . CDCA treatment leads to improvement or normalization of electroencephalographic findings 10 , 11 , 60 , 118) , and epilepsy in CTX seems to respond well to anti-epileptic agents 12 , 114 - 116 , 119) . CDCA treatment could lead to improved seizure control 12 , 60 , 120) , even in patients with drug-resistant epilepsy 121) .
Neuropsychiatric Manifestations/Behavioral manifestations
Psychiatric and behavioral manifestations include personality changes with irritability and aggressivity, depression, delusional syndrome, catatonia, psychosis, attention-deficit hyperactivity disorder, oppositional-defiant disorder, and autism spectrum disorder 13 , 122) . Behavioral disorders and affective/mood disorders associated with learning difficulties or mental retardation appearing during childhood or adolescence should lead to biochemical investigations to exclude CTX.
Radiological, Pathological, and Neurophysiological Examinations
Neuroimaging
The most distinctive neuroradiological findings are signal hyperintensities on T2-weighted and/or FLAIR images in the dentate nuclei and adjacent cerebellar white matter 123 , 124) . Abnormal signal changes in the dentate nuclei can be more clearly detected on FLAIR images than on T2-weighted images 123) . It was found that abnormal hyperintensities on T2-weighted and/or FLAIR images could be detected in the globus pallidus, internal capsule, substantia nigra, cerebral peduncles, inferior olive, and periventricular white matter, with a tendency to spare the U-fibers and corpus callosum 125) . Supratentorial and/or infratentorial atrophy are also observed 123 , 124 , 126) ( Fig.4 ) . Cortical volume, rather than white matter volume, was correlated with clinical status and cortical atrophy could be detected in all neocortical regions, with a preference for the fronto-parietal cotrtex 126) . In addition, cerebellar vacuolation, which is detected as hypointense lesions on both T1-weighted and FLAIR images, has been recently indicated as a marker of a poor prognosis in CTX 127 , 128) , while absence of dentate nuclei signal alteration is considered an indicator of a better prognosis 128) . Furthermore, calcifications were detected in the dentate nuclei in a subgroup of patients 128) and the hot cross bun sign in the pons, a characteristic finding of multiple system atrophy, has been reported 69) .
Fig.4. Brain magnetic resonance imaging (MRI).
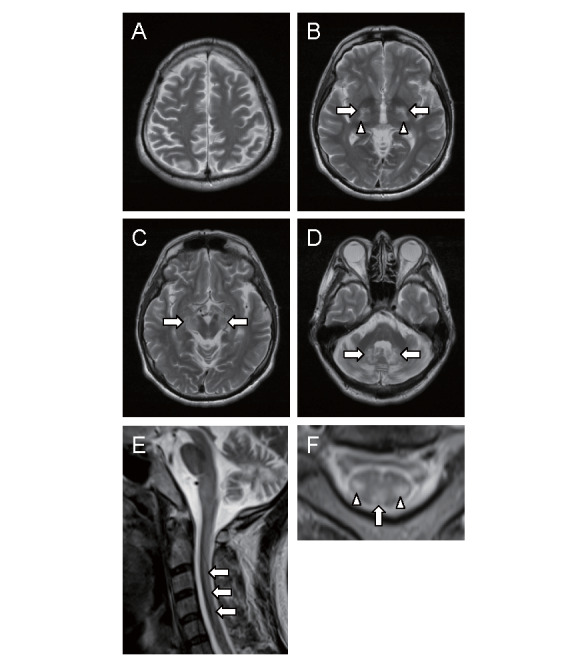
Axial T2-weighted images of the brain showing abnormal hyperintensities in the globus pallidus (arrows in B), internal capsules (arrowheads in B), cerebral peduncles (arrows in C), and dentate nuclei (arrows in D). Diffuse cerebral (A) and cerebellar (D) atrophy are evident. Sagittal T2-weighted image of the spinal cord exhibiting longitudinally extensive hyperintense lesions (arrows in E). Axial T2-weighted image at the C3 level showing involvement of lateral corticospinal tracts (arrowheads in F) and gracile tracts (arrow in F).
In patients with spinal cord involvement, a spinal cord MRI demonstrated longitudinally extensive hyperintense lesions involving lateral corticospinal tracts and gracile tracts on T2-weighted images 40 , 44 - 46) . It is noteworthy that absence of signal changes on spinal cord MRI cannot rule out the possibility of spinal form CTX 42 , 43) .
On magnetic resonance spectroscopy (MRS), decreases in N-acetylaspartate (NAA) intensities and increases in lactate signals point to axonal damage and brain mitochondrial dysfunction, respectively 123) . In addition to decreased NAA intensities, lipid peaks were evident on MRS using a short TE 129) . Increased levels of myo-inositol indicate gliosis and astrocytic proliferation 129 , 130) .
Cerebellar glucose hypometabolism in 18 F-2-deoxy-2-fluoro-glucose positron emission tomography (FDG-PET) and cerebellar hypoperfusion in single photon emission computed tomography (SPECT) with 99m Tc-ethylcysteinate dimer (ECD) have been reported despite normal cerebellar morphology 88 , 131 , 132) . In addition to in the cerebellum, SPECT using 99m Tc- ECD and 123 I- N -isopropyl- p -iodoamphetamine ( 123 I-IMP) revealed cerebral hypoperfusion, predominantly in the fronto-parietal lobes 88 , 89 , 132) . Gray matter atrophy patterns were correlated with hypoperfusion in SPECT using 99m Tc- ECD 133) .
Diffusion tensor imaging (DTI) revealed that fractional anisotropy (FA) reduction preceded structural alterations detected by voxel-based morphometry and correlated with cognitive function 133) . Widespread reductions of FA and decreased track-density were demonstrated 120 , 134) .
Neuropathology
At macroscopic examination, nonspecific brain and cerebellar atrophy and a yellowish soft tissue in the cerebellum, cerebrum, choroid plexus, cerebral peduncles, and globus pallidus were observed 135 - 138) . In the cerebral peduncles, cystic necrosis of the corticospinal tracts has been reported 139) . Microscopic examinations have revealed lipid crystal clefts, neuronal loss, demyelination, reactive astrocytosis, and foamy macrophages in the affected regions, especially in the dentate nucleus and surrounding area, as well as in the cerebrum, basal ganglia, brainstem, and, spinal cord 65 , 124 , 135 - 139) . In patients with spinal cord involvement, extensive symmetric loss of myelin and axons was detected particularly in the lateral corticospinal tracts and gracile tracts of the spinal cord 40) .
Neurophysiological Examinations
In addition to NCV studies and electroencephalography, abnormalities have been found in neurophysiological examinations. The P100 peak latency of visual evoked potentials (VEPs) was delayed 109 , 140 , 141) and the Ⅰ to Ⅲ, Ⅲ to Ⅴ, and Ⅰ to Ⅴ interpeak latencies of brain stem evoked potentials (BAEPs) were prolonged 109 , 140 , 141) . Central conduction time in somatosensory evoked potentials (SSEPs) 109 , 142) and motor evoked potentials (MEPs) 89 , 91 , 141) were increased, with lower extremity predominance.
Diagnosis
Importance of Early Diagnosis and Treatment
CTX is a treatable metabolic disorder; however, once significant neurological symptoms are established, clinical deterioration can occur despite normalization of cholestanol levels after treatment with CDCA 3) . Even with therapy, only 28% of the patients remained stable, whereas 60% continued to deteriorate and 20% died, in a cohort of 25 patients with CTX in Spain 3) . Duell et al. reported that clinical deterioration during follow up was observed in patients who had significant neurological symptoms when they were diagnosed at the age of 25 years or older 8) . Yahalom et al. and Stelten et al. have shown that the age of diagnosis and initiation of CDCA treatment correlates with the prognosis of patients with CTX 12 , 13) . Berginer et al. reported two siblings with CTX who began CDCA treatment from 2 and 7 years of age, respectively, and did not develop any neurological manifestations during a 14-year follow-up period 142) . These findings strongly suggest that early diagnosis and treatment are crucial in CTX. However, retrospective cohort studies on CTX have revealed a substantial diagnostic delay of 15–25 years 2 - 4 , 9) .
Juvenile cataracts are usually the earliest clinical sign that precedes tendon xanthomas and neurological symptoms. Cruysberg et al. emphasized that the combination of juvenile cataracts and chronic diarrhea is noteworthy in the early diagnosis of CTX 143) . It is recommended that all patients with cataracts before the age of 30 years are screened for CTX, especially if they also have CTX-related conditions such as chronic diarrhea, tendon xanthomas, and/or neuropsychiatric symptoms 8) . Verrips et al. emphasized that presence of tendon xanthomas is not obligatory for a diagnosis of CTX and recommended that presence of two of the four clinical features of premature cataracts, intractable diarrhea, progressive neurological signs and symptoms, and tendon xanthomas prompt thorough biochemical screening for CTX 68) . It is also important to consider intellectual disability, usually presenting at school age, for early diagnosis of CTX 4) . In addition, because affected relatives may be asymptomatic, biochemical examination of all siblings of a patient with CTX is recommended 2 , 4) .
To identify and treat CTX patients at an initial stage of the disease, Mignarri et al. created a suspicion index and developed a diagnostic algorithm for early diagnosis of CTX 4) . Their suspicion index comprised weighted scores assigned to indicators such as family history characteristics and common systemic and neurological symptoms. They suggested that their proposed algorithm would be useful for early diagnosis, even in patients before the onset of disabling neurological symptoms including ataxia, spasticity, and psychiatric disturbances 4) .
