

Abstract
Mitochondrial respiration in mammalian cells not only generates ATP to meet their own energy needs but also couples with biosynthetic pathways to produce metabolites that can be exported to support neighboring cells. However, how defects in mitochondrial respiration influence these biosynthetic and exporting pathways remains poorly understood. Mitochondrial dysfunction in retinal pigment epithelium (RPE) cells is an emerging contributor to the death of their neighboring photoreceptors in degenerative retinal diseases including age-related macular degeneration. In this study, we used targeted-metabolomics and 13C tracing to investigate how inhibition of mitochondrial respiration influences the intracellular and extracellular metabolome. We found inhibition of mitochondrial respiration strikingly influenced both the intracellular and extracellular metabolome in primary RPE cells. Intriguingly, the extracellular metabolic changes sensitively reflected the intracellular changes. These changes included substantially enhanced glucose consumption and lactate production; reduced release of pyruvate, citrate, and ketone bodies; and massive accumulation of multiple amino acids and nucleosides. In conclusion, these findings reveal a metabolic signature of nutrient consumption and release in mitochondrial dysfunction in RPE cells. Testing medium metabolites provides a sensitive and noninvasive method to assess mitochondrial function in nutrient utilization and transport.
Keywords: mitochondrial respiration, metabolism, retinal pigment epithelium, metabolites, glucose, amino acids, nucleotides, ketone bodies
Graphical Abstract
INTRODUCTION
The retinal pigment epithelium (RPE) maintains the health of the neural retina by performing several critical functions, including nutrient transport, phagocytosis of the outer segments and secretion of cytokines.1 These RPE-specific processes rely on active energy metabolism. Recent evidence indicates that dysfunctional RPE mitochondrial bioenergetics is a crucial factor for many retinal degenerative diseases, including age-related macular degeneration (AMD), one of the leading causes of blindness.2–4 Inhibition of mitochondrial metabolism specifically in RPE is sufficient to induce AMD-like retinal degeneration in mice.5–7 RPE cells from AMD donors have impaired mitochondrial metabolism.8,9 However, the detailed mechanism of how impaired mitochondrial metabolism in RPE results in photoreceptor degeneration remains elusive.
Mitochondrial respiration is the central process in energy metabolism. Reducing equivalents NADH and FADH2, generated by the tricarboxylic acid (TCA) cycle, donate electrons to complex I to II in the electron transport chain (ETC), and oxygen is finally consumed to produce ATP through ATP synthase (complex V). Besides being a primary generator of energy, mitochondrial respiration through the TCA cycle also provide critical biosynthetic pathways to generate glucose, fatty acid, ketone bodies, and nonessential amino acids.10,11 Growing evidence shows that RPE mitochondria are crucial for the metabolic communication between RPE and outer retina by synthesizing nutrients to support retinal health.12–14 Each day, approximately 10% of photoreceptor outer segments are shed and phagocytosed by RPE.15 RPE mitochondria are capable of converting the phagocytosed lipids into ketone bodies and recycling them back to fuel photoreceptors.16,17 Furthermore, the retina generates a massive amount of lactate by the Warburg effect.18,19 A recent study indicated that RPE mitochondria may use lactate released from photoreceptors as an energy source to maintain their function, thus preserving the majority of glucose for photoreceptors.20 Moreover, we recently showed that RPE mitochondria prefer to metabolize nutrients such as proline into intermediates that are exported toward the retinal side.21 Nevertheless, how mitochondrial dysfunction impacts RPE nutrient utilization and secretion of metabolites toward retina is not well-defined.
Mitochondrial function is typically assessed by measuring oxygen consumption in isolated mitochondria or live cells by sequentially adding substrates and inhibitors of mitochondrial complexes. The major drawbacks of these techniques are (1) cells cannot be reused after the assay, and (2) the activities of mitochondrial biosynthetic pathways are not measured. Primary human fetal or adult RPE cultures are excellent disease models since they maintain many properties similar to native RPE. These primary cultures are expensive and labor-intensive, so a noninvasive method to quantify mitochondrial function for these cells is highly desirable.
In the present study, we used targeted-metabolomics approaches to investigate how inhibition of mitochondrial complexes influences the intracellular and extracellular metabolome in primary cultured human RPE cells. Our findings demonstrate that inhibition of mitochondrial complexes causes early and unique changes in medium metabolites, RPE mitochondrial function is critical for nutrient synthesis, and quantification of metabolites in the media may provide a novel method for measuring mitochondrial metabolism.
EXPERIMENTAL PROCEDURES
Reagents
Mitochondrial respiration inhibitors, culture media, and other reagents are listed in detail in Table S5 in the Supporting Information (SI).
Primary RPE Cell Culture
Human RPE culture was generated from human donors without known ocular diseases as previously described.22 The protocols for this culture and sample preparation were approved by the Institutional Review Board from both the University of Washington and the West Virginia University. RPE was initially plated in a 12-well or 35 mm tissue culture plates coated with Growth Factor Reduced (GFR) Matrigel Matrix (Corning). The cells were cultured in Minimum Essential Medium Alpha (MEMα) medium, supplemented with 5% (v/v) fetal bovine serum (FBS), N1Medium Supplement, hydrocortisone, triiodo-thyronine, taurine, nonessential amino acids, and a penicillin-streptomycin solution (see details in Table S5 in the SI). Then 10 μM Y-27632 dihydrochloride was included for the first 1–2 weeks of culturing. At confluency, the media was changed to a 1% (v/v) FBS supplemented media without Y-27632 dihydrochloride. RPE cells were passaged to a 12-well plate at a density of 400 000–550 000 cells/well for experiments. The RPE cells were cultured for at least 3 weeks to attain maturity before use in experiments.
Mitochondrial Oxygen Consumption Assay
Human RPE cells were seeded on Agilent Seahorse culture plates and cultured for 3–4 weeks to attain maturity. The assay was performed according to the Agilent Seahorse User Guide. Specifically, the day before the assay, the sensor cartridge was hydrated with sterile water overnight in a non-CO2 incubator. Then 1 h before starting, the water was removed and replaced with the XF Calibrant buffer and placed in a non-CO2 incubator for at least 1 h. Before running, the cells were washed twice with the Agilent Base media. The base media was supplemented with 5.5 mM glucose, 1 mM sodium pyruvate, and 2 mM l-glutamine, and the pH was adjusted to 7.2–7.4. Drugs were prepared at the following concentrations: standard treatment of oligomycin 25 μM stock (port A), FCCP 5 μM stock (port B), and rotenone/antimycin 20 μM stock (port C), to yield 2.5, 0.5, and 2 μM, respectively, in the well. For individual mitochondrial inhibitor treatment, piericidin was prepared at 10 μM, antimycin at 10 μg/mL, and oligomycin at 50 μM to yield 1 μM, 1 μg/mL, and 5 μM in the well, respectively. Each of the single drugs tests were loaded in port A with media blanks in the other ports to control for volume. The settings of the “Mito Stress Test” were used as a template. It included 24 min of three cycles: mixing for 3 min, waiting for 2 min, and measuring for 3 min. The oxygen consumption rates (OCR) and extracellular acidification rates (ECAR) measurements were taken at these time points. All of the reagents used for this assay are detailed in Table S5, SI.
Cell Treatment and Sample Preparation
The mature RPE cells were treated with 1 μM piericidin A, 1 μg/mL antimycin A, or 5 μM oligomycin in regular 1% supplemented RPE media described above. An equivalent volume of diluent DMSO was added as vehicle control (1 μL/mL). A total of 50 μL of media was collected at 1, 6, and 24 h. Cells were harvested at 24 h by quickly rinsing with a cold 0.9% NaCl solution and adding 100 μL of 80% methanol precooled at −20 °C. Cells were scraped on dry ice and rinsed with an additional 100 μL of 80% methanol. The cells were transferred to a microcentrifuge, homogenized, and centrifuged to extract metabolites as described.23
For labeling with [13C6]-glucose, piericidin, antimycin, and oligomycin or control DMSO were added at the same concentrations to clear DMEM media (Gibco A144301) without and then supplemented with 1% FBS and penicillin-streptomycin and 5 mM [13C6]-glucose. RPE cells were changed into the DMEM media with DMSO or inhibitors. The media and cells were collected in the same way as mentioned above.
Metabolite Analysis with Liquid Chromatography–Mass Spectrometry (LC-MS) and Gas Chromatography–Mass Spectrometry (GC-MS)
Metabolite preparation and analysis with LC-MS and GC-MS were performed as previously reported.14,23 Medium samples were centrifuged to remove debris, and 10 μL of the supernatant was mixed with 40 μL of cold methanol to extract the metabolites. Supernatants containing aqueous metabolites from both media and cells were dried at 4 °C and analyzed by LC-MS or GC MS. LC-MS used a Shimadzu LC Nexera X2 UHPLC coupled with a QTRAP 5500 LC MS/MS (AB Sciex). An ACQUITY UPLC BEH Amide analytic column (2.1 × 50 mm, 1.7 μm, Waters) was used for chromatographic separation. Each metabolite was tuned with standards for optimal transitions. The extracted multiple reaction monitoring (MRM) peaks were integrated using MultiQuant 3.0.2 software (AB Sciex). Table S6 lists the detailed parameters for the measured metabolites. For GC-MS, the samples were derivatized by methoxyamine and N-tertbutyldimethylsilyl-N-methyltrifluoroacetamide and analyzed by the Agilent 7890B/5977B GC-MS system with a DB-5MS column (30 m × 0.25 mm × 0.25 μm film). Mass spectra were collected from 80–600 m/z under selective ion monitoring mode. The detailed parameters are listed in Table S6, SI. The data was analyzed by Agilent MassHunter Quantitative Analysis Software and natural abundance was corrected by ISOCOR software.24
Statistical Analysis
Multivariate analysis was performed with a supervised classification model partial least-squares discriminant analysis (PLS-DA) after auto scaling using MetaboAnalyst 4.0 (https://www.metaboanalyst.ca/). The comparison of specific metabolites was analyzed with One Way ANOVA and false discovery rate (FDR) correction using Graphpad Prism 8.0 (GraphPad Software, Inc. La Jolla, CA). Data are presented as mean ± standard error unless specified. Values of p < 0.05 were considered significantly different.
