

Abstract
The prevalence of periodontal disease increases with age. Systemic inflammatory dysregulation also increases with age and has been reported to contribute to the myriad of diseases and conditions that become more prevalent with advanced age. As periodontal disease involves a dysregulated host inflammatory response, the age-related inflammatory dysregulation may contribute to the pathogenesis of periodontal disease in aging populations. However, our understanding of what drives the age-related inflammatory dysregulation is limited. Here, we investigate the macrophage and its contribution to periodontal disease in old and young mice using a ligature-induced periodontal disease model. We demonstrate that control old mice present with an aged periodontal phenotype, characterized by increased alveolar bone loss and increased local inflammatory cytokine expression compared to young mice. Macrophages were demonstrated to be present in the periodontium of old and young mice in equal numbers in controls, during disease induction, and during disease recovery. However, it appears age may have a detrimental effect on macrophage activity during disease recovery. Depletion of macrophages during disease recovery in old mice resulted in decreased inflammatory cytokines within the gingiva and decreased bone loss as measured by micro–computed tomography. In young mice, macrophage depletion during disease recovery had no beneficial or detrimental effect. Macrophage depletion during disease induction resulted in decreased disease severity similarly in young and old mice. Findings from this work support the diverse roles of macrophages in disease induction as well as the active roles of disease recovery, including the resolution of inflammation. Here, we conclude that age-related changes to the macrophage appear to be detrimental to the recovery from disease and may explain, in part, the age-related increase in prevalence of periodontal disease. Future studies examining the specific intrinsic age-related changes to the macrophage will help identify therapeutic targets.
Keywords: periodontitis, aging, innate immunity, cell biology, gerontology, cytokines
Introduction
Periodontal disease results from a dysbiosis between the microbial plaque and the host inflammatory response (Van Dyke et al. 2020). In disease, the host inflammatory response results in the destruction of soft and hard tissues supporting the dentition (Papapanou et al. 2018) Management of periodontal disease is largely focused on microbial plaque; however, adequate management of the host inflammatory response remains a clinical challenge.
The prevalence of periodontal disease increases with age, affecting 60% of adults in the United States over the age of 65 y (Eke et al. 2015, 2016). A chronically elevated and dysregulated inflammatory response is also associated with increased age. The term inflamm-aging describes the age-related inflammatory perturbations and has been implicated in many diseases that increase in prevalence with age, including Alzheimer disease, type II diabetes, and atherosclerosis (Franceschi et al. 2000; Xia et al. 2016). As inflammatory dysregulation is implicated in the pathogenesis of periodontal disease, it has been suggested that inflamm-aging may contribute, in part, to the age-related increase of periodontal disease (Ebersole et al. 2016).
The mechanisms responsible for inflamm-aging are not fully understood; however, multiple cellular and molecular processes, including cellular senescence, dysregulated autophagy, and accumulation of danger/damage-associated molecular patterns (DAMPS), have been implicated (Oishi and Manabe 2016; Xia et al. 2016). Similarly, age-related changes to the macrophage are thought to contribute to inflamm-aging (Oishi and Manabe 2016; Valbuena Perez et al. 2020). The macrophage is an important regulator of the innate immune response during periodontal disease and can broadly acquire proinflammatory (M1) or anti-inflammatory (M2) phenotypes (Sima and Glogauer 2013; Wynn et al. 2013). The M1 phenotype is present in response to microbial plaque and propagates the inflammatory response (Mosser and Edwards 2008; Zhou et al. 2019). The M2 phenotype is present to aid in the resolution of inflammation and promote healing of damaged tissues (Mosser and Edwards 2008; Zhou et al. 2019). Control of the polarization of M1 and M2 macrophage phenotypes is important to regulate inflammation and prevent disease, as the presence of M1 and M2 phenotypes has been shown to be related to increased and decreased severity of periodontal disease, respectively (Lappin et al. 2001; Garlet 2010). Macrophage phenotypes are complex and demonstrate disease and tissue specificity. Therefore, the use of M1 and M2 in this article serves only to describe general pro- and anti-inflammatory activities of macrophages.
Our previous work has focused on age-related changes to the macrophage and the impact on inflammatory dysregulation during bone healing. Using an RNA sequencing approach in mice, we have demonstrated significant transcriptional age-related changes to macrophages within bone of old mice compared to young mice, with the macrophages from old mice demonstrating a more proinflammatory and M1-like phenotype (Clark et al. 2020). In addition, the aged macrophage phenotype was associated with delays in fracture healing (Clark et al. 2020). As proper regulation of inflammation is critical in the management of periodontal disease, we hypothesized that similar age-related changes to macrophages may contribute to inflammatory dysregulation and to the pathogenesis of periodontal disease in older populations.
In this work, we examine the differential activity of macrophages during the induction and resolution of inflammation in periodontal disease in old and young mice. We further evaluate the extent to which age-related changes in macrophages contribute to inflammatory dysregulation by examining the effect of a treatment to deplete macrophages in old and young mice on periodontal disease severity.
Materials and Methods
See the Appendix for expanded description of experimental procedures.
Periodontal Disease Model
All animal procedures were approved by the UCSF Institutional Animal Care and Use Committee and were carried out in strict accordance with the guidelines of the National Institutes of Health (NIH) for the care and use of laboratory animals and the guidelines of the ARRIVE 2.0 checklist. Periodontal disease was induced in old (24 mo) and young (3 mo) male C57BL/6 mice with a suture inoculated with Porphyromonas gingivalis (ATCC 33277) tied around the second maxillary molars bilaterally, as previously described (Abe and Hajishengallis 2013). The sutures remained in place for 7 d. At the end of the 7-d induction period, mice were euthanized, or, in a second arm of the study, the sutures were removed, and the animal was allowed to recover for an additional 7 d. At the end of disease induction or disease recovery period, animals were euthanized, and maxillae were collected for analysis.