Diagnostic Criteria
In the absence of generally accepted diagnostic criteria for CTX, we recently proposed new diagnostic criteria with emphasis on early diagnosis ( Table 1 ) 9) . They include clinical symptoms, biochemical findings, genetic analysis, and differential diagnosis. We established three diagnostic categories in accordance with levels of certainty: definite, probable, and possible CTX. The diagnosis of possible CTX is made when there is at least one CTX-related clinical symptom and elevated levels of serum cholestanol (≥ 4.5 µg/mL, mean±SD: 2.35±0.73 µg/mL). Excluding other conditions with elevated levels of cholestanol is necessary for diagnosis of probable CTX. A definite diagnosis of CTX is confirmed by the presence of biallelic mutations in the CYP27A1 gene.
Table 1. Diagnostic criteria for cerebrotendinous xanthomatosis (Sekijima et al. 9) ) .
|
A. Symptoms 1. Tendon xanthoma 2. Progressive neurological dysfunction a or mental retardation 3. Juvenile cataract 4. Juvenile coronary artery disease 5. Chronic unexplained diarrhea 6. Juvenile osteoporosis 7. Prolonged neonatal cholestasis B. Biochemical finding Elevated serum cholestanol level C. Genetic testing Pathogenic mutation in CYP27A1 gene (homozygosity or compound heterozygosity) D. Differential diagnosis Increased serum cholestanol level due to following diseases should be excluded ・Familial hypercholesterolemia ・Sitosterolemia ・Obstructive biliary tract disease ・Hypothyroidism Diagnostic category Definite: At least one of symptom in A and B+C+D Probable: At least one of symptom in A and B+D Possible: At least one of symptom in A and B |
a Representative progressive neurological dysfunction includes cognitive dysfunction, cerebellar symptoms, pyramidal symptoms, extrapyramidal symptoms, seizure, peripheral neuropathy, and sensory disturbance attributed to spinal cord.
Differential Diagnosis
Differential diagnosis of CTX differs substantially according to presenting symptoms. Inborn errors of bile acid metabolism including CTX lead to neonatal cholestasis or hepatitis 144 , 145) , which can be the first manifestation in this disease. In patients with juvenile bilateral cataracts and/or progressive mental deterioration, CTX should be considered 2 , 57) . When xanthomas are evident, differential diagnosis includes familial hypercholesterolemia (FH) and sitosterolemia. FH is characterized by elevated levels of LDL cholesterol, the presence of tendon xanthomas, and premature coronary artery disease, and mutations in LDLR , APOB , and PCSK9 have been reported to cause FH 146) . Sitosterolemia is an autosomal recessive sterol storage disorder characterized by elevated levels of LDL cholesterol and plant sterols such as sitosterol and campesterol, tendinous and tuberous xanthomas, and premature atherosclerosis. It is caused by biallelic mutations in either ABCG5 or ABCG8 147) . The presence of juvenile cataracts, chronic unexplained diarrhea, and progressive neuropsychiatric manifestations can distinguish CTX from these two disorders. Other conditions with elevated levels of cholestanol include obstructive biliary tract diseases and hypothyroidism. In patients with cerebellar ataxia, CTX patients might be misdiagnosed as spinocerebellar atrophy, multiple system atrophy, or Marinesco-Sjögren syndrome 68) . CTX should be included in the differential diagnosis of spastic paraplegia 42 , 44) .
Clinical Management
CDCA has been approved as first-line treatment for CTX. In a landmark study published in 1984, Berginer et al. demonstrated the long-term efficacy of oral CDCA treatment 10) . In addition to a decrease in serum cholestanol and elimination of abnormal urinary and biliary excretion of bile alcohols, CDCA treatment led to an improvement in electroencephalographic findings and neurological manifestations including intellectual impairment, pyramidal and cerebellar signs, and peripheral neuropathy 10 , 11) . CDCA treatment is recommended at a dose of 750 mg/day for adults and 15 mg/kg/day for children in three divided oral doses 10 , 11) . It has been shown to result in a gradual decline in serum cholestanol during the first 2 years 148 , 149) . Assessment of cholestanol levels may be useful in monitoring patient adherence to treatment. However, it should be noted that a decreased level of cholestanol does not necessarily suggest a good prognosis 3) . In Japan, CDCA has been approved for dissolution of gallstones, but not for the treatment of CTX.
Although CDCA is a relatively safe drug, gastrointestinal manifestations and drug-induced liver damage may occur 44 , 60 , 150) . Huidekoper et al. reported an infantile patient with CTX who developed jaundice with hepatomegaly within 6 weeks after initiating CDCA administration at a dosage of 15mg/kg/day 151) . After treatment with CDCA was stopped, liver size and function rapidly normalized. CDCA supplementation was then restarted and maintained at 5 mg/kg/day with no further evidence of liver dysfunction and adequate metabolic control. Duell et al. reported that 9% of patients required dose adjustment for CDCA owing to moderate drug-induced liver damage 8) . These findings suggest that clinical and laboratory monitoring and dosage adjustment for CDCA are essential in the treatment of CTX, especially in infants and young children 8 , 151) .
CDCA was initially preferred to cholic acid because it was more effective in reducing cholesterol 7α-hydroxylase and had a stronger negative feedback effect on it 127 , 152) . Cholic acid has been shown to be effective in the treatment of other genetic defects in bile acid synthesis 153) . Since CDCA is intrinsically hepatotoxic, cholic acid is considered the safer option in CTX, especially in infancy 49) . Mandia et al. reported potential efficacy for cholic acid in adult patients with CTX, including individuals whose CDCA treatment was discontinued due to supply difficulties 127) . Treatment with cholic acid not only significantly reduced cholestanol levels in all patients but also led to improvement or stabilization of systemic and/or neurological manifestations 127) . No adverse effects were reported in patients undergoing cholic acid treatment, suggesting that cholic acid may be a suitable alternative treatment, especially in patients with adverse effects related to CDCA, such as drug-induced liver damage 60 , 127) .
Treatment with ursodeoxycholic acid, which does not inhibit cholesterol 7α-hydroxylase, has been shown to be ineffective 10 , 154) . When ursodeoxycholic acid was substituted for CDCA, plasma cholestanol returned to pretreatment levels 10) .
The effectiveness of 3-hydroxy-3-methylglutaryl (HMG)-CoA reductase inhibitors (statins) remains controversial. Lewis et al. reported that mevinolin normalized serum cholestanol and reduced the size of xanthomas 155) , whereas Batta et al. found that lovastatin did not affect abnormal bile acid synthesis or reduce plasma cholestanol levels 154) . Although synergistic effects of combination therapy with CDCA and HMG-CoA reductase inhibitors on serum cholestanol or urine bile alcohols have been observed 71 , 81 , 149) , absence of an additive effect has also been reported 148) . After switching from combined therapy of CDCA and HMG-CoA reductase inhibitors to HMG-CoA reductase inhibitor monotherapy, clinical symptoms such as xanthomas and neurological manifestations, and electroencephalographic findings were re-exacerbated with reappearance of abnormal bile alcohol excretion or elevated plasma cholestanol 71 , 156) . Therefore, HMG-CoA reductase inhibitors could be beneficial when combined with CDCA, but long-term clinical benefits should be proven.
Low-density lipoprotein (LDL) is a major carrier of serum cholestanol. LDL-apheresis, usually combined with CDCA and HMG-CoA reductase inhibitors, has been performed to reduce serum cholestanol 157 - 160) Levels of serum cholestanol or 7α-hydroxy-4-cholesten-3-one decreased after each LDL-apheresis, but returned to their initial levels within 1–2 weeks 159 , 161) , suggesting that LDL-apheresis at a frequency of at least once every 2 weeks is necessary. The effects of LDL-apheresis on clinical manifestations are still controversial despite the decrease in cholestanol. In addition, the invasiveness of this procedure and its necessity for the long-term management of the disease should be taken into account 161) .
Symptomatic treatments for epilepsy 115 , 120) , psychiatric manifestations 122) , and movement disorders such as dystonia 97 , 98) and parkinsonism 90 , 95) should be considered. Cataract extraction is also usually required 57) .
After treatment with CDCA, improvements in neurophysiological examinations including NCV studies 78) , VEP 72 , 78) , SSEP 72) , MEP 72 , 91) , and EEG 10 , 11) have been reported. Besides conventional MRI, DTI and tractography, MRS, and SPECT imaging might have potential as neuroimaging modalities for monitoring treatment response 120 , 132 - 134 , 162 , 163) .
Conclusions and Perspectives
CTX is considered a rare inherited metabolic disorder. However, it may be under- or misdiagnosed, although effective treatment is available. There is a crucial “point of no return” in CTX, after which treatment initiation can no longer prevent the progression of the disease 12) . The earlier the diagnosis is made and the sooner treatment is started, the more likely it is that the significant neurological manifestations that diminish the quality of life of patients with CTX can be improved or even prevented. Neonatal jaundice, chronic unexplained diarrhea, developmental delays, mental retardation, and learning difficulties are non-specific symptoms, but they could be diagnostic cues for pediatricians. Ophthalmologists have an opportunity to diagnose CTX, because bilateral cataracts are one of the earliest clinical symptoms and juvenile-onset bilateral cataracts could be useful as a screening marker for CTX 57) . Furthermore, it could be beneficial to screen newborns for CTX in the future 164) .
Acknowledgments and Notice of Grant Support
This work has been supported by Health, Labour and Welfare Sciences Research Grant for Research on Rare and Intractable Diseases (H30-nanji-ippan-003).
