

Abstract
Identifying gene regulatory targets of nuclear proteins in tissues is a challenge. Here we describe intranuclear Cellular Indexing of Transcriptomes and Epitopes (inCITE-seq), a scalable method that measures multiplexed intranuclear protein levels and the transcriptome in parallel across thousands of nuclei, enabling joint analysis of transcription factor (TF) levels and gene expression in vivo. We apply inCITE-seq to characterize cell state-related changes upon pharmacological induction of neuronal activity in the mouse brain. Modeling gene expression as a linear combination of quantitative protein levels revealed genome-wide associations of each TF and recovered known gene targets. TF-associated genes were co-expressed as distinct modules that each reflected positive or negative TF levels, showing that our approach can disentangle relative putative contributions of TFs to gene expression and add interpretability to inferred gene networks. InCITE-seq can illuminate how combinations of nuclear proteins shape gene expression in native tissue contexts, with direct applications to solid or frozen tissues and clinical specimen.
Single nucleus RNA-seq is an essential tool for profiling the heterogeneity of solid tissues, particularly in those that are frozen or are challenging to dissociate, or for samples that require preserving cellular activity states by avoiding non-specific activation induced by enzymatic cellular dissociation1–7. The nucleus is also a key site of gene regulation by a wide array of proteins, whose presence and levels shape gene expression. For example, nuclear concentrations of transcription factors (TFs) are mechanistic determinants of gene expression by influencing the dynamics of TF target binding8,9. Simultaneously measuring quantitative protein levels and the transcriptome inside individual nuclei would enable integrating rich phenotypic and genomic information in tissues, and leverage the nuclear localization information of proteins. In addition, cellular proteins also provide a stable, time-integrated information over mRNA which are often rapidly degraded10–12.
Methods that use DNA-conjugated antibodies to measure surface proteins and RNA at single cell resolution, such as CITE-seq13 and REAP-seq14, with recent applications to cytoplasmic protein targets15–19, have been applied to circulating immune cells20,21, but these methods are less suited for non-immune cells and tissues where dissociation disrupts the integrity of cellular membranes. It remains a challenge to quantitatively measure protein levels in the nucleus with the transcriptome in individual nuclei, as DNA-conjugated antibodies are “sticky” inside the nucleus due to ubiquitous non-specific binding22.
Aberrations in nuclear levels of certain TFs can be hallmarks of disease and even be used to predict patient outcomes23–25. Simultaneously measuring nuclear proteins and RNA in single cells would enable relating nuclear proteins levels and newly transcribed RNA to reveal genes and pathways involved in cell state changes2,10–12,26–28, and how gene networks regulated by TFs vary across context and in disease29. Furthermore, nuclear localization of TFs can indicate a change in cellular activity states, e.g. in the case of TFs that shuttle in and out of the nucleus in response to external signals30,31. For example, the activity-regulated TF complexes NF-κB and AP-1, and their components p65 and c-Fos, transiently localize to the nucleus downstream of signal transduction, where they regulate diverse pathways related to inflammation, oncogenesis, apoptosis, cell proliferation, and synaptic remodeling30,32–36. Single nucleus profiling is best suited for studying these activity-regulated TFs because it minimize spurious expression of these pathways due to technical artifacts such as cellular dissociation4.
Current studies that monitor nuclear TF levels and gene expression typically rely on live cell imaging, in situ measurement of proteins and RNA in tissue by staining and hybridization, or cell sorting and profiling based on fluorescent reporters. Such studies have shown that nuclear localization can vary between individual cells stimulated together due to asynchrony in responses and dynamic shuttling37,38. However, methods relying on reporters or a handful of probes are limited in their ability to relate changes in protein localization to their genome-wide impacts on transcription.
Here, we report intranuclear CITE-seq (“inCITE-seq”), a method that enables multiplexed and quantitative intranuclear protein measurements using DNA-conjugated antibodies coupled with RNA-seq on a droplet-based profiling platform (Fig. 1a). To allow antibody diffusion across the nuclear membrane, nuclei are lightly fixed with formaldehyde and permeabilized, blocked under optimized conditions (see blocking buffer, Methods) to minimize non-specific binding of DNA-conjugated antibodies inside the nucleus. Nucleus hashing antibodies are simultaneously added to each sample for multiplexing39 (Methods). We then sequence antibody-stained nuclei with droplet-based snRNA-seq for simultaneous capture of antibody DNA tags and the transcriptome. We demonstrate the utility of inCITE-seq for profiling the response to environmental stimuli in cells and tissues, first in a HeLa cell line responding to cytokine stimulation, then in the mouse brain after pharmacological treatment.
Figure 1. InCITE-seq simultaneously measures intranuclear protein and RNA levels at single nucleus resolution.
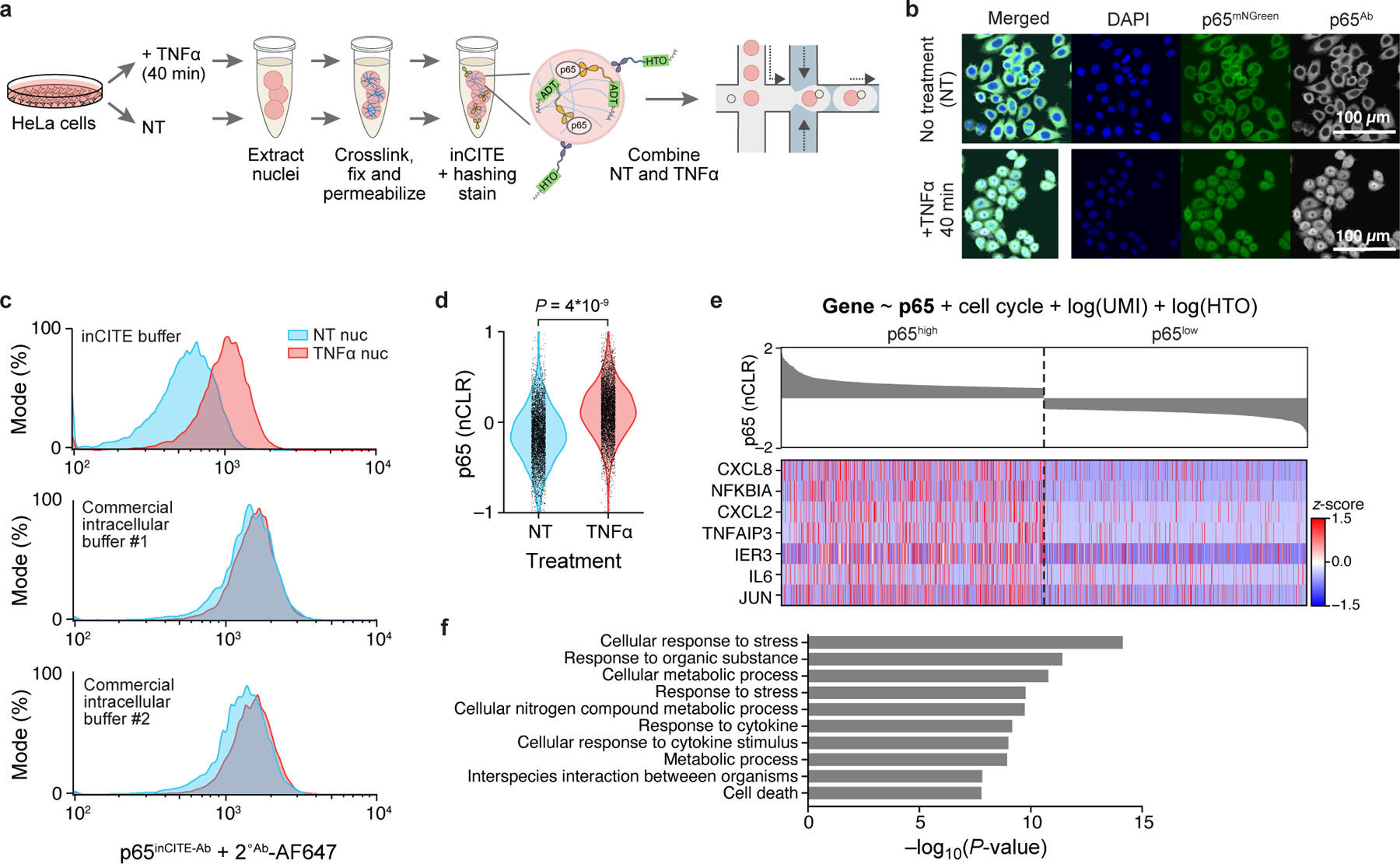
a. Overview of inCITE-seq for droplet-based profiling of nuclear proteins with nucleus hashing in HeLa cells. b. In situ fluorescent images of HeLa cells expressing a p65-mNeonGreen reporter (p65mNGreen) stained with anti-p65 antibody (p65Ab followed by Alexa Fluor 657 conjugated secondary), sampled without treatment (no treatment, “NT”; top) or 40 min after TNFα treatment (bottom); representative of 4 independently conducted experiments. Scale bar, 100μm. c. Flow cytometry of HeLa nuclei stained with p65inCITE-Ab followed by Alexa Fluor 647 secondary (x axis) sampled from NT (blue) or 40 min after TNFα treatment (red). Buffers, from top to bottom: optimized inCITE buffer with dextran sulfate, commercial buffer #1, commercial buffer #2 (Methods). d. Distribution of p65 levels (nCLRs) in NT (blue) and TNFα treated (red) nuclei profiled by inCITE-seq (P=4*10−9, two-sided Kolmogorov-Smirnov test). e. Expression (Z score, color bar) of the top 7 genes (rows) positively associated with p65 levels identified by a linear model (top, Methods) across nuclei (columns), visualized for the top decile (p65high) and bottom decile (p65low) of p65 nuclear protein levels by inCITE-seq (bar plot, top, nCLR). f. Top 10 Gene Ontology terms (y axis) significantly enriched (−log10(P-value), x axis, hypergeometric test) in 142 genes positively associated with p65 levels.
Results
InCITE-seq detects nuclear translocation of a TF induced by an extracellular signal
We first developed inCITE-seq to detect cell state changes in HeLa cells, defined as elevated nuclear levels of a TF that translocates into the nucleus in response to an external stimulus. We used a HeLa cell line expressing a p65-mNeonGreen reporter construct40,41, where p65 is localized to the cytoplasm in untreated cells and translocates to the nucleus upon TNFα stimulation (Fig. 1b; Methods). At peak nuclear translocation (~40 min post-stimulation41), total p65 levels in the whole cell are constant, with no discernible difference between untreated and TNFα stimulated cells in mNeonGreen signal measured by flow cytometry, but nuclear p65 levels are highly elevated (Extended Data Fig. 1a,b).
To resolve p65 TF levels in the nuclei of untreated vs TNFα stimulated cells, we first optimized intranuclear antibody staining conditions using unconjugated anti-p65 antibody (p65Ab), such that flow cytometry detected a clear signal separation, and in agreement with the mNeonGreen reporter signal (Fig. 1c; Extended Data Fig. 1d; Methods). We chose initial fixation and permeabilization parameters based on prior methods42, and validated that antibodies successfully diffused across the nuclear membrane by imaging smears of nuclei stained in suspensions (Extended Data Fig. 1c). However, the same staining conditions to the DNA-conjugated p65 antibody (p65inCITE-Ab) could not resolve p65 signal between NT and TNFα treated nuclei (Extended Data Fig. 1e), underscoring the challenges of non-specific binding of DNA-conjugated antibodies in the nucleus that occurs even in situ22. Adding dextran sulfate to the blocking and staining buffers improved signal separation14,22,43, clearly resolving NT and TNFα populations, even compared to two commercial intracellular staining buffers used for cytoplasmic targets (Fig. 1c; Methods).
We used inCITE-seq to profile 10,014 single nuclei from untreated and TNFα treated HeLa cells that were stained with p65inCITE-Ab (without sorting) and barcoded for multiplexing with nucleus hashing39 (Methods). Antibody levels estimated as counts of antibody-derived tags (ADTs)13 were normalized by the counts of the nucleus hashtag (HTO; hashtag oligos) to yield nuclear ADT (“nADT”) units, accounting for differences in poly-dT capture on beads (Extended Data Fig. 1f), that were then log-scaled to nuclear centered log ratios (“nCLRs”)13,44 (Methods). Sequencing-derived levels of nuclear p65 differed significantly across untreated and TNFα treated populations (Fig. 1d; P=4*10−9, two-sided Kolmogorov-Smirnov test), confirming that quantitative protein detection via inCITE-seq can distinguish altered cell state due to treatment. The quality of the associated snRNA-seq profiles was comparable to that of snRNA-seq data from human-derived HEK cell line45, as reflected by the number of unique transcripts (UMIs) and genes (Extended Data Fig. 1g; median number of UMIs and genes: 2,655 and 1,158 for inCITE-seq; 1,159 and 812 for HEK snRNA-seq).
Relating genome-wide expression to p65 protein levels
To identify genes whose RNA expression was associated with p65, we used a linear model to fit each gene’s expression as a function of continuous p65 levels, while controlling for cell cycle and technical variates (UMIs and HTOs), which identified 142 genes positively associated with p65 (FDR 1%; Methods). These genes included well-known NF-κB targets, such as CXCL8, NFKBIA, and TNFAIP3 (Fig. 1e), and were enriched for pathways such as cytokine response (Fig. 1f, P<3*10−9, two-sided hypergeometric test, Methods).
Notably, the levels of p65 protein and its encoding transcript RELA were not well-correlated (Pearson r2=0.0008, P=0.004; Extended Data Fig. 1h). This was expected at our time scale of stimulation, as RELA expression peaks at ~4 hrs after induction of the NF-kB pathway by LPS but are largely unchanged at 40 min post induction, in contrast to other p65 target genes such as NFKBIA11 (Extended Data Fig. 1i). RELA and p65 were also uncorrelated in untreated cells at steady state (NT: r2=0.001, P=0.032) which confirmed known differences in mRNA and protein kinetics for this gene. At baseline, RELA transcripts are reported to be produced at 12 copies per min and degraded at 102.4 copies per min10, while p65 is translated at 0.9 proteins per mRNA per min and degraded at 4*10−4 per min11. Our own observation supports prior works showing fast mRNA degradation but slow protein degradation kinetics, which further underscores the importance of obtaining protein measurements. Taken together, we show that inCITE-seq accurately quantifies nuclear protein and RNA levels that can be integrated to identify putative targets of a TF.
InCITE-seq profiling of the mouse brain after in vivo induction of seizure
Next, we turned to an in vivo setting and applied inCITE-seq to characterize the mouse hippocampus during early response to seizure, which involves neuroinflammation and oxidative stress affecting multiple cell types and pathways46. Seizures were induced by treatment with kainic acid (KA), a glutamatergic agonist used in models of temporal lobe epilepsy47. As the complexes NF-kB and AP-1 are involved in neuroinflammation, synaptic remodeling, and signal transduction of glutamate receptors48–50, we characterized how their components p65 and c-Fos are linked to gene expression changes in early response to seizure. In addition, we added two nuclear cell type markers, the pan-neuronal marker and regulator of alternative splicing NeuN51, and the TF and microglial lineage marker PU.152. Altogether, we used inCITE-seq to profile single nuclei from the hippocampus two hours after KA treatment, with multiplexed measurements of the proteins p65, c-Fos, NeuN, and PU.1 (Fig. 2a; Methods).
Figure 2. In vivo application of inCITE-seq shows cell type-specific protein expression in the mouse hippocampus.
