

Abstract
Bone loss caused by trauma, neoplasia, congenital defects, or periodontal disease is a major cause of disability and human suffering. Skeletal progenitor cell–extracellular matrix interactions are critical for bone regeneration. Discoidin domain receptor 2 (DDR2), an understudied collagen receptor, plays an important role in skeletal development. Ddr2 loss-of-function mutations in humans and mice cause severe craniofacial and skeletal defects, including altered cranial shape, dwarfing, reduced trabecular and cortical bone, alveolar bone/periodontal defects, and altered dentition. However, the role of this collagen receptor in craniofacial regeneration has not been examined. To address this, calvarial subcritical-size defects were generated in wild-type (WT) and Ddr2-deficient mice. The complete bridging seen in WT controls at 4 wk postsurgery was not observed in Ddr2-deficient mice even after 12 wk. Quantitation of defect bone area by micro–computed tomography also revealed a 50% reduction in new bone volume in Ddr2-deficient mice. Ddr2 expression during calvarial bone regeneration was measured using Ddr2-LacZ knock-in mice. Expression was restricted to periosteal surfaces of uninjured calvarial bone and, after injury, was detected in select regions of the defect site by 3 d postsurgery and expanded during the healing process. The impaired bone healing associated with Ddr2 deficiency may be related to reduced osteoprogenitor or osteoblast cell proliferation and differentiation since knockdown/knockout of Ddr2 in a mesenchymal cell line and primary calvarial osteoblast cultures reduced osteoblast differentiation while Ddr2 overexpression was stimulatory. In conclusion, Ddr2 is required for cranial bone regeneration and may be a novel target for therapy.
Keywords: craniofacial biology/genetics, cell-matrix interactions, collagen receptor, extracellular matrix, osteoblasts, cell differentiation
Introduction
Bone loss caused by trauma, neoplasia, congenital defects, or periodontal disease is a major cause of human disability and represents a significant clinical challenge for craniomaxillofacial and orthopedic surgeons (Bostrom et al. 1999). Bone regeneration procedures are necessary for treating nonunion fractures and other refractory bone defects. Bone grafts, consisting mainly of bone extracellular matrix (ECM) and associated cells, are often used to manage such conditions. Recent research has unveiled many unique characteristics of the ECM that are critical for tissue regeneration. The ECM affects cell function by interacting with specific cell surface receptors, which trigger cellular functions such as adhesion, migration, proliferation, and differentiation (Legate et al. 2009). Therefore, a clear understanding of these ECM receptors will facilitate the development of new bone regeneration strategies.
Collagen receptors are of particular interest in skeletal regeneration since they bind the most abundant ECM protein in bone. The most studied collagen receptors are the β1 integrins (Brunner et al. 2013). Studies showed that stimulation of β1 integrins, using scaffolds that selectively promote integrin binding, significantly accelerated and increased bone formation in nonhealing femoral defects (Reyes and Garcia 2004; Clark et al. 2020). However, while integrins have clear roles in bone formation and regeneration, they cannot account for the entire response of bone cells to ECM since bone-specific knockout of β1 integrins or focal adhesion kinase, a component of integrin signaling, only partially prevents bone formation and regeneration at both cranial and long bone sites (Ekholm et al. 2002; Kim et al. 2007; Shekaran et al. 2014).
Discoidin domain receptor 2 (DDR2) is a second collagen receptor that preferentially binds fibrillar collagens and modulates integrin activity (Shrivastava et al. 1997; Vogel et al. 1997; Bayer et al. 2019). DDR2 has demonstrated roles in cell adhesion, proliferation, migration, and matrix remodeling and has also been associated with cancer progression (Valiathan et al. 2012; Itoh 2018). In humans, DDR2 mutations cause spondylo-meta-epiphyseal dysplasia (SMED), a skeletal disorder associated with dwarfism, bowing of long bones, craniofacial abnormalities, short fingers, and abnormal calcifications (Bargal et al. 2009). Consistent with the human phenotype, mice with a spontaneous Ddr2 mutation, called smallie mice (Ddr2slie/slie), have a SMED-like phenotype characterized by dwarfism and reduction in total bone mass (Kano et al. 2008). Furthermore, detailed analysis of Ddr2slie/slie mice revealed a dramatic reduction in bone volume of the cranial, axial, and appendicular skeleton due to reduction in osteoblast activity and bone formation in the absence of changes in resorption (Ge et al. 2016). Consistent with the preferential effects of Ddr2 deficiency on in vivo bone growth and development, specific functions for this molecule have also been demonstrated in isolated osteoblasts and chondrocytes (Zhang et al. 2011). Ddr2 is also expressed in craniofacial structures, including temporomandibular joint (TMJ), dental follicle/sac, odontoblasts, and the periodontal ligament (PDL), and is required for the formation and maintenance of these structures (Ge et al. 2018; Mohamed et al. 2020).
