

SUMMARY
CRISPR screens have accelerated the discovery of important cancer vulnerabilities. However, single-gene knockout phenotypes can be masked by redundancy among related genes. Paralogs constitute two-thirds of the human protein-coding genome, so existing methods are likely inadequate for assaying a large portion of gene function. Here, we develop paired guide RNAs for paralog genetic interaction mapping (pgPEN), a pooled CRISPR-Cas9 single- and double-knockout approach targeting more than 2,000 human paralogs. We apply pgPEN to two cell types and discover that 12% of human paralogs exhibit synthetic lethality in at least one context. We recover known synthetic lethal paralogs MEK1/MEK2, important drug targets CDK4/CDK6, and other synthetic lethal pairs including CCNL1/CCNL2. Additionally, we identify ten tumor suppressor paralog pairs whose compound loss promotes cell proliferation. These findings nominate drug targets and suggest that paralog genetic interactions could shape the landscape of positive and negative selection in cancer.
Graphical Abstract

In brief
Parrish et al. present pgPEN, a method for identifying paralog genetic interactions, and find that human duplicate genes are enriched for functional redundancy and essentiality. These findings provide a framework for identifying synthetic lethal gene pairs and nominate cancer drug targets.
INTRODUCTION
CRISPR-Cas9 technology has revolutionized functional genomics by enabling high-fidelity, genome-scale, multiplexed loss-of-function screens in human cells. Because of high specificity and ease of application, genome-wide CRISPR screens are increasingly used to identify cancer drug targets and to determine mechanisms of drug resistance (Bartha et al., 2018; Blomen et al., 2015; Hart et al., 2015; Tsherniak et al., 2017; Wang et al., 2015, 2017). However, single-gene knockout (KO) studies have a major blind spot: they are unable to assay the function of paralogs—ancestrally duplicated genes that frequently retain at least partially overlapping functions. The human genome exhibits a high degree of redundancy as a result of diploidy, gene duplication, and functional overlap of metabolic and signaling pathways (Dean et al., 2008; Harrison et al., 2007; Lavi, 2015; Ohno, 1970). Remarkably, paralogs constitute two-thirds of the human genome, making this blind spot the rule, not an exception, and paralogous genes are less likely to be essential for cell growth than non-paralogous (“singleton”) genes in CRISPR KO screens (Wang et al., 2015). This paralog blind spot therefore obscures our understanding of normal human genome function and impedes the identification of new cancer drug targets.
Genetic interactions (GIs) between paralogs have been extensively characterized in yeast, revealing fundamental insights about the differences between whole-genome and small-scale duplicates, functional groups that are enriched for interacting paralogs, and paralog mRNA expression patterns (Dean et al., 2008; Diss et al., 2017; Guan et al., 2007; Harrison et al., 2007). Essential paralogs that compensate for one another’s function exhibit “synthetic lethality,” a GI in which elimination of the entire family is deleterious but individual loss is tolerated. Yeast geneticists have defined quantitative measures of GIs, which can capture both positive (buffering) and negative (synthetic lethal) interactions (Collins et al., 2006). While paralog GIs are still poorly characterized in mammalian cells, the extensive degree of duplication in the human genome is similar to that seen in yeast (Dennis and Eichler, 2016; Lan and Pritchard, 2016; Singh et al., 2012), so experimental evaluation of human cells is likely to also reveal complex GIs.
Querying the GI space of the human genome has been limited by current technology; to survey even every possible pairwise interaction, let alone higher-order interactions, would involve ~200 million unique genetic perturbations. Moreover, the GI landscape among randomly selected genes is exceedingly sparse; existing studies of much smaller sets of gene pairs in human cells identified GIs in fewer than 0.1% of unrelated gene pairs (Han et al., 2017). To proactively identify these rare but functionally important interactions, research should therefore focus on high-value sets of genes likely to be enriched for functional interactions, such as paralogs.
Interestingly, the same duplication that makes paralogs difficult to study provides a tactical advantage for cancer therapy: the highly rearranged genomes typical of cancer often harbor paralog deletions and inactivating mutations. Cancer-associated loss-of-function of one paralog can confer a dependency on the continued activity of a duplicated pair (De Kegel and Ryan, 2019; Lord et al., 2020; Viswanathan et al., 2018), and this phenomenon has been used to identify synthetic lethal relationships of paralogs such as MAGOH/MAGOHB (Viswanathan et al., 2018), ARID1A/ARID1B (Helming et al., 2014), and SMARCA2/SMARCA4 (Hoffman et al., 2014). If the remaining actively expressed paralog could be targeted in tumors with loss of its pair, then tumor cells may show a selective therapeutic window compared to the surrounding normal cells with expression of both paralog members. A successful example of a therapy based on a synthetic lethal interaction is the enhanced sensitivity to poly(ADP-ribose) polymerase (PARP) inhibitors in BRCA1- and BRCA2-mutant tumors (Bryant et al., 2005; Farmer et al., 2005; Lord and Ashworth, 2017). We hypothesized that paralogs could provide a rich source of GIs and that direct experimental identification of synthetic lethal paralogs could therefore enable future drug discovery efforts.
Recently, several groups have developed innovative methods for assessing human GIs at scale (Boettcher et al., 2018; Dede et al., 2020; DeWeirdt et al., 2021; Gier et al., 2020; Gonatopoulos-Pournatzis et al., 2020; Han et al., 2017; Horlbeck et al., 2018; Najm et al., 2018; Shen et al., 2017). Consistently, while the overall rate of GI among gene pairs is low, many of the interactions identified were in paralogous genes. To comprehensively identify GIs between human paralogs, we here report our direct experimental evaluation of GIs among 1,030 paralog pairs (2,060 genes) in two human cell contexts. Our analysis not only revealed an extraordinarily high rate of paralog synthetic lethality, but also identified positive interactions that nominate 10 paralog pairs as tumor suppressor gene families.
RESULTS
A paralog blind spot limits discovery of essential genes and cancer dependencies
The human genome is highly duplicated, with paralogous genes constituting over two-thirds of protein coding genes (Figure 1A). Like other groups (Dandage and Landry, 2019; Dede et al., 2020; Wang et al., 2015), we noticed that paralogous genes are less likely to be essential for cell growth than non-paralogous “singleton” genes in single-gene CRISPR KO screening data (p < 2.20e–16 by one-tailed Kolmogorov-Smirnov [K-S] test, Figure 1B; data from Vichas et al., 2021). Given the utility of targeting cancer-essential genes for therapy, we reasoned that this paralog blind spot may prevent detection of important druggable cancer dependencies.
Figure 1. Paralog dependencies are missed in single-gene CRISPR KO screens.
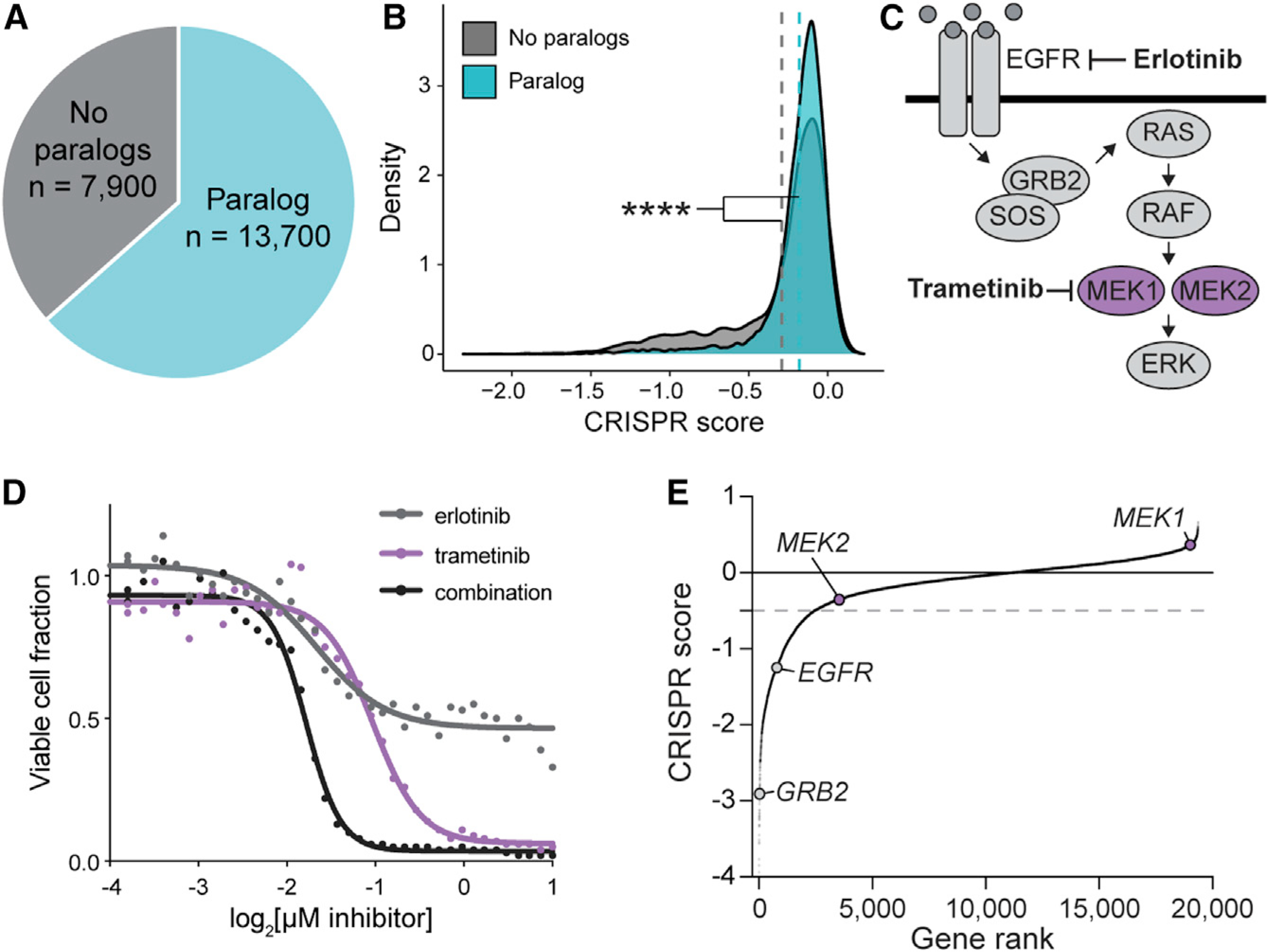
(A) Pie chart of human genes classified based on whether they are part of a paralog gene family with 10%–99% amino acid sequence identity.
(B) Density plot of CRISPR scores for a single-gene CRISPR KO screen in PC9 lung adenocarcinoma cells. Data are from Vichas et al. (2021). Dashed lines indicate the mean CRISPR score of genes in each group. **** indicates p < 2.20e–16 by one-tailed K-S test.
(C) Schematic of the EGFR/Ras/MAPK signalling pathway.
(D) Dose response curve of PC9-Cas9-EGFRT790M/L858R lung adenocarcinoma cells treated with erlotinib, trametinib, or a 1:1 combination of both drugs. The fraction of viable cells was determined by CellTiterGlo luminescence after 96 h of treatment. Data were re-analyzed from a larger drug screen from Berger et al. (2016).
(E) Rank plot of CRISPR scores from an erlotinib sensitization screen in PC9-Cas9-EGFRT790M/L858R cells (Vichas et al., 2021). MEK1 or MEK2 single-gene KO does not result in significantly decreased cell growth. Gray dashed line indicates the threshold for negative selection (−0.5).
