

Abstract
In animals, endocytosis of a 7-transmembrane GPCR is mediated by arrestins to propagate or arrest cytoplasmic G-protein-mediated signaling, depending on the bias of the receptor or ligand, which determines how much one transduction pathway is used compared to another. In Arabidopsis thaliana, GPCRs are not required for G protein-coupled signaling because the heterotrimeric G protein complex spontaneously exchanges nucleotide. Instead, the 7-transmembrane protein AtRGS1 modulates G protein signaling through ligand-dependent endocytosis, which initiates de-repression of signaling without the involvement of canonical arrestins. Here, we found that endocytosis of AtRGS1 initiated from two separate pools of plasma membrane: Sterol-dependent domains and a clathrin-accessible neighborhood, each with a select set of discriminators, activators, and candidate arrestin-like adaptors. Ligand identity (either the pathogen-associated molecular pattern flg22 or the sugar glucose) determined the origin of AtRGS1 endocytosis. Different trafficking origins and trajectories led to different cellular outcomes. Thus, in this system, compartmentation with its associated signalosome architecture drives biased signaling.
In animals, activation of G protein signaling results from GDP exchange for GTP on the Gα subunit, wherein this nucleotide exchange is the rate-limiting step (1). Desensitization to the signal occurs through endocytosis of the GPCR mediated by arrestins (2, 3). However, in Arabidopsis, the Gα subunit AtGPA1 spontaneously exchanges nucleotides without a GPCR and is thus self-activating with the intrinsic GTPase activity being the rate-limiting step (4, 5). AtRGS1 accelerates the intrinsic GTPase activity of AtGPA1 (6) and as a result, inactive Gα remains bound to GDP until AtRGS1 is endocytosed, de-repressing AtGPA1 and permitting activation through nucleotide exchange and subsequent downstream signal transduction (7). AtRGS1 endocytosis is a well-established proxy for sustained G protein activation and the proportion of endocytosed AtRGS1 is linearly related to the proportion of Gα in the GTP-bound state (8).
To date, there are two well-studied signals that directly and rapidly activate the Arabidopsis heterotrimeric G protein signaling pathway through AtRGS1 endocytosis. One signal is flg22, a 22-amino acid pathogen-associated molecular pattern (PAMP) from the plant pathogen Pseudomonas syringe that is recognized by plant cells to initiate the innate immunity pathway (9–11). It is already established that flg22 is perceived extracellularly (12) by the co-receptors BAK1 and FLS2 as part of a larger G protein complex (13, 14)). Another signal is glucose or a glucose metabolite which are products of photosynthesis (15) and metabolism of starch stores (16).
Many sugars, including sucrose, are transported extracellularly in the apoplastic space where they are potentially further metabolized to a signal such as glucose by cell wall localized invertases or UDP-glucose by cell wall sucrose synthases. Many monosaccharides and disaccharides are rapidly taken up through a large family of transporters (17, 18) where they are detected in the cytoplasm, but some sugars may also be detected extracellularly (19). d-glucose and some other glucose containing sugars induce rapid endocytosis of AtRGS1-dependent sugar signaling (7, 8) and because AtRGS1 shares the membrane topology of GPCRs which perceive extracellular signals, extracellular glucose or metabolite has been assumed to be perceived by the AtRGS1/G protein complex. However, neither an extracellular site for perception nor direct evidence for agonist binding to AtRGS1 has been shown.
De-repression of AtRGS1 occurs through physically de-coupling from the heterotrimeric G protein complex by endocytosis, at least for sustained activation. Clathrin-mediated endocytosis (CME) and sterol-dependent endocytosis (SDE) (20) are two modes of endocytosis in plants. However, neither the mode nor the attending adaptors that directly couples to AtRGS1 and responsible for endocytosis are known. In animal and yeast systems, many molecular components in the CME pathway are well characterized, including the ADAPTOR PROTEIN COMPLEX-2 (AP-2), which is required for recognition and binding of cargo, recruiting clathrin to the plasma membrane (PM), and the subsequent formation of clathrin-coated vesicles (21–24). The AP-2 complex is a complex consisting of 2 large subunits (α2 and β2), 1 medium subunit (μ2), and 1 small subunit (σ2) (25). SDE is a clathrin-independent mechanism for internalizing membrane-associated proteins. Sometimes referred to as lipid raft endocytosis, SDE of these microdomains requires flotilin1 (Flot1) and possibly the microdomain-associated protein remorin (26) to internalize sterol-rich vesicles (27). The membrane proteins PIP2;1 and respiratory burst oxidase homolog D (RbohD) are selectively internalized by SDE under salt stress conditions (28, 29).
Here, we present data illustrating a biased system in which two distinct extracellular ligands induced endocytosis of AtRGS1 from separate plasma membrane origins, one of which might be mediated by an arrestin-fold-containing protein, vacuolar protein sorting 26 (VPS26). Flg22 initiated AtRGS1 endocytosis through CME, whereas the monosaccharide d-glucose or its metabolite activated both CME and SDE. Phosphorylation of AtRGS1, involvement of individual subunits of the heterotrimeric G protein complex, and downstream signaling were also ligand specific. From the CME-mediated-AtRGS1 origin, flg22 induced a MAPK cascade that drove transcriptional reprogramming (30), whereas from the SDE-mediated-AtRGS1 origin, glucose induced transcriptional changes that were independent of the MAPK cascade.
Results
Glucose is perceived extracellularly and initiates AtRGS1 endocytosis.
We first sought to determine if d-glucose is perceived extracellularly. The d-glucose-fructose dissacharide 6-0-α-D-glucopyranosyl-D-fructose (also known as isomaltulose or pallatinose) is may not be transported across the plant cell membrane although it acts as an active glucose precursor if synthesized intracellularly (31, 32). It has been suggested that isomaltulose may have lower permeability that glucose (33). Isomaltulose is not hydrolyzed extracellularly (34, 35). To determine if isomaltulose is impermeant to the plasma membrane, we chemically synthesized [14C] isomaltulose (Fig. S1A) and tested for uptake into Arabidopsis seedlings. Isomaltulose was at least 9-fold less permeant to cells than glucose (Fig. S1B). Extracellular isomaltulose induced AtRGS1 internalization more effectively than d-glucose (Fig. 1A and 1B, Fig. S1C).) despite being transported ~10-fold less suggesting that isomaltulose activated AtRGS1 extracellularly. The 5% d-glucose impurity in the isomaltulose preparation would induce at most 5% AtRGS1 internalization and therefore does not account for the observed level of activation. Turanose is another disaccharide that is thought to be perceived extracellularly (31) and is impermeant (36). Similar to both glucose and isomaltulose, turanose activated G signaling (Fig. 1C). Isomaltulose and turanose are disaccharides that share a glucose ring (Fig. 1D), suggesting that glucose or a glucose metabolite is the discriminated structure or is important for a metabolic agonist, leading us to test lactulose, a dissacharide like isomaltulose except that the glucose is replaced by galactose. Lactulose was slightly less effective at inducing AtRGS1 endocytosis (Fig. S1D and E), suggesting that the structural requisite around the 6-carbon ring is less stringent than hypothesized.
Fig. 1. An AtRGS1 complex perceives extracellular flg22 and glucose or glucose metabolite.
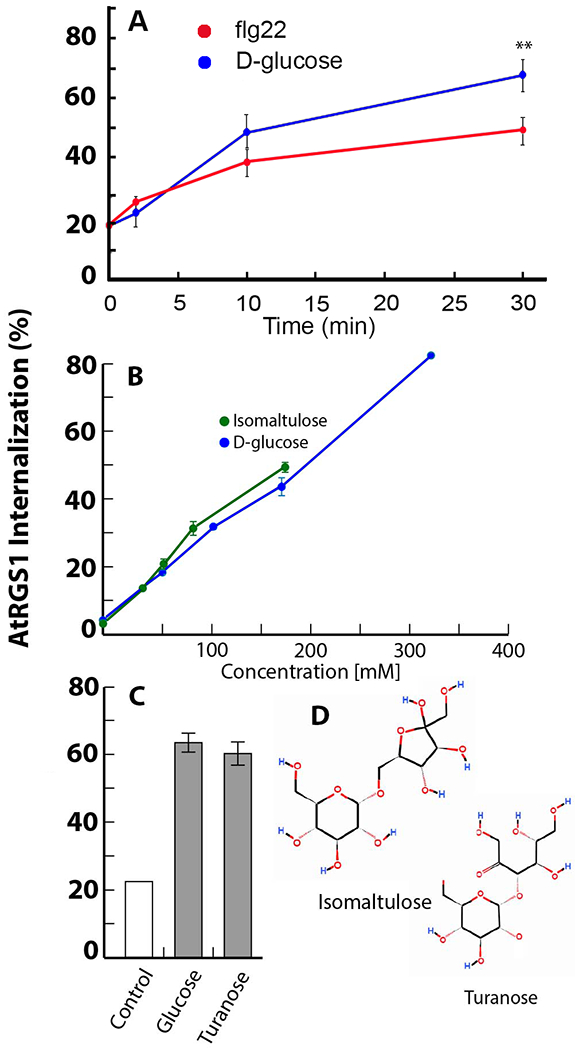
(A) Endocytosis of AtRGS1-YFP induced by flg22 (red) or D-glucose (blue). Values represent the amount of AtRGS1 that is internal to the cell as a percent of the total YFP fluorescence. The type of raw data used to generate these values is shown in Supplemental Fig. S1C. **, P < 0.01. N=4-24 biological replicates per group. (B) Endocytosis of AtRGS1-YFP induced by isomaltulose (green) or d-glucose (blue). Supporting data is in Fig. S1C to E,N=5 biological replicates per group (C) Endocytosis of AtRGS1-YFP induced by turanose or d-glucose. The purity of turanose was >98%. N=12 biological replicates per group. (D) Structures of isomaltulose and turanose, which both contain a glucose ring moiety.
Ligand-specific G protein signaling outputs.
Two rapid events of the flg22 response is the induction of MITOGEN ACTIVATED PROTEIN KINASE 6 (MPK6) activity and Ca2+ signaling (37). To test the impact of flg22 and d-glucose on MPK6 activity in etiolated hypocotyls, we developed a FRET-based sensor that measures kinase activity specifically for MPK6, called sensor of MAPK activity (SOMA) (38), and that was tagged with either the human immunodeficiency virus 1 (HIV-1) nuclear export signal (SOMA-NES) or the SV40 nuclear localization signal (SOMA-NLS) (39, 40) to measure MPK6 activity in the cytosol or nucleus, respectively. These reporters were validated in hypocotyl epidermal cells (Supplemental Fig. S2A). Rapid FRET gains were observed in both SOMA-NES and SOMA-NLS plants within 2-4 minutes after treatment with flg22 (Fig. 2A–F). 6% d-glucose did not induce changes in FRET efficiency, suggesting that d-glucose did not induce activity of MPK6 (Fig. 2G and H).
Fig. 2. MAPK activation and Ca2+ signaling in response to flg22 and gene expression in response to d-glucose.
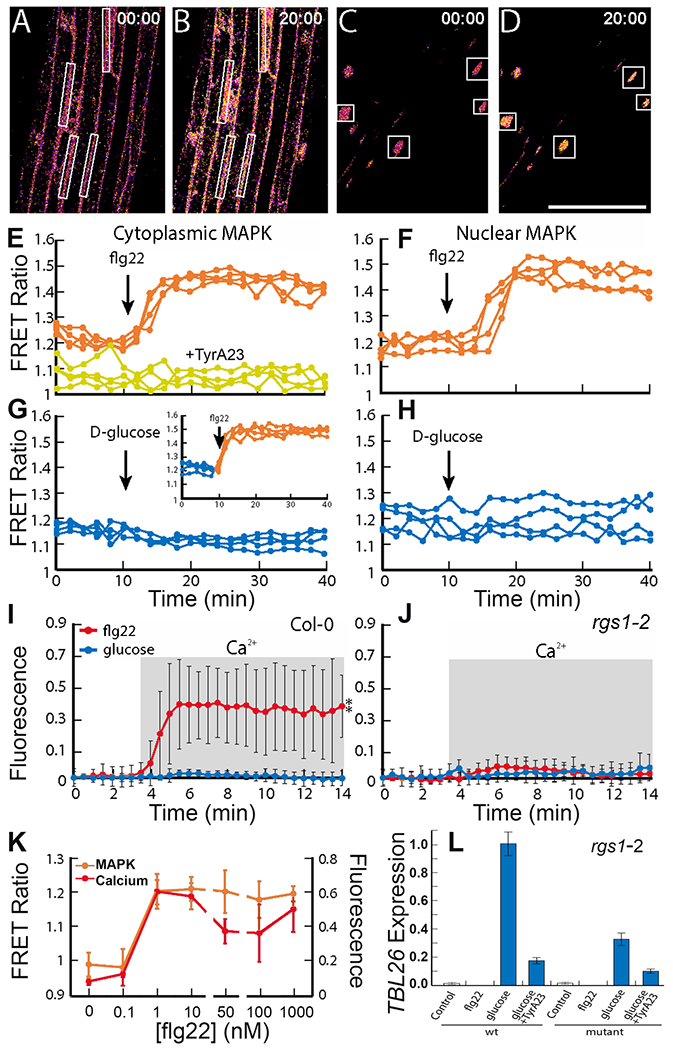
(A-D) Representative processed confocal images of SOMA-NES (A, B) and SOMA-NLS (C, D) etiolated hypocotyl epidermis depicting the ratio of YPet to Turquoise GL emission produced by exciting Turquoise GL. Scale bar, 100 μm. Images with the time stamp 00:00 were collected before treatment, and those with the time stamp 20:00 were collected 5 minutes after treatment with 1 μM flg22. White rectangle represent regions of interest (ROIs) used to measure YPet and Turquoise GL emission. (E-H) The ratio of YPet to Turquoise GL emission produced by exciting Turquoise GL over time was determined using the ROIs shown in (A-D). During the first 10 minutes of each experiment, samples were incubated in water. The arrow indicates the time at which 1 μM flg22 (E, F) or 6% D-glucose (G, H) was added to the sample. Where indicated in (E), SOMA-NES lines were pretreated with water (orange) or 50 μM TyrA23 (yellow) prior to imaging. Inset graph in (G) shows SOMA-NES line pretreated with 6% D-glucose for 30 minutes prior to imaging (flg22 was added after two minutes of imaging). N=4 biological replicates per group. (I, J) R-GECO1-expressing Col-0 (I) or rgs1-2 (J) lines were treated with flg22 or d-glucose. Fluorescence intensity changes of R-GECO1 in ~20 ROIs in Col-0 plants. Fractional fluorescence changes (ΔF/F) for R-GECO1 were calculated from background corrected intensity values of R-GECO1 as (F − F0)/F0, where F0 represents the average fluorescence intensity of the baseline of a measurement of each genotype. Error bars show standard deviation. **, P < 0.01) between treatment and water as determined by 2-way ANOVA. N=5 biological replicates per group. (K) flg22 dose response curves in R-GECO1 and SOMA-NES lines after treatment for two or ten minutes respectively. N=7 biological replicates per group. (L) Quantification of TBL26 gene expression by RT-qPCR in Col-0 and rgs1-2 plants pretreated with TyrA23 for 1 hour as indicated and treated with water, flg22, or D-glucose. N=3 biological replicates per group.