Although the requirement for Ddr2 in bone growth and development is well established, its potential role in bone regeneration has not been examined. In this study, we investigate the consequences of germline Ddr2 deletion on healing of a subcritical-sized calvarial defect that can spontaneously heal in wild-type (WT) mice, define the expression pattern of Ddr2 during calvarial bone regeneration, and relate these findings to cell-autonomous functions of Ddr2 in isolated osteoblasts. As will be shown, Ddr2 deletion almost totally blocks calvarial bone regeneration, a response that may be mediated through altered osteoblast or skeletal progenitor function.
Materials and Methods
Animals
Ddr2slie/slie mice, which contain a spontaneous 150-kb deletion in the Ddr2 locus (deletion of 17 of 18 total exons) to produce an effective null (Kano et al. 2008), were bred into a C57BL6 background for at least 10 generations (Ge et al. 2016). Ddr2-LacZ (Ddr2+/LacZ) knock-in mice, in which the bacterial LacZ gene is knocked into the Ddr2 locus, and Ddr2flox/flox mice were derived from ES cell clone Ddr2tm1a(EUCOMM)Wtsi (EPD0607__B01) obtained from the European Mutant Mouse Repository and were previously described (Ge et al. 2018; Mohamed et al. 2020). Ddr2slie/slie mice were used to measure the effect of global Ddr2 deficiency on regeneration. Ddr2+/LacZ mice were used to assess Ddr2 expression while calvarial cells from Ddr2flox/flox mice with or without adenovirus-Cre treatment were used to measure the effect of Ddr2 inactivation on osteoblast differentiation in cell culture. All animal studies were approved by the University of Michigan Committee on the Use and Care of Animals (UCUCA) and conformed to all guidelines and regulations for the protection of animal subjects. Mice were housed in specific pathogen-free Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC)–certified facilities. This study conforms to the Animal Research: Reporting In Vivo Experiments (ARRIVE) guidelines.
Calvarial Subcritical-Size Defect Surgery
Eight-week-old mice of both sexes were used for experiments. Mice were maintained with general anesthesia (2% isoflurane/100% O2) during the procedure and received buprenorphine (0.02 mg/kg intraperitoneally) for analgesia. Under sterile conditions, a sagittal incision was made, parietal bones were exposed, and a 0.5-mm subcritical size defect (SSD) was created with a carbide bur at low speed in WT, Ddr2slie/slie, and Ddr2+/LacZ mice. Incisions were closed using 4-0 black silk interrupted sutures. The day after surgery, another buprenorphine injection was given to the mice. Bone regeneration of the SSD was measured 2, 4, 6, and 12 wk postsurgery.
Statistical Analysis
All statistical analyses were performed using SPSS version 16.0 software (SPSS, Inc.). Values are presented as mean ± SD. For in vivo studies, 6 to 8 animals with approximately equal numbers of each sex were used per experimental group except where otherwise indicated. In all cases, experimental transgenic animals were compared with littermate controls. For cell culture studies, triplicate independent samples were used. Statistical significance was assessed using Student’s t test for comparisons between 2 experimental groups. A value of P < 0.05 was considered significant.
Detailed materials and methods for the following are included in the Appendix:
Micro–computed tomography analysis of bone regeneration
Tissue preparation and histopathological analysis
Vector construction, cell cultures, and in vitro differentiation
Results
Ddr2 Is Necessary for Regeneration of a Subcritical-Size Calvarial Defect
To investigate the possible role of Ddr2 in bone regeneration, we created 0.5-mm diameter SSDs in both WT and Ddr2slie/slie mice and examined new bone formation for up to 12 wk using histology and micro–computed tomography (CT) analysis. In WT mice, cranial defects in this size range completely heal without any intervention, making this a good model system to evaluate the requirement for Ddr2 in calvarial regeneration. In contrast, defects greater than 2 mm do not spontaneously heal in the absence of external interventions and are considered critical-sized defects (Cowan et al. 2004). To show that initial defects were similar regardless of genotype, WT and Ddr2slie/slie samples were harvested 3 d and 1 wk postsurgery before substantial healing had occurred and evaluated with hematoxylin and eosin (H&E) staining. Calvarial defects were initially identical in both groups (Fig. 1A). However, beginning at 2 wk postsurgery and continuing throughout the entire 12-wk period of the experiment, a clear reduction in defect healing was seen in Ddr2-deficient mice relative to WT controls (Fig. 1B, C). In agreement with previous reports (Park et al. 2016; Wilk et al. 2017), in WT animals, initial bridging of defects was seen after as little as 4 wk, with defects appearing fully healed between 6 and 12 wk (Fig. 1C). Interestingly, defects in Ddr2slie/slie mice did not exhibit any significant increase in bone volume/total defect volume between the 6- and 12-wk time points. This suggests that healing may have been arrested by this time, although further studies would be required to prove that defects would not heal even after extended periods.
