

Abstract
Vitamin A and its derivative retinol are essential for the development of intestinal adaptive immunity. Retinoic acid (RA)-producing myeloid cells are central to this process, but how myeloid cells acquire retinol for conversion to RA is unknown. Here, we show that serum amyloid A (SAA) proteins — retinol-binding proteins induced in intestinal epithelial cells by the microbiota — deliver retinol to myeloid cells. We identify LDL receptor-related protein 1 (LRP1) as an SAA receptor that endocytoses SAA–retinol complexes and promotes retinol acquisition by RA-producing intestinal myeloid cells. Consequently, SAA and LRP1 are essential for vitamin A-dependent immunity, including B and T cell homing to the intestine and immunoglobulin A production. Our findings thus identify a key mechanism by which vitamin A promotes intestinal immunity.
One sentence summary:
Serum amyloid A delivers retinol to retinoic acid-producing myeloid cells through the endocytic receptor LRP1, thus promoting gut immunity.
Vitamin A is a lipid-soluble nutrient that is absorbed from the diet by intestinal epithelial cells and converted to retinol. Retinol is essential for intestinal adaptive immunity by directing the development of intestinal B and T cells and promoting their recruitment to the intestine (1, 2). Consequently, vitamin A deficiency confers an increased susceptibility to infectious diseases, including those of the intestine (3).
Myeloid cells are central to the development of vitamin A-dependent intestinal immunity. Myeloid cells include dendritic cells (DCs) and macrophages, which circulate in the intestine and present antigens to naïve B and T cells to promote their differentiation into functional immune cells (4, 5). Certain intestinal DCs and macrophages can enzymatically convert retinol to RA (6–10). These myeloid cells pass their RA to T cells where it complexes with retinoic acid receptors (RARs), transcription factors that activate RA-dependent gene expression programs (11, 12). These programs include the expression of homing receptors, such as CCR9 and integrin α4β7, which direct T cells to the intestine (1, 13). Similarly, RA-producing myeloid cells imprint homing receptors on developing B cells and induce the production of immunoglobulin A (IgA) (2).
A major unanswered question is how intestinal myeloid cells acquire retinol for conversion to RA. Retinol is lipophilic, necessitating its transport by retinol-binding proteins that shield it from the aqueous environment. However, proteins that deliver retinol to myeloid cells remain unidentified.
Serum amyloid A (SAA) proteins are a family of retinol-binding proteins expressed in the intestinal epithelium and liver (14, 15). SAAs form a trimeric assembly with a central hydrophobic retinol binding pocket (14, 15). Epithelial expression of SAAs is induced by the intestinal microbiota through a signaling circuit involving DCs, innate lymphoid cells, and epithelial STAT3 (16), and by dietary vitamin A through epithelial RARβ (17). SAAs are also produced in the liver following acute bacterial infection and circulate in the serum with bound retinol (14, 15). Thus, SAAs are retinol binding proteins that transport retinol in response to microbial signals, including the microbiota and acute systemic infection. However, the cellular targets of SAA–retinol complexes are unclear.
Here, we identify a molecular basis for retinol acquisition by intestinal myeloid cells. We show that SAAs deliver retinol to RA-producing myeloid cells in the intestine and that low density lipoprotein (LDL) receptor-related protein 1 (LRP1) mediates myeloid cell uptake of SAA–retinol complexes. Together, LRP1 and SAA facilitate myeloid cell retinol acquisition and are essential for the development of vitamin A-dependent adaptive immunity in the intestine.
Identification of LDL receptor-related protein 1 (LRP1) as a cell surface receptor for SAA proteins
As a first step in understanding how SAAs deliver retinol to target cells, we identified a cell surface receptor for SAA. We first screened cultured cell lines for evidence of receptor-mediated cell surface binding of SAAs. We used SAA1 for these studies, since Saa1 expression is highest among the four Saa isotypes in the intestine (fig. S1A). Differentiated 3T3-L1 mouse adipocytes, which are a major extrahepatic retinol storage site (18), bound SAA1 on their cell surface (fig. S1, B and C). Furthermore, the binding was saturable (fig. S1D), suggesting a specific receptor-mediated interaction. Finally, adipocytes acquired retinol more readily from SAA1–retinol complexes than from free retinol (fig. S1E), suggesting that SAAs deliver retinol to receptor-bearing target cells.
To identify the adipocyte SAA receptor(s), we used the 6X histidine tag (His-tag)–nickel resin system and a bifunctional cross-linker, allowing stabilization of the SAA–receptor interaction and purification of the complex (Fig. 1A). Recombinant His-tagged SAA1 was conjugated with the cross-linker, complexed with retinol, and added to adipocytes. After ultraviolet cross-linking, membrane solubilization, and nickel resin purification, we identified a >250-kD covalent protein complex that contained SAA1 (Fig. 1B). Mass spectrometry identified the other major protein as the transmembrane receptor LRP1 (table S1).
Fig. 1. LRP1 is a cell surface receptor for SAAs.
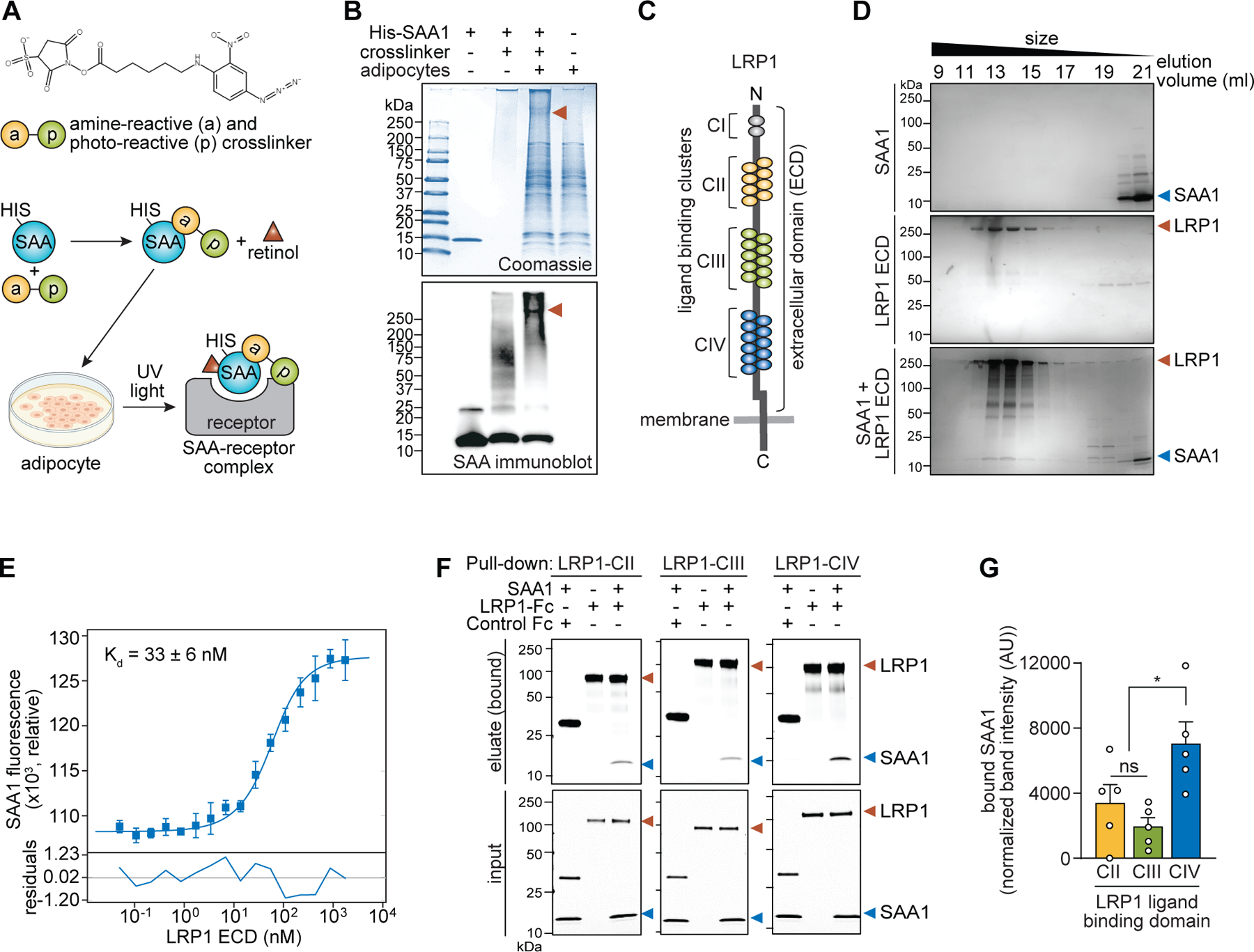
(A) Schematic diagram of the strategy used to identify the SAA receptor. The structure of the amine-reactive and photo-reactive cross-linker is shown at top. The SAA–receptor complex was conjugated by the cross-linker as shown in the diagram and then purified and further analyzed. The cell culture image was generated in BioRender. (B) Protein complexes were resolved by SDS-PAGE and detected with Coomassie blue (top) or immunoblot for SAA (bottom). Red arrowheads indicate the protein complex that was subjected to mass spectrometry for receptor identification. (C) Schematic of the domain organization of LRP1 showing the ectodomain (ECD) and ligand binding clusters (CI-CIV). (D) Formation of a complex between SAA1 and the LRP1 ectodomain (LRP1-ECD) was confirmed by size-exclusion chromatography. (E) Binding affinity between SAA1 and the LRP1-ECD was determined using microscale thermophoresis. Specific LRP1-dependent enhancement of fluorescence of SAA1 was used for the determination of a dissociation constant (Kd). Residuals between the fit and the data are depicted in the bottom panel. Data represent means±SEM from three independent assays. (F and G) Binding of SAA1 to the LRP1 ligand binding clusters (LRP1-CII, LRP1-CIII, and LRP1-CIV) was assessed by pull-down assay. Each Fc-tagged LRP1 protein and Fc control was incubated with SAA1, and Protein G magnetic beads were used to capture the Fc-tagged proteins (top). (G) Bound SAA1 in (F) was quantified by scanning densitometry and normalized to SAA1 bound to control Fc. Results from five independent pull-down assays are shown. In all experiments, SAA1 was pre-incubated with retinol (3:1 retinol:SAA1 molar ratio) at 4°C for 2 hours to allow SAA1–retinol complex formation. *P<0.05 by paired Student’s t test.
LRP1 is a member of the LDL receptor family that binds several distinct ligands, including lipoproteins, and facilitates their cellular uptake through endocytosis (19). It is composed of two non-covalently associated fragments: a long N-terminal extracellular α-fragment containing ligand-binding regions and a short membrane-anchored C-terminal β-fragment (Fig. 1C). To test whether LRP1 directly binds to SAA1, we expressed and purified the full-length ectodomain of human LRP1 (LRP1-ECD). SAA1and LRP1-ECD eluted together during size-exclusion chromatography (Fig. 1D), indicating a direct interaction. We confirmed binding by microscale thermophoresis using a fluorescently-labeled SAA1 and LRP1-ECD. We observed LRP1-dependent enhancement of SAA1 fluorescence and calculated a dissociation constant (Kd) of ~33 nM (Fig. 1E). This Kd is consistent with those of other known LRP1 ligands (20, 21) and indicates high-affinity binding of SAA1 to LRP1.
We next determined which LRP1 domain(s) bind to SAA1. The ectodomain of LRP1 encompasses four ligand binding clusters containing cysteine-rich repeats: CI to CIV (Fig. 1C). Among these, CII, CIII, and CIV directly bind ligands (19). We therefore tested SAA1 binding to these three clusters. Pull-down assays revealed that SAA1 preferentially bound to LRP1-CIV (Fig. 1F and G). We then confirmed that mouse SAA3 also binds to LRP1-CIV (fig. S2). Thus, LRP1 directly binds SAAs with high affinity.
LRP1 mediates cell surface binding and cellular uptake of SAA1–retinol complexes
To determine if LRP1 functions as a SAA receptor that promotes cellular retinol uptake, we examined wild-type (Lrp1+/+) and Lrp1-deficient (Lrp1−/−) fibroblasts. We first assessed whether SAA binding to the cell surface was LRP1-dependent. His-SAA1–retinol complexes were incubated with Lrp1+/+ and Lrp1−/− cell cultures at 4°C to allow binding of SAA1 to the cell surface while preventing endocytosis (22). SAA1 binding was then measured by flow cytometry. More SAA1 bound to Lrp1+/+ cells than Lrp1−/− cells (Fig. 2, A and B), indicating that LRP1 mediates cell surface binding of SAA1. LRP1-dependent binding of SAA1 to the cell surface was saturable (Fig. 2C) and could be blocked by SAA1 that lacked a His tag (Fig. 2D), arguing for a specific binding interaction. Finally, receptor-associated protein (RAP), a chaperone that binds to LRP1 in the endoplasmic reticulum (23, 24), competed for cell surface binding by SAA1 (Fig. 2E), providing further evidence of specificity. Thus, LRP1 is a specific receptor for SAA1.