We tested the role of d-glucose and flg22 in triggering an increased Ca2+ transient response using the intensity-based Ca2+ sensor R-GECO1, a red-shifted intensity-based Ca2+ reporter (41). We measured D-glucose and flg22-induced Ca2+ changes in etiolated hypocotyls of Col-0 WT and rgs1–2 plants stably expressing cytosolic- and nuclear-localized R-GECO1 (Fig. 2I and J). Ca2+ levels in response to flg22 and represented by fractional fluorescence changes increased to a greater extent in WT plants than in rgs1-2 mutant plants. D-glucose treatments did not significantly alter fractional fluorescence in WT or rgs1-2 mutant plants, suggesting that d-glucose does not induce Ca2+ signaling downstream of AtRGS1. Activation of MAPK and the increase in Ca2+ both occurred at a threshold of 1 nM flg22 (Fig. 2K). Despite both d-glucose and flg22 inducing AtRGS1 internalization, these results show that only flg22 can induce MPK6 activity and Ca2+ changes.
Although d-glucose signaling mediated by AtRGS1 did not induce MAPK or Ca2+ signaling, d-glucose elicits a dose- and time-dependent change in the expression of a small set of genes (7, 42), including TRICHOMELESS LIKE 26 (TBL26) in an AtRGS1/G protein complex-dependent manner (42) and which is used as a reporter for activation (7, 8, 43). However, there was a small increase in TBL26 expression in rgs1-2 mutant plants upon D-glucose treatment. Because plants have only one 7TM-RGS protein encoded in their genome (44) , this low amount of TBL26 expression in rgs1-2 mutant plants indicated that secondary D-glucose signaling was independent of the RGS1 pathway. flg22 did not induce TBL26 expression, indicating separate outputs for d-glucose and flg22 (Fig. 2L).
Paradoxically, the loss of AtRGS1 inhibited TBL26 expression indicating that AtRGS1 genetically acts as a positive modulator of signaling which contradicts our understanding of RGS proteins as negative modulators of G protein activation. One solution to this paradox is that AtRGS1 has a positive regulatory role in endosomal complexes, which would require AtRGS1 endocytosis. Glucose-induced TBL26 expression was decreased in the presence of Tyrphostin A23 (TyrA23) (45, 46), an inhibitor of endocytosis that blocks the function of AP2μ, which directly interacts with cargo proteins during CME (23, 47) (Fig. 2L). This result is consistent with an endosomal origin for AtRGS1 signaling, analogous to some GPCRs (48). There was residual upregulation of TBL26 in mutant seedlings upon glucose stimulation in the presence or absence of TyrA23 (Fig. 2L), which may reflect the existence of an RGS1-independent pathway.
The endocytosis of AtRGS1 has two distinct origins.
AtRGS1 endocytosis induced by detection of either flg22 by BAK1/FLS2 (11, 49, 50) or extracellular d-glucose at least partially requires the phosphorylation of AtRGS1 at its C terminal tail (7, 50). We turned to the question of whether these two signals activated the same physical pool of AtRGS1. Both major endocytic pathways in plants, CME and SDE, have been individually associated with the activity of the same proteins and responses (27, 46, 51, 52), such as RbohD in the flg22 pathway (28). To determine if one or both pathways control AtRGS1 activity at the membrane, we induced internalization with d-glucose and flg22 while simultaneously inhibiting the CME or SDE pathway. Suppressing the SDE pathway by sterol solubilization with methyl-β-cyclodextrin (MβCD) (53, 54) did not block flg22–induced AtRGS1 internalization, even when MβCD was used in excess (Fig. 3A). Conversely, at a concentration of 5mM MβCD, AtRGS1 internalization induced by d-glucose was reduced by nearly 50% of the maximal amount when compared to baseline (Fig. 3B). This finding suggests that roughly half of the pool of AtRGS1 undergoes d-glucose-endocytosis through SDE. To determine if AtRGS1 endocytoses by the CME pathway, we applied TyrA23 with flg22 or d-glucose. TyrA23 abolished flg22-induced AtRGS1 endocytosis (Fig. 3C) and reduced d-glucose-induced AtRGS1 endocytosis by ~50% (Fig. 3D), suggesting that the effect of TyrA23 was not due to global cytotoxicity. The structurally similar but inactive TyrA23 analog TyrphostinA51 did not significantly affect AtRGS1 internalization (Fig. 3C and 3D), indicating that the inhibitory effect of TyrA23 is chemically specific (45). When both CME and SDE inhibitors were applied simultaneously with glucose, AtRGS1 internalization was ablated (Fig. 3E).
Fig. 3. Two origins of AtRGS1 endocytosis.
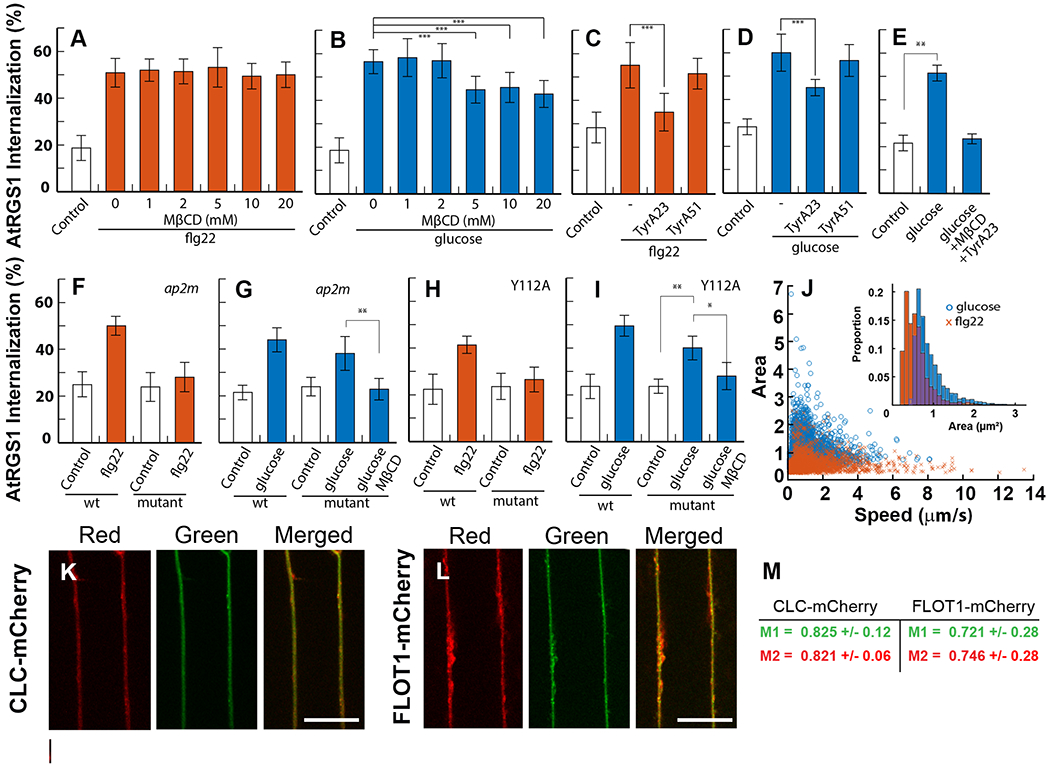
(A, B) AtRGS1-YFP seedlings were treated with increasing concentrations of MβCD at 0 mM (n= 18 biological replicates per group (flg22) and n=15 biological replicates per group (glucose)), 1 mM (n= 14 and 14 biological replicates per group), 2 mM (n= 15 and 16 biological replicates per group), 5 mM (n= 17 and 14 biological replicates per group), 10 mM (n= 12 and 17 biological replicates per group), and 20 mM (n= 15 and 6 biological replicates per group) for 45 min, then incubated in the same solution supplemented with 1 μM flg22 for 10 min (A) or 6% d-glucose (B) for 30 min before epidermal cells were imaged. Internalized AtRGS1-YFP was quantified to determine total endocytosis of AtRGS1. ***, p<0.01. (C-D) Endocytosis of AtRGS1 induced by flg22 (n=14 biological replicates per group) (C) and glucose (n=27 biological replicates per group) (D) in the presence or absence of TyrA23 (n= 8-27 biological replicates per group) or its inactive analog TyrA51 (n=19 biological replicates per group). ***, p<0.01. (E) Glucose-induced endocytosis of AtRGS1 in the absence or presence of both MβCD and TyrA23. (n=8 biological replicates per group) (**, p<0.01). (F, G) Endocytosis of AtRGS1 in WT or a genetic null mutant of ap2μ, which encodes the CME cargo recognition complex, induced by flg22 (n=16 biological replicates per group) (F) or glucose (n=32 biological replicates per group) (G). MβCD was added as indicated (n=18 biological replicates per group) (**, p<0.01). (H, I) Endocytosis of WT or AtRGS1Y112A, which has a mutation in the tyrosine motif recognized by ap2μ, induced by flg22 (n=16 biological replicates per group) (H) or glucose (n=12 biological replicates per group) (I). MβCD was added as indicated (n=9 biological replicates per group) (*, p<0.05, **, p<0.01). (J) The speed and surface area of AtRGS1-GFP particles as tracked and measured by IMARIS applied to 30 second time lapse imaging acquired by TIRF microscopy at 5 minutes after treatment with glucose (n=4619 tracked particles across 3 biological replicates and 2 technical replicates) or flg22 (n=2026 tracked particles across 3 biological replicates and 2 technical replicates). (K, L) Confocal images of AtRGS1-GFP (green) and either CLC-mCherry (red, K) or FLOT1-mCherry (red, L). Individual images for AtRGS1-GFP and mCherry-tagged endocytosis markers are in Supplemental Fig. S4E and F. Scale bar = 20 μm. (M) The average overlap scores with standard deviation.for CLC-mCherry and FLOT1-mCherry with AtRGS1-GFP (n= 10 and 9 biological replicates per group, respectively) are given M1 and M2 represent the overlap of fluorescence in channel 1 (green) with channel 2 (red) and vice versa.
These results suggest that there is a single flg22-induced pool that internalizes through a CME pathway and that there are two distinct glucose-induced pools that internalize separately through the CME and SDE pathways. Because the glucose-induced pool was reduced to a similar extent by CME or SDE inhibition, a rapid equilibrium between the pools is unlikely; otherwise neither inhibitor would have shown efficacy. To test this further, AtRGS1 internalization was induced with flg22 then by d-glucose. The addition of d-glucose slightly increased the percentage of AtRGS1 internalization although the increase was not always statistically different compared to the d- glucose treatment alone (Fig. S2G).
Because the use of TyrA23 as a specific endocytosis inhibitor is controversial (55–57), we added a genetic approach by measuring AtRGS1 internalization in the AP-2μ null mutant, ap2m (58). In ap2m mutant seedlings, flg22-induced AtRGS1 internalization was ablated and d-glucose induced about half maximal AtRGS1 internalization compared to WT (Fig. 3F). These results were consistent with the effect of TyrA23. The addition of MβCD with d-glucose ablated AtRGS1 internalization in ap2m mutant seedlings to basal levels (Fig. 3G). These observations suggests that there are two signal-dependent pools of AtRGS1 on the plasma membrane, a homogenous pool for flg22 signaling and a conglomerate pool for D-glucose signaling.
Because d-glucose utilizes both CME and SDE pathways to induce AtRGS1 internalization, we tested if pretreating with d-glucose to induce AtRGS1 CME would alter flg22-induced MPK6 activation, but did not observe an effect (Fig. 2G inset) suggesting that the pools of AtRGS1 that undergo D-glucose- or flg22-induced CME are distinct. We pretreated SOMA-NES seedlings with TyrA23 and found that inhibition of CME prevented the flg22-induced increase in MPK6 activity (Fig 2E, Fig. S2C). The clathrin heavy-chain inhibitor ES9–17 (59) also inhibited flg22-induced MAPK activation but not consistently, and on occasion, ES9–17 activated MAPKs in the absence of flg22 (Fig. S3, A–D), suggesting that ES9–17 has off-target effects. TyrA51, a negative control for TyrA23, did not block flg22-induced MAPK activation (Fig. S2 D), indicating that the effect of TyrA23 is chemically specific to its role in blocking CME and suggesting that AtRGS1 endocytosis per se is required. Furthermore, these data exclude the possibility that flg22-induced MAPK activation simply requires endocytosis in general, rather than AtRGS1 endocytosis specifically, or some other specific protein that is rate-limiting for activation of the MAPK cascade.
A point mutation in the intracellular loop disrupts ligand-specific internalization.