Figure 1.

Ddr2slie/slie mice failed to heal a calvarial subcritical size defect (SSD). Defects were generated as described in Materials and Methods. (A) Hematoxylin and eosin staining 3 d and 1 wk postsurgery of SSD for wild-type (WT) and Ddr2slie/slie mice showing that defects are initially the same size in WT and mutant mice. (B) Representative micro–computed tomography (µCT) and histological images of the defect area showing amount of bone regeneration. Note that for panels A and B, low-power (left) and high-power images (right) are shown for each histological sample. (C) Measurements are shown for regenerated bone volume/total volume calculated as described in the Materials and Methods. Statistical analysis between WT andDdr2slie/slie mice at each time point. n = 6–8 mice/group. *P < 0.05. **P < 0.001. Scale bars: histological sections = 50 µm, 2-dimensional µCT image = 500 µm, 3-dimensional µCT image = 2 mm.
Ddr2 Is Expressed throughout Calvarial Bone Regeneration
We next examined Ddr2 expression during calvarial bone regeneration using a Ddr2-lacZ reporter mouse line (Ddr2+/lacZ mice; Fig. 2). Prior to injury, LacZ was expressed in periosteum and select marrow cells (Fig. 2A). At 3 d postsurgery, a few LacZ-positive cells were observed near the defect margin (Fig. 2B). By 1 wk, positive cells dramatically increased in the soft tissue at the center of the defect and toward the dura surface (Fig. 2C). As the defect continued to heal and new bone formation was initiated (2 wk postsurgery), strong Ddr2 expression continued to be observed throughout the defect with some LacZ+ cells on the surface of newly formed bone in the defect margin as well as in nonmineralized soft tissue in the defect center (Fig. 2D, arrowheads). After 4 and 6 wk postsurgery (wps), positive cells were restricted to a few cells on the bone surface and in soft tissue areas (Fig. 2E, F). Staining was notably absent from osteocytes (*). Note that unlike WT mice in which the SSD had totally healed after 4 to 6 wk, healing was still incomplete in Ddr2+/lacZ mice at these later times probably due to Ddr2 haploinsufficiency. Also note that no X-gal staining was seen in calvarial defects from WT littermates, confirming the specificity of the X-gal staining (Fig. 2G). From these results, we conclude that Ddr2 expression is low at early times after injury but greatly expands in soft tissue regions after 1 and 2 wk and then becomes preferentially associated with bone surfaces as regeneration proceeds.
Figure 2.

Localization of Ddr2 expression during calvarial bone regeneration. X-gal staining in frozen sections of Ddr2+/LacZ mouse calvaria at the indicated times from 3 d to 6 wk postsurgery (dps, wps). (A) No defect, (B) 3 dps, (C) 1 wps, (D) 2 wps, (E) 4 wps, and (F) 6 wps. Arrowheads indicate positive cells near the forming bone surface. Staining is notably absent from osteocytes (*). (G) No X-gal staining seen in Ddr2+/+ control at 1 wk. For each sample, the boxed area in the left panel is magnified in the right panel. To better visualize tissue morphology, the far-right panel shows hematoxylin and eosin–stained sections adjacent to X-gal–stained sections. Scale bars: 25 µm.
Ddr2 Deficiency Decreases Cell Proliferation and Early Markers of Osteoblast Differentiation
To investigate the initial cellular response to injury, we analyzed cell proliferation in the calvarial SSD region using EdU incorporation analysis. Three days postsurgery, Ddr2slie/slie and WT mice were injected with EdU and sacrificed 4 h later. As shown in Figure 3A, DNA synthesis was reduced in defects from Ddr2slie/slie mice by approximately 65%. To determine if Ddr2 status affected osteoblast differentiation, we measured 2 early markers by immunofluorescence in 1-wk postsurgery SSDs. Osterix/Sp7 (OSX) and RUNX2 are both transcription factors necessary for induction of osteoblast gene expression and subsequent formation of bone-forming cells (Ducy et al. 1997; Nakashima et al. 2002).
Figure 3.