Fig. 2. LRP1 mediates cell surface binding and cellular uptake of SAA1-retinol complexes.
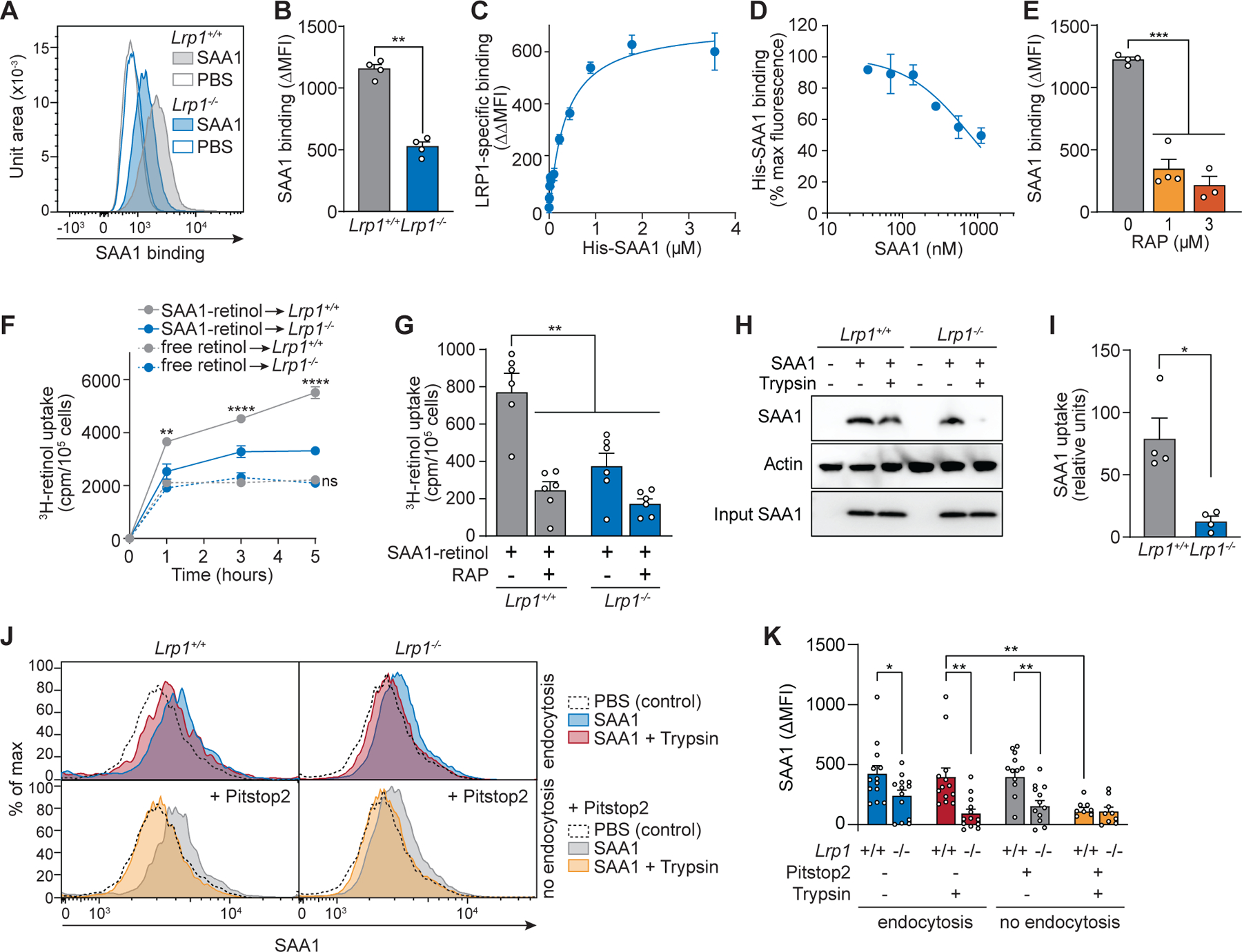
(A–E) Wild-type (Lrp1+/+) and Lrp1-deficient (Lrp1−/−) fibroblasts were incubated with His-SAA1-retinol complexes at 4°C, and the binding of SAA to the cell surface was analyzed by flow cytometry under non-permeabilizing staining conditions using PE-conjugated anti-His tag antibody. (A and B) His-SAA1 (1.8 μM) was added to the cells and representative flow cytometry histograms (A) and ΔMFI (MFISAA-MFIPBS) representing SAA1 binding to the cell surface (B) were calculated. PBS was added instead of SAA1 in control samples. (C) LRP1-specific SAA1 binding to the fibroblast cell surface was calculated and indicates saturable binding. LRP1-specific binding was calculated as the difference of SAA binding (ΔMFI) to Lrp1+/+ and Lrp1−/− fibroblasts (ΔΔMFI = ΔMFILrp1+/+-ΔMFILrp1−/−). (D) Competitive binding assays were conducted by adding to the cells increasing concentrations of non-His tagged SAA1 together with His-SAA1 (1.8 μM). LRP1-specific binding of His-SAA1 to the cell surface was calculated (ΔΔMFI) and is shown as a percentage of the maximum fluorescence. (E) Receptor-associated protein (RAP) was added to wild-type fibroblasts together with His-SAA1 (1 μM). Binding of SAA1 to the cell surface was measured by flow cytometry and is shown as ΔMFI (MFISAA-MFIPBS). (F and G) Cellular 3H-retinol uptake was measured. (F) 3H-retinol was added to the cells either as free retinol or in complex with SAA1. After incubation at 37°C, the counts per minute (cpm) in the cell pellets were determined and normalized to cell number. (G) RAP (3 μM) was added to cells together with 0.6 μM SAA1–3H-retinol complex for competition analyses. (H-K) Cellular SAA uptake was analyzed. Cells were incubated with His-SAA1-retinol complex at 37°C for 30 min and subsequently treated with trypsin for 3 min to remove surface-bound SAA1. (H and I) Cells were lysed and cellular SAA uptake was analyzed by immunoblotting using an anti-His antibody. SAA band intensity in the cell lysates after trypsin digestion was measured, normalized with loading controls, and is shown in relative units in (I). (J and K) LRP1 promotes SAA1 endocytosis. SAA associated with Lrp1+/+ and Lrp1−/− fibroblasts was measured by flow cytometry. Representative histograms are shown in (J) and ΔMFI (MFISAA-MFIPBS) representing SAA1 measurements from three independent experiments are shown (K). Cells were treated with trypsin to remove surface-bound SAA1, and Pitstop2 was added to block endocytosis. In all experiments, SAA1 was pre-incubated with retinol or 3H-retinol (3:1 retinol:SAA1 molar ratio) at 4°C for at least 2 hours to allow SAA-retinol complex formation. Means±SEM are shown from one (A–E) and at least two independent experiments (F–K). All results are representative of at least two independent experiments. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001 by Student’s t test.
Next, we tested whether LRP1 facilitates the delivery of SAA-bound retinol into cells. We added a 3H-retinol tracer to fibroblasts, either as free retinol or in complex with SAA1, and measured the cell-associated retinol by scintillation counting. More retinol was acquired by Lrp1+/+ cells from SAA1-retinol complexes than from free retinol. Furthermore, retinol uptake from SAA1–retinol complexes was reduced in Lrp1−/− cells (Fig. 2F). Addition of RAP as a competitive inhibitor of SAA1 binding to LRP1 reduced retinol uptake by Lrp1+/+ cells (Fig. 2G). Thus, LRP1 facilitates retinol uptake by binding to SAA1–retinol complexes.
LRP1 internalizes many of its known ligands by endocytosis (19, 25), suggesting that it may facilitate retinol uptake through endocytosis of the SAA1–retinol complex. To test this idea, we asked whether cell-associated SAA1 is degraded by added trypsin, which can digest cell surface but not internalized proteins (15). We added His-tagged SAA1–retinol complexes to Lrp1+/+ and Lrp1−/− fibroblasts at 4°C to allow cell surface binding, incubated at 37°C to initiate endocytosis, and then exposed the cells to trypsin. When assessed by immunoblot, SAA1 was mostly protected from trypsin proteolysis when added to Lrp1+/+ fibroblasts, whereas the SAA1 added to Lrp1−/− fibroblasts was mostly digested (Fig. 2, H and I). Similarly, flow cytometry revealed that most of the SAA1 associated with Lrp1+/+ fibroblasts was protected from trypsin digestion but became susceptible when the endocytosis inhibitor Pitstop2 was added to the cells (Fig. 2, J and K). By contrast, SAA1 added to Lrp1−/− cells was susceptible to trypsin proteolysis even in the absence of Pitstop2 (Fig. 2, J and K). Thus, LRP1 promotes the endocytosis of SAA1–retinol complexes.
LRP1 is expressed on intestinal CD11c+ myeloid cells
We next sought to identify intestinal cells that express LRP1 and thus might be targets of SAA–retinol complexes. We first compared expression of Saa1 transcripts to the expression of transcripts encoding retinol-binding protein 4 (RBP4), which delivers retinol from the liver to peripheral tissues under non-inflammatory conditions (26). Whereas Rpb4 transcripts were more abundant than Saa1 transcripts in the liver, Saa1 transcripts were more abundant in the small intestine (Fig. 3A). Likewise, SAA protein levels were higher than RBP4 levels in the small intestine (Fig. 3, B and C). At the same time, transcripts encoding the SAA receptor LRP1 were more abundant in the small intestine than transcripts encoding two known RBP4 receptors, stimulated by retinoic acid 6 (STRA6) (27) and RBP receptor 2 (RBPR2) (28) (Fig. 3D). Thus, SAA1 and its receptor LRP1 are more highly expressed in the intestine than RBP4 and its receptors, suggesting that SAAs might play a predominant role in mobilizing retinol in the intestine.
Fig. 3. LRP1 is expressed on intestinal CD11c+ myeloid cells.