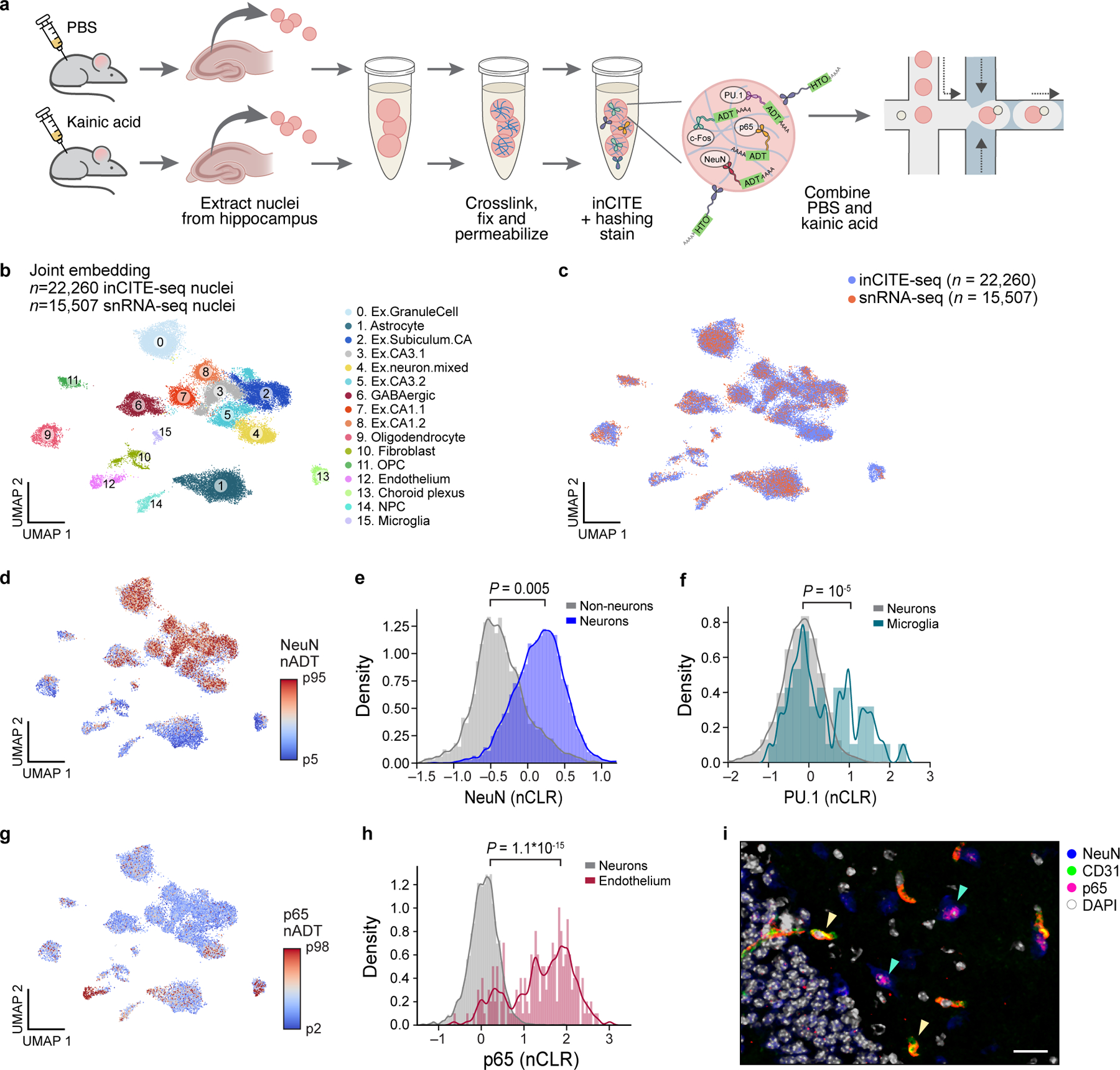
a. InCITE-seq of the mouse hippocampus after kainic acid or PBS (control) treatment, with nucleus hashing. b. Cell types from the adult mouse hippocampus identified by joint embedding of inCITE-seq and snRNA-seq. UMAP embedding of single nucleus RNA profiles from two batches of inCITE-seq (n=22,260) and two snRNA-seq experiments (this study and Habib et al.54, n=15,507) of the mouse hippocampus, after regressing out treatment and batch (Methods), colored by cluster and annotated post hoc (color legend). “Ex”: excitatory neurons clusters. c. Integration of inCITE-seq and snRNA-seq profiles. UMAP embedding as in (b), colored by assay type (inCITE-seq, blue; snRNA-seq, pink). d,g. UMAP embeddings as in (b), but showing only inCITE-seq nuclei profiles colored by protein levels (nADT) for NeuN (d, 5th to 95th percentile) and p65 (g, color scale from 2th to 98th percentile). e,f,h. Distribution of protein levels (nCLR, x axis) for NeuN in neuronal (blue) and non-neuronal (gray) nuclei (e; P=0.005, two-sided KS test), PU.1 in microglial (turquoise) and neuronal (gray) nuclei (f; P=10−5, two-sided KS test), and p65 in endothelial (fuchsia) and neuronal (gray) nuclei (h; P=1.1*10−15, two-sided KS test), from one batch. Curve: kernel density estimate. i. Immunofluorescence stain of the hippocampus with endothelial marker CD31 (green), NeuN (blue), p65 (pink), and DAPI (white); representative of 3 independently conducted experiments. Yellow arrowheads: co-localization of CD31 and p65. Green arrowheads: lowly expressed p65 in neurons. Scale bar, 50μm.
Antibody validation and optimization for inCITE-seq in mouse brain
InCITE-conjugated antibodies were first validated with flow cytometry on stained nuclei that were extracted from frozen mouse hippocampus. Cell type markers NeuN and PU.1 labeled subpopulations at the expected proportions (58.3% and 2.9% of all nuclei, respectively), with PU.1 specific to microglia (PU.1high in CD11bhighCX3CR1high populations but not in CD4high cells; Extended Data Fig. 2a–c). Levels of p65 were bimodal, and c-Foshigh nuclei subsets were elevated in KA treated samples as anticipated from neuronal activity, from 0.21% to 48.7% (Extended Data Fig. 2d–f). Antibody signals varied across a wide range of concentrations, underscoring the importance of choosing an appropriate concentration regime53 (Extended Data Fig. 3).
Antibodies suitable for inCITE-seq should detect epitopes in frozen tissue after minimal fixation, we therefore expect antibodies that are optimized for flow cytometry, immunocytochemistry, or even immunoprecipitation to be compatible with inCITE-seq, while those optimized for heavily fixed tissues (e.g., formalin-fixed paraffin-embedded (FFPE)) may not be suitable. As an example, we compared two different antibodies targeting each of NeuN, PU.1, and c-Fos in tissues that were frozen immediately vs. after overnight fixation in 4% PFA revealed a stark contrast in epitope detection (Extended Data Fig. 4); for example, one version of a c-Fos antibody was unable to detect epitopes in frozen tissue, while another version used for inCITE-seq exhibited clean epitope detection in frozen but not in overnight fixed tissue. To validate antibodies for inCITE-seq, we recommend conducting flow cytometry of antibody stains on nuclei isolated from frozen tissues of interest as well as in situ immunofluorescence in frozen sections (post-fixed) to help determine the cell types and features that express protein targets (Extended Data Figs. 3,4).
RNA profiles from inCITE-seq reveals key cell subsets of the mouse hippocampus
Profiling 24,444 nuclei from control (PBS) and KA treated mice with inCITE-seq yielded RNA profiles that captured all major cell types of the hippocampus. Compared to snRNA-seq of the mouse hippocampus from a matched experiment and from another study using the same nuclei extraction protocol54, RNA profiles from inCITE-seq were reduced in quality, with a 6.2- and 4.8- fold reduction in the median UMI and gene counts, respectively (Extended Data Fig. 5a). Despite the loss in complexity, unsupervised clustering using snRNA-seq profiles from inCITE-seq alone still discerned major cell types of the hippocampus by post hoc annotation with known cell type markers54,55 (after addressing ambient RNA56, batch correction, and regressing out treatment; Extended Data Fig. 5b,c; Methods).
Cell cluster separation was substantially improved by jointly embedding the single nucleus RNA profiles from inCITE-seq and snRNA-seq, including data from a published study54. Unsupervised clustering with jointly variable genes of 37,767 high quality nuclei (22,260 inCITE-seq and 15,507 snRNA-seq nuclei) showed 15 well-delineated clusters, each with contributions from both assays, with robust mixing across batches and treatment, and well-annotated post hoc using known cell type markers (Fig. 2b,c; Extended Data Fig. 5d–j; Methods).
Protein levels match cell type- and condition-specific expression in RNA-based clusters
Nuclear protein levels measured by inCITE-seq differed across RNA-defined cell types as expected. As before, protein counts were normalized by nucleus hashtag oligos (HTOs) counts, then scaled to centered log ratios within each batch (i.e. using batch-specific geometric means) to account for systematic batch differences (Extended Data Fig. 6). NeuN levels were elevated in neuronal clusters as expected (Fig. 2d,e; P=0.005, two-sided KS test); low NeuN in subpopulations of other clusters may reflect background signal (such as ambient RNA or doublets that were missed in filtering), as NeuN is highly expressed in this tissue. PU.1, the microglial marker and lineage-specifying TF, was significantly higher in microglia compared to neurons (Fig. 2f; P=10−5, two-sided KS test). Expression of p65 was enriched in endothelial nuclei (Fig. 2g,h; P=1.1*10−15, two-sided KS test), which was confirmed by immunohistochemistry with the endothelial marker CD31 (Fig. 2i, CD31+p65+, yellow arrowheads), and also expressed at lower levels in neurons (Fig. 2i, NeuN+p65+, green arrowheads).
KA treatment elevated the level of c-Fos, with variations across neuronal subtypes. InCITE-seq derived nuclear c-Fos levels showed widespread expression across multiple cell types (Fig. 3a), with significant upregulation in neurons of KA-treated mice compared to PBS (Fig. 3b; P=10−15, two-sided KS test). Subsets of neurons differed in c-Fos levels, such that nuclei from CA neurons had lower levels than from granule cells (GC) of the dentate gyrus (DG) (Fig. 3c; P=1.7*10−7, two-sided KS test). In contrast, p65 did not change after KA treatment at this time scale (Fig. 3d), as expected49. These patterns were confirmed by immunofluorescence, where c-Fos is expressed in multiple neuronal types, including DG granule cells, CA neurons, and somatostatin (SST+) interneurons (IN), with higher c-Fos intensity in granule cells compared to CA neurons (Fig. 3e,f), and no change in p65 due to treatment (Fig. 3g). Overall, inCITE-seq quantitatively measured nuclear protein levels that reflected diverse levels of activity-regulated TFs across cell types and treatment.
Figure 3. InCITE-seq measures changes in nuclear TF levels after stimulation of the mouse hippocampus.

a. UMAP embedding of inCITE-seq nuclei (as in Fig. 2b) colored by c-Fos protein levels (nADT, color scale from 5th to 95th percentile). Distribution of c-Fos (b,c) or p65 (d) protein levels (nCLR, x axis) shown as a kernel density estimate in neurons of KA vs. PBS treated mice (b, P=10−15, two-sided KS test), in granule cells (GC) vs. cornu ammonis (CA) neurons in KA treated mice (c, P=1.7*10−7, two-sided KS test), or in neurons of KA vs. PBS treated mice (d, not significant; P=0.15 two-sided KS test). e,f,g. Immunofluorescence stain of the hippocampus after PBS (gray border) or KA (green border) treatment; representative of 3 independent experiments. Major hippocampal features denoted: dentate gyrus (DG), cornu ammonis (CA). e. Stain of c-Fos (red), NeuN (green), and DAPI (blue). Left: scale bar, 600μm. Right: close-up of the DG (dashed box) shows heterogeneity in c-Fos intensity; scale bar, 100μm. f. Stain of SST (red), c-Fos (green), and DAPI (blue). Left: scale bar, 100μm. Right: close-up of the DG (dashed area box); scale bar, 30μm. g. Immunofluorescence stains of p65 (red), NeuN (green), and DAPI (blue), PBS or KA treatment. Left: all stains. Right: p65 stain. Scale bar, 100μm. h. Distribution of mRNA levels (Z score of log-normalized counts, y axis) in nuclei with high or low levels (defined in Extended Data Fig. 6) of the encoded protein (x axis) under PBS (gray) or KA (green) treatment. Boxplot: centre line indicates median, box bounds represent first and third quartiles, whiskers span from each quartile to the minimum or the maximum (1.5 interquartile range below 25% or above 75% quartiles). Dots: nuclei with non-zero mRNA levels measured across n=2 biologically independent samples, with 1,696 nuclei, 214 nuclei, and 653 nuclei shown for Fos, Rela, and Rbfox3, respectively. Significance, from bottom-left to top-right: P=2*10−6, P=9*10−5, P=4*10−6, P=9.7*10−3, two-sided Mann-Whitney test. NS – not significant.
Relating protein and mRNA levels of inCITE target genes
Relating the mRNA and protein levels of inCITE target genes revealed a wide range of regulatory dynamics. We compared transcript and protein levels across nuclei with “high” or “low” protein levels that were categorized using batch-specific thresholds (Extended Data Fig. 6a-d), and assessed how treatment impacted their relationship (Fig. 3h). Transcript levels of the activity-regulated gene Fos were elevated in populations with high protein levels (c-Foshigh) compared to c-Foslow (P=9*10−5, two-sided Mann-Whitney test), but only under PBS treatment, whereas for Fos mRNA was highly expressed regardless of its protein levels after KA treatment. This is consistent with a model where c-Fos positively regulates its own transcript, whose expression is already saturated after treatment at this time scale57 and when the downstream effect of mRNA induction on protein levels is not yet observable. In contrast, NeuN and its encoding transcript Rbfox3 were inversely associated (P<0.01, two-sided MW test), although unspliced, intron-retaining pre-mRNA levels of Rbfox3 (estimated by Velocyto27, Methods) were upregulated by KA treatment (Extended Data Fig. 7a). This is consistent with other RNA-binding proteins that negatively regulate their own expression via intron retention58. Rela mRNA levels did not differ across p65 protein levels (Fig. 3h, P=1, two-sided MW test), and Spi1 transcripts (encoding PU.1) were not detected, underscoring the importance of protein measurements as a complement to RNA, particularly for TFs whose RNAs are often lowly expressed59.
Modeling genome-wide association with each protein recovers known TF targets
We devised an approach to infer the putative genome-wide impact of each TF on gene expression based on nuclear protein levels. As concentrations of TFs shape gene expression in the regulatory context of the nucleus8,9, we modeled gene expression as a function of protein levels. To first identify global impacts of TFs, we modeled each gene’s RNA as a linear combination of the four proteins – c-Fos, p65, PU.1, NeuN – after regressing out the contributions of cell type (cluster), treatment, and their interaction, to account for collinearity (especially between KA treatment and c-Fos; Methods). Genes significantly associated with the three TFs were interpreted as putative TF-regulated genes, with the effect size estimated by their coefficients. TF-associated genes comprised of known targets and pathways (Fig. 4a–c; Extended Data Fig. 7b), with PU.1 associated genes including direct regulatory targets and known microglia markers, e.g. Trem2, Tyrobp and C1qa60, and c-Fos associated genes including known targets Npas4, Nr4a1, Homer1, and its own transcript Fos, that reflect activity-induced upregulation35. Although our model also identified NeuN-associated genes (Extended Data Fig. 7c), we reasoned that these genes could reflect direct changes in transcript levels via splicing or indirect effects of a generally transcriptionally active state of the nucleus, as NeuN is associated with decondensed chromatin and enlarged nuclei61.
Figure 4. Inferring TF effects on gene and module expression using joint protein and transcriptome measurements.
