

Abstract
Psoriasis is a common inflammatory skin disease characterized by aberrant inflammation and epidermal hyperplasia. Molecular mechanisms that regulate psoriasis-like skin inflammation remain to be fully understood. Here, we show that the expression of Ovol1 (encoding ovo-like 1 transcription factor) is upregulated in psoriatic skin, and its deletion results in aggravated psoriasis-like skin symptoms following stimulation with imiquimod. Using bulk and single-cell RNA sequencing, we identify molecular changes in the epidermal, fibroblast, and immune cells of Ovol1-deficient skin that reflect an altered course of epidermal differentiation and enhanced inflammatory responses. Furthermore, we provide evidence for excessive full-length IL-1α signaling in the microenvironment of imiquimod-treated Ovol1-deficient skin that functionally contributes to immune cell infiltration and epidermal hyperplasia. Collectively, our study uncovers a protective role for OVOL1 in curtailing psoriasis-like inflammation and the associated skin pathology.
INTRODUCTION
Psoriasis is one of the most common inflammatory skin diseases, affecting 2–3% of the world population (Ayala-Fontanez et al., 2016). The etiology of the disease is multi-factorial, encompassing both genetic and environmental components that cause dysregulations of the skin barrier and immune cell function (Benhadou et al., 2019; Bergboer et al., 2012; Lowes et al., 2014; Roberson and Bowcock, 2010). Central to skin barrier function is the epidermis, a self-renewing epithelium composed of a basal layer containing stem and progenitor cells and multiple suprabasal layers containing progressively differentiating cells that produce stratum corneum, the actual physical barrier at the skin’s outmost surface (Gonzales and Fuchs, 2017). In psoriasis and other aberrant skin inflammatory processes, epidermal cells both serve as the target of immune cells and play a crucial role in shaping the immune responses (Kobayashi et al., 2019; Pasparakis et al., 2014). The molecular mechanisms that regulate the complex cross-talk between epidermal cells and immune cells in psoriasis remain to be fully elucidated.
SNPs in OVOL1 have been shown to associate with several inflammatory skin diseases, namely atopic dermatitis, atopic march, and acne (Hirota et al., 2012; Kang et al., 2015; Marenholz et al., 2015; Navarini et al., 2014; Paternoster et al., 2011). OVOL1 encodes a zinc finger transcription factor, and its mouse homolog Ovol1 is functionally required for proper embryonic epidermal development (Dai et al., 1998; Lee et al., 2014; Nair et al., 2006; Teng et al., 2007). Specifically, Ovol1 is expressed in the transiently proliferative epidermal intermediate and spinous cells, and its germline ablation results in a thickened epidermis with expanded early differentiating cells (spinous layers) and a transient delay in barrier acquisition that is resolved by birth. Studies using cultured human keratinocytes (KCs) and clinical samples have implicated a possible functional involvement of OVOL1 in atopic dermatitis (Furue et al., 2019; Tsuji et al., 2020, 2017). Specifically, OVOL1 was shown to regulate the expression of epidermal terminal differentiation genes FLG and LOR. However, the in vivo function of Ovol1/OVOL1 in skin homeostasis and inflammation has not been addressed.
Despite the long-held view of distinct mechanisms underlying each inflammatory skin disorder, overlapping molecular mechanisms that affect both psoriasis and atopic dermatitis exist (Guttman-Yassky and Krueger, 2017). In this work, we report elevated OVOL1/Ovol1 expression in lesional psoriatic human skin and in epidermal cells of mouse skin with psoriasis-like inflammation. We show that Ovol1 is largely dispensable for adult epidermal homeostasis, but its deletion significantly aggravates imiquimod (IMQ)-induced psoriasis-like skin defects featuring epidermal hyperplasia and neutrophil accumulation. Using bulk and single-cell (sc) RNA sequencing (RNA-seq) coupled with functional studies, we identify aberrant IL-1α expression and signaling as an important mediator of the exacerbated inflammation and epidermal hyperplasia in Ovol1-deficient mice.
RESULTS
OVOL1/Ovol1 expression is upregulated in psoriatic skin
To seek clues for a functional involvement of OVOL1/Ovol1 in psoriasis, we examined our previously published RNA-seq data on human psoriasis skin samples (Yu et al., 2020). OVOL1 expression was found to be upregulated by >2-fold in psoriatic skin compared with normal skin (Figure 1a). We also interrogated a public dataset on paired nonlesional and lesional skin from human patients with psoriasis (Correa da Rosa et al., 2017; Suárez-Fariñas et al., 2012) and observed significantly higher OVOL1 expression in the lesional skin (Figure 1b). To determine if this is the case in an animal model of psoriasis, we turned to IMQ-treated mouse back skin. IMQ is a toll-like receptor 7/8 agonist and its short-term (5- to 7-day-long) application induces dermatitis with phenotypic and histological resemblance to human psoriasis lesions, including hyperkeratosis, erythema, scaling, neutrophil microabscesses in epidermis, and infiltration of γδ T cells and T helper type 17 cells (Gilliet et al., 2004; Schön et al., 2006; Sumida et al., 2014; Swindell et al., 2017; van der Fits et al., 2009; Wu et al., 2004). We generated Ovol1-LacZ mice that express β-galactosidase downstream of the Ovol1 promoter and found that IMQ treatment results in visibly increased β-galactosidase activity in both interfollicular epidermis and hair follicles (Figure 1c). Thus, OVOL1/Ovol1 expression is upregulated in psoriatic skin of both human and mouse.
Figure 1. OVOL1/Ovol1 expression is upregulated in psoriatic skin.