To determine whether known therapeutic vulnerabilities are missed in CRISPR KO screens, we compared our previous drug sensitivity profiling of PC9-EGFRL858R/T790M cells (Berger et al., 2016) to recent genetic vulnerabilities identified in the same system (Vichas et al., 2021). These cells exhibit resistance to the epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor, erlotinib, which can be reversed by treatment with trametinib, a kinase inhibitor of MEK1 and MEK2—protein kinases encoded by the paralogous genes MEK1 and MEK2 (also known as MAP2K1/MAP2K2) that are part of the Ras/MAPK pathway (Figures 1C and 1D). The Ras/MAPK pathway is frequently activated in lung cancer by mutation of upstream receptor tyrosine kinases such as EGFR or mutation of the Kirsten rat sarcoma virus (KRAS) gene or MEK1 itself (Arcila et al., 2015; Sanchez-Vega et al., 2018; Cancer Genome Atlas Research Network, 2014). We noted in single-gene CRISPR KO data in the same cellular context that while EGFR and other Ras pathway members such as GRB2 were essential as expected, neither MEK1 nor MEK2 was essential when knocked out individually (Figure 1E). We reasoned that paralog redundancy might underlie the apparent disconnect between the small molecule and genetic assays. We therefore sought to develop a multiplexed CRISPR approach to directly probe paralog compensation on a genome scale, enabling the discovery of many more paralogous drug targets that may be missed in current CRISPR-based target discovery efforts.
The pgPEN library enables single KO and DKO of 1,030 human paralog families
To identify synthetic lethal paralogs that could serve as potential lung cancer drug targets, we focused on duplicated genes—paralog families of only two genes. We identified paralog families from Ensembl (Vilella et al., 2009) and then selected families in which a maximum of two genes shared 50%–99% amino acid identity (Figure S1A). Next, we designed a paired-guide RNA (pgRNA) CRISPR library to knock out each paralog alone or in combination with its respective pair. Using single-guide RNA (sgRNA) sequences from the Brunello CRISPR library (Doench et al., 2016), we designed 16 four-by-four pairwise double KO (DKO) pgRNAs for each paralog pair. In addition, we designed single KO pgRNAs containing one targeting sgRNA paired with a non-targeting control sgRNA having no match to the human reference genome. This was done for both paralogs to generate a total of 16 single KO pgRNAs. Five hundred double non-targeting pgRNAs were included as a control. This “paired guide RNAs for paralog GI mapping (pgPEN)” library (Table S1) was synthesized and cloned at 1000-fold coverage using previously developed methods (Gasperini et al., 2017; Thomas et al., 2020). Next-generation sequencing confirmed that >99.99% of pgRNAs were present in the cloned plasmid pool. The final pgPEN library consists of 33,170 pgRNAs targeting 1,030 paralog pairs (2,060 genes) in single KO and DKO combinations. Over half of the paralogs in the pgPEN library are unique to this study, while the remainder were also assayed in recent GI maps (Dede et al., 2020; Gonatopoulos-Pournatzis et al., 2020; Thompson et al., 2021; Figure S1B). In total, 554 of the gene products of pgPEN-targeted genes are considered “druggable” by recent criteria (Finan et al., 2017; Figure S1C).
To map GIs between paralogs, we applied the pgPEN library to PC9 lung adenocarcinoma cells previously engineered to constitutively express Cas9 (Thomas et al., 2020; Vichas et al., 2021) using standard pooled CRISPR screening methodology in triplicate (Figure 2A). pgRNAs that were positively or negatively selected were identified by Illumina sequencing of pgRNA abundance after ~12 population doublings in vitro compared to the starting abundance in the plasmid pool (Figure 2A). The sequencing strategy used for the pgPEN method is outlined in Figure S1D, and pgRNA library coverage and pairing statistics are included in Table S2. Only properly paired pgRNAs were included in our CRISPR screen analysis. Plasmid pgRNA abundance was highly correlated with early time point samples taken immediately following lentiviral transduction and puromycin selection (mean Pearson’s r = 0.93; Figure S2A). Endpoint samples exhibited expected changes in pgRNA abundance (Figure S2B) that were highly correlated across replicates (mean Pearson’s r = 0.82; Figure S2C). Single KO pgRNAs targeting pan-essential genes (Meyers et al., 2017) showed the expected dropout in late timepoint samples (Figure 2B). These data indicate that the screen was performed without significant bottlenecking and that the pgRNAs performed as expected for known essential genes. Similar to previously established CRISPR screen analysis methods (Meyers et al., 2017), we generated normalized CRISPR scores (CS) by scaling pgRNA log2(fold change) values such that the median CRISPR score of double non-targeting constructs was zero and the median CRISPR score of pan-essential single KO constructs was −1.
Figure 2. The pgPEN CRISPR library enables GI mapping of 1,030 human paralog pairs.
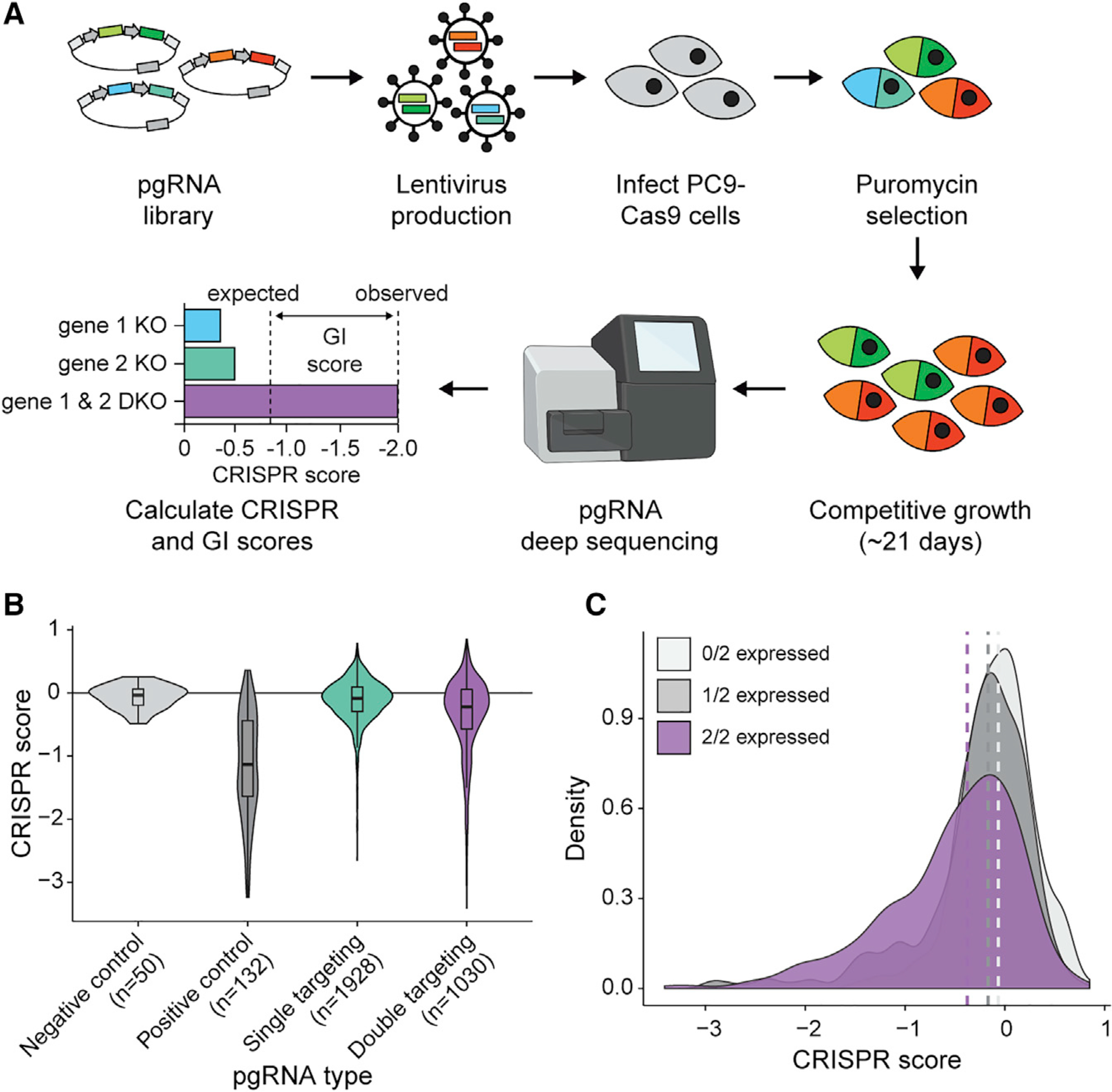
(A) Schematic of pgPEN screening approach for paralog GI mapping.
(B) Violin plots of target-level CRISPR scores for negative control (double non-targeting control), positive control (single KO pgRNAs targeting known essential genes), all other single KO pgRNAs, and DKO pgRNAs in the PC9 screen. The double-targeting pgRNA group had significantly lower CRISPR scores than did the single-targeting pgRNA group (p < 2.20e–16 by one-tailed K-S test).
(C) Density plot of target-level CRISPR scores for DKO pgRNAs grouped by whether zero, one, or both targeted genes are expressed (TPM ≥ 2) in PC9 cells. Dashed lines indicate the median CRISPR score for each group. pgRNAs targeting expressed genes had significantly lower CRISPR scores than those targeting two unexpressed genes for both the 2/2 genes expressed (p < 2.20e–16 by one-tailed K-S test) and 1/2 genes expressed (p = 4.03e–03 by one-tailed K-S test) groups.
See also Figures S1 and S2 and Tables S1, S2, and S3.
CRISPR-Cas9 gene KO involves the generation of double-strand breaks that can inhibit cell proliferation rate (Aguirre et al., 2016). One concern in targeting multiple loci with Cas9 is that the increased generation of double-strand breaks could, independent of any specific gene effect, result in enhanced negative selection of DKO compared to single KO pgRNAs. To control for this possibility, we further normalized data such that the median CRISPR score of all single KO pgRNAs targeting non-expressed genes would be zero and the median CRISPR score of all DKO pgRNAs targeting two non-expressed genes would be zero, with unexpressed genes defined as having an abundance of less than two transcripts per million (TPM) in PC9 RNA-sequencing (RNA-seq) data (Method details; Figures S2D, S2E, and 2C). After this normalization, DKO constructs still had significantly lower CRISPR scores than did single KO constructs (p = 2.20e–16 by one-tailed K-S test), indicative of possible GIs in the DKO group. As expected, constructs targeting expressed genes had significantly lower CRISPR scores than those targeting unexpressed genes for both single-targeting (p < 1.96e–10 by one-tailed K-S test; Figure S2E) and double-targeting (p < 2.20e–16 by one-tailed K-S test; Figure 2C) constructs. After normalization, only a minimal effect of paralog copy number (Figure S2F) on CRISPR score was observed (Figures S2G and S2H). Scaled CRISPR scores for pgRNAs in the PC9 screen can be found in Table S3.
Direct identification of paralog GIs in human lung cancer cells
Using the PC9 CRISPR scores, we calculated GI scores for each paralog pair under a multiplicative model following recently developed methods for human GI mapping (DeWeirdt et al., 2021; Han et al., 2017; Method details). Comparison of the expected and observed CRISPR scores for each paralog pair enabled identification of interacting paralogs (Figures 3A and 3B) and calculation of GI scores for each paralog pair (Figures 3C and 3D; Table S4). This approach identified 87 synthetic lethal and 68 buffering GIs among the 1,030 paralog pairs. Synthetic lethal interactions (GI < −0.5 and false discovery rate [FDR] < 0.1) included top pairs CCNL1/CCNL2, CDK4/CDK6, GSK3A/GSK3B, G3BP1/G3BP2, CNOT7/CNOT8, and OXSR1/STK39 (Figures 3B–3D). Interestingly, CCNL1/CCNL2 code for cyclins L1 and L2, which activate the paralogous proteins CDK11A/CDK11B. Active CDK11 is involved in regulating pre-mRNA splicing and may also play a role in cell-cycle regulation (Loyer and Trembley, 2020; Loyer et al., 2008). We found CDK11A/CDK11B were also synthetic lethal in PC9 cells (Figure 3B), and recent work has shown that CDK11A/CDK11B are targeted by the small-molecule inhibitor OTS964 (Lin et al., 2019). OXSR1 and STK39 encode evolutionarily conserved kinases involved in the oxidative stress response, and STK39 has a possible role in promoting apoptosis (Balatoni et al., 2009; Gagnon and Delpire, 2012). We also found a significant synthetic lethal interaction for MEK1/MEK2 (Figures 3B–3D), confirming that the discrepancy between genetic and drug data in Figure 1 was indeed due to paralog redundancy. Known synthetic lethal paralogs such as ARID1A/ARID1B (Helming et al., 2014) and MAPK1/MAPK3 (Dede et al., 2020; DeWeirdt et al., 2021) were also identified (Table S4).