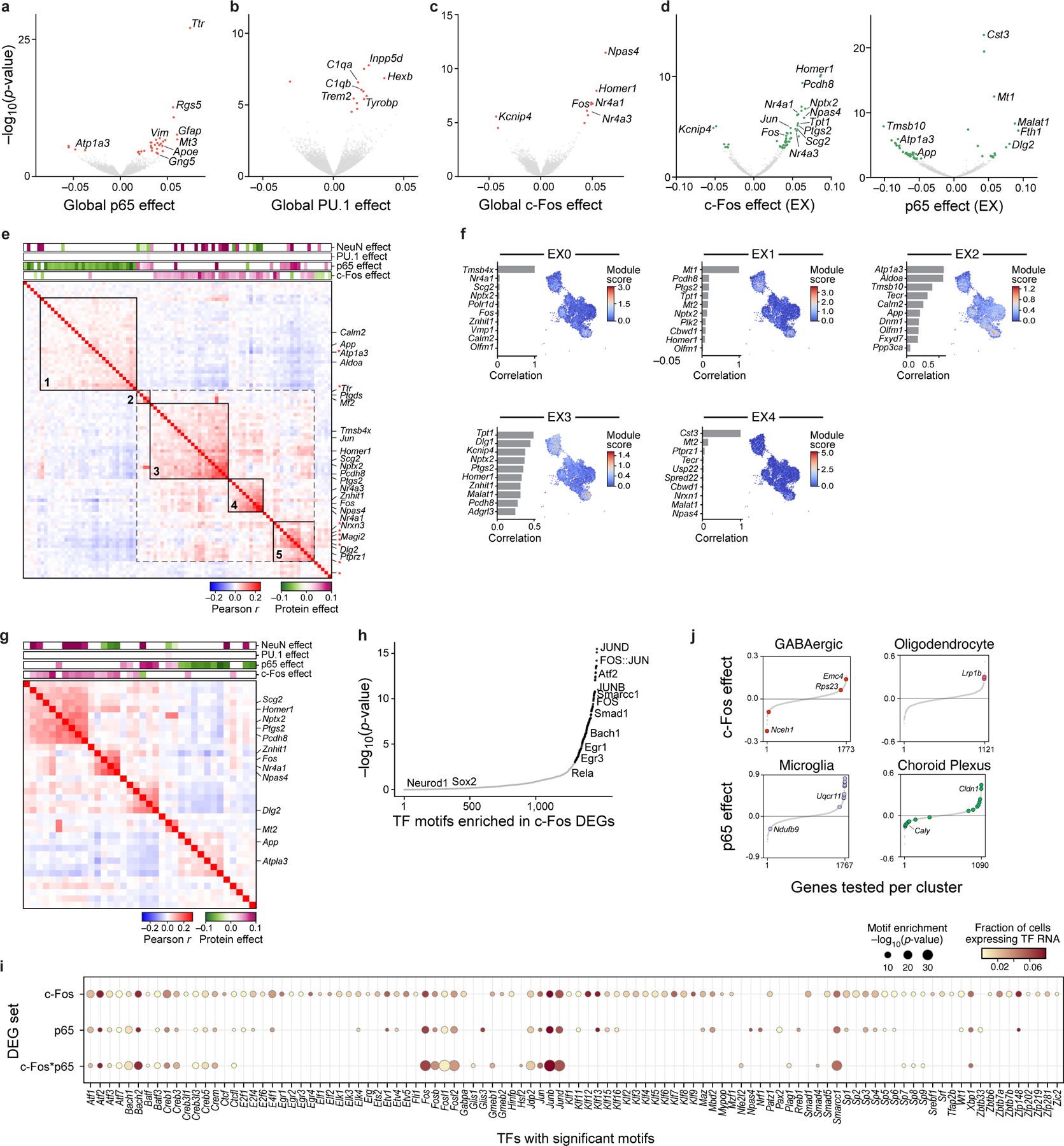
a-c. Global association of TFs to genes. Significance (y axis, −log10(P-value)) and effect size (x axis) for genes (dots) associated with p65 (a), PU.1 (b), and c-Fos (c) protein levels across all nuclei, by a model of gene expression as a linear combination of TFs/proteins. Colored dots: Benjamini-Hochberg FDR<5%; select genes labeled. d. Genes associated with each TF within excitatory neurons (EX). Volcano plot axes for c-Fos (left) or p65 (right) are the same as in (a-c). Colored dots: Benjamini-Hochberg FDR<5%; select genes labeled. e. Pearson correlation coefficient (red/blue colorbar) of pairwise gene expression across excitatory neurons (rows and columns), for genes that are positively (purple) or negatively (green) associated with c-Fos or p65, ordered by hierarchical clustering. Top bars: Effect size of each protein. Black boxes: co-expression modules. Red asterisk: DEGs associated with c-Fos*p65 in the interaction model (see Extended Data Fig. 8b). f. NMF programs of excitatory neurons. Right: UMAP embedding of the excitatory neuron subset (as in Fig. 2b), colored by the NMF program score. Left: Top 10 program genes (y axis) and their Pearson correlation with program scores (x axis). g. Pearson correlation coefficient (red/blue colorbar) of pairwise gene expression across EX neurons (rows and columns) using the top 10 genes of each program, ordered by hierarchical clustering. Top bars (purple/green): significant effect sizes of each protein from the linear model. h. Significance (−log10(P-value), y axis) and rank order (x axis) of TF motifs (dots) in enhancers of c-Fos DEGs in excitatory neurons. Black: significant motifs (P<10−3, hypergeometric test); Gray: not significant. i. Enriched TF motifs (columns; dot size, −log10(P-value)) and their corresponding RNA expression in EX neurons (dot color), identified in the enhancers of DEGs associated with each of the following (rows): c-Fos (additive model), p65 (additive model), or c-Fos*p65 (interaction model). j. Cell type-specific DEGs of c-Fos or p65 after KA treatment. Effect size (y axis) of c-Fos (top) or p65 (bottom), sorted by rank order (x axis), in select cell types (top). Color: significant genes (Benjamini-Hochberg FDR<5%).
TF-associated genes are co-expressed as distinct modules within excitatory neurons
Next, we probed how the expression profiles of TF-associated genes relate to each other by their co-expression patterns. Focusing on the broad type of excitatory neurons (EX neurons), we again modeled each gene as a linear combination of the four proteins after regressing out treatment, within only EX neurons (Methods; Fig. 4d). We then clustered the differentially expressed genes (DEGs) associated with c-Fos and p65 based on the correlation of their co-expression across all nuclei of EX neurons. Genes associated with c-Fos or p65 were co-expressed as distinct modules, such that each module reflected a unique set of inferred TF effects (Fig. 4e). Specifically, modules 3 and 4 corresponded to a positive c-Fos effect, modules 1 and 2 to a negative and positive p65 effect, respectively, and module 5 to a mixture of both c-Fos and p65 effects.
We hypothesized that the widespread weak correlations between c-Fos and p65- associated genes (Fig. 4e, dotted gray box) may reflect interactions between these two TFs, whose complexes AP-1 and NF-κB are known to interact synergistically62,63. To test for protein-protein interactions, we reimplemented our model with interaction terms (c-Fos*p65, c-Fos*NeuN, and p65*NeuN), which uncovered 56 genes associated with c-Fos*p65 that were also co-expressed as a module (Extended Data Fig. 8a,b; Methods). Of these 56 genes, only 11 were associated with c-Fos or p65 alone in the previous model (genes marked by a red asterisk in Fig. 4e), suggesting that these genes may uniquely reflect direct or indirect regulation that require both c-Fos and p65 to be highly expressed within a cell. Although we caution against overinterpretation of the specific genes due to the limited size of our data and subsequently underpowered analyses, we demonstrate that as a proof-of-concept, we can use protein combination measurements to infer gene modules reflecting TF combinations vs individual TFs.
The association between gene modules and inferred TF effect types prompted us to ask whether gene programs that are normally identified from expression alone64 align with protein effects. Using non-negative matrix factorization (NMF) on RNA profiles alone, we identified 5 gene programs within excitatory neurons (Fig. 4f; Methods), some of which were upregulated in response to treatment (Extended Data Fig. 8c). Co-expression patterns of the top 10 genes of each program revealed that each program also coincided with the different types of TF effects (Fig. 4g). Altogether, our approach allows quantifying the associations between TFs and gene expression modules or pathways, and even disentangling contributions from TF combinations that can increase the interpretability of gene expression programs.
To assess whether TFs could play a direct or indirect role in regulating their associated DEGs, and to predict other TFs that may be involved in co-regulation, we identified cis regulatory TF motifs enriched in the enhancer regions of c-Fos- and p65- associated genes. Using enhancers defined by differentially accessible regions (DARs) previously profiled in the mouse hippocampus of saline- and KA- treated mice65, we identified TF motifs found significantly in each gene set (c-Fos and p65 DEGs from the additive model; c-Fos*p65 DEGs from the interaction model; Methods). The observed TF motifs reflected components of the AP-1 complex (Fos, JunB, and JunD), activity-regulated TFs Egr1 and Egr3, and Atf2 which is phosphorylated by c-Jun in KA-induced seizure models66 (Fig. 4h; Extended Data Fig. 9a). Additionally, the enrichment of Smarcc1, part of the chromatin remodeling BAF (SWI/SNF) complex, among genes associated with c-Fos was consistent with the key role of AP-1 components in recruiting the BAF complex for chromatin remodeling67. The RNA transcripts of these TFs were lowly expressed in our data (Fig. 4i). As the widespread presence of AP-1 motifs likely reflected the role of AP-1 itself in chromatin remodeling following KA treatment65,67, we additionally examined TF motifs enriched in DEGs compared to other enhancers under KA treatment (Extended Data Fig. 9b,c). Notably, motifs of NF-κB components (Rel, Rela, Relb, Nfkb1, Nfkb2) were not significant. This suggests that TF-associated genes in our analysis may also reflect indirect effects of each TF, or that direct effects occur at a different time scale in the case of p65, or depend on the direction of the TF effect (i.e. downregulated vs. upregulated).
Inferred TF impact on genes depends on treatment context and cell type
Finally, we analyzed whether TF impact on individual gene expression depended on the treatment context of the mouse hippocampus, at a global scale and within individual cell types. To assess the global treatment impact, we modeled RNA levels as a linear combination of the four proteins separately within each treatment (PBS or KA) and compared their effects on each gene across treatment (Methods; Extended Data Fig. 9d). The impact of p65 on gene expression was largely consistent across PBS and KA treatment, as reflected by correlated effect sizes (R2=0.03, P=6*10−82). In contrast, c-Fos effects varied by treatment (R2=0.0001, P=0.21). Genes specifically associated with c-Fos only under KA treatment included Ptgs2 (encoding cyclooxygenase-2, COX-2), a responder to oxidative stress after traumatic brain injury68. TF effects under KA treatment were also cell type-specific, as expected33,36 (Methods; Fig. 4j). Altogether, our results demonstrate that the direct or indirect regulatory impact of TFs on individual genes can depend on cell type and environmental context.
Discussion
InCITE-seq reliably measures quantitative protein and RNA levels in individual nuclei, offering a way to better understand the relationship between regulatory proteins and their target genes during dynamic response, in the native context of tissues. Nuclear protein levels can be utilized as an interpretable and mechanistic link bridging regulatory proteins and their genome-wide effects, enabling cell type-specific studies of signaling pathways in complex tissues in vivo. While numerous multimodal methods measure intracellular protein targets15,16,19, proteins in the cytoplasm carry different information than those in the nucleus, such as in the case of regulatory proteins and translocating TFs; buffers used for intracellular and cytoplasmic targets may result in high background for nuclear targets (Fig. 1c). Furthermore, nucleus-based multimodal profiling surmounts key technical challenges to enable characterization of cells from solid tissues that are either hard to dissociate or archived in frozen form, especially clinical specimens from human disease studies such as cancer and neurodegeneration. InCITE-seq is particularly well-suited for studying proteins in pathways that are affected by cellular dissociation protocols, e.g. the activity-regulated TF c-Fos.
Antibodies compatible with inCITE-seq should recognize epitopes in frozen tissues after light fixation. We recommend validating antibodies by flow cytometry and in situ immunofluorescence on frozen tissues (Extended Data Figs. 3,4). Jointly embedding RNA profiles from inCITE-seq and snRNA-seq should help in discerning clusters despite the currently lower complexity of inCITE-seq data. In the future, computational methods that tackle ambient RNA and batch effects through principled modeling, such as cellbender56 and totalVI69 could be adapted to denoise ambient nuclear protein expression; transfer learning from inCITE-seq to snRNA-seq may be used to predict protein levels in snRNA-seq datasets collected without protein measurements.
Future studies can also combine inCITE-seq with metabolic labeling70–72, antibodies targeting phosphorylated forms of TFs, joint RNA and chromatin accessibility profiles73 coupled with spatial inference74, as well as samples collected across time, for refined understanding of gene regulation in tissues. Our intranuclear staining conditions could also be readily adapted to enrich for snRNA-seq of subpopulations based on marker proteins75. By measuring multiple TFs simultaneously, inCITE-seq opens the way to decipher complex phenotypes and regulatory mechanisms in development when TF combinations are key to define cell type diversity. Multiplexed profiling of signaling proteins across several pathways will enable deciphering changes in activity states in vivo, which could be used to recover the impact of ligands acting on multiple receptors across cell types in tissues, where GWAS highlights the role of variants in regulatory regions, by monitoring protein targets alongside gene expression changes.
METHODS
Mice.
C57BL/6J (Jax 000664) mice were purchased from The Jackson Laboratory and bred in-house. Male mice were used at ~8 weeks of age. All mice were maintained under SPF conditions on a 12-h light–dark cycle, at ambient temperature 21.5 ± 1°C and relative humidity between 30% and 70%, and provided food and water ad libitum. All mouse experiments were approved by, and performed in accordance with, the Institutional Animal Care and Use Committee guidelines at Weill Cornell Medicine.
Cell culture.
HeLa-TetR-Cas9 cells expressing a p65-mNeonGreen reporter construct (gift from Jonathan Schmid-Burgk, original source: Iain Cheeseman, MIT40,41) were cultured at 37°C and 5% CO2 in DMEM with high glucose, pyruvate, GlutaMax™ (ThermoFisher Scientific 10569010), heat-inactivated fetal bovine serum (ThermoFisher Scientific 16000044) and 100 U/mL penicillin-streptomycin (ThermoFisher Scientific 15140163). For immunohistochemistry, cells were seeded on poly-L-lysine treated #1.5 glass coverslips (Thomas Scientific 1217N81) in 6-well plates at a density of ~5×104 cells/mL, 24 hours prior to TNFα stimulation. For inCITE-seq, HeLa cells were seeded in 10cm Petri dishes at least 24 hours prior to TNFα stimulation, and were assayed at 70–80% confluence.
Stimulation of HeLa cells with TNFα.
HeLa cells were stimulated by adding media containing TNFα at final concentration of 30 ng/mL and incubated at 37°C for 40 min to induce translocation of p65 into the nucleus. TNFα containing media was aspirate, and cells were washed with 1x PBS. We then added 2mL EZ lysis buffer (Sigma Aldrich N3408), scraped cells, and moved the lysates into a 15mL falcon tube for nucleus extraction.
Kainic acid injection of mice.
8-week-old male mice were acclimated in the procedure room for 1h prior, then injected i.p. with either PBS or 20mg/kg kainic acid (KA; Sigma K0250) dissolved in PBS. All animals were observed continuously for two hours and scored using a modified Racine scale (stages 0–6)76; any mice not reaching at least stage 1 (immobility and rigidity) by 30 minutes post-KA were given an additional injection of 10mg/kg KA to facilitate seizure activity. After 2 hours, mice were euthanized by CO2, perfused through the left ventricle with 20mL of PBS, and the entire brain removed. The hippocampus was then dissected on ice before freezing on dry ice.
Immunohistochemistry of HeLa cells.
Coverslips (#1.5, 18mm, Thomas Scientific 1217N81) treated with poly-L-lysine were seeded with HeLa cells in 6-well plates and stimulated with TNFα as described. Wells were washed with PBS, fixed with 4% PFA at RT for 15 min, then washed 3x with PBS. For subsequent steps, coverslips placed with cells facing down on a sheet of Parafilm, such solution volumes (100μL) were sandwiched between the coverslip and Parafilm. Cells were blocked and permeabilized at RT for 30 min (1X PBS, 5% normal goat serum, 0.3% Triton X-100), incubated with 1:200 p65Ab (BioLegend cat #622601) in antibody solution (5% BSA, 0.02% Tween 20 in 1X PBS) for 1 hr at RT, and washed 3x with PBST (1X PBS, 0.02% Tween 20). Anti-rabbit Alexa Fluor 647 secondary (Invitrogen A27040) was added at 1:1000 in PBST for 1 hr in dark at RT, then washed 4x with PBST, with the final wash containing 1:1000 DAPI. Coverslips were mounted onto SuperFrost slides (Fisher Scientific 22-037-246) with antifade (ThermoFisher Scientific S36937) and sealed with nail polish. Slides were stored at 4°C until imaging.
Immunohistochemistry of mouse hippocampus.
Mice were euthanized 2 hours after PBS or KA injection and perfused through the left ventricle with 20mL of PBS. The entire brain tissue (CNS) was removed, then immersed in OCT and quickly frozen on dry ice. Tissue was sectioned at 10 μm using a cryotome and collected on slides, which were frozen on dry ice and stored at −30°C until further use. Slides were removed from storage and fixed with 4% PFA for 10 min at RT, washed in PBS, then blocked (PBS containing 0.1% Triton-X 100, 5% normal donkey serum and 5% normal goat serum) for 30 min at RT. Sections were incubated with the following primary antibodies at the indicated dilution in blocking buffer overnight at 4°C: NeuN (BioLegend 1B7, cat #834502), p65 (BioLegend Poly6226, cat #622601), c-Fos (BioLegend Poly6414, cat #641401), CD31 (eBioscience 390, cat #14-0311-82). Sections were then washed in PBS 3x before incubating in secondary antibodies (Jackson Immunoresearch: donkey anti-rabbit AF647 #711-605-152, donkey anti-rabbit AF594 #711-585-152, donkey anti-mouse AF488 #715-545-150, donkey anti-rat AF647 #712-605-153) at 1:500 in blocking buffer for 1 hr at RT. Sections were washed 1x with PBS, 1x with PBS containing DAPI, and a final time in PBS before mounting (Prolong Diamond Antifade, ThermoFisher). Slides covered with coverslip were dried overnight, sealed with clear nail polish, and imaged.
