

Abstract
Rationale: Airway macrophages (AMs) are key regulators of the lung environment and are implicated in the pathogenesis of idiopathic pulmonary fibrosis (IPF), a fatal respiratory disease with no cure. However, knowledge about the epigenetics of AMs in IPF is limited.
Objectives: To assess the role of epigenetic regulation of AMs during lung fibrosis.
Methods: We undertook DNA methylation (DNAm) profiling by using Illumina EPIC (850k) arrays in sorted AMs from healthy donors (n = 14) and donors with IPF (n = 30). Cell-type deconvolution was performed by using reference myeloid-cell DNA methylomes.
Measurements and Main Results: Our analysis revealed that epigenetic heterogeneity was a key characteristic of IPF AMs. DNAm “clock” analysis indicated that epigenetic alterations in IPF AMs were not associated with accelerated aging. In differential DNAm analysis, we identified numerous differentially methylated positions (n = 11) and differentially methylated regions (n = 49) between healthy and IPF AMs, respectively. Differentially methylated positions and differentially methylated regions encompassed genes involved in lipid (LPCAT1 [lysophosphatidylcholine acyltransferase 1]) and glucose (PFKFB3 [6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3]) metabolism, and importantly, the DNAm status was associated with disease severity in IPF.
Conclusions: Collectively, our data identify that changes in the epigenome are associated with the development and function of AMs in the IPF lung.
Keywords: pathogenesis, monocytes, epigenetics, DNA methylation, interstitial lung disease
At a Glance Commentary
Scientific Knowledge on the Subject
Airway macrophages (AMs) are key in the maintenance and defense of the airways and are implicated in the pathogenesis of idiopathic pulmonary fibrosis (IPF), a deadly respiratory disease with no cure. However, little is known of the epigenetics of AMs in health and disease.
What This Study Adds to the Field
We investigated the epigenetics of AMs by profiling DNA methylation (DNAm) in primary AMs obtained from patients with IPF and healthy donors. Our work revealed that AM epigenetic heterogeneity was a key feature of AMs from IPF donors, and “epigenetic clock” analysis revealed that these epigenetic changes were not associated with accelerated aging (a finding in contrast to those on many other age-related morbidities). Furthermore, we found IPF-associated changes in AM DNAm that encompassed genes and pathways pertinent to macrophage biology and IPF pathogenesis. Importantly, epigenetic changes in genes involved in lipid and glucose metabolism were related to clinical features of IPF severity. Thus, our study establishes a link among the epigenome, AM metabolism, and disease severity during IPF.
Airway macrophages (AMs) are sentinel innate cells of the lungs that contribute to homeostasis and the immune response (1). The ontogeny of AMs is complex, encompassing both self-renewing, fetal- derived “resident” cells and monocyte-derived “recruited” cells (2). The understanding of AM ontogeny in disease states and aging is contentious and has relied heavily on murine models. However, we recently helped clarify the AM ontogeny in humans by identifying that 1 year after lung transplantation, AMs in adults are derived exclusively from recruited peripheral monocytes (3).
The influence of the local microenvironment in shaping macrophage development and function is increasingly being appreciated (4). Responses to growth factors or inflammatory mediators can skew macrophage development as exemplified by the proinflammatory “M1” and pro–wound-healing “M2” paradigm. However, in vivo macrophages exhibit tremendous heterogeneity in both healthy and diseased states (5–7), indicating a remarkable plasticity.
Key processes in macrophage development are reflected in changes to the epigenome, including DNA methylation (DNAm) (8). Occurring in the context of cytosine–guanine dinucleotides (CpGs), DNAm influences chromatin accessibility, transcription factor binding, and gene expression (9, 10). DNAm represents one of the most stable epigenetic marks and can be measured as a means of assessing the influence of development and disease on the epigenome. In AMs, DNAm is altered in genetically and environmentally induced chronic airway diseases (11–13). Regional differences in the lung anatomy also influence DNAm in AMs (14), suggesting that the shaping of AM development in the lung microenvironment comprises an epigenetic component. However, despite recent advances in our understanding of AM ontogeny, knowledge about the epigenetics of monocyte-to-macrophage development in the lung and the influence of disease on these processes remains limited.