Figure 3. pgPEN uncovers synthetic lethal and buffering GIs.

(A) Scatterplot of target-level observed versus expected CRISPR scores in the PC9 screen. The solid line is the linear regression line for the negative control (single KO) pgRNAs, while dashed lines indicate ± 2 residuals.
(B) CRISPR scores for representative synthetic lethal paralog pairs. Data shown are the mean CRISPR score for each single KO or DKO target across three biological replicates with replicate data shown in overlaid points.
(C) Rank plot of target-level GI scores in PC9 cells. Table insert, top synthetic lethal paralogs based on GI score.
(D) Volcano plot of target-level GI scores in PC9 cells. FDR indicates the multiple hypothesis-adjusted p values from a two-tailed t test (Method details). Blue, synthetic lethal paralog GIs with GI < −0.5 and FDR < 0.1; red, buffering paralog GIs with GI > 0.25 and FDR < 0.1.
See also Table S4.
To experimentally validate these findings, we developed a competitive fitness assay in red (mCherry) and green (GFP) labeled PC9-Cas9 cells (Figures S3A–S3D). In designing the validation experiment for top synthetic lethal paralog pairs, we used safe-targeting sgRNAs (Morgens et al., 2017) in place of non-targeting sgRNAs to account for the growth effects observed by generating one versus two double-strand breaks. We transduced PC9-Cas9-GFP-NLS cells with a double safe-targeting pgRNA, while PC9-Cas9-mCherry-NLS cells were transduced with paralog-targeting pgRNAs designed to target each paralog individually or both paralogs together (Table S5). The cells were then pooled at a 1:1 ratio of GFP:mCherry cells.
Using this approach, we determined the effects of targeting four top synthetic lethal paralog pairs from the PC9 screen: CCNL1/CCNL2, CDK4/CDK6, MEK1/MEK2, and OXSR1/STK39. The results of these competitive fitness assays mirrored the gene KO effects observed in the pooled screen format (Figures 4A and 4B). For CCNL1/CCNL2 and OXSR1/STK39, individual gene KOs showed little effect on cell growth, whereas combined KO of both paralogs resulted in severe growth effects in both the screen (Figure 3B) and the competitive fitness assay (Figure 4A). The CRISPR screen data indicated that CDK4 and MEK2 single KOs were essential on their own, with the DKO causing further negative growth effects for each pair (Figure 3B), and these effects were also observed in the competitive fitness assays for these pairs (Figure 4A). We validated the synthetic lethality of these four pairs by confirming that the observed DKO growth phenotype was significantly less than an expected DKO growth phenotype, which was calculated based on the sum of the two single KO growth effects (p = 9.56e–05 for CCNL1/CCNL2; p = 5.29e–05 for CDK4/CDK6; p = 2.20e–04 for MEK1/MEK2; p = 8.49e–05 for OXSR1/STK39; all by one-tailed t test). Line graphs of growth phenotypes across the entire competitive fitness assay are shown in Figures S3E–S3H. We experimentally validated two additional pairs with non-significant GIs, MAGOH/MAGOHB and PSMB5/PSMB8, confirming a slightly negative but non-significant interaction for MAGOH/MAGOHB and that the PSMB5/PSMB8 DKO growth phenotype was not significantly less than the expected, summed DKO growth phenotype (Figures S3I–S3L). Overall, there was good concordance between the GIs observed in the pgPEN PC9 CRISPR screen and in the validation competitive growth assays (Figure S3M).
Figure 4. CRISPR validation experiments confirm top PC9 synthetic lethal interactions.

(A) Boxplots of growth phenotypes for PC9-Cas9-mCherry cells expressing the indicated pgRNA compared to PC9-Cas9-GFP cells expressing a double-safe-targeting control pgRNA. Boxes indicate mean ± SEM of six biological replicates, which are shown as overlaid points. Growth phenotype is defined as the log2-scaled ratio of mCherry:GFP cell counts at the late time point compared to the day 1 mCherry:GFP cell counts. Expected DKO phenotypes are the sum of single KO growth phenotypes. The expected and observed DKO phenotypes were compared using a one-tailed t test. Data shown are for the time point with the most extreme difference between expected and observed DKO growth phenotypes, termed the late time point: CCNL1/CCNL2 (day 12), CDK4/CDK6 (day 7), MEK1/MEK2 (day 11), and OXSR1/STK39 (day 10). Full time course data are shown in Figure S3.
(B) Fluorescence microscopy images of competitive fitness assays on early (day 1) and late time points as indicated above for (A). Scale bar, 100 μM.
(C) Western blot validation of single KO and DKO pgRNA-induced gene inactivation. For CCNL1, pie charts of percent mutant alleles based on next-generation sequencing are shown due to lack of a suitable CCNL1 antibody for western blotting. Additional genomic DNA-level validation data are presented in Figure S4.
See also Figures S3 and S4 and Table S5.
We also confirmed the successful generation of genomic DNA CRISPR edits at sgRNA target sites and the loss of target protein expression for the paralog pairs shown in Figures 4A and 4B. Confirmation of genomic DNA edits was done by next-generation sequencing for CCNL1/CCNL2, as we were unable to identify a suitable antibody for CCNL1 (Figures 4C and S4A–S4D), and by Sanger sequencing for CDK4/CDK6, MEK1/MEK2, and OXSR1/STK39 (Figures S4E–S4I). Western blots for CCNL2, CDK4/CDK6, MEK1/MEK2, and OXSR1/STK39 showed expected patterns of protein loss based on the pgRNAs expressed in each cell line (Figure 4C). Taken together, these data suggest that pgPEN is an effective strategy for uncovering synthetic lethal paralog interactions.
A second pgPEN screen identifies shared versus cell-line-specific paralog synthetic lethal interactions
Next, we applied the pgPEN approach to a different tissue context, HeLa cervical carcinoma cells, using similar methodology, with the exception of using a doxycycline-inducible Cas9 system (Cao et al., 2016). Quality control analyses of HeLa screening data again indicated successful generation of expected gene KO phenotypes (Figures S5A–S5J; Table S6). Calculation of GI scores identified 70 significant synthetic lethal interactions and 44 significant buffering interactions (Figures 5A, 5B, and S5K; Table S4). Many of the top synthetic lethal pairs were shared between HeLa and PC9 cells, including CCNL1/CCNL2, GSK3A/GSK3B, and MEK1/MEK2 (FDR < 0.1 in both cell lines; Figures 5C and 5D). Other paralog families were synthetic lethal in only one of the cell lines (FDR < 0.1 in PC9 or HeLa only). These included SOS1/SOS2, which were highly essential and synthetic lethal only in HeLa cells (Figures 5C and 5D), and CDK4/CDK6, which were only required in PC9 cells (Figures 5C and 5D). In total, 122 paralog pairs were identified as synthetic lethal in at least one context. Surprisingly, we noted that cell-line-specific synthetic lethal interactions were often not explained by expression differences (Figure 5D), demonstrating that paralog dependencies, like other cancer dependencies, are modified by cellular context or other biological factors besides gene expression.
Figure 5. Identification of cell-line-specific and shared synthetic lethal paralog pairs.

(A) Rank plot of target-level GI scores in HeLa cells. Table insert, top synthetic lethal paralogs based on GI score.
(B) Volcano plot of target-level GI scores in HeLa cells. FDR indicates the multiple hypothesis-adjusted p values from a two-tailed t test (Method details). Blue, synthetic lethal paralog GIs with GI < −0.5 and FDR < 0.1; red, buffering paralog GIs with GI > 0.25 and FDR < 0.1.
(C) Scatterplot of target-level GI scores for paralog pairs in PC9 versus HeLa cells. Blue, synthetic lethal paralog pairs with GI < −0.5 and FDR < 0.1 in either PC9 or HeLa cells; gray, all paralog pairs with GI ≥ −0.5 or FDR ≥ 0.1.
(D) CRISPR scores for representative synthetic lethal paralog pairs identified in the PC9 and HeLa cell screens. Top row: data shown are the mean CRISPR score for each single KO or DKO target across three biological replicates with replicate data shown in overlaid points. Shared synthetic lethal paralogs (e.g., CCNL1/CCNL2 and MEK1/MEK2) have FDR < 0.1 in both cell lines; PC9-specific paralogs (e.g., CDK4/CDK6 and OXSR1/STK39) have FDR < 0.1 in PC9 only; and HeLa-specific paralogs (e.g., GFTP1/GFPT2 and SOS1/SOS2) have FDR < 0.1 in HeLa only. Dashed lines indicate CRISPR score < −0.5. Bottom row: paralog gene expression in PC9 and HeLa cells from RNA-seq analysis. Dashed lines indicate log2(TPM) = 1, the threshold for gene expression.
(E) Boxplots comparing the effect of CRISPR-mediated KO of the indicated gene in DepMap cell lines with high (top quartile) compared to low (bottom quartile) copy number of its paralogous gene. For boxplots, the middle line, hinges, notches, and whiskers indicate the median, 25th/75th percentiles, 95% confidence interval, and data points within 1.5× the interquartile range from the hinge, respectively. p values were computed using a two-tailed Wilcoxon rank-sum test. CRISPR score and copy number data were obtained from DepMap.
(F) As in (E), but for gene expression.
(G) Bar plot indicating the p values (computed using a two-tailed Wilcoxon rank-sum test) obtained by comparing the effect of a single paralog KO to the copy number (as in E) or gene expression (as in F) of its pair across human cancer cell lines profiled by DepMap. Bar color indicates whether each pair was synthetic lethal in PC9 only, HeLa only, or both cell lines in the pgPEN screens. Dashed line indicates p = 0.05.
Some synthetic lethal paralog pairs, including SEC24A/SEC24B, COQ10A/COQ10B, CNOT7/CNOT8, TIA/TIAL, and VPS4/VPS4B, have been highlighted in previous studies (Dede et al., 2020; Gonatopoulos-Pournatzis et al., 2020; Lord et al., 2020; Neggers et al., 2020; Szymańska et al., 2020). However, to our knowledge, many of the synthetic lethal paralogs identified in the pgPEN screens were not previously known to be functionally redundant in human cells. These include CCNL1/CCNL2 and OXSR1/STK39, along with eukaryotic translation initiation factors EIF1/EIF1B; DNA and RNA helicase and cGAS/STING pathway members G3BP1/G3BP2; hexosamine biosynthesis pathway members GFPT1/GFPT2; and PDS5A/PDS5B, which regulate sister chromatid cohesion during mitosis. Individual members of many of these synthetic lethal paralog families have been previously implicated in cancer; for instance, high GFPT2 expression has been linked to tumor metabolic reprogramming in lung adenocarcinoma (Zhang et al., 2018), and PDS5B is a negative regulator of cell proliferation and has been highlighted as a possible tumor suppressor gene in prostate cancer (Maffini et al., 2008).