For immunohistochemistry of mouse hippocampus sections from fixed brains, mice were perfused with 20mL of PBS followed by 20mL of 4% PFA. The entire brain was removed, and brain tissue was incubated in 4% PFA overnight before washing 3x in 10mL of PBS. Brain tissue was dehydrated in 30% sucrose in PBS overnight, then embedded and frozen in OCT. Sections were processed in the same manner as described above, but without the 10 min post-cut fixation in 4% PFA. Antibodies used for fixed vs. frozen brain comparison in Extended Data Fig. 4: c-Fos (Abcam ab190289), NeuN (Abcam ab190565), PU.1 (Cell Signaling Technology #2258).
Microscopy.
HeLa cells and mouse hippocampal sections were imaged on an Olympus Fluoview FV1200 biological confocal scanning microscope at 20X or 40X (Olympus, LUCPLFLN) with sequential laser emission and Kalman filtering. Images were processed with ImageJ.
FACS analysis of PU.1 stain in microglia suspensions.
Mice were euthanized with CO2 and perfused through the left ventricle with 20mL of PBS. Whole brain was removed and placed in 2.5 mL of digestion buffer (PBS, 5% FCS, 1mM HEPES) before finely chopping. 400U of Collagenase D (Roche) was added to the mixture, then incubated at 37°C for 30 min before adding 50 μL of 0.5M EDTA followed by a 5 min incubation. Digested tissue was mashed through a 40μm cell strainer, pelleted at 700g in a swinging bucket centrifuge, then resuspended in 10mL of 38% isotonic Percoll, and centrifuged at 2000 RPM for 30 min with no brake. The myelin debris layer was removed by aspiration and the pellet washed with PBS. Cells were then blocked with 1:100 FcX (BioLegend 156604) before a 15 min incubation with the following antibodies at a 1:200 dilution in PBS: CD45.2-FITC (eBioscience 104 cat #11-0454-82), CD4-BUV395 (BD GK1.5 cat #563790), CD11b-BV421 (BioLegend M1/70 cat #101235, CX3CR1-APC (BioLegend SA011F11 cat #149008). Cells were washed in PBS, then fixed and permeabilized using the Foxp3/Transcription Factor Staining Buffer Set (eBioscience) before staining with PU.1-PE (BioLegend 7C2C34 cat #681307) or rat IgG2a-PE isotype (BioLegend RTK2758, cat #400507) for 30 minutes at room temperature. Cells were washed once, resuspended, run on a LSRFortessa cytometer (BD Biosciences) and analyzed with FlowJo software (Tree Star).
Nucleus extraction.
Nuclei from tissue or cell lines were extracted using EZ Prep (Sigma-Aldrich N3408) and Glass Dounce Kit (Sigma Aldrich D8938) as previously described2. Briefly, cells or frozen tissue were placed in 2mL of EZ lysis buffer containing Recombinant RNase Inhibitor (Takara Bio 2313A), and dounced 24 times with pestle A then 24 times with pestle B. Nucleus suspensions were transferred to a 15mL Falcon tube, added with an additional 3mL of EZ lysis buffer, incubated on ice for 5 min, pelleted (500g for 5 min at 4°C) with a swinging bucket centrifuge, resuspended in 5mL EZ lysis buffer with a P1000 pipette, incubated on ice for 5 min, and pelleted as in previous step. Nuclei were then resuspended in 1mL pre-chilled buffer (1X PBS, 3mM MgCl2, Recombinant RNase Inhibitor (Takara Bio #2313A)) and filtered through a 35μm FACS tube (Falcon 352235).
Intranuclear antibody stain of nucleus suspensions.
Nuclei were simultaneously fixed and permeabilized by adding 3mL of 1.33% FA-NT (1.33% formaldehyde, 0.2% NP-40, 0.1% Tween 20, 3μL glacial acetic acid) to 1mL of nuclei suspended in PBS with 3mM MgCl2. Samples were incubated for 10 min at 4°C with rocking. Fixation was quenched by adding 3μL of 1M glycine, then immediately filtering through a 20μm strainer (pluriSelect 431002040). Nuclei were pelleted in a swinging bucket centrifuge at 850g, 5 min, 4°C (centrifuge conditions for all subsequent spins), then resuspended in 500μL of blocking buffer (see below) and incubated for 15 min at 4°C with rocking, and pelleted. Pellets were resuspended in 200μL blocking buffer containing primary antibodies and incubated at 4°C for 1 hour with rocking. Primary antibody concentrations were as follows: p65Ab (raised in rabbit) at 1:400, p65inCITE-Ab (raised in rabbit) at 1:400, c-FosinCITE-Ab (raised in rabbit) at 1:400, NeuNinCITE-Ab (raised in mouse) at 1:500, PU.1inCITE-Ab (raised in rat) at 1:200. Nucleus hashing antibodies (BioLegend cat #682213, 682215) were simultaneously added to each sample at 1:200. After incubation, nuclei were pelleted, washed 2x with 500μL of 0.2% PBST + Recombinant RNase Inhibitor, incubated for 5 min, and re-pelleted. Nuclei were then either resuspended in 300μL of 1X PBS in preparation of loading on the 10x Genomics Chromium instrument (below), or resuspended in 200μL of secondary antibodies at 1:1000 dilution in blocking buffer and 10x DAPI, incubated in the dark at 4°C for 30 min, washed twice as previously described, resuspended in 0.2% PBST + Recombinant RNase Inhibitor and filtered through a 20μm strainer (pluriSelect 431002040) for flow cytometry.
Antibodies used: p65Ab or p65inCITE-Ab (BioLegend Poly6226, #622601), c-FosinCITE-Ab (BioLegend Poly6414, cat# 641401), NeuNinCITE-Ab (BioLegend 1B7, cat #834502), PU.1inCITE-Ab (BioLegend 7C2C34, cat #681307).
Blocking buffers used:
Optimized inCITE-seq buffer: 1:100 FcX (BioLegend 156604), 1% UltraPure BSA (ThermoFisher Scientific AM2618), 0.05% dextran sulfate, 0.2% Tween20, Recombinant RNase Inhibitor, in 1x PBS. Dextran sulfate may be substituted with a 1:200 dilution of HCR probe hybridization buffer (tissue section format) from Molecular Instruments.
Intranuclear stain buffer used for anti-p65 antibody in Extended Data Fig. 1c: 1:100 FcX (BioLegend 156604), 1% UltraPure BSA (ThermoFisher Scientific AM2618), 0.2% Tween20, Recombinant RNase Inhibitor, in 1x PBS.
Commercial intracellular buffer #1 in Fig. 1c: Intracellular Staining Permeabilization Wash Buffer (Biolegend 42100), used according to the manufacturer’s instructions.
Commercial intracellular buffer #2 in Fig. 1c: eBioscience Permeabilization Buffer (ThermoFisher 00-8333-56), used according to the manufacturer’s instructions.
InCITE antibodies.
Pure clones of all antibodies were conjugated with the TotalSeq™-A format (BioLegend).
InCITE-seq.
Antibody stained nuclei were resuspended in PBS with 3mM MgCl2, filtered through a 10μm filter, counted in a hemocytometer, and promptly loaded onto a Chromium single-cell V3 3’ chip (10X Genomics) according to the manufacturer’s protocol for GEM formation. For the HeLa experiment, a single V3 3’ 10x channel was loaded with 10,000 NT nuclei and 10,000 TNFα treated nuclei with nucleus hashing. For the mouse hippocampus experiment, two V3 3’ 10x channels were loaded, each channel with 30,000 nuclei of a 1:1 mix of nucleus-hashed PBS and kainic acid sample, such that in total, n=2 PBS sample and n=2 kainic acid treated sample were loaded across two channels.
After GEM formation, simultaneous reverse crosslinking and reverse transcription were conducted by incubating GEMs at 53°C for 45 min followed by 85°C for 5 min. Samples were stored at −20°C until GEM recovery according to the manufacturer’s instructions. cDNA was amplified using the standard 10X Genomics single cell 3’ V3 protocol (10X Genomics) with both HTO and ADT PCR additive primers included in the AMP mix at 0.1μM and 0.2μM, respectively. After amplification, antibody-oligo derived cDNA fragments were separated from mRNA derived cDNA through SPRI-based size selection by incubating cDNA amplification product in 0.6x SPRIselect (Beckman Coulter B23319) for 5 min at RT. At this stage, antibody-oligo derived cDNA is contained in the supernatant, while mRNA derived cDNA remains on SPRIselect beads. Supernatant containing antibody-oligo derived cDNA was removed and separately stored for HTO/ADT library construction. SPRIselect containing mRNA derived cDNA (WTA) were washed two times in 80% ethanol, eluted into 40μL of Elution Buffer and stored for gene expression library construction.
Gene expression libraries were constructed from cleaned WTA (10μL) using manufacturer-specific enzymatic fragmentation, adaptor ligation, and sample index attachment, then eluted in 30μL of Elution Buffer according to the standard 10X Genomics single cell 3’ V3 protocol. Samples were stored at −20°C until library quantification and sequencing.
For HTO/ADT library construction, antibody-oligo derived cDNA was mixed with 1.4x SPRIselect, incubated at RT for 5 min, incubated on magnet for 5 min, washed 2x with standard 80% ethanol washes, and eluted into 24μL of sterile ddH2O. Afterwards, 6μL of eluted solution was added to one of two PCR solutions containing either a unique HTO or ADT index primer mix and NEBNext 2x Mastermix (New England BioLabs M0541L) in order to construct separate HTO and ADT libraries. Libraries were constructed via PCR amplification with the following conditions: 98°C for 5 min, 21 cycles at 98°C for 2 sec and 72°C for 15 sec, and a final 72°C for 1 min. PCR products were purified with 2.0x SPRI beads with 10 min incubation, 5 min magnetic separation, and two 80% ethanol washes. Purified products were eluted into 20μL of EB. Samples were stored at 4°C until library quantification and sequencing.
Gene expression, HTO, and ADT libraries were quantified using standard Qubit (ThermoFisher Q32853) and Agilent TapeStation (Agilent G2991AA) to check for library size. Optimal library size was ~420bp for gene expression and ~180bp for ADT/HTO. Libraries were pooled and sequenced on the NextSeq500 platform (Illumina) using a 75-cycle kit (Read 1: 28 cycles, Index 1: 8 cycles, Read 2: 55 cycles).
snRNA-seq of mouse hippocampus.
Nuclei were extracted from the hippocampus of PBS (n=1) and kainic acid (n=1) treated mice as described above, resuspended in 1mL chilled 1x PBS with 3mM MgCl2, filtered through a 10μm filter, nucleus hashed per treatment as previously described, and promptly loaded onto a Chromium single-cell V3.1 3’ chip (10X Genomics) according to the manufacturer’s protocol. A single V3.1 3’ 10x channel was loaded with 10,000 nuclei in a 1:1 mix of nucleus-hashed PBS sample and a kainic acid sample. cDNA and HTO libraries were generated and sequenced as previously described39.
InCITE-seq and snRNA-seq data pre-processing.
Sequencing data were processed with Cellranger v4.0.0 on Cumulus v1.077. Reads from demultiplexed FASTQ files were aligned to pre-mRNA annotated genomes of the mouse mm10 or human GRCh38 reference genome as previously described78. Hashed nuclei were demultiplexed using DemuxEM39 with min_num_genes=10, min_num_umis=1, min_signal_hashtag=1; nuclei with ambiguous treatment assignment (i.e., nuclei not assigned as NT or TNFα among HeLa nuclei, or as PBS or KA among mouse hippocampus nuclei) were discarded (5.9% and 2.7% for HeLa and mouse, respectively); barcode collision rates are discussed elsewhere13,39,79. For mouse hippocampus data, raw counts across both channels were combined. Unspliced pre-mRNA and spliced mRNA counts were generated from BAM files using Velocyto v0.17.1727. All gene expression matrices were analyzed by Scanpy (v1.6.0)80. For inCITE-seq data, protein ADT counts corresponding to each nucleus/cell barcode were also added.
Analysis of gene expression.
For HeLa data, genes in at least 10 nuclei, and nuclei with at least 500 genes and at most 5,000 UMI counts were retained, resulting in a matrix of 10,014 nuclei with 13,942 genes across both NT and TNFα. For mouse hippocampus data, genes in at least 3 nuclei, and nuclei with at least 50 genes and at most 900 or 1,200 UMI counts for replicate/batch 1 or 2, respectively were retained; we then removed nuclei with mitochondrial gene content >5%, hashtag oligo counts (HTO counts) greater than 5,000, and c-Fos antibody counts exceeding 300 ADT counts were removed, resulting in a matrix of 41,332 nuclei with 20,679 genes. Gene counts were normalized within each nucleus then log normalized as ln(X+1).
Normalizing protein expression.
Protein abundances measured by antibody-derived tag (ADT) were normalized by nucleus hashtag oligo counts (HTO) after adding a pseudo-count as nADT = (ADT + 1)/HTO then scaled to centered log ratios (nuclear; “nCLR”) as nCLR = nADT/(Πi nADTi)1/n, where the denominator is the geometric mean. For mouse hippocampus data, nCLR scaling was conducted within each batch, such that batch-specific geometric means were used in the denominator.
Clustering mouse hippocampus data using single nucleus RNA profiles of inCITE-seq.
5,194 variable genes on log normalized counts were selected using Scanpy’s highly_variable_genes function (min_mean=0.004, max_min=0.08, min_disp=0.3), log counts were scaled, and UMI counts and mitochondrial content were regressed out using Scanpy’s regress_out function. Dimensionality reduction was performed with PCA on variable genes in Scanpy, followed by a pytorch implementation of Harmony81 to correct for batch based on 10x channels and to regress out differences due to treatment (PBS and KA). Nearest-neighbor graph was constructed with k=10 neighbors and top 40 principal components, clustered with the Leiden algorithm, and embedded using UMAP82, all in Scanpy.
Joint embedding of inCITE-seq and snRNA-seq mouse hippocampus profiles.
snRNA-seq profiles of fixed nuclei with inCITE-seq (n=24,444 nuclei) were jointly embedded with snRNA-seq data from this study (n=1,887) and from a published study54 (n=15,001; WT only). 7,541 variable genes were identified in a single joint analysis from log normalized counts across all datasets using Scanpy’s highly_variable_genes function (min_mean=0.004, max_min=0.08, min_disp=0.3). Log counts were then scaled; UMI counts, mitochondrial content, and sequencing assay type were regressed out with Scanpy’s regress_out function. Dimensionality reduction was performed on variable genes via PCA in Scanpy, followed by a Python implementation of Harmony81 to correct for batch based on 10x channels, to regress out differences due to treatment (PBS and KA), and to further correct for assay type. A k-nearest-neighbor (k-NN) graph was constructed with k=10 neighbors and top 40 principal components, clustered with the Leiden algorithm83 in Scanpy, and embedded using UMAP82 in Scanpy. Unsupervised clusters were identified after accounting for treatment, batch, and sequencing assay type. A cluster with high mitochondrial content (n=327 nuclei) and a subcluster with low transcript abundance (n=179) were removed; 3,059 individual suspected doublets identified by Scrublet84 was removed. The final embedding consisted of n=22,260 inCITE-seq and n=15,507 snRNA-seq nuclei RNA profiles.
Genes associated with p65 protein levels in HeLa.
Gene expression was modeled using a generalized linear model with a negative binomial fit, as follows: Yi ~ p65 + G2M_score + S_score + log(UMI) + log(HTO), where Yi is the log normalized, unscaled ln(X+1) counts for gene i, p65 is the p65 protein level in units of nCLR, G2M_score and S_score are cell cycle scores calculated with score_genes_cell_cycle in Scanpy using previously defined genes85, and log(UMI) and log(HTO) are natural log counts of unique RNA molecular identifiers and nucleus hashtag oligos, respectively. Significance was established at a false discovery rate (FDR) of 1% after Benjamini-Hochberg correction using the statsmodels package in python.
Gene Ontology analysis.
Gene sets were queried using scanpy’s queries.enrich module, a wrapper around gprofiler86, to identify Gene Ontology (GO) Biological Processes. P-values were calculated using a two-sided hypergeometric test, corrected for term and query size.
Genes globally associated with protein levels in mouse hippocampus.
We implemented a two-step mixed linear model using the statsmodel package in Python to account for collinearity, specifically between treatment and c-Fos protein levels. First, we modeled gene expression as the following to regress out effects of treatment and cell type: Yi ~ C(cluster) + C(treatment) + C(cluster)*C(treatment) + log(UMI) + log(HTO) + (1|B), where Yi is the scaled z-score of the ln(X+1) counts for gene i across all nuclei, C(cluster) is a categorical variable indicating cluster membership (cell type), C(treatment) is a categorical variable indicating PBS or KA treatment, log(UMI) and log(HTO) are natural log counts of unique RNA molecular identifiers and nucleus hashtag oligos, and B is a categorical variable denoting the 10x channel batch. We then fit the residuals of each gene, rYi, as a linear combination of the four proteins, accounting for batch: rYi ~ NeuN + c-Fos + p65 + PU.1 + (1|B). Genes in at least 15 nuclei were used for analysis; and significance was established at FDR 5% after Benjamini-Hochberg correction. For identifying treatment-dependent global effects, we implemented the two-step model separately for PBS or KA treated nuclei.