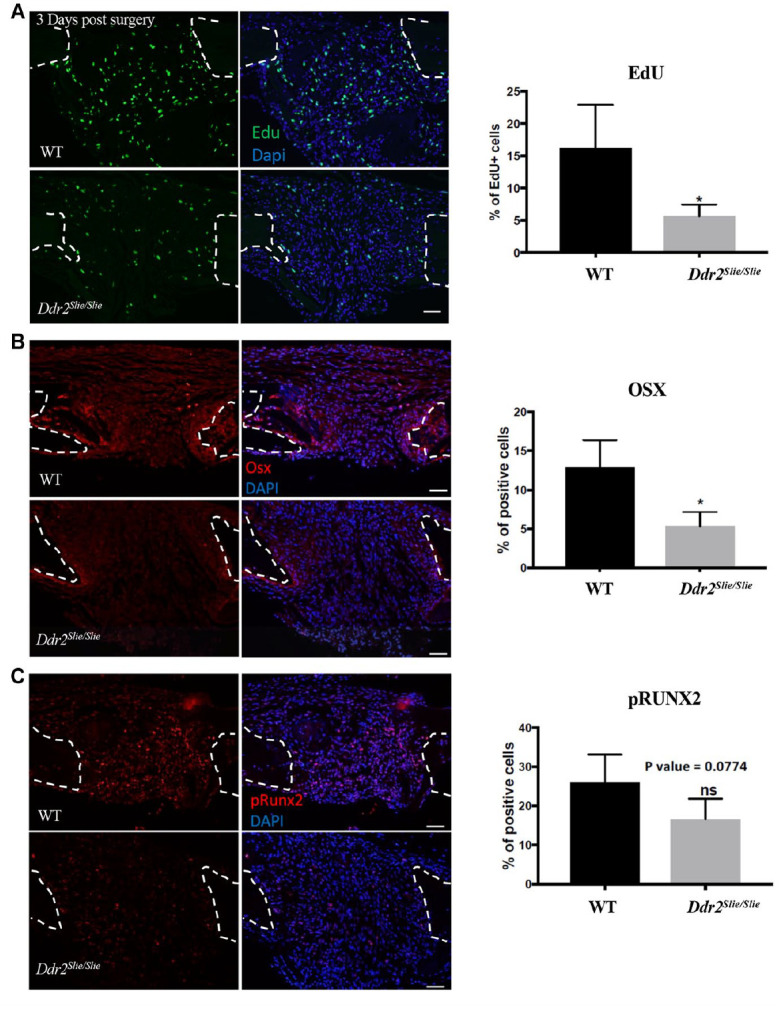
Ddr2 deficiency results in decreased cell proliferation and osteogenic marker protein levels during subcritical size defect (SSD) regeneration. (A) Paraffin sections of calvarial defects from wild-type (WT) (top) and Ddr2slie/slie mice (bottom) at 3 d postsurgery showing EdU+ cells in green (left panel) and merged with nuclear stain (middle panel). Quantification of percent EdU+ cells is shown at right. (B, C) Immunofluorescence (IF) staining for OSX (B) and p-RUNX2 (C) at 1 wk postsurgery. IF, red; DAPI, blue. Quantitation of percentage positive cells is shown at right. Dashed line, margins of bone defect. *P < 0.05. n = 4.
As shown by immunofluorescence analysis, OSX was significantly decreased in Ddr2-deficient mice (Fig. 3B). In addition, S319 phosphorylation of RUNX2, which is associated with increased transcriptional activity and bone formation (Ge et al. 2009, 2012), also trended toward a decrease in Ddr2-deficient mice (P = 0.074; Fig. 3C). Together, these results suggest that DDR2 is required for in vivo proliferation of progenitor cells and subsequent induction of osteoblast differentiation during cranial regeneration.
Effect of Ddr2 Deficiency on Osteoclasts
Overexpression of Ddr2 has been reported to suppress bone resorption markers in vivo and osteoclastogenesis in cell culture (Zhang et al. 2015). Also, Ddr2-deficient mice exhibit increase alveolar bone loss and osteoclast surface adjacent to the dentition (Mohamed et al. 2020). To determine if Ddr2 deficiency affected osteoclasts during calvarial defect regeneration, tartrate-resistant acid phosphatase (TRAP) staining was performed on sections from WT and Ddr2-deficient mice at 6 wk postsurgery when bone remodeling would be expected to occur. TRAP-positive cells were detected in association with regenerated bone in both groups (delineated by dashed lines), but the number of osteoclasts/defects was actually lower in Ddr2slie/slie mice versus controls (Appendix Fig. 1). Thus, the reduction in defect healing in Ddr2-deficient mice cannot be explained by increased osteoclastic resorption.
Ddr2 Regulates Differentiation of Skeletal Progenitors
Ddr2 is expressed in mesenchyme-derived cells, including osteoblasts and chondrocytes. It is also present in marrow stromal cells known to have skeletal progenitor activity (Zhang et al. 2011; Leitinger 2014; Ge et al. 2016). As shown in Figure 2, Ddr2 is also induced in select cells within calvarial defects at early times after injury and continues to be expressed throughout the healing process. Although the identity of these Ddr2-expressing cells is not known, it is likely that some of them have skeletal progenitor activity since Ddr2 knockout reduced osteoblast markers within the defect while also blocking regeneration (also see Discussion). Two cell culture approaches were taken to determine if Ddr2 can regulate skeletal progenitor activity. In the first, lentivirus-mediated Ddr2 overexpression and knockdown were used to evaluate Ddr2 activity in the ST2 mesenchymal cell line (Fig. 4). In this case, stable transduction with a Ddr2-expressing lentivirus (Ddr2(OE)) increased DDR2 protein levels approximately 2-fold compared to the control group (Fig. 4B) and also increased osteoblast differentiation as measured by Alizarin Red staining and induction of the osteoblast marker messenger RNAs (mRNAs) encoding Bglap2 and Ibsp (Fig. 4A, C, D). In contrast, lentivirus-expressing Ddr2 short hairpin RNA (shRNA) decreased DDR2 protein by approximately 60% compared with the scrambled shRNA control and decreased differentiation/mineralization. DDR2 knockdown also decreased cell proliferation while overexpression was stimulatory (Appendix Fig. 2).