As a complementary approach to validate our PC9 and HeLa screens, we used single gene CRISPR screening data to determine the essentiality of one paralog in the context of low expression or spontaneous copy number loss of its pair in hundreds of cancer cell lines profiled by DepMap (Tsherniak et al., 2017). For this analysis, we grouped cell lines according to whether “gene 1” of a paralog pair was highly (top quartile) or lowly (bottom quartile) expressed and calculated the median CRISPR score for “gene 2” in these groups (Table S7). This strategy was also used to determine the influence of gene copy number, and the inverse analysis (i.e., “gene 1” dependency in the context of low “gene 2” expression/copy number) was also performed. We found that for VPS4A/VPS4B, CCNL1/CCNL2, and TLK1/TLK2, reduced copy number of one family member was significantly associated with greater dependency of the paralogous genes (Figure 5E). We observed similar dependencies when considering gene expression for EIF1/EIF1B, TIA1/TIAL1, and AP2A1/AP2A2 (Figure 5F). The top 10 shared synthetic lethal paralogs identified in both PC9 and HeLa screens displayed evidence of GIs using these approaches (Figure 5G; Table S7).
Identification of tumor suppressor paralog pairs
In addition to synthetic lethal interactions, pgPEN screens can identify positive GIs. We noticed that these positive interactions include both buffering interactions, where loss of one paralog prevents the deleterious phenotype of loss of the other—we identified 108 such interactions in at least one cell context—as well as cases where the combined loss of both genes synergistically promotes cell growth. The latter are likely to be paralog families with tumor suppressor functions that require complete loss of the family to reveal the cellular phenotype. To identify these tumor suppressor paralogs, we restricted our analysis to significant buffering interactions (GI > 0.25, FDR < 0.1) between expressed paralogs in which the DKO was positively enriched in each CRISPR screen (CS > 0.25). Under these relatively stringent criteria, four tumor suppressor interactions were identified in PC9 and six in HeLa cells (Figure 6A). None of the 10 interactions were shared across cell lines, potentially reflecting the differing biology of HeLa and PC9 cells and the difficulty in achieving positive selection in basal culture conditions of rapidly proliferating cancer cell lines.
Figure 6. Paralog buffering interactions include tumor suppressor paralogs.
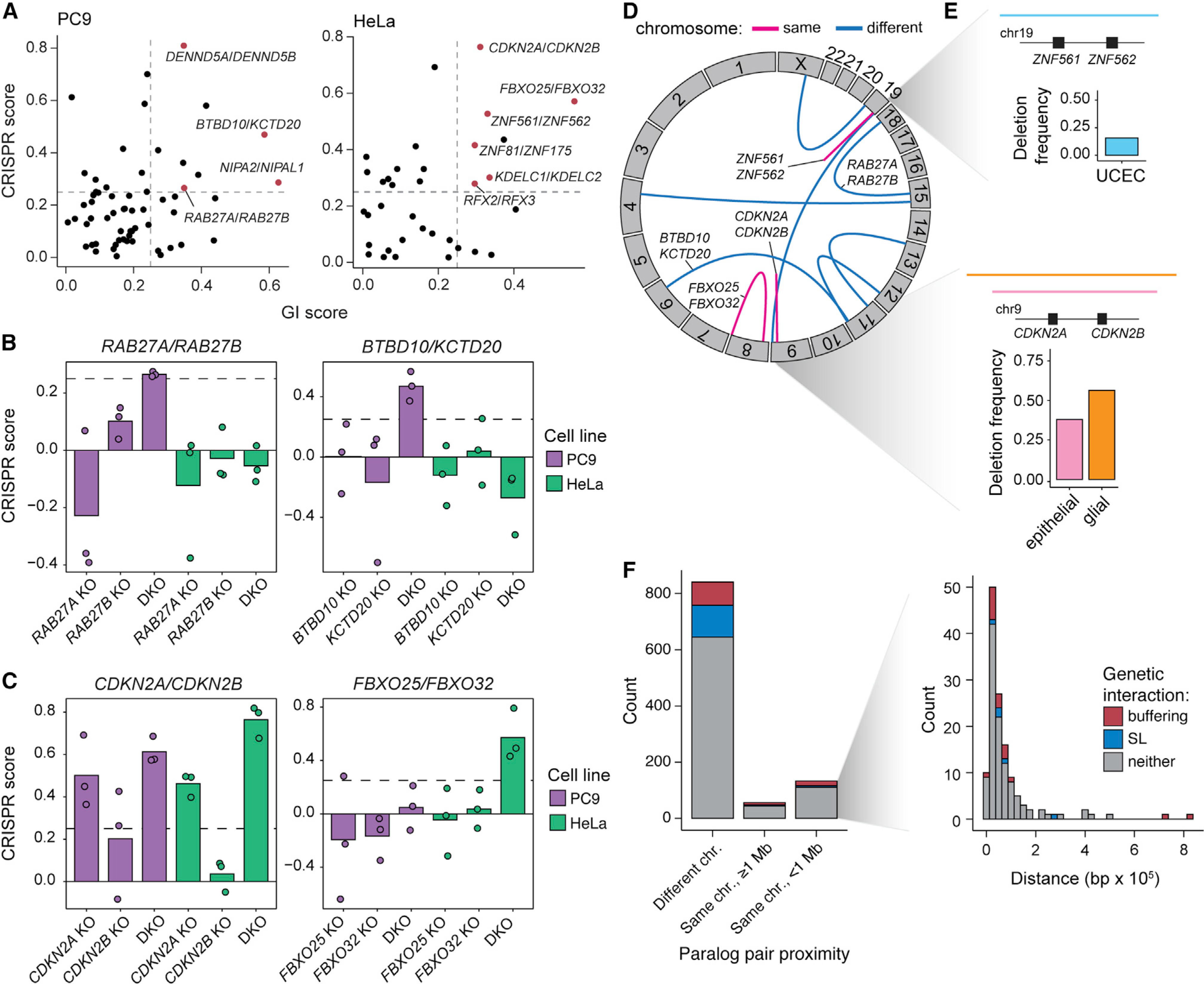
(A) Identification of tumor suppressor paralog interactions (GI score > 0.25; FDR < 0.1; CRISPR score > 0.25).
(B) CRISPR scores of PC9-specific tumor suppressor paralog pairs. Data shown are the mean of three biological replicates with replicate data shown in overlaid points. Dashed lines indicate CRISPR score = 0.25.
(C) CRISPR scores of HeLa-specific tumor suppressor paralog pairs. Data shown as in (B).
(D) Circos plot showing the genomic locations of tumor suppressor paralog pairs. Blue arcs indicate paralog pairs located on different chromosomes, while pink arcs represent paralog pairs located on the same chromosome.
(E) Top: diagram of a recurrent deletion seen in uterine corpus endometrial carcinoma (UCEC) data from The Cancer Genome Atlas (TCGA) that spans the genomic locus containing ZNF561 and ZNF562 and a bar plot indicating the deletion frequency. Bottom: diagram of recurrent deletions in epithelial and glial cancers that span the genomic locus containing CDKN2A and CDKN2B and a bar plot showing the deletion frequency in each cancer subtype.
(F) Genomic distance between paralogs for the 1,030 paralogs pairs included in the pgPEN library in three proximity categories: on different chromosomes, on the same chromosome but ≥ 1 Mb apart, and on the same chromosome within 1 Mb. Inset: histogram of paralog distance for pairs that are within 1 Mb of one another.
Tumor suppressor pairs identified in PC9 cells include RAB27A/RAB27B, encoding Rab-family GTPases involved in vesicle trafficking (Li et al., 2018), and the BTB/POZ-domain genes BTBD10/KCTD20 (Figure 6B). In HeLa cells, one of the top pairs identified was CDKN2A/CDK2NB (Figure 6C), frequently deleted tumor suppressors that encode the CDK4/6 inhibitors INK4A/ARF and INK4B (Kim and Sharpless, 2006). Another top tumor suppressor paralog pair in HeLa cells was FBXO25/FBXO32 (Figure 6C), which encode SCF-type E3 ligase proteins. Although little is known about the function of these proteins and their substrates, the GI between FBXO25 and FBXO32 suggests that these two proteins may share similar functions or substrates. FBXO25 and FBXO32 have been individually proposed to have tumor suppressor function in previous studies (Xue et al., 2012; Zhou et al., 2017).
While this direct identification of tumor suppressor paralog pairs has merit for understanding basic genome function, spontaneous loss of two unlinked genes in cancer should be rare, and therefore it is unlikely that DKO of paralogs contributes to tumorigenesis for most paralog families. Interestingly, however, we noted that two tumor suppressor pairs contained genes co-located in the same chromosomal locus (Figure 6D). In addition to CDKN2A/CDKN2B, whose combined loss is well known to promote tumorigenesis, ZNF561 and ZNF562 are co-located and reside on chromosome 19p13.2, a frequently deleted region in uterine corpus endometrial cancer (Berger et al., 2018; Cherniack et al., 2017; Figure 6E). Beyond the tumor suppressor paralogs, 13% of all the paralog pairs in the pgPEN library are located within 1 megabase (Mb) of each other in the human genome (Figure 6F), which raises the possibility that the cell fitness consequences of DKO of human paralogs could contribute to the selective forces that drive aneuploidy patterns in human cancer (Ben-David and Amon, 2020; Taylor et al., 2018).
DISCUSSION
This work provides, to our knowledge, the largest direct experimental assessment of paralog GIs in the human genome to date. The pgPEN library we developed uses two Cas9-type sgRNAs driven from independent promoters to enable KO of two paralogs simultaneously and targets 2,060 duplicate human paralogs. Complementing three other recent studies of human paralog GIs (Dede et al., 2020; Gonatopoulos-Pournatzis et al., 2020; Thompson et al., 2021), our library adds over 1,000 unique paralogs and brings the total set of human paralogs assayed to date to just over 3,900. Both Thompson et al. (2021) and the present study use Cas9-type CRISPR systems, whereas the other two studies include Cas12a-derived enzymes. Cas12a systems have the benefit of using an array of sgRNAs on a single transcript that is processed by Cas12a, enabling programmable delivery of multiple sgRNAs to the same cell (DeWeirdt et al., 2021). Continued application of Cas12a for CRISPR screening will enable the experimental identification of higher-order combinatorial GIs in human cells. However, for pairwise interactions of paralogs, the pgPEN library may provide an ease of application to investigators with Cas9-expressing cell systems already developed.
Remarkably, our pgPEN screens revealed that 12% of duplicate paralogs exhibit synthetic lethality, demonstrating that paralogs are a rich source of GIs. A recent meta-analysis of multiplexed paralog CRISPR screens revealed that this hit rate was consistent with work published by other groups, and pairwise comparisons of the four studies showed that many shared paralogs (i.e., those that were screened by both groups) were consistently classified as synthetic lethal (De Kegel et al., 2021). These findings strengthen our conclusions, underscoring the importance of simultaneously targeting redundant genes and demonstrating that a large fraction of cancer dependencies is missed by current single-gene KO approaches. Like others (De Kegel and Ryan, 2019; Viswanathan et al., 2018), we propose that synthetic lethal interactions among paralogs could be harnessed for cancer therapy, since the aneuploid genomes typical of cancer cells commonly harbor deletions and inactivating mutations in one or more paralogs. Targeting lineage-specific essential paralogs or paralog families with partial loss in cancer could provide an orthogonal approach for cancer therapy to be applied in combination with existing therapies to provide durable cancer control and improved patient outcomes. In addition, even paralogs that are not lost in cancer may represent tractable cancer targets; the same homology and redundancy that complicate genetic identification of paralogs as cancer dependencies could enable simultaneous targeting of each protein with ease. This strategy is exemplified by the current use of small molecules targeting several of the top synthetic lethal paralogs we identified, such as CDK4/CDK6 and GSK3A/GSK3B. Indeed, a recent study demonstrated in cell lines and retrospective clinical analyses that tumors with low CDK6 levels rely on CDK4 expression and show increased sensitivity to CDK4/6 inhibitors (Wu et al., 2021).