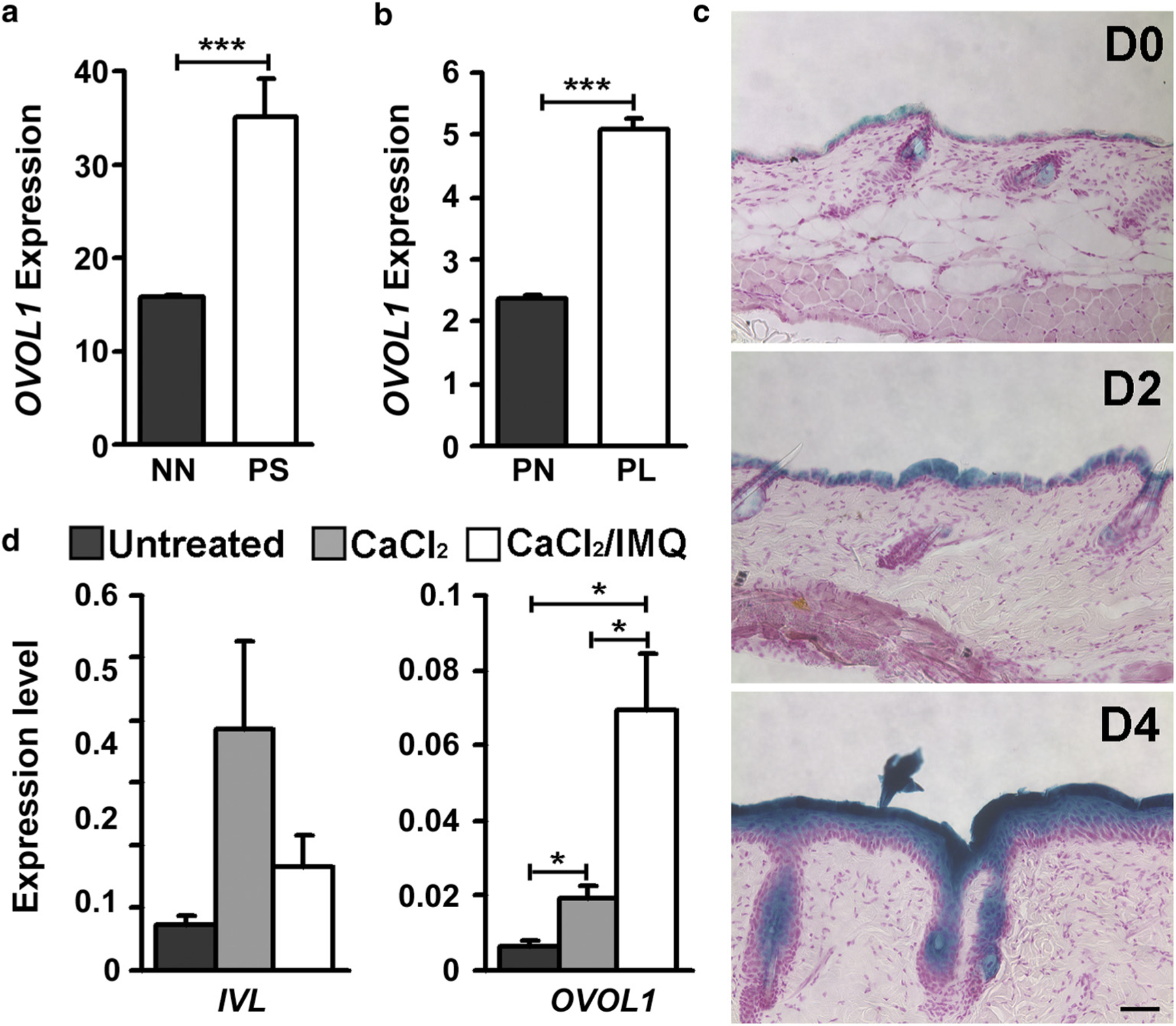
(a) OVOL1 expression in psoriatic PS taken from each patient and control NN skin samples obtained from surgical discard specimens of healthy people. n = 5. (b) OVOL1 expression in PL and PN skin of human patients with psoriasis. n = 81. (c) X-gal staining of β-galactosidase activity in skin of Ovol1-LacZ mice. n = 2–3 (shown are images from 8-week-old mice). Bar = 100 µm. (d) RT-qPCR analysis of the indicated genes in human foreskin keratinocytes following calcium (1.2 mM for 30 hours) and IMQ (50 µg/ml for 6 hours) treatment. Data are from three independent experiments. ***P < 0.005, *P < 0.05. D, day after IMQ treatment; IMQ, imiquimod; IVL, involucrin; NN, normal nonpsoriatic; PL, paired lesional; PN, paired nonlesional; PS, plaque skin.
We previously showed that calcium, which is known to induce epidermal differentiation, induces Ovol1 expression in mouse keratinocytes (Dai et al., 1998). Using human keratinocytes isolated from neonatal foreskin, we found that calcium increased the level of OVOL1 transcript by ~3-fold, but calcium together with IMQ elicited a ~10-fold increase (Figure 1d). This finding suggests that IMQ can directly upregulate OVOL1 expression in epidermal cells.
Ovol1 deficiency aggravates IMQ-induced epidermal hyperplasia and inflammatory response
We next investigated Ovol1 function in adult skin by analyzing Ovol1−/− mice on a CD1 outbred background because of perinatal lethality in a C57BL/6 strain background (Dai et al., 1998; Nair et al., 2006). Histology of, and number of Ki67+ proliferative cells in, Ovol1−/− epidermis were largely similar to those in the littermate control epidermis (Figure 2a–c). However, a slight increase in the ratio between suprabasal and basal cells in Ovol1−/− mice compared with the controls (Figure 2d–f), suggesting a very mild alteration in KC differentiation.
Figure 2. Epidermal cellular defects in Ovol1-deficient mice.

(a) H&E images of skin from 7-week-old Ovol1+/+ and Ovol1−/− littermates. Bar = 10 µm. (b, c) Ki67 immunostaining. Shown are (b) representative images and (c) quantification of percentage of proliferating basal cells (n = 4 pairs of Ovol1−/− and control littermates). (d) K1/K14 and (e) loricrin/K14 double immunostaining. (f) Quantitative analysis of the ratio between suprabasal (K1+ spinous and K14−K1− granular) and K14+ basal cells (n = 3 pairs of Ovol1−/− and control littermates). The number of basal cells was set as 100. DAPI stains the nuclei. (b), (d), and (e) Bar = 50 µm. K, keratin.