The μ subunit of the AP-2 complex is a cargo recognition and interaction component in the CME pathway. It binds to tyrosine-containing motifs YXXΦ, where X is any amino acid and Φ is an amino acid with a hydrophobic side chain (60). We generated a mutant with a tyrosine to alanine mutation (AtRGS1Y112A) in the Y112FIF motif, which fits the AP-2μ consensus motif and is located in the second intracellular loop of AtRGS1 (61). flg22 failed to induce endocytosis of the AtRGS1Y112A mutant (Fig. 3H). D-glucose-induced internalization of the AtRGS1Y112A was reduced to half and was further reduced to the unstimulated level with the addition of MβCD (Fig. 3I). Although both TyrA23 and genetic ablation of AP-2μ may have off-target effects, the effect of the point mutation in the AtRGS1 cargo recognition motif provides strong evidence for flg22- and glucose-induced CME of distinct pools of AtRGS1.
Physically distinct, dynamic populations of AtRGS1 suggest two different AtRGS1 signalosomes.
We showed that two pools of AtRGS1 undergo sterol or clathrin-mediated endocytosis dependent on the stimulus, raising the possibility that the two AtRGS1 pools are physically distinct on the cell membrane. To test this hypothesis using physical property differences, we imaged AtRGS1 with a C-terminal GFP tag using total internal reflection microscopy (TIRF) and surface tracking (Fig. S4A). Although we could not distinguish membrane bound AtRGS1-GFP from AtRGS1-GFP located at the extreme membrane periphery, we can otherwise quantitate the intrinsic properties of AtRGS1-GFP populations under different ligand conditions. We calculated the average size and speed of GFP fluorescence during time-lapse imaging at 5 and at 15 minutes post treatment with glucose and flg22. Two clearly distinct, signal-dependent populations of AtRGS1 were observed (Fig. 3J). After 5 minutes of glucose stimulation, the surface area of AtRGS1-GFP clusters were significantly larger than those in flg22-treated cells (Fig. 3J inset). At 15 minutes, the area of clusters in flg22-treated cells increased slightly to 0.6733 μm2, and the area of the glucose-treated clusters increased to 1.0072 μm2 (Fig. S4, B–D). For flg22-treated cells, the average velocity of the clusters were 1.53μm/s and 1.63μm/s at 5 and 15 minutes, respectively and for the d-glucose-treated cells, the average velocities were 0.98μm/s and 1.01μm/s at 5 and 15 minutes, respectively. Two populations of differently-sized AtRGS1 protein clusters moving at different speeds provides further support two origins of endocytosis.
AtRGS1 is associated with both CME and SDE markers.
The clathrin light chain (CLC) and FLOT1 proteins are associated with CME and SDE, respectively (20, 27). We generated transgenic lines expressing AtRGS1-GFP with either CLC-mCherry (as a marker for CME) or Flot1-mCherry (as a marker for SDE) to investigate the localization of AtRGS1 in relation to both markers. AtRGS1-GFP and our endocytosis markers showed a high average overlap (Fig. 3K, 3L, and 3M and Fig. S4, E and F), suggesting that AtRGS1 colocalizes with both CLC and FLOT1 under basal conditions (tonic cycling).
Different AtRGS1 trafficking paths involve phosphorylation from different kinases.
Phosphorylation of AtRGS1 is a necessary requisite for both d-glucose- and flg22-induced endocytosis. The C-terminus of AtRGS1 contains multiple di-serine residues that could function as potential phosphorylation sites for signal transduction. A truncated version of AtRGS1 lacking the 43 C-terminal residues, AtRGS1∆Ct, did not internalize in response to flg22 (Fig. 4A). However, D-glucose application resulted in the internalization of the AtRGS1∆Ct mutant, but at 50% the level of WT AtRGS1. MβCD inhibited internalization of the AtRGS1∆Ct mutant (Fig. 4B). These results show that the C-terminus is necessary for flg22- and glucose-induced endocytosis of AtRGS1 and support two subpopulations of AtRGS1 among the glucose-induced pool.
Fig. 4. CME of AtRGS1 is phosphorylation dependent and G protein specific.
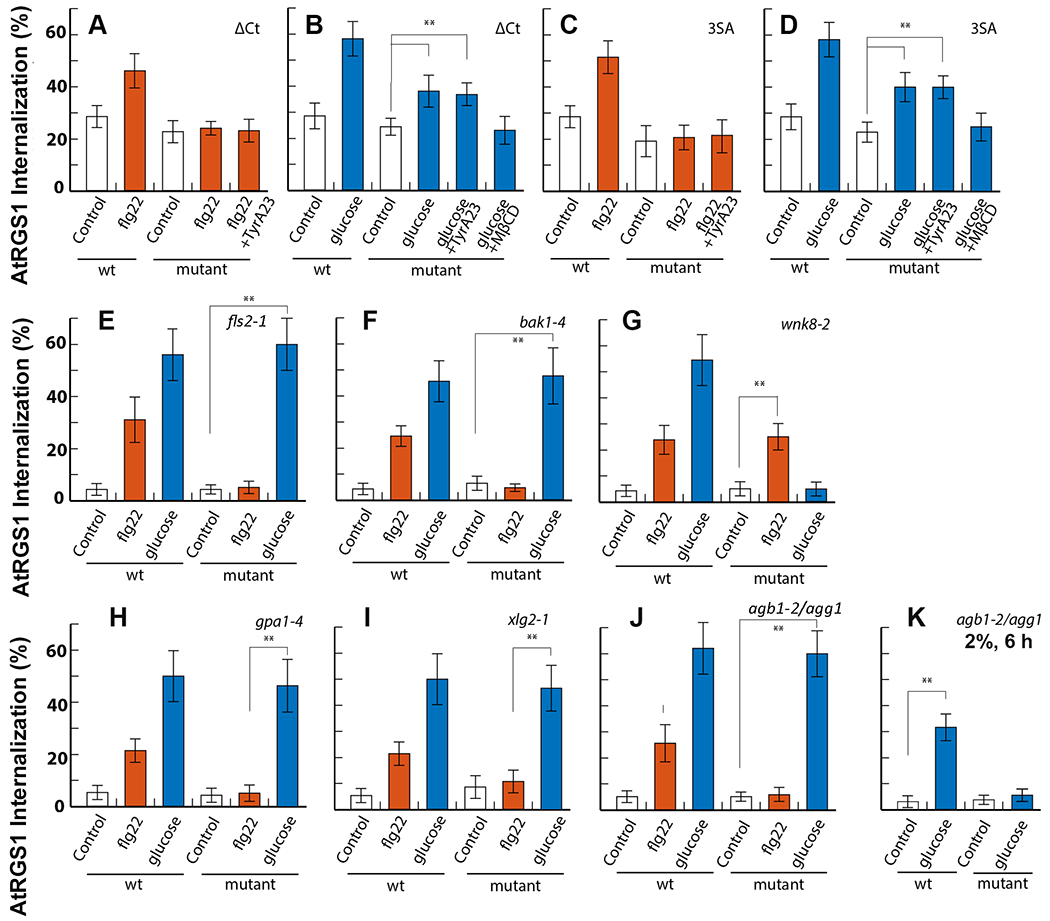
(A and B) Endocytosis of WT or AtRGS1∆Ct (ΔCt), which lacks the 43 most C-terminal residues (including 8 serine residues), induced by flg22 (n=11 biological replicates per group) or glucose (n=12 biological replicates per group). TyrA23 (n= 12 biological replicates per group) or MβCD were added as indicated (n=13 biological replicates per group). (C and D) Endocytosis of WT or AtRGS13SA, in which serine residues were mutated to alanine at positions 428, 435, and 436, induced by flg22 (n=10 biological replicates per group) or glucose (n=22 biological replicates per group). (E) Endocytosis of AtRGS1 in WT or a genetic null mutant of fls2, which encodes the flg22 receptor, induced by flg22 (n=12 biological replicates per group) or glucose (n=9 biological replicates per group). (F) Endocytosis of AtRGS1 in WT or a genetic null mutant of bak1-4, which encodes of the flg22 co-receptor, induced by flg22 (n=16 biological replicates per group) or glucose (n=17 biological replicates per group). (G) Endocytosis of AtRGS1 in WT or a genetic null mutant of wnk8-2, which encodes the kinase WNK8, induced by flg22 (n=10 biological replicates per group) or glucose (n=13 biological replicates per group). (H) Endocytosis of AtRGS1 in WT or a genetic null mutant of gpa1-4, which encodes Gα, induced by flg22 (n=15 biological replicates per group) or glucose (n=22 biological replicates per group). (I) Endocytosis of AtRGS1 in WT or a genetic null mutant of xlg2-1, which encodes the extra-large Gα protein XLG2,, induced by flg22 (n=14 biological replicates per group) or glucose (n=19 biological replicates per group). (J and K) Endocytosis of AtRGS1 in WT or a genetic null mutant of agb1-2/agg1, which encodes the AGB1/AGG1 heterodimer, induced by flg22 (n=15 biological replicates per group) (J), high dose, low duration glucose (n=17 biological replicates per group) (J), or low dose, long duration glucose (n=25 biological replicates per group) (K). Error bars represent standard deviation. **, p<0.01.
Two phosphorylated serine residues in the C-terminus of AtRGS1 at positions 428 and 435 or 436 are necessary for at least partial endocytosis of AtRGS1 induced by both d-glucose and flg22. These three serines were mutated to alanine (AtRGS13SA) to determine if these specific residues were necessary to induce internalization by either agonist. Upon treatment with flg22, AtRGS13SA internalization remained at the basal level (Fig. 4C), confirming previous results (50) and suggesting that the CME pathway depends on the phosphorylation of Ser428, Ser435 and/or Ser436. d-glucose-induced endocytosis was only partially impaired by the AtRGS13SA mutant (Fig. 4D). MβCD completely blocked glucose-induced endocytosis of the AtRGS13SA mutant (Fig. 4D), consistent with our previous results suggesting that glucose-induced internalization utilizes both CME and SDE pathways and that the CME pathway requires phosphorylation at Ser428/435/436. Furthermore, these various genetic results enabled us to exclude an osmotic effect that effectively sequesters a portion of the plasma membrane pool of AtRGS1 into a sterol-dependent microdomain that cannot internalize its components.
Because the cluster of phosphorylated serine residues on the C-terminus of AtRGS1 was required for AtRGS1 internalization, we hypothesized that a mechanism for selectivity is to functionally-sequester cognate kinases for d-glucose and flg22 into their respective ligand-delineated pools. To test this hypothesis, we quantified AtRGS1 internalization in FLS2 kinase (fls2) and BAK1 co-receptor (bak1) and the d-glucose-activated kinase With No Lysine (wnk8-2, wnk1-1) null mutants. AtRGS1 endocytosis induced by flg22 was ablated in fls2 and bak1-4 mutants, whereas d-glucose-induced internalization in these mutants was unaffected (Fig. 4E and 4F). In contrast, d-glucose-induced internalization in the wnk8-2 mutant was ablated without affecting flg22-induced AtRGS1 endocytosis (Fig. 4G). Our results suggest that FLS2 and BAK1 are specific to the CME pathway and WNK8 is specific to the SDE pathway.
Gβγ is required for the recruitment of WNK1 and WNK8 for the phosphorylation of AtRGS1, leading to endocytosis of and activation of downstream G signaling (7, 8). To test the hypothesis that individual components of the G protein heterotrimer may be required for biased signaling, we quantified AtRGS1 endocytosis in G protein mutant backgrounds after flg22 and glucose activation. In the gpa1-4 mutant (which is null for Gα), glucose-induced AtRGS1 endocytosis was not affected whereas flg22-induced AtRGS1 endocytosis was reduced to basal levels, indicating that the Gα subunit is essential for this pathway (Fig. 4H). We additionally tested AtRGS1 in the absence of the atypical G protein XLG2. Loss of XLG2 in the xlg2-1 mutant inhibited AtRGS1 endocytosis induced flg22, but not that induced by D-glucose (Fig. 4I). In the agb1-2/agg1 double null mutant, which lacks the Gβγ dimer, AtRGS1 endocytosis was diminished after addition of flg22 but not after 30 minutes of treatment with 6% d-glucose (Fig. 4J). However, activation of CME by a lower dose (2%) of d-glucose over a longer duration (6 hour) required AGB1/AGG1 (Fig. 4K). Kinases are recruited to AtRGS1 at the plasma membrane by different paths. WNK8 associates with the Gβγ dimer quickly after the addition of D-glucose (62), whereas the receptor-like kinase BAK1 physically interacts with AtRGS1 upon flg22 addition (14).
VPS26 is necessary for AtRGS1 endocytosis in the CME pathway.
Higher plant genomes do not encode visual or nonvisual arrestins (63). Therefore, we sought candidate adaptors for AtRGS1 that may function like β-arrestins to recruit AP2/clathrin to AtRGS1 for endocytosis by querying 3-D structure databases. We identified three Arabidopsis VACUOLAR PROTEIN SORTING 26 (VPS26) proteins that may contain arrestin folds (64) and are orthologous to human VPS26 (Fig. S5A, B, and E–L). Arabidopsis VPS26A and VPS26B amino acid sequences are 91% identical whereas VPS26-like is 20% identical to VPS26A and VPS26B. In mammals and plants, VPS26 operates with VPS29 and VPS35 in the retromer complex on the endosome (65, 66). To compare the arrestin and AtVPS26A protein structures, we created five models of AtVPS26A based on human VPS26A 2FAU because there is no crystal structure of AtVPS26A available. Discrete optimized protein energy (DOPE) scores calculated for each residue between bovine arrestin-3 and the five atvps26a models (Fig. S5D) were dissimilar in only three positions (residues around 60, 240, and 260). The atvps26a2 model was selected based on the lowest root mean square deviation (RMSD) value (Fig. S5D) and plotted DOPE per-residue score curve (Fig. S4C). With an RMSD of 0.17 Ȧ, atvps26a-2 differed from bovine arrestin-3 by less than the length of a C-C bond. Although the model showed that plant VPS26 proteins are not arrestin homologs, it suggested that Arabidopsis VPS26 proteins have classical arrestin folds.