Conflicts of Interest
Atsushi Nohara has nothing to disclose. Hayato Tada has nothing to disclose. Masatsune Ogura has received honoraria from Amgen Inc., Astellas Pharma Inc. Sachiko Okazaki has received scholarship grants from Minophagen Pharmaceutical Co., Ltd., Kowa Company, Ltd. Koh Ono has nothing to disclose. Hitoshi Shimano has nothing to disclose. Hiroyuki Daida has received honoraria from Amgen Inc., Daiichi-Sankyo Co., Ltd., Kowa Co., Ltd., and MSD K.K., Novartis Pharma K.K., Bayer Yakuhin, Ltd. and received clinical research funding from Canon Medical Systems Corporation, Philips Japan, Ltd., Toho Holdings Co., Ltd., Asahi Kasei Corporation, and Inter Reha Co., Ltd. HD has also received scholarship grants from Nippon Boehringer Ingelheim Co., Ltd., Otsuka Pharmaceutical Co., Ltd., Sanofi K.K., MSD K.K., Daiichi-Sankyo Co., Ltd., Pfizer Co., Ltd., Mitsubishi Tanabe Pharma Corp., Astellas Pharma Inc., Takeda Pharmaceutical Co., Ltd., Teijin Pharma, Ltd., Shionogi & Co., Ltd., Actelion Pharmaceuticals, Ltd., Actelion Ltd., Kowa Co., Ltd., Bayer Yakuhin, Ltd. HD has also courses endowed by companies, including Philips Japan, Ltd., ResMed, Fukuda Denshi Co., Ltd., and Paramount Bed Co., Ltd. Kazushige Dobashi has nothing to disclose. Toshio Hayashi has nothing to disclose. Mika Hori has nothing to disclose. Kota Matsuki has nothing to disclose. Tetsuo Minamino has nothing to disclose. Shinji Yokoyama has nothing to disclose. Mariko Harada-Shiba has received stock holdings or options from Liid Pharma, honoraria from Amgen Inc., Astellas Pharma Inc., Sanofi, and scholarship grants from Aegerion Pharmaceuticals, Inc., Recordati Rare Diseases Japan, and Kaneka Corporation. Katsunori Ikewaki has nothing to disclose. Yasushi Ishigaki has nothing to disclose. Shun Ishibashi has received honoraria from Kowa Co., Ltd., and a scholarship grant from Ono Pharmaceutical Co., Ltd. Kyoko Inagaki has nothing to disclose. Hirotoshi Ohmura has nothing to disclose. Hiroaki Okazaki has received scholarship grants from Minophagen Pharmaceutical Co., Ltd., Kowa Company, Ltd. Masa-aki Kawashiri has nothing to disclose. Masayuki Kuroda has nothing to disclose. Masahiro Koseki has received clinical research funding from Kowa Company, Ltd., Rohto Pharmaceutical Co., Ltd. Takanari Gotoda has nothing to disclose. Shingo Koyama has nothing to disclose. Yoshiki Sekijima has nothing to disclose. Manabu Takahashi has nothing to disclose. Yasuo Takeuchi has nothing to disclose. Misa Takegami has nothing to disclose. Kazuhisa Tsukamoto has received honoraria from Bayer Yakuhin, Ltd., MSD Ltd., Takeda Pharmaceutical Company Ltd., and scholarship grants from Mitsubishi Tanabe Pharma Corporation., Bayer Yakuhin, Ltd., Sanofi K.K. Atsuko Nakatsuka has nothing to disclose. Kimitoshi Nakamura has nothing to disclose. Satoshi Hirayama has nothing to disclose. Hideaki Bujo has nothing to disclose. Daisaku Masuda has received clinical research funding from MSD K.K., Ono Pharmaceutical Co., Ltd., Takeda Pharmaceutical Co., Ltd., Kowa Co., Ltd. Takashi Miida has nothing to disclose. Yoshihiro Miyamoto has nothing to disclose. Takeyoshi Murano has nothing to disclose. Takashi Yamaguchi has nothing to disclose. Shizuya Yamashita has received honoraria from Kowa Company, Ltd., MSD K.K. Masashi Yamamoto has nothing to disclose. Koutaro Yokote has received honoraria from Kowa Company, Ltd., MSD K.K., Astellas Pharma Inc., Mitsubishi Tanabe Pharma Corp., Amgen K.K., Takeda Pharmaceutical Company Limited, Sanofi K.K., Ono Pharmaceutical Co., Ltd., AstraZeneca K.K., Daiichi-Sankyo Co., Ltd., Novartis Pharma K.K., Sumitomo Dainippon Pharma Co., Ltd., Kyowa Kirin Co., Ltd., Pfizer Japan Inc., Novo Nordisk Pharma Ltd., Nippon Boehringer Ingelheim Co., Ltd., Eli Lilly Japan K.K., Taisho Pharmaceutical Co., Ltd., Janssen Pharmaceutical K.K., and received clinical research funding from Taisho Pharmaceutical Co., Ltd. KY has also received scholarship grants from Mitsubishi Tanabe Pharma Corp., Takeda Pharmaceutical Co., Ltd., MSD K.K., Pfizer Japan Inc., Novo Nordisk Pharma Ltd., Taisho Pharmaceutical Co., Ltd., Kao Corporation, Ono Pharmaceutical Co., Ltd., Eli Lilly Japan K.K., Sumitomo Dainippon Pharma Co., Ltd., Nippon Boehringer Ingelheim Co., Ltd., Daiichi-Sankyo Co., Ltd., Teijin Pharma, Ltd., Shionogi Co., Ltd., Bayer Yakuhin, Ltd. Jun Wada has nothing to disclose.
References
- 1).Cali JJ, Hsieh CL, Francke U, Russell DW: Mutations in the bile acid biosynthetic enzyme sterol 27-hydroxylase underlie cerebrotendinous xanthomatosis. J Biol Chem, 1991; 266: 7779-7783 [PMC free article] [PubMed] [Google Scholar]
- 2).Verrips A, Hoefsloot LH, Steenbergen GC, Theelen JP, Wevers RA, Gabreëls FJ, van Engelen BG, van den Heuvel LP: Clinical and molecular genetic characteristics of patients with cerebrotendinous xanthomatosis. Brain, 2000; 123: 908-919 [DOI] [PubMed] [Google Scholar]
- 3).Pilo-de-la-Fuente B, Jimenez-Escrig A, Lorenzo JR, Pardo J, Arias M, Ares-Luque A, Duarte J, Muñiz-Pérez S, Sobrido MJ: Cerebrotendinous xanthomatosis in Spain: clinical, prognostic, and genetic survey. Eur J Neurol, 2011; 18: 1203-1211 [DOI] [PubMed] [Google Scholar]
- 4).Mignarri A, Gallus GN, Dotti MT, Federico A: A suspicion index for early diagnosis and treatment of cerebrotendinous xanthomatosis. J Inherit Metab Dis, 2014; 37: 421-429 [DOI] [PubMed] [Google Scholar]
- 5).Nie S, Chen G, Cao X, Zhang Y: Cerebrotendinous xanthomatosis: a comprehensive review of pathogenesis, clinical manifestations, diagnosis, and management. Orphanet J Rare Dis, 2014; 9: 179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6).Salen G, Steiner RD: Epidemiology, diagnosis, and treatment of cerebrotendinous xanthomatosis (CTX). J Inherit Metab Dis, 2017; 40: 771-781 [DOI] [PubMed] [Google Scholar]
- 7).Wong JC, Walsh K, Hayden D, Eichler FS: Natural history of neurological abnormalities in cerebrotendinous xanthomatosis. J Inherit Metab Dis, 2018; 41: 647-656 [DOI] [PubMed] [Google Scholar]
- 8).Duell PB, Salen G, Eichler FS, DeBarber AE, Connor SL, Casaday L, Jayadev S, Kisanuki Y, Lekprasert P, Malloy MJ, Ramdhani RA, Ziajka PE, Quinn JF, Su KG, Geller AS, Diffenderfer MR, Schaefer EJ: Diagnosis, treatment, and clinical outcomes in 43 cases with cerebrotendinous xanthomatosis. J Clin Lipidol, 2018; 12: 1169-1178 [DOI] [PubMed] [Google Scholar]
- 9).Sekijima Y, Koyama S, Yoshinaga T, Koinuma M, Inaba Y: Nationwide survey on cerebrotendinous xanthomatosis in Japan. J Hum Genet, 2018; 63: 271-280 [DOI] [PubMed] [Google Scholar]
- 10).Berginer VM, Salen G, Shefer S: Long-term treatment of cerebrotendinous xanthomatosis with chenodeoxycholic acid. N Engl J Med, 1984; 311: 1649-1652 [DOI] [PubMed] [Google Scholar]
- 11).van Heijst AF, Verrips A, Wevers RA, Cruysberg JR, Renier WO, Tolboom JJ: Treatment and follow-up of children with cerebrotendinous xanthomatosis. Eur J Pediatr, 1998; 157: 313-316 [DOI] [PubMed] [Google Scholar]
- 12).Yahalom G, Tsabari R, Molshatzki N, Ephraty L, Cohen H, Hassin-Baer S: Neurological outcome in cerebrotendinous xanthomatosis treated with chenodeoxycholic acid: early versus late diagnosis. Clin Neuropharmacol, 2013; 36: 78-83 [DOI] [PubMed] [Google Scholar]
- 13).Stelten BML, Huidekoper HH, van de Warrenburg BPC, Brilstra EH, Hollak CEM, Haak HR, Kluijtmans LAJ, Wevers RA, Verrips A: Long-term treatment effect in cerebrotendinous xanthomatosis depends on age at treatment start. Neurology, 2019; 92: e83-e95 [DOI] [PubMed] [Google Scholar]