Macrophage Depletion
During disease induction or disease recovery, PLX3397 (Plexxikon) was administered to half the mice in each age group to deplete macrophages. PLX3397 is a small-molecule inhibitor that antagonizes the macrophage colony stimulating factor 1 receptor (CSF1R) (Butowski et al. 2016). Inhibition of CSF1R prevents monocyte differentiation into mature macrophages. Inhibition of FMS-like tyrosine kinase 3 (FLT3) and proto-oncogene receptor c-KIT by PLX3397 has also been reported (Butowski et al. 2016). PLX3397 was delivered ad libitum throughout the 7-d induction or recovery period.
Micro–Computed Tomography Analysis
Bone volume/total volume (BV/TV) and bone mineral density (BMD) of the alveolar bone were quantified as a measure of disease severity. The region of interest was delineated along transverse slices isolating the alveolar bone supporting the teeth and excluding tooth structure.
Quantitative Real-Time Polymerase Chain Reaction
Gingiva was isolated from half of the maxillae and homogenized in Trizol, and messenger RNA (mRNA) was isolated. Complementary DNA (cDNA) was reverse transcribed using Superscript III (Invitrogen). Gene expression was normalized to the housekeeping gene GAPDH and presented as relative gene expression (ΔΔCT) or fold change (2−ΔΔCT). GAPDH demonstrated stable expression across age groups (Appendix Table 1).
Tissue Embedding
Half of the maxilla was isolated, fixed, and decalcified over 28 d. Samples were embedded in optimal cutting temperature compound (Tissue-Tek; Sakura Finetek). Frozen serial sections were cut at a thickness of 8 µm.
Immunohistochemistry
Quantification of F4/80+ macrophages was performed on frozen sections incubated with the primary antibody, rat anti-mouse F4/80 (BD Biosciences). A secondary antibody, horseradish peroxidase (HRP)–conjugated goat anti-mouse IgG, and VectaStain ABC Kit (Vector) were applied, and sections were stained with 3,3′-diaminobenzidine (DAB) substrate. Regions of interest (ROIs) were imaged and F480+ macrophages were quantified using ImageJ (National Institutes of Health) by a blinded examiner.
Tartrate-Resistant Acid Phosphatase Staining
Quantification of osteoclasts within the periodontium was performed via tartrate-resistant acid phosphatase (TRAP) staining using the acid phosphatase leukocyte kit (Sigma-Aldrich). ROIs were imaged and TRAP+ multinucleated osteoclast numbers (N.Oc/BS) and surface (Oc.S/BS) were measured using ImageJ by a blinded examiner according to the histomorphometric analysis standards of the American Society for Bone and Mineral Research (Dempster et al. 2013).
Linear Bone Loss Measurements
In control young and old mice, half of the maxillae were isolated and defleshed. Magnified images of the maxillae were captured using a dissecting microscope, and ImageJ was used for analysis. Bone loss was measured from the cementoenamel junction (CEJ) to the alveolar bone crest (ABC) at 6 sites per tooth.
Statistical Analysis
The BV/TV, BMD, linear bone loss, and cell quantification were calculated per sample and presented as mean ± standard deviation (SD). Groups were first analyzed via analysis of variance (ANOVA) for significant differences, and between-group differences were analyzed using a 2-tailed t test. Quantitative real-time polymerase chain reaction (qRT-PCR) used technical triplicates and the mean QT value was calculated. Relative gene expression (ΔΔCT) was calculated and presented as mean ± standard error of the mean (SEM) and analyzed using an ANOVA followed by between-group comparisons using a 2-tailed t test. Significance for all analysis was determined at P < 0.05. All statistical analysis was performed using GraphPad Prism v.7 software (GraphPad Software).
Results
Age-Related Changes Are Present in the Periodontium of Old Mice
Linear alveolar bone loss, as measured by the distance from CEJ to ABC, was compared in old (24 mo) and young (3 mo) healthy control mice (n = 5/group) (Fig. 1A, B). Old mice had significantly increased bone loss (0.19 ± 0.007 mm) compared to young (0.09 ± 0.015 mm) (P < 0.01) (Fig. 1C). Increased bone loss in control old mice was associated with inflammatory dysregulation within the gingiva. Expression of interleukin (IL)–1β, tumor necrosis factor α (TNFα), IL-6, and IL-17 was significantly increased in the gingiva of old mice compared to young (P < 0.05) (Fig. 1D). The increased bone loss and inflammatory dysregulation was not associated with increased quantities of macrophages or osteoclasts, as the number of macrophages and TRAP+ osteoclasts within the periodontium of old and young control mice were not significantly different (Fig. 1E–G). Indeed, both cell types were present within the periodontium at basal levels in healthy controls of both age groups (Fig. 1H–J).
Figure 1.