Figure 4.

DDR2 regulates ST2 mesenchymal cell differentiation. (A) Alizarin red staining (top) and quantification (bottom) for ST2 cells transduced with lentivirus-expressing green fluorescent protein (GFP), wild-type Ddr2 (Ddr2(OE)), scrambled short hairpin RNA (shRNA), or Ddr2-specific shRNA (shDdr2). (B) Western blot and quantification of DDR2 protein levels. (C, D) Real-time quantitative reverse transcription polymerase chain reaction detection of osteoblast differentiation markers bone gamma carboxyglutamate protein 2 (Bglap2, C) and bone sialoprotein (Ibsp, D). *P < 0.05. **P < 0.01. n = 3.
In a second approach, primary calvarial osteoblast cultures (COB cells) were established from 3-mo-old Ddr2flox/flox mice using sequential digestion. These cells, which are largely derived from the periosteum and sutures, are a major source of skeletal progenitors that participate in the regeneration of calvarial defects (Behr et al. 2010; Quarto et al. 2010). As shown in Figure 2A, Ddr2-expressing cells are present in the periosteum before surgical injury, and these cells or their progeny likely contribute to calvarial healing. After reaching confluence, Ddr2flox/flox calvarial cells were transduced with control (adLacZ) or Cre-expressing adenovirus (adCre), and osteoblast differentiation was then measured over 14 d (Fig. 5). As previously reported (Ge et al. 2016), Ddr2 mRNA increased over time in control cultures. In contrast, Adeno-Cre treatment severely reduced Ddr2 expression at all time points examined (e.g., 90% reduction at day 14; Fig. 5B). Ddr2 knockout also dramatically blocked osteoblast differentiation as assessed by inhibition of mineralization (Fig. 5A) and expression of osteoblast marker mRNAs (Fig. 5B). Taken together, these studies show that Ddr2 is necessary for osteoblast differentiation of skeletal progenitors, including calvarial progenitors.
Figure 5.

Ddr2 knockout blocks osteoblast differentiation of calvarial cells. Primary cell cultures were established from 8-wk-old Ddr2flox/flox mice. After treatment with a control (adLacZ) or Cre adenovirus (adCre), cells grown in osteogenesis medium for up to 14 d. (A) Alizarin red staining after growth in osteogenic medium for 14 d. (B) Real-time quantitative reverse transcription polymerase chain reaction detection of Ddr2, Runx2, bone gamma carboxyglutamate protein 2 (Bglap2), and integrin binding sialoprotein (Ibsp). *P < 0.05. n = 3.
Discussion
This study shows for the first time that the nonintegrin collagen receptor, DDR2, is required for cranial bone regeneration and further emphasizes the importance of ECM receptors as mediators of regenerative events. In WT mice, the subcritical size 0.5-mm calvarial defect used in our studies showed initial bridging with new bone after as little as 2 wk and complete healing without intervention after approximately 4 to 6 wk (Fig. 1F). In Ddr2-deficient mice, a small amount of new bone initially accumulated at the margins of the defect for up to 6 wk, but no further significant increase in bone volume occurred between 6 and 12 wk, and no bridging of the defect was ever observed. This is similar to what is seen in critical-size defects where the size of the injury (normally 5 mm in mouse skull) is too large to heal in the absence of added osteogenic factors or bone progenitor cells (Zhao et al. 2007). Thus, bone regeneration is severely compromised in the absence of DDR2.
The identity, origin, and fate of Ddr2-positive cells associated with cranial defect healing are not currently known. Ddr2 expression, measured in Ddr2+/LacZ mice, was quite low at early times after injury (3 d) but dramatically increased after 1 wk when it was mainly located in the center of the defect. Positive cells then expanded throughout the defect with some being associated with growing bone surfaces at later times (Fig. 2). Since most cells initially filling the defect were Ddr2 negative, the positive cells detected at later times were either derived from this initial infiltrate or migrated into the defect (possibly from the periosteum or suture). In previous studies, Ddr2 was detected in skeletal progenitor cells from marrow and calvaria, where it was necessary for osteoblast differentiation (Zhang et al. 2011; Ge et al. 2016). It is not currently known if the Ddr2-positive cells in calvarial defects are also skeletal progenitors. Molecular characterization of Ddr2-expressing cells and lineage tracing before and at early times postinjury will be required to define this cell population and determine if it contains osteoprogenitors involved in calvarial defect healing.