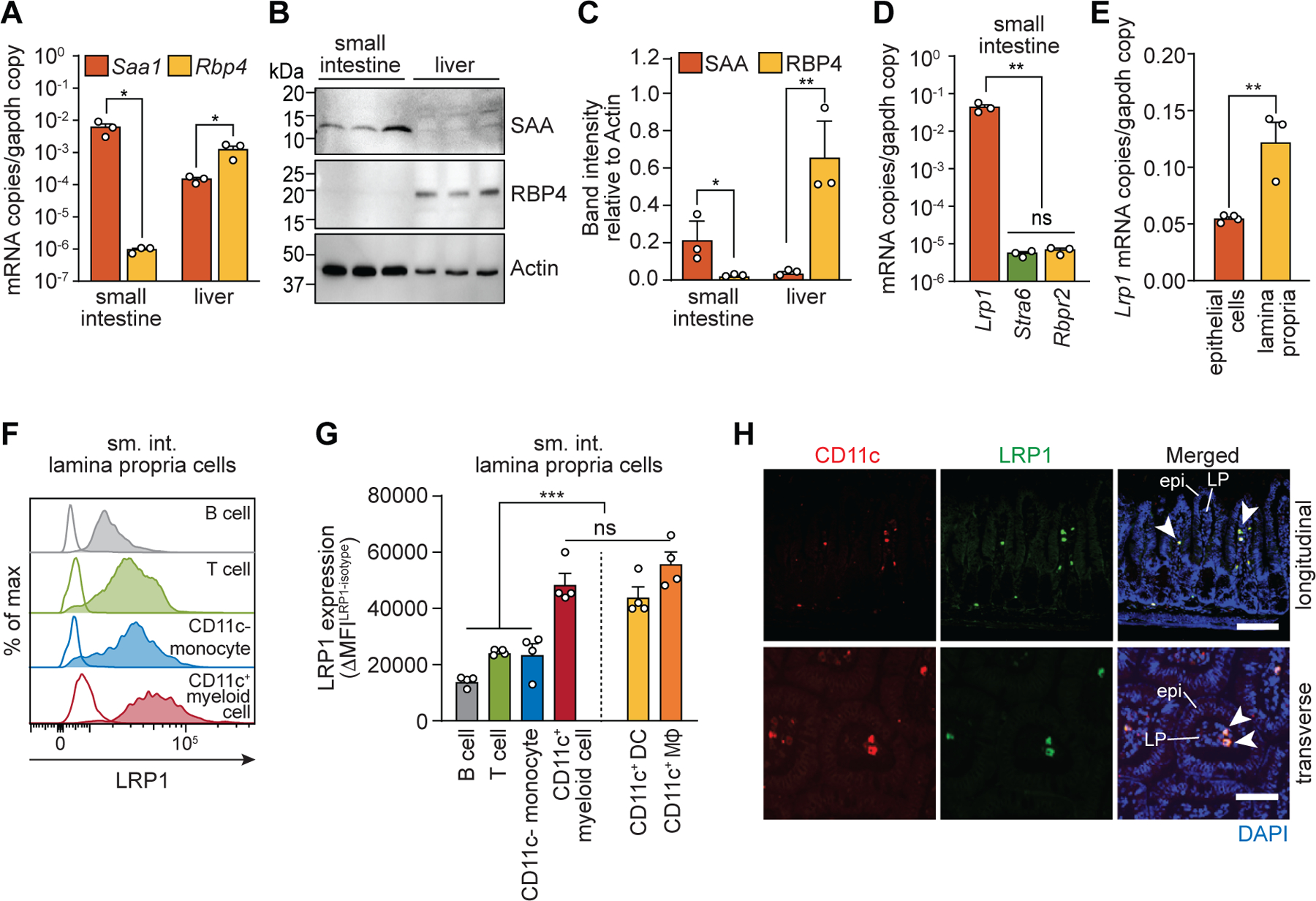
(A) qPCR analysis of Saa1 and Rbp4 transcripts in ileum and liver. (B and C) Immunoblotting of SAA, RBP4, and actin (loading control) in mouse ileum and liver. SAA and RPB4 band intensities were measured, normalized to actin, and are shown in relative units in (C). (D) qPCR analysis of Lrp1, Stra6, and Rbpr2 transcripts in mouse ileum. (E) qPCR analysis of Lrp1 transcripts in small intestinal epithelial cells and immune cells isolated from the small intestinal lamina propria. (F and G) Flow cytometry analysis of LRP1 expression on B cells, T cells, monocytes, and CD11c+ myeloid cells. (F) Representative histograms showing LRP1 expression. Empty histograms are from isotype controls. (G) LRP1 expression (ΔMFI) is shown as the difference in MFI between isotype-matched control and anti-LRP1 antibody staining (MFILRP1-MFIisotype). (H) Immunofluorescence detection of CD11c (red), LRP1 (green), and nuclei (blue) in the mouse small intestine. Longitudinal (top) and transverse (bottom) section of villi are shown. Examples of LRP1+ and CD11c+ cells are indicated with arrowheads. Epi, epithelium; LP, lamina propria. Scale bars indicate 100 μm (upper panel) and 50 μm (lower panel), respectively. Detection with isotype control antibody is shown in fig. S5.
sm. int., small intestine; MΦ, macrophage. At least three wild-type C57BL/6 mice were analyzed in each experiment. For qPCR, mRNA copy numbers were calculated from the standard and normalized to Gapdh copy numbers. Means±SEM are plotted. *P<0.05; **P<0.01; ***P<0.001; ns, not significant by Student’s t test.
To identify intestinal cells expressing LRP1, we measured LRP1 expression in various intestinal cell populations. Lrp1 transcripts were less abundant in epithelial cells than in lamina propria cells, which include various immune cells (Fig. 3E). To determine whether LRP1 is expressed on immune cells, we measured LRP1 expression on various lamina propria immune cell populations by flow cytometry (fig. S3 and S4). LRP1 was highest on CD11c+MHCII+ myeloid cells when compared to other immune cells, including B cells, T cells, and CD11c− monocytes. CD11c+MHCII+ myeloid cells include F4/80− DCs and F4/80+ macrophages, which both expressed LRP1 (Fig. 3, F and G). Immunofluorescence analysis of mouse small intestine confirmed LRP1 expression by intestinal lamina propria CD11c+ cells (Fig. 3H and fig. S5). When His-SAA1–retinol complexes were added to lamina propria immune cells, CD11c+ myeloid cells bound more SAA relative to other immune cell populations (fig. S6). Thus, CD11c+ myeloid cells express LRP1 and capture SAA–retinol complexes.
LRP1 promotes myeloid cell uptake of SAA-bound retinol
Next, we examined whether LRP1 facilitates SAA binding and retinol uptake in CD11c+ myeloid cells. We generated mice having a CD11c+ myeloid cell-specific deletion of the Lrp1 gene (Lrp1ΔCd11c mice) and investigated bone marrow-derived myeloid cells (BM-MCs). CD11c+ cells were isolated by magnetic sorting from cells grown in GM-CSF, yielding BM-MCs that included both DCs and macrophages (fig. S7, A and B) (29, 30). Bone marrow-derived DCs and macrophages isolated from Lrp1ΔCd11c mice showed lowered expression of LRP1 (fig. S7C) and supported less binding of His-SAA1–retinol than BM-MCs from Lrp1fl/fl mice (Fig. 4, A and B, and fig. S7, D and E). Likewise, 3H-retinol uptake from SAA1–3H-retinol complexes was lower in BM-MCs from Lrp1ΔCd11c mice than from Lrp1fl/fl mice (Fig. 4C). Furthermore, 3H-retinol uptake in Lrp1fl/fl BM-MCs was higher with the addition of SAA1–3H-retinol than with free 3H-retinol (Fig. 4C). Thus, LRP1 facilitates myeloid cell retinol uptake from SAA1–retinol complexes.
Fig. 4. LRP1 promotes myeloid cell uptake of SAA-bound retinol.
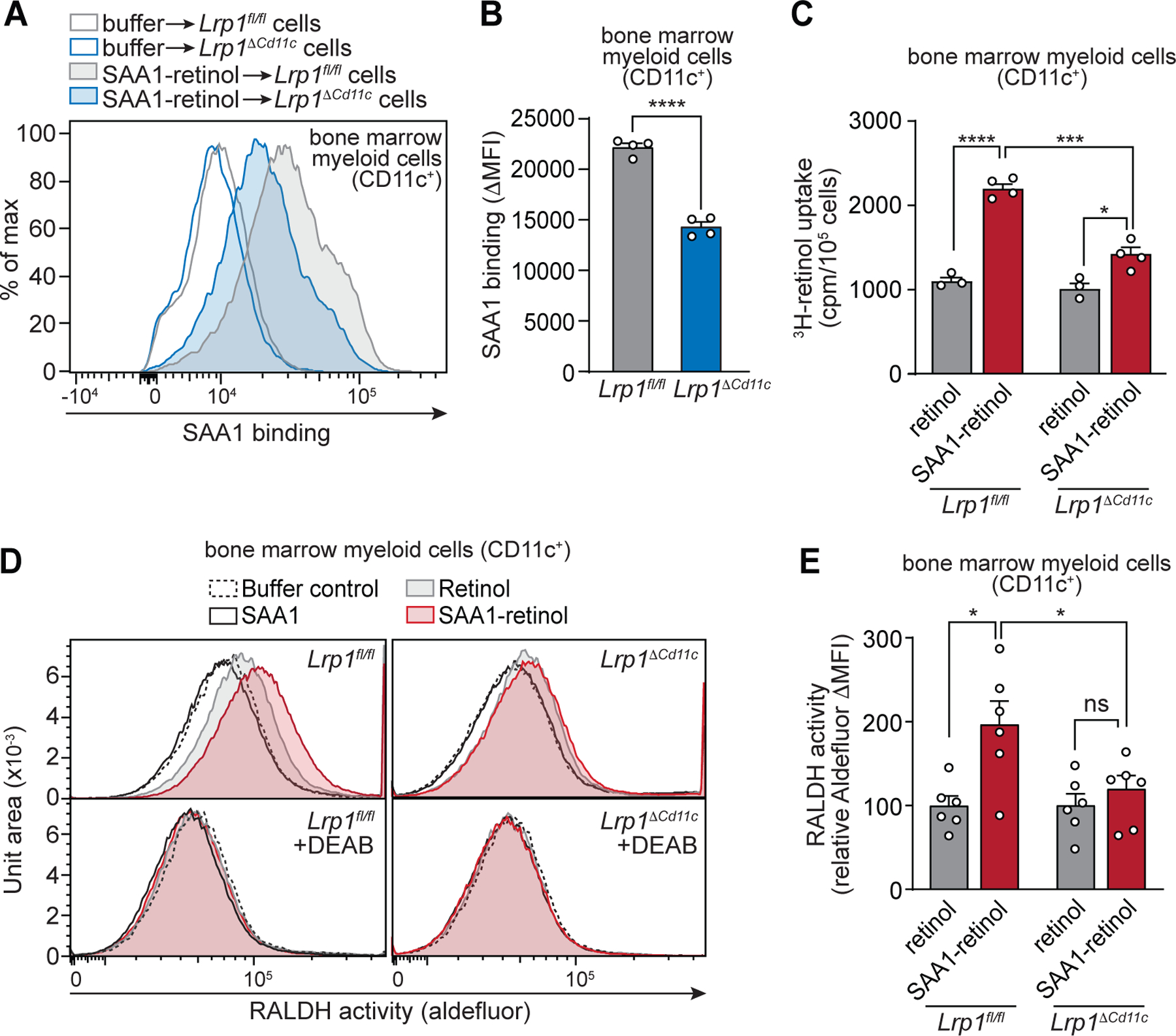
CD11c+ myeloid cells were isolated from bone marrow myeloid cells that were acquired from Lrp1fl/fl and Lrp1ΔCd11c mice and grown in GM-CSF. The CD11c+ myeloid cells were used to measure SAA binding (A and B), 3H-retinol uptake (C), and retinaldehyde dehydrogenase (RALDH) activity (D and E). (A and B) Binding of SAA to CD11c+ bone marrow myeloid cells was analyzed by flow cytometry under non-permeabilizing staining conditions. 1.5×105 cells were incubated with 1.8 μM His-SAA1–retinol complex for 1 hour at 4°C. For controls, PBS was added instead of SAA1. (A) Representative histograms are shown with empty lines representing the controls. (B) SAA binding was determined as SAA ΔMFI (MFISAA–MFIcontrol) and normalized to Lrp1fl/fl set at 100. (C) 3H-retinol (200 nM) was added to the cells either as free retinol or in complex with SAA1. After a 2 hour incubation at 37°C, the counts per minute (cpm) in the cell pellets were determined and normalized to cell number, and Lrp1fl/fl were set at 100. (D and E) Measurement of cellular RALDH activity. Cells were incubated with SAA1-retinol complexes, retinol only, SAA1 only, or buffer (control) for 1 hour at 4°C. After washing and incubation for 1.5 hours at 37°C, the cells were analyzed by Aldefluor assay. The aldehyde dehydrogenase inhibitor DEAB was added to each sample as a control. (D) Representative histograms showing Aldefluor fluorescence. (E) To measure RALDH activity, the MFI of the DEAB treatment control was first subtracted from the Aldefluor MFI of each sample (ΔMFI =MFIsample – MFIcontrol) and then converted to relative values with the ΔMFI for the buffer control set at 0 and the retinol only ΔMFI set at 100. Means±SEM are plotted from one (A–C) and two (D and E) independent experiments. All results are representative of at least two independent experiments. *P<0.05; ***P<0.001; ****P<0.0001; ns, not significant by Student’s t test.
CD11c+ myeloid cells convert retinol to RA through a two-step enzymatic oxidation. First, retinol is converted to retinaldehyde by alcohol dehydrogenases (ADHs) or retinol dehydrogenases (RDHs). Second, retinaldehyde is irreversibly converted to RA by members of the aldehyde dehydrogenase family (also known as retinaldehyde dehydrogenases (RALDHs)) (31). RALDH expression in intestinal myeloid cells is induced by vitamin A and RA (32). The reduced retinol uptake in Lrp1ΔCd11c myeloid cells therefore suggested that these cells might show reduced RALDH activity. We assessed RALDH activity by Aldefluor assay, which measures conversion of a fluorescent RALDH substrate by flow cytometry. Addition of SAA1–retinol increased RALDH activity in Lrp1fl/fl but not Lrp1ΔCd11c myeloid cells (Fig. 4, D and E). The increase in RALDH activity was higher with SAA1–retinol than with free retinol. This activity was suppressed by the RALDH inhibitor N,N-diethylaminobenzaldehyde (DEAB) (Fig. 4, D and E). Thus, LRP1 facilitates retinol uptake from SAA1–retinol complexes and this uptake increases RALDH activity in myeloid cells.