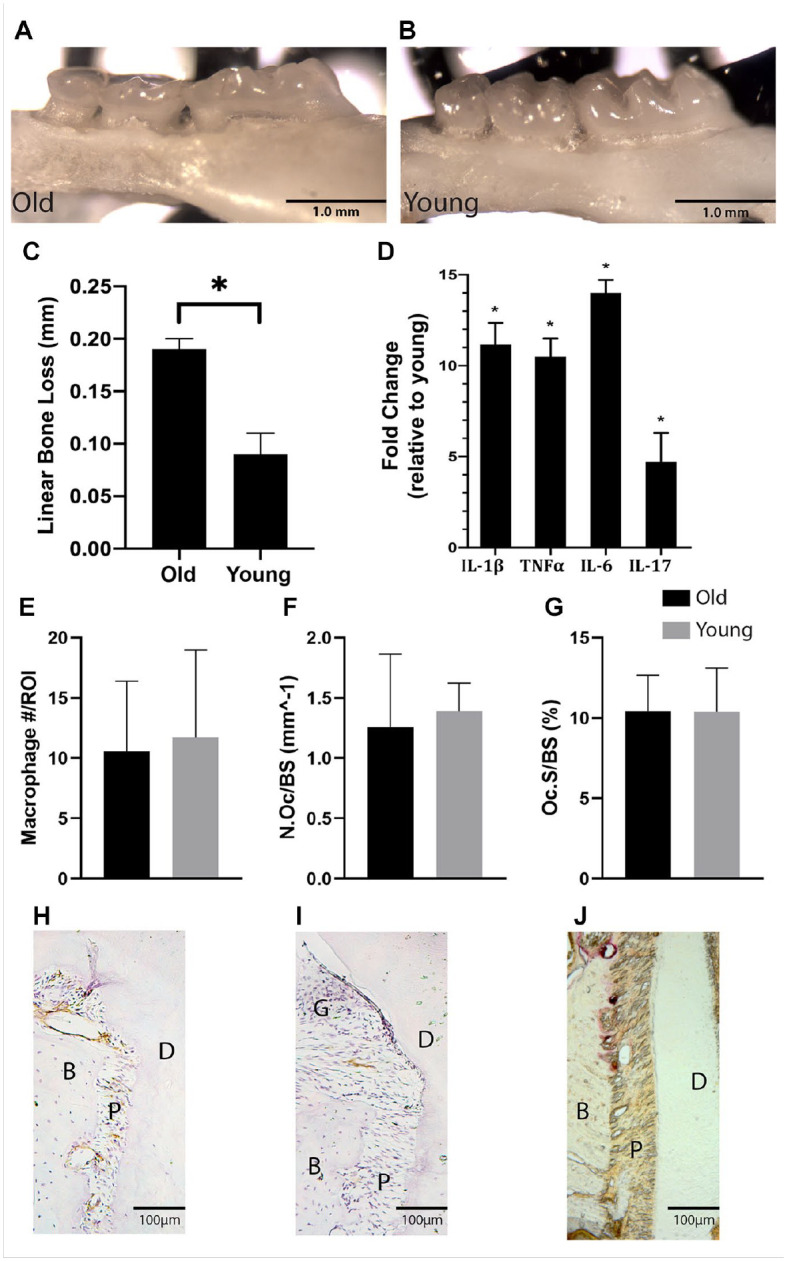
Old mice demonstrate an aged periodontal phenotype. The maxillae of control old (24 mo) and young (3 mo) male mice (C57BL/6J) were collected and analyzed (A, B). Significantly increased linear bone loss, measured from the cementoenamel junction to the alveolar bone crest, was demonstrated in old mice (C). The increased bone loss was associated with a significant increase in proinflammatory cytokine expression within the gingiva as measured by quantitative real-time polymerase chain reaction (D). Frozen histological sections were stained for F4/80 and tartrate-resistant acid phosphatase (TRAP), and F4/80+ macrophages, TRAP+ cell surface/bone surface, and TRAP+ osteoclast number per bone surface were quantified. Macrophage and osteoclast quantity in the periosteum were similar in old and young mice (E–G). Histomorphometry demonstrates the presence of F4/80+ macrophages (DAB, brown) (H, I) and TRAP+ osteoclasts (red) (J) within the periodontium in controls. B, bone; D, dentin; G, gingival tissue; P, periodontal ligament. Data presented as mean ± SD. *P < 0.05.
Macrophage Number within the Periodontium Is Similar in Old and Young Mice
Macrophages within the periodontium were quantified in old and young mice (n = 6/group) using immunohistochemistry to visualize macrophages. At baseline, the quantity of macrophages within the periodontium of old and young control mice was not significantly different (P > 0.05) (Fig. 1E–G). Indeed, macrophages were present throughout the periodontium at basal levels in healthy mice of both age groups (Fig. 1H, I). It appears the increased bone loss and inflammatory dysregulation demonstrated in old mice were not associated with increased numbers of macrophages within the periodontium.
Macrophages were also quantified after 7 d of periodontal disease induction and after 7 d of disease recovery. A ligature model was used and confirmed to adequately induce periodontal disease with significant bone loss in old and young mice (Fig. 2). Macrophages were present within the periodontium at baseline, during disease induction, and during recovery (Fig. 3A–C). The number of macrophages was similar in old and young mice (P > 0.05) (Fig. 3G). Macrophages were present within the gingival epithelium/connective tissue, alveolar bone, and periodontal ligament (PDL) (Fig. 3D–F). High numbers of macrophages were present during both disease induction and disease recovery, demonstrating the diverse role of macrophages throughout the development and resolution of periodontal disease.
Figure 2.