RESULTS
Inhibition of Mitochondrial Respiration Severely Disrupts Nutrient Consumption and Utilization in RPE Cells
To study how mitochondrial dysfunction affects metabolites, we used piericidin, antimycin, and oligomycin to inhibit mitochondrial complex I, complex III, and complex V, respectively, in fully differentiated primary RPE cells. We first tested the efficiency of these inhibitors on mitochondrial respiration by measuring oxygen consumption with the Seahorse Analyzer. Similar to other cells, primary RPE cells had a robust mitochondrial oxygen consumption rate (OCR) in basal and maximal respiration (Figure 1A). Inhibition of mitochondrial respiration with any of the three inhibitors efficiently inhibited basal respiration and enhanced glycolysis as indicated by the OCR and extracellular acidification rate (ECAR) (Figure 1B–D). We incubated human RPE cells with these inhibitors at the same concentrations that were used in oxygen consumption assays. The culture media at 1, 6, and 24 h and cells at 24 h were then collected to analyze metabolites by mass spectrometry (Figure 2A). To evaluate the toxicity of these inhibitors, we analyzed lactate dehydrogenase (LDH) activity in the media since dead cells release LDH into the culture media. We did not observe changes in LDH activity at 1, 6, and 24 h for all inhibitors except for antimycin, which increased LDH activity only at 24 h (Figure S1). To study whether the overall metabolites in the media were different between groups treated with control (dimethyl sulfoxide, DMSO) or inhibitors, we performed a multivariate analysis of the metabolomics data with PLS-DA. The scores plot showed that the metabolites with three inhibitors in the media at 1 h overlapped with those from the control, but the metabolites with inhibitors were well separated from the control in media and cells at 6 and 24 h after treatment (Figure 2B–E). Although the oligomycin group had a distinctive profile in the cells, it was not separate from the groups with the other two inhibitors in the media. Among the 126 metabolites we detected, 11–43 extracellular metabolites were significantly changed from 1 to 24 h, and 118 intracellular metabolites were significantly altered (Figure 2F and Tables S1–S4). Pathway analysis showed that the changed extracellular metabolites were enriched in amino acid metabolism, glycolysis and TCA cycle, nucleotide metabolism, ketone bodies and acylcarnitines, NAD metabolism, and urea cycle (Figure 2G). Mitochondrial complexes are critical for the production of ATP, which could be stored as phosphocreatine (PCr). As expected, high-energy metabolites, including ATP, ADP, and PCr, were significantly attenuated by these inhibitors, confirming that mitochondrial energy metabolism was severely impaired in the primary RPE cells (Figure 2H).
Figure 1.
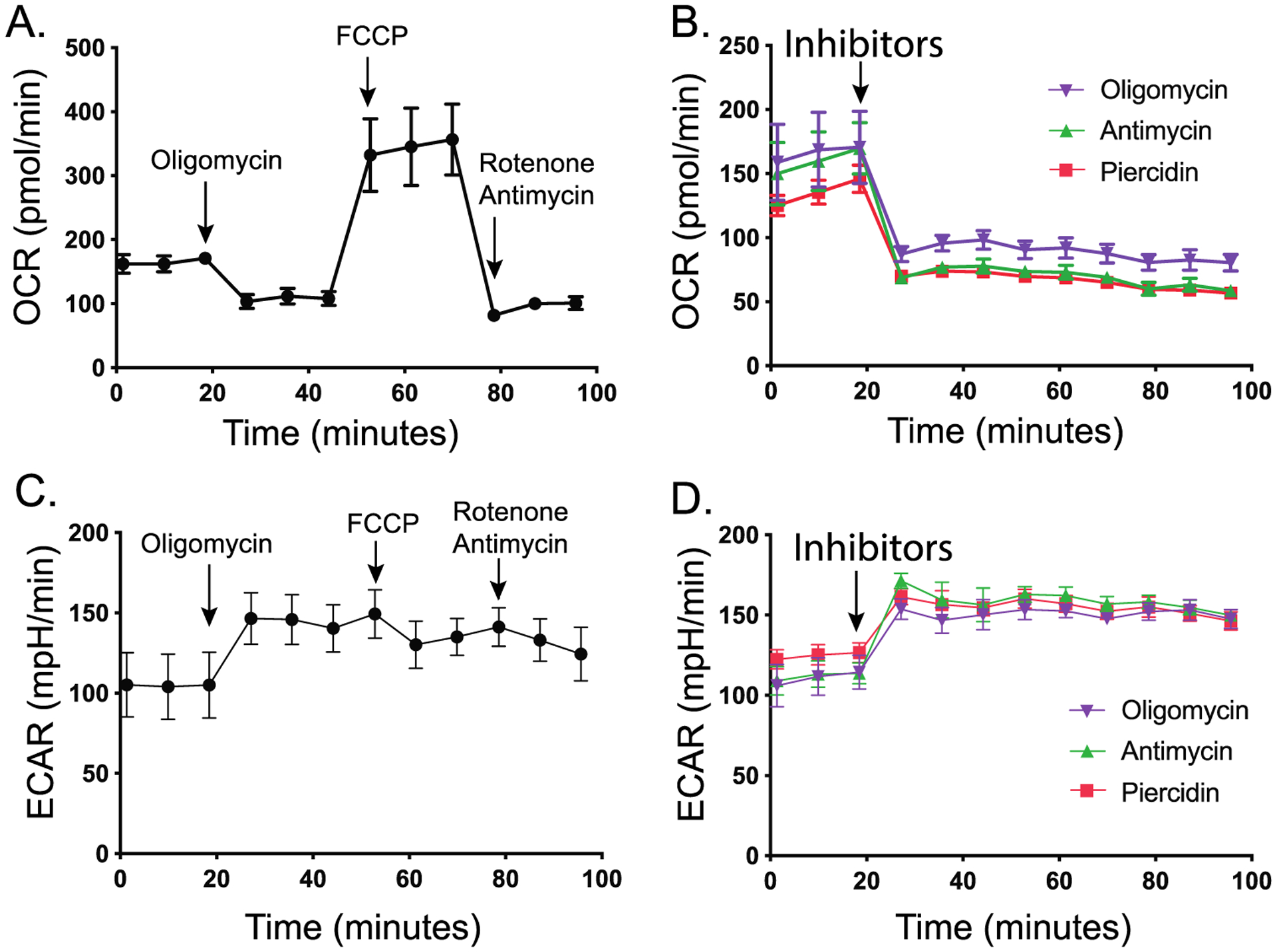
Mitochondrial complex inhibitors limit oxygen consumption and increase glycolysis in human RPE cells. (A–D) Primary RPE cells were measured for oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) using the Seahorse Analyzer. Panels A and C were standard treatments by sequential addition of oligomycin (2.5 μM), FCCP (0.5 μM), and rotenone and antimycin (2 μM) as indicated by arrows. Panels B and D were treated with different inhibitors (oligomycin at 5 μM, antimycin at 1 μg/mL, piericidin at 1 μM) at time points indicated by arrows. Error bars indicate standard error of the mean (SEM). N = 4. p < 0.0001 vs cells without inhibitors.
Figure 2.
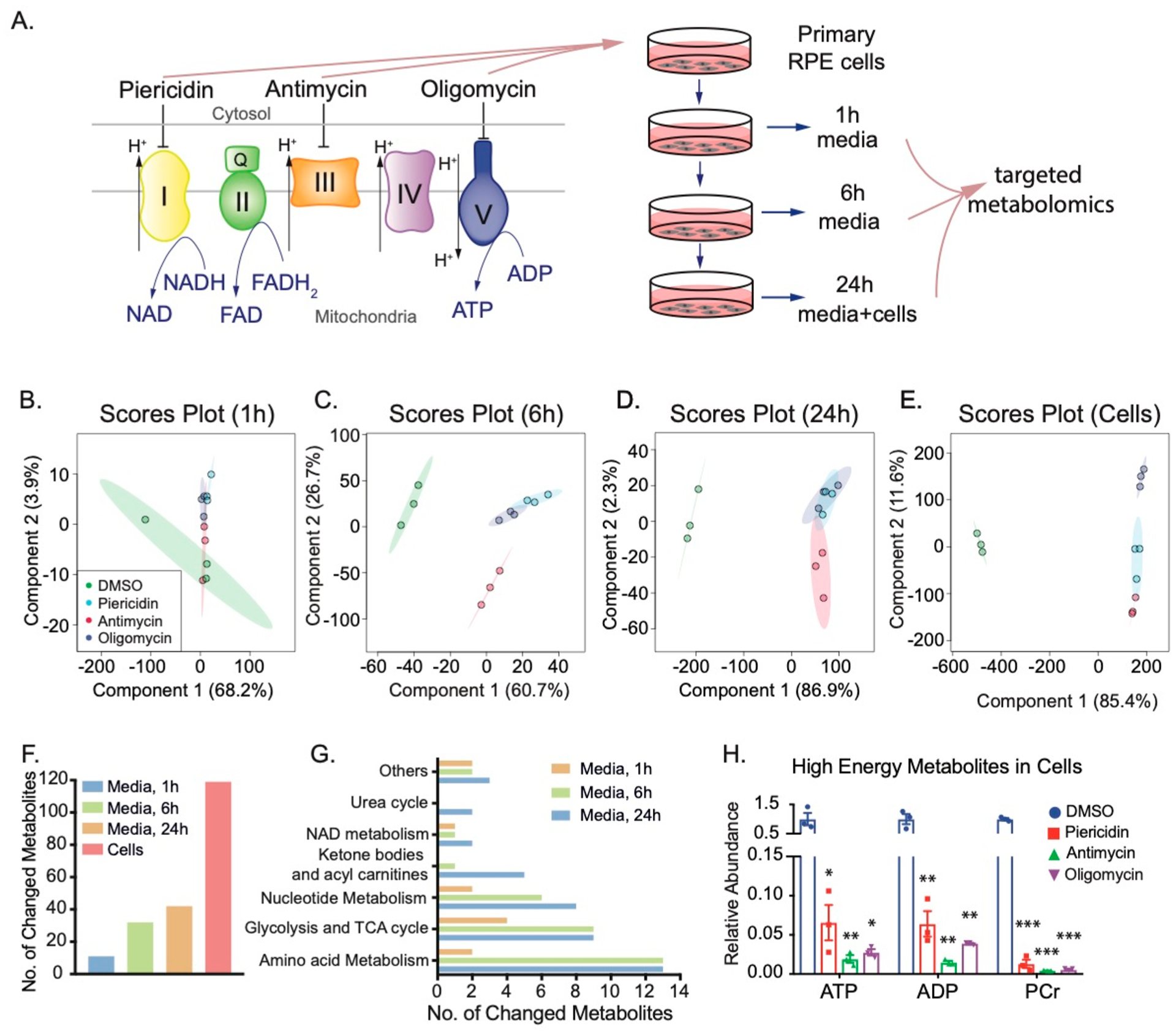
Inhibition of mitochondrial metabolism changes both intracellular and extracellular metabolites in human RPE cells. (A) Experimental design for quantifying intracellular and extracellular metabolites caused by mitochondrial dysfunction. Mitochondrial electron transport chain shuttles electrons from NADH and FADH to O2 to produce ATP. We inhibited complexes I, III, or V with their specific inhibitors, piericidin, antimycin, or oligomycin, respectively, in primary cultured RPE cells to investigate the impact of mitochondrial dysfunction on intracellular metabolites. Media and cells were collected at different time points as indicated and quantified by targeted metabolomics. (B–E) Scores plots of metabolites in cells and media at 1, 6, and 24 h by PLS-DA. (F) The number of changed metabolites in the medium at 1, 6, and 24 h, and in RPE cells. (G) The number of changed metabolites in different pathways in the medium at 1, 6, and 24 h. (H) Mitochondrial inhibitors decreased the levels of high-energy metabolites in cells. N = 3. *P < 0.05, **P < 0.01, *P < 0.001 vs the cells treated with DMSO. PCr, phosphocreatine.