- 14).DeBarber AE, Luo J, Star-Weinstock M, Purkayastha S, Geraghty MT, Chiang JP, Merkens LS, Pappu AS, Steiner RD: A blood test for cerebrotendinous xanthomatosis with potential for disease detection in newborns. J Lipid Res, 2014; 55: 146-154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15).Salen G: Cholestanol deposition in cerebrotendinous xanthomatosis. A possible mechanism. Ann Intern Med, 1971; 75: 843-851 [DOI] [PubMed] [Google Scholar]
- 16).Björkhem I, Skrede S, Buchmann MS, East C, Grundy S: Accumulation of 7 alpha-hydroxy-4-cholesten-3-one and cholesta-4,6-dien-3-one in patients with cerebrotendinous xanthomatosis: effect of treatment with chenodeoxycholic acid. Hepatology, 1987; 7: 266-271 [DOI] [PubMed] [Google Scholar]
- 17).Bhattacharyya AK, Lin DS, Connor WE: Cholestanol metabolism in patients with cerebrotendinous xanthomatosis: absorption, turnover, and tissue deposition. J Lipid Res, 2007; 48: 185-192 [DOI] [PubMed] [Google Scholar]
- 18).DeBarber AE, Connor WE, Pappu AS, Merkens LS, Steiner RD: ESI-MS/MS quantification of 7alpha-hydroxy-4-cholesten-3-one facilitates rapid, convenient diagnostic testing for cerebrotendinous xanthomatosis. Clin Chim Acta, 2010; 41: 43-48 [DOI] [PubMed] [Google Scholar]
- 19).DeBarber AE, Luo J, Giugliani R, Souza CF, Chiang JP, Merkens LS, Pappu AS, Steiner RD: A useful multi-analyte blood test for cerebrotendinous xanthomatosis. Clin Biochem, 2014; 47: 860-863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20).Menkes JH, Schimschock JR, Swanson PD: Cerebrotendinous xanthomatosis. The storage of cholestanol within the nervous system. Arch Neurol, 1968; 19: 47-53 [DOI] [PubMed] [Google Scholar]
- 21).Panzenboeck U, Andersson U, Hansson M, Sattler W, Meaney S, Björkhem I: On the mechanism of cerebral accumulation of cholestanol in patients with cerebrotendinous xanthomatosis. J Lipid Res, 2007; 48: 1167-1174 [DOI] [PubMed] [Google Scholar]
- 22).Båvner A, Shafaati M, Hansson M, Olin M, Shpitzen S, Meiner V, Leitersdorf E, Björkhem I: On the mechanism of accumulation of cholestanol in the brain of mice with a disruption of sterol 27-hydroxylase. J Lipid Res, 2010; 51: 2722-2730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23).Salen G, Berginer V, Shore V, Horak I, Horak E, Tint GS, Shefer S: Increased concentrations of cholestanol and apolipoprotein B in the cerebrospinal fluid of patients with cerebrotendinous xanthomatosis. Effect of chenodeoxycholic acid. N Engl J Med, 1987; 316: 1233-1238 [DOI] [PubMed] [Google Scholar]
- 24).Batta AK, Salen G, Shefer S, Tint GS, Batta M: Increased plasma bile alcohol glucuronides in patients with cerebrotendinous xanthomatosis: effect of chenodeoxycholic acid. J Lipid Res, 1987; 28: 1006-1012 [PubMed] [Google Scholar]
- 25).Mast N, Anderson KW, Lin JB, Li Y, Turko IV, Tatsuoka C, Bjorkhem I, Pikuleva IA: Cytochrome P450 27A1 Deficiency and Regional Differences in Brain Sterol Metabolism Cause Preferential Cholestanol Accumulation in the Cerebellum. J Biol Chem, 2017; 292: 4913-4924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26).Babiker A, Andersson O, Lund E, Xiu RJ, Deeb S, Reshef A, Leitersdorf E, Diczfalusy U, Björkhem I: Elimination of cholesterol in macrophages and endothelial cells by the sterol 27-hydroxylase mechanism. Comparison with high density lipoprotein-mediated reverse cholesterol transport. J Biol Chem, 1997; 272: 26253-26261 [DOI] [PubMed] [Google Scholar]
- 27).von Bahr S, Movin T, Papadogiannakis N, Pikuleva I, Rönnow P, Diczfalusy U, Björkhem I: Mechanism of accumulation of cholesterol and cholestanol in tendons and the role of sterol 27-hydroxylase (CYP27A1). Arterioscler Thromb Vasc Biol, 2002; 22: 1129-1135 [DOI] [PubMed] [Google Scholar]
- 28).Björkhem I, Andersson O, Diczfalusy U, Sevastik B, Xiu RJ, Duan C, Lund E: Atherosclerosis and sterol 27-hydroxylase: evidence for a role of this enzyme in elimination of cholesterol from human macrophages. Proc Natl Acad Sci U S A, 1994; 91: 8592-8596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29).Fu X, Menke JG, Chen Y, Zhou G, MacNaul KL, Wright SD, Sparrow CP, Lund EG: 27-Hydroxycholesterol Is an Endogenous Ligand for Liver X Receptor in Cholesterol-loaded Cells. J Biol Chem, 2001; 276: 38378-38387 [DOI] [PubMed] [Google Scholar]
- 30).Tall AR, Costet P, Wang N: Regulation and mechanisms of macrophage cholesterol efflux. J Clin Invest, 2002; 110: 899-904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31).Marengo B, Bellora F, Ricciarelli R, De Ciucis C, Furfaro A, Leardi R, Colla R, Pacini D, Traverso N, Moretta A, Pronzato MA, Bottino C, Domenicotti C: Oxysterol mixture and, in particular, 27-hydroxycholesterol drive M2 polarization of human macrophages. Biofactors, 2016; 42: 80-92 [DOI] [PubMed] [Google Scholar]
- 32).Lorincz MT, Rainier S, Thomas D, Fink JK: Cerebrotendinous xanthomatosis: possible higher prevalence than previously recognized. Arch Neurol, 2005; 62: 1459-1463 [DOI] [PubMed] [Google Scholar]
- 33).Appadurai V, DeBarber A, Chiang PW, Patel SB, Steiner RD, Tyler C, Bonnen PE: Apparent underdiagnosis of Cerebrotendinous Xanthomatosis revealed by analysis of ~60,000 human exomes. Mol Genet Metab, 2015; 116: 298-304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34).Berginer VM, Abeliovich D: Genetics of cerebrotendinous xanthomatosis (CTX): an autosomal recessive trait with high gene frequency in Sephardim of Moroccan origin. Am J Med Genet, 1981; 10: 151-157 [DOI] [PubMed] [Google Scholar]
- 35).Falik-Zaccai TC, Kfir N, Frenkel P, Cohen C, Tanus M, Mandel H, Shihab S, Morkos S, Aaref S, Summar ML, Khayat M: Population screening in a Druze community: the challenge and the reward. Genet Med, 2008; 10: 903-909 [DOI] [PubMed] [Google Scholar]
- 36).Cali JJ, Russell DW: Characterization of human sterol 27-hydroxylase. A mitochondrial cytochrome P-450 that catalyzes multiple oxidation reaction in bile acid biosynthesis. J Biol Chem, 1991; 266: 7774-7778 [PubMed] [Google Scholar]
- 37).Leitersdorf E, Reshef A, Meiner V, Levitzki R, Schwartz SP, Dann EJ, Berkman N, Cali JJ, Klapholz L, Berginer VM: Frameshift and splice-junction mutations in the sterol 27-hydroxylase gene cause cerebrotendinous xanthomatosis in Jews or Moroccan origin. J Clin Invest, 1993; 91: 2488-2496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38).Gallus GN, Dotti MT, Federico A: Clinical and molecular diagnosis of cerebrotendinous xanthomatosis with a review of the mutations in the CYP27A1 gene. Neurol Sci, 2006; 27: 143-149 [DOI] [PubMed] [Google Scholar]
- 39).Chen SF, Tsai NW, Chang CC, Lu CH, Huang CR, Chuang YC, Chang WN: Neuromuscular abnormality and autonomic dysfunction in patients with cerebrotendinous xanthomatosis. BMC Neurol, 2011; 11: 63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40).Verrips A, Nijeholt GJ, Barkhof F, Van Engelen BG, Wesseling P, Luyten JA, Wevers RA, Stam J, Wokke JH, van den Heuvel LP, Keyser A, Gabreëls FJ: Spinal xanthomatosis: a variant of cerebrotendinous xanthomatosis. Brain, 1999; 122: 1589-1595 [DOI] [PubMed] [Google Scholar]
- 41).Bartholdi D, Zumsteg D, Verrips A, Wevers RA, Sistermans E, Hess K, Jung HH: Spinal phenotype of cerebrotendinous xanthomatosis--a pitfall in the diagnosis of multiple sclerosis. J Neurol, 2004; 251: 105-107 [DOI] [PubMed] [Google Scholar]
- 42).Nicholls Z, Hobson E, Martindale J, Shaw PJ: Diagnosis of spinal xanthomatosis by next-generation sequencing: identifying a rare, treatable mimic of hereditary spastic paraparesis. Pract Neurol, 2015; 15: 280-283 [DOI] [PubMed] [Google Scholar]