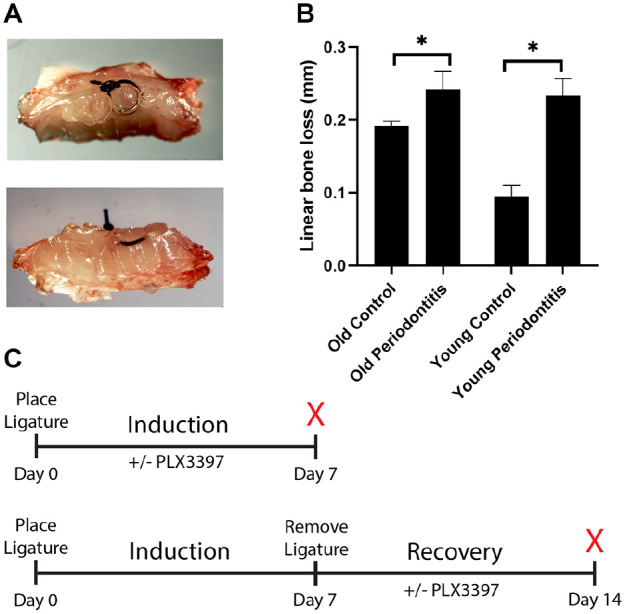
Ligature model of periodontal disease induces bone loss in old and young mice. A 6.0 silk suture inoculated with Porphyromonas gingivalis was tied around the second maxillary molar bilaterally (A). Sutures remained in place for 7 d to induce disease. The inoculated ligature induced significant linear bone loss, as measured from the cementoenamel junction to the alveolar bone crest, in both old and young mice (B). Mice of each age group were divided between 2 arms of the study, a disease induction arm and disease recovery arm (C). In the induction arm, the animal was euthanized after 7 d of disease induction. In the recovery arm, the ligature was removed at day 7 and the mice were allowed to recover for an additional 7 d before the animal was euthanized and tissues analyzed. PLX3397 was administered to deplete macrophages during the induction or recovery period. Non-PLX3397 groups received control chow. n = 6/group. Data presented as mean ± SD. *P < 0.05.
Figure 3.
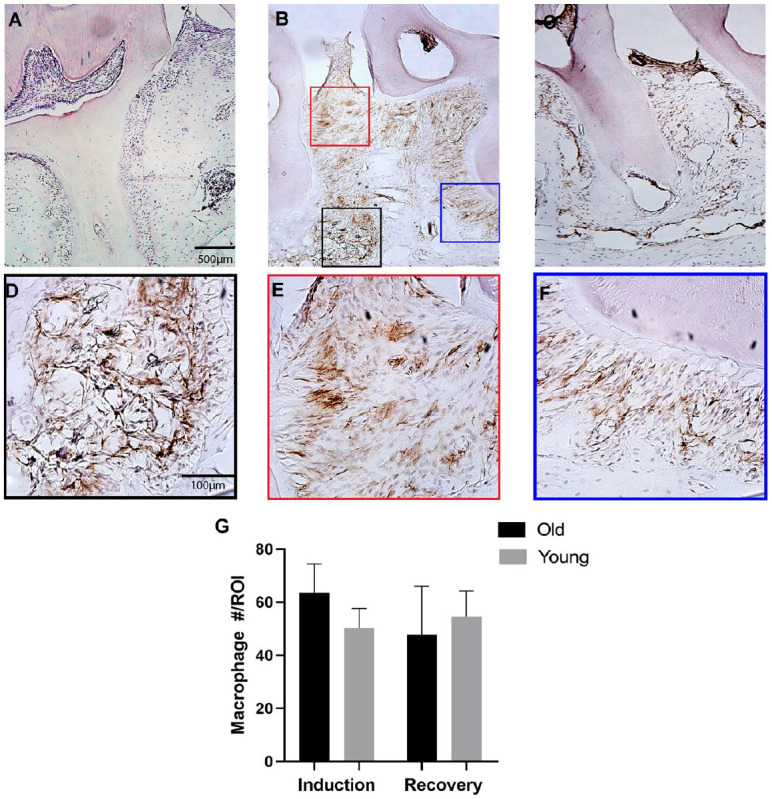
Macrophage quantity within the periodontium is similar in young and old mice during disease induction and recovery. Immunohistochemistry staining for F4/80+ macrophages was performed on frozen histological sections of the periodontium of old and young mice. F4/80+ macrophages (DAB, brown) were present in healthy controls (A), after 7 d of disease inductions (B), and after 7 d of disease recovery (C). Macrophages were quantified within 3 distinct regions of interest (ROIs) per histological section. ROIs were identified within an osseous region (D), gingival tissue region (E), and periodontal ligament (PDL) region (F). Macrophage quantity within the periodontium during disease induction and recovery was similar in old and young mice (n = 6/group) (G). Data presented as mean ± SD.
Depletion of Macrophages during the Disease Induction Period Attenuates Disease Severity in Old and Young Mice
To better understand the differential contribution of macrophages from old and young mice in the pathogenesis of periodontal disease, macrophages were depleted from the periodontium during the induction period. The resulting inflammatory profile and disease severity were evaluated as a function of macrophage depletion.