Genes associated with protein levels in mouse hippocampus within excitatory neurons.
Similar to the global approach, we first regressed out treatment: Yi ~ C(treatment) + log(UMI) + log(HTO) + (1|B), where all variates are the same as previously described. We then modeled the residuals of each gene as a linear combination of the four proteins, accounting for batch. In the additive model, we used: rYi ~ NeuN + c-Fos + p65 + PU.1 + (1|B); in the interaction model, we used rYi ~ NeuN + c-Fos + p65 + PU.1 + c-Fos*p65 + c-Fos*NeuN + p65*NeuN + (1|B). Genes found in ≥3% of excitatory neuronal nuclei were used for analysis (1,757 genes tested across 15,404 excitatory neuronal nuclei); significance was established at FDR 5% with Benjamini-Hochberg correction.
Identifying cell type-specific gene programs with non-negative matrix factorization.
Gene programs of excitatory neurons were identified via non-negative matrix factorization (Python sklearn package, NMF function, random_state=0, L1 regularization with l1_ratio=1, alpha=0) of their RNA profiles on a subset of genes (a combination of highly variable genes identified for clustering and c-Fos and p65-associated genes, with manual removal of highly expressed and variable genes Ttr, Fth1, Ptgds). Five programs were identified for excitatory neurons.
Cell type-specific genes associated with protein levels in mouse hippocampus after KA treatment.
Similar to before, we implemented the following mixed linear model for each cell cluster c using the statsmodels package in Python: Yi,c ~ NeuN + c-Fos + p65 + PU.1 + log(UMI) + log(HTO) + (1|B), where: Yi.c is the scaled z-score of the ln(X+1) counts for gene i in cluster c, NeuN, c-Fos, p65, PU.1 are protein levels in units of nCLR, log(UMI) and log(HTO) are natural log counts of unique RNA molecular identifiers and nucleus hashtag oligos, and B is a categorical variable denoting the 10x channel batch. Protein levels (nCLR) were first scaled using the Python package sklearn preprocessing. Genes found in ≥3% nuclei of each cluster were used for analysis; significance was established at FDR 5% with Benjamini-Hochberg correction.
Transcription factor motif enrichment in DEGs.
We used differentially accessible regions (DARs) of the mouse hippocampus profiled in saline treated and kainic acid treated (1hr post treatment) previously defined by Fernandez-Albert et al65. Nearest DARs located more than 1kb from the transcriptional start site (upstream) or transcriptional termination site (downstream) of DEGs associated with c-Fos or p65 (additive model) or c-Fos*p65 (interactional model) were considered as enhancers and were used for motif enrichment analysis. To find enriched motifs, we scanned a given set of differentially accessible peaks for all DNA-binding motifs in the cis-bs (http://cisbp.ccbr.utoronto.ca) and JASPAR2018_CORE_vertebrates_non-redundant (http://jaspar2018.genereg.net) databases. We then tested for the probability of our observed motif frequency among the kainic acid treated DARs using a hypergeometric test, with a null model based on random sampling of all ATAC-seq peaks matching for GC content. Motifs with P<10−3 were considered significant.
Extended Data
Extended Data Fig. 1. Optimization of intranuclear antibody staining in HeLa cells.
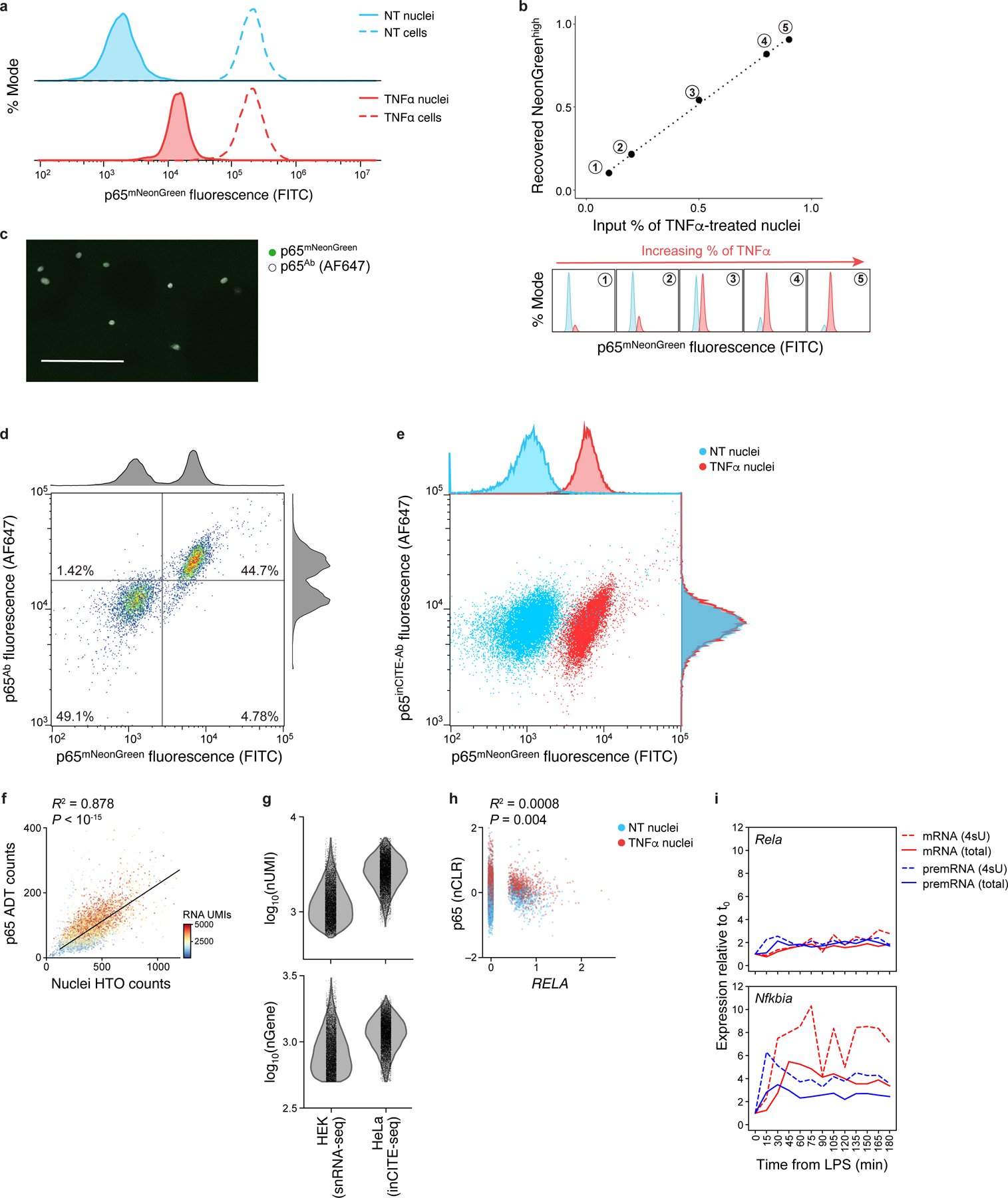
a. Nuclear p65 levels change after TNFα treatment, while total p65 in cells remains unchanged. Distribution of p65-mNeonGreen reporter fluorescence (x axis; % mode of singlet nuclei, y axis) measured by flow cytometry of nuclei (solid line) vs. cells (dashed line) from untreated (“NT”, blue) or TNFα treated cells (red). b. Flow cytometry distinguishes p65-mNeonGreen signals across mixtures of NT and TNFα. Top: Flow cytometry measures of mNeonGreenhigh fraction (x axis) match the input fraction of TNFα nuclei (x axis). Bottom: Corresponding high (red) and low (blue) mNeonGreen distributions. c. Immunofluorescence of nuclei smeared onto a slide after intranuclear p65 stain in suspension, showing complete antibody diffusion into the nucleus; representative of 3 experiments. Scale: 100μm. d,e. Comparing antibody- and fluorescence reporter-derived p65 levels. Antibody (from Alexa Fluor 647 secondary, y axis) and mNeonGreen (x axis) signal of p65 in an equal mixture of NT and TNFα stimulated nuclei. Histograms: marginal distributions. d. Agreement between unconjugated p65 antibody and mNeonGreen signal. e. No relationship between DNA-conjugated p65inCITE-Ab and mNeonGreen signal using standard intranuclear staining buffer (pre-optimization). f. Relation between nuclei hashtag oligonucleotide (HTO; x axis) counts and p65 antibody-derived tag (ADT; y axis) counts, shown across 10,014 NT and TNFα nuclei, colored by the number of RNA UMIs. Top left: Pearson R2 and associated P-value (two-sided t-test). To control for this relation, we normalize protein ADT counts by nuclei HTO counts (Methods). g. Comparing RNA complexity from inCITE-seq (fixed HeLa nuclei) and MULTI-seq (unfixed HEK nuclei, from McGinnis et al.45) by the distribution of the number of detected transcripts (UMIs; top) and genes (bottom). h. Low correlation between p65 protein (y axis, nCLR) and RELA RNA levels (x axis, log normalized), with Pearson R2 and associated P-value (two-sided t-test). Dots: nuclei colored by treatment (NT, blue; TNFα, red). i. Dynamics of gene expression after LPS stimulation in mouse dendritic cells, from Rabani et al.10, measured across time (x axis). Relative expression to steady state, t0 (y axis): pre-mRNA precursor (blue) and mRNA (red) for total (solid) vs. 4sU labeled (dashed) RNA, shown for Rela (top) and Nfkbia (bottom), a p65 target as in Fig. 1e.
Extended Data Fig. 2. Flow cytometry of inCITE targets on nuclei or cells extracted from frozen mouse hippocampus.
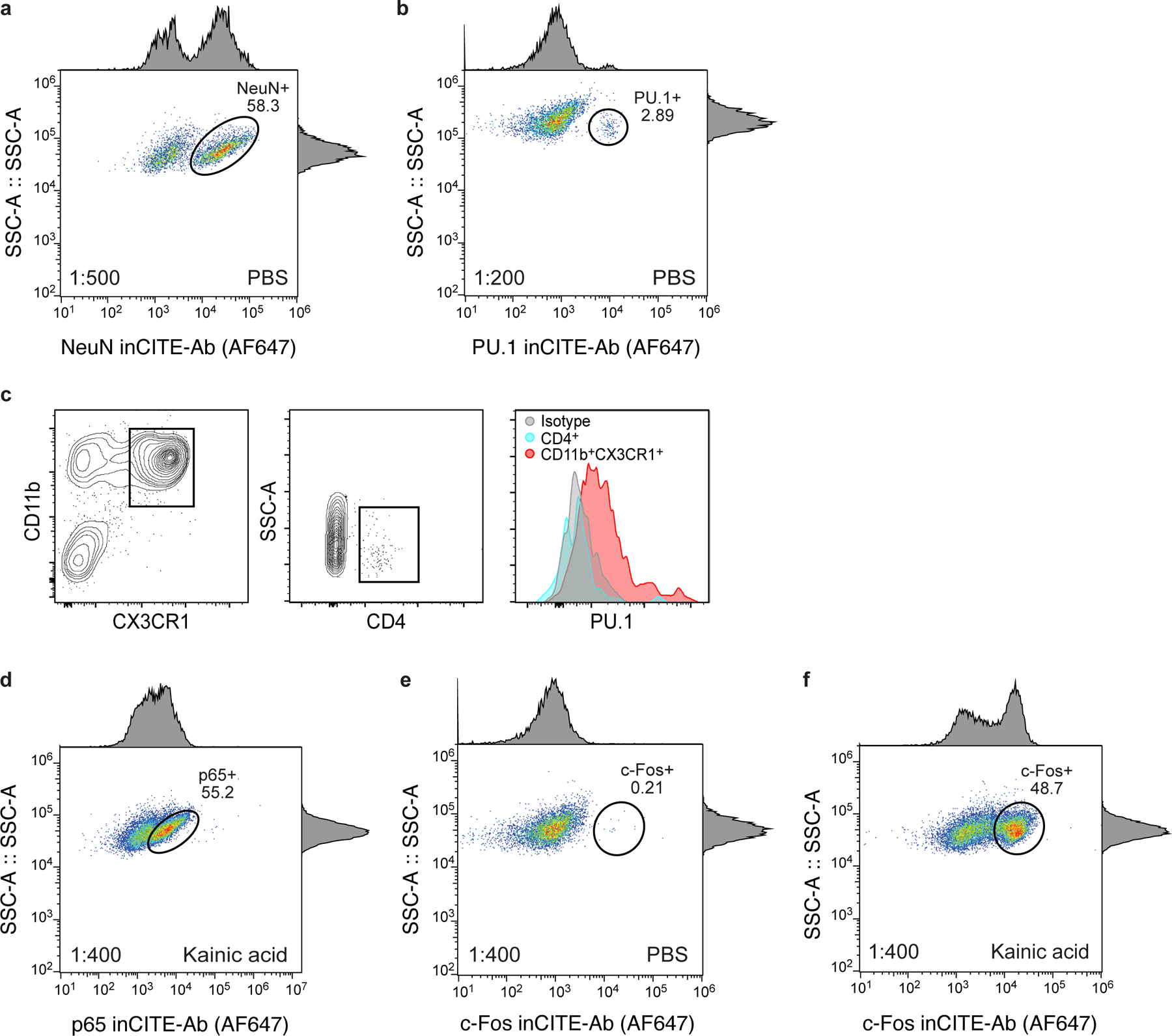
Flow cytometry of nuclei populations from the mouse hippocampus after intranuclear stains with inCITE antibodies, followed by Alexa Fluor 647-conjugated secondary stain: NeuN in PBS (a), PU.1 in PBS (b), p65 in kainic acid (KA) (d), and c-Fos in PBS (e) and kainic acid (KA) (f) treated mice. Axes show fluorescence signal (x axis) and side scatter (y axis) of singlet nuclei (dots); histograms show marginal distributions. Oval gates show NeuNhigh (a, 58.3%), PU.1high (b, <3%), p65high (d, 55.2%), c-Foshigh (0.21% in PBS (e), and 48.7% after KA treatment (f)). c. Right: Distribution of PU.1 in microglia (CD11b+ CX3CR1+, red), CD4+ cells (blue) and isotype (gray) cells measured by flow cytometry (left and middle panels) after simultaneous surface protein and intracellular protein stains (Methods).
Extended Data Fig. 3. Antibody signal varies across concentration regimes.