We tested the hypothesis that AtVPS26 is a candidate adaptor for CME of AtRGS1 or, in any case, necessary for the observed signaling bias. We isolated null alleles of the three VPS26 genes in Arabidopsis: VPS26A, VPS26B, and VPS26-like (also known as VPS26C) (66, 67) and phenotyped the hypocotyl length at 5 days, the same age stage used for the previous assays (Fig. 1–4). There were no major qualitative differences between VPS26 null mutants and wild type phenotype at this young etiolated stage (3-5 days) (Fig. S5M), precluding any developmental basis for alterations in AtRGS1 activation in these mutants. However, the light-grown 10-d old seedling and adult plants had profound phenotypes (Fig. S5 N, O) as previously reported 129. Loss of either VPS26A or VPS26B reduced the flg22-induced internalization of AtRGS1 to levels that are not statistically different from the baseline level whereas loss of VPS26-like had no statistical effect on AtRGS1 internalization by flg22 (Fig. 5A). Loss of any of the three VPS26 proteins had no statistical effect on d-glucose-induced internalization of AtRGS1 (p<0.001) suggesting that VPS26A/B are involved in flg22, but not glucose-induced AtRGS1 endocytosis. To confirm that VPS26 is not involved in d-glucose-induced AtRGS1 internalization, we quantitated the CME and SDE portions of this trafficking pathway in the vps26 mutants. In each of the vps26 mutant backgrounds, both the TyrA23A and MβCD-inhibitable segments of the d-glucose-induced internalization of AtRGS1 remained intact (Fig. 5B). This suggests that the TyrA23-dileneated (CME) pathway used by AtRGS1 when activated by flg22 differs from the TyrA23A-dileneated pathway activated by d-glucose.
Fig. 5. Arabidopsis VPS26 subunits of the retromer complex dimerizes and facilitates AtRGS1 internalization.
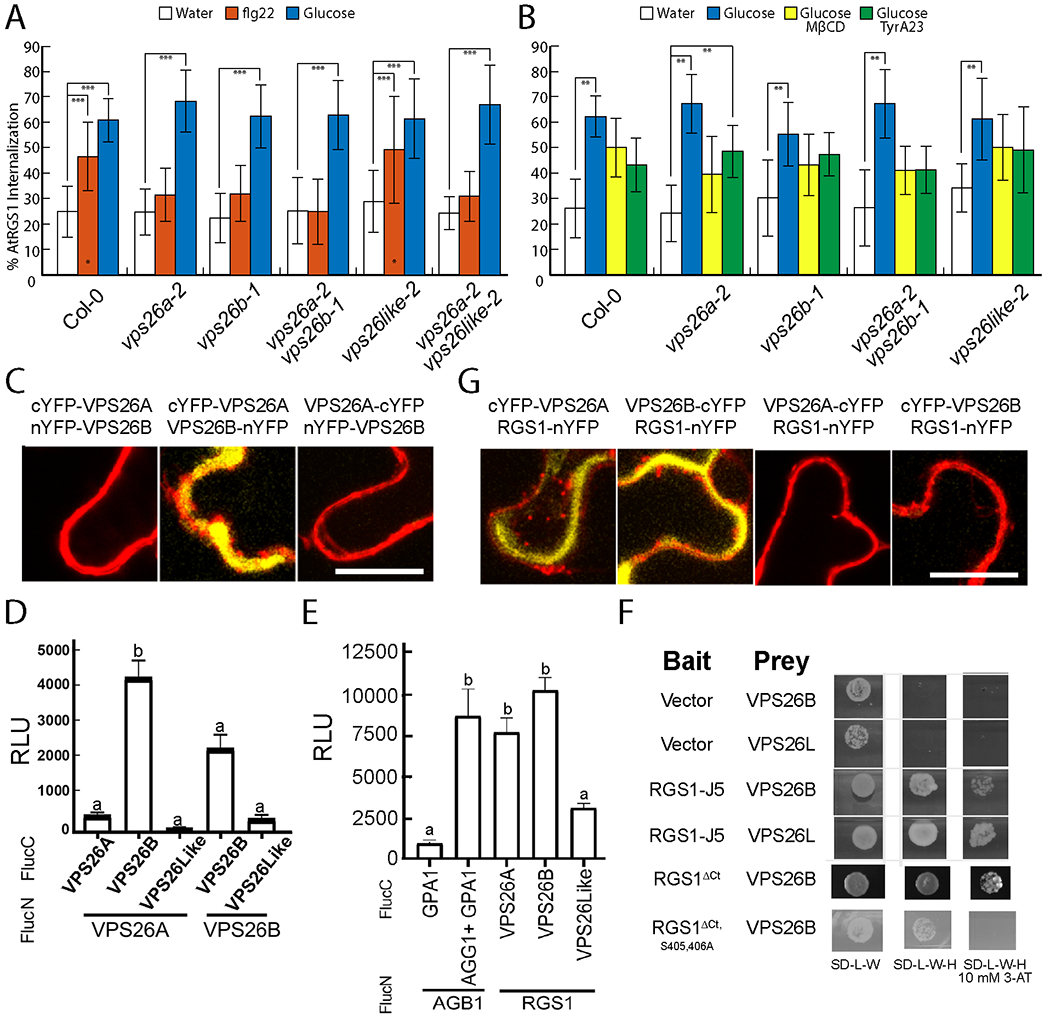
(A) Quantification of AtRGS1-YFP internalization in WT and vps26 null mutants after treatment with water, 1 μM flg22 or 6% d-glucose. N=29-58 biological replicates per group. ***, P < 0.001 as determined by a two-way ANOVA with post-hoc Tukey HSD analysis. (B) AtRGS1-YFP internalization in seedlings pretreated with MβCD, TyrA23, or both prior to treatment with d-glucose. Error bars represent standard deviation. Data was averaged across three separate experiments. **, P < 0.01 as determined by a two-way ANOVA with post-hoc Tukey HSD analysis between treatments within genotype. (C, G) Bifluorescence complementation (BiFC) of VPS26A and VPS26B measuring dimerization and interaction. Representative cells shown. n=5 biological replicates. Experiment repeated 2 times. (D) Split luciferase complementation by VPS26A and VPS26B heterodimers and VPS26B homodimers. Error bars are standard error of the mean (SEM). Means with different letters indicate significantly different (Tukey’s Honestly Significant Difference test, p<0.05). n=64 leaf discs from 4 individual tobacco plants. (E) Split-luciferase complementation by VPS26A and AtRGS1. Positive control is complementation by the heterotrimeric G protein complex (AtGPA1/AGB1/AGG1). Negative control is AtGPA1 and AGB1 in the absence of AGG1. Error bars are SEM. n=64 biological replicates. Means with different letters indicate significantly different (one-way ANOVA with post-hoc Tukey’s HSD test, p<0.05). (F)Yeast two-hybrid complementation between the cytoplasmic domain of AtRGS1 (RGS1-J5) and VPS26B. RGS1-J5 contains the linker between the 7TM domain and the RGS box, the RGS box and a C-terminal tail (Ct). RGS1ΔCt lacks the C-terminal tail containing the phosphorylation cluster required for AtRGS1 endocytosis. RGS1ΔCt S405,406A lacks the Ct and has two additional phosphosites mutated. –LW is the leucine/tryptophan drop out media and -LWH is the leucine/tryptophan/histidine drop out media. 10 mM 3-AT indicates the addition of 10 mM 3-amino-1,2,4-triazole. Scale bars for C and G are 20 μm.
Self-association of arrestin family members occurs and may be part of a regulatory mechanism for arrestin activation (68). The genetic data that both the single null mutations each of VPS26A and VPS26B ablate flg22-induced AtRGS1 endocytosis suggest that VPS26A and B form obligate dimers. To test if VPS26A and B subunits heterodimerize in vivo, irreversible BiFC analysis was conducted and showed that heterodimers can form from ectopically-expressed monomers and suggested that the orientation is head-to-tail (Fig. 5C). For relative quantitation of in vivo interaction, reversible split luciferase was performed and found that the strongest interaction was between VPS26A and VPS26B (Fig. 5D). VPS26B dimers formed under these conditions but not VPS26A dimers or any oligomer with VPS26-like subunits. We have yet to find conditions that allow stable expression of VPS26 subunits tagged with a full-length auto-fluorescent protein in Arabidopsis suggesting that the additional mass of the tag prevents dimer formation and that monomers are unstable.
We next tested whether the full-length AtRGS1 protein interacted with the VPS26 subunits in planta. The split luciferase assay confirmed that this interaction was similar in strength as that between AtGPA1 and its AGB1/AGG1 partner (Fig. 5E). Yeast two-hybrid analysis showed that the interaction between AtRGS1 and VPS26 was direct. The C-terminal half of AtRGS1 (thus lacking the 7 TM domain) interacts with VPS26B (Fig. 5F). Removal of the post RGS-box C-terminal tail which includes the Ser428,435,436 phosphorylation cluster (Fig. 5F) did not ablate this interaction; however, the loss of two other phosphorylated Ser residues located between helices VII and VIII of the RGS box completely abolished the interaction (Fig. 5F). Fluorescent complementation of YFP by AtRGS1-nYFP with cYFP-VPS26A and with VPS26B-cYFP was observed (Fig. 5G). Finally, in planta analysis using BiFC confirmed the interaction by fluorescent complementation of VPS26A and VPS26B in one orientation but not in the other (Fig. 5C.
To summarize our findings on these predicted, arrestin-fold-containing VPS26 proteins (Fig. 5, A to F and S5, A to O), genetic and other in vivo data are consistent with VPS26A/VPS26B heterodimers moonlighting as physically-direct adaptors in the flg22-initiated, CME of AtRGS1. We do not exclude the possibility that VPS26A and B proteins are adaptors to a larger set of targets. Due to difficulty in resolving the periphery of the plant cell cytoplasm (equivalent to the cortex in an animal cell), we also acknowledge the possibility that the lack of apparent internalization of AtRGS1 in the vps26 mutants may be due only to loss of retrograde movement from the endosome to the Golgi.
Discussion
Different ligands discriminated by the same receptor or utilizing the same core of signaling elements can set in motion a cascade of events that lead to multiple outcomes. When subsets of outcomes are ligand-specific, we label this biased signaling. This bias can be encoded in the ligand structure (biased ligands), in the receptors (biased receptors), or in the interactions between the signaling components of the system (biased systems) (69). Ligand bias occurs when a ligand stabilizes one or a subset of conformations of a receptor protein which then preferentially recruits or activates signaling elements such as arrestin and the heterotrimeric G protein complex that lead toward one cellular outcome over another (70, 71). Receptor bias occurs through recognition of the same ligand by multiple receptor types (72) including so-called decoy receptors (73, 74), some of which may in fact be functional receptors that use non-classical signaling pathways (73, 75). System bias involves a cell-autonomous shift to one pathway over another by some unknown mechanism but one possibility is through mass action, for example by making arrestin more abundant at the receptor than Gα or a particular kinase (76). This last category of bias signaling is the least understood and was the subject of the present work.
Phosphorylation patterns are the chemical bar codes for biased signaling at least for arrestin-dependent outcomes (77–81). G-protein coupled Receptor Kinases (GRKs) phosphorylate the C-terminus or other cytoplasmic elements of GPCRs in response to agonist binding (82). The subsequent coupling of arrestin to phosphorylated GPCRs does not end G-protein-independent signaling. Arrestin-bound GPCRs further activate some kinase pathways including MAPK and tyrosine kinases (83–85) but not all (86). Although plants lack GRKs, the Arabidopsis WNK family of kinases phosphorylate the C-terminal tail of AtRGS1 in response to extracellular glucose (7, 8). AtRGS1 is also phosphorylated by other kinases including BAK1, a co-receptor for the PAMP flg22 (50, 87, 88) and loss of AtRGS1 and other G protein components affect resistance to multiple pathogens (89). System-biased signaling may be particularly relevant in plants where AtRGS1 modulates intracellular signaling transduction through G protein activation. AtRGS1 de-represses G protein signaling with the ligand discrimination likely facilitated by membrane bound receptor like kinases (RLKs) of which there are more than 400 members with receptor topology in Arabidopsis (90) raising the possibility that signal discrimination through the heterotrimeric G protein pathway in plants is as diverse as in animals.
Our study uncovered system bias such that a single receptor-like RGS protein modulated different quantifiable signaling outputs from two distinct signal inputs (Fig. 6A). All of the measured signal outputs corresponded to a specific ligand input. flg22 induced Ca2+ release (Fig. 2I–K) and MAPK signaling (Fig. 2E–H, K) whereas d-glucose or its metabolite affected gene expression (Fig. 2L). For flg22, all members of the G protein heterotrimer, along with XLG2 and VPS26A/B were necessary for CME of AtRGS1. Additionally, specific phosphorylation of di-serines at the AtRGS1 C-terminus was required, which was also required for transduction of D-glucose signaling through CME of AtRGS1. This phosphorylation event did not require VPS26 for high dose, low duration exposure to sugar (Fig. 6B). This phenomenon of signaling through AtRGS1 mediated by the same mechanism of phosphorylation-dependent endocytosis with different G proteins and adapters raises the important question of how extracellular ligands are discriminated for downstream signaling events. Although AtRGS1 endocytosis was necessary for G protein activation it would seem that AtRGS1 is not the discriminator. Instead, an RLK protein that directly phosphorylates AtRGS1 (50, 87), AtGPA1 (91), and potentially VPS26 provides the requisite information for ligand-specific downstream signal transduction in cells.
Fig. 6. Overview of flg22- and D-glucose-biased signaling.