Periodontal disease was induced in old and young mice (n = 6/group). Administration of PLX3397 throughout the induction period resulted in a significant decrease in macrophage quantity within the periodontium of old and young mice (P < 0.05) (Fig. 4A). Old mice, treated with PLX3397, had significantly higher BV/TV compared to nontreated controls (61% ± 3% vs. 53% ± 3%) (P < 0.01) and significantly higher BMD compared to nontreated controls (822 ± 51 mg HA/mm3 vs. 707 ± 58 mg HA/mm3) (P < 0.01) (Fig. 4F). In addition, treated old mice demonstrated significantly decreased expression of IL-1β, TNFα, and IL-6 (P < 0.05) (Fig. 4D). In young mice, a similar trend was also observed. Depletion of macrophages resulted in significantly higher BV/TV (58% ± 0% vs. 45% ± 2%) (P < 0.01) and significantly higher BMD compared to nontreated controls (776 ± 7 mg HA/mm3 vs. 598 ± 19 mg HA/mm3) (P < 0.01) (Fig. 4G). Relative mRNA expression of inducible nitric oxide synthase (iNOS) was significantly reduced in young mice with treatment compared to nontreated controls (P < 0.05) (Fig. 4E). Administration of PLX3397 also resulted in a significant decrease in TRAP+ osteoclasts within the periodontium in old and young mice (P < 0.05) (Fig. 4B, C).
Figure 4.

Macrophage depletion during disease induction attenuates disease severity in old and young mice. Periodontal disease was induced in old and young mice (n = 6/group) for a period of 7 d. Macrophages were depleted via treatment with PLX3397 throughout the induction period. After the 7-d induction period, the maxillae were isolated and analyzed. PLX3397 treatment resulted in a significant decrease in macrophage quantity within the periodontium of old and young mice (A). PLX3397 treatment also resulted in significant decrease in tartrate-resistant acid phosphatase–positive (TRAP+) osteoclast cell surface/bone surface and TRAP+ osteoclast number per bone surface in the periodontium of old and young mice (B, C). Attenuation of disease severity was demonstrated in both old and young mice via treatment with PLX3397. Similarly, across both age groups, PLX3397 resulted in significantly decreased proinflammatory cytokine expression within the gingiva of old mice as measured by quantitative real-time polymerase chain reaction (D, E) and significantly greater bone volume fraction (BV/TV) and bone mineral density (BMD) as measured by micro–computed tomography (CT) (F, G). Representative micro-CT images demonstrate the effect of PLX3397 treatment in old and young mice (H–K). Data presented as mean ± SD and mean ± SEM for D and E. *P < 0.05. **P < 0.01.
Depletion of Macrophages in Old Mice Improves Disease Resolution
The differential contribution of macrophages from old and young mice during the resolution period of periodontal disease was also evaluated. Administration of PLX3397 throughout the recovery period of the disease model resulted in a significant decrease in macrophages within the periodontium of old and young mice (P < 0.05) (Fig. 5A). Depleting macrophages in old mice resulted in significantly higher BV/TV compared to nontreated controls (62% ± 2% vs. 56% ± 2%) (P < 0.01) and significantly higher BMD compared to nontreated controls (816 ± 37 mg HA/mm3 vs. 750 ± 33 mg HA/mm3) (P < 0.01) (Fig. 5F). The expression of IL-1β was also significantly decreased within the gingiva of old treated mice compared to nontreated controls (P < 0.05) (Fig. 5D). However, in young mice, no benefit of PLX3397 administrated was demonstrated during disease recovery. There was no significant difference in BV/TV, BMD, and inflammatory gene expression in young treated mice compared to nontreated controls (P > 0.05) (Fig. 5E, G). During disease recovery, the quantity of TRAP+ osteoclasts in old and young mice was not affected by PLX3397 administration (Fig. 5B, C).
Figure 5.
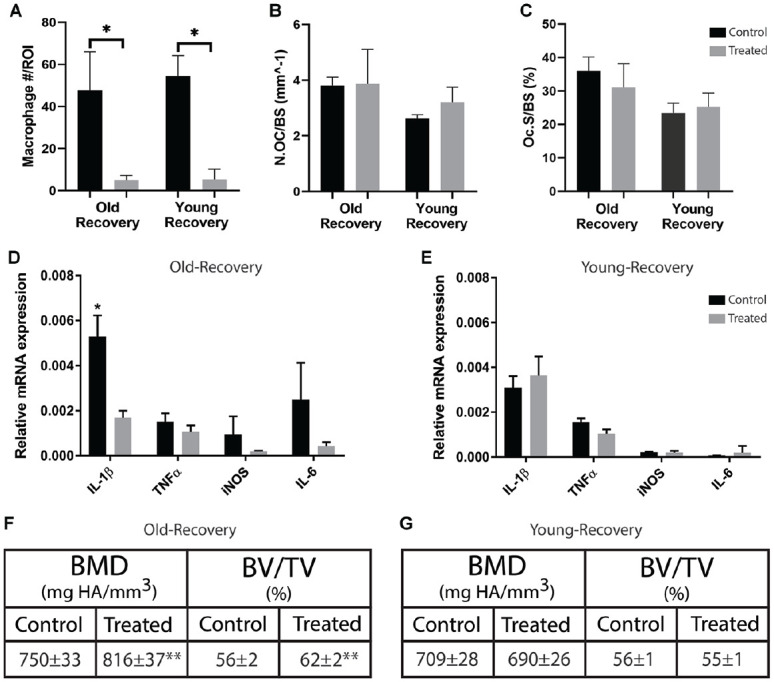
Macrophage depletion in old mice improves disease recovery. Periodontal disease was induced in old and young mice (n = 6/group) for a period of 7 d. At the end of the induction period, the ligatures were removed and the mice were allowed to recover for 7 additional days. Macrophages were depleted via treatment with PLX3397 throughout the recovery period. After the 7-d recovery period, the maxillae were isolated and analyzed. PLX3397 treatment resulted in a significant decrease in macrophage quantity within the periodontium at the end of the recovery period (A). Treatment with PLX3397 did not have an effect on tartrate-resistant acid phosphatase–positive (TRAP+) osteoclast cell surface/bone surface or TRAP+ osteoclast number per bone surface in the periodontium of old or young mice (B, C). Treatment resulted in significantly decreased proinflammatory cytokine expression within the gingiva of old mice as measured by quantitative real-time polymerase chain reaction (D). In addition, treatment with PLX3397 resulted in significantly greater bone volume fraction (BV/TV) and bone mineral density (BMD) as measured by micro–computed tomography (F) compared to untreated controls. No treatment effect was demonstrated in young mice using the same analysis (E, G). Data presented as mean ± SD and mean ± SEM for D and E. *P < 0.05. **P < 0.01.