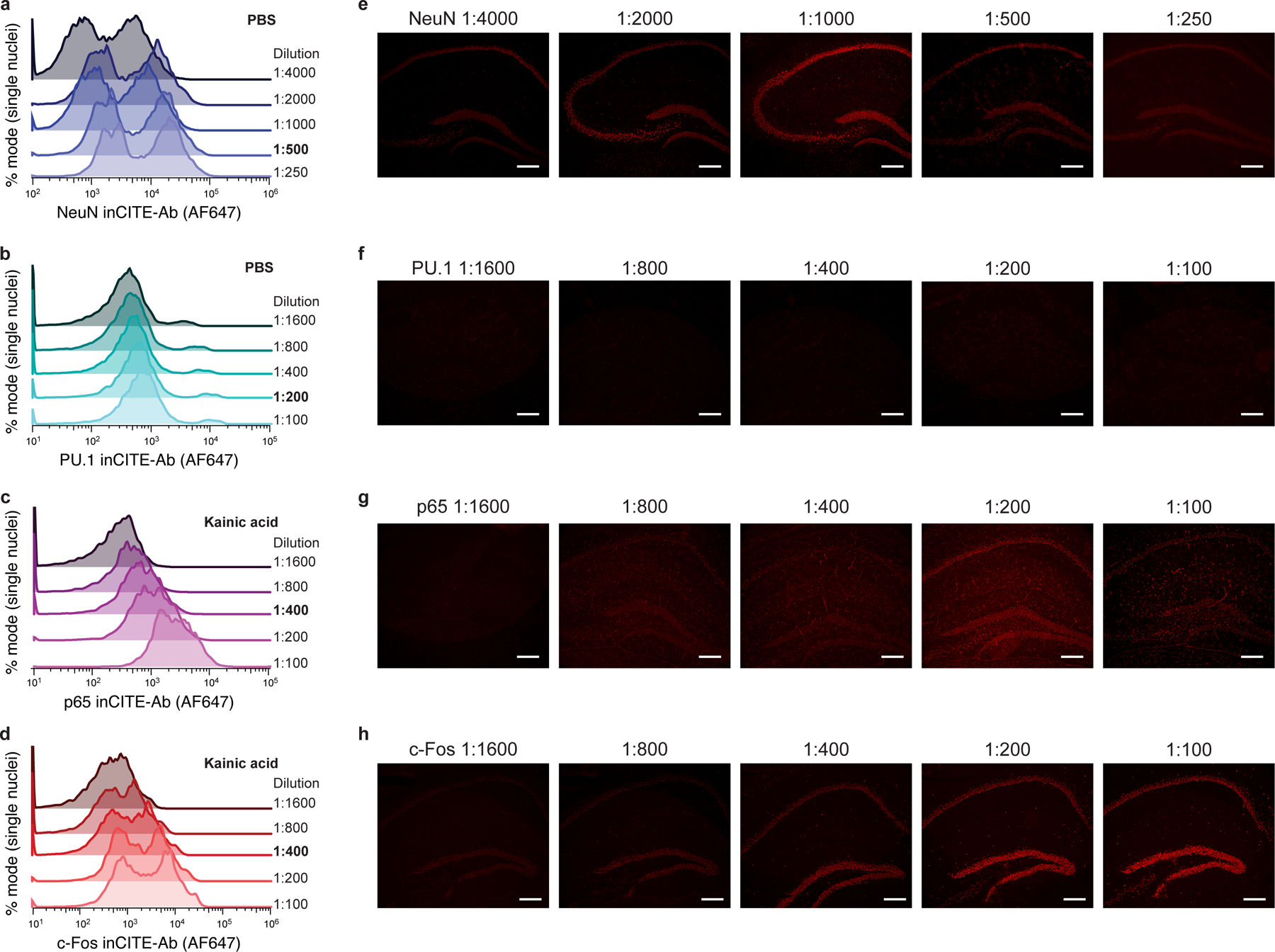
Antibody stains of the mouse hippocampus (extracted nuclei or in situ) with inCITE antibodies across a wide range of dilutions, targeting NeuN in PBS (a,e), PU.1 in PBS (b,f), p65 in kainic acid (c,g), and c-Fos in kainic acid (d,h) treated mice. Antibody-derived fluorescence measured by Alexa Fluor 647-conjugated secondary antibody stain. a-d. Histograms are normalized as % mode of nuclei singlets. Antibody dilutions are indicated to the right of each axis, with dilutions used for inCITE-seq in bold (NeuN 1:500, PU.1 1:200, p65 1:400, c-Fos 1:400). e-h. In situ immunofluorescence of frozen mouse hippocampus with inCITE antibodies across different dilutions, matching the concentrations used in flow cytometry; representative of 2 independently conducted experiments. Scale bars, 100μm.
Extended Data Fig. 4. Impact of tissue preparation on epitope detection by antibodies.
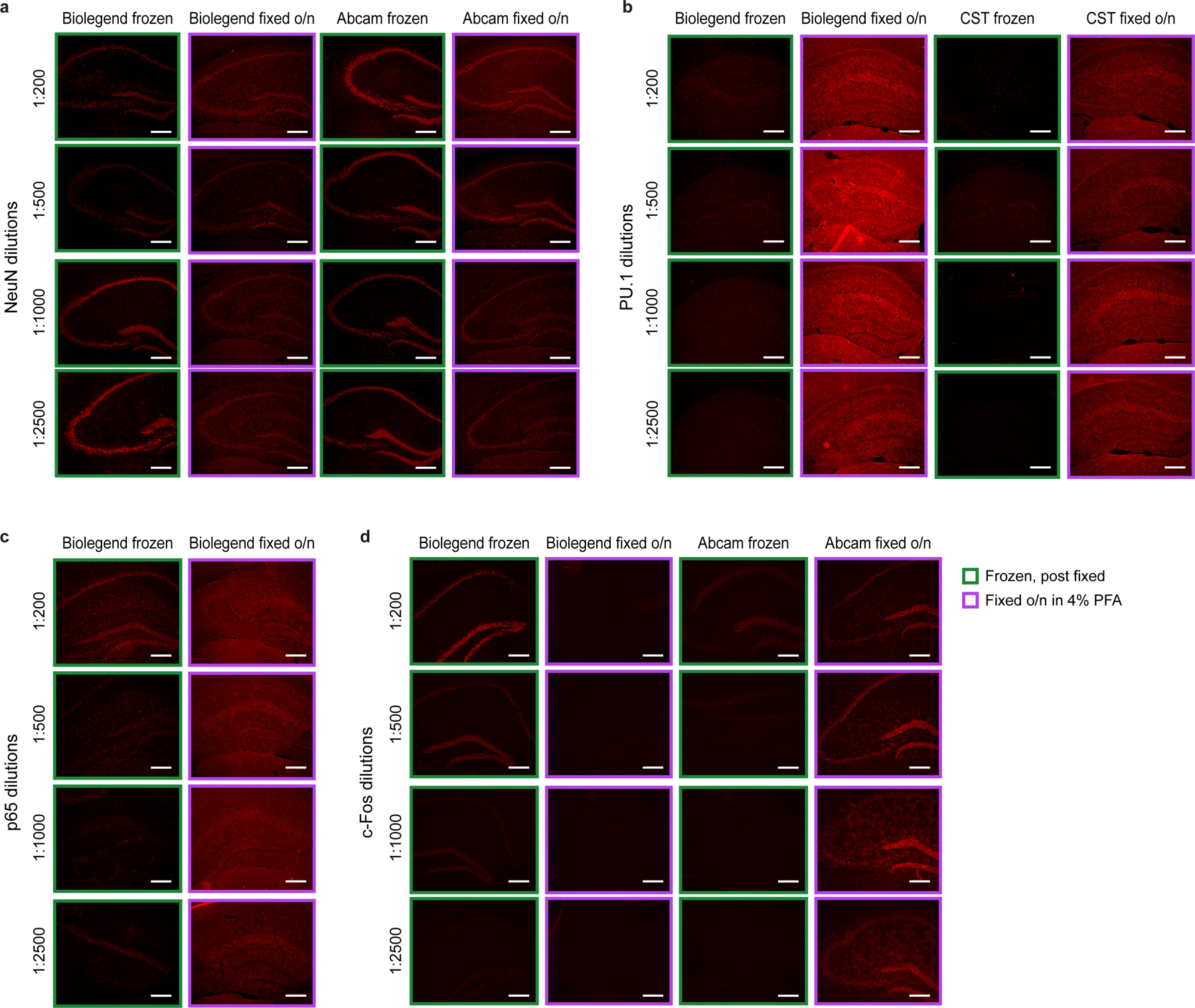
Comparing in situ immunofluorescence of antibody stains (followed by Alexa Fluor 647-conjugated secondary stain) in mouse hippocampus tissue that were immediately frozen (green box) or frozen after overnight fixation in 4% PFA (purple box, Methods) across a wide range of antibody dilutions. Images are representative of 2 independent experiments. a. NeuN in PBS. Biolegend NeuN antibody (clone 1B7) used for inCITE and Abcam NeuN antibody (clone EPR12763). b. PU.1 in PBS. Biolegend PU.1 antibody (clone 7C2C34) used for inCITE and Cell Signaling Technology PU.1 antibody (clone 9G7). c. p65 in KA. Biolegend p65 antibody (clone Poly6226) used for inCITE. d. c-Fos in KA treated mice. Biolegend c-Fos antibody (clone Poly6414) used for inCITE and Abcam c-Fos antibody (ab190289). Scale bars, 100μm.
Extended Data Fig. 5. Comparing and combining single nucleus RNA profiles from inCITE-seq and snRNA-seq of mouse hippocampus.
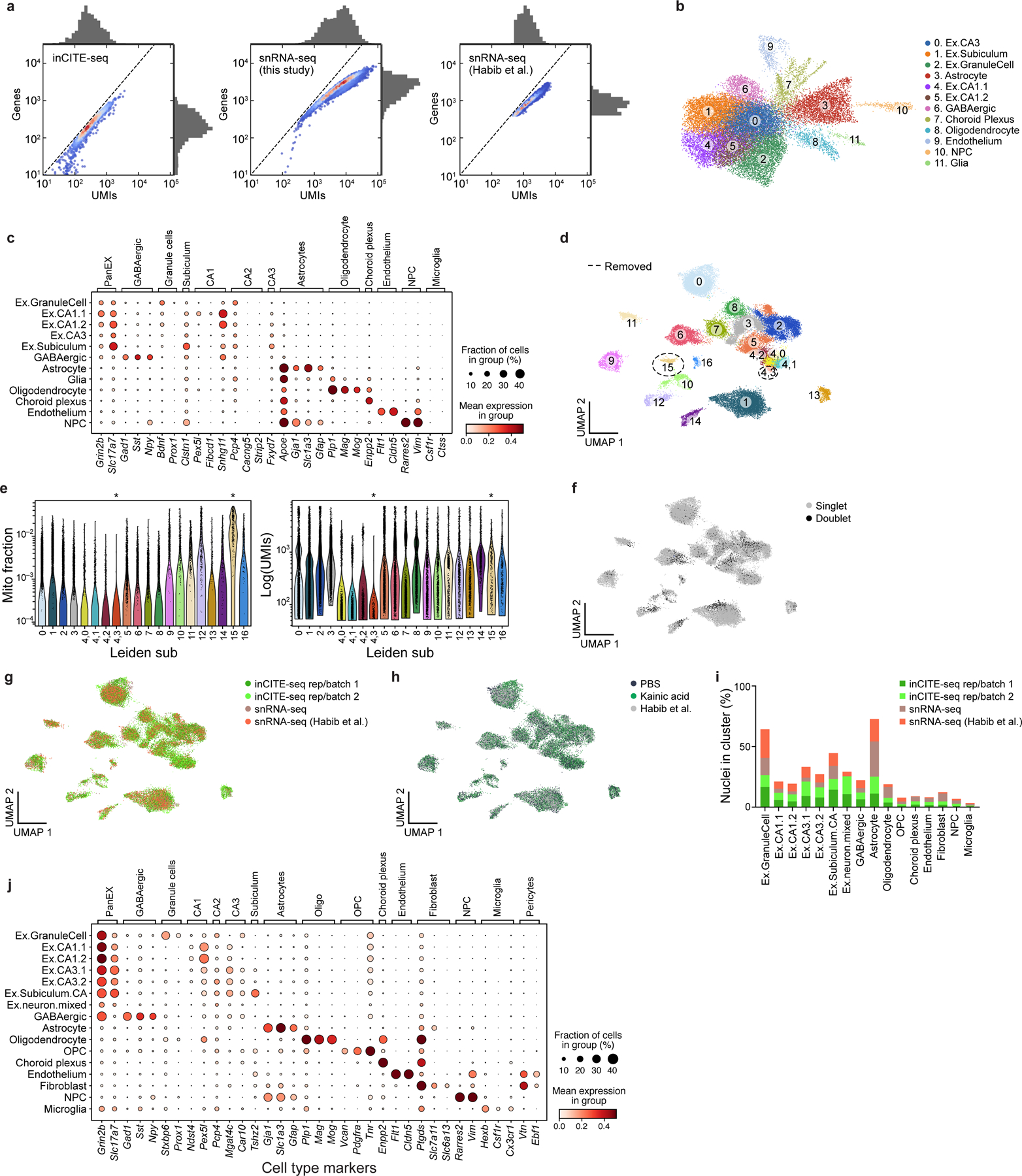
a. Comparing the complexity of RNA profiles from inCITE-seq and standard snRNA-seq of the mouse hippocampus. Distributions (marginals) of the number of UMIs (x axis) and genes (y axis) from inCITE-seq (left), matching mouse hippocampus snRNA-seq in this study (middle), and previously published snRNA-seq (right). Scatter plot shows the density of individual nuclei (dots) calculated with a Gaussian kernel estimate. b,c. Major cell types from the adult mouse hippocampus identified from inCITE-seq RNA profiles alone. b. UMAP embedding of 24,444 single nucleus inCITE-seq RNA profiles (dots) colored by annotated cluster (number). c. Expression of marker genes (columns) used for annotating cell type clusters (rows), showing mean expression of log normalized counts (dot color) and proportion of expressing cells (dot size). d-j. Enhanced cell type distinctions and annotation by combining RNA profiles from inCITE-seq and snRNA-seq. Joint UMAP embedding of 22,260 inCITE-seq and 15,507 snRNA-seq RNA profiles (dots) colored by unsupervised leiden clusters or subcluster of leiden group 4 (numbers) (Methods). e. Distribution of mitochondrial fraction of total gene content (y axis, left) and total transcript counts (y axis, right) in each leiden cluster or subcluster of leiden group 4 (x axis, both). Asterisks indicate cluster 15 (n=327 nuclei) and subcluster 4,3 (n=179 nuclei) that were removed for high mitochondrial content and for low RNA complexity, respectively. f-h. UMAP embedding as in Fig. 2d colored by doublets that were removed from subsequent analyses (n=3,059 doublets, (f)), batch and assay (g), or condition (h). i. Percent of nuclei (y axis) from each batch/assay (color) in each cluster (x axis). j. Mean expression of log normalized counts (dot color) and proportion of expressing cells (dot size) of marker genes (columns) used for annotating cell type clusters in d (rows).
Extended Data Fig. 6. Protein levels by inCITE-seq batch (replicate).
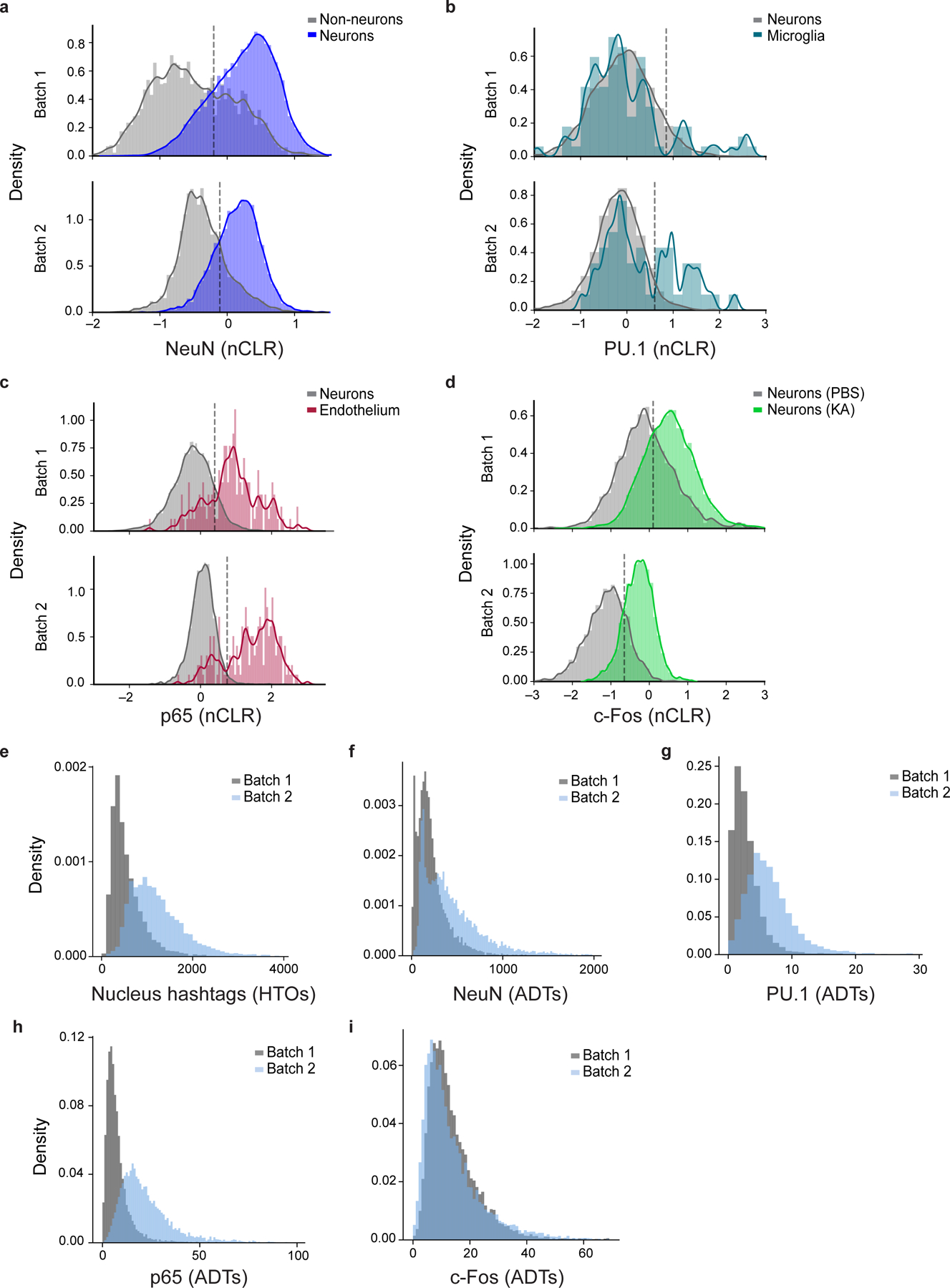
a-d. Distribution of protein levels (x axis, nCLR) shown as kernel density estimates of NeuN (a), PU.1 (b), p65 (c), or c-Fos (d) in each batch (top: batch 1; bottom: batch 2) in biologically relevant subsets as foreground (color) and appropriate background set of nuclei (grey). Dashed line: Batch-specific threshold used to partition protein level as high vs. low. e-i. Density distribution of (e) nucleus hashtag counts (x axis, HTOs) or (f-i) antibody-derived tags (x axis, ADTs) of inCITE target proteins, colored by batch (batch 1, gray; batch 2, blue).