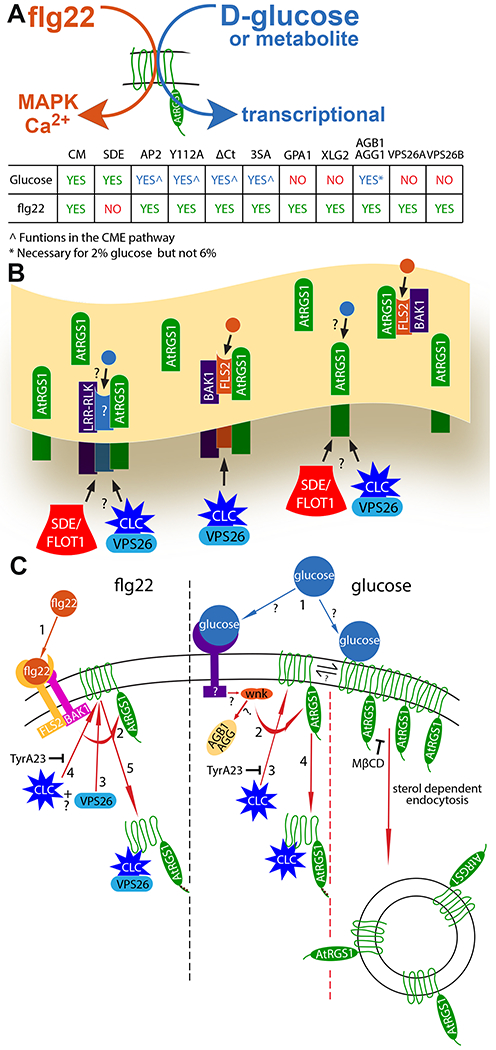
(A) Simple model illustrating the biased signaling output through AtRGS1 leading from flg22 and glucose or metabolite input, and a chart summarizing the origins of endocytosis (CME and SDE), recognition motif and phosphorylation requirements, and individual proteins necessary for glucose- and flg22-induced endocytosis of AtRGS1 where green “yes” indicates a requirement for signaling, red “no” indicates not required, and blue “yes” indicates a requirement with some restrictions. (B) Membrane overview illustrating proposed AtRGS1 microdomain clusters with common RLK neighbors. flg22 (orange circle) binds to FLS2 to initiate signaling through AtRGS1. The mechanism of glucose (blue circle) perception is unknown as indicated by the question marks. After ligand perception, SDE or CME of AtRGS1 occurs to permit downstream signaling. (C) A diagram of the individual components involved in the mechanism of endocytosis initiated by flg22 and glucose or metabolite and the presumed sequence of events. For flg22: (step 1) perception of ligand, (step 2) phosphorylation of AtRGS1 by a RLK, (steps 3 and 4) binding of clathrin complex and/or VPS26 in an unknown order, and (step 5) internalization of AtRGS1. For glucose or metabolite: (Step 1) ligand perception by a RLK or direct interaction with AtRGS1, (step 2) receptor interaction with WNK (for SDE, step 2 indicates immediate endocytosis because other key components of the pathway are unknown), (step 3) phosphorylation of AtRGS1 by WNK, (step 4) binding of clathrin complex to AtRGS1, and (step 5) internalization of AtRGS1. TyrA23 inhibits CME in both pathways, and MβCD inhibits AtRGS1 microdomain formation at the membrane.
The two mechanisms of AtRGS1 endocytosis induced by d-glucose or its metabolites implies a bipartite pool of AtRGS1, prompting the question of an equilibrium or steady state between the populations. The evidence so far suggests that it is not. If these AtRGS1 pools were dynamically exchanging within the membrane, we would expect to induce internalization of nearly all membrane bound AtRGS1 by inhibiting one mechanism of endocytosis thereby causing a shift entirely to the other. For example, inhibiting CME with TyrA23 did not force AtRGS1 endocytosis entirely to the SDE mode. Inhibiting one mechanism of endocytosis only partly reduced AtRGS1 internalization by half, indicating the AtRGS1 populations may be physically isolated and static in the cell membrane (Fig. 6C). Some AtRGS1 proteins may be grouped into so-called microdomains or clusters surrounded by receptor/co-receptor RLK proteins whereas other AtRGS1 proteins may be distributed throughout the membrane without common-neighbor RLKs (Fig. 6C). Similarly, the question exists whether an equilibrium between the flg22-mediated and D-glucose-mediated CME pools exists and the conclusion is again that an equilibrium is not likely, consistent with a genuine system bias in signaling. We defined as the “CME pools” the portion of either the flg22- or d-glucose-activated AtRGS1 population that was inhibited by TyrA23, by loss of the tyrosine-binding motif, and by loss of the AP2μ subunit of the clathrin complex (Fig. 3 C to G). Although the two pools share these properties, they do not share the requirement of the candidate adapter, VPS26 monomer or dimer (Fig. 5A and B). Moreover, depletion of the AtRGS1 pool at the plasma membrane by D-glucose stimulation did not affect the amplitude of flg22 activation (Fig. 2G inset) as would be expected if these pools were in equilibrium. The two mechanisms in the composite glucose-induced AtRGS1 endocytosis may be a result of two distinct mechanisms of sugar perception: one through direct interaction of AtRGS1 with a sugar or sugar metabolite and the other through sugar binding to a RLK in the membrane similar to flg22::FLS2 binding (Fig. 6D).
What the present work revealed about each origin of endocytosis including phosphorylation, binding of VPS26, formation of clathrin coated vesicles, and endocytosis of AtRGS1 either through clathrin coated vesicles or sterol dependent rafts (glucose) is shown in the context of the ordered steps of signal transduction from ligand perception to internalization of AtRGS1 (Fig. 6D). If D-glucose or a metabolite is perceived by two distinct sensing mechanisms, each mechanism may operate exclusively through one origin of endocytosis with different subsets of G protein and internalization signaling components. Within the glucose model, a system bias may favor one origin of endocytosis that results from architectural differences in the membrane surrounding AtRGS1. A high density of d-glucose-binding RLKs may favor d-glucose-induced CME over SDE of AtRGS1.
The division of G protein involvement in d-glucose-induced CME and SDE challenges previously published reports on the necessity for subunits of the G protein heterotrimer in d-glucose signaling through AtRGS1, specifically the abrogation of AtRGS1 endocytosis in the Gβ null mutant and partial reduction of d-glucose-induced endocytosis with the loss of Gα (7). Our results confirmed that Gβ was required for AtRGS1 endocytosis, but only at a low concentration (2%, ~110 nM). Neither Gβ nor Gα was required at a higher concentration (6%, ~330 nm). Higher sugar concentrations, those typically found at or around vascular unloading areas, may elicit a quicker signaling response than low dose sugar likely found in or near epidermal cells where we quantified AtRGS1 internalization. The importance of sugar signaling in cell growth, division, and maintenance may necessitate multiple dose-dependent mechanisms of signal transduction encoded in different origins of endocytosis and the individual G protein associated components.
Both flg22- and d-glucose outputs required AtRGS1 (14, 42, 61, 87) and an intact heterotrimeric G protein complex (Fig. 4 A to K S4 A to H) but d-glucose responsiveness depended on both the concentration and the duration of d-glucose stimulation. We previously designated this non-threshold-based activation phenomenon as dose-duration reciprocity (DDR) in which a low dose of d-glucose stimulation for a long period reaches the same output amplitude as a high dose pulse (8). The proposed mechanism is recruitment of WNK8 and WNK1 kinases to the membrane by the Gβγ dimer AGB1/AGG as a function of DDR to phosphorylate AtRGS1 for endocytosis. However, our higher resolution analyses here challenge some aspects of that original model and provide deeper mechanistic insight. AGB1 was essential for low but not high d-glucose DDR. WNK8 was required as previously shown but WNK1 was not required at longer, low-glucose treatments (6 hours) in contrast to previously published data (8). This discrepancy is most likely due to differences in expression level between the two studies (transient in (62) compared to stable here) because glucose responsiveness is sensitive to the pool size of AtRGS1 (92).
We acknowledge the possibility that insufficient spatial resolution precludes us from concluding that VPS26 functions solely as a retromer subunit and not also as an arrestin-like adaptor, meaning that VPS26 has both a retromer and adaptor function. Specifically, blocked endocytosis (arrestin function) may have a similar phenotype as a block in retrograde trafficking (retromer function). However, some of our observations point toward VPS26 acting as an arrestin-like adaptor. Contrary to a prior report (93), we found that VPS26 proteins dimerized (Fig. 5C and D,), similar to arrestins. Cargo recognition by VPS26 within the retromer is physically indirect; specifically, it strictly requires a sorting nexin to complete the required protein interface (93), yet we showed here that VPS26 bound directly to AtRGS1, indicating a potentially new role for VPS26.
Besides the two visual and nonvisual β-arrestins, the arrestin super family in humans also has 6 α-arrestin members. These α–arrestins may be closest to the ancestral arrestin from which the visual/non-visual arrestins evolved because they are found in stromenopiles basal to the opistokont lineage containing metazoans and fungi (63). Using Mus ARRDC3/TLIMP (BAC65781) as the search query, we found α–arrestin orthologs in the 4-cell alga Tetrabaena (PNH03120) and in the green alga Chara (GBG71752), both basal to land plants, but none in Arabidopsis (eudicot), rice (monocot), Amborella (base of angiosperms), and Physcomitrella (basal to higher land plants). This finding suggests that α–arrestins were lost in Plantae, which possibly allowed expansion of the related VPS26 family to evolve another function (63). Because many GPCR-containing organisms divergent to humans lack arrestin proteins, another important finding from this study was the suggestion that the retromer subunits AtVPS26A and AtVPS26B moonlight as arrestin-like adaptors for phosphorylation-induced, clathrin-dependent AtRGS1 endocytosis in Arabidopsis. Could this function be ubiquitous? Even in humans, some GPCR endocytosis occurs independently of β-arrestins yet still requires C-terminal phosphorylation, a tyrosine motif, AP2, and clathrin (94, 95) raising the possibility that VPS26 orthologs in humans may have a similar adaptor function for a subset of GPCRs.
This study focuses on biased signaling launched from different architectures of two AtRGS1-centered, physically distinct pathways. We genetically showed that different sets of kinases encoded this bias. However, we have not addressed how the architecture is established or maintained but we speculate that this too is based on a phosphorylation bar code on the AtRGS1/G protein complex. Therefore, establishing the dynamics of both the pre- and post-signaling phosphorylation bar codes is important for our understanding of biased system signaling.
Why both cytoplasmic and extracellular glucose pools are monitored by plant cells remains unclear. We speculate that the concentration of the extracellular pool of glucose is more dynamic than the cytoplasmic pool due to its rapid metabolic flux in the cytoplasm compared to the apoplast. Therefore, an extracellular glucose/metabolite detection system may be more appropriate for monitoring sugars produced by photosynthesis for real time information. This is consistent with the finding that AtRGS1 is important for detecting fluctuations in CO2-fixed sugar in the minute time range over the diel cycle (15, 61).
It is conceivable that this biased system sits at the crux of the “defense vs. growth trade-off” dilemma that plants face. Specifically, pathogen attack compels the plant to shift its utilization of fixed sugars from building cell walls to synthesizing defense molecules (96). The AtRGS1/G protein complex may be the fulcrum for this balance because AtRGS1 is involved in detecting fixed sugars (15, 97), establishing cell wall composition (98–100) and serving as a sentinel in innate immunity (89).
In conclusion, our data provide evidence for biased-system signaling through AtRGS1 and identified an arrestin-like adaptor. We also showed the importance of system architecture in bias in G-protein complex signaling. We showed that although phosphorylation was a requisite for AtRGS1 internalization, the signal discriminator was not the phosphorylation of di-serines in AtRGS1, but is likely to be the interaction of RLKs with other components in the signaling pathway. Subsequent analysis of protein clustering in the membrane and the phosphorylation barcode are necessary to understand the full extent of the system bias in signal transduction through AtRGS1.
Materials and Methods
Chemicals.
Methyl-β-cyclodextrin was purchased from Frontier Scientific, tyrphostin A51 and turanose were purchased from Sigma-Aldrich, and tyrphostin A23 was purchased from Santa Cruz Biotechnology. All chemicals were indicated by the vendors to be >98% pure. Other chemicals were synthesized.
Genetic material.
The ecotype for all the Arabidopsis used in this study is Columbia-0 (Col-0). The rgs1-2 protein-null, T-DNA-insertion mutant was created as previously described (61) . Expression of AtRGS1-YFP using the native reporter for RGS1-YFP was not detectable so this study used the 35S-AtRGS1-YFP reporter in Col-0 and in rgs1-2 as previously described (7, 62). The MAPK reporters used here (SOMA) were generated as previously described (38) and crossed into the AtRGS1-YFP reporter, rgs1-2 line. The R-GECO calcium reporter lines in wild type and rgs1-2 mutant were created as previously described (14). The ap2m-1 mutant generated for this study is a T-DNA insertion located at the 4th exon-intron border (SALK 083693) in gene At5g46630. The AtRGS1(Y112A)-YFP/rgs1-2 generated for this study is an alanine replacement of tyrosine 112 located in the tyrosine motif on the second intracellular loop of AtRGS1 tagged with YFP. The promoter was cauliflower mosaic viral (CaMV) promoter 35S. The AtRGS1-ΔCT and AtRGS1(3SA)-expressing lines and the wnk mutants were generated as previously described (7) . The gpa1-4 mutant was generated as previously described (101). The agb1-2 mutant was screened from a T-DNA-insertion population as previously described (102). The xlg2-1 mutants are as previously described (103). The agb1-2, agg1-1 mutant was generated by genetic cross of agb1-2 with agg1-1 (introgressed from WS) which is a T-DNA insertion mutant in the second intron of AGG1 (At3g63420). The AtRGS1-YFP, rgs1-2 reporter was combined with vps26a-2 (GABI 076F06), vps26b-1 (SALK 142592), and vps26like-2 (SALK 057991) mutant backgrounds through generating stable transformants, homozygous at all these loci. For this study, FLOT1 and CLC cDNAs behind the CaMV 35S promoter were tagged with m-Cherry at the C terminus and stably transformed into Arabidopsis. Each of these marker lines was crossed into a line expressing AtRGS1-GFP and made homozygous.
Plant growth conditions.
WT and mutant Arabidopsis seeds (all Col-0 ecotype) were surface sterilized with 80% EtOH for 10 seconds while vortexing followed by a 10-second vortex with 30% bleach. Seeds were subsequently washed 3X with ultrapure H20 and suspended in 12-well cell culture plates with ¼ MS with no sugar at pH 5.7 with 10-12 seeds per well. Plates were wrapped in aluminum foil, cold-treated at 4°C for 2 days, and treated with light for 2 hours to induce germination. After light treatment, plates were again wrapped in aluminum foil and placed on a horizontal shaker at ambient temperature for 5 days before imaging. All genotypes used in this study have been deposited into the Arabidopsis Biological Resource Center.
AtRGS1 internalization assay, proxy for G protein activation.