Inhibition of Mitochondrial Respiration Disrupts Glucose Utilization and Metabolism
Glucose is an important nutrient to produce energy and intermediates through glycolysis and the TCA cycle (Figure 3A). After 24 h, mitochondrial inhibitors almost completely depleted intracellular glucose but significantly elevated intracellular lactate. However, most intracellular glycolytic intermediates upstream of lactate were decreased, especially by piericidin and antimycin (Figure 3B). Intracellular TCA cycle intermediates including citrate, isocitrate, and α-KG were decreased, but oxaloacetate (OAA) accumulated, indicating that citrate synthesis is inhibited (Figure 3C). Antimycin and oligomycin, but not piericidin, substantially increased intracellular succinate, confirming that the inhibition of complexes is specific because succinate does not need complex I for its oxidation (Figure 3C). Similar to the intracellular changes, glucose, pyruvate, citrate, and isocitrate dropped 2–10 fold as early as 6 h in the media, while lactate and OAA were increased by all inhibitors (Figure 3D–H,M). Similarly, extracellular succinate was increased by antimycin and oligomycin at 24 h (Figure 3J). These results support the concept that medium metabolites are important indicators for the metabolic status inside cells. However, the extracellular levels of α-KG, fumarate, and malate had different patterns of change (Figure 3I, 3K–L). This may be caused by different reaction rates of multiple metabolic pathways that are involved in their synthesis and catabolism.
Figure 3.
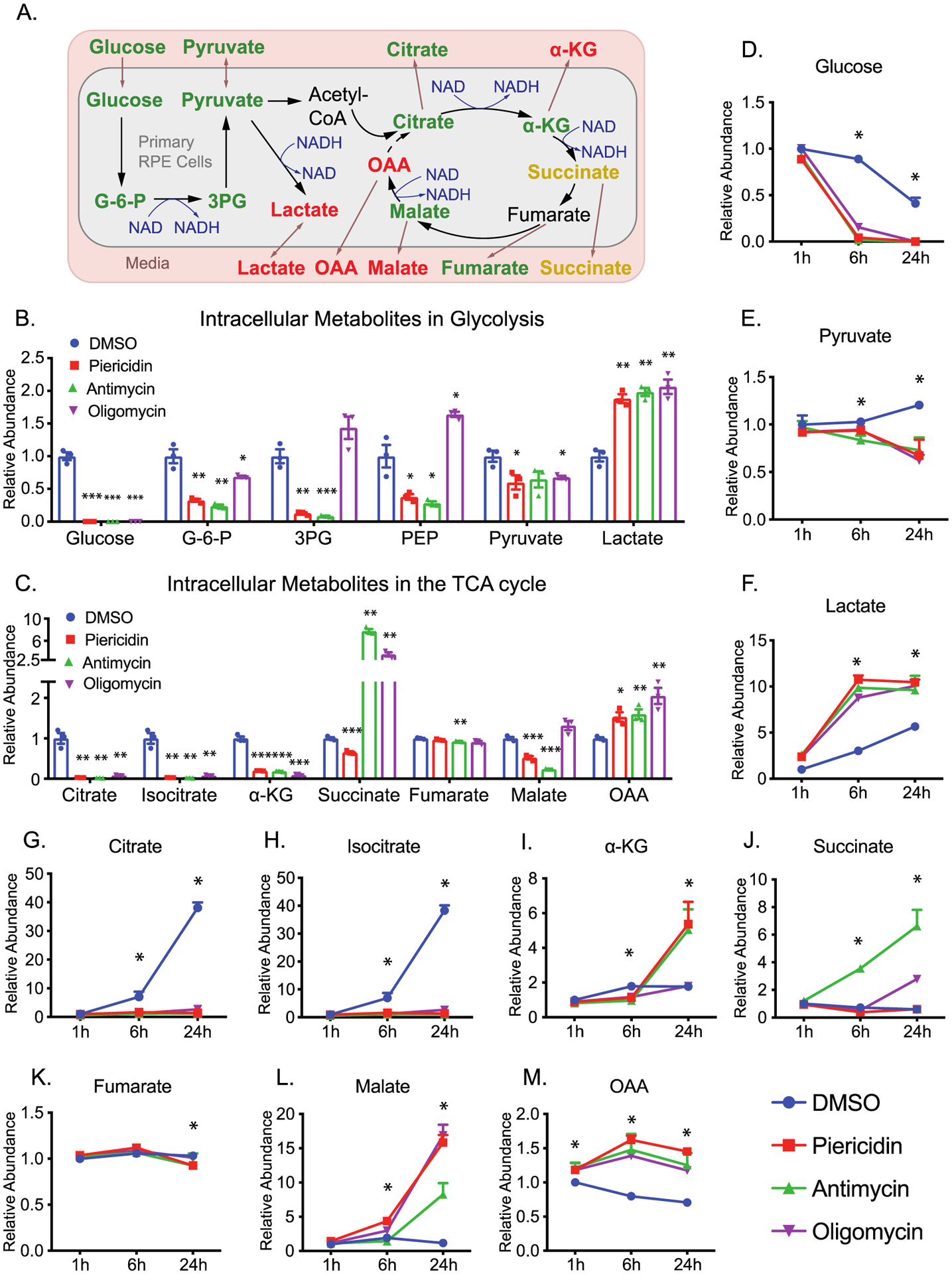
Inhibition of mitochondrial metabolism impairs intracellular and extracellular metabolites in glycolysis and the TCA cycle. (A) A schematic of the impact of mitochondrial dysfunction on glucose metabolism. Green represents a decrease, red represents an increase, and orange represents mixed changes by different inhibitors. (B–C) Significantly changed intracellular metabolites in glycolysis and the TCA cycle in the RPE cells. N = 3. *P < 0.05, **P < 0.01, *P < 0.001 vs the cells treated with DMSO. (D–M) The changed extracellular metabolites in glucose metabolism. N = 3. *P < 0.05 vs DMSO. G-6-P, glucose-6-phosphate; 3PG, 3-phosphoglycerate; PEP, phosphoenolpyruvate.
Inhibition of Mitochondrial Respiration Impairs the Utilization of 13C Glucose
The intermediates in glycolysis and the TCA cycle can be derived from multiple nutrients. To ask whether the changes come from glucose, we used [13C6]-glucose to trace glucose metabolism and analyzed 13C-labeled metabolites with GC-MS (Figure 4A). To avoid the interference of supplements such as glucose and pyruvate, we switched the media to DMEM without glucose. After the addition of 5 mM [13C6]-glucose in DMEM, more than 90% of intracellular pyruvate and lactate and 25–80% of intracellular TCA intermediates were 13C-labeled (Figure S2). Except for lactate, the enrichment (percentage of labeling) of all of the intermediates was reduced when mitochondrial metabolism was inhibited (Figure S2). Similarly, the levels of 13C-labeled intracellular pyruvate and mitochondrial intermediates were substantially diminished by mitochondrial inhibitors, while lactate was slightly increased or unchanged (Figure 4B,C). These results indicate that the inhibition of mitochondrial complexes may block the oxidation of NADH and activate the conversion of pyruvate into lactate. As expected, the ratio of total lactate to pyruvate, an indicator of cytosolic NADH, was increased 5–10 fold by these mitochondrial inhibitors (Figure S3). Consistently, the exported 13C-labeled lactate in the media increased as early as 1 h and had increased further at 6 and 24 h, but 13C pyruvate release decreased (Figure 4D,E), suggesting that the medium lactate and pyruvate are sensitive indicators of mitochondrial metabolism. We found RPE cells had high efflux of glucose-derived citrate and α-KG, but the inhibition of mitochondrial respiration entirely blocked this efflux (Figure 4F,G), consistent with the intracellular changes. However, intracellular and extracellular succinate, fumarate and malate changed in different patterns after mitochondrial inhibition (Figure 4H–J), indicating other factors or mechanisms are involved in regulating their release.
Figure 4.
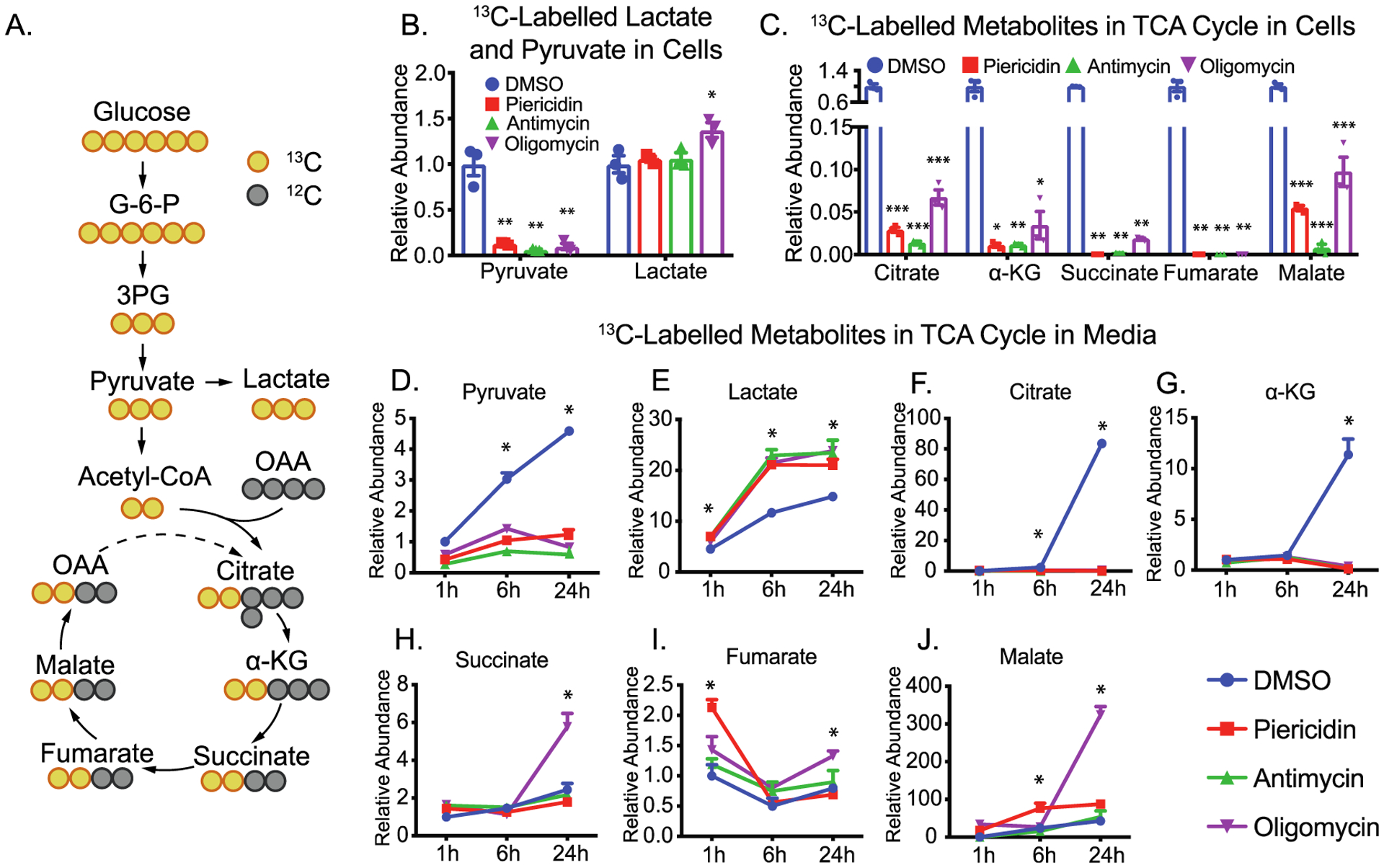
Inhibition of mitochondrial metabolism impairs glucose utilization. (A) A schematic for 13C labeling in intermediates in glycolysis and TCA cycle from [13C6]-glucose (yellow circles). OAA, oxaloacetate. (B and C) The relative abundance of 13C-labeled intracellular metabolites over the group treated with DMSO. (D–J) The relative abundance of 13C-labeled extracellular metabolites over the group treated with DMSO. N = 3. *P < 0.05, **P < 0.01, ***P < 0.001 vs the groups treated with DMSO.