Next, we asked whether Ovol1 deletion impacts IMQ-induced skin inflammation and the associated epidermal hyperplasia. Over a 6-day course of IMQ treatment, psoriasis-like features including erythema and scaling were significantly more pronounced in Ovol1−/− mice than in control littermates (Figure 3a and b; Supplementary Figure S1a). H&E and immunostaining analysis revealed that IMQ-treated Ovol1−/− epidermis launched a hyperproliferative response faster than control littermates (Figure 3c–e). IMQ-induced defects in terminal differentiation were also more prominent in Ovol1−/− skin, shown by lower levels of the differentiation marker K10 (Figure 3f). Moreover, the aberrant upregulation of LM332 protein production as an indicator of tissue repair (Iorio et al., 2015) was detected earlier in the skin of IMQ-treated Ovol1−/− mice compared with control littermate skin (Figure 3g; Supplementary Figure S1b). Flow cytometry performed on whole skin revealed a 3.5-fold increase in CD45+ (immune) cells, a 16.5-fold increase in CD45+/Ly6G+/CD11b+ neutrophils, and a trending increase in CD45+/F4/80+/CD11b+/Ly6G− macrophages in IMQ-treated Ovol1−/− skin compared with control littermate skin (Figure 3h; Supplementary Figure S1c). Together, these data indicate that Ovol1 loss leads to rapid and severe psoriasis-like symptoms in response to IMQ.
Figure 3. IMQ-induced skin phenotypes in Ovol1−/− and control littermates.

(a) External appearance, (b) cumulative scoring, and (c) skin histology before and at different days after IMQ treatment. n = 4–5 pairs. (d) Quantification of epidermal thickness at different time points after a single IMQ application. n = 3–5 per time point. (e–g) Immunostaining at 24 hours after the (e, g) first and (f) third IMQ applications. (h) Summary of flow analysis data collected at 24 hours after the second IMQ application. Left: number of the indicated cell types per 1 × 105 living cells. Right: percentages of CD45+ cells per living cells, neutrophils (CD45+/Ly6G+/CD11b+) per CD45+ cells, and macrophages (CD45+/Ly6G−/CD11b+/F4/80+) per CD45+ cells. n = 4 pairs. (c), (e–g) Bar = 50 µm. ***P < 0.005; **P < 0.01; *P < 0.05. D, day; IMQ, imiquimod.
Significantly elevated inflammatory gene expression in IMQ-treated Ovol1−/− epidermis
To elucidate the molecular nature of Ovol1 deficiency‒induced changes in skin, we performed RNA-seq analysis on dispase-isolated epidermis from Ovol1−/− mice and control littermates that were either untreated or 6 hours after a single IMQ treatment (when epidermal hyperproliferation was not yet apparent; Figure 3d). Principle component analyses revealed that IMQ treatment and sex of the mice analyzed are major sources of variation in gene expression (Figure 4a; Supplementary Figure S2a). Dramatic differences in gene expression between IMQ-treated Ovol1−/− and control littermate epidermis were observed (Figure 4a and b; Supplementary Table S1). Molecular differences between untreated Ovol1−/− and control littermate epidermis were also detected but were considerably less prominent than those with IMQ treatment (Figure 4a and b; Supplementary Table S1).
Figure 4. Gene expression in Ovol1−/− and Ctrl littermate epidermis.

(a) PCA showing epidermal gene expression differences between untreated and IMQ-treated Ovol1−/− and Ctrl littermates. (b) Volcano plots showing DE genes between Ctrl and Ovol1−/− epidermis from untreated and IMQ-treated mice (n = 2 per genotype: 1 male and 1 female). Genes were filtered for subsequent analysis using the following cut-off: fold change > 2; adjusted P-value < 0.05. (c, d) Functional annotation of genes that were upregulated in IMQ-treated Ovol1−/− epidermis using Hallmark gene sets provided by (c) MSigDB and (d) GO terms in Biological Process 2018. (e) GO terms associated with genes upregulated in untreated Ovol1−/− epidermis. Shown are the top 10 functional terms and the vertical lines correspond to p = 0.05. Ctrl, control; DE, differentially expressed; GO, Gene Ontology; IMQ, imiquimod; MSigDB, Molecular Signatures Database; PCA, principal component analysis.
To identify the associated biological processes and pathways, we annotated genes differentially expressed between Ovol1−/− and control littermate epidermis using Molecular Signatures Database to reveal the hallmark gene sets and using Enrichr to probe the GO_Biological_Process_2018 library (Chen et al., 2013; Kuleshov et al., 2016) (Supplementary Table S2). Gene signatures related to “inflammatory response” and “regulation of inflammatory response” were upregulated in IMQ-treated and untreated Ovol1−/− epidermis, respectively, relative to control counterparts (Figure 4c–e). Also enriched in IMQ-treated Ovol1−/− epidermis were Gene Ontology (GO) terms of “TNF-α signaling”, “cytokine-mediated signaling pathway”, “chemokine-mediated signaling pathway”, “regulation of cell proliferation”, “skin development”, and “keratinocyte differentiation” (Figure 4d). Specific upregulated genes in these GO terms (e.g., Tgfa, Slpi, Aqp3, Ccl20, Krt16, and Lce3c/d/e) are also upregulated in human psoriatic skin (Supplementary Table S3; Figure 1a). A number of inflammatory cytokine and chemokine genes, such as Il1a (3.6×, P < 10−16) and Tgfa (2.3×, P = 0.0001), were upregulated in IMQ-treated Ovol1−/− epidermis, whereas the expression of known psoriasis-associated cytokine genes Il17, Il23, Il1b, and Il6 was not affected at this early time point. Moreover, the expression of Flg and Lor, whose human counterparts are known to be regulated by OVOL1 (Furue et al., 2019; Tsuji et al., 2017), was not significantly affected by Ovol1 deletion in untreated and IMQ-treated epidermis. GO terms that were more enriched in IMQ-treated control littermate epidermis compared with Ovol1−/− epidermis included “interferon alpha response”, “cholesterol homeostasis”, “interferon gamma response”, “skin development”, and “keratinocyte differentiation” (Supplementary Figure S2b and c). GO terms that were more enriched in untreated control epidermis than Ovol1−/− epidermis included “positive regulation of cAMP metabolic process” and “interleukin-18-mediated signaling pathway” (Supplementary Figure S2d). Taken together, our bulk RNA data identify enhanced inflammation and altered KC differentiation as major effects of Ovol1 loss in epidermis.
scRNA-seq analysis reveals altered gene expression in epidermal basal, suprabasal, fibroblast, and immune cells of the IMQ-treated Ovol1−/− skin
Next, we molecularly profiled the skin of Ovol1−/− mice and control littermates at 24 hours after IMQ treatment, but this time using the 10X Genomics scRNA-seq platform to achieve a single-cell resolution. After quality control (Supplementary Materials and Methods; Supplementary Figure S3a), a total of 23,395 cells from four distinct conditions (6,289, 5,553, 5,857, and 5,696 cells from untreated control littermate, untreated Ovol1−/−, IMQ-treated control littermate, and IMQ-treated Ovol1−/− mice, respectively) were aggregated for subsequent analysis (Supplementary Figure S3b). In the overall t-distributed stochastic neighbor embedding plot, the major cell types identified were epithelial cells (Krt14+ or Krt1+; 42.4%) and fibroblasts (Col1a2high; 46.6%) (Figure 5a, top, and 5b; Supplementary Figure S3b and c). Epithelial cells or fibroblasts from untreated Ovol1−/− skin intermingled with the same cell type from untreated control littermate skin, whereas IMQ elicited differential gene expression resulting in the formation of clusters of epithelial cells or fibroblasts that were distinct from the IMQ-treated Ovol1−/− and control littermate skin (Figure 5a, bottom).