Last, we provide a systematic identification of tumor suppressor paralog pairs. We identified 10 paralog pairs whose combined loss significantly promotes cancer cell line growth. Although combined loss of some of these pairs is likely to be rare, 2 of the 10 pairs we identified are located in the same chromosomal locus. Many paralog loci are frequently deleted in cancer. These data, therefore, shed light on the basis for the positive selection of these genome deletions and suggest that combined paralog loss may shape the landscape of positive and negative selection in human cancer.
STAR★METHODS
Detailed methods are provided in the online version of this paper and include the following:
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Rabbit polyclonal anti-CCNL2 (Cyclin L2) | Novus Biologicals | Cat# NB100–87009; RRID: ABJ201144 |
| Rabbit polyclonal anti-OXSR1 (OSR1) | Cell Signaling Technology | Cat #3729; RRID: AB_2157610 |
| Rabbit polyclonal anti-STK39 (SPAK) | Cell Signaling Technology | Cat# 2281; RRID: AB_2196951 |
| Mouse monoclonal anti-MEK1 | Cell Signaling Technology | Cat# 2352; RRID: AB_10693788 |
| Rabbit monoclonal anti-MEK2 | Cell Signaling Technology | Cat# 9147; RRID: AB_2140641 |
| Rabbit monoclonal anti-CDK4 | Cell Signaling Technology | Cat# 12790; RRID: AB_2631166 |
| Rabbit monoclonal anti-CDK6 | Cell Signaling Technology | Cat# 13331; RRID: AB_2721897 |
| Mouse monoclonal anti-vinculin | Sigma | Cat# V9264; RRID: AB_10603627 |
|
| ||
| Deposited data | ||
|
| ||
| Raw and analyzed CRISPR screen data | This paper | GEO: GSE178179 |
| DepMap | Tsherniak et al., 2017 | https://depmap.org/portal/ |
| TCGA Copy Number Portal | Beroukhim et al., 2010 | https://portals.broadinstitute.org/tcga/home |
| PC9 and HeLa RNA-seq data | Thomas et al., 2020 | GEO: GSE120703 |
| EnsemblCompara GeneTrees | Vilella et al., 2009 | https://www.ensembl.org/info/genome/compara/homology_method.html |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| Human: PC9-Cas9 cells | Thomas et al., 2020 | N/A |
| Human: iCas9/HeLa cells | Cao et al., 2016 | N/A |
| Human: PC9-Cas9-mCherry-NLS cells | This paper | N/A |
| Human: PC9-Cas9-GFP-NLS cells | This paper | N/A |
| Human: HEK293T cells | ATCC | CRL-3216 |
|
| ||
| Oligonucleotides | ||
|
| ||
| All oligos used, see Table S5 | This paper | N/A |
|
| ||
| Recombinant DNA | ||
|
| ||
| pgPEN plasmid library | This paper | Addgene: 171172 |
| pLentiGuide-Puro | Sanjana et al., 2014 | Addgene: 52963 |
| pRRLSIN.cPPT.PGK-GFP.WPRE | N/A (unpublished) | Addgene: 12252 |
| psPAX2 | N/A (unpublished) | Addgene: 12260 |
| pCMV-VSV-G | (Stewart et al., 2003) | Addgene: 8454 |
|
| ||
| Software and algorithms | ||
|
| ||
| pgRNA_sequencing_analysis | This paper | Zenodo: https://doi.org/10.5281/zenodo.5081113 |
| MAGeCK v0.5.9.2 | Li et al., 2014 | https://sourceforge.net/projects/mageck/ |
| Bowtie v1.2.2 | Langmead et al., 2009 | https://sourceforge.net/projects/bowtie-bio/; RRID: SCR_005476 |
| SAMtools v1.9 | Li et al., 2009 | https://www.htslib.org; RRID: SCR_002105 |
| Tidyverse v1.3.0 | Wickham et al., 2019 | https://cran.r-project.org/web/packages/tidyverse/index.html; RRID: SCR_019186 |
| Bioconductor v3.1.0 | Huber et al., 2015 | https://bioconductor.org/install; RRID: SCR_006442 |
| Gen5 v3.02 | BioTek Instruments | https://www.biotek.com/products/software-robotics-software/gen5-microplate-reader-and-imager-software/; RRID: SCR_017317 |
| TIDE | Brinkman et al., 2014 | http://shinyapps.datacurators.nl/tide/ |
| CRISPResso | Clement et al., 2019 | https://github.com/pinellolab/CRISPResso2 |
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dr. Alice Berger (ahberger@fredhutch.org).
Materials availability
The pgPEN CRISPR plasmid library has been deposited to Addgene as the Human Paralog Knockout Library (pgPEN), Addgene: 171172. All other plasmids and cell lines generated for this study will be shared by the lead contact upon request.
Data and code availability
PC9-Cas9 and HeLa/iCas9 RNA-seq data (GEO: GSE120703) and raw and processed CRISPR screen GSE120703 sequencing data for the PC9-Cas9 and HeLa/iCas9 pgPEN CRISPR screens (GEO: GSE178179) has been deposited to GEO. All GEO-deposited data is listed in the Key resources table and is publicly available as of the date of publication. This paper also analyzes existing, publicly available datasets. The accession numbers for these datasets are listed in the Key resources table. All other data reported in this paper will be shared by the lead contact upon request.
Original pgRNA counting code is publicly available (Zenodo: https://doi.org/10.5281/zenodo.5081113) as of the date of publication and is listed in the Key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
PC9 cells were originally derived from a metastatic lung adenocarcinoma from a 45 year old male patient. PC9-Cas9 cells were previously generated (Thomas et al., 2020) and cultured in RPMI-1640 (GIBCO) supplemented with 10% Fetal Bovine Serum (FBS, Sigma). PC9-Cas9-GFP-NLS and PC9-Cas9-mCherry-NLS cells were generated by transducing PC9-Cas9 cells with lentivirus containing GFP-NLS or mCherry-NLS-encoding vectors (parental backbone was a gift from Dider Trono, Addgene: 12252). mCherry or GFP positive cells were selected using flow cytometry. HeLa cells were originally derived from a cervical carcinoma from a 31 year old female patient. HeLa/iCas9 cells were previously generated (Cao et al., 2016) and cultured in Dulbecco’s Modified Eagle’s Medium (DMEM, Genesee Scientific) supplemented with 10% FBS. The HEK293T cell line was originally derived from kidney tissue from a female fetus. HEK293T cells were obtained from ATCC (CRL-3216) and cultured in DMEM supplemented with 10% FBS. All cells were maintained at 37°C in 5% CO2 and confirmed mycoplasma-free.
METHOD DETAILS
Human paralog analysis and selection
For analysis of human paralog versus singleton essentiality (Figures 1A and 1B), a list of human protein-coding genes was obtained from Ensembl (Vilella et al., 2009). Mitochondrial genes and splice variants were removed from the analysis. The remaining genes were divided into two groups: (1) paralogous genes with >10% amino acid sequence identity and (2) singleton genes.
For the pgPEN library, the list of human paralogs was further filtered to include only those with >50% reciprocal amino acid sequence identity with only one other gene. Genes encoding components of olfactory signaling and T cell receptors were also excluded. As shown in Figure S1A, a total of 2,060 paralogous genes (1,030 pairs) were included in the pgPEN library. Note that paralogs MEK1/MEK2 may also be referred to as MAP2K1/MAP2K2.
PC9 single-gene CRISPR screen
PC9-Cas9 and PC9-Cas9-EGFRT790M/L858R single-gene CRISPR knockout screen data was re-analyzed from previously published data (Vichas et al., 2021). The relative essentiality of singletons versus paralogs in the PC9-Cas9 CRISPR knockout screen using the Brunello library (Doench et al., 2016) was assessed via a two-tailed Kolmogorov-Smirnov test (Figure 1B).
PC9 drug sensitivity profiling
PC9-Cas9 and PC9-Cas9-EGFRT790M/L858R erlotinib/trametinib drug sensitivity data was re-analyzed from previously published data (Berger et al., 2016). For combination dosing, erlotinib and trametinib were delivered to cells in a 1:1 molar ratio.
pgPEN library design and cloning
The pgPEN library was designed using sgRNA sequences selected from the Brunello library (Doench et al., 2016). sgRNAs containing BsmBI restriction target sequences and U6 termination signals were excluded from the library. Given that previous data demonstrated no position effects using the pgRNA approach (Gasperini et al., 2017), the sgRNA targeting a given gene was located at the same site in every pgRNA.
pgRNA oligonucleotides were synthesized by Twist Biosciences and cloned per published protocols (Gasperini et al., 2017; Thomas et al., 2020). Briefly, the pgRNA oligonucleotides were amplified (primers RKB1169 and RKB1170, Table S5) using NEBNext High Fidelity 2X Ready Mix (New England Biolabs) and purified via a 1.8X Ampure XP SPRI bead (Beckman Coulter) clean-up. Amplified oligonucleotides were then cloned into BsmBI (FastDigest Esp3I, Thermo Fisher Scientific)-digested lentiGuide-Puro (Addgene: 52963) (Sanjana et al., 2014) plasmid backbone via the NEBuilder HiFi (New England Biolabs) assembly system. Cloned plasmids were purified using a 0.8X Ampure bead clean-up and transformed into Endura ElectroCompetent E. coli cells (Lucigen) via electroporation to generate the pLGP-2xSpacer vector. The pLGP-2xSpacer vector was isolated using the NucleoBond Xtra Maxiprep kit (Macherey-Nagel) and linearized by BsmBI digest. A GBlock (synthesized by Integrated DNA Technologies) containing a second sgRNA backbone and H1 promoter sequence was digested with BsmBI, purified via a 1.8X Ampure bead clean-up, and ligated into the pLGP-2xSpacer backbone using NEB Quick Ligase (New England Biolabs). The reaction product was purified using an 0.8X Ampure bead cleanup and transformed into Endura Electrocompetent E. coli via electroporation to propagate the final pLGP-pgRNA vectors. The pLGP-pgRNA plasmids were again isolated using the NucleoBond Xtra Maxiprep kit, and the cloned library was amplified and sequenced as described below to confirm high coverage. At each cloning step, individual E. coli colonies were sequence verified via colony PCR and Sanger sequencing with primer RKB1148 (Table S5). Over 1000X coverage of each pgRNA was maintained throughout plasmid library cloning, amplification, and sequencing; coverage depth was selected based on our previous screen experience as well as published recommendations (Doench, 2018; Joung et al., 2017).