SAAs and LRP1 promote retinol uptake by intestinal CD11c+ myeloid cells in vivo.
We next examined the role of SAA and LRP1 in retinol acquisition in vivo by small intestinal CD11c+ myeloid cells. We previously used CRISPR/Cas9-mediated gene targeting to delete the entire mouse Saa locus (encompassing Saa1 to Saa4) (15), and verified that the numbers and composition of CD11c+ myeloid cells were not altered in Saa−/− mice (fig. S8, A and B). We measured retinol uptake by small intestinal CD11c+ myeloid cells by gavaging mice with a 3H-retinol tracer and tracking it to lamina propria myeloid cells. Fifteen hours after gavage, CD11c+ cells from Saa−/− mice had acquired less 3H-retinol than cells from wild-type mice (Fig. 5A and fig. S9, A and B), indicating that SAAs promote retinol delivery to intestinal CD11c+ cells. Overall 3H-retinol levels were similar in the small intestines of wild-type and Saa−/− mice (fig. S10A), arguing that the lowered retinol uptake by CD11c+ cells from Saa−/− mice did not arise from a decrease in intestinal retinol absorption. 3H-retinol levels were also similar in lamina propria CD11c− cells from wild-type and Saa−/− mice, which demonstrated that SAAs selectively promote retinol uptake in CD11c+ cells.
Fig. 5. SAA and LRP1 promote retinol uptake by intestinal CD11c+ myeloid cells in vivo.
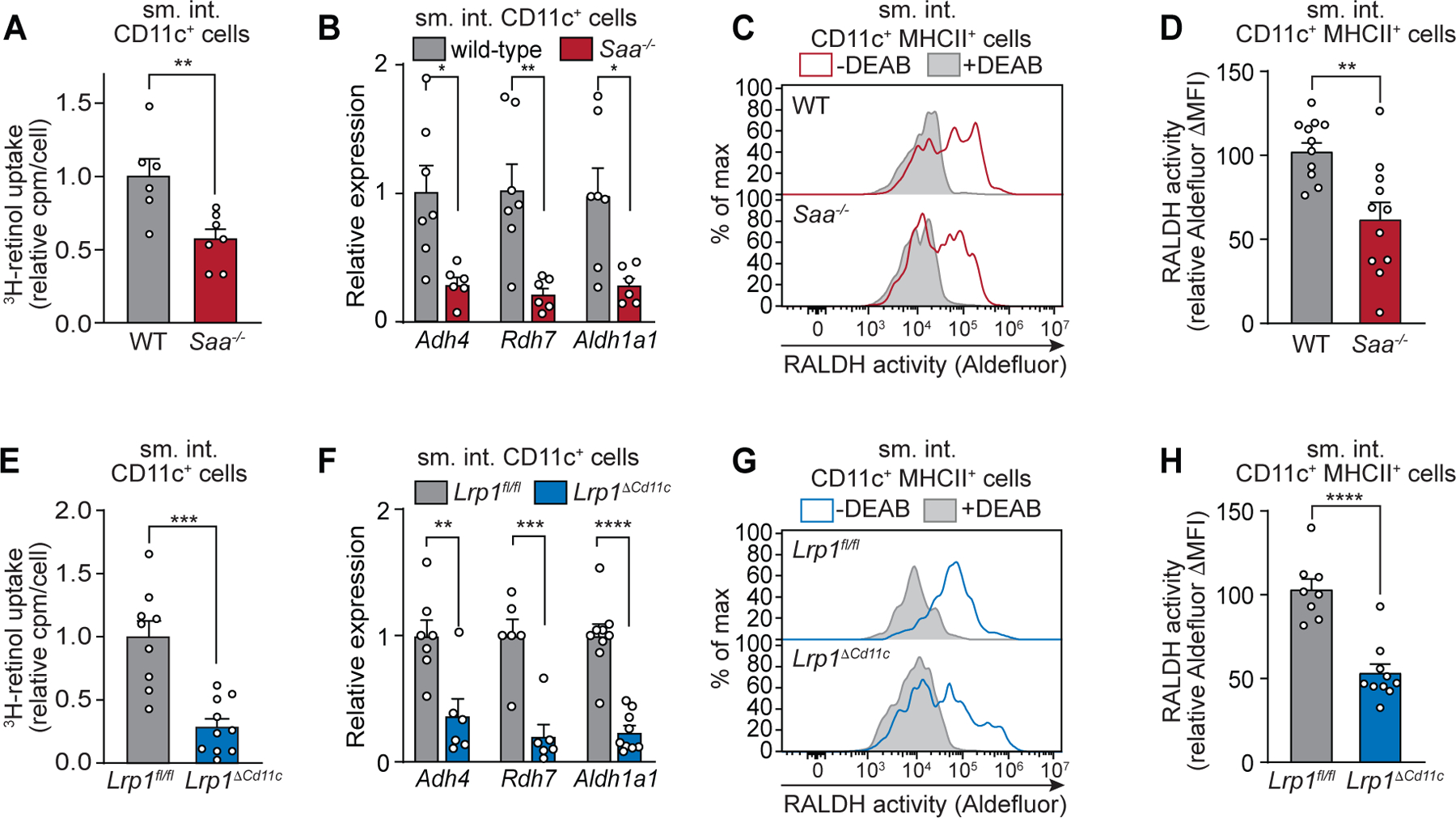
Wild-type (WT) and Saa−/− mice (A–D) or Lrp1fl/fl and Lrp1ΔCd11c (E–H) mice were analyzed. (A and E) Mice were gavaged with 3H-retinol and CD11c+ cells were isolated from the small intestinal (sm. int.) lamina propria. Counts per minute (cpm) were determined and normalized to cell number, and wild-type (A) and Lrp1fl/fl (E) were set at 1. (B and F) qPCR analysis of transcripts encoding proteins involved in conversion of retinol to retinoic acid in small intestinal lamina propria CD11c+ cells. Transcript abundances were normalized to Gapdh transcript abundance and expressed relative to wild-type (B) and Lrp1fl/fl mice (F). (C and G) Retinaldehyde dehydrogenase (RALDH) activity of CD11c+MHCII+ myeloid cells (live CD45+CD11c+MHCII+CD3−B220−) was assessed by Aldefluor assay. Representative histograms showing Aldefluor fluorescence are shown. Filled gray histograms are from controls incubated with the ALDH inhibitor DEAB, and empty lines show samples without DEAB. (D and H) RALDH activity was measured as Aldefluor ΔMFI (MFIsample–MFIcontrol). Means±SEM from at least three independent experiments with n≥5 mice/group are plotted. *P<0.05, **P<0.01, ***P<0.001, **** P<0.0001 by Student’s t test.
CD11c+ cells from Saa−/− mice had reduced expression of Adh4, Rdh7, and Aldh1a1 (Fig. 5B). These genes encode enzymes that convert retinol to RA (33–35) and their expression is induced by retinol or RA (fig. S10B) (32). In contrast, expression of these genes was similar in CD11c− cells from wild-type and Saa−/− mice (fig. S10C), consistent with the idea that SAAs selectively deliver retinol to CD11c+ cells. Likewise, RALDH activity was lower in CD11c+MHCII+ myeloid cells from Saa−/− mice than from wild-type mice (Fig. 5, C and D). Thus, SAAs facilitate myeloid cell retinol uptake that promotes the expression of enzymes that convert retinol to RA.
Next, we compared Lrp1fl/fl and Lrp1ΔCd11c mice to determine if LRP1 also facilitates retinol uptake by intestinal CD11c+ myeloid cells. We first verified that the numbers and composition of CD11c+ myeloid cells did not differ between Lrp1fl/fl and Lrp1ΔCd11c mice (fig. S11, A and B). As expected, Lrp1 transcripts and protein were less abundant in small intestinal CD11c+ cells from Lrp1ΔCd11c mice than from Lrp1fl/fl mice while remaining similar in CD11c− cells (fig. S12, A and B). Following 3H-retinol gavage, CD11c+ cells from Lrp1ΔCd11c mice acquired less 3H-retinol than cells from Lrp1fl/fl mice (Fig. 5E), indicating that LRP1 facilitates retinol uptake by intestinal CD11c+ cells. Consistent with our findings in wild-type and Saa−/− mice (fig. S10A), 3H-retinol levels in lamina propria CD11c− cells from Lrp1fl/fl and Lrp1ΔCd11c mice were similar (fig. S13A), indicating that LRP1 selectively promotes retinol uptake in CD11c+ cells. Finally, in agreement with our findings in Saa−/− mice, CD11c+ cells from Lrp1ΔCd11c mice showed reduced expression of Adh4, Rdh7, and Aldh1a1 (Fig. 5F and fig. S13B) and lower RALDH activity as compared to Lrp1fl/fl mice (Fig. 5, G and H). Thus, SAAs and their LRP1 receptor promote retinol uptake by CD11c+ myeloid cells and increase the expression of enzymes that convert retinol to RA.
SAA and LRP1 promote vitamin A-dependent adaptive immunity in the intestine.
We next assessed whether SAA and LRP1 promote vitamin A-dependent adaptive immunity in the intestine. RA-producing myeloid cells induce expression of the gut-homing receptor CC-chemokine receptor 9 (CCR9) on B cells and CD4+ T cells in mesenteric lymph nodes (mLN) and gut-associated lymphoid tissue, which directs the lymphocytes to the intestine (1, 2, 13). Consistent with the reduced retinol uptake in their RA-producing myeloid cells, the small intestines of Saa−/− mice had reduced frequencies of CD4+ T cells imprinted with CCR9 (Fig. 6, A and B), reduced numbers of CCR9+CD4+ T cells (Fig. 6B), and reduced overall numbers of CD4+ T cells (Fig. 6C) relative to wild-type mice. CCR9+ B cell frequencies (Fig. 6, D and E) and numbers (Fig. 6E), as well as overall B cell numbers (Fig. 6F) were also reduced in the small intestines of Saa−/− mice. Although RA promotes the development of FOXP3+ Treg (T regulatory) (6, 9, 12), Treg cell frequencies among CD4+ T cells were similar when comparing the small intestines (fig. S14, A and B) and mLN (fig. S14, C and D) of wild-type and Saa−/− mice. Thus, SAAs regulate B cell and CD4+ T cell homing to the intestine, but do not appear to be required for the generation of intestinal Treg cells.
Fig. 6. SAA and LRP1 promote vitamin A-dependent immunity in the intestine.