AtRGS1-YFP internalization was induced with d-glucose and flg22 as described (7, 8, 50). Briefly, Arabidopsis seeds expressing 35S:AtRGS1-YFP were sterilized and then sown on 1-mL liquid 1/4 X Murashige and Skoog (MS) medium without sugar in 12-well plates, stratified at 5°C for 2 days, treated for 2 hours with light, and grown in darkness at RT for 3-5 days. For optimal results, the plates were kept in darkness but moved to the microscope room on the third day to acclimate. 6% d-glucose or 1μm flg22 were applied to seedlings for 30 and 10 minutes respectively before imaging. Image acquisition was done on either a Zeiss LSM710 (Zeiss Microscopy, Oberkochen, Germany) with a C-Apochromat 40X 1.2NA water immersion objective (for Figs. 1A–B, 3A–3I, and 4A–D) or a Zeiss LSM880 with a C-Apochromat 40x/1.2NA water immersion objective (for Figs. 4E–K, 5A–B, and S1C–E). YFP excitation was at 514nm and emission collection 525-565nm. Emission collection on the LSM880 was done with a GaAsP detector. For RGS internalization assays a z-stack series was acquired at 0.5μm intervals between images. Image processing and RGS internalization measurements were done with the Fiji distribution of ImageJ (104) by calculating the ratio of internal intensity to total intensity including cell membrane as described (7) with the following modification: Internalized YFP fluorescence was measured and subtracted from total YFP fluorescence of individual cells. Images were acquired on the hypocotyl epidermis 2-4 mm below the cotyledons of seedlings treated with water, glucose, and flg22 in addition to the pharmacological inhibitors. Seedling exposure to light was minimized as much as was practical while imaging to avoid light induced internalization of AtRGS1. Statistical analysis was performed using analysis of variance. Differences in basal and treatment induced levels of RGS1-YFP internalization are a result of switching image acquisition from the LSM710 (Fig. 3A–I, 4A–D, & 5H and I) to the LSM880 (Fig. 4E–K) to improve image resolution and accuracy of internalization measurements.
Pharmacological inhibition of RGS internalization.
3-day old seedlings were preincubated with TyrA23 for 60 minutes at the specified concentrations, treated with TyrA23 and 6% d-Glucose for 30 minutes, and immediately imaged. For flg22, TyrA23 and 1μm flg22 were applied to seedlings for 10 minutes after the pre-incubation with TyrA23. Seedlings were preincubated with MβCD, for 45 minutes at the specified concentrations. When both inhibitors were simultaneously applied, the pre-incubation period was 60 minutes.
TIRF imaging and area/speed measurements of AtRGS1-GFP.
Five-day-old seedlings expressing 35S-RGS1-GFP were transferred to a solution of either 6% d-glucose or 1μm flg22 and imaged at 5, 10, and 15 minutes while immersed in the ligand solution. Imaging was performed on a Nikon Ti Eclipse with SR Apo TIRF 100x lens (NA 1.5, WD 120μm). GFP excitation occurred at 488nm and emission collection at 515-555nm with an Andor iXon3 EMCCD camera. 60 second time-lapse imaging was initiated at the beginning of each time point with 200ms acquisition speed. Time-lapse sequences were normalized for fluorescence over time using IMARIS (v9.2.2, Bitplane AG, Zurich, Switzerland). The IMARIS Surface feature was used to track and calculate the area and speed of individually identifiable AtRGS1 proteins/clusters (labeled as tracks in IMARIS) over time. The average speed and area of each unique track for a 30 second interval between 5:15-5:45 or 15:15-15:45 was calculated using a script in Matlab (Fig. S7).
Mander’s Coefficient Analysis.
Z-stacks were acquired with a Zeiss LSM880 with a C-Apochromat 40x/1.2NA water immersion objective at 0.5μm intervals between images. ROIs for Mander’s correlation coefficient analysis were placed on cells based on substantial RGS1-GFP and endocytosis marker-mCherry expression as well as visible internalized GFP and mCherry fused proteins. This was done to include internalized cytosolic proteins and membrane bound proteins to avoid analysis of only one distinct pool of proteins within the cell. To reduce bias introduced by the placement of ROIs on individual cells, the Mander’s correlation coefficient for the entire sample area in respective images was averaged into the analysis with the smaller individual ROIs. We used Manders correlation coefficient instead of Pearson’s correlation coefficient because Manders provides a quantitative measure of the change in the overlap of the two signals, not the change in signal intensity (as would be measured by Pearson’s correlation coefficient) that could arise due to AtRGS1 internalization (for example change in compartment pH) that is not correlated to direct interaction with either endocytosis marker.
Statistical analysis.
In all figures, the errors bars represent the standard deviation. In Figs. 1, 3, 4, and 5D and E, one-way ANOVA with post-hoc Tukey HSD analysis was performed. In all other comparisons made in which P values for confidence were provided, Student T tests were applied.
Synthesis of [14C] isomaltulose.
Sucrose isomerase (SI) from Pantoea dispersa UQ68J (GenBank AY223549) was cloned into the expression vector pET24b (Novagen) by performing PCR of genomic DNA using the PCR primers 5′-GGA TCC AAC AAT GGC AAC GAA TAT ACA AAA GTC C-3′ (which included a BamHI restriction site and a start codon) and 5′-ATA GGT ACC TCA GTT CAG CTT ATA GAT CCC-3′ (which included a KpnI restriction site and a stop codon). SI was expressed in E. coli BL21(DE3) (Novagen) grown at 37°C and 225 rpm. When the optical density at 600nm reached 1.00, protein expression was induced by adding isopropyl-D-thiogalactopyranoside to a final concentration of 0.5 mM. The cultures were continued for another 3 h at 28°C. Cells were harvested by centrifugation (3,000 ×g, 4°C, 10 min), resuspended in 50 mM Tris-HCl (pH 8.0)-2 mM EDTA, and then re-centrifuged. The cell pellet was immediately frozen in liquid nitrogen and stored at −75°C. Cells were suspended in extraction buffer (20 mM Tris-HCl (pH 7.4), 200 mM NaCl, 1 mM EDTA, 1 mM azide, 10 mM β-mercaptoethanol) and lysed by sonication (nine 15-s pulses at 50 W with a Branson Sonifier 450 microprobe), centrifuged (10,000 ×g, 4°C, 10 min), and filtered through a 0.45-μm-pore-size membrane (Gelman Acrodisc). The pET24b vector introduced a carboxy-terminal six-His tag into expressed proteins, which were purified by adsorption to nitrilotriacetic acid (NTA) agarose (QIAGEN) and elution with 25 mM NaH2PO4-150 mM NaCl-125 mM imidazole buffer (pH 8.0) by following the manufacturer’s instructions. The purity of SI proteins was verified by SDS-PAGE as a single band on Coomassie Blue R-250 staining. [14C] isomaltulose was prepared using 1.48MBq [U-14C]Sucrose (Amersham, UK) in 200 μl water with 3% ethanol (equivalent to 0.3379 mM). This was reacted with 30 μl purified UQ68J SI for at 30 °C 60 min. The converted [14C] isomatulose concentration by UQ68J SI was estimated by three-replicate parallel conversions of unlabelled sucrose (S7903, Sigma) in the same concentration of 0.3379 mM with 3% ethanol by the same enzyme. BioLC DX600 (Dionex, USA) determinations showed 84.0±0.106% (Mean±SE) was converted into isomaltulose; 3.5±0.197% into trehalose; 4.6±0.072% and 7.8±0.237% into the by-products of glucose and fructose, respectively and there was no sucrose left after the reaction was stopped (Fig. S1A).
Synthesis of 5-bromo-N-phenylthiophene-2-sulfonamide (ES9-17)
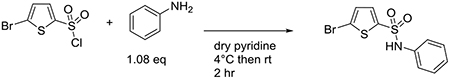
All chemical reagents and solvents were obtained from chemical suppliers (Acros, Fisher Scientific, or Sigma-Aldrich) and used without further purification, unless otherwise noted. Aniline (0.385 g, 4.13 mmol) was added to an oven dried flask and dissolved in 10 mL of freshly distilled pyridine. The mixture was cooled to 4°C by placing it into an ice water bath. 5-bromothiophene-2-sulfonyl chloride (1.00 g, 3.82 mmol) was then added in small portions over the course of 5 minutes. The reaction mixture was left to stir at room temperature for 2 hours. The resulting crude solution was concentrated under vacuum and dissolved with ethyl acetate. The ethyl acetate extract was washed with saturated sodium bicarbonate and brine. The organic extract was dried with anhydrous magnesium sulfate and concentrated under vacuum. The resulting red oily crude mixture was then purified using silica gel chromatography (50:50 n-hexane:ethyl acetate, Rf = 0.6) or recrystallized from isopropanol to yield the desired yellowish-beige crystalline sulfonamide product (0.856 g, 70% yield). Reaction progress was monitored by thin layer chromatography (EMD Millipore TLC silica gel 60 F254). Silica gel chromatography was performed using SiliaFlash Irregular Silica Gel, P60, grit size 40–60 μm, 60Å. LC-MS analyses were obtained using an Agilent Rapid Resolution LC-MSD system equipped with online degasser, binary pump, autosampler, heated column compartment, and diode array detector. A Halo C18 column (4.6 x 100 mm, 2.7 μm) was used for all separations with 0.2% formic acid H2O – acetonitrile gradients. Mass spectra (ESI+) were acquired on a single quad mass spectrometer using ultrascan mode by using a drying temperature of 350°C, a nebulizer pressure of 45 psi, a drying gas flow of 10 L/min and a capillary voltage of 3000 V. The experimental NMR spectra (Fig. S6, A to E) agree with a previously reported characterization (59). Proton magnetic resonance spectra were acquired on 400 MHz Bruker instrument in the Department of Chemistry NMR facility at the University of North Carolina – Chapel Hill. NMR spectra were processed with Mnova (Mestrelab).
Sugar uptake assay.
One-week-old seedlings grown on a filter disc overlaying 1/2X Murashige and Skoog Basal Salts, 0.7% phytogel 23°C, pH 5.8, 8h/d of 100 mole/m2/s1 were lifted off the plate and overlaid with 6 mL of water containing approx. 25,000 cpm of [14C] sugars as indicated. The specific activity of the sugars was 12GBq/mmol. At the indicated times, triplicate sets of 10 seedling were gently rinsed and placed in a 1.7-mL microfuge tube with 1 mL of scintillation fluid (Perkin Elmer Inc) and radioactivity was quantitated by liquid scintillation counting. CPM from time zero (typically 80-150 cpm) was subtracted from the average of the 3 samples. The CPM for [14C] isomaltulose uptake into seedlings at each time was corrected for its 84% purity.
Live cell imaging of MAPK reporter (SOMA) lines.
Detached etiolated hypocotyls were prepared for imaging on the confocal microscope using the HybriWell™ method as previously described (38, 105). A hypocotyl from a dark grown 5-day-old seedling was placed on top of the droplet, and a HybriWell™ (Grace BioLabs, cat. no. 611102) was gently placed on the cover slide with the hypocotyl in the center to form a 150-μm deep imaging chamber with a volume of 30 μl. Ultrapure water (300 μl) was injected through one of the HybriWell™ ports using a pipette to fill the 30-μl chamber with water and to expel any air bubbles. A 200-μl droplet of ultrapure water was then placed on one of the ports to prevent the chamber from drying out. The HybriWells containing the mounted hypocotyls were then placed in covered Petri dishes and equilibrated by incubating at 20–23°C under constant light for 6–8 hours prior to imaging.
Confocal microscopy was performed using a Zeiss LSM 710 with a C-Apochromat 40x/1.20 water immersion objective lens. Samples were excited at 458nm with 3% power, and emission was measured between 463 and 517nm for Turquoise GL and between 534 and 570nm for YPet. Z-stacks were collected every 2 min with an optical slice thickness of 1.2 μm. Chemical treatments were added to the samples during imaging by pipetting 200 μl of solution containing the treatment onto one port of the HybriWell. For experiments involving tyrphostin A23 and tyrphostin A51, hypocotyls were pretreated with 50 μM of these compounds for 30 min prior to imaging.
Post-processing of the raw image data was performed using Fiji (104). The ‘Z-projection’ function was performed on an image stack using the ‘Max Intensity’ setting. The resulting projection was separated into two images, one for the Turquoise GL emission channel and one for the YPet emission channel. The ‘Subtract Background’ function was performed on both images, with the ‘rolling-ball radius’ set as the default 50 pixels. A mask was created from the YPet channel using the ‘Convert to Mask’ function. The background subtracted YPet and Turquoise GL images were converted into 32-bit images, which were multiplied by the Mask file. The resulting YPet image was divided by the resulting Turquoise GL image using the ‘Image Calculator’ function to create a ratio image representing the ratio of YPet to Turquoise emission. Finally, the ‘Threshold’ function was performed using the default values, with the ‘NaN background’ option enabled. The ‘Fire’ lookup table was applied to the final ratio image. To measure the ratio of YPet to Turquoise GL emission, a region of interest (ROI) was selected within the ratio image using Fiji and the average ratio value within that ROI was measured.
Live cell Ca2+ imaging with R-GECO1.
5-day-old, etiolated hypocotyls expressing the Ca2+ reporter R-GECO1 were grown in aqueous media containing ¼ MS. Hypocotyls were excised and mounted in HybriWells 6-8 hours prior to imaging with a Zeiss LSM710 confocal laser scanning microscope equipped with a C-Apochromat 40×/1.20 water immersion objective. R-GECO1 was excited using 561nm laser with 7.0 % laser power, and emission was measured between 620 and 650 nm. Z-stacks were collected 2 min after chemical treatment with an optical slice thickness of 1.5μm. Chemical treatments were added to the samples during imaging by pipetting 200 μl of solution containing the treatment onto one port of the HybriWell. The digital images were analyzed with Fiji (104). R-GECO1 fluorescence was quantified and normalized using ΔF/F where ΔF is the difference between the fluorescence intensity before and after flg22 application divided by initial fluorescence intensity.
Modeling AtVPS26.