Lentivirus production and titration
With our cloned library, we produced lentivirus via a large-format transfection in HEK293T cells using a protocol adapted from Joung et al. (2017). Briefly, we used TransIT-LT1 (Mirus Bio) as a transfection reagent, with packaging plasmid psPAX2 (Addgene: 12260; plasmid was a gift from Didier Trono) and envelope plasmid pCMV-VSV-G (Addgene: 8454) (Stewart et al., 2003) and Opti-MEM (Thermo Fisher Scientific). Plasmids were added at a 4:2:1 ratio of transfer to packaging to envelope plasmid. At 18 hours post-transfection, media was changed to high-serum DMEM (30% FBS). Lentivirus was harvested 48 hours post-transfection. Over 500X coverage of each pgRNA was maintained throughout; coverage depth was selected based on our previous screen experience as well as published recommendations (Doench, 2018; Joung et al., 2017).
pgPEN CRISPR screens
PC9-Cas9 and HeLa/iCas9 cells were transduced with the pgPEN library at low multiplicity of infection (~0.3) to ensure the integration of a single pgRNA construct into >95% of transduced cells (Doench, 2018). Transduced cells were then selected using puromycin (1.0 μg/mL, Sigma) for 48–72 hours until all uninfected control cells were dead. For the PC9-Cas9 screen, cells were split into three biological replicates after infection but before puromycin selection, and genomic DNA (gDNA) was harvested from each replicate after puromycin selection for an early time point sample. For the HeLa/iCas9 screen, cells were kept in the pooled format until puromycin selection was complete, resulting in a single early time point sample. HeLa/iCas9 cells were then induced using doxycycline (1.0 μg/mL, Sigma) and split into three biological replicates. For both screens, cells were then passaged for approximately 12 population doublings while maintaining over 500X coverage of each pgRNA at every step. An endpoint gDNA sample was harvested from each biological replicate and stored at −80°C. Genomic DNA was extracted using the QIAamp DNA Blood Maxi Kit (QIAGEN).
pgPEN library preparation and sequencing
Plasmid and gDNA samples were amplified and sequenced at >500X coverage per pgRNA according to our previously established methods (Thomas et al., 2020). All primer sequences used for library preparation are included in Table S5. First, 2.5 μg of gDNA was used as input for each reaction, with a total of 48 reactions (120 μg total input gDNA) to ensure >500X coverage per sample. Input DNA was amplified using NEBNext High Fidelity 2X Ready Mix with primers RKB2713/RKB2714 followed by 1.8X Ampure bead clean-up. Second, the amplicon from PCR #1 was used as input for PCR #2, with 10 ng input DNA in one reaction per sample. The input DNA was amplified using primers RKB2715/RKB2716 followed by 1X Ampure bead clean-up. Third, 10 ng of the amplicon from PCR #2 was used as input for PCR #3 and was amplified using a common forward primer (RKB2717) and a sample-specific barcoded reverse primer (see Table S5) to allow for multiplexed sequencing. Product from PCR #3 was purified using a 1X Ampure bead clean-up, quantified by a Qubit assay (Thermo Fisher Scientific), and pooled at equimolar amounts prior to Illumina sequencing. The custom sequencing strategy used for pgPEN is outlined in Figure S1D.
pgRNA cloning for validation
Validation pgRNA oligonucleotides consisted of two sgRNA sequences separated by the H1 promoter and were synthesized by Genewiz (Brooks Life Sciences). All pgRNA sequences used for validation experiments are available in Table S5. Each validation pgRNA was cloned into a BsmBI-digested LentiGuide-Puro backbone using a one-step Gibson reaction using the NEBuilder HiFi (New England Biolabs) DNA assembly system, as described above for pgPEN library cloning. Cloned plasmids were transformed into One Shot Stbl3 Chemically Competent E. coli (Invitrogen). Individual colonies were sequence verified via colony Sanger sequencing with primer RKB1148 (Table S5), then the final pgRNA vector was isolated using the Plasmid Plus Midi Kit (QIAGEN).
Competitive fitness assay
For the bichromatic competitive fitness assay, PC9-Cas9-GFP-NLS cells were transduced with a control pgRNA and PC9-Cas9-mCherry-NLS cells with either a paralog single KO pgRNA or a paralog DKO pgRNA. After 48–72 hours of selection with puromycin (1 μg/mL), cells were pooled at an equal ratio and seeded in tissue culture-treated plates (Corning). The day cells were pooled was termed Day 0. For the MAGOH/MAGOHB paralog pair, non-targeting control (NTC) gRNAs were used as controls and each competition (double NTC versus MAGOH single KO, double NTC versus MAGOHB single KO, double NTC versus MAGOH/MAGOHB DKO) was carried out in triplicate. For all other competitive fitness assays (CCNL1/CCNL2, CDK4/CDK6, MEK1/MEK2, OXSR1/STK39, and PSMB5/PSMB8), safe-targeting gRNAs that target intergenic regions (Morgens et al., 2017) were used as controls to account for the different number of double-strand breaks generated by single KO versus DKO pgRNAs and each competition was carried out in six biological replicates. After pooling (Day 0), cells were imaged 24 hours later (Day 1) using a Cytation 5 imager (BioTek Instruments). Raw counts of mCherry- and GFP-expressing cells were computed using Gen5 v3.02 software (BioTek Instruments) to determine the initial paralog-targeting and safe-targeting pgRNA abundance. Cells were imaged and mCherry and GFP cell counts were taken every 1–3 days.
Genomic DNA sequencing for validation
Genomic DNA was extracted from validation cell lines using the DNeasy Blood & Tissue Kit (QIAGEN). On-target editing efficiencies for each gene target were determined via PCR and Sanger or next-generation sequencing. sgRNA target regions were amplified using NEBNExt High-Fidelity 2X PCR Master Mix (New England Biolabs) and custom primers designed for each target; primer sequences are available in Table S5. PCR products were then purified via Ampure bead clean-up and submitted for sequencing by Genewiz. For Sanger sequencing, results were analyzed using the online tool Tracking of Indels by Deconvolution (TIDE, http://shinyapps.datacurators.nl/tide/) which uses Sanger traces to approximate CRISPR editing efficiencies (Brinkman et al., 2014). Next-generation sequencing results were mapped, aligned and CRISPR indels were quantified using the CRISPREsso v2 pipeline (Clement et al., 2019).
Cell lysis and western blotting for validation
Whole-cell extracts for immunoblotting were prepared by incubating cells on a rocker at 4°C in RTK lysis buffer (20 mM Tris (pH 8.0), 2 mM EDTA (pH 8), 137 mM NaCl, 1% IGEPAL CA-630, 10% Glycerol) plus Pierce protease and phosphatase inhibitors (Thermo Scientific) for 20 minutes. Following centrifugation (> 15,000 × g for 20 minutes at 4°C), protein lysates were quantified using the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific). Lysates were separated by SDS–PAGE and transferred to PVDF membranes using the Trans-blot Turbo Transfer System (BioRad). Membranes were blocked in Intercept Blocking Buffer (LiCOR) with 0.1% Tween 20 Solution (BioRad) for 1 hour at room temperature followed by overnight incubation at 4°C with primary antibodies diluted in blocking buffer. IRDye (LiCOR) secondary antibodies were used for detection and were imaged on Odyssey CLx Imaging system (LiCOR). Loading control and experimental protein were probed on the same membrane in all cases. For clarity, loading control is cropped and shown below experimental condition in all panels regardless of the relative molecular weights of the two proteins.
Primary antibodies used for western blotting: CCNL2 (Novus Biologicals #NB100–87009, 1:2000), MEK1 (Cell Signaling Technology #2352, 1:1000), MEK2 (Cell Signaling Technology #9147, 1:1000), OXSR1 (alias OSR1, Cell Signaling Technology #3729, 1:1000), STK39 (alias SPAK, Cell Signaling Technology #2281, 1:500), CDK4 (Cell Signaling Technology #12790, 1:1000), CDK6 (Cell Signaling Technology #13331, 1:1000), vinculin (Sigma #V9264, 1:10,000).
QUANTIFICATION AND STATISTICAL ANALYSIS
Unless otherwise noted, results were analyzed for statistical significance with Rv3.6.3 in an Rstudio v1.2.5 environment. Statistical details of all experiments can be found below or in the corresponding figure legends.
pgPEN CRISPR screen sequencing analysis
Sequencing, image analysis, and base calling for pgPEN screens were performed on the Illumina HiSeq 2500 with RTA 1.18.66.3 software. FASTQ files were generated using Illumina’s bcl2fastq v2.20 conversion software. Reads were trimmed using FASTX-Toolkit v0.014, and samples were demultiplexed using idemp (https://github.com/yhwu/idemp). Sequencing reads for each pgRNA were mapped separately to the pgPEN library annotation using Bowtie v1.2.2 (Langmead et al., 2009). Aligned SAM files were converted to BAM format and sorted using SAMtools v1.9 (Li et al., 2009). pgRNA counts were obtained using a custom R script with R v3.6.2 and R packages Rsamtools v1.34.1 (accessed via Bioconductor v1.3.0; Huber et al., 2015) and Tidyverse v1.2.1 (Wickham et al., 2019). Based on the reference set, correctly-paired pgRNAs were retained while incorrectly-paired gRNAs were discarded. pgRNAs with < 2 reads per million (RPM) in the plasmid pool or with a read count of zero at any time point were also removed. The log2-scaled fold change (LFC) of each pgRNA was then computed using the MAGeCK v0.5.9.2 (Li et al., 2014) test command to compare initial abundance in the plasmid pool to abundance at early and late time points.
LFC values were scaled so that the median of negative control (double non-targeting) pgRNAs was set to zero, while the median of positive control (single-targeting pgRNAs targeting Project Achilles pan-essential genes; Meyers et al., 2017) pgRNAs was set to −1 (Figures 2B and S5D). We also used RNA-seq data from each cell line (Thomas et al., 2020) to control for growth defects caused by the double-strand break generation and repair process. To do this, we adjusted pgRNA LFCs so that the median LFC of single- and double-targeting pgRNAs targeting unexpressed genes (TPM < 2) was set to zero (Figures 2C, S2D, S2E, and S5E–S5G). Finally, we analyzed copy number effects using data from DepMap, accessed via Bioconductor v1.3.0 (Huber et al., 2015) package depmap v1.0.0. We grouped pgRNAs by the combined copy number of targeted genes for each construct and analyzed the CRISPR scores of each copy number group (Figures S2F–S2H and S5H–S5J). Given that the copy number of the vast majority of paralogs included in our library was close to 2, we did not adjust for copy number effects. The scaled and normalized LFC for each pgRNA was termed a CRISPR score (CS). Target-level CRISPR scores were calculated by taking the mean across pgRNAs with the same single KO or DKO paralog target. Final CRISPR scores were computed by taking the mean across the three biological replicates for each screen.
Genetic interaction score calculations
To compute a genetic interaction (GI) score for each paralog pair, we combined two previously published methods for genetic interaction mapping in human cells (DeWeirdt et al., 2021; Han et al., 2017). We first calculated an expected and observed CS for each pgRNA. For DKO pgRNAs (pgRNA-Paralog1_Paralog2), we calculated the expected CS by first taking the mean CRISPR scores of each single KO pgRNA with the same targeting sgRNA sequence paired with a non-targeting control (NTC) sgRNA sequence (i.e., mean(pgRNA-Paralog1_NTC1, pgRNA-Paralog1_NTC2) and mean(pgRNA-NTC1_Paralog2, pgRNA-NTC2_Paralog2)). We summed these two single KO mean CS values to calculate an expected CS for each paralog pair, and compared this expected CS to the observed DKO CS (pgRNA-Paralog1_Paralog2). To establish a distribution of non-interacting GI scores, we used single KO pgRNAs as a negative control. We calculated an expected CS for single KO pgRNAs by computing the sum of (1) the CS for the other single KO pgRNA containing the same targeting sgRNA sequence paired with a different NTC sgRNA sequence (pgRNA-Paralog1_NTC2) and (2) the mean CS of double NTC pgRNAs (pgRNA-NTC1_NTC2) containing the same NTC sgRNA sequence (i.e., mean(pgRNA-NTC1_NTC2, pgRNA-NTC1_NTC3)). This single KO expected CS was then compared to the observed single KO CS (pgRNA-Paralog1_NTC1 or pgRNA-NTC1_Paralog2). Target-level single KO and DKO expected and observed CRISPR scores were calculated by taking the mean across pgRNAs.
We then obtained the distribution of CRISPR scores for control (single KO) pgRNAs by calculating the linear regression of control expected versus observed CS values (Figure 3A for PC9 and Figure S5K for HeLa). GI scores were determined by calculating the residual of each observed CS value for each paralog pair from the control regression line. Statistical significance of DKO GI scores was determined using a t test compared to the distribution of control (single KO) GI scores. A Benjamini-Hochberg false discovery rate (FDR) correction (Benjamini and Hochberg, 1995) was then applied, and FDR < 0.1 was considered significant.