- 43).Saute JA, Giugliani R, Merkens LS, Chiang JP, DeBarber AE, de Souza CF: Look carefully to the heels! A potentially treatable cause of spastic paraplegia. J Inherit Metab Dis, 2015; 38: 363-364 [DOI] [PubMed] [Google Scholar]
- 44).Abe R, Sekijima Y, Kinoshita T, Yoshinaga T, Koyama S, Kato T, Ikeda SI: Spinal form cerebrotendinous xanthomatosis patient with long spinal cord lesion. J Spinal Cord Med, 2016; 39: 726-729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45).Yanagihashi M, Kano O, Terashima T, Kawase Y, Hanashiro S, Sawada M, Ishikawa Y, Shiraga N, Ikeda K, Iwasaki Y: Late-onset spinal form xanthomatosis without brain lesion: a case report. BMC Neurol, 2016; 16: 21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46).Mutlu D, Tuncer A, Gocmen R, Yalcin-Cakmakli G, Saygı S, Elibol B: Diagnostic challenge: A case of late-onset spinal form cerebrotendinous xanthomatosis. Neurology, 2019; 92: 438-439 [DOI] [PubMed] [Google Scholar]
- 47).Clayton PT, Verrips A, Sistermans E, Mann A, Mieli-Vergani G, Wevers R: Mutations in the sterol 27-hydroxylase gene (CYP27A) cause hepatitis of infancy as well as cerebrotendinous xanthomatosis. J Inherit Metab Dis, 2002; 25: 501-513 [DOI] [PubMed] [Google Scholar]
- 48).von Bahr S, Björkhem I, Van’t Hooft F, Alvelius G, Nemeth A, Sjövall J, Fischler B: Mutation in the sterol 27-hydroxylase gene associated with fatal cholestasis in infancy. J Pediatr Gastroenterol Nutr, 2005; 40: 481-486 [DOI] [PubMed] [Google Scholar]
- 49).Pierre G, Setchell K, Blyth J, Preece MA, Chakrapani A, McKiernan P: Prospective treatment of cerebrotendinous xanthomatosis with cholic acid therapy. J Inherit Metab Dis, 2008; 31: S241-245 [DOI] [PubMed] [Google Scholar]
- 50).Gong JY, Setchell KDR, Zhao J, Zhang W, Wolfe B, Lu Y, Lackner K, Knisely AS, Wang NL, Hao CZ, Zhang MH, Wang JS: Severe Neonatal Cholestasis in Cerebrotendinous Xanthomatosis: Genetics, Immunostaining, Mass Spectrometry. J Pediatr Gastroenterol Nutr, 2017; 65: 561-568 [DOI] [PubMed] [Google Scholar]
- 51).Sundaram SS, Bove KE, Lovell MA, Sokol RJ: Mechanisms of disease: Inborn errors of bile acid synthesis. Nat Clin Pract Gastroenterol Hepatol, 2008; 5: 456-468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52).Edwards PA, Kast HR, Anisfeld AM: BAREing it all: the adoption of LXR and FXR and their roles in lipid homeostasis. J Lipid Res, 2002; 43: 2-12 [PubMed] [Google Scholar]
- 53).Degos B, Nadjar Y, Amador Mdel M, Lamari F, Sedel F, Roze E, Couvert P, Mochel F: Natural history of cerebrotendinous xanthomatosis: a paediatric disease diagnosed in adulthood. Orphanet J Rare Dis, 2016; 11: 41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54).van Heijst AF, Wevers RA, Tangerman A, Cruysberg JR, Renier WO, Tolboom JJ: Chronic diarrhoea as a dominating symptom in two children with cerebrotendinous xanthomatosis. Acta Paediatr, 1996; 85: 932-936 [DOI] [PubMed] [Google Scholar]
- 55).McKenna P, Morgan SJ, Bosanquet RC, Laker MF: A case of cerebrotendinous xanthomatosis. II: The sterol content of a cataractous lens. Br J Ophthalmol, 1990; 74: 629-630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56).Tibrewal S, Duell PB, DeBarber AE, Loh AR: Cerebrotendinous xanthomatosis: early diagnosis on the basis of juvenile cataracts. J AAPOS, 2017; 21: 505-507 [DOI] [PubMed] [Google Scholar]
- 57).Freedman SF, Brennand C, Chiang J, DeBarber A, Del Monte MA, Duell PB, Fiorito J, Marshall R: Prevalence of Cerebrotendinous Xanthomatosis Among Patients Diagnosed With Acquired Juvenile-Onset Idiopathic Bilateral Cataracts. JAMA Ophthalmol, 2019; Epub ahead of print [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58).Dotti MT, Rufa A, Federico A: Cerebrotendinous xanthomatosis: heterogeneity of clinical phenotype with evidence of previously undescribed ophthalmological findings. J Inherit Metab Dis, 2001; 24: 696-706 [DOI] [PubMed] [Google Scholar]
- 59).Miyamoto M, Ishii N, Mochizuki H, Shiomi K, Kaida T, Chuman H, Nakazato M: Optic Neuropathy with Features Suggestive of Optic Neuritis in Cerebrotendinous Xanthomatosis. Case Rep Neurol Med, 2019; 2019: 2576826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60).Waterreus RJ, Koopman BJ, Wolthers BG, Oosterhuis HJ: Cerebrotendinous xanthomatosis (CTX): a clinical survey of the patient population in The Netherlands. Clin Neurol Neurosurg, 1987; 89: 169-175 [DOI] [PubMed] [Google Scholar]
- 61).Dotti MT, Salen G, Federico A: Cerebrotendinous xanthomatosis as a multisystem disease mimicking premature ageing. Dev Neurosci, 1991; 13: 371-376 [DOI] [PubMed] [Google Scholar]
- 62).Tian D, Zhang ZQ: 2 Novel deletions of the sterol 27-hydroxylase gene in a Chinese Family with Cerebrotendinous Xanthomatosis. BMC Neurol, 2011; 11: 130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63).Varman KM, Dunbar K, Usifo K, Stevens CA: Cerebrotendinous Xanthomatosis: A Treatable Genetic Disease Not to Be Missed. J Clin Rheumatol, 2016; 22: 92-93 [DOI] [PubMed] [Google Scholar]
- 64).Koopal C, Visseren FL, Marais AD, Westerink J, Spiering W: Tendon xanthomas: Not always familial hypercholesterolemia. J Clin Lipidol, 2016; 10: 1262-1265 [DOI] [PubMed] [Google Scholar]
- 65).Sperhake JP, Matschke J, Orth U, Gal A, Püschel K: Sudden death due to cerebrotendinous xanthomatosis confirmed by mutation analysis. Int J Legal Med, 2000; 113: 110-113 [DOI] [PubMed] [Google Scholar]
- 66).Vanrietvelde F, Lemmerling M, Mespreuve M, Crevits L, De Reuck J, Kunnen M: MRI of the brain in cerebrotendinous xanthomatosis (van Bogaert-Scherer-Epstein disease). Eur Radiol, 2000; 10: 576-578 [DOI] [PubMed] [Google Scholar]
- 67).Brienza M, Fiermonte G, Cambieri C, Mignarri A, Dotti MT, Fiorelli M: Enlarging brain xanthomas in a patient with cerebrotendinous xanthomatosis. J Inherit Metab Dis, 2015; 38: 981-982 [DOI] [PubMed] [Google Scholar]
- 68).Verrips A, van Engelen BG, Wevers RA, van Gee Mondelli l BM, Cruysberg JR, van den Heuvel LP, Keyser A, Gabreëls FJ: Presence of diarrhea and absence of tendon xanthomas in patients with cerebrotendinous xanthomatosis. Arch Neurol, 2000; 57: 520-524 [DOI] [PubMed] [Google Scholar]
- 69).Jain RS, Sannegowda RB, Agrawal A, Hemrajani D, Jain R, Mathur T: ‘Hot cross bun’ sign in a case of cerebrotendinous xanthomatosis: a rare neuroimaging observation. BMJ Case Rep, 2013; doi: 10.1136/bcr-2012-006641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70).Okada J, Oonishi H, Tamada H, Kizaki T, Yasumi K, Matuo T: Gallium uptake in cerebrotendinous xanthomatosis. Eur J Nucl Med, 1995; 22: 1069-1072 [DOI] [PubMed] [Google Scholar]
- 71).Nakamura T, Matsuzawa Y, Takemura K, Kubo M, Miki H, Tarui S: Combined treatment with chenodeoxycholic acid and pravastatin improves plasma cholestanol levels associated with marked regression of tendon xanthomas in cerebrotendinous xanthomatosis. Metabolism, 1991; 40: 741-746 [DOI] [PubMed] [Google Scholar]
- 72).Mondelli M, Sicurelli F, Scarpini C, Dotti MT, Federico A: Cerebrotendinous xanthomatosis: 11-year treatment with chenodeoxycholic acid in five patients. An electrophysiological study. J Neurol Sci, 2001; 190: 29-33 [DOI] [PubMed] [Google Scholar]
- 73).Berginer VM, Shany S, Alkalay D, Berginer J, Dekel S, Salen G, Tint GS, Gazit D: Osteoporosis and increased bone fractures in cerebrotendinous xanthomatosis. Metabolism, 1993; 42: 69-74 [DOI] [PubMed] [Google Scholar]