Inhibition of Mitochondrial Respiration Impairs the Amino Acid Metabolism
The regular RPE culture medium is typically supplemented with abundant amino acids. The inhibition of mitochondrial respiration caused widespread changes of 44 intracellular amino acids and 13 extracellular amino acids (Figures 5, S3, and S4 and Tables S1–S4). Choline and proline were the two most sensitive and dramatically changed amino acids. With mitochondrial inhibition, both amino acids in the media were significantly increased at 1 h and had accumulated 5–20 fold by 24 h (Figure 5C,D). Consistently with changes in the medium, intracellular choline and proline also accumulated 5–12 fold (Figure 5A). These results suggest that choline and proline are sensitive amino acid markers for RPE mitochondrial metabolism. We found healthy RPE cells time-dependently consumed branch-chain amino acids (BCAAs) including leucine, isoleucine and valine from the media. However, both intracellular and extracellular BCAAs were elevated with the inhibition of mitochondrial metabolism (Figure 5A,E–G). These data suggest mitochondrial dysfunction blocks the utilization of BCAAs.
Figure 5.
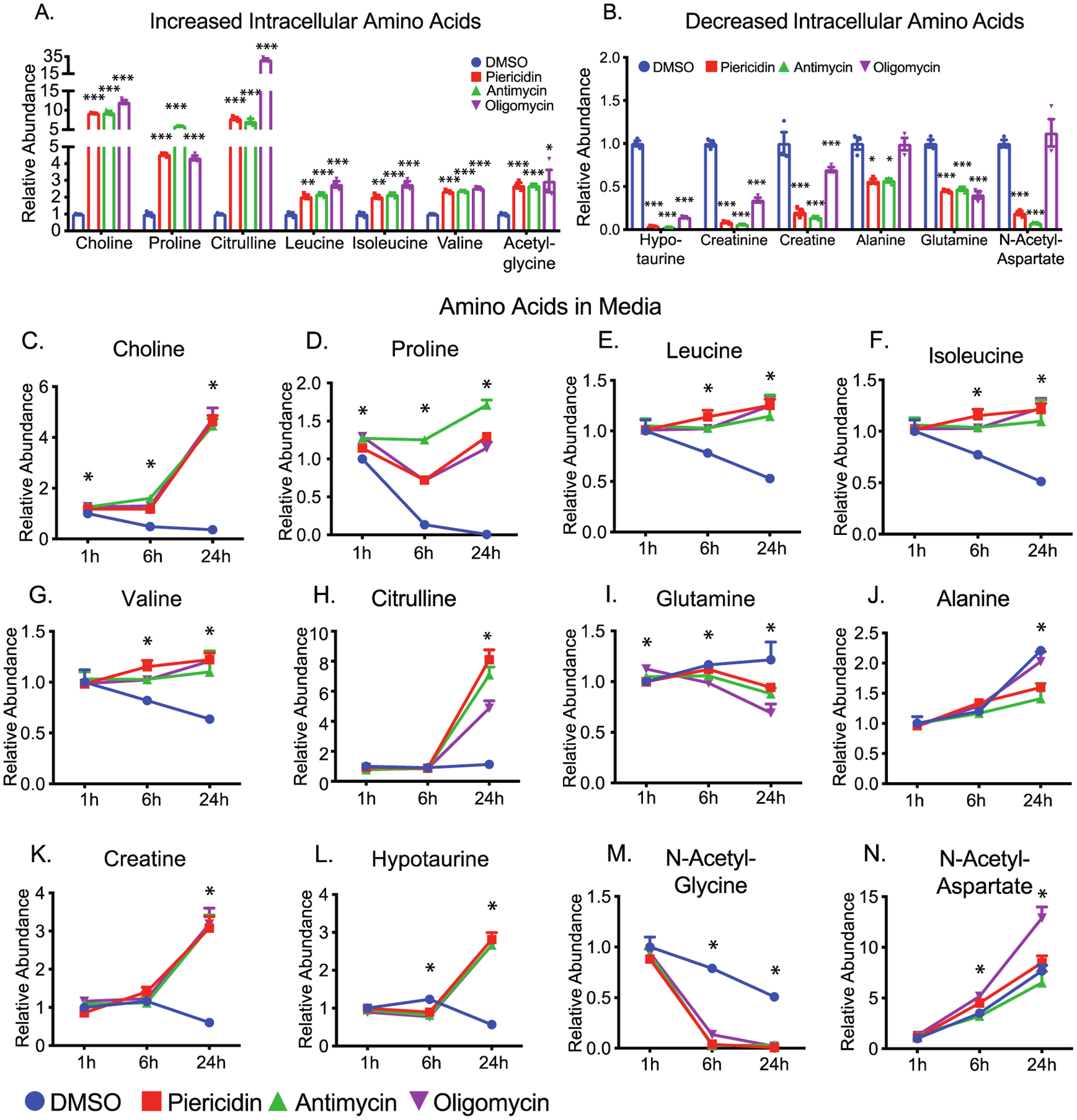
Inhibition of mitochondrial metabolism impairs amino acid metabolism and the urea cycle. (A and B) The relative abundance of significantly increased or decreased intracellular amino acids. (C–N) The relative abundance of amino acids in the media at different time points. N = 3. *P < 0.05, **P < 0.01, ***P < 0.001 vs the groups treated with DMSO. Gln, glutamine.
Although the urea cycle is restricted to the liver, several enzymes in the urea cycle are ubiquitous in mammalian cells to regulate aspartate, citrulline and arginine metabolism (Figure S4). Both intracellular and extracellular citrulline accumulated substantially (Figure 5A, 4H). Ornithine and aspartate (Figure S4) were increased or unchanged, but arginosuccinate was decreased by both piericidin and antimycin (Figure S4). These results suggest arginosuccinate synthase, the enzyme that catalyzes the conversion of citrulline into arginosuccinate, might be inhibited by the lack of ATP.
Glutamine and alanine decreased in both cells and media by mitochondrial inhibitors, suggesting that their consumption is enhanced, or their release is inhibited (Figures 5B and 4I,J). Changes of most other amino acids in media were opposite or inconsistent with their changes in cells such as creatine, hypotaurine, N-acetyl-glycine, and N-acetyl-aspartate (Figures 5A,B,K–N, S3, and S4). These disparities suggest that efflux or influx of these amino acids are affected differently by the inhibition of mitochondrial respiration.
Inhibition of Mitochondrial Respiration Impairs the Metabolism of Ketone Bodies and Acylcarnitines
RPE has active lipid metabolism to produce and release ketone bodies such as 3-hydroxybutyrate (3-HB; Figure 6A).16,17 Consistently, RPE cells time-dependently released 3-HB into the media. However, all three inhibitors almost completely shut down the production and release of 3-HB (Figure 6A–C), indicating that fatty acid oxidation is inhibited. Fatty acids use acylcarnitine shuttle into mitochondria for β-oxidation through forming acyl-CoAs. The intracellular levels of acetyl-carnitine (C2), propionyl-carnitine (C3), butyryl-carnitine (C4), hexanoyl-carnitine (C6), and myristoyl-carnitine (C14) were significantly decreased, supporting that inhibition of mitochondrial respiration resulted in impaired fatty oxidation (Figure 6D). Propionyl-carnitine, isobutyryl-carnitine, and methylbutyroyl-carnitine could be produced by the oxidation of BCAAs. The reduction of these acyl-carnitines confirms our findings in Figure 5 that the oxidation of BCAAs is impaired by mitochondrial inhibition. Paradoxically, extracellular acylcarnitines remained unchanged or accumulated by mitochondrial inhibitors, especially, oligomycin (Figure 6E–G). In sum, these data suggest that mitochondrial dysfunction inhibits fatty acid oxidation and ketone body production.
Figure 6.
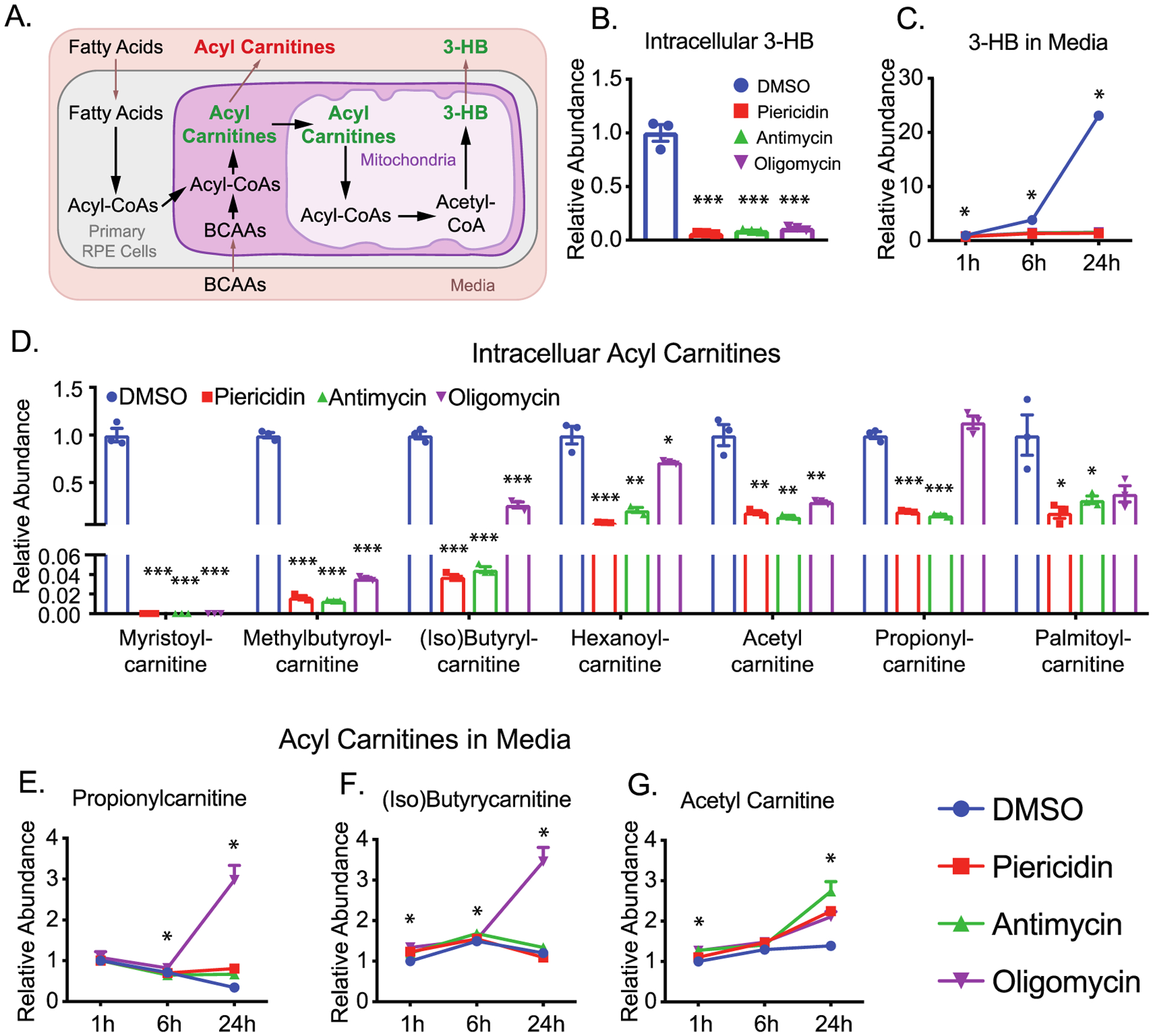
Inhibition of mitochondrial metabolism impairs the metabolism of ketone body production and acylcarnitines. (A) A schematic for the metabolism of ketone body and acylcarnitines. (B and C) Both intracellular and extracellular 3-hydroxybutyrate (3-HB) was significantly reduced after the inhibition of mitochondrial metabolism. (D) Inhibition of mitochondrial metabolism decreased the levels of intracellular acylcarnitines. (E-H) The relative abundance of extracellular acylcarnitines. N = 3. *P < 0.05, **P < 0.01, ***P < 0.001 vs the groups treated with DMSO.