It has been reported that skeletal progenitor cells involved in cranial repair mainly reside in the suture mesenchyme, periosteum, and dura (Doro et al. 2017). The suture mesenchyme is of particular interest since this anatomical region contains unique cell populations having stem cell properties. In particular, cells expressing Gli1, Axin2, or Prx1 genes have all been reported to have stem cell properties and participate in calvarial defect repair (Zhao et al. 2015; Maruyama et al. 2016; Wilk et al. 2017). Interestingly, we also detected Ddr2 expression in calvarial sutures and found that at least some Ddr2-positive cells also express Gli1 (studies to be reported elsewhere). However, the role of Ddr2 in Gli1-positive cells during cranial regeneration is not currently known.
The basis for the defective cranial defect healing in Ddr2 deficiency may be related to defects in both the proliferation and differentiation of cells that occupy the defect after injury. By 3 d postinjury, it was already clear that Ddr2-deficient cells did not proliferate as well as WT cells. By 1 wk, they also exhibited defects in early osteoblast differentiation, as measured by the reduced ability to express the osteogenic transcription factor OSX and activate (phosphorylate) RUNX2 (Fig. 3). Although it is still not resolved if these in vivo effects of Ddr2 deficiency represent cell-autonomous actions of Ddr2 on a specific cell population, we presented evidence that this receptor has direct actions on a mesenchymal stem cell line (ST2 cells) and primary calvarial cells. Ddr2 shRNA knockdown in ST2 cells reduced proliferation and suppressed osteoblast differentiation while Ddr2 overexpression stimulated proliferation and differentiation (Fig. 4, Appendix Fig. 2). In primary calvarial cell cultures that are known to be enriched in skeletal progenitors, adCre-mediated knockout of Ddr2 in cells from from Ddr2loxp/loxp mice also strongly inhibited osteoblast differentiation (Fig. 5). These findings are similar to previous results from our group and others where bone marrow stromal cells and calvarial cells from globally Ddr2-deficient mice exhibited reduced osteoblast differentiation (Zhang et al. 2011; Ge et al. 2016). However, studies where Ddr2 is selectively inactivated in cranial stem cells will be required to prove Ddr2 functions in this cell population during in vivo regeneration.
The mechanism(s) used by DDR2 to alter cell function during regeneration are also not well understood. After exposure of osteoprogenitor cells to fibrillar collagens, DDR2 undergoes autophosphorylation with a characteristic slow and sustained kinetics, leading to activation of ERK1/2 MAP kinases. These, in turn, phosphorylate and activate the osteogenic transcription factor, RUNX2, which stimulates osteoblast differentiation (Zhang et al. 2011; Ge et al. 2016). This may at least in part explain the defective calvarial bone regeneration seen in Ddr2-deficient mice where defect-associated RUNX2 phosphorylation was reduced (Fig. 3). However, DDR2 is also known to have other important actions, including effects on collagen fibrillogenesis, metalloproteinase activation, and ECM remodeling, all of which could be important for regeneration (Blissett et al. 2009; Flynn et al. 2010; Itoh 2018).
In summary, our studies show that DDR2 is necessary for calvarial bone regeneration by regulating osteoblast proliferation and differentiation. Based on our studies, it may be speculated that DDR2 is a possible target for stimulating bone formation and regeneration and that approaches to either activate DDR2 or enhance its expression could be the basis for future therapies.
Author Contributions
A. Binrayes, C. Ge, contributed to conception, design, data acquisition, analysis, and interpretation, drafted and critically revised the manuscript; F.F. Mohamed, contributed to design, data acquisition, analysis, and interpretation, critically revised the manuscript; R.T. Franceschi, contributed to conception, design, data analysis, and interpretation, drafted and critically revised the manuscript. All authors gave final approval and agree to be accountable for all aspects of the work.
Supplemental Material
Supplemental material, sj-pdf-1-jdr-10.1177_00220345211007447 for Role of Discoidin Domain Receptor 2 in Craniofacial Bone Regeneration by A. Binrayes, C. Ge, F.F. Mohamed and R.T. Franceschi in Journal of Dental Research
Footnotes
A supplemental appendix to this article is available online.
Declaration of Conflicting Interests: The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by National Institute of Dental and Craniofacial Research (NIDCR) grants DE11723 and DE29012, Department of Defense Contract W81XWH2010571 (to R.T. Franceschi), the Michigan Musculoskeletal Health Core Center (National Institutes of Health/NIAMS P30 AR069620) and NIDCR Career Development Award K12 DE023574 (C. Ge; PI: S. Kapila), and scholarships from King Saud University (A. Binreyes) and the Ministry of Higher Education and Scientific Research, Libyan Transitional Government (F.F. Mohamed).