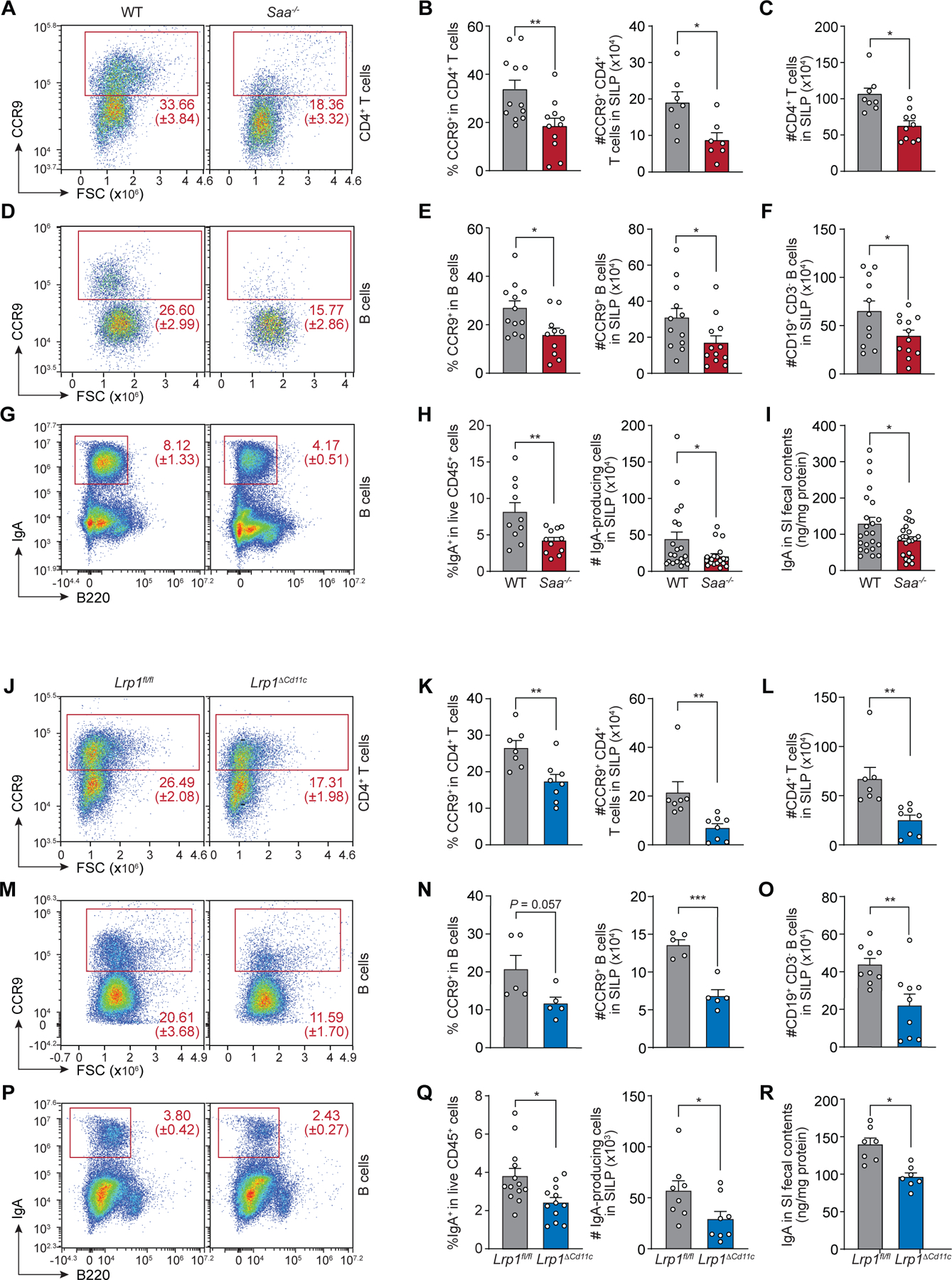
Small intestinal lamina propria (SILP) cells from wild-type (WT) and Saa−/− mice (A–I), and Lrp1fl/fl and Lrp1ΔCd11c mice (J–R) were analyzed. (A and J) Representative flow cytometry plots of CD4+ T cells (live CD45+CD3+CD4+CD19−CD8−). (B and K) Frequencies of CCR9+ cells in CD4+ T cells (left) and absolute numbers of CCR9+CD4+ T cells (right). (C and L) Total numbers of CD4+ T cells (CD45+CD3+CD4+CD19−CD8−). (D and M) Representative flow cytometry plots of B cells (live CD45+CD19+CD3−). (E and N) Frequencies of CCR9+ cells in B cells (left) and absolute numbers of CCR9+ B cells (right). (F and O) Total numbers of B cells (CD45+CD19+CD3−). (G and P) Representative flow cytometry plots of IgA+ cells (live CD45+CD3−). (H and Q) Frequencies of IgA+ cells in total live CD45+ cells (left) and numbers of IgA-producing cells (IgA+CD45+CD3−) (right). (I and R) Quantification of IgA from small intestinal fecal contents by ELISA. Data represent n≥5 mice/group from at least two independent experiments. Means±SEM are plotted. *P<0.05, **P<0.01 by Student’s t test.
Lrp1ΔCd11c mice were similar to Saa−/− mice, with reduced frequencies and numbers of CCR9+CD4+ T cells (Fig. 6, J and K) and reduced total numbers of CD4+ T cells (Fig. 6L) in the small intestine. Lrp1ΔCd11c mice had decreased frequencies and numbers of CCR9+ B cells (Fig. 6, M and N) and decreased total numbers of B cells (Fig. 6O) in the small intestine. Treg frequencies among CD4+ T cells were also similar in the small intestines (fig. S15, A and B) and mLN (fig. S15, C and D) of Lrp1fl/fl and Lrp1ΔCd11c mice. Thus, LRP1 also regulates B cell and CD4+ T cell homing to the intestine, but is not necessary for the generation of intestinal Treg cells.
TH17 (T helper 17) cells are specialized CD4+ T cells that secrete a distinct set of cytokines, including IL-17. Although SAAs are not required for TH17 cell lineage commitment, they boost TH17 cell effector function by stimulating IL-17A production (16). Accordingly, TH17 frequencies among CD4+ T cells were similar in the small intestines (fig. S14, A and B) and mLN (fig. S14, C and D) of wild-type and Saa−/− mice, but IL-17A production was reduced in TH17 cells from Saa−/− mice (fig. S14, E and F). Similarly, TH17 cell frequencies were similar in Lrp1fl/fl and Lrp1ΔCd11c mice (fig. S15, A to D), but IL-17A production was reduced in TH17 cells from Lrp1ΔCd11c mice (fig. S15, E and F). Thus, SAAs and myeloid cell LRP1 both regulate TH17 cell effector function.
Finally, Saa−/− and Lrp1ΔCd11c mice were deficient in the production of IgA, which defends against invading intestinal pathogens and maintains homeostasis with the microbiota (36–38). Saa−/− mice had reduced frequencies and numbers of small intestinal IgA+ cells (Fig. 6, G and H) and decreased fecal IgA concentrations (Fig. 6I) when compared with wild-type mice. Similarly, frequencies and numbers of IgA-producing cells (Fig. 6, P and Q) and fecal IgA concentrations (Fig. 6R) were decreased in the small intestines of Lrp1ΔCd11c when compared to Lrp1fl/fl mice. These findings are consistent both with the overall reduction in intestinal B cells due to reduced homing and with the role of RA-producing myeloid cells in promoting the development of IgA-producing cells from B cells (2). The differences in immune cell frequencies did not arise from altered microbiota taxonomic composition, as this was similar when comparing wild-type to Saa−/− mice, and Lrp1fl/fl to Lrp1ΔCd11c mice (fig. S16). Thus, SAA and LRP1 promote intestinal lymphocyte homing and IgA production, thus implicating SAA-mediated retinol delivery in the development of vitamin A-dependent intestinal immunity.
SAAs and LRP1 promote immunity to enteric bacterial infection.
The importance of SAAs and LRP1 for intestinal adaptive immunity suggested that these proteins might promote immunity to enteric bacterial infection. We tested this idea using the gastrointestinal pathogen Salmonella enterica Serovar Typhimurium (S. Typhimurium). Prior to infection, mice were twice immunized by oral gavage with heat-killed S. Typhimurium (Fig. 7A). This resulted in higher levels of S. Typhimurium-specific fecal IgA in wild-type mice as compared to Saa−/− mice and in Lrp1fl/fl mice as compared to Lrp1ΔCd11c mice (Fig 7, B and D). Accordingly, Saa−/− and Lrp1ΔCd11c mice showed higher mortality when orally infected with S. Typhimurium following immunization (Fig. 7, C and E). Thus, SAAs and LRP1 promote specific immunity after oral immunization and resistance to enteric pathogen infection.
Fig. 7. SAA and LRP1 promote immunity to enteric bacterial infection.
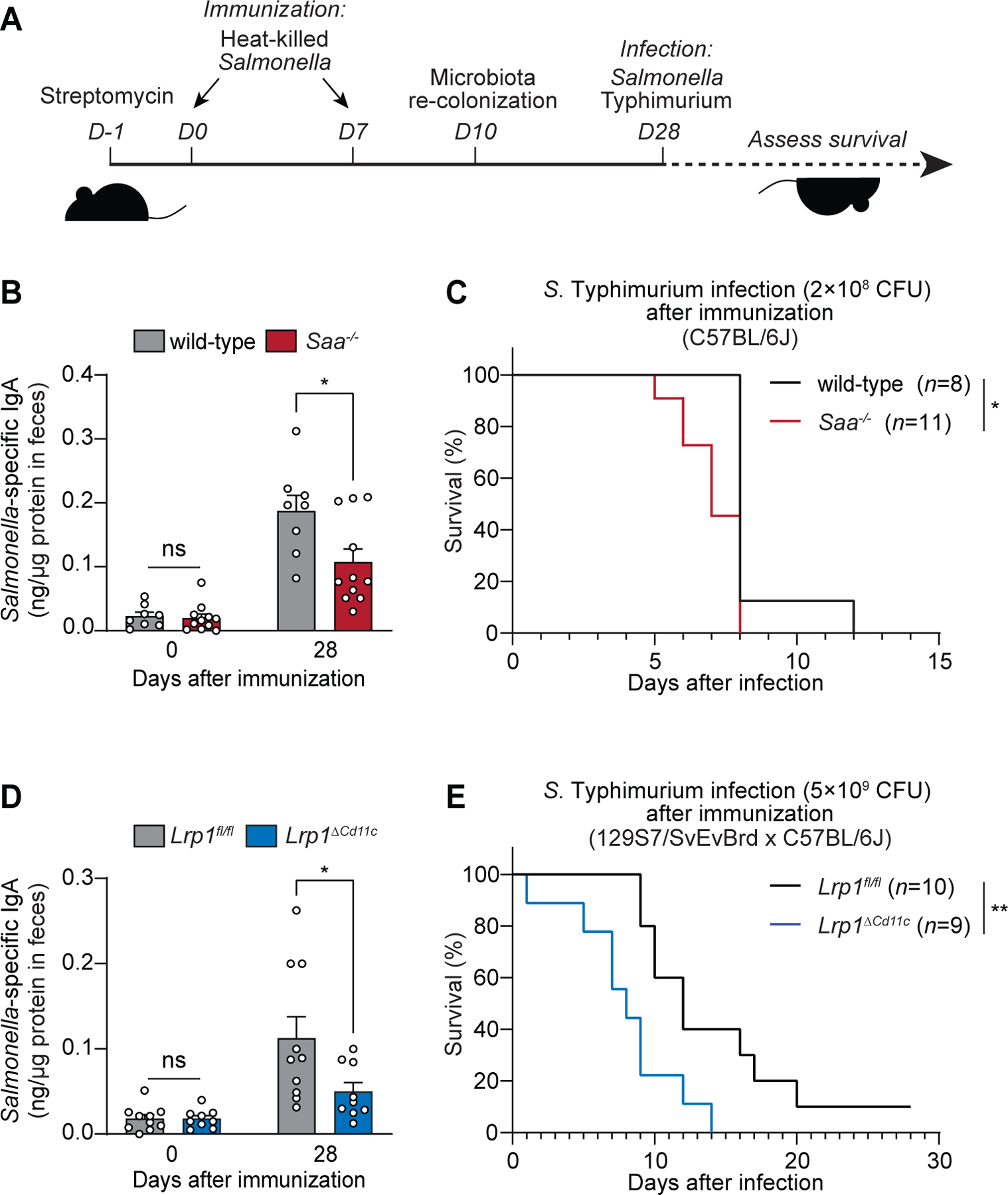
(A) Wild-type and Saa−/− mice (B and C), and Lrp1fl/fl and Lrp1ΔCd11c mice (D and E) were immunized with 1010 CFU of heat-killed Salmonella Typhimurium twice through oral gavage. Four weeks after the first immunization, the mice were orally infected with 2×108 CFU (C) and 5×109 CFU (E) of log-phase S. Typhimurium (note that the infection dose differs because resistance to infection differs according to mouse background). (B and D) S. Typhimurium-specific fecal IgA was measured by ELISA. Means±SEM are plotted. *P<0.05 by Student’s t test. (C and E) Survival rates were monitored after infection. Data represent results from five co-housed cages of wild-type and Saa−/− littermates (B and D) and five co-housed cages of Lrp1fl/fl and Lrp1ΔCd11c littermates (D and E). *P<0.05, **P<0.01 by the log-rank test.
Discussion
We have identified SAAs as retinol-binding proteins that deliver retinol to RA-producing intestinal myeloid cells. SAA–retinol complexes bind to LRP1 on the myeloid cell surface and are endocytosed, thus providing retinol for the enzymatic generation of RA. Consequently, SAAs and LRP1 promote the development of vitamin A-dependent adaptive immunity, including B and T cell homing to the intestine and IgA production, and enhance resistance to enteric infection after immunization. Our findings therefore identify a molecular basis for retinol acquisition by intestinal myeloid cells and illuminate a mechanism that is essential for vitamin A-dependent immunity (fig. S17).
Previously, we found that vitamin A promotes epithelial cell Saa expression through the transcription factor RARβ, which is activated by RA and binds directly to Saa promoters (17). RarbΔIEC mice, which selectively lack RARβ in intestinal epithelial cells, have defects in intestinal vitamin A-dependent immunity, including reduced gut homing of CD4+ T cells and lowered IgA production (17). Our finding that SAAs deliver retinol to myeloid cells thus provides a molecular explanation for reduced vitamin A-dependent immunity in RarbΔIEC mice. Furthermore, RarbΔIEC mice show reduced SAA expression that is restricted to epithelial cells (17), arguing that epithelial SAAs, rather than SAAs produced by the liver, deliver retinol to intestinal myeloid cells.