Inhibition of Mitochondrial Respiration Impairs Purine and Pyrimidine Metabolism
Purine and pyrimidine could synthesize from de novo or salvage pathways using glucose and amino acids as precursors or catabolized nucleosides as precursors (Figure 7A,B). The inhibition of mitochondrial respiration massively accumulated metabolites from purine degradation such as guanine, guanosine, hypoxanthine, and xanthine (Figures 7C,E–J and S5). These purine metabolites increased by 5–20 fold in both cells and media, whereas some upstream purine intermediates such as ribulose-5-phosphate (R-5-P), inosine monophosphate (IMP), and adenosine monophosphate (AMP) were substantially diminished (Figure 7A,C–J). These results strongly suggest that inhibition of mitochondrial respiration accelerates the catabolism of purine metabolites and blocks purine synthesis. In a similar pattern, we found metabolites in pyrimidine catabolism including 3-aminoisobutyric acid (BAIBA), cytidine, cytosine, uracil, and β-alanine were accumulated in cells or media, but UDP was severely depleted (Figures 7B–D,K–N and S5). Impressively, BAIB accumulation in the media started at 1 h and reached more than five folds over control at 24 h (Figure 7M). Cytidine was not detected in the control media at 1 h but significantly increased in the media with inhibitors at 6 and 24 h (Figure 7K). Taken together, we found inhibition of mitochondrial metabolism enhances the catabolism of purine and pyrimidine. Nucleotides such as hypoxanthine and BAIBA may serve as sensitive and robust medium indicators for mitochondrial dysfunction.
Figure 7.
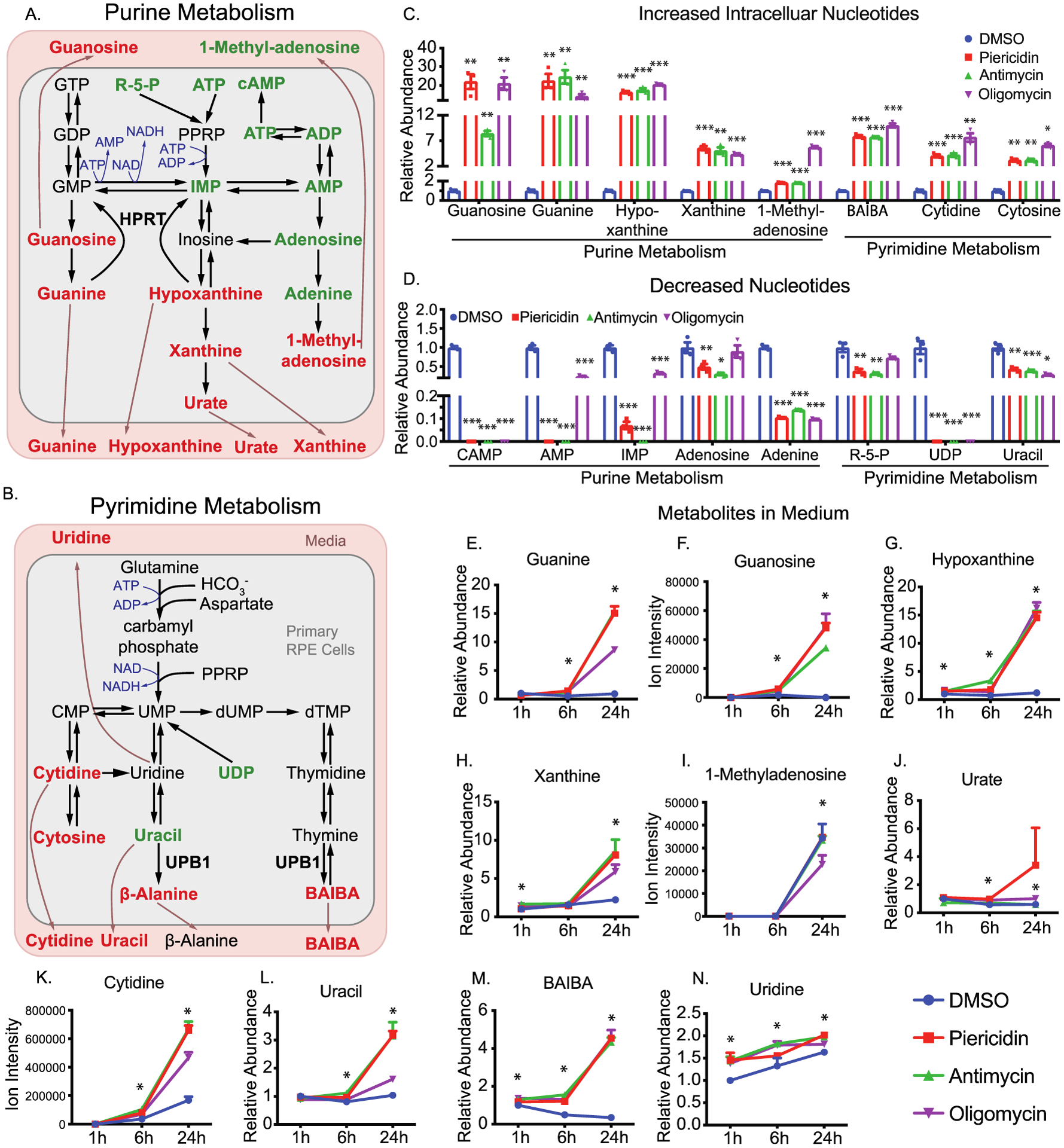
Inhibition of mitochondrial metabolism impairs nucleotide metabolism. (A and B) Schematics for purine and pyrimidine metabolism. The metabolites are colored to represent relative abundance by the inhibition of mitochondrial metabolism (red for the increase, green for decrease, and black for no change or not detected). (C–D) The relative abundance of significantly changed nucleotides by the inhibition of mitochondrial metabolism. (E–M) The relative abundance of significantly changed medium metabolites in nucleotide metabolism. N = 3. *P < 0.05, **P < 0.01, *P < 0.001 vs the groups treated with DMSO.
Inhibition of Mitochondrial Respiration Impairs NAD Metabolism
NAD is essential in mitochondrial energy metabolism by transferring protons from glycolysis, TCA cycle, and fatty acid oxidation. NAD could be synthesized from tryptophan or nicotinamide (Figure 8A). Upon the inhibition of mitochondrial respiration, intracellular NAD and its derivatives, NADH and NADP(H), were depleted. (Figure 8A,B). Because NAD(H) and NADP(H) are intracellularly produced and impermeable to cell membranes, they could not be detected in the media. Their precursors, tryptophan and nicotinamide, were significantly decreased in cells (Figure 8B). Medium tryptophan was not changed, while medium nicotinamide was increased ~5 fold at 24 h (Figure 8C). These findings demonstrate that nicotinamide consumption is blocked by mitochondrial inhibitors. The methylation of nicotinamide into 1-methylnicotinamide (MNA) for excretion is an important degradation pathway to regulate nicotinamide homeostasis (Figure 8A). We found healthy RPE cells released a large amount of MNA, ~20-fold increase in 24 h. Interestingly, extracellular MNA was significantly deceased, starting from 1 h, consistent with intracellular changes except for oligomycin which increased intracellular NMA (Figures 8B and 7D). These results indicate that NAD metabolism is impaired by mitochondrial inhibition, and nicotinamide and MNA are useful media markers for NAD metabolism.
Figure 8.
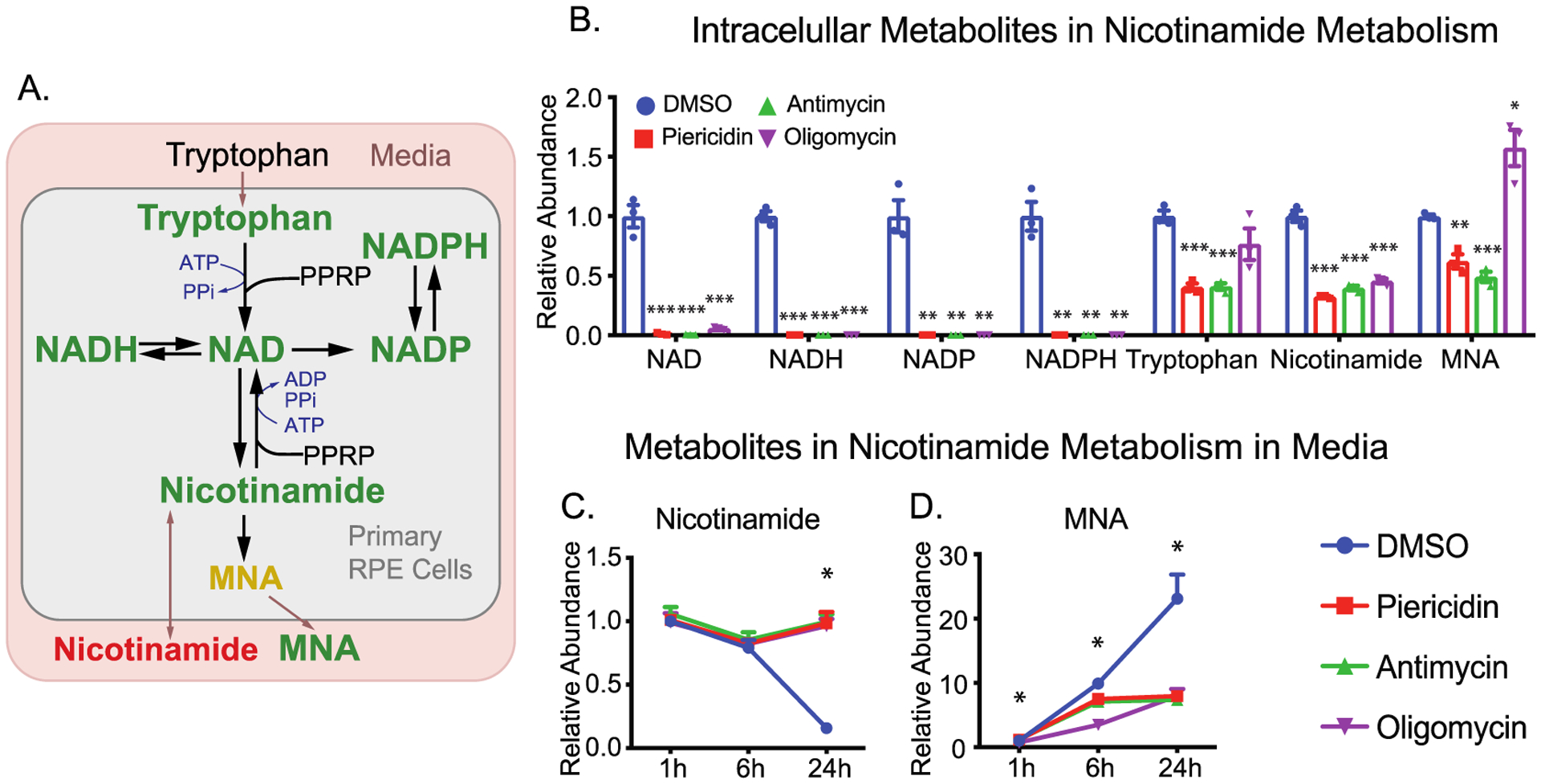
Inhibition of mitochondrial metabolism impairs NAD metabolism. (A) A schematic for the impaired NAD metabolism with mitochondria inhibitors. MNA, 1-methylnicotinamide. (B) The relative abundance of intracellular NAD intermediates over the groups with DMSO. (C and D) The relative abundance of extracellular metabolites in NAD metabolism. N = 3. *P < 0.05, **P < 0.01, ***P < 0.001 vs the groups treated with DMSO.