Figure 5. scRNA-seq analysis of homeostatic versus inflamed Ovol1−/− and Ctrl littermate skin.

(a) tSNE plots showing all cells under analysis that has passed quality control, labeled by cell type (top) and condition (bottom). Ctrl-UT and Ctrl-IMQ, untreated and IMQ-treated Ctrl littermate (Ovol1+/−) skin, respectively. MUT-UT and MUT-IMQ, untreated and IMQ-treated Ovol1−/− skin, respectively. (b) Feature plots showing expression of the indicated marker genes as in (a). (c) tSNE map of aggregated epidermal suprabasal cells, labeled by clusters (top) and conditions (bottom). (d) Quantification of Krt14/Krt1 single- or double-positive epidermal cells under the four conditions. (e) Heatmap for the top 10 genes enriched in each cluster from (c). All marker genes are listed in Supplementary Table S4. (f) Violin plots showing expression of the indicated pathway gene signatures or genes in suprabasal keratinocytes across the four conditions. Ctrl, control; IMQ, imiquimod; MUT, mutant; RNA-seq, RNA sequencing; sc, single-cell; tSNE, t-distributed stochastic neighbor embedding; UT, untreated.
Given that Ovol1 is predominantly expressed in epidermal suprabasal cells, Krt1high (suprabasal spinous cell) clusters from each sample (untreated or IMQ-treated control and Ovol1−/−) were computationally isolated, and those from all four conditions were aggregated together in a combined dataset (Supplementary Figure S3b). Suprabasal cells from the two untreated genotypes were well mixed in cluster 0, but those from IMQ-treated control skin were enriched in cluster 1 and those from IMQ-treated Ovol1−/− skin were enriched in cluster 2 (Figure 5c), indicating that IMQ induced distinct gene expression changes in control and mutant suprabasal cells. We also noted that IMQ-treated Ovol1−/− skin contained a higher number of epidermal cells that expressed both Krt1 and Krt14 than the other conditions, a finding validated by immunofluorescence detecting appreciable presence of keratin 1+/keratin 14+ cells in intact Ovol1−/− skin (Figure 5d; Supplementary Figure S3d). Top marker genes of the IMQ-treated Ovol1−/− suprabasal cells included proinflammatory genes S100a8 and S100a9 (Ryckman et al., 2003; Wang et al., 2018) and keratin-encoding genes Krt6a, Krt6b, and Krt16 that have been shown to be upregulated in hyperproliferative or injured epidermis (Chen et al., 2019; Esaki et al., 2015; Lessard et al., 2013) (Figure 5e; Supplementary Table S4). Notably, the same S100a and keratin genes have been shown by scRNA-seq to be upregulated in the suprabasal cells of human psoriatic skin (Cheng et al., 2018). Epidermal basal cell clusters (Krt14high/Krt17−/CD34−/Sostdc1−) were also computationally isolated and aggregated together for analysis (Supplementary Figure S3b and e). Again, IMQ elicited distinct gene expression changes in Ovol1−/− basal cells and control littermate counterparts, featuring upregulation of inflammation- and hyperproliferation-associated genes, including S100a8, S100a9, and Krt6a in the former (Supplementary Figure S3e and f; Supplementary Table S5).
We next analyzed the epidermal expression of select gene signatures associated with proliferation, differentiation, metabolism, and inflammation (Supplementary Table S6). Specifically, G2/M cell cycle checkpoint genes were enriched in basal cells, whereas the epidermal differentiation process was enriched in suprabasal cells, of the IMQ-treated Ovol1−/− skin (Figure 5f; Supplementary Figure S3g). Of note, expression of the early differentiation gene Krtdap was slightly upregulated in basal cells of untreated, but dramatically upregulated in basal cells of IMQ-treated, Ovol1−/− skin. Genes involved in metabolic processes (glycolysis, oxidative phosphorylation) were considerably more active in IMQ-treated Ovol1−/− epidermal cells in both basal and suprabasal compartments. IMQ-treated Ovol1−/− skin scored the highest for an inflammatory response‒associated gene set in their suprabasal spinous cells, whereas IMQ-treated control skin scored the highest for this gene set in their basal keratinocytes, raising the possibility that Ovol1 deletion switches the major site of KC inflammatory response from basal cells to spinous cells. Overall, the molecular changes revealed by scRNA-seq analysis suggest significantly elevated rates of cellular flux characterized by increased basal cell proliferation and early differentiation, inefficient basal-to-mature spinous transition, and enhanced metabolism- and inflammation-associated gene expression in IMQ-treated Ovol1−/− skin epidermis.
To further investigate how Ovol1 loss impacts IMQ-induced changes in skin, fibroblasts from the four conditions were computationally aggregated and analyzed (Supplementary Figure S3b). A total of four clusters were observed: clusters 0 and 3 contained fibroblasts from untreated control and Ovol1−/− skin, whereas clusters 2 and 1 were enriched with fibroblasts from IMQ-treated control and Ovol1−/− skin, respectively (Supplementary Figure S3h). Top markers in IMQ-treated Ovol1−/− fibroblasts included inflammation-associated genes such as Saa3 and monocyte chemoattractant Ccl2 (Kratofil et al., 2017; Sack, 2018) (Supplementary Figure S3i; Supplementary Table S7). Moreover, IMQ induced a slight increase in the inflammatory response gene set score in control fibroblasts but a dramatic increase in Ovol1−/− fibroblasts (Supplementary Figure S3j).