Five models designated atvps26a-1 through 5 were created by MODELLER (106) using the automodel script based on the human VPS26A template 2FAU. We calculated the objective function (molpdf), DOPE score, GA341 assessment score and RMSD between the model and the template. The best model was defined as the one with the lowest value of the molpdf, overall DOPE assessment scores, per-residue DOPE scores between the template and the models compared using GNUPLOT (Fig. S5C and S4D), and the lowest RMSD value (Fig. S5D).
Bimolecular Fluorescence Complementation (BiFC)
Transient expression for BiFC was performed in N. benthamiana. nYFP- and cYFP-tagged proteins were generated by subcloning the genes of interest into pENTR/D-TOPO and recombining them into one or more of the BiFC vectors pBatTL–sYFP-N or pBatTL–sYFP-C (for C-terminal tagged nYFP and cYFP halves, respectively) and pCL112_JO or pCL113_JO (for N-terminal tagged nYFP and cYFP halves, respectively). The mitochondrial RFP marker Mt-rk, which was obtained from the ABRC (CD3-991), was used as an internal positive transformation control. On the 4th to 6th day post-infiltration, confocal images of BiFC samples were acquired using a Zeiss LSM 880 confocal laser scanning microscope equipped with a C-Apochromat 40X/1.20NA water immersion objective. A 514-nm argon laser was tuned to excite YFP and emission was detected at 526–555 nm by a photomultiplier tube detector. mt-Rk data were collected from 585–615 nm range after the sample was excited with a 561-nm diode laser. Agrobacterium carrying a plasmid of interest and P19, which is a viral RNA silencing suppressor (107) were grown in 3 ml LB medium with 50 μg/ml rifampicin, 50 μg/ml gentamycin, and 100 μg/ml kanamycin (or spectinomycin based on the plasmid selection marker) overnight in 28°C incubator at 220 rpm. Fifty ml fresh LB with 50 μg/ml rifampicin, 50 μg/ml gentamycin, 100 μg/ml kanamycin/spectinomycin, and 20 μM acetosyringone was inoculated with 200 μl of overnight culture. Bacteria cells were harvested by centrifugation at 5,900 × g for 15 min at 20°C–24°C after the culture was grown overnight at 28°C and 220 rpm. Cells were resuspended and diluted in infiltration buffer (10 mM MgCl2, 10 mM 2-(N-morpholino) ethanesulfonic acid and 200 μM acetosyringone) buffer. Agrobacterium carrying the plasmids of interest or the gene silencing suppressor protein P19 were cultured to an OD600 of 1.0, mixed in a 3:1 ratio and infiltrated into the abaxial side of 4–5 week-old N. benthamiana leaves with a needleless syringe.
Split firefly-luciferase assays.
pCAMBIA/des/cLuc and pCAMBIA/des/nLuc (108) were used to generate the following plasmids: AtVPS26A-nLUC, AtVPS26B-nLUC, cLUC-AtVPS26A, cLUC-AtVPS26B, cLUC-AtVPS26like, cLUC-AtGPA1, AtAGB1-nLUC and AtRGS1-nLUC. pART27H-mCherry-AtAGG1 plasmid was obtained from Dr. Jose R Botella (University of Queensland, Brisbane, Australia). All plasmids were transformed into A. tumefaciens strain GV3101. nLUC and cLUC fusion partners were co-expressed in N. benthamiana leaves by agroinfiltration following protocols in (109). 48 hours after infiltration, 6mm leaf discs were collected to 96-well plate and 40μl 0.4mM D-luciferin was added to each well. Luminescence was measured by a spectraMax L microplate reader (Molecular Devices).
Yeast two-hybrid assays.
Constructs used in yeast two-hybrid assays were pAS–RGS-J5 (amino acids 284–459), RGS-∆Ct (amino acids 284–416), RGS-∆CtS405,406A, pACTGW–AtVPS26B and AtVPS26like. Yeast two-hybrid-tested pairs were co-transformed into AH109 yeast cells and plated on SD-LW media (110). Colonies were then inoculated into SD-LW liquid media and grew overnight at 30 °C. Yeast cells diluted 1/10 were spotted on SD-LW, SD-LWH, and SD-LWH 10mM 3-AT (3-amino-1,2,4-triazole) plates. The plates were incubated at 30°C for 4 days and were imaged by gel documentation system (Axygen) under white light.
Supplementary Material
Acknowledgments
We would like to thank Dr. N Phan for creating the AtRGS1Y112A plasmid and transgenic lines and for experiments testing the involvement of adaptors in AtRGS1 endocytosis and data in Fig. 1A and B. We thank Dr. V Gurevich, Dr. M Garcia-Marcos and Dr. CE Alvarez for helpful comments and Dr. K Ghusinga for assistance with writing MatLab code. We thank Dr. J Huang for help in the turanose experiment. We thank Dr. Tony Perdue from the UNC Dept. of Biology Microscopy Core and Dr. Michelle Itano from the UNC Neuroscience Center Microscopy Core for assistance with TIRF image acquisition and analysis.
Funding
This work was supported by grants from the NIGMS (GM065989) and NSF (MCB-1713880) to A.M.J. The Division of Chemical Sciences, Geosciences, and Biosciences, Office of Basic Energy Sciences of the US Department of Energy through the grant DE-FG02-05er15671 to A.M.J. funded the biochemical aspects of this project. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. This research was supported by NIGMS Institutional Research and Academic Career Development Award (GM000678).
Footnotes
Competing interests
The authors declare that they have no competing interests.
Data and materials availability
All data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials.
References and Notes
- 1.Ferguson KM, Higashijima T, Smigel MD, Gilman AG, The influence of bound GDP on the kinetics of guanine nucleotide binding to G proteins. J. Biol. Chem 261, 7393–7399 (1986). [PubMed] [Google Scholar]
- 2.Benovic J et al. , Functional desensitization of the isolated beta-adrenergic receptor by the beta-adrenergic receptor kinase: potential role of an analog of the retinal protein arrestin (48-kDa protein). Proceedings of the National Academy of Sciences of the United States of America 84, 8879–8882 (1987). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lohse M, Benovic J, Codina J, Caron M, Lefkowitz R, beta-Arrestin: a protein that regulates beta-adrenergic receptor function. Science 248, 1547–1550 (1990). [DOI] [PubMed] [Google Scholar]
- 4.Johnston CA et al. , GTPase acceleration as the rate-limiting step in Arabidopsis G protein-coupled sugar signaling. Proceedings of the National Academy of Sciences 104, 17317–17322 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jones J et al. , The crystal structure of a self-activating G protein α subunit reveals its distinct mechanism of signal initiation. Sci. Signal. 4, ra8 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jones JC, Temple BRS, Jones AM, Dohlman HG, Functional reconstitution of an atypical G protein heterotrimer and regulator of G protein signaling protein (RGS1) from Arabidopsis thaliana. Journal of Biological Chemistry 286, 13143–13150 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Urano D et al. , Endocytosis of the seven-transmembrane RGS1 protein activates G-protein-coupled signalling in Arabidopsis. Nat Cell Biol 14, 1079–1088 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fu Y et al. , Reciprocal encoding of signal intensity and duration in a glucose-sensing circuit Cell 156, 1084–1096 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Asai T et al. , MAP kinase signalling cascade in Arabidopsis innate immunity. Nature 415, 977–983 (2002). [DOI] [PubMed] [Google Scholar]
- 10.Felix G, Duran JD, Volko S, Boller T, Plants have a sensitive perception system for the most conserved domain of bacterial flagellin. The Plant Journal 18, 265–276 (1999). [DOI] [PubMed] [Google Scholar]
- 11.Gómez-Gómez L, Boller T, FLS2: An LRR receptor–like kinase involved in the perception of the bacterial elicitor flagellin in Arabidopsis. Molecular Cell 5, 1003–1011 (2000). [DOI] [PubMed] [Google Scholar]
- 12.Jelenska J, Davern SM, Standaert RF, Mirzadeh S, Greenberg JT, Flagellin peptide flg22 gains access to long-distance trafficking in Arabidopsis via its receptor, FLS2. Journal of Experimental Botany 68, 1769–1783 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sun Y et al. , Structural basis for flg22-induced activation of the Arabidopsis FLS2-BAK1 immune complex. Science 342, 624–628 (2013). [DOI] [PubMed] [Google Scholar]
- 14.Tunc-Ozdemir M, Jones AM, Ligand-induced dynamics of heterotrimeric G protein-coupled receptor-like kinase complexes. PLOS ONE 12, e0171854 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tunc-Ozdemir M, Liao K-L, Ross-Elliott TJ, Elston TC, Jones AM, Long-distance communication in Arabidopsis involving a self-activating G protein. Plant Direct 2, e00037 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pharr D, Keller F, in Photoassimilate distribution plants and crops source-sink relationships. (Routledge, 2017), pp. 157–183. [Google Scholar]
- 17.Chen L-Q et al. , Sugar transporters for intercellular exchange and nutrition of pathogens. Nature 468, 527 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Williams LE, Lemoine R, Sauer N, Sugar transporters in higher plants – a diversity of roles and complex regulation. Trends in Plant Science 5, 283–290 (2000). [DOI] [PubMed] [Google Scholar]
- 19.Li L, Sheen J, Dynamic and diverse sugar signaling. Current Opinion in Plant Biology 33, 116–125 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fan L, Li R, Pan J, Ding Z, Lin J, Endocytosis and its regulation in plants. Trends in Plant Science 20, 388–397 (2015). [DOI] [PubMed] [Google Scholar]
- 21.Jackson LP et al. , A large-scale conformational change couples membrane recruitment to cargo binding in the AP2 clathrin adaptor Complex. Cell 141, 1220–1229 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kelly BT et al. , A structural explanation for the binding of endocytic dileucine motifs by the AP2 complex. Nature 456, 976 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Krauss M, Kukhtina V, Pechstein A, Haucke V, Stimulation of phosphatidylinositol kinase type I-mediated phosphatidylinositol (4,5)-bisphosphate synthesis by AP-2μ–cargo complexes. Proceedings of the National Academy of Sciences 103, 11934–11939 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Boucrot E, Saffarian S, Zhang R, Kirchhausen T, Roles of AP-2 in clathrin-mediated endocytosis. PLoS One 5, e10597 (2010. ). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Collins BM, McCoy AJ, Kent HM, Evans PR, Owen DJ, Molecular architecture and functional model of the endocytic AP2 complex. Cell 109, 523–535 (2002). [DOI] [PubMed] [Google Scholar]
- 26.Raffaele S et al. , Remorin, a solanaceae protein resident in membrane rafts and plasmodesmata, impairs potato virus X movement. The Plant Cell 21, 1541–1555 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li R et al. , A membrane microdomain-associated protein, Arabidopsis Flot1, is involved in a clathrin-independent endocytic pathway and is required for seedling development. The Plant Cell 24, 2105–2122 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hao H et al. , Clathrin and membrane microdomains cooperatively regulate RbohD dynamics and activity in Arabidopsis. The Plant Cell 26, 1729–1745 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li X et al. , Single-molecule analysis of PIP2;1 dynamics and partitioning reveals multiple modes of Arabidopsis plasma membrane aquaporin regulation. The Plant Cell 23, 3780–3797 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee J, Eschen-Lippold L, Lassowskat I, Böttcher C, Scheel D, Cellular reprogramming through mitogen-activated protein kinases. Frontiers in Plant Science 6, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Loreti E, Alpi A, Perata P, Glucose and disaccharide-sensing mechanisms modulate the expression of alpha -amylase in barley embryos. Plant Physiol. 123, 939–948 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sinha AK et al. , Metabolizable and non-metabolizable sugars activate different signal transduction pathways in tomato. Plant Physiol. 128, 1480–1489 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Börnke F et al. , High-level production of the non-cariogenic sucrose isomer palatinose in transgenic tobacco plants strongly impairs development. Planta 214, 356–364 (2002). [DOI] [PubMed] [Google Scholar]
- 34.Fernie AR, Roessner U, Geigenberger P, The sucrose analog palatinose leads to a stimulation of Sucrose degradation and starch synthesis when supplied to discs of growing potato tubers. Plant Physiol. 125, 1967–1977 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wu L, Birch R, Isomaltulose is actively metabolized in plant cells. Plant Physiol 157, 2094–2101 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rolland F, Moore B, Sheen J, Sugar sensing and signaling in plants. The Plant Cell 14, S185–S205 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ranf S, Eschen-Lippold L, Pecher P, Lee J, Scheel D, Interplay between calcium signalling and early signalling elements during defence responses to microbe- or damage-associated molecular patterns. The Plant Journal 68, 100–113 (2011). [DOI] [PubMed] [Google Scholar]
- 38.Zaman N et al. , A Förster resonance energy transfer sensor for live-cell imaging of mitogen-activated protein kinase activity in Arabidopsis. The Plant Journal 97, 970–983 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kalderon D, Roberts BL, Richardson WD, Smith AE, A short amino acid sequence able to specify nuclear location. Cell 39, 499–509 (1984). [DOI] [PubMed] [Google Scholar]
- 40.Wen W, Meinkotht JL, Tsien RY, Taylor SS, Identification of a signal for rapid export of proteins from the nucleus. Cell 82, 463–473 (1995). [DOI] [PubMed] [Google Scholar]
- 41.Keinath NF et al. , Live cell imaging with R-GECO1 sheds light on flg22- and chitin-induced transient [Ca2+]cyt patterns in Arabidopsis. Molecular Plant 8, 1188–1200 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Grigston JC, Osuna D, Scheible WR, Stitt M, Jones AM, D-glucose sensing by a plasma membrane regulator of G signaling protein, AtRGS1. . FEBS Lett. 582, 3577–3584 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Urano D et al. , Saltatory evolution of the heterotrimeric G protein signaling mechanisms in the plant kingdom. Science Signaling 9, ra93 (2016). [DOI] [PubMed] [Google Scholar]
- 44.Urano D, Chen J-G, Botella JR, Jones AM, Heterotrimeric G protein signalling in the plant kingdom. Open Biology 3, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Banbury DN, Oakley JD, Sessions RB, Banting G, Tyrphostin A23 inhibits internalization of the transferrin Receptor by perturbing the interaction between tyrosine motifs and the medium chain subunit of the AP-2 adaptor complex. J. Biol. Chem 278, 12022–12028 (2003). [DOI] [PubMed] [Google Scholar]