DISCUSSION
In this study, we found signature changes of metabolites in the media caused by the inhibition of mitochondrial respiration. These changes include (1) enhanced glucose consumption and lactate production, (2) reduced release of pyruvate and citrate, (3) accumulated choline, proline and BCAAs, (4) decreased release of 3-HB, (5) accumulated nucleosides from nucleotide degradation, and (6) elevated nicotinamide and MNA (Figure 9). Our findings demonstrate that mitochondrial dysfunction disrupts nutrient consumption and secretion, strongly supporting the model that RPE mitochondrial metabolism may actively synthesize nutrients to support the outer retinal metabolism (Figure 9).
Figure 9.
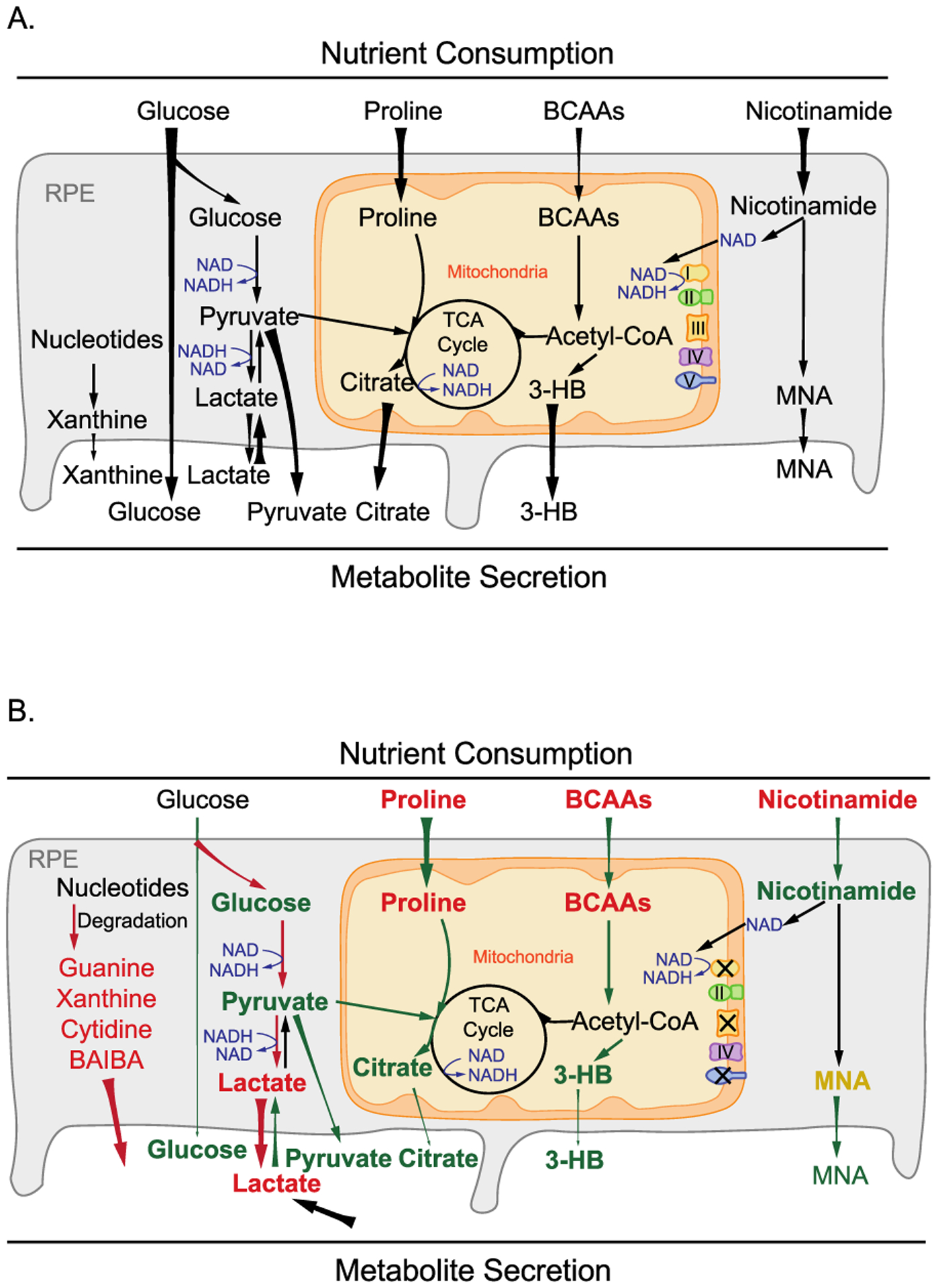
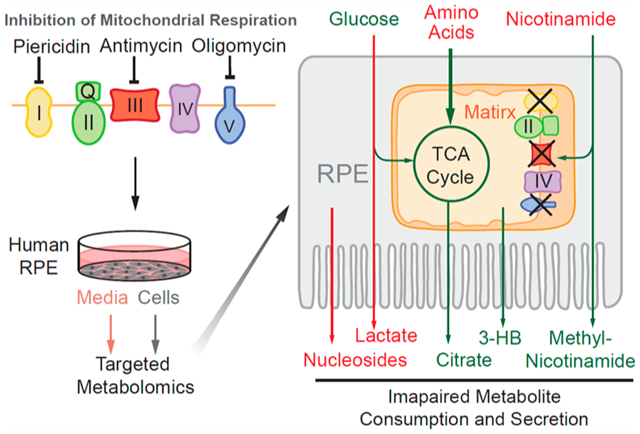
Schematic for the role of mitochondrial metabolism in nutrient consumption and metabolite export. (A) Healthy RPE mitochondria use different nutrients such as glucose, amino acids, lipids, and nicotinamide and export mitochondria-derived metabolites including citrate, isocitrate, and 3-HB to the outer retina. (B) When mitochondrial respiration is inhibited, RPE cells consume more glucose into lactate but use fewer other fuels, leading to a substantial reduction in exporting glucose, citrate, and 3-HB. Moreover, large amounts of lactate and nucleosides such as guanine, guanosine, hypoxanthine, xanthine, and BAIBA were exported out of RPE cells.
Why does the inhibition of mitochondrial respiration cause such a broad spectrum of metabolic changes? Mitochondrial respiration accepts electrons from NADH and FADH2 to drive the pump of hydrogen to generate ATP and H2O by consuming oxygen. The inhibition of mitochondrial respiration by blocking complexes could substantially reduce ATP supply to cause bioenergetics deficit and accumulation of reducing equivalents including NADH/FADH2.25–28 ATP deficiency would slow or halt many metabolic reactions that require ATP to overcome energy barriers or serve as a cofactor. For example, as ATP is required for the reactions by creatine kinase, arginosuccinate synthase and choline kinase, inhibition of mitochondrial respiration led to depletion of phosphocreatine and accumulation of citrulline and choline (Figures 2 and 5). NADH needs to be recycled into NAD to sustain the reactions in glycolysis, TCA cycle, and fatty acid oxidation. The ETC is one of the major electron acceptors to regenerate NAD. Both pharmacological inhibitors and genetic deletions of mitochondrial complexes are known to increase NADH/NAD.25–29 To regenerate NAD from NADH, the cells may use pyruvate to generate lactate through LDH and inhibit NAD-dependent dehydrogenases. Consistently, we have found a substantial increase in lactate and OAA, but a decrease of pyruvate, citrate, isocitrate. The decreased citrate production and enhanced lactate formation could stimulate glycolysis to increase glucose consumption in the RPE cells. Recent studies in other cells also show that inhibition of mitochondrial complex I is sufficient to increase glucose uptake.30,31 Photoreceptors could uptake citrate and pyruvate to support their mitochondrial metabolism and export lactate to facilitate glucose transport through RPE.14,20,21,32 The reduced release of citrate and pyruvate from RPE, and accumulation of lactate may interfere with photoreceptor metabolism and disrupt the metabolic ecosystem.33–35
The TCA cycle enzyme succinate dehydrogenase, which is also mitochondrial complex II, accepts electrons from FADH2. Succinate decreased from 13C glucose (Figures 3 and 4), confirming that mitochondrial oxidation of pyruvate is inhibited. The accumulated succinate in unlabeled RPE culture should come from other sources such as amino acids, fatty acids, and pyrimidine. Similar to NAD, FAD needs to be regenerated from FADH2 for FAD-dependent dehydrogenases such as acyl-CoA dehydrogenases in fatty acid oxidation and BCAA catabolism.36,37 Consistently, our findings showed inhibition of mitochondrial respiration reduced acylcarnitines but accumulated BCAAs (Figure 6).
Besides being recycled from NADH, NAD needs to be replenished by biosynthesis because multiple pathways consume it such as poly(ADP-ribose) polymerases (PARPs) and sirtuins.38–40 Activated PARPs can consume large amounts of NAD by transferring ADP-ribose to target proteins.41 We reported previously in primary RPE cells that oxidative stress could deplete cellular NAD, and the inhibition of PARP partially restores NAD to protect oxidative damage.22 Mitochondrial dysfunction could activate PARPs,42 contributing to the depletion of NAD and its derivatives. Furthermore, either de novo synthesis from tryptophan or salvage synthesis from nicotinamide requires multiple ATP molecules (Figure 8A).40 The ATP deficiency from inhibition of mitochondrial metabolism could worsen the depletion of cellular NAD. Consistently, nicotinamide was decreased in cells but accumulated in the media (Figure 8B and C), confirming that NAD biosynthesis is inhibited. Intriguingly, RPE cells exported large amounts of MNA (Figure 8D), methylated nicotinamide by nicotinamide N-methyltransferase (NNMT). Recent studies show NNMT is a novel regulator of energy homeostasis in adipose tissue.43,44 We reported that MNA is one of several metabolites that are commonly increased in aged ocular tissues.45 The sensitive changes of MNA in the media in our study suggest that medium MNA might be a promising indicator of NAD metabolism and mitochondrial metabolism.
We have reported recently that differentiated mature RPE cells prefer to consume proline as a nutrient and export proline-derived intermediates to be used by the retina.14,21 In agreement with these findings, healthy RPE cells almost completely consumed proline within 24 h. However, complex inhibitors block proline consumption, especially the complex III inhibitor that further elevated proline in the media. To be oxidized, proline is first converted into pyrroline-5-carboxylate (P5C) through a FAD-dependent proline dehydrogenase, and P5C is further oxidized into glutamate by a NAD-dependent P5C dehydrogenase to enter the TCA cycle.46 The ETC inhibitors, especially the inhibitor of complex III that accepts electrons from both FADH2 and NADH, could accumulate reducing equivalents and decrease the synthesis of NAD as aforementioned, resulting in the accumulation of proline in the media. As only differentiated RPE that relies on mitochondrial metabolism could utilize proline,14 the medium proline level could be a sensitive marker for RPE mitochondrial metabolism.