Discussion
The proper balance of the pro- and anti-inflammatory activities of the macrophage is necessary in the management of periodontal disease. In this work, we demonstrated the presence of macrophages in the periodontium throughout the induction and recovery phases of our periodontal disease model. While we did not see a difference in macrophage numbers accumulated in the periodontium, a central finding was the differential effect of macrophage depletion in old and young mice during disease resolution compared to disease induction. During disease induction, depletion of macrophages had a beneficial effect and attenuated inflammation and disease severity in young and old mice (Fig. 4). However, during disease resolution, depletion of macrophages was beneficial only in old mice while having no effect in young mice (Fig. 5). These findings are interesting because they demonstrate how distinct macrophage functions, initiation and resolution of inflammation, may be differentially affected by age.
We suspect the reason that inhibiting macrophages from participating in disease recovery was beneficial in old mice and not young was due to transcriptional differences intrinsic to macrophages from old mice. In our previous work using a fracture model in old and young mice, age-related inflammatory dysregulation in the healing bone could be explained, in part, by an increased proinflammatory transcriptional prolife in macrophages from old mice compared to young (Clark et al. 2020). Isolated macrophages from old mice demonstrated significantly increased expression of M1 markers and proinflammatory cytokines and chemokines as analyzed via RNAseq (Clark et al. 2020). In this study, depleting macrophages in old mice may have reduced a source of cytokines and chemokines that propagate the inflammatory response. However, during the recovery phase, macrophages in young mice may have already made the phenotypic switch toward an anti-inflammatory phenotype and, therefore, depleting the macrophages in young mice did not have the same effect in reducing proinflammatory chemokines and cytokines. In this context, age-related changes may have affected the phenotypic conversion in macrophages and contributed to the inflammatory dysregulation observed in old mice. Future work is needed to evaluate the age-related transcriptional changes of macrophages unique to the periodontium to elucidate functional changes key to the pathogenesis of periodontal disease.
In further support that age-related changes in macrophages contribute to inflammatory dysregulation and disease severity, we found that the quantity of macrophages was not affected as a function of age. The number of macrophages within the periodontium during disease induction or resolution was similar in both age groups (Fig. 3). Given the changes observed in cytokine production as a function of age (Fig. 1D), differences in macrophage function, rather than quantity, may be more relevant in the pathogenesis of periodontal disease. These findings were similar to our previous work that demonstrated the quantity of macrophages infiltrating the fracture site was similar in old and young mice (Clark et al. 2020). Other studies investigating the effect of age on chemokinesis and chemotaxis of innate immune cells have been inconsistent, and more work is needed on the subject (Shaw et al. 2013).
We demonstrated an aged periodontal phenotype in old mice characterized by increased bone loss and increased proinflammatory cytokine expression (Fig. 1). Similar findings have been demonstrated by other groups examining the aging periodontium of mice (Liang et al. 2010). Interestingly, our study highlights that macrophages were present throughout the periodontium in healthy controls (Fig. 1). The oral biome may elicit a basal innate immune response within peripheral zones of epithelium and connective tissue, as macrophages and neutrophils are present in healthy gingival tissues (Hajishengallis and Hajishengallis 2014; Garaicoa-Pazmino et al. 2019). However, the presence of macrophages lining alveolar bone and the PDL space may represent a tissue-resident population of macrophage, as has been described in other tissues (Chang et al. 2008).
Limitations of the ligature model of periodontal disease may exist in the study. Ligature models induce a robust osteolytic inflammatory response that is more acute compared to the natural chronic inflammatory processes in periodontal disease. This robust inflammatory response may have masked any age-related differences during disease induction and could explain, in part, why age-related differences are present in the recovery arm when the ligature was no longer the inflammatory stimulus.
The administration of PLX3397 significantly reduced the number of macrophages within the periodontium. Decreased macrophage quantity was associated with decreased inflammatory cytokine expression and decreased disease severity. Other groups have shown similar improvements in periodontal disease severity with the administration of compounds that target inflammatory mediators. Alveolar bone loss was significantly reduced in a mouse model of periodontal disease by local delivery of CL22, which increased the recruitment of T regulatory cells (Glowacki et al. 2013). Similarly, promoting the resolution of inflammation in a mouse model of periodontitis with the administration of resolvins resulted in decreased bone loss (Hasturk et al. 2006). We did not directly show the cellular source of cytokines measured before and after administration of PLX3397. It is possible other immune cells produced similar inflammatory cytokines that contributed to the pathogenesis of periodontal disease. Macrophages interact with other immune cells in the propagation of inflammation. Macrophage depletion may have resulted in decreased activation of other immune cells in their production of cytokines and ultimately had the same effect of reducing cytokine expression and disease severity.