Synthetic lethal paralogs were defined as those with a GI score < −0.5 and FDR < 0.1, while buffering paralogs were defined as those with GI score > 0.25 and FDR < 0.1. Tumor suppressor paralogs were defined as buffering paralogs with an additional filter for DKO CS > 0.25 in either PC9 or HeLa cells. Cancer deletion data for paralog tumor suppressor analysis shown in Figure 6E were obtained from The Cancer Genome Atlas Copy Number Portal (Beroukhim et al., 2010).
Competitive fitness assay analysis
For each competition, the ratio of mCherry (targeting) to GFP (control) cells was computed for each sample replicate at each time point. The log2-fold enrichment of pgRNAs relative to Day 1 was calculated by dividing each subsequent day’s mCherry:GFP ratio by the Day 1 ratio. The expected DKO growth effect under a null model of no interaction was calculated by arbitrarily pairing single KO target (i.e., Gene1_Safe and Gene2_Safe) replicates to calculate the sum of single KO growth effects. For each paralog-targeting pgRNA and for the previously calculated expected (single KO sum) growth phenotypes, the mean and standard error of the mean (SEM) of across replicates were calculated at each time point. Failed replicates were excluded from the analysis. The expected growth effect was compared to the observed DKO growth effects using a one-tailed t test (Figures 4A, S3E–S3H, S3K, and S3L).
DepMap validation analysis
For each paralog pair, we determined the effect of CRISPR-mediated knockout of one gene in cell lines with high (top quartile) compared to low (bottom quartile) expression or copy number of its paralogous gene (Figures 5E–5G). CRISPR score, expression, and copy number data was obtained from DepMap via the Bionconductor package depmap v1.0.0. We compared the effect of paralog 1 knockout in paralog 2 low versus high cell lines using a two-tailed Wilcoxon Rank-Sum test.
Supplementary Material
Highlights.
Human paralog genetic interaction mapping via a double-knockout CRISPR-Cas9 approach
Duplicated paralogs are highly enriched for genetic interactions
Synthetic lethal paralogs include CCNL1/CCNL2, CDK4/CDK6, and OXSR1/STK39
Tumor suppressor paralog pairs include CDKN2A/CDKN2B and FBXO25/FBXO32
ACKNOWLEDGMENTS
We thank Dr. Athea Vichas (Fred Hutchinson Cancer Research Center) for advice on CRISPR screens in PC9 cells and Madeleine Duran (University of Washington) for assistance with improving the efficiency and reproducibility of custom scripts. This work was funded with support from the Lung Cancer Research Foundation. P.C.R.P. was supported by NSF DGE-1762114 and NIH T32-HG000035. J.D.T. is a Washington Research Foundation Postdoctoral Fellow. A.H.B. was supported in part by the NIH/NCI (R00 CA197762 and R37 CA252050); the Devereaux Outstanding Investigator Award from the Prevent Cancer Foundation; the Stephen H. Petersdorf Lung Cancer Research Award; the Seattle Translational Tumor Research program; and the Innovators Network Endowed Chair. R.K.B. was supported in part by the NIH/NIDDK (R01 DK103854); NIH/NHLBI (R01 HL128239 and R01 HL151651); NIH/NCI (R01 CA251138); Edward P. Evans Foundation; Blood Cancer Discoveries Grant program through the Leukemia & Lymphoma Society, Mark Foundation For Cancer Research, and Paul G. Allen Frontiers Group (8023–20); Department of Defense Breast Cancer Research Program (W81XWH-20-1-0596); and the McIlwain Family Endowed Chair in Data Science. R.K.B. is a Scholar of The Leukemia & Lymphoma Society (1344–18). Sequencing was performed by the Fred Hutch Genomics Shared Resource (supported by NIH/NCI Cancer Center Support Grant P30 CA015704). Computational studies were supported in part by FHCRC’s Scientific Computing Infrastructure (ORIP S10 OD028685).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109597.
REFERENCES
- Aguirre AJ, Meyers RM, Weir BA, Vazquez F, Zhang C-Z, Ben-David U, Cook A, Ha G, Harrington WF, Doshi MB, et al. (2016). Genomic Copy Number Dictates a Gene-Independent Cell Response to CRISPR/Cas9 Targeting. Cancer Discov. 6, 914–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arcila ME, Drilon A, Sylvester BE, Lovly CM, Borsu L, Reva B, Kris MG, Solit DB, and Ladanyi M (2015). MAP2K1 (MEK1) Mutations Define a Distinct Subset of Lung Adenocarcinoma Associated with Smoking. Clin. Cancer Res 21, 1935–1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balatoni CE, Dawson DW, Suh J, Sherman MH, Sanders G, Hong JS, Frank MJ, Malone CS, Said JW, and Teitell MA (2009). Epigenetic silencing of Stk39 in B-cell lymphoma inhibits apoptosis from genotoxic stress. Am. J. Pathol 175, 1653–1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartha I, di Iulio J, Venter JC, and Telenti A (2018). Human gene essentiality. Nat. Rev. Genet 19, 51–62. [DOI] [PubMed] [Google Scholar]
- Ben-David U, and Amon A (2020). Context is everything: aneuploidy in cancer. Nat. Rev. Genet 21, 44–62. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, and Hochberg Y (1995). Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Series B Stat. Methodol 57, 289–300. [Google Scholar]
- Berger AH, Brooks AN, Wu X, Shrestha Y, Chouinard C, Piccioni F, Bagul M, Kamburov A, Imielinski M, Hogstrom L, et al. (2016). High-throughput Phenotyping of Lung Cancer Somatic Mutations. Cancer Cell 30, 214–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger AC, Korkut A, Kanchi RS, Hegde AM, Lenoir W, Liu W, Liu Y, Fan H, Shen H, Ravikumar V, et al. ; Cancer Genome Atlas Research Network (2018). A Comprehensive Pan-Cancer Molecular Study of Gynecologic and Breast Cancers. Cancer Cell 33, 690–705.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, Barretina J, Boehm JS, Dobson J, Urashima M, et al. (2010). The landscape of somatic copy-number alteration across human cancers. Nature 463, 899–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blomen VA, Májek P, Jae LT, Bigenzahn JW, Nieuwenhuis J, Staring J, Sacco R, van Diemen FR, Olk N, Stukalov A, et al. (2015). Gene essentiality and synthetic lethality in haploid human cells. Science 350, 1092–1096. [DOI] [PubMed] [Google Scholar]
- Boettcher M, Tian R, Blau JA, Markegard E, Wagner RT, Wu D, Mo X, Biton A, Zaitlen N, Fu H, et al. (2018). Dual gene activation and knockout screen reveals directional dependencies in genetic networks. Nat. Biotechnol 36, 170–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkman EK, Chen T, Amendola M, and van Steensel B (2014). Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 42, e168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, and Helleday T (2005). Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 434, 913–917. [DOI] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network (2014). Comprehensive molecular profiling of lung adenocarcinoma. Nature 511, 543–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao J, Wu L, Zhang S-M, Lu M, Cheung WKC, Cai W, Gale M, Xu Q, and Yan Q (2016). An easy and efficient inducible CRISPR/Cas9 platform with improved specificity for multiple gene targeting. Nucleic Acids Res. 44, e149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherniack AD, Shen H, Walter V, Stewart C, Murray BA, Bowlby R, Hu X, Ling S, Soslow RA, Broaddus RR, et al. ; Cancer Genome Atlas Research Network (2017). Integrated Molecular Characterization of Uterine Carcinosarcoma. Cancer Cell 31, 411–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clement K, Rees H, Canver MC, Gehrke JM, Farouni R, Hsu JY, Cole MA, Liu DR, Joung JK, Bauer DE, and Pinello L (2019). CRISPResso2 provides accurate and rapid genome editing sequence analysis. Nat. Biotechnol 37, 224–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins SR, Schuldiner M, Krogan NJ, and Weissman JS (2006). A strategy for extracting and analyzing large-scale quantitative epistatic interaction data. Genome Biol. 7, R63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dandage R, and Landry CR (2019). Paralog dependency indirectly affects the robustness of human cells. Mol. Syst. Biol 15, e8871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Kegel B, and Ryan CJ (2019). Paralog buffering contributes to the variable essentiality of genes in cancer cell lines. PLoS Genet. 15, e1008466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Kegel B, Quinn N, Thompson NA, Adams DJ, and Ryan CJ (2021). Comprehensive prediction of robust synthetic lethality between paralog pairs in cancer cell lines. BioRxiv. 10.1101/2020.12.16.423022. [DOI] [PubMed] [Google Scholar]
- Dean EJ, Davis JC, Davis RW, and Petrov DA (2008). Pervasive and persistent redundancy among duplicated genes in yeast. PLoS Genet. 4, e1000113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dede M, McLaughlin M, Kim E, and Hart T (2020). Multiplex enCas12a screens detect functional buffering among paralogs otherwise masked in monogenic Cas9 knockout screens. Genome Biol. 21, 262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis MY, and Eichler EE (2016). Human adaptation and evolution by segmental duplication. Curr. Opin. Genet. Dev 41, 44–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeWeirdt PC, Sanson KR, Sangree AK, Hegde M, Hanna RE, Feeley MN, Griffith AL, Teng T, Borys SM, Strand C, et al. (2021). Optimization of AsCas12a for combinatorial genetic screens in human cells. Nat. Biotechnol 39, 94–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diss G, Gagnon-Arsenault I, Dion-Coté A-M, Vignaud H, Ascencio DI, Berger CM, and Landry CR (2017). Gene duplication can impart fragility, not robustness, in the yeast protein interaction network. Science 355, 630–634. [DOI] [PubMed] [Google Scholar]
- Doench JG (2018). Am I ready for CRISPR? A user’s guide to genetic screens. Nat. Rev. Genet 19, 67–80. [DOI] [PubMed] [Google Scholar]
- Doench JG, Fusi N, Sullender M, Hegde M, Vaimberg EW, Donovan KF, Smith I, Tothova Z, Wilen C, Orchard R, et al. (2016). Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol 34, 184–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farmer H, McCabe N, Lord CJ, Tutt ANJ, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, et al. (2005). Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434, 917–921. [DOI] [PubMed] [Google Scholar]
- Finan C, Gaulton A, Kruger FA, Lumbers RT, Shah T, Engmann J, Galver L, Kelley R, Karlsson A, Santos R, et al. (2017). The druggable genome and support for target identification and validation in drug development. Sci. Transl. Med 9, eaag1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagnon KB, and Delpire E (2012). Molecular physiology of SPAK and OSR1: two Ste20-related protein kinases regulating ion transport. Physiol. Rev 92, 1577–1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasperini M, Findlay GM, McKenna A, Milbank JH, Lee C, Zhang MD, Cusanovich DA, and Shendure J (2017). CRISPR/Cas9-Mediated Scanning for Regulatory Elements Required for HPRT1 Expression via Thousands of Large, Programmed Genomic Deletions. Am. J. Hum. Genet 101, 192–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gier RA, Budinich KA, Evitt NH, Cao Z, Freilich ES, Chen Q, Qi J, Lan Y, Kohli RM, and Shi J (2020). High-performance CRISPR-Cas12a genome editing for combinatorial genetic screening. Nat. Commun 11, 3455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonatopoulos-Pournatzis T, Aregger M, Brown KR, Farhangmehr S, Braunschweig U, Ward HN, Ha KCH, Weiss A, Billmann M, Durbic T, et al. (2020). Genetic interaction mapping and exon-resolution functional genomics with a hybrid Cas9-Cas12a platform. Nat. Biotechnol 38, 638–648. [DOI] [PubMed] [Google Scholar]