Extended Data Fig. 7. Protein effects on global gene expression.
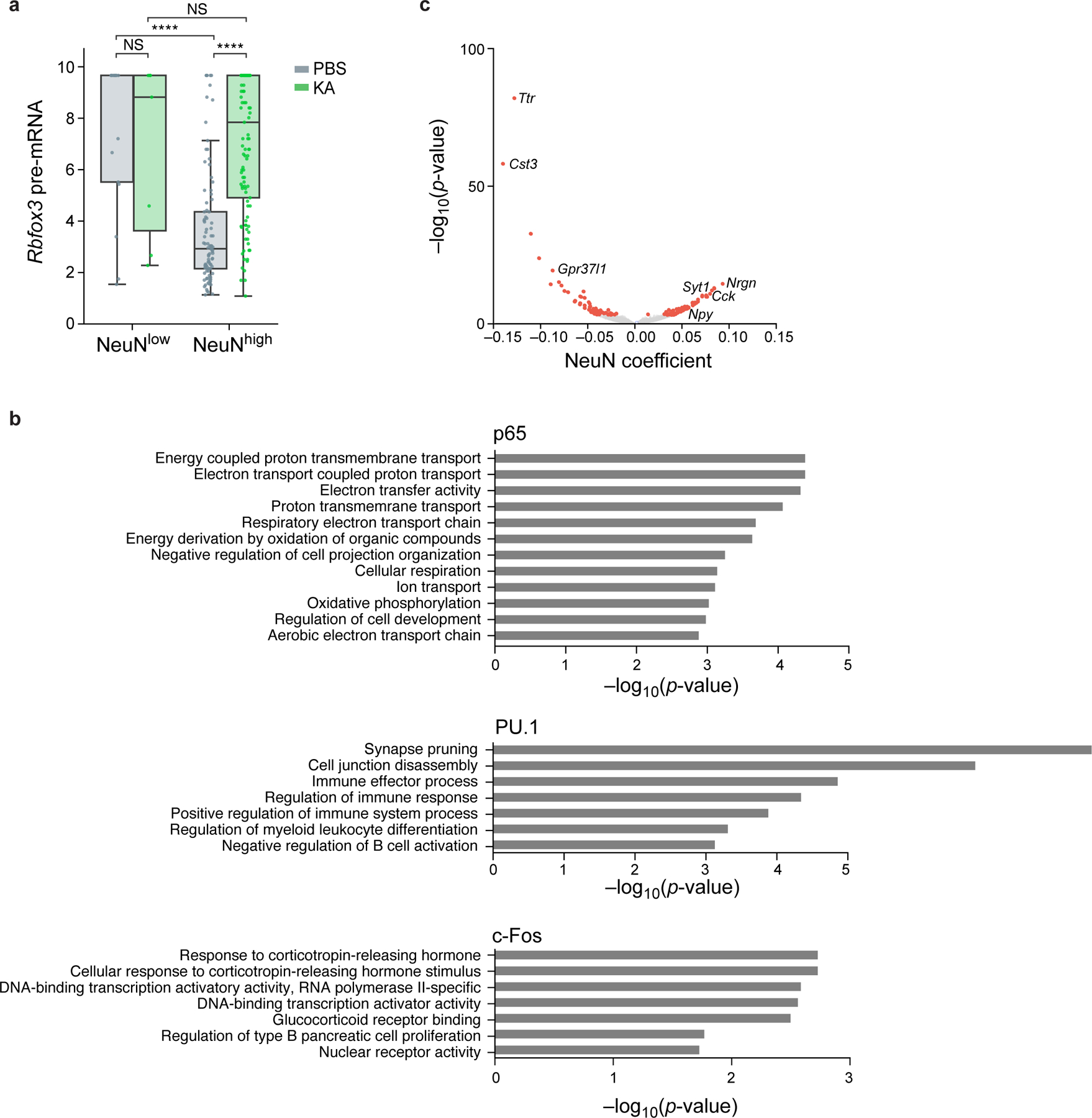
a. Relation between unspliced pre-mRNA expression of Rbfox3 and nuclear protein levels of NeuN. Distribution of pre-mRNA levels (Z score of log-normalized counts, y axis) in nuclei with high or low levels of NeuN (x axis) after PBS (gray) or KA (green) treatment (NeuN thresholds in Extended Data Fig. 6). Boxplots show the median (centre line), box bounds represent first and third quartiles, and whiskers span from each quartile to the minimum or the maximum (1.5 interquartile range below 25% or above 75% quartiles). Dots correspond to 227 individual nuclei with non-zero pre-mRNA levels measured across n=2 biologically independent samples. Significance, from left: P=5*10−15, P=9*10−5 two-sided Mann-Whitney test. NS – not significant. b. Functional gene sets enriched in TF associated genes. Enrichment (−log10(P-value), x axis, hypergeometric test) of Gene Ontology (GO) terms (y axis) in genes significantly associated (from top to bottom) with p65 (33 genes), PU.1 (13 genes), and c-Fos (10 genes). c. Genes associated with NeuN. Effect size (x axis) and associated significance (y axis, −log10(P-value)) for the association of each gene (dots) with NeuN by a model of gene expression as a linear combination of the four inCITE-seq target proteins after regressing out treatment and cell type (Methods). Select genes are labeled. Colored dots: Benjamini-Hochberg FDR <5%.
Extended Data Fig. 8. Genes and modules associated with TFs within excitatory (EX) neurons.
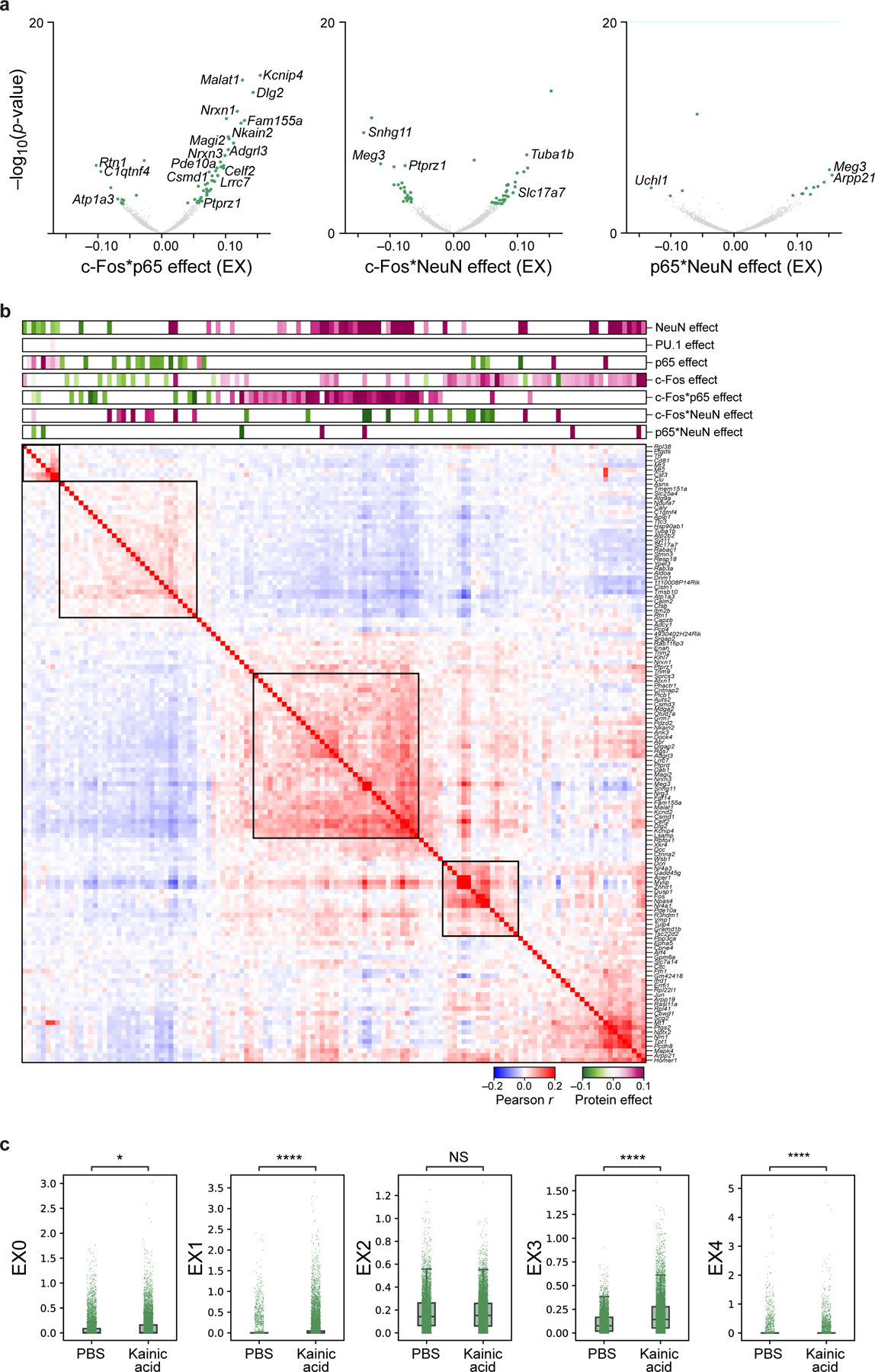
a. Genes associated with protein-protein pairs in the interaction model, identified by modeling gene expression across excitatory neurons as a linear combination of individual proteins and their pairwise interactions after regressing out treatment. Effect size (x axis) and significance (y axis, −log10(P-value)) for DEGs (dots) associated with each protein-protein interaction term: p65 and c-Fos (left), c-Fos and NeuN (middle), and p65 and NeuN (right). Select genes are labeled. Colored dots: Benjamini-Hochberg FDR<5%. b. Pearson correlation coefficient (red/blue colorbar) of pairwise gene expression profiles (rows and columns) significantly (FDR<5%) associated positively (purple) or negatively (green), with c-Fos (additive model), p65 (additive model), or c-Fos*p65 (interaction model), ordered by hierarchical clustering. Top bars: Effect size of each protein or protein-protein pair. c. Treatment effect on gene programs. Program scores (y axis) for 5 EX programs (in Fig. 4f) of 15,226 individual nuclei (dots) from PBS or KA treated mice (x axis) measured across 2 biologically independent experiments. Boxplots show the median (centre line), box bounds represent first and third quartiles, and whiskers span from each quartile to the minimum or the maximum (1.5 interquartile range below 25% or above 75% quartiles). Significance, from left: P=0.049, P=2.7*10−271, P=2.2*10−199, P=6.1*10−7, two-sided Mann-Whitney test. NS – not significant.
Extended Data Fig. 9. Treatment-dependent cis-regulatory elements and TF-associated genes.
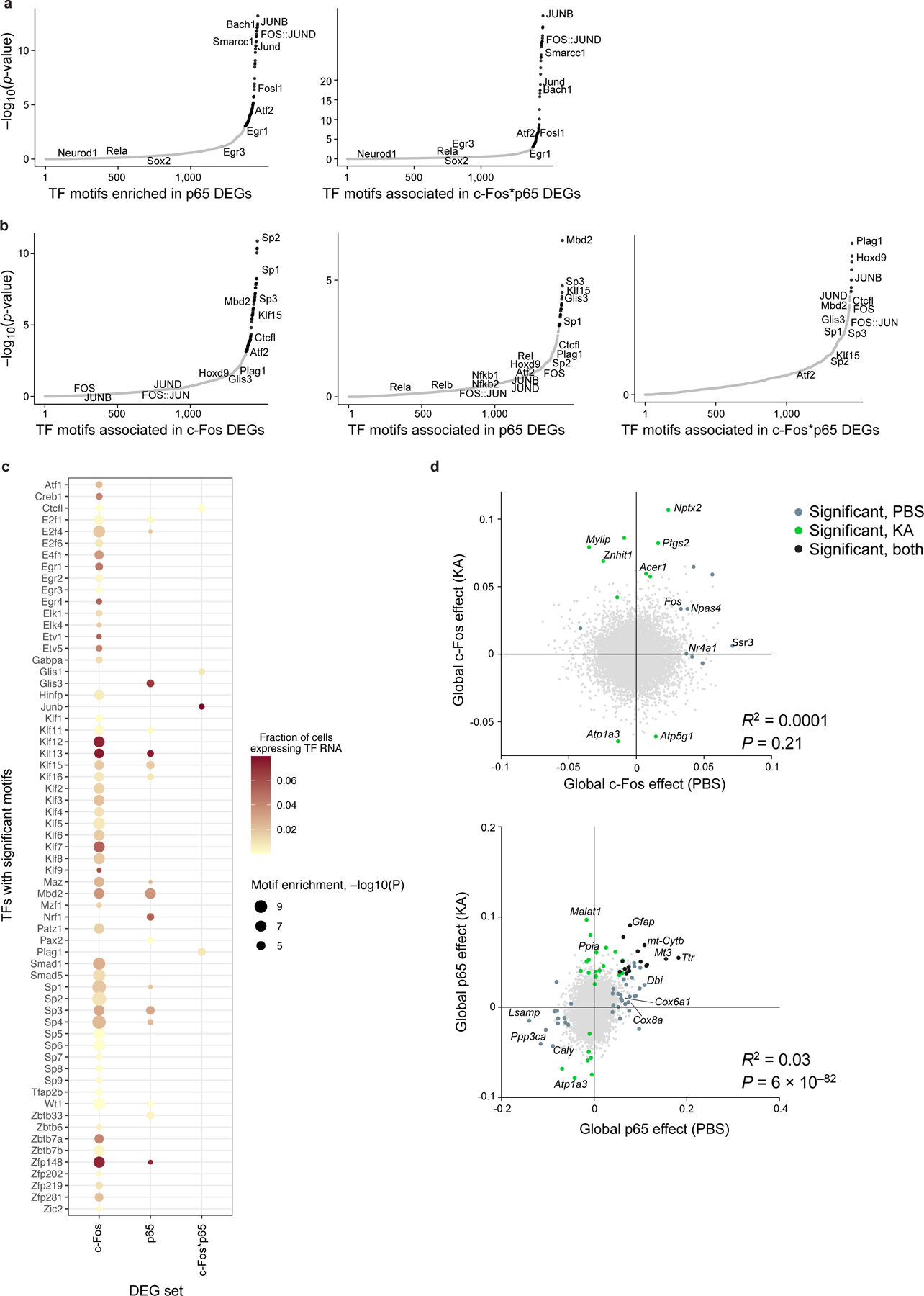
a-c. Prediction of co-regulatory patterns by TF motif enrichment in DEGs associated with c-Fos or p65 (additive model), or their interaction c-Fos*p65 (interaction model). a,b. Significance (−log10(P-value), y axis) and rank order (x axis) of TF motifs (dots) enriched in enhancers of DEGs associated with each protein (additive model) or protein-protein (interaction model) term in excitatory neurons, using enhancers of PBS (a) or KA (b) treated sample as background. Black: significant motifs (P<10−3, hypergeometric test); gray: not significant. c. TF motif enrichment (columns; dot size, −log10(P-value)) and proportion of excitatory neuron nuclei expressing the RNA (color) of significant TFs (rows) in the enhancers of c-Fos (additive model), p65 (additive model), or c-Fos*p65 (interaction model) DEGs, compared to other enhancers within the KA treated sample. d. Treatment-dependence of gene association with c-Fos and p65. Global effect size of genes (dots) associated with c-Fos (left) and p65 (right), after PBS (x axis) or KA treatment (y axis) (Methods). Colored dots: genes with significant coefficients (Benjamini-Hochberg FDR<5%) in PBS (gray), KA (green), or both (black). Select genes are labeled. Bottom right: linear correlation R2 and associated P value (two-sided t-test).