Our results provide molecular insight into how the intestinal microbiota shapes intestinal adaptive immunity. Expression of SAAs by intestinal epithelial cells requires the microbiota (39, 40), which activates Saa expression through an immunological signaling circuit (16). Our findings suggest that gut microbes promote vitamin A-dependent adaptive immunity by regulating epithelial SAA expression. These findings help explain long-standing observations of reduced numbers of B cells and CD4+ T cells and lowered IgA production in the intestines of germ-free mice (41).
A key question for future research is whether SAAs deliver retinol to other target cells. For example, our finding that 3T3-L1 adipocytes express LRP1 suggests that SAAs may deliver retinol to adipose tissue, which is a major extrahepatic retinol storage site (18). Additionally, SAAs may deliver retinol to TH17 cells thereby promoting TH17 cell effector functions (16). Supporting this idea, TH17 cell function requires vitamin A, as mice given a vitamin A-deficient diet show reduced intestinal IL-17 production (12) and TH17 cells from mice lacking T cell RARα produce less IL-17 (12). In vitro experiments suggest that SAAs impact TH17 cells through a direct interaction with the T cells (16, 42). It is possible that the expression of LRP1 on T cells, although lower than that on myeloid cells, is sufficient to support SAA binding and retinol uptake. Alternatively, T cells may express a distinct SAA receptor. However, our finding that TH17 cell effector functions are reduced in Lrp1ΔCd11c mice suggests the involvement of LRP1-expressing myeloid cells. This is consistent with a previously reported requirement for myeloid cells in IL-17 production by T cells in response to commensal bacteria (43, 44).
SAAs also carry retinol in the circulation and protect against systemic bacterial infection (14). Thus, it appears likely that SAAs also deliver retinol to cells that promote systemic immunity. This is a topic of ongoing and future research that should clarify whether SAAs represent a general mechanism for mobilizing retinol to the immune system during a microbiological or immunological challenge.
Finally, by illuminating how vitamin A is mobilized to the intestinal immune system, our results may offer additional approaches for enhancing resistance to infection and for increasing the efficacy of mucosal vaccines, which depend on adequate dietary vitamin A (45). Our findings also suggest the possibility of selectively regulating the availability of retinol to the immune system during inflammatory disease, perhaps by deploying small-molecule inhibitors of the SAA–retinol or SAA–LRP1 binding interactions.
Materials and Methods
Mice.
C57BL/6 wild-type, Saa−/−, Lrp1fl/fl, and Lrp1ΔCd11c mice were bred and maintained in the barrier facility at the University of Texas Southwestern Medical Center. Saa−/− mice were generated using CRISPR/Cas9 genome editing on a C57BL/6J background (15). Lrp1fl/fl mice (46) on a 129S7/SvEvBrd genetic background were used to generate a CD11c+ cell-specific Lrp1 knockout (Lrp1ΔCd11c) mouse by crossing with a mouse expressing Cre recombinase under the control of the CD11c promoter (Jackson Laboratories; B6.Cg-Tg (Itgax-cre)1-1Reiz/J). Mice 8–12 weeks of age were used for all experiments and co-housed littermates were used as controls. Experiments were performed using protocols approved by the Institutional Animal Care and Use Committees of the UT Southwestern Medical Center.
Cell culture.
Cell lines were purchased from ATCC and cultured at 37°C under 5% CO2. 3T3-L1 preadipocytes (ATCC CL-173) were cultured in DMEM containing 10% bovine calf serum, 100 U/ml of penicillin, and 100 μg/ml of streptomycin until confluence. Two days post-confluence, the cells were differentiated with 1.5 μg/ml of insulin, 1 μM dexamethasone, 500 μM 3-isobutyl-1-methylxanthine (IBMX), and 1 μM rosiglitazone in DMEM/F12 (1:1) containing 10% fetal bovine serum (FBS). After 48 hours of differentiation, the cells were treated with 1.5 μg/ml insulin in DMEM/F12 (1:1) with 10% (v/v) FBS, 100 U/ml of penicillin, and 100 μg/ml of streptomycin. The culture medium was changed every 2 days for 7–9 days. Wild-type and Lrp1-deficient murine embryonic fibroblasts, MEF1 (ATCC CRL-2214) and PEA13 (ATCC CRL-2216) respectively, were cultured in DMEM containing 10% FBS, 100 U/ml of penicillin, and 100 μg/ml of streptomycin. Isolated bone marrow cells from femurs and tibiae were cultured for 6 days in RPMI containing 10% FBS, 50 μM β-mercaptoethanol, 100 U/ml of penicillin, 100 μg/ml of streptomycin, and 20 ng/ml of GM-CSF (29). Adherent and loosely adherent cells were harvested by scraping and subjected for further experiments.
Proteins, antibodies, and reagents.
Recombinant mouse SAA1 and SAA3 were expressed and purified as described previously (14, 15). Recombinant Fc-tagged human LRP1-CII, -CIII, and –CIV containing ligand binding cluster II, III, and IV of LRP1 were purchased from R&D Biosystems. 6X histidine-tagged human RAP was purchased from Molecular Innovations and was used in binding assays after histidine-tag removal as described previously (15). Briefly, RAP protein was digested with thrombin (Roche), incubated overnight at 4°C, and purified using size-exclusion chromatography on a Superdex 75 10/300 GL column (GE Life Sciences) in PBS with 10% glycerol. Rabbit anti-SAA antiserum was produced previously (14). Armenian hamster monoclonal anti-CD11c (Clone N418) and rabbit monoclonal anti-LRP1 antibody (Clone EPR3724) recognizing 85-kDa light chain of LRP1 were purchased from Abcam. Mouse monoclonal anti-6X His tag (Clone HIS.H8) (Abcam) and PE-conjugated mouse monoclonal anti-6X His tag (Clone AD1.1.10) (R&D Biosystems) were used for the detection of His-tagged SAA by immunoblotting and flow cytometry respectively. Antibodies used for flow cytometry analysis are listed in table S3. All-trans retinol was purchased from Sigma-Aldrich.
Immunofluorescence assay.
Mouse ileum was washed with PBS, fixed in Bouin’s fixative overnight at 4°C and embedded in paraffin. Sections were washed in xylene twice and rehydrated in decreasing concentrations of ethanol (100%, 95%, 70%, 50%, and 0%). Slides were boiled in 10 mM sodium citrate (pH 6.0) for 25 min and washed twice in PBS. For immunofluorescence assay of adipocytes, cells were seeded and differentiated on cover glasses, fixed with 4% paraformaldehyde for 15 min at room temperature, and washed with PBS-T (PBS, 0.05% Tween-20). Samples were incubated with with blocking buffer (10% FBS, 1% BSA, 1% Triton X-100 in PBS) for 1 hour at room temperature and incubated with primary antibodies (anti-LRP1, 1:200 dilution; anti-CD11c, 1:200 dilution) in the blocking buffer at 4°C overnight. Cy3 anti-rabbit secondary antibody (Thermo Fisher Scientific) and AlexaFluor 647-conjugated anti-Armenian hamster secondary antibody (Abcam) were diluted 1:400 and applied to slides for 1 hour at room temperature in the dark. Samples were washed and mounted with DAPI Fluoromount-G (Southern Biotechnology). For a non-permeabilizing staining condition, buffers without Triton X-100 were used. Images were captured using a Zeiss AxioImage MI microscope with Zeiss AxioCam MRm Rev3 and Zeiss EC Plan-Neofluar 10x/0.30 M27 objective lens.
Analysis of cell-surface binding of SAA by flow cytometry.
Wild-type (Lrp1+/+) and Lrp1-deficient (Lrp1−/−) fibroblasts were detached from culture plates with PBS with 1.5 mM EDTA and washed twice with serum-free media. Suspensions containing 2×105 cells in 0.2 ml of serum-free culture media were incubated with His-SAA1. For competition studies, non-tagged SAA1 proteins were added as indicated together with His-tagged SAA1 proteins. Following incubation for 1 hour at 4°C, cells were washed twice with ice-cold DPBS, fixed with 4% paraformaldehyde, stained with PE-conjugated anti-His-tag antibody, and subjected to flow cytometry by using LSRII (BD Bioscience) or Novocyte 3005 (ACEA Biosciences). To assess LRP1-specific SAA binding, the differences in SAA binding (ΔMFI=MFISAA-MFIPBS) between wild-type cells and Lrp1-deficient cells were analyzed, expressed as ΔΔMFI (ΔMFILrp1+/+-ΔMFILrp1−/−), and fit with GraphPad Prism 7 using the nonlinear regression, one site-binding equation. For adipocytes, His-SAA1 proteins were added to 2×105 differentiated adipocytes grown in 96-well plates in DMEM/F12 (1:1) and processed with the same staining procedures. To analyze SAA binding to lamina propria cells, one million small intestinal lamina propria (SILP) cells were isolated and incubated with the His-SAA1–retinol complex (7 μM) for 20 min at 4°C in serum-free RPMI. The cells were then washed and fixed as described above and stained for His-tag to detect His-SAA1 alongside immune cell markers.
Cellular retinol uptake assay.
Radioactive 11,12-3H(N)-retinol was purchased from PerkinElmer. For the in vitro retinol uptake assay, SAA1 was incubated with 3H-retinol at a 3:1 molar ratio at 4°C for at least 2 hours in serum-free media containing protease inhibitors (Roche). The SAA1-retinol complex was purified using PD-10 desalting column (GE Healthcare). Cells were incubated with 480 nM of SAA1-3H-retinol complex or free 3H-retinol in the serum-free media at 37°C and 5% CO2 (Fig. 2F). Cells were incubated with 600 nM SAA1–3H-retinol complex with and without RAP in serum-free media at 4°C for 1 hour, washed twice with cold media, and incubated at 37°C and 5% CO2 for 3 hours (Fig. 2G) and for 2 hours (Fig. 4C). The reactions were stopped by washing the cells with ice-cold DPBS and solubilizing the cells in Biosol solution (National Diagnostics). For the in vivo retinol uptake assay, 3H-retinol was diluted in corn oil and fed to mice (1 μCi/mouse). Fifteen to eighteen hours later, immune cells were isolated from the small intestine and CD11c+ cells were isolated by magnetic sorting. Cells (2×105) were solubilized in 0.5 ml of Biosol solution. A 3.5-ml volume of Biosinct solution (National Diagnostics) was mixed with each sample and radioactivity was measured with a liquid scintillation analyzer (PerkinElmer) and normalized to cell number.
SAA-receptor purification and identification.
A hetero-bifunctional crosslinker, Sulfo-SANPAH (Thermo Fisher Scientific), was used to crosslink SAA with its putative receptor on the adipocyte surface, similar to a previously described procedure (27). The amine-reactive group on the crosslinker was first covalently linked to the purified His-SAA1 by incubating 25 μM of purified His-SAA1 protein with 2.3 mM sulfo-SANPAH in PBS for 1 hour on ice. The reaction was stopped by adding glycine to a final concentration of 80 mM and SAA1-crosslinker conjugate was purified using a PD-10 desalting column (GE Healthcare) in DPBS. The conjugate was mixed with retinol and then added to adipocytes in serum-free medium. After 20 min of incubation at 4°C, the mixture was UV-irradiated to crosslink SAA1 and its receptor using UV transilluminator (Syngene). Cell pellets were collected and lysed by sonication in PBS containing 1% Triton X-100 and 0.1% SDS to solubilize membrane proteins. The proteins were bound to Ni-NTA resin (Qiagen) overnight at 4°C. The resin was washed twice with PBS and three times with PBS containing 25 mM imidazole. SAA1-receptor complexes were eluted in PBS containing 250 mM imidazole, mixed with SDS-loading buffer, and subjected to SDS-PAGE. The protein band identified by immunoblot as containing SAA, which was absent in the negative controls, was excised, digested with trypsin, and run on an Orbitrap Fusion Lumos mass spectrometer for LC-MS/MS analysis at the University of Texas Southwestern Medical Center Proteomics Core. Proteins were identified using Proteome Discover 2.2.
Immunoblotting assay.
Tissue or cell lysates were separated using a 4 to 20% gradient SDS-PAGE gel then transferred to a PVDF membrane. Membranes were blocked with 5% nonfat milk in TBS-T buffer (0.1% Tween-20 in Tris-buffered saline), and then were sequentially incubated with primary antibodies and HRP-conjugated secondary antibodies. Membranes were visualized using a Bio-Rad ChemiDoc™ Touch system.
Recombinant expression and purification of full-length LRP1 ectodomain.