References
- Bargal R, Cormier-Daire V, Ben-Neriah Z, Le Merrer M, Sosna J, Melki J, Zangen DH, Smithson SF, Borochowitz Z, Belostotsky R, et al. 2009. Mutations in DDR2 gene cause SMED with short limbs and abnormal calcifications. Am J Hum Genet. 84(1):80–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayer SV, Grither WR, Brenot A, Hwang PY, Barcus CE, Ernst M, Pence P, Walter C, Pathak A, Longmore GD. 2019. DDR2 controls breast tumor stiffness and metastasis by regulating integrin mediated mechanotransduction in CAFs. Elife. 8:e45508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behr B, Panetta NJ, Longaker MT, Quarto N. 2010. Different endogenous threshold levels of fibroblast growth factor-ligands determine the healing potential of frontal and parietal bones. Bone. 47(2):281–294. [DOI] [PubMed] [Google Scholar]
- Blissett AR, Garbellini D, Calomeni EP, Mihai C, Elton TS, Agarwal G. 2009. Regulation of collagen fibrillogenesis by cell-surface expression of kinase dead DDR2. J Mol Biol. 385(3):902–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bostrom MP, Saleh KJ, Einhorn TA. 1999. Osteoinductive growth factors in preclinical fracture and long bone defects models. Orthop Clin North Am. 30(4):647–658. [DOI] [PubMed] [Google Scholar]
- Brunner M, Jurdic P, Tuckerman JP, Block MR, Bouvard D. 2013. New insights into adhesion signaling in bone formation. Int Rev Cell Mol Biol. 305:1–68. [DOI] [PubMed] [Google Scholar]
- Clark AY, Martin KE, Garcia JR, Johnson CT, Theriault HS, Han WM, Zhou DW, Botchwey EA, Garcia AJ. 2020. Integrin-specific hydrogels modulate transplanted human bone marrow–derived mesenchymal stem cell survival, engraftment, and reparative activities. Nat Commun. 11(1):114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowan CM, Shi YY, Aalami OO, Chou YF, Mari C, Thomas R, Quarto N, Contag CH, Wu B, Longaker MT. 2004. Adipose-derived adult stromal cells heal critical-size mouse calvarial defects. Nat Biotechnol. 22(5):560–567. [DOI] [PubMed] [Google Scholar]
- Doro DH, Grigoriadis AE, Liu KJ. 2017. Calvarial suture-derived stem cells and their contribution to cranial bone repair. Front Physiol. 8:956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducy P, Zhang R, Geoffroy V, Ridall AL, Karsenty G. 1997. Osf2/cbfa1: a transcriptional activator of osteoblast differentiation. Cell. 89(5):747–754. [DOI] [PubMed] [Google Scholar]
- Ekholm E, Hankenson KD, Uusitalo H, Hiltunen A, Gardner H, Heino J, Penttinen R. 2002. Diminished callus size and cartilage synthesis in alpha 1 beta 1 integrin-deficient mice during bone fracture healing. Am J Pathol. 160(5):1779–1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn LA, Blissett AR, Calomeni EP, Agarwal G. 2010. Inhibition of collagen fibrillogenesis by cells expressing soluble extracellular domains of DDR1 and DDR2. J Mol Biol. 395(3):533–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge C, Mohamed F, Binrayes A, Kapila S, Franceschi RT. 2018. Selective role of discoidin domain receptor 2 in murine temporomandibular joint development and aging. J Dent Res. 97(3):321–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge C, Wang Z, Zhao G, Li B, Liao J, Sun H, Franceschi RT. 2016. Discoidin receptor 2 controls bone formation and marrow adipogenesis. J Bone Miner Res. 31(12):2193–2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge C, Xiao G, Jiang D, Yang Q, Hatch NE, Roca H, Franceschi RT. 2009. Identification and functional characterization of ERK/MAPK phosphorylation sites in the Runx2 transcription factor. J Biol Chem. 284(47):32533–32543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge C, Yang Q, Zhao G, Yu H, Kirkwood KL, Franceschi RT. 2012. Interactions between extracellular signal-regulated kinase 1/2 and P38 Map kinase pathways in the control of RUNX2 phosphorylation and transcriptional activity. J Bone Miner Res. 27(3):538–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh Y. 2018. Discoidin domain receptors: microenvironment sensors that promote cellular migration and invasion. Cell Adh Migr. 12(4):378–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kano K, Marin de, Evsikova C, Young J, Wnek C, Maddatu TP, Nishina PM, Naggert JK. 2008. A novel dwarfism with gonadal dysfunction due to loss-of-function allele of the collagen receptor gene, Ddr2, in the mouse. Mol Endocrinol. 22(8):1866–1880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JB, Leucht P, Luppen CA, Park YJ, Beggs HE, Damsky CH, Helms JA. 2007. Reconciling the roles of fak in osteoblast differentiation, osteoclast remodeling, and bone regeneration. Bone. 41(1):39–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legate KR, Wickstrom SA, Fassler R. 2009. Genetic and cell biological analysis of integrin outside-in signaling. Genes Dev. 23(4):397–418. [DOI] [PubMed] [Google Scholar]