Few immune cells were recovered in our 24-hour post-IMQ scRNA-seq analysis (Figure 5a and b). However, when these cells were computationally aggregated for analysis, it was still apparent that IMQ induced distinct gene expression changes in immune cells from Ovol1−/− and control littermate skin (Supplementary Figures S3b and S4a). Cd3g+ T cells in IMQ-treated Ovol1−/− skin showed elevated expression of Il17a and Il17f compared with counterparts from the other three conditions (Supplementary Figure S4b–d; Supplementary Table S8), suggesting a heightened T helper type 17 response in the Ovol1 mutant. Together, these findings show that Ovol1 deficiency results in an overall more inflammatory skin microenvironment affecting the responses of not only epidermal cells but also fibroblasts and immune cells.
Inhibiting IL-1α signaling suppresses IMQ-induced skin inflammation in Ovol1-deficient mice
IL-1 signaling represents a key proinflammatory pathway in myriad skin inflammatory processes (Cai et al., 2019; Murphy et al., 2000). The scRNA-seq data enabled us to specifically assess IL-1 pathway activity through examining the expression of ligands (Il1a, Il1b) and receptors (Il1r1, Il1r2) in different skin cell types. Expression of Il1a was barely detectable in the single-cell dataset, but Il1b expression was seen in the non-T immune cell cluster that are likely resident macrophage and dendritic cells (Supplementary Figure S5a). Il1r1 was expressed in most of the cell types with a slight fibroblast enrichment, whereas Il1r2 expression was highly enriched in the fibroblasts (Supplementary Figure S5a and b). Il1r2 expression responded differently to IMQ treatment in control and Ovol1−/− skin, characterized by increased expression in control epidermal basal and suprabasal cells and fibroblasts but decreased expression in Ovol1−/− epidermal cells and no detectable change in Ovol1−/− fibroblasts. In contrast, changes in Il1r1 expression were nonremarkable (Supplementary Figure S5b). Additionally, IMQ induced overall similar changes in the expression of Il1b and Ilr1/2 in immune cells of control and Ovol1−/− mice (Supplementary Figure S5c). Because Il1r2 encodes an IL-1 decoy receptor that inhibits IL-1α/β signaling (Schlü ter et al., 2018), our findings suggest that the absence of Ovol1 results in a skin microenvironment that is no longer able to efficiently suppress IL-1 signaling when chemically challenged.
Despite the inability of scRNA-seq to detect Il1a transcripts, RNAscope analysis confirmed the bulk RNA-seq‒revealed upregulation of Il1a mRNA in granular cells of Ovol1−/−, but not control, skin at 6 hours after IMQ treatment (Figure 6a). Furthermore, IL-1α protein was detected at greatly enhanced levels in loricrin-positive epidermal granular cells of Ovol1−/− skin compared with control skin at 24 hours after IMQ treatment (Figure 6b and c). Western blotting showed that the full-length IL-1α precursor protein, but not the mature form, was predominantly elevated (Figure 6d). These results identify epidermal granular cells as the major source of IL-1α in inflamed Ovol1-deficient skin.
Figure 6. Expression and function of Il1a/IL-1α in Ovol1-deficient skin.

(a) RNAscope of Ovol1−/− and control skin at 6 hours after IMQ using the indicated probes (color-matched). Loricrin antibody stains granular cells. Arrows: Il1a signals. Bar = 25 µm. (b, c) Immunostaining of Ovol1−/− and control skin at 24 hours after IMQ treatment. Bar = 50 µm. (d) Western blotting for IL-1α (top) and β-tubulin (bottom). (e) External appearance and (f) cumulative scoring of IMQ-treated Ovol1−/− skin after IL-1Ra or PBS administration (n = 5 each). (g) Histology, (h) epidermal thickness, and (i) immune cell quantification of IMQ-treated Ovol1−/− skin after IL-1Ra or PBS administration (n = 4 each). (g) Bar =100 µm. **P < 0.01; *P < 0.05. Ctrl, control; IMQ, imiquimod; UT, untreated.
IL-1α is a dual function cytokine; its secreted and membrane-bound forms signal through the IL-1R1 (Il1r1) receptor, and its N-terminal calpain cleavage product translocates into the nucleus to regulate chromatin structure and transcription (Rider et al., 2013; Werman et al., 2004). To ask whether the excessively produced, full-length IL-1α in IMQ-treated Ovol1−/− skin epidermis acts through signaling, we pharmacologically inhibited the IL-1 signaling pathway. Specifically, we administered recombinant IL-1Ra, an antagonist of IL-1α/β through competitive receptor binding (Petrasek et al., 2012), or PBS via daily topical skin applications at 30 minutes before IMQ application and every 24 hours until the end of experiments (24 hours after the second IMQ application). IL-1Ra treatment resulted in the normalization of psoriasis-like phenotypes of IMQ-treated Ovol1−/− skin, manifested by reductions in erythema, scaling, epidermal thickness, and number of immune cells (neutrophils in particular) (Figure 6e–i; Supplementary Figure S6). Together, these data show that the excessive full-length IL-1α protein produced by the epidermal granular cells in inflamed Ovol1−/− skin functions via signaling to mediate the exaggerated inflammation, immune cell infiltration, and associated epidermal hyperproliferation.
DISCUSSION
Our work uncovers an important function of the OVOL1 transcription factor in regulating psoriasis-like skin inflammation in vivo. During normal epidermal homeostasis, Ovol1 function is largely dispensable, possibly reflecting the existence of compensatory mechanisms. However, Ovol1 loss predisposes skin to exacerbated psoriasis-like inflammation and epidermal hyperplasia in response to IMQ. Based on our mouse model data suggesting a protective role of Ovol1, we speculate that OVOL1 may also play a protective role in human skin, such that its expression is upregulated in psoriatic skin to dampen disease progression and/or severity. Future work outside the scope of this study will be needed to address this intriguing possibility.
Ovol1 expression is upregulated in the epidermal spinous cells of IMQ-treated skin. Hence, our gene expression analysis focused on the early changes, which likely represent the more immediate effects of Ovol1 loss in the epidermis rather than secondary consequences of disease progression. Both our bulk and scRNA-seq experiments revealed increased expression of inflammation-associated genes and epidermal differentiation genes in the absence of Ovol1 shortly after IMQ treatment. Some of the molecular changes (e.g., Krt16, Lce3c/d/e, S100a8/9) are shared by human psoriatic skin, implicating the potential clinical relevance of our mouse model findings. Moreover, genes upregulated in IMQ-treated Ovol1−/− skin at the early time points (6–24 hours after IMQ in our analysis) also show some overlap (e.g., Lce3c/d/e, S100a8/9, Slpi) with those upregulated in skin at 6 days after IMQ treatment (Swindell et al., 2017). This said, our data sets present unique changes that likely reflect the specific effects of Ovol1 loss and/or the early acute events in psoriasis-like skin inflammation.