The full-length ectodomain of human LRP1 (residues 1-4414, NCBI accession: NM_002332.3) and human RAP (NCBI accession: NM_002337.4) containing a C-terminal 6xHis tag were cloned into a pEZT-BM vector (47). The baculovirus was generated in Sf9 cells (Thermo Fisher Scientific) following the standard protocol. In order to boost the protein yield, the full-length ectodomain of human LRP1 was co-expressed with RAP in HEK293F cells (Life Technologies) at a ratio of 1:40 (virus:cells, v:v) and supplemented with 10 mM sodium butyrate. Cells were cultured in suspension at 37°C and the medium was harvested after 48 hr of infection. The medium containing the secreted LRP1–RAP protein complex was incubated with Ni-NTA resin (Qiagen) overnight with gentle agitation at 4°C. To purify LRP1–RAP protein complex, the resin was collected on a disposable gravity column (Bio-Rad), washed three times with buffer A (15 mM Tris-HCl pH 8.0, 150 mM NaCl, and 30 mM imidazole), and the His-tagged RAP protein together with the nontagged LRP1 protein was eluted with buffer B (15 mM Tris-HCl pH 8.0, 150 mM NaCl, and 300 mM imidazole). Size-exclusion chromatography (Superose 6 Increase 10/300 GL, GE Healthcare) was used to remove the excess free RAP protein and purify the LRP1–RAP protein complex in buffer containing 10 mM Tris-HCI pH 7.2 and 100 mM NaCl. In order to dissociate the LRP1–RAP protein complex and purify the free LRP1 protein, fractions containing the LRP1–RAP protein complex were pooled together and subjected to size-exclusion chromatography in buffer containing 10 mM MES (pH 5.5) and 100 mM NaCl. Fractions from size-exclusion chromatography were analyzed by Coomassie staining after SDS-PAGE, and the fractions containing the full-length ectodomain of human LRP1 protein were pooled and concentrated for future experiments. The fractions containing RAP protein were pooled separately and used in experiments.
Pull-down assay.
Fc-tagged human LRP1-CII, LRP1-CIII, LRP1-CIV, and control IgG-Fc were used (R&D Systems). LRP1 proteins (250 nM) and IgG-Fc were individually attached to 40 μl of Dynabead protein G magnetic beads (Thermo Fisher Scientific) using Fc-tags. After unbound proteins were removed by washing twice with PBS, SAA1–retinol complex (800 nM) was added to the beads and incubated for overnight at 4°C. The beads were washed five times with PBS containing 0.025% Tween-20 and the bound protein complexes were isolated by magnetic separation.
Size exclusion chromatography and purification of the SAA-retinol complex.
Binding of SAA1 to the LRP1 ectodomain was confirmed by size exclusion chromatography. Purified SAA1 was incubated with retinol at a molar ratio of 1:1.5 at 4°C in the dark for 20 min to allow formation of the SAA1–retinol complex, followed by passage over a Superdex 75 10/300 GL size exclusion chromatography column (GE Healthcare) to remove the excess retinol. Next, the SAA1-retinol complex was mixed with the purified full-length ectodomain of LRP1 at a molar ratio of 10:1 in buffer containing 10 mM MES pH 5.5 and 100 mM NaCl. The mixture was incubated on ice for 1 hour and then loaded onto a Superose 6 increase 10/300 GL column (GE Healthcare). As controls, LRP1 ectodomain and SAA1–retinol were individually analyzed in the same way.
Microscale thermophoresis.
The dissociation constant for the LRP1–SAA1 interaction was determined by microscale thermophoresis (48). Purified SAA1 was labeled with AlexaFluor 594 using the Microscale Protein Labeling Kit (Invitrogen). Labeled SAA1 was incubated with retinol (or DMSO) at a 3:1 molar ratio for 2 hours at 4°C in MST buffer (150 mM NaCl, 5 mM CaCl2, 0.05% (v/v) Tween 20, and 20 mM HEPES, pH 6.5). Next, a 16-fold titration series of purified LRP1 ectodomain (0 to 1.75 μM) diluted 1:1 with the MST buffer was mixed with SAA1 at 50 nM. The mixtures containing diluted LRP1 and SAA1 were incubated for 30 min at room temperature in the dark to enable binding. Each titration sample was then loaded into a premium capillary (NanoTemper) and analyzed on a Monolith NT.115 Pico (NanoTemper). LRP1-dependent fluorescence enhancement was observed, and absolute fluorescence was used to calculate the Kd. The specificity of the binding event was carefully validated by the SDS-denaturation test according to the manufacturer’s protocol. Binding isotherms were deduced by using PALMIST software (49) and the graph was created in GUSSI (50).
Trypsin protection assay.
Wild-type and Lrp1-deficient fibroblasts (2.5×105) were incubated with His-SAA1–retinol complex (1 μM) at 4°C in serum-free DMEM for 1 hour to allow SAA to bind to cell surface LRP1. Unbound SAAs in the media were removed and cells were washed twice with cold-PBS. Pre-warmed DMEM was added and the cells were moved to 37°C to initiate endocytosis. In some cases, 25 μM Pitstop2 (Abcam) was added to the cells to block clathrin-mediated endocytosis before the 37°C incubation. After 30 min, cells were treated with 0.0125% Trypsin-EDTA (Thermo Fisher Scientific) for 3 min at 37°C. Cells were then lysed for immunoblotting analyses or fixed with 4% PFA for flow cytometry analyses, and SAA1 was detected using the anti-His-antibody.
Quantitative real-time PCR (qPCR).
cDNA was synthesized from purified RNA using M-MLV Reverse Transcriptase (Thermo Fisher Scientific). qPCR was performed using TaqMan assays (Thermo Fisher Scientific) on a QuantStudio 7 Flex Real-Time PCR System (Applied Biosystems). Relative expression values were determined using the comparative Ct (ΔΔCt) method (51), and transcript abundances were normalized to Gapdh transcript abundance. Absolute RNA transcript copy numbers were determined from standards and the results are reported as copies of transcript of interest/copies of Gapdh. TaqMan probe assay IDs are provided in table S2.
Lamina propria immune cell isolation and flow cytometry analysis.
Immune cells from small intestinal lamina propria were isolated as previously described with minor modification (17). Briefly, the small intestine was flushed with 20 ml of ice-cold PBS with 3% FBS, opened longitudinally, cut into 1.5-cm pieces, and washed three times with vortexing for 1 min in 10 ml of ice-cold PBS with 3% FBS. The pieces were incubated with 15 ml of PBS with 3% FBS, 1 mM EDTA, and 1 mM DTT for 15 min at 37°C with rotation on a platform shaker (250 rpm). The epithelial cells and intraepithelial lymphocytes were collected by passing through a 100-μm cell strainer. The intestine pieces were washed in PBS with 3% FBS, and placed in 20 ml of digestion solution (RPMI, 3% FBS, 0.2 mg/ml of collagenase IV (Sigma), 0.15 mg/ml of DNase I (Sigma), and 0.75 U/ml of Dispase II (Sigma)). After incubation at 37°C for 25 min with rotation on a platform shaker (250 rpm), the solution was vortexed and passed through a 40-μm cell strainer. Cells from EDTA treatment and enzyme digestion were combined and filtered through a glass wool column (Thermo Fisher Scientific) to remove epithelial cells. The immune cells were further isolated by Percoll gradient separation collected at the 40%–80% interphase of the Percoll gradient. After washing twice with PBS(3% FBS), the cells were either used immediately for experiments or fixed overnight in 1 ml of PBS with 3% FBS, 2 mM EDTA, and 0.2% paraformaldehyde at 4°C. For the analyses shown in Fig. 6 and fig. S4, Peyer’s patches were removed before the immune cell isolation process. For intracellular staining (LRP1, IgA, FOXP3, RORγt, and IL-17A), cells were first stained with surface markers, and then fixed, permeabilized, and stained using eBioscience™ Foxp3/Transcription factor staining buffer set (Thermo Fisher Scientific). Flow cytometry was performed with a LSRII or a Novocyte 3005 and data were analyzed with FlowJo (Tree Star) and NovoExpress (ACEA Biosciences) softwares. For CD11c+ cell isolation, dead cells were first removed by using dead cell removal kit following manufacturer’s protocol (Miltenyi Biotec). Cells were incubated with anti-CD11c microbeads and then immunomagnetically sorted by sequential passage over two MACS separation columns (Miltenyi Biotec), resulting in about 90% purity (fig. S9, A and B). To analyze IL-17 production in fig. S14 and S15, one million cells were treated with 50 ng/ml of phorbol myristate acetate, 1 mM ionomycin, and 1 mg/ml of brefeldin A for 4 hours at 37°C (17). Antibodies used for flow cytometry analysis are listed in table S3.
Measurement of RALDH activity by Aldefluor assay.
RALDH activity was measured by Aldefluor assay (Stemcell Technologies) following the manufacturer’s protocol with minor modifications. One million immune cells recovered from the small intestine were resuspended in 1 ml of assay buffer. Aldefluor reagent was added to each sample and incubated at 37°C for 30 min. The brightly fluorescent RALDH-expressing cells were detected by flow cytometry in the green channel (520–540 nm). As a control, the ALDH/RALDH inhibitor diethylaminobenzaldehyde (DEAB) was added to half of each sample.
16S rRNA gene sequencing and data analysis.
Fecal contents from the ileum and colon were collected from wild-type and Saa−/− littermates, and Lrp1fl/fl and Lrp1ΔCd11c littermates, that were co-caged. DNA was extracted and purified using the FastDNA Spin Kit (MP Biomedicals) and the FastPrep-24 Homogenizer (MP Biomedicals) following the manufacturer’s protocol (52). 16S rRNA gene sequencing and data analysis were done by the UT Southwestern Microbiome Research Laboratory. Briefly, sequencing libraries were prepared following the Illumina Nextera protocol (Part # 15044223 Rev. B) with primers flanking the hypervariable regions V3-V4. Sequencing was performed on MiSeqDX using the PE300 (Paired end 300 bp) v3 kit. Taxonomic classification and Operational Taxonomic Unit (OTU) abundance were analyzed using the CLC Bio microbial genomics module. Principle component analysis was performed using the “prcomp” function in R and differential abundance analysis was performed using DESeq2 (53).
Salmonella Typhimurium infection.
Mouse survival after infection with Salmonella enterica Serovar Typhimurium (Salmonella Typhimurium) was measured after immunization with heat-killed bacteria as described elsewhere (54, 55). S. Typhimurium strain SL1344 (streptomycin-resistant) was grown in Luria-Bertani (LB) broth containing 50 μg/ml of streptomycin at 37°C. For immunization, heat-killed S. Typhimurium was prepared in PBS by incubation at 65°C for 3 hours. Mice were treated with streptomycin (20 mg per mouse) by oral gavage a day before immunization, and then orally immunized with heat-killed S. Typhimurium (1010 CFU equivalents per mouse) twice at seven-day intervals. Three days after the second immunization, mice were colonized with a normal gut microbiota by inoculation with a fecal slurry prepared from the intestinal contents of conventional wild-type mice reared in the UT Southwestern mouse facility. Four weeks after the first immunization, mice were inoculated by oral gavage with 2×108 bacteria for wild-type and Saa−/− mice (on a C57BL/6J background) or with 5×109 bacteria for Lrp1fl/fl and Lrp1ΔCd11c mice (on a 129S7/SvEvBrd × C57BL/6J background). The dose varied because resistance to S. Typhimurium infection varies according to mouse background.
S. Typhimurium-specific IgA was measured in feces as previously described (56). Briefly, LPS was purified from S. Typhimurium SL1344 cultures using the LPS extraction kit (Abcam) and used as antigen for ELISA. 96-well microplates (Greiner Bio-one) were coated with LPS (1 μg per well) in carbonate coating buffer (Thermo Scientific). The plates were then blocked with 5% w/v skim milk in TBST and the fecal extracts were added to the plates. After washing, bound IgA was detected using HRP-conjugated goat anti-mouse IgA antibody (Invitrogen) and SuperSignal ELISA Pico Chemiluminescent Substrate (Thermo Scientific).
Supplementary Material
Acknowledgments:
We thank B. Hassell for assistance with mouse experiments and the UT Southwestern Proteomics Core for assistance for proteomics analysis.
Funding:
This work was supported by NIH Grant R01 DK070855 (L.V.H.), Welch Foundation Grant I-1874 (L.V.H.), the Walter M. and Helen D. Bader Center for Research on Arthritis and Autoimmune Diseases (L.V.H.), and the Howard Hughes Medical Institute (L.V.H.). J.H. was supported by NIH grants R37 HL63762, R01 NS108115, R01 NS093382, RF1 AG053391, and BrightFocus Foundation A2016396S. Y.-J.B. was supported by a Crohn’s and Colitis Foundation of America Research Fellowship Award (509845).