We suspected that antagonizing CSF1R via administration of PLX3397 may also affect osteoclasts, as CSF1R stimulation leads to monocyte differentiation toward macrophages and osteoclasts (Boyce 2013). PLX3397 administration significantly decreased the quantity of osteoclasts during disease induction (Fig. 4) but had no effect on osteoclast quantity during disease recovery in both age groups (Fig. 5). As osteoclasts contribute to the pathogenesis of periodontal disease, continued osteoclast recruitment and activity are present throughout the induction phase of this study. However, osteoclasts likely have a less substantial role in the resolution of inflammation. Thus, inhibiting osteoclast activation during the recovery phase had no measurable effect on osteoclast quantity, as osteoclasts were likely not actively differentiating and recruited during the resolution period under normal conditions. In contrast, the macrophage is active throughout initiation and resolution of inflammation. Thus, we can measure a significant decrease in quantity of macrophages during both stages. Other strategies for macrophage depletion exist, including administration of clodronate liposomes or using the transgenic Mac Fas–induced apoptosis (MaFIA) mouse. However, each method lacks specificity in their targeted depletion and can similarly result in depletion of osteoclasts (McCubbrey et al. 2017; Michalski et al. 2019).
Conclusion
The work presented here demonstrates the diverse role of macrophages during periodontal disease initiation and resolution, and the findings provide support that the age-related changes to the macrophage contribute to the pathogenesis of periodontal disease. Age appears to perturb macrophage activity most noticeably during disease resolution. Such age-related changes to the macrophage may explain, in part, the increased prevalence of periodontal disease in elderly populations. By depleting the macrophage infiltration during disease recovery, inflammation and disease severity were reduced in old mice. Interestingly, the beneficial outcome demonstrated from macrophage depletion could serve as a therapeutic model for treatment directed at the specific age-related perturbations that contribute to the pathogenesis of periodontal disease in the elderly population.
Author Contributions
D. Clark, contributed to conception, design, data acquisition, analysis, and interpretation, drafted and critically revised the manuscript; B. Halpern, contributed to data acquisition, analysis, and interpretation, critically revised the manuscript; T. Miclau, contributed to conception, critically revised the manuscript; M. Nakamura, contributed to design, data acquisition, and interpretation, critically revised the manuscript; Y. Kapila, contributed to design, data analysis, and interpretation, critically revised the manuscript; R. Marcucio, contributed to conception, design, and data interpretation, critically revised the manuscript. All authors gave final approval and agree to be accountable for all aspects of the work.
Supplemental Material
Supplemental material, sj-pdf-1-jdr-10.1177_00220345211009463 for The Contribution of Macrophages in Old Mice to Periodontal Disease by D. Clark, B. Halpern, T. Miclau, M. Nakamura, Y. Kapila and R. Marcucio in Journal of Dental Research
Acknowledgments
We thank Gina Baldoza for laboratory support.
Footnotes
A supplemental appendix to this article is available online.
Declaration of Conflicting Interests: The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Institutes of Health (NIA) grant number R01AG046282, National Institutes of Health (NIDCR) grant number K08DE029505, the UCSF Core Center for Musculoskeletal Biology and Medicine of the NIAMS (P30AR066262), and the Russel/Engleman Arthritis Center at UCSF and the Department of Veterans Affairs Health Care System.
ORCID iD: D. Clark  https://orcid.org/0000-0003-3453-6498
https://orcid.org/0000-0003-3453-6498
References
- Abe T, Hajishengallis G. 2013. Optimization of the ligature-induced periodontitis model in mice. J Immunol Methods. 394(1–2):49–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyce BF. 2013. Advances in the regulation of osteoclasts and osteoclast functions. J Dent Res. 92(10):860–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butowski N, Colman H, De Groot JF, Omuro AM, Nayak L, Wen PY, Cloughesy TF, Marimuthu A, Haidar S, Perry A, et al. 2016. Orally administered colony stimulating factor 1 receptor inhibitor PLX3397 in recurrent glioblastoma: an Ivy Foundation Early Phase Clinical Trials Consortium phase II study. Neuro Oncol. 18(4):557–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang MK, Raggatt L-J, Alexander KA, Kuliwaba JS, Fazzalari NL, Schroder K, Maylin ER, Ripoll VM, Hume DA, Pettit AR. 2008. Osteal tissue macrophages are intercalated throughout human and mouse bone lining tissues and regulate osteoblast function in vitro and in vivo. J Immunol. 181(2):1232–1244. [DOI] [PubMed] [Google Scholar]
- Clark D, Brazina S, Yang F, Hu D, Hsieh CL, Niemi EC, Miclau T, Nakamura MC, Marcucio R. 2020. Age-related changes to macrophages are detrimental to fracture healing in mice. Aging Cell. 19(3):e13112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dempster DW, Compston JE, Drezner MK, Glorieux FH, Kanis JA, Malluche H, Meunier PJ, Ott SM, Recker RR, Parfitt AM. 2013. Standardized nomenclature, symbols, and units for bone histomorphometry: a 2012 update of the report of the ASBMR Histomorphometry Nomenclature Committee.J Bone Miner Res. 28(1):2–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebersole JL, Graves CL, Gonzalez OA, Dawson D, Morford LA, Huja PE, Hartsfield JK, Huja SS, Pandruvada S, Wallet SM. 2016. Aging, inflammation, immunity and periodontal disease. Periodontol 2000. 72(1):54–75. [DOI] [PubMed] [Google Scholar]