- 46.Dhonukshe P et al. , Clathrin-mediated constitutive endocytosis of PIN auxin efflux carriers in Arabidopsis. Current Biology 17, 520–527 (2007). [DOI] [PubMed] [Google Scholar]
- 47.Bashline L, Li S, Anderson CT, Lei L, Gu Y, The endocytosis of cellulose synthase in Arabidopsis Is dependent on μ2, a clathrin-mediated endocytosis adaptin. Plant Physiology 163, 150–160 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Eichel K, von Zastrow M, Subcellular organization of GPCR signaling. Trends in Pharmacological Sciences 39, 200–208 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chinchilla D, Bauer Z, Regenass M, Boller T, Felix G, The Arabidopsis receptor kinase FLS2 binds flg22 and determines the specificity of flagellin perception. The Plant Cell 18, 465–476 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tunc-Ozdemir M, Urano D, Jaiswal DK, Clouse SD, Jones AM, Direct modulation of heterotrimeric G protein-coupled signaling by a receptor kinase complex. Journal of Biological Chemistry 291, 13918–13925 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Adam T et al. , Constitutive expression of clathrin hub hinders elicitor-induced clathrin-mediated endocytosis and defense gene expression in plant cells. FEBS Letters 586, 3293–3298 (2012). [DOI] [PubMed] [Google Scholar]
- 52.Irani NG et al. , Fluorescent castasterone reveals BRI1 signaling from the plasma membrane. Nature chemical biology 8, 583 (2012). [DOI] [PubMed] [Google Scholar]
- 53.Ilangumaran S, Hoessli DC, Effects of cholesterol depletion by cyclodextrin on the sphingolipid microdomains of the plasma membrane. Biochemical Journal 335, 433–440 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ohtani Y, Irie T, Uekama K, Fukunaga K, Pitha J, Differential effects of alpha-, beta- and gamma-cyclodextrins on human erythrocytes. Eur J Biochem 186, 17–22 (1989). [DOI] [PubMed] [Google Scholar]
- 55.Dejonghe W et al. , Mitochondrial uncouplers inhibit clathrin-mediated endocytosis largely through cytoplasmic acidification. Nature Communications 7, 11710 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang C et al. , Differential regulation of cathrin and its adaptor proteins during membrane recruitment for endocytosis. Plant Physiol. 171, 215–229 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Smith J, Salamango D, Leslie M, Collins C, Heese A, Sensitivity to Flg22 is modulated by ligand-induced degradation and de novo synthesis of the endogenous flagellin-receptor FLAGELLIN-SENSING2. Plant Physiol. 164, 440–454 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kim SY et al. , Adaptor protein complex 2–mediated endocytosis is crucial for male reproductive organ development in Arabidopsis. The Plant Cell 25, 2970–2985 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dejonghe W et al. , Disruption of endocytosis through chemical inhibition of clathrin heavy chain function. Nature chemical biology 15, 641–649 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ohno H et al. , Interaction of tyrosine-based sorting signals with clathrin-associated proteins. Science 269, 1872–1875 (1995). [DOI] [PubMed] [Google Scholar]
- 61.Chen J-G et al. , A seven-transmembrane RGS protein that modulates plant cell proliferation. Science 301, 1728–1731 (2003). [DOI] [PubMed] [Google Scholar]
- 62.Fu Y et al. , Reciprocal encoding of signal intensity and duration in a glucose-sensing circuit. Cell 156, 1084–1095 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Alvarez CE, On the origins of arrestin and rhodopsin. BMC Evolutionary Biology 8, 222 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Oliviusson P et al. , Plant retromer, Localized to the prevacuolar compartment and microvesicles in Arabidopsis, may interact with vacuolar sorting receptors. The Plant Cell 18, 1239–1252 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shi H, Rojas R, Bonifacino JS, Hurley JH, The retromer subunit Vps26 has an arrestin fold and binds Vps35 through its C-terminal domain. Nature Structural & Molecular Biology 13, 540–548 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zelazny E et al. , Mechanisms governing the endosomal membrane recruitment of the core retromer in Arabidopsis. Journal of Biological Chemistry 288, 8815–8825 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jha SG et al. , Vacuolar Protein Sorting 26C encodes an evolutionarily conserved large retromer subunit in eukaryotes that is important for root hair growth in Arabidopsis thaliana. The Plant Journal 94, 595–611 (2018). [DOI] [PubMed] [Google Scholar]
- 68.Chen Q et al. , in Arrestins - Pharmacology and Therapeutic Potential, Gurevich VV, Ed. (Springer Berlin Heidelberg, Berlin, Heidelberg, 2014), pp. 205–223. [Google Scholar]
- 69.Smith J, Lefkowitz R, Rajagopal S, Biased signalling: from simple switches to allosteric microprocessors. . Nature Reviews Drug Discovery 17, 243 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bohn LM, Gainetdinov RR, Lin F-T, Lefkowitz RJ, Caron MG, μ-Opioid receptor desensitization by β-arrestin-2 determines morphine tolerance but not dependence. . Nature 720–723, (2000). [DOI] [PubMed] [Google Scholar]
- 71.DeWire SM et al. , A G protein-biased Ligand at the <em>μ</em>-Opioid receptor is potently analgesic with reduced gastrointestinal and respiratory dysfunction compared with morphine. Journal of Pharmacology and Experimental Therapeutics 344, 708–717 (2013). [DOI] [PubMed] [Google Scholar]
- 72.Géhin M et al. , Structural basis for engineering of retinoic acid receptor isotype-selective agonists and antagonists. Chem Biol. 6, 519–529 (1999. ). [DOI] [PubMed] [Google Scholar]
- 73.Lin W-W, Hsieh S-L, Decoy receptor 3: A pleiotropic immunomodulator and biomarker for inflammatory diseases, autoimmune diseases and cancer. Biochemical Pharmacology 81, 838–847 (2011). [DOI] [PubMed] [Google Scholar]
- 74.Pan G et al. , An Antagonist Decoy Receptor and a Death Domain-Containing Receptor for TRAIL. Science 277, 815–818 (1997). [DOI] [PubMed] [Google Scholar]
- 75.Rajagopal S et al. , β-arrestin- but not G protein-mediated signaling by the “decoy” receptor CXCR7. Proceedings of the National Academy of Sciences 107, 628–632 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Urs NM et al. , Distinct cortical and striatal actions of a β-arrestin–biased dopamine D2 receptor ligand reveal unique antipsychotic-like properties. Proceedings of the National Academy of Sciences 113, E8178–E8186 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Butcher AJ et al. , Differential G-protein-coupled receptor phosphorylation provides evidence for a signaling bar code. Journal of Biological Chemistry 286, 11506–11518 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Nobles KN et al. , Distinct phosphorylation sites on the β2-adrenergic receptor establish a barcode that encodes differential functions of β-arrestin. Science Signaling 4, ra51–ra51 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Shiraishi Y et al. , Phosphorylation-induced conformation of β2-adrenoceptor related to arrestin recruitment revealed by NMR. Nature Communications 9, 194 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yang F et al. , Phospho-selective mechanisms of arrestin conformations and functions revealed by unnatural amino acid incorporation and 19F-NMR. Nature Communications 6, 8202 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yang Z et al. , Phosphorylation of G protein-coupled receptors: from the barcode hypothesis to the flute model. Molecular pharmacology 92, 201–210 (2017). [DOI] [PubMed] [Google Scholar]
- 82.Choi M et al. , G protein–coupled receptor kinases (GRKs) orchestrate biased agonism at the β2-adrenergic receptor. Science Signaling 11, eaar7084 (2018). [DOI] [PubMed] [Google Scholar]
- 83.DeFea KA et al. , β-Arrestin–dependent endocytosis of proteinase-activated receptor 2 is required for intracellular targeting of activated Erk1/2. The Journal of Cell Biology 148, 1267–1282 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Luttrell LM et al. , Activation and targeting of extracellular signal-regulated kinases by β-arrestin scaffolds. Proceedings of the National Academy of Sciences 98, 2449–2454 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Rajagopal S, Rajagopal K, Lefkowitz RJ, Teaching old receptors new tricks: biasing seven-transmembrane receptors. Nature Reviews Drug Discovery 9, 373 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.O’Hayre M et al. , Genetic evidence that β-arrestins are dispensable for the initiation of β2 adrenergic receptor signaling to ERK. Science Signaling 10, eaal3395 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Liang X et al. , Ligand-triggered de-repression of Arabidopsis heterotrimeric G proteins coupled to immune receptor kinases. Cell research 28, 529–543 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Tunc-Ozdemir M et al. , Predicted functional implications of phosphorylation of regulator of G protein signaling protein in plants. Frontiers in Plant Science 8, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhong C-L, Zhang C, Liu J-Z, Heterotrimeric G protein signaling in plant immunity. Journal of Experimental Botany 70, 1109–1118 (2018). [DOI] [PubMed] [Google Scholar]
- 90.Shiu SH, Bleecker AB, Plant receptor-llike gene family: diversity, function, and signaling. Science STKE Dec. 18th, RE22 (2001). [DOI] [PubMed] [Google Scholar]
- 91.Xue J, Gong B-Q, Yao X, Huang X, Li J-F, BAK1-mediated phosphorylation of canonical G protein alpha during flagellin signaling in Arabidopsis. Journal of Integrative Plant Biology 0. [DOI] [PubMed] [Google Scholar]
- 92.Liao K-L et al. , Dose-Duration Reciprocity for G protein activation: Modulation of kinase to substrate ratio alters cell signaling. PLoS ONE 12, e0190000 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lucas M et al. , Structural mechanism for cargo Recognition by the retromer complex. Cell 167, 1623–1635.e1614 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Paing MM, Stutts AB, Kohout TA, Lefkowitz RJ, Trejo J, β-arrestins regulate protease-activated receptor-1 desensitization but not internalization or down-regulation. Journal of Biological Chemistry 277, 1292–1300 (2002). [DOI] [PubMed] [Google Scholar]
- 95.Smith TH et al. , Protease-activated receptor-4 signaling and trafficking is regulated by the clathrin adaptor protein complex-2 independent of β-arrestins. Journal of Biological Chemistry 291, 18453–18464 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Huot B, Yao J, Montgomery BL, He SY, Growth–defense tradeoffs in plants: A balancing act to optimize fitness. Molecular Plant 7, 1267–1287 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Mudgil Y et al. , Photosynthate regulation of the root system architecture mediated by the heterotrimeric G protein complex in Arabidopsis. Frontiers in Plant Science 7, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Delgado-Cerezo M et al. , Arabidopsis heterotrimeric G-protein regulates cell wall defense and resistance to necrotrophic fungi. Molecular Plant 5, 98–114 (2012). [DOI] [PubMed] [Google Scholar]
- 99.Escudero V et al. , Alteration of cell wall xylan acetylation triggers defense responses that counterbalance the immune deficiencies of plants impaired in the β-subunit of the heterotrimeric G-protein. The Plant Journal 92, 386–399 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Klopffleisch K et al. , Arabidopsis G-protein interactome reveals connections to cell wall carbohydrates and morphogenesis. Mol Syst Biol 7, 532 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Botto JF, Ibarra S, Jones AM, The heterotrimeric G-protein complex modulates light sensitivity in Arabidopsis thaliana seed germination. Photochemistry and Photobiology 85, 949–954 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ullah H et al. , The β subunit of the Arabidopsis G protein negatively regulates auxin-induced cell division and affects multiple developmental processes. Plant Cell 15, 393–409 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Urano D et al. , Saltational evolution of the heterotrimeric G protein signaling mechanisms in the plant kingdom. Science Signaling 9, ra93–ra93 (2016). [DOI] [PubMed] [Google Scholar]
- 104.Schindelin J et al. , Fiji: an open-source platform for biological-image analysis. Nature Methods 9, 676 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Vang S, Seitz K, Krysan P, A simple microfluidic device for live cell imaging of Arabidopsis cotyledons, leaves, and seedlings. Biotechniques 64, 255–261 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Martí-Renom MA et al. , Comparative protein structure modeling of genes and genomes. Annual Review of Biophysics and Biomolecular Structure 29, 291–325 (2000). [DOI] [PubMed] [Google Scholar]
- 107.Au-Shamloul M, Au-Trusa J, Au-Mett V, Au-Yusibov V, Optimization and utilization of Agrobacterium-mediated transient protein production in Nicotiana. JoVE, e51204 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lin Z-JD, Liebrand TWH, Yadeta KA, Coaker G, PBL13 is a serine/threonine protein kinase that negatively regulates Arabidopsis immune responses. Plant Physiology 169, 2950–2962 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zhou Z, Bi G, Zhou J-M, Luciferase complementation assay for protein-protein interactions in plants. Current Protocols in Plant Biology 3, 42–50 (2018). [DOI] [PubMed] [Google Scholar]
- 110.Wagemans J, Lavigne R, in Protein-Protein Interactions: Methods and Applications, Meyerkord CL, Fu H, Eds. (Springer New York, New York, NY, 2015), pp. 409–431. [Google Scholar]
- 127.Ullah H, Chen J-G, Temple B, Boyes DC, Alonso JM, Ecker JR, Jones AM The beta-subunit of the Arabidopsis G protein negatively regulates auxin-induced cell division and affects multiple developmental processes. Plant Cell 15, 393–409. doi: 10.1105/tpc.006148 (2003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Trusov Y, Rookes JE, Tilbrook K, Chakravorty D, Mason MG, Anderson D, Chen J-G, Jones AM, Botella JR Heterotrimeric G protein γ subunits provide functional selectivity in Gβγ dimer signaling in Arabidopsis. Plant Cell 19, 1235–1250 doi: 10.1105/tpc.107.050096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Zelazny E, Santambrogio M, Pourcher M, Chambrier P, Berne-Dedieu A, Fobis-Loisy I, Miège C, Jaillais Y, Gaude T Mechanisms governing the endosomal membrane recruitment of the core retromer in Arabidopsis. J. Biol Chem 288, 8815–8825 doi: 10.1074/jbc.M112.440503 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.