- Leitinger B. 2014. Discoidin domain receptor functions in physiological and pathological conditions. Int Rev Cell Mol Biol. 310:39–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruyama T, Jeong J, Sheu TJ, Hsu W. 2016. Stem cells of the suture mesenchyme in craniofacial bone development, repair and regeneration. Nat Commun. 7:10526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohamed FF, Ge C, Binrayes A, Franceschi RT. 2020. The role of discoidin domain receptor 2 in tooth development. J Dent Res. 99(2):214–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakashima K, Zhou X, Kunkel G, Zhang Z, Deng JM, Behringer RR, de Crombrugghe B. 2002. The novel zinc finger-containing transcription factor osterix is required for osteoblast differentiation and bone formation. Cell. 108(1):17–29. [DOI] [PubMed] [Google Scholar]
- Park S, Zhao H, Urata M, Chai Y. 2016. Sutures possess strong regenerative capacity for calvarial bone injury. Stem Cells Dev. 25(23):1801–1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quarto N, Wan DC, Kwan MD, Panetta NJ, Li S, Longaker MT. 2010. Origin matters: differences in embryonic tissue origin and Wnt signaling determine the osteogenic potential and healing capacity of frontal and parietal calvarial bones. J Bone Miner Res. 25(7):1680–1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes CD, Garcia AJ. 2004. Alpha2beta1 integrin-specific collagen-mimetic surfaces supporting osteoblastic differentiation. J Biomed Mater Res A. 69(4):591–600. [DOI] [PubMed] [Google Scholar]
- Shekaran A, Shoemaker JT, Kavanaugh TE, Lin AS, LaPlaca MC, Fan Y, Guldberg RE, Garcia AJ. 2014. The effect of conditional inactivation of beta 1 integrins using twist 2 Cre, Osterix Cre and osteocalcin Cre lines on skeletal phenotype. Bone. 68:131–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrivastava A, Radziejewski C, Campbell E, Kovac L, McGlynn M, Ryan TE, Davis S, Goldfarb MP, Glass DJ, Lemke G, et al. 1997. An orphan receptor tyrosine kinase family whose members serve as nonintegrin collagen receptors. Mol Cell. 1(1):25–34. [DOI] [PubMed] [Google Scholar]
- Valiathan RR, Marco M, Leitinger B, Kleer CG, Fridman R. 2012. Discoidin domain receptor tyrosine kinases: new players in cancer progression. Cancer Metastasis Rev. 31(1–2):295–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel W, Gish GD, Alves F, Pawson T. 1997. The discoidin domain receptor tyrosine kinases are activated by collagen. Mol Cell. 1(1):13–23. [DOI] [PubMed] [Google Scholar]
- Wilk K, Yeh SA, Mortensen LJ, Ghaffarigarakani S, Lombardo CM, Bassir SH, Aldawood ZA, Lin CP, Intini G. 2017. Postnatal calvarial skeletal stem cells expressing PRX1 reside exclusively in the calvarial sutures and are required for bone regeneration. Stem Cell Reports. 8(4):933–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Su J, Wu S, Teng Y, Yin Z, Guo Y, Li J, Li K, Yao L, Li X. 2015. DDR2 (discoidin domain receptor 2) suppresses osteoclastogenesis and is a potential therapeutic target in osteoporosis. Sci Signal. 8(369):ra31. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Su J, Yu J, Bu X, Ren T, Liu X, Yao L. 2011. An essential role of discoidin domain receptor 2 (DDR2) in osteoblast differentiation and chondrocyte maturation via modulation of Runx2 activation. J Bone Miner Res. 26(3):604–617. [DOI] [PubMed] [Google Scholar]
- Zhao H, Feng J, Ho TV, Grimes W, Urata M, Chai Y. 2015. The suture provides a niche for mesenchymal stem cells of craniofacial bones. Nat Cell Biol. 17(4):386–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z, Wang Z, Ge C, Krebsbach P, Franceschi RT. 2007. Healing cranial defects with AdRunx2-transduced marrow stromal cells. J Dent Res. 86(12):1207–1211. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material, sj-pdf-1-jdr-10.1177_00220345211007447 for Role of Discoidin Domain Receptor 2 in Craniofacial Bone Regeneration by A. Binrayes, C. Ge, F.F. Mohamed and R.T. Franceschi in Journal of Dental Research