- Guan Y, Dunham MJ, and Troyanskaya OG (2007). Functional analysis of gene duplications in Saccharomyces cerevisiae. Genetics 175, 933–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han K, Jeng EE, Hess GT, Morgens DW, Li A, and Bassik MC (2017). Synergistic drug combinations for cancer identified in a CRISPR screen for pairwise genetic interactions. Nat. Biotechnol 35, 463–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison R, Papp B, Pál C, Oliver SG, and Delneri D (2007). Plasticity of genetic interactions in metabolic networks of yeast. Proc. Natl. Acad. Sci. USA 104, 2307–2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart T, Chandrashekhar M, Aregger M, Steinhart Z, Brown KR, MacLeod G, Mis M, Zimmermann M, Fradet-Turcotte A, Sun S, et al. (2015). High-Resolution CRISPR Screens Reveal Fitness Genes and Genotype-Specific Cancer Liabilities. Cell 163, 1515–1526. [DOI] [PubMed] [Google Scholar]
- Helming KC, Wang X, Wilson BG, Vazquez F, Haswell JR, Manchester HE, Kim Y, Kryukov GV, Ghandi M, Aguirre AJ, et al. (2014). ARID1B is a specific vulnerability in ARID1A-mutant cancers. Nat. Med 20, 251–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman GR, Rahal R, Buxton F, Xiang K, McAllister G, Frias E, Bagdasarian L, Huber J, Lindeman A, Chen D, et al. (2014). Functional epigenetics approach identifies BRM/SMARCA2 as a critical synthetic lethal target in BRG1-deficient cancers. Proc. Natl. Acad. Sci. USA 111, 3128–3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horlbeck MA, Xu A, Wang M, Bennett NK, Park CY, Bogdanoff D, Adamson B, Chow ED, Kampmann M, Peterson TR, et al. (2018). Mapping the Genetic Landscape of Human Cells. Cell 174, 953–967.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber W, Carey VJ, Gentleman R, Anders S, Carlson M, Carvalho BS, Bravo HC, Davis S, Gatto L, Girke T, et al. (2015). Orchestrating high-throughput genomic analysis with Bioconductor. Nat. Methods 12, 115–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joung J, Konermann S, Gootenberg JS, Abudayyeh OO, Platt RJ, Brigham MD, Sanjana NE, and Zhang F (2017). Genome-scale CRISPR-Cas9 knockout and transcriptional activation screening. Nat. Protoc 12, 828–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim WY, and Sharpless NE (2006). The regulation of INK4/ARF in cancer and aging. Cell 127, 265–275. [DOI] [PubMed] [Google Scholar]
- Lan X, and Pritchard JK (2016). Coregulation of tandem duplicate genes slows evolution of subfunctionalization in mammals. Science 352, 1009–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, and Salzberg SL (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavi O (2015). Redundancy: a critical obstacle to improving cancer therapy. Cancer Res. 75, 808–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R; 1000 Genome Project Data Processing Subgroup (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Xu H, Xiao T, Cong L, Love MI, Zhang F, Irizarry RA, Liu JS, Brown M, and Liu XS (2014). MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol. 15, 554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Fang R, Fang J, He S, and Liu T (2018). Functional implications of Rab27 GTPases in Cancer. Cell Commun. Signal 16, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin A, Giuliano CJ, Palladino A, John KM, Abramowicz C, Yuan ML, Sausville EL, Lukow DA, Liu L, Chait AR, et al. (2019). Off-target toxicity is a common mechanism of action of cancer drugs undergoing clinical trials. Sci. Transl. Med 11, eaaw4412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lord CJ, and Ashworth A (2017). PARP inhibitors: Synthetic lethality in the clinic. Science 355, 1152–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lord CJ, Quinn N, and Ryan CJ (2020). Integrative analysis of large-scale loss-of-function screens identifies robust cancer-associated genetic interactions. eLife 9, e58925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loyer P, and Trembley JH (2020). Roles of CDK/Cyclin complexes in transcription and pre-mRNA splicing: Cyclins L and CDK11 at the cross-roads of cell cycle and regulation of gene expression. Semin. Cell Dev. Biol 107, 36–45. [DOI] [PubMed] [Google Scholar]
- Loyer P, Trembley JH, Grenet JA, Busson A, Corlu A, Zhao W, Kocak M, Kidd VJ, and Lahti JM (2008). Characterization of cyclin L1 and L2 interactions with CDK11 and splicing factors: influence of cyclin L isoforms on splice site selection. J. Biol. Chem 283, 7721–7732. [DOI] [PubMed] [Google Scholar]
- Maffini M, Denes V, Sonnenschein C, Soto A, and Geck P (2008). APRIN is a unique Pds5 paralog with features of a chromatin regulator in hormonal differentiation. J. Steroid Biochem. Mol. Biol 108, 32–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyers RM, Bryan JG, McFarland JM, Weir BA, Sizemore AE, Xu H, Dharia NV, Montgomery PG, Cowley GS, Pantel S, et al. (2017). Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nat. Genet 49, 1779–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgens DW, Wainberg M, Boyle EA, Ursu O, Araya CL, Tsui CK, Haney MS, Hess GT, Han K, Jeng EE, et al. (2017). Genome-scale measurement of off-target activity using Cas9 toxicity in high-throughput screens. Nat. Commun 8, 15178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Najm FJ, Strand C, Donovan KF, Hegde M, Sanson KR, Vaimberg EW, Sullender ME, Hartenian E, Kalani Z, Fusi N, et al. (2018). Orthologous CRISPR-Cas9 enzymes for combinatorial genetic screens. Nat. Biotechnol 36, 179–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neggers JE, Paolella BR, Asfaw A, Rothberg MV, Skipper TA, Yang A, Kalekar RL, Krill-Burger JM, Dharia NV, Kugener G, et al. (2020). Synthetic Lethal Interaction between the ESCRT Paralog Enzymes VPS4A and VPS4B in Cancers Harboring Loss of Chromosome 18q or 16q. Cell Rep. 33, 108493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno S (1970). Evolution by Gene Duplication (Springer-Verlag; ). [Google Scholar]
- Sanchez-Vega F, Mina M, Armenia J, Chatila WK, Luna A, La KC, Dimitriadoy S, Liu DL, Kantheti HS, Saghafinia S, et al. ; Cancer Genome Atlas Research Network (2018). Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 173, 321–337.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjana NE, Shalem O, and Zhang F (2014). Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen JP, Zhao D, Sasik R, Luebeck J, Birmingham A, Bojorquez-Gomez A, Licon K, Klepper K, Pekin D, Beckett AN, et al. (2017). Combinatorial CRISPR-Cas9 screens for de novo mapping of genetic interactions. Nat. Methods 14, 573–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh PP, Affeldt S, Cascone I, Selimoglu R, Camonis J, and Isambert H (2012). On the expansion of “dangerous” gene repertoires by whole-genome duplications in early vertebrates. Cell Rep. 2, 1387–1398. [DOI] [PubMed] [Google Scholar]
- Stewart SA, Dykxhoorn DM, Palliser D, Mizuno H, Yu EY, An DS, Sabatini DM, Chen ISY, Hahn WC, Sharp PA, et al. (2003). Lentivirus-delivered stable gene silencing by RNAi in primary cells. RNA 9, 493–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szymańska E, Nowak P, Kolmus K, Cybulska M, Goryca K, Derezińska-Wołek E, Szumera-Ciećkiewicz A, Brewińska-Olchowik M, Grochowska A, Piwocka K, et al. (2020). Synthetic lethality between VPS4A and VPS4B triggers an inflammatory response in colorectal cancer. EMBO Mol. Med 12, e10812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor AM, Shih J, Ha G, Gao GF, Zhang X, Berger AC, Schumacher SE, Wang C, Hu H, Liu J, et al. (2018). Genomic and Functional Approaches to Understanding Cancer Aneuploidy. Cancer Cell. 33, 676–689.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas JD, Polaski JT, Feng Q, De Neef EJ, Hoppe ER, McSharry MV, Pangallo J, Gabel AM, Belleville AE, Watson J, et al. (2020). RNA isoform screens uncover the essentiality and tumor-suppressor activity of ultraconserved poison exons. Nat. Genet 52, 84–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson NA, Ranzani M, van der Weyden L, Iyer V, Offord V, Droop A, Behan F, Gonçalves E, Speak A, Iorio F, et al. (2021). Combinatorial CRISPR screen identifies fitness effects of gene paralogues. Nat. Commun 12, 1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsherniak A, Vazquez F, Montgomery PG, Weir BA, Kryukov G, Cowley GS, Gill S, Harrington WF, Pantel S, Krill-Burger JM, et al. (2017). Defining a Cancer Dependency Map. Cell 170, 564–576.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vichas A, Riley AK, Nkinsi NT, Kamlapurkar S, Parrish PCR, Lo A, Duke F, Chen J, Fung I, Watson J, et al. (2021). Integrative oncogene-dependency mapping identifies RIT1 vulnerabilities and synergies in lung cancer. Nat. Commun 12 (1), 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilella AJ, Severin J, Ureta-Vidal A, Heng L, Durbin R, and Birney E (2009). EnsemblCompara GeneTrees: Complete, duplication-aware phylogenetic trees in vertebrates. Genome Res. 19, 327–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viswanathan SR, Nogueira MF, Buss CG, Krill-Burger JM, Wawer MJ, Malolepsza E, Berger AC, Choi PS, Shih J, Taylor AM, et al. (2018). Genome-scale analysis identifies paralog lethality as a vulnerability of chromosome 1p loss in cancer. Nat. Genet 50, 937–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Birsoy K, Hughes NW, Krupczak KM, Post Y, Wei JJ, Lander ES, and Sabatini DM (2015). Identification and characterization of essential genes in the human genome. Science 350, 1096–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Yu H, Hughes NW, Liu B, Kendirli A, Klein K, Chen WW, Lander ES, and Sabatini DM (2017). Gene Essentiality Profiling Reveals Gene Networks and Synthetic Lethal Interactions with Oncogenic Ras. Cell 168, 890–903.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham H, Averick M, Bryan J, Chang W, McGowan L, François R, Grolemund G, Hayes A, Henry L, Hester J, et al. (2019). Welcome to the tidyverse. J. Open Source Softw 4, 1686. [Google Scholar]
- Wu X, Yang X, Xiong Y, Li R, Ito T, Ahmed TA, Karoulia Z, Adamopoulos C, Wang H, Wang L, et al. (2021). Distinct CDK6 complexes determine tumor cell response to CDK4/6 inhibitors and degraders. Nat. Cancer 2, 429–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue W, Kitzing T, Roessler S, Zuber J, Krasnitz A, Schultz N, Revill K, Weissmueller S, Rappaport AR, Simon J, et al. (2012). A cluster of cooperating tumor-suppressor gene candidates in chromosomal deletions. Proc. Natl. Acad. Sci. USA 109, 8212–8217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Bouchard G, Yu A, Shafiq M, Jamali M, Shrager JB, Ayers K, Bakr S, Gentles AJ, Diehn M, et al. (2018). GFPT2-Expressing Cancer-Associated Fibroblasts Mediate Metabolic Reprogramming in Human Lung Adenocarcinoma. Cancer Res. 78, 3445–3457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, Liu Y, Zhu R, Ding F, Wan Y, Li Y, and Liu Z (2017). FBXO32 suppresses breast cancer tumorigenesis through targeting KLF4 to proteasomal degradation. Oncogene 36, 3312–3321. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
PC9-Cas9 and HeLa/iCas9 RNA-seq data (GEO: GSE120703) and raw and processed CRISPR screen GSE120703 sequencing data for the PC9-Cas9 and HeLa/iCas9 pgPEN CRISPR screens (GEO: GSE178179) has been deposited to GEO. All GEO-deposited data is listed in the Key resources table and is publicly available as of the date of publication. This paper also analyzes existing, publicly available datasets. The accession numbers for these datasets are listed in the Key resources table. All other data reported in this paper will be shared by the lead contact upon request.
Original pgRNA counting code is publicly available (Zenodo: https://doi.org/10.5281/zenodo.5081113) as of the date of publication and is listed in the Key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.