- 74).Martini G, Mignarri A, Ruvio M, Valenti R, Franci B, Del Puppo M, Federico A, Nuti R, Dotti MT: Long-term bone density evaluation in cerebrotendinous xanthomatosis: evidence of improvement after chenodeoxycholic acid treatment. Calcif Tissue Int, 2013; 92: 282-286 [DOI] [PubMed] [Google Scholar]
- 75).Zübarioğlu T, Bilen İP, Kıykım E, Doğan BB, Enver EÖ, Cansever MŞ, Zeybek AÇA: Evaluation of the effect of chenodeoxycholic acid treatment on skeletal system findings in patients with cerebrotendinous xanthomatosis. Turk Pediatri Ars, 2019; 54: 113-118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76).Federico A, Dotti MT, Loré F, Nuti R: Cerebrotendinous xanthomatosis: pathophysiological study on bone metabolism. J Neurol Sci, 1993; 115: 67-70 [DOI] [PubMed] [Google Scholar]
- 77).Zubarioglu T, Kiykim E, Yesil G, Demircioglu D, Cansever MS, Yalcinkaya C, Aktuglu-Zeybek C: Early diagnosed cerebrotendinous xanthomatosis patients: clinical, neuroradiological characteristics and therapy results of a single center from Turkey. Acta Neurol Belg, 2019; 119: 343-350 [DOI] [PubMed] [Google Scholar]
- 78).Ginanneschi F, Mignarri A, Mondelli M, Gallus GN, Del Puppo M, Giorgi S, Federico A, Rossi A, Dotti MT: Polyneuropathy in cerebrotendinous xanthomatosis and response to treatment with chenodeoxycholic acid. J Neurol, 2013; 260: 268-274 [DOI] [PubMed] [Google Scholar]
- 79).Kuriyama M, Fujiyama J, Yoshidome H, Takenaga S, Matsumuro K, Kasama T, Fukuda K, Kuramoto T, Hoshita T, Seyama Y, Okatu Y, Osame M: Cerebrotendinous xanthomatosis: clinical and biochemical evaluation of eight patients and review of the literature. J Neurol Sci, 1991; 102: 225-232 [DOI] [PubMed] [Google Scholar]
- 80).Fujiyama J, Kuriyama M, Arima S, Shibata Y, Nagata K, Takenaga S, Tanaka H, Osame M: Atherogenic risk factors in cerebrotendinous xanthomatosis. Clin Chim Acta, 1991; 200: 1-11 [DOI] [PubMed] [Google Scholar]
- 81).Burnett JR, Moses EA, Croft KD, Brown AJ, Grainger K, Vasikaran SD, Leitersdorf E, Watts GF: Clinical and biochemical features, molecular diagnosis and long-term management of a case of cerebrotendinous xanthomatosis. Clin Chim Acta, 2001; 306: 63-69 [DOI] [PubMed] [Google Scholar]
- 82).Valdivielso P, Calandra S, Durán JC, Garuti R, Herrera E, González P: Coronary heart disease in a patient with cerebrotendinous xanthomatosis. J Intern Med, 2004; 255: 680-683 [DOI] [PubMed] [Google Scholar]
- 83).Tada H, Inaba S, Pozharitckaia D, Kawashiri MA: Prominent Tendon Xanthomas and Abdominal Aortic Aneurysm Associated with Cerebrotendinous Xanthomatosis Identified Using Whole Exome Sequencing. Intern Med, 2018; 57: 1119-1122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84).Souto MJS, Almeida-Santos MA, Ferreira EJP, Gonçalves LFG, Oliveira JLM, Sousa ACS: Spontaneous Coronary Artery Dissection in a Patient with Cerebrotendinous Xanthomatosis. Arq Bras Cardiol, 2020; 115: 18-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85).Potkin BN, Hoeg JM, Connor WE, Salen G, Quyyumi AA, Brush JE Jr, Roberts WC, Brewer HB Jr: Aneurysmal coronary artery disease in cerebrotendinous xanthomatosis. Am J Cardiol, 1988; 61: 1150-1152 [DOI] [PubMed] [Google Scholar]
- 86).Dotti MT, Mondillo S, Plewnia K, Agricola E, Federico A: Cerebrotendinous xanthomatosis: evidence of lipomatous hypertrophy of the atrial septum. J Neurol, 1998; 245: 723-726 [DOI] [PubMed] [Google Scholar]
- 87).Kawabata M, Kuriyama M, Mori S, Sakashita I, Osame M: Pulmonary manifestations in cerebrotendinous xanthomatosis. Intern Med, 1998; 37: 922-926 [DOI] [PubMed] [Google Scholar]
- 88).Guyant-Maréchal L, Verrips A, Girard C, Wevers RA, Zijlstra F, Sistermans E, Vera P, Campion D, Hannequin D: Unusual cerebrotendinous xanthomatosis with fronto-temporal dementia phenotype. Am J Med Genet A, 2005; 139A: 114-117 [DOI] [PubMed] [Google Scholar]
- 89).Mukaino A, Tsuda M, Yamashita S, Kosaka T, Wada K, Ando Y: Cerebrotendinous xanthomatosis presenting with extensive cerebral cortex symptoms: A case report. Clin Neurol Neurosurg, 2018; 174: 217-219 [DOI] [PubMed] [Google Scholar]
- 90).Rubio-Agusti I, Kojovic M, Edwards MJ, Murphy E, Chandrashekar HS, Lachmann RH, Bhatia KP: Atypical parkinsonism and cerebrotendinous xanthomatosis: report of a family with corticobasal syndrome and a literature review. Mov Disord, 2012; 27: 1769-1774 [DOI] [PubMed] [Google Scholar]
- 91).Mignarri A, Rossi S, Ballerini M, Gallus GN, Del Puppo M, Galluzzi P, Federico A, Dotti MT: Clinical relevance and neurophysiological correlates of spasticity in cerebrotendinous xanthomatosis. J Neurol, 2011; 258: 783-790 [DOI] [PubMed] [Google Scholar]
- 92).Siebner HR, Berndt S, Conrad B: Cerebrotendinous xanthomatosis without tendon xanthomas mimicking Marinesco-Sjoegren syndrome: a case report. J Neurol Neurosurg Psychiatry, 1996; 60: 582-585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93).Ly H, Bertorini TE, Shah N: An adult male with progressive spastic paraparesis and gait instability. J Clin Neuromuscul Dis, 2014; 16: 98-103 [DOI] [PubMed] [Google Scholar]
- 94).Alhariri A, Hamilton K, Oza V, Cordoro K, Sobreira NL, Malloy M, Slavotinek A: Clinical report: A patient with a late diagnosis of cerebrotendinous xanthomatosis and a response to treatment. Am J Med Genet A, 2017; 173: 2275-2279 [DOI] [PubMed] [Google Scholar]
- 95).Su CS, Chang WN, Huang SH, Lui CC, Pan TL, Lu CH, Chuang YC, Huang CR, Tsai NW, Hsieh MJ, Chang CC: Cerebrotendinous xanthomatosis patients with and without parkinsonism: clinical characteristics and neuroimaging findings. Mov Disord, 2010; 25: 452-458 [DOI] [PubMed] [Google Scholar]
- 96).Kirmi O, Murphy E, Carecchio M, Sulkin T, Rankin J, Robertson F: Teaching neuroimages: progressive asymmetric parkinsonism and tendon xanthomas. Neurology, 2011; 77: e97-98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97).Posada IJ, Ramos A: Botulinum toxin-responsive oromandibular dystonia in cerebrotendinous xanthomatosis. Parkinsonism Relat Disord, 2011; 17: 570-572 [DOI] [PubMed] [Google Scholar]
- 98).Lagarde J, Roze E, Apartis E, Pothalil D, Sedel F, Couvert P, Vidailhet M, Degos B: Myoclonus and dystonia in cerebrotendinous xanthomatosis. Mov Disord, 2012; 27: 1805-1810 [DOI] [PubMed] [Google Scholar]
- 99).Yoshinaga T, Sekijima Y, Koyama S, Maruyama K, Yoshida T, Kato T, Ikeda S: Clinical and radiological findings of a cerebrotendinous xanthomatosis patient with a novel p.A335V mutation in the CYP27A1 gene. Intern Med, 2014; 53: 2725-2729 [DOI] [PubMed] [Google Scholar]
- 100).Szlago M, Gallus GN, Schenone A, Patiño ME, Sfaelo Z, Rufa A, Da Pozzo P, Cardaioli E, Dotti MT, Federico A: The first cerebrotendinous xanthomatosis family from Argentina: a new mutation in CYP27A1 gene. Neurology, 2008; 70: 402-404 [DOI] [PubMed] [Google Scholar]
- 101).Dutta AK, Danda S, Muthusamy K, Alexander M, Sudhakar SV, Hansdak S, Bandyopadhyay R, Bakhya Shree GB, Rekha L: Cerebrotendinous xanthomatosis: Possibility of founder mutation in CYP27A1 gene (c.526delG) in Eastern Indian and Surinamese population. Mol Genet Metab Rep, 2015; 3: 33-35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102).Isenhardt K, Schmitt R, Nagel A, Drach L, Schlote W: Inherited cholesterol lipidosis: cerebrotendinous xanthomatosis (van Bogaert Scherer Epstein disease). A clinicopathological study. Clin Neuropathol, 2005; 24: 276-283 [PubMed] [Google Scholar]
- 103).Stelten BML, van de Warrenburg BPC, Wevers RA, Verrips A: Movement disorders in cerebrotendinous xanthomatosis. Parkinsonism Relat Disord, 2019; 58: 12-16 [DOI] [PubMed] [Google Scholar]