Our work also presents a snapshot of the molecular and cellular changes of IMQ-induced skin inflammation at a single-cell resolution. Not surprisingly, inflammatory responses to IMQ are reprogrammed by loss of Ovol1 not only in epidermal cells but also in fibroblasts and immune cells. Of particular interest, although epidermal cells and fibroblasts in IMQ-treated skin are capable of suppressing IL-1 signaling, these cells in Ovol1-deficient skin are in a state that is highly conductive to the excessive IL-1α signals produced by KCs. Our data provide a useful resource for dissecting the complex signaling cross-talk among the various resident cell types in inflamed mouse skin.
We have previously shown that Ovol1 is required for efficient cell cycle exit of embryonic epidermal progenitor cells (Nair et al., 2006). Ovol1 may function in a similar, cell-autonomous manner in IMQ-treated skin to help prevent excessive epidermal proliferation. Additionally, or alternatively, altered KC‒immune cell cross-talk may contribute to the aggravated epidermal hyperplasia in IMQ-treated Ovol1-deficient mice through feedback mechanisms. Ovol1-deficient epidermal cells in IMQ-treated mice show an expansion of the Krt14/Krt1 double-positive subpopulation and molecular features of increased G2/M cell cycle transition, differentiation, and metabolism, suggesting that loss of Ovol1 in an inflammatory or stressed environment traps the KC in a state between active cycling and terminal differentiation.
The phenotypic rescue of Ovol1 mutant mice by IL-1Ra administration supports the functional importance of excessive full-length IL-1α signaling in mediating the aggravated skin inflammation and epidermal hyperplasia in these mice. Moreover, the finding that topical application of a recombinant IL-1Ra protein can be effective at symptom relief raises the possibility of such use in disease treatment. Given the association of OVOL1 SNPs with multiple inflammatory skin diseases, our study lays the groundwork for future investigation into the functional contribution of OVOL1 to disease pathogenesis and the underlying molecular mechanisms.
MATERIALS AND METHODS
Mice
Ovol1-LacZ (Ovol1tm1a(KOMP)Wtsi) mice in a C57BL/6N background, where a cassette composed of an Frt site, a LacZ sequence, and loxP sites is inserted into the Ovol1 locus, were purchased from UC Davis KOMP Repository (https://www.komp.org/). Ovol1+/− mice in a C57BL/6 strain background (Dai et al., 1998; Nair et al., 2006) were outcrossed onto a CD1 strain background and then intercrossed to produce homozygous mutant (Ovol1−/−) progeny for study. CD1 Ovol1−/− mice survive to adulthood but are sometimes smaller than control littermates, so sex- and weight-matched control and mutant littermates were used for all analyses. Information for all genotyping primers and control littermate genotypes used in each analysis is provided in Supplementary Tables S9 and S10. All animal studies have been approved and abide by regulatory guidelines of the Institutional Animal Care and Use Committee of the University of California, Irvine.
IMQ-induced psoriasis model
Mice at 7–8 weeks of age received a daily topical dose of 62.5 mg 5% IMQ cream (Perrigo, Allegan, MI) on shaved backs for six consecutive days or as indicated. Based on a previously described objective scoring system called PASI (van der Fits et al., 2009), erythema (redness of skin) and scaling (approximated by dry, white cracks and patches on skin surface) were blindly scored independently by one or more investigators, on a score from 0 (none) to 4 (most severe). The cumulative score (erythema plus scaling) served as a measure of the severity of clinical signs (score, 0–8).
Flow cytometry
To obtain single-cell suspensions, minced samples were digested with 10 ml of a solution containing 0.25% collagenase (Sigma, St. Louis, MO; C9091), 0.01 M HEPES (Thermo Fisher Scientific, Waltham, MA; BP310), 0.001 M sodium pyruvate (Thermo Fisher Scientific; BP356), and 0.1 mg/ml DNase (Sigma; DN25) at 37 °C for 1 hour with rotation, and then filtered through a 70-µm filter, spun down, and resuspended in 2% fetal bovine serum. A total of 5 × 105 cells were stained by incubation for 30 minutes at room temperature with the following antibodies diluted in PBS/2% fetal bovine serum: Alexa Fluor 488-conjugated anti-CD11b (BioLegend, San Diego, CA; 101217), phycoerythrin-conjugated anti-F4/80 (BioLegend; 123110), allophycocyanin-conjugated anti-CD45 (Tonbo Biosciences, San Diego, CA; clone 30-F11, 20–0451), allophycocyanin-Cy7-conjugated anti-Ly6G (Tonbo Biosciences; clone 1A8, 25–1276), 7-aminoactinomycin (BD Biosciences, San Jose, CA; 559925), and phycoerythrin-conjugated anti-CD49f (BD Biosciences; 555736).
Bulk RNA-Seq and scRNA-Seq
These experiments were performed as previously described (Haensel et al., 2020; Lee et al., 2014). For scRNA-seq, single cells were isolated from normal and inflamed back skin of adult mice, and live cells were FACS-sorted for analysis. Additional details are described in the Supplementary Materials and Methods.
In vivo administration of recombinant IL-1Ra protein
Purified IL-1Ra recombinant protein was produced as previously described (Zhang et al., 2009) and was dissolved in sterilized water to a concentration of 15 mg/ml. Same-sex/same-weight Ovol1−/− littermates received topical applications of IL-1Ra (40 mg/kg) or PBS on shaved back skin once at 30 minutes before IMQ treatment and then once every 24 hours until the end of experiments. Skin samples were fixed in 4% paraformaldehyde for H&E staining or embedded in OCT and frozen for immunostaining analysis.
Statistics and reproducibility
Most experiments were performed on at least three biological replicates or repeated at least twice. The sample size and number of independent experiments are indicated in the relevant figure legends. Disease pathology scoring was performed blindly. No data were excluded. For analysis of differences between groups, Student’s unpaired t-test was performed with 2-tailed in Excel. P-values of 0.05 or less were considered statistically significant. Error bars represent mean 士 SEM. For bulk RNA-seq analysis, genes with a log2 fold change ≥1 and an adjusted P-value < 0.05 (generated by the negative binomial generalized models and Wald test employed by DESeq2) were considered significant and used to generate GO terms.