One of the striking changes in this study is the massive accumulation of nucleosides in the media from nucleotide metabolism including guanine, guanosine, hypoxanthine, BAIBA, and cytidine. Nucleotide metabolism is finely regulated through synthesis and degradation to maintain DNA integrity and RNA production.47 Nucleosides are also the basic building blocks for high-energy metabolites including ATP, GTP, CTP, and UTP. The energy deficit caused by the inhibition of mitochondrial respiration may accelerate the degradation of ADP into AMP, which can activate AMP deaminase to degrade AMP into IMP and hypoxanthine.31 Xanthine oxidoreductase can further oxidize hypoxanthine into xanthine and urate. In this study, AMP and IMP were depleted while hypoxanthine, xanthine, urate, guanine, and guanosine were substantially elevated. The salvage pathway through hypoxanthine-guanine phosphoribosyltransferase (HPRT) using hypoxanthine, guanine, and xanthine as substrates is the primary pathway for purine synthesis (Figure 7A).48 However, HPRT requires 5-phosphoribosyl 1-pyrophosphate (PRPP) to transfer the 5-phosphoribosyl group. Inhibition of mitochondrial respiration depleted ATP and ribose-5-phosphate, which might decrease PRPP and thus block salvage synthesis. This will ultimately cause further depletion of AMP and IMP. Therefore, our results suggest that inhibition of mitochondrial metabolism increased purine degradation and inhibited its synthesis. Consistently, we have reported that nutrient deprivation causes energy deficient and time-dependent accumulation of purine metabolites including hypoxanthine and xanthine in mouse RPE and retina tissues.45 In photoreceptors, purine metabolism is tightly regulated by light to control the cellular cyclic GMP (cGMP) level.49,50 Impaired purine metabolism causes inherited retinal degeneration.51,52 The excessive excretion of purine metabolites from RPE with mitochondrial dysfunction might disturb photoreceptor metabolism and result in retinal degeneration.
We have found a similar accumulation of metabolites in pyrimidine degradation, especially BAIBA. β-ureidopropionase catalyzes the last step in the pyrimidine degradation pathway, producing BAIBA and β-alanine from thymine and uracil, respectively (Figure 7B). Both intracellular BAIBA and β-alanine accumulated in the RPE, but in the media, only BAIBA substantially accumulated in a time-dependent manner (Figures 7M and S5). This might be attributed to different pathways in their catabolism. BAIBA is catabolized in the mitochondria into propionyl-CoA, which can be converted into succinyl-CoA to be oxidized through the TCA cycle.53 The accumulation of succinate by the inhibition of mitochondrial respiration should block the degradation of BAIBA to cause its elevation. However, β-alanine can be metabolized into multiple metabolites including acetate, malonate, pantothenic acid, and carnosine.54,55
In conclusion, we demonstrate that inhibition of mitochondrial respiration strikingly influences both the intracellular and extracellular metabolome of primary RPE cells, and the extracellular metabolic changes consistently reflect intracellular changes. These findings provide important information on how RPE mitochondrial dysfunction disturbs nutrient consumption and release. Future studies on coculture and in vivo models will be essential to elucidate the impacts of these changed metabolites on photoreceptor viability and their contribution to the pathogenesis of AMD and other mitochondria-related retinal diseases.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by NIH Grants EY026030 (to J.R.C. and J.D.), the Retina Research Foundation (to J.D.), BrightFocus Foundation (to J.D.), funds for Core facilities P20 GM103434 (NIH WV INBRE grant), WVCTSI Grant GM104942, Research to Prevent Blindness Sybil B. Harrington Physician-Scientist Award for Macular Degeneration (J.R.C.), and an unrestricted Grant from Research to Prevent Blindness (J.R.C.). All raw mass spectrometry data have been deposited to MassIVE (Data set identifier: MSV000086157 and MSV000086159).
Footnotes
The authors declare no competing financial interest.
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.jproteome.0c00690
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jproteome.0c00690.
Supplemental Methods: LDH Activity Assay Figure S1. The impact of mitochondrial inhibitors on LDH activity. Figure S2. The impact of the inhibition of mitochondrial respiration on glycolysis and TCA cycle intermediates. Figure S3. The impact of mitochondrial inhibition on the metabolism of amino acids. Figure S4. The impact of mitochondrial inhibition on the urea cycle. Figure S5. The impact of mitochondrial inhibition on the metabolism of nucleotides. Table S1. Significantly changed metabolites in 1 h media after inhibition of mitochondrial respiration. Table S2. Significantly changed metabolites in 6 h media after inhibition of mitochondrial respiration. Table S3. Significantly changed metabolites in 24 h media after inhibition of mitochondrial respiration. Table S4. Significantly changed metabolites in cells at 24 h after inhibition of mitochondrial respiration. Table S5. The reagents and key resources. Table S6. The list of metabolites and parameters for LC MS and GC MS. (PDF)
Contributor Information
Rui Zhang, Department of Ophthalmology and Department of Biochemistry, West Virginia University, Morgantown, West Virginia 26506, United States; Save Sight Institute, Sydney Medical School, University of Sydney, Sydney, NSW 2000, Australia.
Abbi L. Engel, Department of Ophthalmology, University of Washington, Seattle, Washington 98109, United States.
Jennifer R. Chao, Department of Ophthalmology, University of Washington, Seattle, Washington 98109, United States;
Jianhai Du, Department of Ophthalmology and Department of Biochemistry, West Virginia University, Morgantown, West Virginia 26506, United States;.
REFERENCES
- (1).Strauss O The retinal pigment epithelium in visual function. Physiol. Rev 2005, 85 (3), 845–81. [DOI] [PubMed] [Google Scholar]
- (2).Ferrington DA; Fisher CR; Kowluru RA Mitochondrial Defects Drive Degenerative Retinal Diseases. Trends Mol. Med 2020, 26 (1), 105–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Brown EE; Lewin AS; Ash JD Mitochondria: Potential Targets for Protection in Age-Related Macular Degeneration. Adv. Exp. Med. Biol 2018, 1074, 11–17. [DOI] [PubMed] [Google Scholar]
- (4).Gong J; Cai H; Noggle S; Paull D; Rizzolo LJ; Del Priore LV; Fields MA Stem cell-derived retinal pigment epithelium from patients with age-related macular degeneration exhibit reduced metabolism and matrix interactions. Stem Cells Transl. Med 2020, 9 (3), 364–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Zhao C; Yasumura D; Li X; Matthes M; Lloyd M; Nielsen G; Ahern K; Snyder M; Bok D; Dunaief JL; LaVail MM; Vollrath D mTOR-mediated dedifferentiation of the retinal pigment epithelium initiates photoreceptor degeneration in mice. J. Clin. Invest 2011, 121 (1), 369–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Kurihara T; Westenskow PD; Gantner ML; Usui Y; Schultz A; Bravo S; Aguilar E; Wittgrove C; Friedlander M; Paris LP; Chew E; Siuzdak G; Friedlander M Hypoxia-induced metabolic stress in retinal pigment epithelial cells is sufficient to induce photoreceptor degeneration. eLife 2016, 5, 14319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Felszeghy S; Viiri J; Paterno JJ; Hyttinen JMT; Koskela A; Chen M; Leinonen H; Tanila H; Kivinen N; Koistinen A; Toropainen E; Amadio M; Smedowski A; Reinisalo M; Winiarczyk M; Mackiewicz J; Mutikainen M; Ruotsalainen AK; Kettunen M; Jokivarsi K; Sinha D; Kinnunen K; Petrovski G; Blasiak J; Bjorkoy G; Koskelainen A; Skottman H; Urtti A; Salminen A; Kannan R; Ferrington DA; Xu H; Levonen AL; Tavi P; Kauppinen A; Kaarniranta K Loss of NRF-2 and PGC-1alpha genes leads to retinal pigment epithelium damage resembling dry age-related macular degeneration. Redox Biol. 2019, 20, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Ferrington DA; Ebeling MC; Kapphahn RJ; Terluk MR; Fisher CR; Polanco JR; Roehrich H; Leary MM; Geng Z; Dutton JR; Montezuma SR Altered bioenergetics and enhanced resistance to oxidative stress in human retinal pigment epithelial cells from donors with age-related macular degeneration. Redox Biol. 2017, 13, 255–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Golestaneh N; Chu Y; Cheng SK; Cao H; Poliakov E; Berinstein DM Repressed SIRT1/PGC-1alpha pathway and mitochondrial disintegration in iPSC-derived RPE disease model of age-related macular degeneration. J. Transl. Med 2016, 14 (1), 344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Owen OE; Kalhan SC; Hanson RW The key role of anaplerosis and cataplerosis for citric acid cycle function. J. Biol. Chem 2002, 277 (34), 30409–12. [DOI] [PubMed] [Google Scholar]
- (11).Gibala MJ; Young ME; Taegtmeyer H Anaplerosis of the citric acid cycle: role in energy metabolism of heart and skeletal muscle. Acta Physiol. Scand 2000, 168 (4), 657–65. [DOI] [PubMed] [Google Scholar]
- (12).Xu R; Ritz BK; Wang Y; Huang J; Zhao C; Gong K; Liu X; Du J The retina and retinal pigment epithelium differ in nitrogen metabolism and are metabolically connected. J. Biol. Chem 2020, 295 (8), 2324–2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Wang W; Kini A; Wang Y; Liu T; Chen Y; Vukmanic E; Emery D; Liu Y; Lu X; Jin L; Lee SJ; Scott P; Liu X; Dean K; Lu Q; Fortuny E; James R; Kaplan HJ; Du J; Dean DC Metabolic Deregulation of the Blood-Outer Retinal Barrier in Retinitis Pigmentosa. Cell Rep. 2019, 28 (5), 1323–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Yam M; Engel AL; Wang Y; Zhu S; Hauer A; Zhang R; Lohner D; Huang J; Dinterman M; Zhao C; Chao JR; Du J Proline mediates metabolic communication between retinal pigment epithelial cells and the retina. J. Biol. Chem 2019, 294 (26), 10278–10289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).LaVail MM Rod outer segment disk shedding in rat retina: relationship to cyclic lighting. Science 1976, 194 (4269), 1071–4. [DOI] [PubMed] [Google Scholar]