Supplementary Material
Acknowledgments
We thank J. Schmid-Burgk and I. Cheeseman for the HeLa p65-mNeonGreen reporter line, L. Gaffney for assistance with figure preparation, P. Thakore for coining the acronym inCITE-seq, A. Rubin for critical feedback on the manuscript, C. McGinnis for helpful sharing of data, the Broad Institute Flow Cytometry Core facility, and all members of the Regev lab for helpful discussions. This research was supported by NIH/NHGRI CEGS grant 5RM1 HG006193. A.R. was a Howard Hughes Medical Institute Investigator (until July 31, 2020). The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Footnotes
Code Availability. Code used for analyses are available on https://github.com/klarman-cell-observatory/inCITE-seq.
Competing Interests Statement
A.R. is a founder and equity holder of Celsius Therapeutics, an equity holder in Immunitas Therapeutics, and until August 31, 2020 was an SAB member of Syros Pharmaceuticals, Neogene Therapeutics, Asimov and ThermoFisher Scientific. From August 1, 2020, A.R. is an employee of Genentech. From May 2021, D.P. is an employee of Genentech. B.Y. was formerly an employee of BioLegend and is now an employee of Spatial Genomics. The remaining authors declare no competing interests.
Data Availability Statement.
Raw gene expression count matrices of all inCITE-seq data, BAM files of mapped reads, and matrix of mouse hippocampus inCITE-seq data jointly embedded with snRNA-seq data are available on Gene Expression Omnibus under the accession GSE163480. Data from Habib et al54. is available under GSE143758. Data from MULTI-seq used to compare RNA complexity in HEK cells is available under GSE129578. Databases of transcription factor motifs (cis-bs and JASPAR2018_CORE_vertebrates_non-redundant) are available at http://cisbp.ccbr.utoronto.ca and http://jaspar2018.genereg.net, respectively.
References
- 1.Habib N et al. Div-Seq: Single-nucleus RNA-Seq reveals dynamics of rare adult newborn neurons. Science 353, 925–928 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Habib N et al. Massively parallel single-nucleus RNA-seq with DroNc-seq. Nat. Methods 14, 955–958 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Slyper M et al. A single-cell and single-nucleus RNA-Seq toolbox for fresh and frozen human tumors. Nat. Med 26, 792–802 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lacar B et al. Nuclear RNA-seq of single neurons reveals molecular signatures of activation. Nat. Commun 7, 11022 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van den Brink SC et al. Single-cell sequencing reveals dissociation-induced gene expression in tissue subpopulations. Nat. Methods 14, 935–936 (2017). [DOI] [PubMed] [Google Scholar]
- 6.Drokhlyansky E et al. The human and mouse Enteric nervous system at single-cell resolution. Cell (2020) doi: 10.1016/j.cell.2020.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hwang WL et al. Single-nucleus and spatial transcriptomics of archival pancreatic cancer reveals multi-compartment reprogramming after neoadjuvant treatment. bioRxiv (2020) doi: 10.1101/2020.08.25.267336. [DOI] [Google Scholar]
- 8.Chen J et al. Single-molecule dynamics of enhanceosome assembly in embryonic stem cells. Cell 156, 1274–1285 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu Z & Tjian R Visualizing transcription factor dynamics in living cells. J. Cell Biol 217, 1181–1191 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rabani M et al. High-resolution sequencing and modeling identifies distinct dynamic RNA regulatory strategies. Cell 159, 1698–1710 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jovanovic M et al. Immunogenetics. Dynamic profiling of the protein life cycle in response to pathogens. Science 347, 1259038 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rabani M, Pieper L, Chew G-L & Schier AF A massively parallel reporter assay of 3′ UTR sequences identifies in vivo rules for mRNA degradation. Mol. Cell 68, 1083–1094.e5 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stoeckius M et al. Simultaneous epitope and transcriptome measurement in single cells. Nat. Methods 14, 865–868 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Peterson VM et al. Multiplexed quantification of proteins and transcripts in single cells. Nat. Biotechnol 35, 936–939 (2017). [DOI] [PubMed] [Google Scholar]
- 15.Gerlach JP et al. Combined quantification of intracellular (phospho-)proteins and transcriptomics from fixed single cells. Sci. Rep 9, 1469 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reimegård J et al. Combined mRNA and protein single cell analysis in a dynamic cellular system using SPARC. bioRxiv (2019) doi: 10.1101/749473. [DOI] [Google Scholar]
- 17.Katzenelenbogen Y et al. Coupled scRNA-seq and intracellular protein activity reveal an immunosuppressive role of TREM2 in cancer. Cell 182, 872–885.e19 (2020). [DOI] [PubMed] [Google Scholar]
- 18.Rivello F et al. Single-cell intracellular epitope and transcript detection revealing signal transduction dynamics. bioRxiv (2020) doi: 10.1101/2020.12.02.408120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mimitou EP et al. Scalable, multimodal profiling of chromatin accessibility, gene expression and protein levels in single cells. Nat. Biotechnol (2021) doi: 10.1038/s41587-021-00927-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mimitou EP et al. Multiplexed detection of proteins, transcriptomes, clonotypes and CRISPR perturbations in single cells. Nat. Methods 16, 409–412 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Frangieh CJ et al. Multi-modal pooled Perturb-CITE-Seq screens in patient models define novel mechanisms of cancer immune evasion. bioRxiv (2020) doi: 10.1101/2020.09.01.267211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang Y et al. Multiplexed in situ protein imaging using DNA-barcoded antibodies with extended hybridization chain reactions. bioRxiv (2018) doi: 10.1101/274456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weichert W et al. High expression of RelA/p65 is associated with activation of nuclear factor-kappaB-dependent signaling in pancreatic cancer and marks a patient population with poor prognosis. Br. J. Cancer 97, 523–530 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lehmann A et al. High class I HDAC activity and expression are associated with RelA/p65 activation in pancreatic cancer in vitro and in vivo. BMC Cancer 9, 395 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang S-H et al. Nuclear expression of glioma-associated oncogene homolog 1 and nuclear factor-κB is associated with a poor prognosis of pancreatic cancer. Oncology 85, 86–94 (2013). [DOI] [PubMed] [Google Scholar]
- 26.Kim HD, Shay T, O’Shea EK & Regev A Transcriptional regulatory circuits: predicting numbers from alphabets. Science 325, 429–432 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.La Manno G et al. RNA velocity of single cells. Nature 560, 494–498 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Buccitelli C & Selbach M mRNAs, proteins and the emerging principles of gene expression control. Nat. Rev. Genet 21, 630–644 (2020). [DOI] [PubMed] [Google Scholar]
- 29.Lee TI & Young RA Transcriptional regulation and its misregulation in disease. Cell 152, 1237–1251 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nelson DE et al. Oscillations in NF-kappaB signaling control the dynamics of gene expression. Science 306, 704–708 (2004). [DOI] [PubMed] [Google Scholar]
- 31.Hafner A et al. p53 pulses lead to distinct patterns of gene expression albeit similar DNA-binding dynamics. Nat. Struct. Mol. Biol 24, 840–847 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.O’Neill LAJ & Kaltschmidt C NF-kB: a crucial transcription factor for glial and neuronal cell function. Trends Neurosci. 20, 252–258 (1997). [DOI] [PubMed] [Google Scholar]
- 33.Spiegel I et al. Npas4 regulates excitatory-inhibitory balance within neural circuits through cell-type-specific gene programs. Cell 157, 1216–1229 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hu P et al. Dissecting cell-type composition and activity-dependent transcriptional state in mammalian brains by massively parallel single-nucleus RNA-seq. Mol. Cell 68, 1006–1015.e7 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hrvatin S et al. Single-cell analysis of experience-dependent transcriptomic states in the mouse visual cortex. Nat. Neurosci 21, 120–129 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yap E-L et al. Bidirectional perisomatic inhibitory plasticity of a Fos neuronal network. Nature (2020) doi: 10.1038/s41586-020-3031-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sigal A et al. Dynamic proteomics in individual human cells uncovers widespread cell-cycle dependence of nuclear proteins. Nat. Methods 3, 525–531 (2006). [DOI] [PubMed] [Google Scholar]
- 38.Purvis JE et al. P53 dynamics control cell fate. Science 336, 1440–1444 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gaublomme JT et al. Nuclei multiplexing with barcoded antibodies for single-nucleus genomics. Nat. Commun 10, 2907 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McKinley KL & Cheeseman IM Large-scale analysis of CRISPR/Cas9 cell-cycle knockouts reveals the diversity of p53-dependent responses to cell-cycle defects. Dev. Cell 40, 405–420.e2 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Feldman D et al. Optical pooled screens in human cells. Cell 179, 787–799.e17 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rosenberg AB et al. Single-cell profiling of the developing mouse brain and spinal cord with split-pool barcoding. Science 360, 176–182 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Saka SK et al. Immuno-SABER enables highly multiplexed and amplified protein imaging in tissues. Nat. Biotechnol 37, 1080–1090 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Aitchison J Measures of location of compositional data sets. Math. Geol 21, 787–790 (1989). [Google Scholar]
- 45.McGinnis CS et al. MULTI-seq: sample multiplexing for single-cell RNA sequencing using lipid-tagged indices. Nat. Methods 16, 619–626 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vezzani A, French J, Bartfai T & Baram TZ The role of inflammation in epilepsy. Nat. Rev. Neurol 7, 31–40 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lévesque M & Avoli M The kainic acid model of temporal lobe epilepsy. Neurosci. Biobehav. Rev 37, 2887–2899 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lerea LS & McNamara JO Ionotropic glutamate receptor subtypes activate c-fos transcription by distinct calcium-requiring intracellular signaling pathways. Neuron 10, 31–41 (1993). [DOI] [PubMed] [Google Scholar]
- 49.Shen W, Zhang C & Zhang G Nuclear factor kappaB activation is mediated by NMDA and non-NMDA receptor and L-type voltage-gated Ca(2+) channel following severe global ischemia in rat hippocampus. Brain Res. 933, 23–30 (2002). [DOI] [PubMed] [Google Scholar]
- 50.Kaltschmidt B, Widera D & Kaltschmidt C Signaling via NF-kappaB in the nervous system. Biochim. Biophys. Acta 1745, 287–299 (2005). [DOI] [PubMed] [Google Scholar]
- 51.Dammer EB et al. Neuron enriched nuclear proteome isolated from human brain. J. Proteome Res 12, 3193–3206 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kierdorf K et al. Microglia emerge from erythromyeloid precursors via Pu.1- and Irf8-dependent pathways. Nat. Neurosci 16, 273–280 (2013). [DOI] [PubMed] [Google Scholar]
- 53.Buus TB et al. Improving oligo-conjugated antibody signal in multimodal single-cell analysis. Elife 10, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Habib N et al. Disease-associated astrocytes in Alzheimer’s disease and aging. Nat. Neurosci 23, 701–706 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Saunders A et al. Molecular diversity and specializations among the cells of the adult mouse brain. Cell 174, 1015–1030.e16 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fleming SJ, Marioni JC & Babadi M CellBender remove-background: a deep generative model for unsupervised removal of background noise from scRNA-seq datasets. bioRxiv (2019) doi: 10.1101/791699. [DOI] [Google Scholar]
- 57.Morgan JI, Cohen DR, Hempstead JL & Curran T Mapping patterns of c-fos expression in the central nervous system after seizure. Science 237, 192–197 (1987). [DOI] [PubMed] [Google Scholar]
- 58.Bergeron D, Pal G, Beaulieu YB, Chabot B & Bachand F Regulated intron retention and nuclear pre-mRNA decay contribute to PABPN1 autoregulation. Mol. Cell. Biol 35, 2503–2517 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hughes TK et al. Second-Strand Synthesis-based massively parallel scRNA-seq reveals cellular states and molecular features of human inflammatory skin pathologies. Immunity 53, 878–894.e7 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Crotti A et al. Mutant Huntingtin promotes autonomous microglia activation via myeloid lineage-determining factors. Nat. Neurosci 17, 513–521 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lind D, Franken S, Kappler J, Jankowski J & Schilling K Characterization of the neuronal marker NeuN as a multiply phosphorylated antigen with discrete subcellular localization. J. Neurosci. Res 79, 295–302 (2005). [DOI] [PubMed] [Google Scholar]
- 62.Stein B et al. Cross-coupling of the NF-kappa B p65 and Fos/Jun transcription factors produces potentiated biological function. EMBO J. 12, 3879–3891 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fujioka S et al. NF-kappaB and AP-1 connection: mechanism of NF-kappaB-dependent regulation of AP-1 activity. Mol. Cell. Biol 24, 7806–7819 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cleary B, Cong L, Cheung A, Lander ES & Regev A Efficient generation of transcriptomic profiles by random composite measurements. Cell 171, 1424–1436.e18 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fernandez-Albert J et al. Immediate and deferred epigenomic signatures of in vivo neuronal activation in mouse hippocampus. Nat. Neurosci 22, 1718–1730 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gupta S, Campbell D, Dérijard B & Davis RJ Transcription factor ATF2 regulation by the JNK signal transduction pathway. Science 267, 389–393 (1995). [DOI] [PubMed] [Google Scholar]
- 67.Vierbuchen T et al. AP-1 Transcription Factors and the BAF Complex Mediate Signal-Dependent Enhancer Selection. Mol. Cell 68, 1067–1082.e12 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Strauss KI et al. Prolonged cyclooxygenase-2 induction in neurons and glia following traumatic brain injury in the rat. J. Neurotrauma 17, 695–711 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gayoso A et al. Joint probabilistic modeling of single-cell multi-omic data with totalVI. Nat. Methods 18, 272–282 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hartmann FJ et al. Single-cell metabolic profiling of human cytotoxic T cells. Nat. Biotechnol (2020) doi: 10.1038/s41587-020-0651-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Qiu Q et al. Massively parallel and time-resolved RNA sequencing in single cells with scNT-seq. Nat. Methods (2020) doi: 10.1038/s41592-020-0935-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Boon R, Silveira GG & Mostoslavsky R Nuclear metabolism and the regulation of the epigenome. Nat Metab (2020) doi: 10.1038/s42255-020-00285-4. [DOI] [PubMed] [Google Scholar]
- 73.Ma S et al. Chromatin potential identified by shared single-cell profiling of RNA and chromatin. Cell 183, 1103–1116.e20 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Biancalani T et al. Deep learning and alignment of spatially-resolved whole transcriptomes of single cells in the mouse brain with Tangram. bioRxiv (2020) doi: 10.1101/2020.08.29.272831. [DOI] [Google Scholar]
- 75.Amamoto R et al. FIN-Seq: transcriptional profiling of specific cell types from frozen archived tissue of the human central nervous system. Nucleic Acids Res. 48, e4 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Racine RJ Modification of seizure activity by electrical stimulation: II. Motor seizure. Electroencephalogr. Clin. Neurophysiol 32, 281–294 (1972). [DOI] [PubMed] [Google Scholar]
- 77.Li B et al. Cumulus provides cloud-based data analysis for large-scale single-cell and single-nucleus RNA-seq. Nat. Methods 17, 793–798 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bakken TE et al. Single-nucleus and single-cell transcriptomes compared in matched cortical cell types. PLoS One 13, e0209648 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Stoeckius M et al. Cell Hashing with barcoded antibodies enables multiplexing and doublet detection for single cell genomics. Genome Biol. 19, 224 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wolf FA, Angerer P & Theis FJ SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 19, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Korsunsky I et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 16, 1289–1296 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.McInnes L, Healy J & Melville J UMAP: Uniform Manifold Approximation and Projection for Dimension Reduction. arXiv [stat.ML] (2018). [Google Scholar]
- 83.Traag VA, Waltman L & van Eck NJ From Louvain to Leiden: guaranteeing well-connected communities. Sci. Rep 9, 5233 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wolock SL, Lopez R & Klein AM Scrublet: Computational identification of cell Doublets in Single-cell transcriptomic data. Cell Syst. 8, 281–291.e9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tirosh I et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 352, 189–196 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Raudvere U et al. g:Profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 47, W191–W198 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw gene expression count matrices of all inCITE-seq data, BAM files of mapped reads, and matrix of mouse hippocampus inCITE-seq data jointly embedded with snRNA-seq data are available on Gene Expression Omnibus under the accession GSE163480. Data from Habib et al54. is available under GSE143758. Data from MULTI-seq used to compare RNA complexity in HEK cells is available under GSE129578. Databases of transcription factor motifs (cis-bs and JASPAR2018_CORE_vertebrates_non-redundant) are available at http://cisbp.ccbr.utoronto.ca and http://jaspar2018.genereg.net, respectively.