- Eke PI, Dye BA, Wei L, Slade GD, Thornton-Evans GO, Borgnakke WS, Taylor GW, Page RC, Beck JD, Genco RJ. 2015. Update on prevalence of periodontitis in adults in the United States: NHANES 2009 to 2012.J Periodontol. 86(5):611–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eke PI, Wei L, Borgnakke WS, Thornton-Evans G, Zhang X, Lu H, McGuire LC, Genco RJ. 2016. Periodontitis prevalence in adults ≥ 65 years of age, in the USA. Periodontol 2000. 72(1):76–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franceschi C, Bonafè M, Valensin S, Olivieri F, De Luca M, Ottaviani E, De Benedictis G. 2000. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann N Y Acad Sci. 30(7):374–381. [DOI] [PubMed] [Google Scholar]
- Garaicoa-Pazmino C, Fretwurst T, Squarize CH, Berglundh T, Giannobile WV, Larsson L, Castilho RM. 2019. Characterization of macrophage polarization in periodontal disease. J Clin Periodontol. 46(8):830–839. [DOI] [PubMed] [Google Scholar]
- Garlet GP. 2010. Critical reviews in oral biology & medicine: destructive and protective roles of cytokines in periodontitis: a re-appraisal from host defense and tissue destruction viewpoints. J Dent Res. 89(12):1349–1363. [DOI] [PubMed] [Google Scholar]
- Glowacki AJ, Yoshizawa S, Jhunjhunwala S, Vieira AE, Garlet GP, Sfeir C, Little SR. 2013. Prevention of inflammation-mediated bone loss in murine and canine periodontal disease via recruitment of regulatory lymphocytes. Proc Natl Acad Sci USA. 110(46):18525–18530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajishengallis E, Hajishengallis G. 2014. Neutrophil homeostasis and periodontal health in children and adults. J Dent Res. 93(3):231–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasturk H, Kantarci A, Ohira T, Arita M, Ebrahimi N, Chiang N, Petasis NA, Levy BD, Serhan CN, Van Dyke TE. 2006. RvE1 protects from local inflammation and osteoclastmediated bone destruction in periodontitis. FASEB J. 20(2):401–403. [DOI] [PubMed] [Google Scholar]
- Lappin DF, Macleod CP, Kerr A, Mitchell T, Kinane DF. 2001. Anti-inflammatory cytokine IL-10 and T cell cytokine profile in periodontitis granulation tissue. Clin Exp Immunol. 123(2):294–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang S, Hosur KB, Domon H, Hajishengallis G. 2010. Periodontal inflammation and bone loss in aged mice. J Periodontal Res. 45(4):574–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCubbrey AL, Allison KC, Lee-Sherick AB, Jakubzick CV, Janssen WJ. 2017. Promoter specificity and efficacy in conditional and inducible transgenic targeting of lung macrophages. Front Immunol. 8:1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalski MN, Zweifler LE, Sinder BP, Koh AJ, Yamashita J, Roca H, McCauley LK. 2019. Clodronate-loaded liposome treatment has site-specific skeletal effects. J Dent Res. 98(4):459–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosser DM, Edwards JP. 2008. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 8(12):958–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oishi Y, Manabe I. 2016. Macrophages in age-related chronic inflammatory diseases. NPJ Aging Mech Dis. 2:16018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papapanou PN, Sanz M, Buduneli N, Dietrich T, Feres M, Fine DH, Flemmig TF, Garcia R, Giannobile WV, Graziani F, et al. 2018. Periodontitis: consensus report of workgroup 2 of the 2017 World Workshop on the Classification of Periodontal and Peri-Implant Diseases and Conditions.J Periodontol. 89(Suppl 1):S173–S182. [DOI] [PubMed] [Google Scholar]
- Shaw AC, Goldstein DR, Montgomery RR. 2013. Age-dependent dysregulation of innate immunity. Nat Rev Immunol. 13(12):875–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sima C, Glogauer M. 2013. Macrophage subsets and osteoimmunology: tuning of the immunological recognition and effector systems that maintain alveolar bone. Periodontol 2000. 63(1):80–101. [DOI] [PubMed] [Google Scholar]
- Valbuena Perez JV, Linnenberger R, Dembek A, Bruscoli S, Riccardi C, Schulz MH, Meyer MR, Kiemer AK, Hoppstädter J. 2020. Altered glucocorticoid metabolism represents a feature of macroph-aging. Aging Cell. 19(6):e13156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dyke TE, Bartold PM, Reynolds EC. 2020. The nexus between periodontal inflammation and dysbiosis. Front Immunol. 11:511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wynn TA, Chawla A, Pollard JW. 2013. Macrophage biology in development, homeostasis and disease. Nature. 496(7446):445–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia S, Zhang X, Zheng S, Khanabdali R, Kalionis B, Wu J, Wan W, Tai X. 2016. An update on inflamm-aging: mechanisms, prevention, and treatment. J Immunol Res. 2016:8426874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou LN, Bi CS, Gao LN, An Y, Chen F, Chen FM. 2019. Macrophage polarization in human gingival tissue in response to periodontal disease. Oral Dis. 25(1):265–273. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material, sj-pdf-1-jdr-10.1177_00220345211009463 for The Contribution of Macrophages in Old Mice to Periodontal Disease by D. Clark, B. Halpern, T. Miclau, M. Nakamura, Y. Kapila and R. Marcucio in Journal of Dental Research