- (16).Reyes-Reveles J; Dhingra A; Alexander D; Bragin A; Philp NJ; Boesze-Battaglia K Phagocytosis-dependent ketogenesis in retinal pigment epithelium. J. Biol. Chem 2017, 292 (19), 8038–8047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Adijanto J; Du J; Moffat C; Seifert EL; Hurley JB; Philp NJ The retinal pigment epithelium utilizes fatty acids for ketogenesis. J. Biol. Chem 2014, 289 (30), 20570–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Du J; Cleghorn W; Contreras L; Linton JD; Chan GC; Chertov AO; Saheki T; Govindaraju V; Sadilek M; Satrustegui J; Hurley JB Cytosolic reducing power preserves glutamate in retina. Proc. Natl. Acad. Sci. U. S. A 2013, 110 (46), 18501–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Hurley JB; Lindsay KJ; Du J Glucose, lactate, and shuttling of metabolites in vertebrate retinas. J. Neurosci. Res 2015, 93 (7), 1079–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Kanow MA; Giarmarco MM; Jankowski CS; Tsantilas K; Engel AL; Du J; Linton JD; Farnsworth CC; Sloat SR; Rountree A; Sweet IR; Lindsay KJ; Parker ED; Brockerhoff SE; Sadilek M; Chao JR; Hurley JB Biochemical adaptations of the retina and retinal pigment epithelium support a metabolic ecosystem in the vertebrate eye. eLife 2017, 6, 28899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Chao JR; Knight K; Engel AL; Jankowski C; Wang Y; Manson MA; Gu H; Djukovic D; Raftery D; Hurley JB; Du J Human retinal pigment epithelial cells prefer proline as a nutrient and transport metabolic intermediates to the retinal side. J. Biol. Chem 2017, 292 (31), 12895–12905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Du J; Yanagida A; Knight K; Engel AL; Vo AH; Jankowski C; Sadilek M; Tran VT; Manson MA; Ramakrishnan A; Hurley JB; Chao JR Reductive carboxylation is a major metabolic pathway in the retinal pigment epithelium. Proc. Natl. Acad. Sci. U. S. A 2016, 113 (51), 14710–14715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Du J; Linton JD; Hurley JB Probing Metabolism in the Intact Retina Using Stable Isotope Tracers. Methods Enzymol. 2015, 561, 149–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Millard P; Letisse F; Sokol S; Portais J-C IsoCor: correcting MS data in isotope labeling experiments. Bioinformatics 2012, 28 (9), 1294–1296. [DOI] [PubMed] [Google Scholar]
- (25).Titov DV; Cracan V; Goodman RP; Peng J; Grabarek Z; Mootha VK Complementation of mitochondrial electron transport chain by manipulation of the NAD+/NADH ratio. Science 2016, 352 (6282), 231–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Sullivan LB; Gui DY; Hosios AM; Bush LN; Freinkman E; Vander Heiden MG Supporting Aspartate Biosynthesis Is an Essential Function of Respiration in Proliferating Cells. Cell 2015, 162 (3), 552–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Diebold LP; Gil HJ; Gao P; Martinez CA; Weinberg SE; Chandel NS Mitochondrial complex III is necessary for endothelial cell proliferation during angiogenesis. Nat. Metab 2019, 1 (1), 158–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Yam M; Engel AL; Wang Y; Zhu S; Hauer A; Zhang R; Lohner D; Huang J; Dinterman M; Zhao C; Chao JR; Du J Proline mediates metabolic communication between retinal pigment epithelial cells and the retina. J. Biol. Chem 2019, 294 (26), 10278–10289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Birsoy K; Wang T; Chen WW; Freinkman E; Abu-Remaileh M; Sabatini DM An Essential Role of the Mitochondrial Electron Transport Chain in Cell Proliferation Is to Enable Aspartate Synthesis. Cell 2015, 162 (3), 540–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Shum M; Houde VP; Bellemare V; Junges Moreira R; Bellmann K; St-Pierre P; Viollet B; Foretz M; Marette A Inhibition of mitochondrial complex 1 by the S6K1 inhibitor PF-4708671 partly contributes to its glucose metabolic effects in muscle and liver cells. J. Biol. Chem 2019, 294 (32), 12250–12260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Zabielska MA; Borkowski T; Slominska EM; Smolenski RT Inhibition of AMP deaminase as therapeutic target in cardiovascular pathology. Pharmacol. Rep 2015, 67 (4), 682–8. [DOI] [PubMed] [Google Scholar]
- (32).Grenell A; Wang Y; Yam M; Swarup A; Dilan TL; Hauer A; Linton JD; Philp NJ; Gregor E; Zhu S; Shi Q; Murphy J; Guan T; Lohner D; Kolandaivelu S; Ramamurthy V; Goldberg AFX; Hurley JB; Du J Loss of MPC1 reprograms retinal metabolism to impair visual function. Proc. Natl. Acad. Sci. U. S. A 2019, 116 (9), 3530–3535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Sheline CT; Zhou Y; Bai S Light-induced photoreceptor and RPE degeneration involve zinc toxicity and are attenuated by pyruvate, nicotinamide, or cyclic light. Molecular Vision 2010, 16, 2639. [PMC free article] [PubMed] [Google Scholar]
- (34).Kanow MA; Giarmarco MM; Jankowski CS; Tsantilas K; Engel AL; Du J; Linton JD; Farnsworth CC; Sloat SR; Rountree A; Sweet IR; Lindsay KJ; Parker ED; Brockerhoff SE; Sadilek M; Chao JR; Hurley JB Biochemical adaptations of the retina and retinal pigment epithelium support a metabolic ecosystem in the vertebrate eye. eLife 2017, 6, e28899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Zeevalk GD; Nicklas WJ Lactate prevents the alterations in tissue amino acids, decline in ATP, and cell damage due to aglycemia in retina. J. Neurochem 2000, 75 (3), 1027–1034. [DOI] [PubMed] [Google Scholar]
- (36).Olsen RKJ; Konarikova E; Giancaspero TA; Mosegaard S; Boczonadi V; Matakovic L; Veauville-Merllie A; Terrile C; Schwarzmayr T; Haack TB; Auranen M; Leone P; Galluccio M; Imbard A; Gutierrez-Rios P; Palmfeldt J; Graf E; Vianey-Saban C; Oppenheim M; Schiff M; Pichard S; Rigal O; Pyle A; Chinnery PF; Konstantopoulou V; Moslinger D; Feichtinger RG; Talim B; Topaloglu H; Coskun T; Gucer S; Botta A; Pegoraro E; Malena A; Vergani L; Mazza D; Zollino M; Ghezzi D; Acquaviva C; Tyni T; Boneh A; Meitinger T; Strom TM; Gregersen N; Mayr JA; Horvath R; Barile M; Prokisch H Riboflavin-Responsive and -Non-responsive Mutations in FAD Synthase Cause Multiple Acyl-CoA Dehydrogenase and Combined Respiratory-Chain Deficiency. Am. J. Hum. Genet 2016, 98 (6), 1130–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Barile M; Giancaspero TA; Leone P; Galluccio M; Indiveri C Riboflavin transport and metabolism in humans. J. Inherited Metab. Dis 2016, 39 (4), 545–57. [DOI] [PubMed] [Google Scholar]
- (38).Ame JC; Spenlehauer C; de Murcia G The PARP superfamily. BioEssays 2004, 26 (8), 882–93. [DOI] [PubMed] [Google Scholar]
- (39).Belenky P; Bogan KL; Brenner C NAD+ metabolism in health and disease. Trends Biochem. Sci 2007, 32 (1), 12–9. [DOI] [PubMed] [Google Scholar]
- (40).Liu L; Su X; Quinn WJ 3rd; Hui S; Krukenberg K; Frederick DW; Redpath P; Zhan L; Chellappa K; White E; Migaud M; Mitchison TJ; Baur JA; Rabinowitz JD Quantitative Analysis of NAD Synthesis-Breakdown Fluxes. Cell Metab. 2018, 27 (5), 1067–1080 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Alano CC; Garnier P; Ying W; Higashi Y; Kauppinen TM; Swanson RA NAD+ depletion is necessary and sufficient for poly(ADP-ribose) polymerase-1-mediated neuronal death. J. Neurosci 2010, 30 (8), 2967–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Lai YC; Baker JS; Donti T; Graham BH; Craigen WJ; Anderson AE Mitochondrial Dysfunction Mediated by Poly(ADPRibose) Polymerase-1 Activation Contributes to Hippocampal Neuronal Damage Following Status Epilepticus. Int. J. Mol. Sci 2017, 18 (7), 1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Ehebauer F; Ghavampour S; Kraus D Glucose availability regulates nicotinamide N-methyltransferase expression in adipocytes. Life Sci. 2020, 248, 117474. [DOI] [PubMed] [Google Scholar]
- (44).Kraus D; Yang Q; Kong D; Banks AS; Zhang L; Rodgers JT; Pirinen E; Pulinilkunnil TC; Gong F; Wang YC; Cen Y; Sauve AA; Asara JM; Peroni OD; Monia BP; Bhanot S; Alhonen L; Puigserver P; Kahn BB Nicotinamide N-methyltransferase knockdown protects against diet-induced obesity. Nature 2014, 508 (7495), 258–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Wang Y; Grenell A; Zhong F; Yam M; Hauer A; Gregor E; Zhu S; Lohner D; Zhu J; Du J Metabolic signature of the aging eye in mice. Neurobiol. Aging 2018, 71, 223–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Phang JM; Liu W; Zabirnyk O Proline metabolism and microenvironmental stress. Annu. Rev. Nutr 2010, 30, 441–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Lane AN; Fan TW Regulation of mammalian nucleotide metabolism and biosynthesis. Nucleic Acids Res. 2015, 43 (4), 2466–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Fasullo M; Endres L Nucleotide salvage deficiencies, DNA damage and neurodegeneration. Int. J. Mol. Sci 2015, 16 (5), 9431–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Du J; Rountree A; Cleghorn WM; Contreras L; Lindsay KJ; Sadilek M; Gu H; Djukovic D; Raftery D; Satrustegui J; Kanow M; Chan L; Tsang SH; Sweet IR; Hurley JB Phototransduction Influences Metabolic Flux and Nucleotide Metabolism in Mouse Retina. J. Biol. Chem 2016, 291 (9), 4698–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Plana-Bonamaiso A; Lopez-Begines S; Fernandez-Justel D; Junza A; Soler-Tapia A; Andilla J; Loza-Alvarez P; Rosa JL; Miralles E; Casals I; Yanes O; de la Villa P; Buey RM; Mendez A Post-translational regulation of retinal IMPDH1 in vivo to adjust GTP synthesis to illumination conditions. eLife 2020, 9, 56418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Du J; An J; Linton JD; Wang Y; Hurley JB How Excessive cGMP Impacts Metabolic Proteins in Retinas at the Onset of Degeneration. Adv. Exp. Med. Biol 2018, 1074, 289–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Bowne SJ; Sullivan LS; Blanton SH; Cepko CL; Blackshaw S; Birch DG; Hughbanks-Wheaton D; Heckenlively JR; Daiger SP Mutations in the inosine monophosphate dehydrogenase 1 gene (IMPDH1) cause the RP10 form of autosomal dominant retinitis pigmentosa. Hum. Mol. Genet 2002, 11 (5), 559–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Tanianskii DA; Jarzebska N; Birkenfeld AL; O’Sullivan JF; Rodionov RN Beta-Aminoisobutyric Acid as a Novel Regulator of Carbohydrate and Lipid Metabolism. Nutrients 2019, 11 (3), 524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Blancquaert L; Baba SP; Kwiatkowski S; Stautemas J; Stegen S; Barbaresi S; Chung W; Boakye AA; Hoetker JD; Bhatnagar A; Delanghe J; Vanheel B; Veiga-da-Cunha M; Derave W; Everaert I Carnosine and anserine homeostasis in skeletal muscle and heart is controlled by beta-alanine transamination. J. Physiol 2016, 594 (17), 4849–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Hayaishi O; Nishizuka Y; Tatibana M; Takeshita M; Kuno S Enzymatic studies on the metabolism of beta-alanine. J. Biol. Chem 1961, 236, 781–90. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.