Footnotes
Competing interests: Authors declare no competing interests.
Data and materials availability:
16S rRNA gene sequencing data (fig. S16) have been submitted to the Sequence Read Archive (SRA) with BioProject IDs PRJNA727197 and PRJNA727212. Lrp1fl/fl mice are available under a material transfer agreement with UTSW. All other lines in the study are commercially available. All other data are available in the main text or the supplementary materials.
References and Notes
- 1.Iwata M et al. , Retinoic acid imprints gut-homing specificity on T cells. Immunity. 21, 527–538 (2004). [DOI] [PubMed] [Google Scholar]
- 2.Mora JR et al. , Generation of gut-homing IgA-secreting B cells by intestinal dendritic cells. Science. 314, 1157–60 (2006). [DOI] [PubMed] [Google Scholar]
- 3.Bhaskaram P, Micronutrient Malnutrition, Infection, and Immunity: An Overview. Nutr. Rev 60, 40–45 (2002). [DOI] [PubMed] [Google Scholar]
- 4.Sun T, Nguyen A, Gommerman JL, Dendritic Cell Subsets in Intestinal Immunity and Inflammation. J. Immunol 204, 1075–1083 (2020). [DOI] [PubMed] [Google Scholar]
- 5.Koscsó B et al. , Gut-resident CX3CR1hi macrophages induce tertiary lymphoid structures and IgA response in situ. Sci. Immunol 5, 62 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Coombes JL et al. , A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-β -and retinoic acid-dependent mechanism. J. Exp. Med 204, 1757–1764 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Denning TL, Wang Y, Patel SR, Williams IR, Pulendran B, Lamina propria macrophages and dendritic cells differentially induce regulatory and interleukin 17–producing T cell responses. Nat. Immunol 8, 1086–1094 (2007). [DOI] [PubMed] [Google Scholar]
- 8.Mucida D et al. , Reciprocal TH17 and regulatory T cell differentiation mediated by retinoic acid. Science (80-. ). 317, 256–260 (2007). [DOI] [PubMed] [Google Scholar]
- 9.Sun C-M et al. , Small intestine lamina propria dendritic cells promote de novo generation of Foxp3 T reg cells via retinoic acid. J. Exp. Med 204, 1775–85 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Denning TL et al. , Functional specializations of intestinal dendritic cell and macrophage subsets that control Th17 and regulatory T cell responses are dependent on the T cell/APC ratio, source of mouse strain, and regional localization. J. Immunol 187, 733–47 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nolting J et al. , Retinoic acid can enhance conversion of naive into regulatory T cells independently of secreted cytokines. J. Exp. Med 206, 2131–2139 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hall JA et al. , Essential role for retinoic acid in the promotion of CD4+ T cell effector responses via retinoic acid receptor alpha. Immunity. 34, 435–447 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mora JR et al. , Selective imprinting of gut-homing T cells by Peyer’s patch dendritic cells. Nature. 424, 88–93 (2003). [DOI] [PubMed] [Google Scholar]
- 14.Derebe MG et al. , Serum amyloid A is a retinol binding protein that transports retinol during bacterial infection. Elife. 3, 1–18 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hu Z, Bang YJ, Ruhn KA, Hooper LV, Molecular basis for retinol binding by serum amyloid A during infection. Proc. Natl. Acad. Sci. U. S. A 116, 19077–19082 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sano T et al. , An IL-23R/IL-22 Circuit Regulates Epithelial Serum Amyloid A to Promote Local Effector Th17 Responses. Cell. 163, 381–393 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gattu S et al. , Epithelial retinoic acid receptor β regulates serum amyloid A expression and Vitamin A-dependent intestinal immunity. Proc. Natl. Acad. Sci. U. S. A 166, 10911–10916 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Frey SK, Vogel S, Vitamin A metabolism and adipose tissue biology. Nutrients. 3, 27–39 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lillis AP, Van Duyn LB, Murphy-Ullrich JE, Strickland DK, LDL receptor-related protein 1: Unique tissue-specific functions revealed by selective gene knockout studies. Physiol. Rev 88, 887–918 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kounnas MZ, Henkin J, Argraves WS, Strickland DK, Low density lipoprotein receptor-related protein/α2-macroglobulin receptor mediates cellular uptake of pro-urokinase. J. Biol. Chem 268, 21862–21867 (1993). [PubMed] [Google Scholar]
- 21.Lenting PJ et al. , The light chain of factor VIII comprises a binding site for low density lipoprotein receptor-related protein. J. Biol. Chem 274, 23734–23739 (1999). [DOI] [PubMed] [Google Scholar]
- 22.Weigel PH, Oka JA, Temperature dependence of endocytosis mediated by the asialoglycoprotein receptor in isolated rat hepatocytes. Evidence for two potentially rate-limiting steps. J. Biol. Chem 256, 2615–2617 (1981). [PubMed] [Google Scholar]
- 23.Herz J, Goldstein JL, Strickland DK, Ho YK, Brown MS, 39-kDa protein modulates binding of ligands to low density lipoprotein receptor-related protein/α2-macroglobulin receptor. J. Biol. Chem 266, 21232–21238 (1991). [PubMed] [Google Scholar]
- 24.Bu G, Geuze HJ, Strous GJ, Schwartz AL, 39 kDa receptor-associated protein is an ER resident protein and molecular chaperone for LDL receptor-related protein. EMBO J. 14, 2269–2280 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li Y, Marzolo MP, Van Kerkhof P, Strous GJ, Bu G, The YXXL motif, but not the two NPXY motifs, serves as the dominant endocytosis signal for low density lipoprotein receptor-related protein. J. Biol. Chem 275, 17187–17194 (2000). [DOI] [PubMed] [Google Scholar]
- 26.Blaner WS, Retinol-binding protein: The serum transport protein for vitamin a. Endocr. Rev 10, 308–316 (1989). [DOI] [PubMed] [Google Scholar]
- 27.Kawaguchi R et al. , A membrane receptor for retinol binding protein mediates cellular uptake of vitamin A. Science (80-. ). 315, 820–825 (2007). [DOI] [PubMed] [Google Scholar]
- 28.Alapatt P et al. , Liver retinol transporter and receptor for serum retinol-binding protein (RBP4). J. Biol. Chem 288, 1250–1265 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Helft J et al. , GM-CSF Mouse Bone Marrow Cultures Comprise a Heterogeneous Population of CD11c+MHCII+ Macrophages and Dendritic Cells. Immunity. 42, 1197–1211 (2015). [DOI] [PubMed] [Google Scholar]
- 30.Rang Na Y, Jung D, Jeong Gu G, Hyeok Seok S, GM-CSF Grown Bone Marrow Derived Cells Are Composed of Phenotypically Different Dendritic Cells and Macrophages. Mol. Cells 39, 734–741 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Blomhoff R, Blomhoff HK, Overview of retinoid metabolism and function. J. Neurobiol 66, 606–630 (2006). [DOI] [PubMed] [Google Scholar]
- 32.Molenaar R et al. , Expression of Retinaldehyde Dehydrogenase Enzymes in Mucosal Dendritic Cells and Gut-Draining Lymph Node Stromal Cells Is Controlled by Dietary Vitamin A. J. Immunol 186, 1934–1942 (2011). [DOI] [PubMed] [Google Scholar]
- 33.Chou CF, Lai CL, Chang YC, Duester G, Yin SJ, Kinetic mechanism of human class IV alcohol dehydrogenase functioning as retinol dehydrogenase. J. Biol. Chem 277, 25209–25216 (2002). [DOI] [PubMed] [Google Scholar]
- 34.Su J, Chai X, Kahn B, Napoli JL, cDNA cloning, tissue distribution, and substrate characteristics of a cis-retinol/3α-hydroxysterol short-chain dehydrogenase isozyme. J. Biol. Chem 273, 17910–17916 (1998). [DOI] [PubMed] [Google Scholar]
- 35.Gagnon I, Duester G, Bhat PV, Enzymatic characterization of recombinant mouse retinal dehydrogenase type 1. Biochem. Pharmacol 65, 1685–1690 (2003). [DOI] [PubMed] [Google Scholar]
- 36.Boullier S et al. , Secretory IgA-Mediated Neutralization of Shigella flexneri Prevents Intestinal Tissue Destruction by Down-Regulating Inflammatory Circuits. J. Immunol 183, 5879–5885 (2009). [DOI] [PubMed] [Google Scholar]
- 37.Forbes SJ, Bumpus T, McCarthy EA, Corthésy B, Mantis NJ, Transient suppression of shigella flexneri type 3 secretion by a protective O-antigen-specific monoclonal igA. MBio. 2 (2011), doi: 10.1128/mBio.00042-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bunker JJ et al. , Innate and Adaptive Humoral Responses Coat Distinct Commensal Bacteria with Immunoglobulin A. Immunity. 43, 541–553 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ivanov II et al. , Induction of Intestinal Th17 Cells by Segmented Filamentous Bacteria. Cell. 139, 485–498 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Reigstad CS, Lundén GÖ, Felin J, Bäckhed F, Regulation of Serum Amyloid A3 (SAA3) in Mouse Colonic Epithelium and Adipose Tissue by the Intestinal Microbiota. PLoS One. 4, e5842 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cebra JJ, Periwal SB, Lee G, Lee F, Shroff KE, Development and maintenance of the gut-associated lymphoid tissue (GALT): The roles of enteric bacteria and viruses. Dev. Immunol 6, 13–18 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee JY et al. , Serum Amyloid A Proteins Induce Pathogenic Th17 Cells and Promote Inflammatory Disease. Cell. 180, 79–91.e16 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Panea C et al. , Intestinal Monocyte-Derived Macrophages Control Commensal-Specific Th17 Responses. Cell Rep. 12, 1314–1324 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Goto Y et al. , Segmented filamentous bacteria antigens presented by intestinal dendritic cells drive mucosal Th17 cell differentiation. Immunity. 40, 594–607 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Penkert RR et al. , Influences of Vitamin A on vaccine immunogenicity and efficacy. Front. Immunol 10, 1576 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rohlmann A, Gotthardt M, Willnow TE, Hammer RE, Herz J, Sustained somatic gene inactivation by viral transfer of Cre recombinase. Nat. Biotechnol 14, 1562–1565 (1996). [DOI] [PubMed] [Google Scholar]
- 47.Morales-Perez CL, Noviello CM, Hibbs RE, Manipulation of Subunit Stoichiometry in Heteromeric Membrane Proteins. Structure. 24, 797–805 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rainard JM, Pandarakalam GC, McElroy SP, Using Microscale Thermophoresis to Characterize Hits from High-Throughput Screening: A European Lead Factory Perspective. SLAS Discov. 23 (2018), pp. 225–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Scheuermann TH, Padrick SB, Gardner KH, Brautigam CA, On the acquisition and analysis of microscale thermophoresis data. Anal. Biochem 496, 79–93 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brautigam CA, Calculations and Publication-Quality Illustrations for Analytical Ultracentrifugation Data. Methods Enzymol. 562, 109–133 (2015). [DOI] [PubMed] [Google Scholar]
- 51.Schmittgen TD, Livak KJ, Analyzing real-time PCR data by the comparative CT method. Nat. Protoc 3, 1101–1108 (2008). [DOI] [PubMed] [Google Scholar]
- 52.Kuang Z et al. , The intestinal microbiota programs diurnal rhythms in host metabolism through histone deacetylase 3. Science (80-. ). 365, 1428–1434 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Love MI, Huber W, Anders S, Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Waldman RH, Grunspan R, Ganguly R, Oral immunization of mice with killed Salmonella typhimurium vaccine. Infect. Immun 6, 58–61 (1972). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kolodziejczyk AA, Zheng D, Elinav E, Diet-microbiota interactions and personalized nutrition. Nat. Rev. Microbiol, doi: 10.1038/s41579-019-0256-8. [DOI] [PubMed] [Google Scholar]
- 56.Xu C et al. , Live attenuated Salmonella typhimurium vaccines delivering SaEsxA and SaEsxB via type III secretion system confer protection against Staphylococcus aureus infection. BMC Infect. Dis 18, 1–13 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
16S rRNA gene sequencing data (fig. S16) have been submitted to the Sequence Read Archive (SRA) with BioProject IDs PRJNA727197 and PRJNA727212. Lrp1fl/fl mice are available under a material transfer agreement with UTSW. All other lines in the study are commercially available. All other data are available in the main text or the supplementary materials.