- 104).Mignarri A, Falcini M, Vella A, Giorgio A, Gallus GN, Del Puppo M, Vattimo A, Federico A, Dotti MT: Parkinsonism as neurological presentation of late-onset cerebrotendinous xanthomatosis. Parkinsonism Relat Disord, 2012; 18: 99-101 [DOI] [PubMed] [Google Scholar]
- 105).Schotsmans K, De Cauwer H, Baets J, Ceyssens S, van den Hauwe L, Deconinck T, Helsen G: Cerebrotendinous xanthomatosis presenting with asymmetric parkinsonism: a case with I-123-FP-CIT SPECT imaging. Acta Neurol Belg, 2012; 112: 287-289 [DOI] [PubMed] [Google Scholar]
- 106).Ohnishi A, Yamashita Y, Goto I, Kuroiwa Y, Murakami S, Ikeda M: De- and remyelination and onion bulb in cerebrotendinous xanthomatosis. Acta Neuropathol, 1979; 45: 43-45 [DOI] [PubMed] [Google Scholar]
- 107).Argov Z, Soffer D, Eisenberg S, Zimmerman Y: Chronic demyelinating peripheral neuropathy in cerebrotendinous xanthomatosis. Ann Neurol, 1986; 20: 89-91 [DOI] [PubMed] [Google Scholar]
- 108).Verrips A, van Engelen BG, ter Laak H, Gabreëls-Festen A, Janssen A, Zwarts M, Wevers RA, Gabreëls FJ: Cerebrotendinous xanthomatosis. Controversies about nerve and muscle: observations in ten patients. Neuromuscul Disord, 2000; 10: 407-414 [DOI] [PubMed] [Google Scholar]
- 109).Pilo B, de Blas G, Sobrido MJ, Navarro C, Grandas F, Barrero FJ, Moya MA, Jimenez-Escrig A: Neurophysiological study in cerebrotendinous xanthomatosis. Muscle Nerve, 2011; 43: 531-536 [DOI] [PubMed] [Google Scholar]
- 110).Wang Z, Yuan Y, Zhang W, Zhang Y, Feng L: Cerebrotendinous xanthomatosis with a compound heterozygote mutation and severe polyneuropathy. Neuropathology, 2007; 27: 62-66 [DOI] [PubMed] [Google Scholar]
- 111).Hashimoto T, Sennari Y, Okada K, Adachi H: Cerebrotendinous Xanthomatosis with Nodular-hypertrophy of the Lumbosacral Roots. Intern Med, 2018; 57: 1669-1670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112).Federico A, Dotti MT, Volpi N: Muscle mitochondrial changes in cerebrotendinous xanthomatosis. Ann Neurol, 1991; 30: 734-735 [DOI] [PubMed] [Google Scholar]
- 113).Dotti MT, Manneschi L, Federico A: Mitochondrial enzyme deficiency in cerebrotendinous xanthomatosis. J Neurol Sci, 1995; 129: 106-108 [DOI] [PubMed] [Google Scholar]
- 114).Arlazoroff A, Roitberg B, Werber E, Shidlo R, Berginer VM: Epileptic seizure as a presenting symptom of cerebrotendinous xanthomatosis. Epilepsia, 1991; 32: 657-661 [DOI] [PubMed] [Google Scholar]
- 115).Koyama S, Kawanami T, Tanji H, Arawaka S, Wada M, Saito N, Kato T: A case of cerebrotendinous xanthomatosis presenting with epilepsy as an initial symptom with a novel V413D mutation in the CYP27A1 gene. Clin Neurol Neurosurg, 2012; 114: 1021-1023 [DOI] [PubMed] [Google Scholar]
- 116).Pedroso JL, Pinto WB, Souza PV, Santos LT, Abud IC, Avelino MA, Barsottini OG: Early-onset epilepsy as the main neurological manifestation of cerebrotendinous xanthomatosis. Epilepsy Behav, 2012; 24: 380-381 [DOI] [PubMed] [Google Scholar]
- 117).Larson A, Weisfeld-Adams JD, Benke TA, Bonnen PE: Cerebrotendinous Xanthomatosis Presenting with Infantile Spasms and Intellectual Disability. JIMD Rep, 2017; 35: 1-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118).Berginer VM, Radwan H, Korczyn AD, Kott E, Salen G, Shefer S: EEG in cerebrotendinous xanthomatosis (CTX). Clin Electroencephalogr, 1982; 13: 89-96 [DOI] [PubMed] [Google Scholar]
- 119).Wakamatsu N, Hayashi M, Kawai H, Kondo H, Gotoda Y, Nishida Y, Kondo R, Tsuji S, Matsumoto T: Mutations producing premature termination of translation and an amino acid substitution in the sterol 27-hydroxylase gene cause cerebrotendinous xanthomatosis associated with parkinsonism. J Neurol Neurosurg Psychiatry, 1999; 67: 195-198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120).Catarino CB, Vollmar C, Küpper C, Seelos K, Gallenmüller C, Bartkiewicz J, Biskup S, Hörtnagel K, Klopstock T: Brain diffusion tensor imaging changes in cerebrotendinous xanthomatosis reversed with treatment. J Neurol, 2018; 265: 388-393 [DOI] [PubMed] [Google Scholar]
- 121).Kauffman MA, Gonzalez-Morón D, Consalvo D, Kochen S: Cerebrotendinous xanthomatosis revealed in drug-resistant epilepsy diagnostic workup. Am J Med Sci, 2012; 343: 332-333 [DOI] [PubMed] [Google Scholar]
- 122).Fraidakis MJ: Psychiatric manifestations in cerebrotendinous xanthomatosis. Transl Psychiatry, 2013; 3: e302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123).De Stefano N, Dotti MT, Mortilla M, Federico A: Magnetic resonance imaging and spectroscopic changes in brains of patients with cerebrotendinous xanthomatosis. Brain, 2001; 124: 121-131 [DOI] [PubMed] [Google Scholar]
- 124).Barkhof F, Verrips A, Wesseling P, van Der Knaap MS, van Engelen BG, Gabreëls FJ, Keyser A, Wevers RA, Valk J: Cerebrotendinous xanthomatosis: the spectrum of imaging findings and the correlation with neuropathologic findings. Radiology, 2000; 217: 869-876 [DOI] [PubMed] [Google Scholar]
- 125).Ahmed RM, Murphy E, Davagnanam I, Parton M, Schott JM, Mummery CJ, Rohrer JD, Lachmann RH, Houlden H, Fox NC, Chataway J: A practical approach to diagnosing adult onset leukodystrophies. J Neurol Neurosurg Psychiatry, 2014; 85: 770-781 [DOI] [PubMed] [Google Scholar]
- 126).Guerrera S, Stromillo ML, Mignarri A, Battaglini M, Marino S, Di Perri C, Federico A, Dotti MT, De Stefano N: Clinical relevance of brain volume changes in patients with cerebrotendinous xanthomatosis. J Neurol Neurosurg Psychiatry, 2010; 81: 1189-1193 [DOI] [PubMed] [Google Scholar]
- 127).Mandia, Besson G, Lamari F, Castelnovo G, Curot J, Duval F, Giral P, Lecerf JM, Roland D, Pierdet H, Douillard C, Nadjar Y: Cholic acid as a treatment for cerebrotendinous xanthomatosis in adults. J Neurol, 2019; 266: 2043-2050 [DOI] [PubMed] [Google Scholar]
- 128).Mignarri A, Dotti MT, Federico A, De Stefano N, Battaglini M, Grazzini I, Galluzzi P, Monti L: The spectrum of magnetic resonance findings in cerebrotendinous xanthomatosis: redefinition and evidence of new markers of disease progression. J Neurol, 2017; 264: 862-874 [DOI] [PubMed] [Google Scholar]
- 129).Embiruçu EK, Otaduy MC, Taneja AK, Leite CC, Kok F, Lucato LT: MR spectroscopy detects lipid peaks in cerebrotendinous xanthomatosis. Am J Neuroradiol, 2010; 31: 1347-1349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130).Mignarri A, Dotti MT, Del Puppo M, Gallus GN, Giorgio A, Cerase A, Monti L: Cerebrotendinous xanthomatosis with progressive cerebellar vacuolation: six-year MRI follow-up. Neuroradiology, 2012; 54: 649-651 [DOI] [PubMed] [Google Scholar]
- 131).Ragno M, Di Marzio F, Fuccio C, Pianese L, Cozzolino V, Carboni T, Cinti A, D’Andreamatteo G, Trojano L, Mignarri A, Gallus GN, Dotti MT: Cerebellar hypometabolism with normal structural findings in Cerebrotendinous xanthomatosis. A case report. Clin Neurol Neurosurg, 2015; 139: 221-223 [DOI] [PubMed] [Google Scholar]
- 132).Caroppo P, D’Agata F, Mignarri A, Stromillo ML, Dotti MT, Mongini T: Brain metabolism changes after therapy with chenodeoxycholic acid in a case of cerebrotendinous xanthomatosis. Neurol Sci, 2013; 34: 1693-1696 [DOI] [PubMed] [Google Scholar]
- 133).Chang CC, Lui CC, Wang JJ, Huang SH, Lu CH, Chen C, Chen CF, Tu MC, Huang CW, Chang WN: Multi-parametric neuroimaging evaluation of cerebrotendinous xanthomatosis and its correlation with neuropsychological presentations. BMC Neurol, 2010; 10: 59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134).Seidel S, Kasprian G, Prayer D, Krssák M, Sycha T, Auff E: Visualisation of treatment response in a case of cerebrotendinous xanthomatosis. J Neurol Neurosurg Psychiatry, 2011; 82: 703-704 [DOI] [PubMed] [Google Scholar]