Additional details for these methods, as well as methods for β-galactosidase assay, human foreskin KC culture and RT-PCR, histology, immunostaining, western blotting, and RNAscope are described in Supplementary Materials and Methods.
Supplementary Material
ACKNOWLEDGMENTS
We thank Anand Ganesan and Sebastien de Feraudy for inspiration and advice, the Genomics High Throughput Facility and the Institute for Immunology FACS Core Facility at the University of California, Irvine (UCI) for expert service. This work was supported by National Institutes of Health (NIH) grants R01-AR068074 and R01-GM123731 (XD). RV, MD, and DH were partially supported by UCI National Science Foundation (NSF)-Simons Center for Multiscale Cell Fate Research through NSF grant DMS1562176 and Simons Foundation grant 594598 (QN). RV is a recipient of NSF predoctoral fellowship (DGE-1839285), and MD is partially supported by the NIH T32 Immunology Research Training Grant (AI 060573).
Abbreviations:
- GO
Gene Ontology
- IMQ
imiquimod
- RNA-seq
RNA sequencing
- sc
single-cell
Footnotes
CONFLICT OF INTEREST
The authors state no conflicts of interest.
SUPPLEMENTARY MATERIAL
Supplementary material is linked to the online version of the paper at www.jidonline.org, and at https://doi.org/10.1016/j.jid.2020.10.025.
Data availability statement
Datasets related to this article are deposited in the Gene Expression Omnibus database under accession code GSE158112. Code for single-cell analysis will be provided on request.
REFERENCES
- Ayala-Fontánez N, Soler DC, McCormick TS. Current knowledge on psoriasis and autoimmune diseases. Psoriasis (Auckl) 2016;6:7–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benhadou F, Mintoff D, Del Marmol V. Psoriasis: keratinocytes or immune cells - which is the trigger? Dermatology 2019;235:91–100. [DOI] [PubMed] [Google Scholar]
- Bergboer JGM, Zeeuwen PLJM, Schalkwijk J. Genetics of psoriasis: evidence for epistatic interaction between skin barrier abnormalities and immune deviation. J Invest Dermatol 2012;132:2320–31. [DOI] [PubMed] [Google Scholar]
- Cai Y, Xue F, Quan C, Qu M, Liu N, Zhang Y, et al. A critical role of the IL-1β‒IL-1R signaling pathway in skin inflammation and psoriasis pathogenesis. J Invest Dermatol 2019;139:146–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, et al. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 2013;14:128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JG, Fan HY, Wang T, Lin LY, Cai TG. Silencing KRT16 inhibits keratinocyte proliferation and VEGF secretion in psoriasis via inhibition of ERK signaling pathway. Kaohsiung J Med Sci 2019;35:284–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng JB, Sedgewick AJ, Finnegan AI, Harirchian P, Lee J, Kwon S, et al. Transcriptional programming of normal and inflamed human epidermis at single-cell resolution. Cell Rep 2018;25:871–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correa da Rosa J, Kim J, Tian S, Tomalin LE, Krueger JG, Suárez-Fariñ as M. Shrinking the psoriasis assessment gap: early gene-expression profiling accurately predicts response to long-term treatment. J Invest Dermatol 2017;137:305–12. [DOI] [PubMed] [Google Scholar]
- Dai X, Schonbaum C, Degenstein L, Bai W, Mahowald A, Fuchs E. The ovo gene required for cuticle formation and oogenesis in flies is involved in hair formation and spermatogenesis in mice. Genes Dev 1998;12:3452–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esaki H, Ewald DA, Ungar B, Rozenblit M, Zheng X, Xu H, et al. Identification of novel immune and barrier genes in atopic dermatitis by means of laser capture microdissection. J Allergy Clin Immunol 2015;135:153–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furue K, Ito T, Tsuji G, Ulzii D, Vu YH, Kido-Nakahara M, et al. The IL-13‒OVOL1‒FLG axis in atopic dermatitis. Immunology 2019;158:281–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilliet M, Conrad C, Geiges M, Cozzio A, Thürlimann W, Burg G, et al. Psoriasis triggered by toll-like receptor 7 agonist imiquimod in the presence of dermal plasmacytoid dendritic cell precursors. Arch Dermatol 2004;140:1490–5. [DOI] [PubMed] [Google Scholar]
- Gonzales KAU, Fuchs E. Skin and its regenerative powers: an alliance between stem cells and their niche. Dev Cell 2017;43:387–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guttman-Yassky E, Krueger JG. Atopic dermatitis and psoriasis: two different immune diseases or one spectrum? Curr Opin Immunol 2017;48:68–73. [DOI] [PubMed] [Google Scholar]
- Haensel D, Jin S, Sun P, Cinco R, Dragan M, Nguyen Q, et al. Defining epidermal basal cell states during skin homeostasis and wound healing using single-cell transcriptomics. Cell Rep 2020;30:3932–47.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirota T, Takahashi A, Kubo M, Tsunoda T, Tomita K, Sakashita M, et al. Genome-wide association study identifies eight new susceptibility loci for atopic dermatitis in the Japanese population. Nat Genet 2012;44:1222–6. [DOI] [PubMed] [Google Scholar]
- Iorio V, Troughton LD, Hamill KJ. Laminins: roles and utility in wound repair. Adv Wound Care (New Rochelle) 2015;4:250–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang Z, Li Q, Fu P, Yan S, Guan M, Xu J, et al. Correlation of KIF3A and OVOL1, but not ACTL9, with atopic dermatitis in Chinese pediatric patients. Gene 2015;571:249–51. [DOI] [PubMed] [Google Scholar]
- Kobayashi T, Naik S, Nagao K. Choreographing immunity in the skin epithelial barrier. Immunity 2019;50:552–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kratofil RM, Kubes P, Deniset JF. Monocyte conversion during inflammation and injury. Arterioscler Thromb Vasc Biol 2017;37:35–42. [DOI] [PubMed] [Google Scholar]
- Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res 2016;44:W90–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee B, Villarreal-Ponce A, Fallahi M, Ovadia J, Sun P, Yu QC, et al. Transcriptional mechanisms link epithelial plasticity to adhesion and differentiation of epidermal progenitor cells. Dev Cell 2014;29:47–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lessard JC, Piña-Paz S, Rotty JD, Hickerson RP, Kaspar RL, Balmain A, et al. Keratin 16 regulates innate immunity in response to epidermal barrier breach. Proc Natl Acad Sci USA 2013;110:19537–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowes MA, Suarez-Fariñas M, Krueger JG. Immunology of psoriasis. Annu Rev Immunol 2014;32:227–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marenholz I, Esparza-Gordillo J, Rüschendorf F, Bauerfeind A, Strachan DP, Spycher BD, et al. Meta-analysis identifies seven susceptibility loci involved in the atopic march. Nat Commun 2015;6:8804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy JE, Robert C, Kupper TS. Interleukin-1 and cutaneous inflammation: a crucial link between innate and acquired immunity. J Invest Dermatol 2000;114:602–8. [DOI] [PubMed] [Google Scholar]
- Nair M, Teng A, Bilanchone V, Agrawal A, Li B, Dai X. Ovol1 regulates the growth arrest of embryonic epidermal progenitor cells and represses c-myc transcription. J Cell Biol 2006;173:253–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarini AA, Simpson MA, Weale M, Knight J, Carlavan I, Reiniche P, et al. Genome-wide association study identifies three novel susceptibility loci for severe acne vulgaris. Nat Commun 2014;5:4020. [DOI] [PubMed] [Google Scholar]
- Pasparakis M, Haase I, Nestle FO. Mechanisms regulating skin immunity and inflammation. Nat Rev Immunol 2014;14:289–301. [DOI] [PubMed] [Google Scholar]
- Paternoster L, Standl M, Chen CM, Ramasamy A, Bønnelykke K, Duijts L, et al. Meta-analysis of genome-wide association studies identifies three new risk loci for atopic dermatitis. Nat Genet 2011;44:187–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrasek J, Bala S, Csak T, Lippai D, Kodys K, Menashy V, et al. IL-1 receptor antagonist ameliorates inflammasome-dependent alcoholic steatohepatitis in mice. J Clin Invest 2012;122:3476–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rider P, Carmi Y, Voronov E, Apte RN. Interleukin-1α. Semin Immunol 2013;25:430–8. [DOI] [PubMed] [Google Scholar]
- Roberson ED, Bowcock AM. Psoriasis genetics: breaking the barrier. Trends Genet 2010;26:415–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryckman C, McColl SR, Vandal K, de Médicis R, Lussier A, Poubelle PE, et al. Role of S100A8 and S100A9 in neutrophil recruitment in response to monosodium urate monohydrate crystals in the air-pouch model of acute gouty arthritis. Arthritis Rheum 2003;48:2310–20. [DOI] [PubMed] [Google Scholar]
- Sack GH Jr. Serum amyloid A - a review. Mol Med 2018;24:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlüter T, Schelmbauer C, Karram K, Mufazalov IA. Regulation of IL-1 signaling by the decoy receptor IL-1R2. J Mol Med (Berl) 2018;96:983–92. [DOI] [PubMed] [Google Scholar]
- Schön MP, Schön M, Klotz KN. The small antitumoral immune response modifier imiquimod interacts with adenosine receptor signaling in a TLR7- and TLR8-independent fashion. J Invest Dermatol 2006;126:1338–47. [DOI] [PubMed] [Google Scholar]
- Suárez-Fariñas M, Li K, Fuentes-Duculan J, Hayden K, Brodmerkel C, Krueger JG. Expanding the psoriasis disease profile: interrogation of the skin and serum of patients with moderate-to-severe psoriasis. J Invest Dermatol 2012;132:2552–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumida H, Yanagida K, Kita Y, Abe J, Matsushima K, Nakamura M, et al. Interplay between CXCR2 and BLT1 facilitates neutrophil infiltration and resultant keratinocyte activation in a murine model of imiquimod-induced psoriasis. J Immunol 2014;192:4361–9. [DOI] [PubMed] [Google Scholar]
- Swindell WR, Michaels KA, Sutter AJ, Diaconu D, Fritz Y, Xing X, et al. Imiquimod has strain-dependent effects in mice and does not uniquely model human psoriasis. Genome Med 2017;9:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng A, Nair M, Wells J, Segre JA, Dai X. Strain-dependent perinatal lethality of Ovol1-deficient mice and identification of Ovol2 as a downstream target of Ovol1 in skin epidermis. Biochim Biophys Acta 2007;1772:89–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuji G, Hashimoto-Hachiya A, Kiyomatsu-Oda M, Takemura M, Ohno F, Ito T, et al. Aryl hydrocarbon receptor activation restores filaggrin expression via OVOL1 in atopic dermatitis. Cell Death Dis 2017;8:e2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuji G, Hashimoto-Hachiya A, Yen VH, Miake S, Takemura M, Mitamura Y, et al. Aryl hydrocarbon receptor activation downregulates IL-33 expression in keratinocytes via ovo-like 1. J Clin Med 2020;9:891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Fits L, Mourits S, Voerman JSA, Kant M, Boon L, Laman JD, et al. Imiquimod-induced psoriasis-like skin inflammation in mice is mediated via the IL-23/IL-17 axis. J Immunol 2009;182:5836–45. [DOI] [PubMed] [Google Scholar]
- Wang S, Song R, Wang Z, Jing Z, Wang S, Ma J. S100A8/A9 in inflammation. Front Immunol 2018;9:1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werman A, Werman-Venkert R, White R, Lee JK, Werman B, Krelin Y, et al. The precursor form of IL-1alpha is an intracrine proinflammatory activator of transcription. Proc Natl Acad Sci USA 2004;101:2434–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu JK, Siller G, Strutton G. Psoriasis induced by topical imiquimod. Australas J Dermatol 2004;45:47–50. [DOI] [PubMed] [Google Scholar]
- Yu Z, Gong Y, Cui L, Hu Y, Zhou Q, Chen Z, et al. High-throughput transcriptome and pathogenesis analysis of clinical psoriasis. J Dermatol Sci 2020;98:109–18. [DOI] [PubMed] [Google Scholar]
- Zhang J, Xiang D, Zhu S, Mao W, Lu H, Wu M, et al. Interleukin 1 receptor antagonist inhibits normal hematopoiesis and reduces lethality and bone marrow toxicity of 5-fluouracil in mouse. Biomed Pharmacother 2009;63: 501–8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Datasets related to this article are deposited in the Gene Expression Omnibus database under accession code GSE158112. Code for single-cell analysis will be provided on request.