- 135).Soffer D, Benharroch D, Berginer V: The neuropathology of cerebrotendinous xanthomatosis revisited: a case report and review of the literature. Acta Neuropathol, 1995; 90: 213-220 [DOI] [PubMed] [Google Scholar]
- 136).Kato H, Koyabu S, Aoki S, Tamai T, Sugawa M, Watanabe M, Shiraishi T: An autopsy case of gallbladder cancer developing in a Japanese man with cerebrotendinous xanthomatosis: genetic analysis of the sterol 27-hydroxylase and p53 genes. Pathology, 2003; 35: 141-144 [PubMed] [Google Scholar]
- 137).Pilo de la Fuente B, Ruiz I, Lopez de Munain A, Jimenez-Escrig A: Cerebrotendinous xanthomatosis: neuropathological findings. J Neurol, 2008; 255: 839-842 [DOI] [PubMed] [Google Scholar]
- 138).Isenhardt K, Schmitt R, Nagel A, Drach L, Schlote W: Inherited cholesterol lipidosis: cerebrotendinous xanthomatosis (van Bogaert Scherer Epstein disease). A clinicopathological study. Clin Neuropathol, 2005; 24: 276-283 [PubMed] [Google Scholar]
- 139).Wallon D, Guyant-Maréchal L, Laquerrière A, Wevers RA, Martinaud O, Kluijtmans LA, Yntema HG, Saugier-Veber P, Hannequin D: Clinical imaging and neuropathological correlations in an unusual case of cerebrotendinous xanthomatosis. Clin Neuropathol, 2010; 29: 361-364 [DOI] [PubMed] [Google Scholar]
- 140).Tokimura Y, Kuriyama M, Arimura K, Fujiyama J, Osame M: Electrophysiological studies in cerebrotendinous xanthomatosis. J Neurol Neurosurg Psychiatry, 1992; 55: 52-55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141).Mondelli M, Rossi A, Scarpini C, Dotti MT, Federico A: Evoked potentials in cerebrotendinous xanthomatosis and effect induced by chenodeoxycholic acid. Arch Neurol, 1992; 49: 469-475 [DOI] [PubMed] [Google Scholar]
- 142).Berginer VM, Gross B, Morad K, Kfir N, Morkos S, Aaref S, Falik-Zaccai TC: Chronic diarrhea and juvenile cataracts: think cerebrotendinous xanthomatosis and treat. Pediatrics, 2009; 123: 143-147 [DOI] [PubMed] [Google Scholar]
- 143).Cruysberg JR, Wevers RA, Tolboom JJ: Juvenile cataract associated with chronic diarrhea in pediatric cerebrotendinous xanthomatosis. Am J Ophthalmol, 1991; 112: 606-607 [DOI] [PubMed] [Google Scholar]
- 144).Sundaram SS, Bove KE, Lovell MA, Sokol RJ: Mechanisms of disease: Inborn errors of bile acid synthesis. Nat Clin Pract Gastroenterol Hepatol, 2008; 5: 456-468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145).Clayton PT: Disorders of bile acid synthesis. J Inherit Metab Dis, 2011; 34: 593-604 [DOI] [PubMed] [Google Scholar]
- 146).Soutar AK, Naoumova RP: Mechanisms of disease: genetic causes of familial hypercholesterolemia. Nat Clin Pract Cardiovasc Med, 2007; 4: 214-225 [DOI] [PubMed] [Google Scholar]
- 147).Tada H, Nohara A, Inazu A, Sakuma N, Mabuchi H, Kawashiri MA: Sitosterolemia, Hypercholesterolemia, and Coronary Artery Disease. J Atheroscler Thromb, 2018; 25: 783-789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148).Salen G, Batta AK, Tint GS, Shefer S: Comparative effects of lovastatin and chenodeoxycholic acid on plasma cholestanol levels and abnormal bile acid metabolism in cerebrotendinous xanthomatosis. Metabolism, 1994; 43: 1018-1022 [DOI] [PubMed] [Google Scholar]
- 149).Verrips A, Wevers RA, Van Engelen BG, Keyser A, Wolthers BG, Barkhof F, Stalenhoef A, De Graaf R, Janssen-Zijlstra F, Van Spreeken A, Gabreëls FJ: Effect of simvastatin in addition to chenodeoxycholic acid in patients with cerebrotendinous xanthomatosis. Metabolism, 1999; 48: 233-238 [DOI] [PubMed] [Google Scholar]
- 150).Kuriyama M, Tokimura Y, Fujiyama J, Utatsu Y, Osame M: Treatment of cerebrotendinous xanthomatosis: effects of chenodeoxycholic acid, pravastatin, and combined use. J Neurol Sci, 1994; 125: 22-28 [DOI] [PubMed] [Google Scholar]
- 151).Huidekoper HH, Vaz FM, Verrips A, Bosch AM: Hepatotoxicity due to chenodeoxycholic acid supplementation in an infant with cerebrotendinous xanthomatosis: implications for treatment. Eur J Pediatr, 2016; 175: 143-146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152).Ellis E, Axelson M, Abrahamsson A, Eggertsen G, Thörne A, Nowak G, Ericzon BG, Björkhem I, Einarsson C: Feedback regulation of bile acid synthesis in primary human hepatocytes: evidence that CDCA is the strongest inhibitor. Hepatology, 2003; 38: 930-938 [DOI] [PubMed] [Google Scholar]
- 153).Setchell KD, Heubi JE: Defects in bile acid biosynthesis--diagnosis and treatment. J Pediatr Gastroenterol Nutr, 2006; 43: S17-22 [DOI] [PubMed] [Google Scholar]
- 154).Batta AK, Salen G, Tint GS: Hydrophilic 7 beta-hydroxy bile acids, lovastatin, and cholestyramine are ineffective in the treatment of cerebrotendinous xanthomatosis. Metabolism, 2004; 53: 556-562 [DOI] [PubMed] [Google Scholar]
- 155).Lewis B, Mitchell WD, Marenah CB, Cortese C, Reynolds EH, Shakir R: Cerebrotendinous xanthomatosis: biochemical response to inhibition of cholesterol synthesis. Br Med J (Clin Res Ed), 1983; 287: 21-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156).Luyckx E, Eyskens F, Simons A, Beckx K, Van West D, Dhar M: Long-term follow-up on the effect of combined therapy of bile acids and statins in the treatment of cerebrotendinous xanthomatosis: a case report. Clin Neurol Neurosurg, 2014; 118: 9-11 [DOI] [PubMed] [Google Scholar]
- 157).Mimura Y, Kuriyama M, Tokimura Y, Fujiyama J, Osame M, Takesako K, Tanaka N: Treatment of cerebrotendinous xanthomatosis with low-density lipoprotein (LDL)-apheresis. J Neurol Sci, 1993; 114: 227-230 [DOI] [PubMed] [Google Scholar]
- 158).Ito S, Kuwabara S, Sakakibara R, Oki T, Arai H, Oda S, Hattori T: Combined treatment with LDL-apheresis, chenodeoxycholic acid and HMG-CoA reductase inhibitor for cerebrotendinous xanthomatosis. J Neurol Sci, 2003; 216: 179-182 [DOI] [PubMed] [Google Scholar]
- 159).Dotti MT, Lütjohann D, von Bergmann K, Federico A: Normalisation of serum cholestanol concentration in a patient with cerebrotendinous xanthomatosis by combined treatment with chenodeoxycholic acid, simvastatin and LDL apheresis. Neurol Sci, 2004; 25: 185-191 [DOI] [PubMed] [Google Scholar]
- 160).Matysik S, Orsó E, Black A, Ahrens N, Schmitz G: Monitoring of 7α-hydroxy-4-cholesten-3-one during therapy of cerebrotendinous xanthomatosis: a case report. Chem Phys Lipids, 2011; 164: 530-534 [DOI] [PubMed] [Google Scholar]
- 161).Berginer VM, Salen G: LDL-apheresis cannot be recommended for treatment of cerebrotendinous xanthomatosis. J Neurol Sci, 1994; 121: 229-232 [DOI] [PubMed] [Google Scholar]
- 162).Selva-O’Callaghan A, Bardes I, Jacas C, Jubany L, Lorenzo-Bosquet C, Cuberas-Borrós G, Vilardell-Tarres M: SPECT imaging for brain improvement quantification in a patient with cerebrotendinous xanthomatosis. Clin Nucl Med, 2011; 36: 38-39 [DOI] [PubMed] [Google Scholar]
- 163).Amador MDM, Masingue M, Debs R, Lamari F, Perlbarg V, Roze E, Degos B, Mochel F: Treatment with chenodeoxycholic acid in cerebrotendinous xanthomatosis: clinical, neurophysiological, and quantitative brain structural outcomes. J Inherit Metab Dis, 2018; 41: 799-807 [DOI] [PubMed] [Google Scholar]
- 164).DeBarber AE, Kalfon L, Fedida A, Fleisher Sheffer V, Ben Haroush S, Chasnyk N, Shuster Biton E, Mandel H, Jeffries K, Shinwell ES, Falik-Zaccai TC: Newborn screening for cerebrotendinous xanthomatosis is the solution for early identification and treatment. J Lipid Res, 2018; 59: 2214-2222 [DOI] [PMC free article] [PubMed] [Google Scholar]