Idiopathic pulmonary fibrosis (IPF) is a deadly respiratory disease of unknown etiology with heterogeneous cellular and molecular mechanisms (15). The pathobiology of IPF is characterized by a profibrotic wound-healing cascade that does not resolve, leading to progressive scarring, loss of lung function, and ultimately death (16). Although IPF risk involves a strong genetic component (17), the greatest risk factor for IPF is age (median, 65 yr [18]), and the prognosis in IPF is worse than that of some cancers, with a mean survival of 3–5 years (19). In the IPF lung, AMs exhibit transcriptional (7), immunophenotypic (1), and metabolic differences (20, 21). Recent studies employing single-cell RNA sequencing (scRNA-Seq) have indicated a transcriptional spectrum of AMs in the IPF lung that reflects facets of both M1 and M2 macrophage paradigms (5–7). However, despite their emerging role in IPF pathogenesis, the molecular mechanisms underlying transcriptional and other phenotypic characteristics of AMs in IPF are poorly understood.
In the current study, we investigated the epigenetics of AMs by undertaking genome-wide DNAm profiling by using the Illumina EPIC (850k) arrays. By comparing AMs and other myeloid-cell DNA methylomes, we sought to clarify the epigenetics of AM development in the lung. By profiling AMs from healthy donors and donors with IPF, we also sought to determine whether changes in the epigenome characterize features of AMs observed in the IPF lung. Some of the results of these studies have been previously reported in the form of a preprint (https://doi.org/10.1101/2020.12.04.410191).
Methods
Patient Recruitment and AM Enrichment
Study donors underwent bronchoscopy and collection of BAL fluid, as outlined previously (20, 21). All study donors provided written informed consent to participate in the study, which was approved by the research ethics committee (10/HO720/12, 15/LO1399, and 15/SC/0101). The clinical characteristics of the donors are outlined in Table 1. CD206+ AMs were enriched from donor BAL fluid by using the magnetically based MACS system (Miltenyi Biotech) and a fluorescence-activated cell sorter as outlined previously (3, 20, 21).
Table 1.
Study Donor Demographic Data
| Healthy | IPF | P Value | |
|---|---|---|---|
| Number | 14 | 30 | — |
| Age, yr, median (min–max) | 50 (22–67) | 68 (52–82) | <0.0001 |
| Sex, M, n (%) | 7 (50) | 19 (63.3) | 0.40 |
| Smoking, n (%) | |||
| Current | 1 (7.1) | 0 (0) | 0.57 |
| Ex | 3 (21.4) | 17 (56.6) | 0.02 |
| Never | 10 (71.4) | 13 (43.3) | 0.02 |
| Pack-year history* | 3.6 (0.0–20.0) | 14.7 (0.0–45.0) | 0.02 |
| FACS enrichment, n (%) | 7 (50) | 21 (70) | 0.19 |
| FEV1* | 3.2 (2.4–4.2) | 2.2 (1.2–3.8) | <0.0001 |
| FEV1% predicted* | 97.9 (80.1–114.4) | 87.8 (48.0–123.5) | 0.10 |
| FVC* | 4.0 (3.0–5.0) | 2.7 (1.4–4.4) | <0.0001 |
| FVC % predicted* | 98.6 (78.7–114.7) | 84.1 (45.7–124.9) | 0.01 |
| FEV1/FVC %* | 80.2 (70.0–94.0) | 82.2 (71.2–99.0) | 0.23 |
| DlCO* | 107.0 (107.0–107.0) | 51.2 (71.2–99) | <0.0001 |
Definition of abbreviations: IPF = idiopathic pulmonary fibrosis; FACS = fluorescence-activated cell sorter; max = maximum; min = minumum.
Data are shown as the mean (min–max).
DNA/RNA Extraction and EPIC Methylation Arrays
Nucleic acids were extracted from cells by using the AllPrep Mini Kit (QIAGEN), and quality was assessed by using the Genomic DNA ScreenTape and TapeStation System (Agilent). DNA was submitted to the University College London Genomics Core facility for hybridization on Infinium MethylationEPIC BeadChip Arrays (Illumina). We employed the RnBeads 2.0 pipeline (RnBeads Development Team) (22) for methylation array preprocessing. Briefly, quality control metrics were generated, and samples passing quality control were normalized by using the Dasen function (23). After preprosessing, n = 784,669 probes for each n = 44 samples remained for downstream analysis.
Myeloid-Marker CpGs and Deconvolution Analysis
Whole-genome bisulfite sequencing (WGBS) data for BLUEPRINT methylomes (2016 release) (24) were accessed through the RnBeads methylome resource (https://rnbeads.org/methylomes.html). Samples representing the myeloid-cell compartment (venous blood monocytes and macrophage subsets) and derived from donors of ages comparable with those of our study population (i.e., >50 yr) were selected (n = 13; see Table E4 in the online supplement). EPIC array data and WGBS data were merged, resulting in n = 298,945 CpGs across each n = 44 CD206+ and n = 13 reference methylomes. The top 500 most variable CpGs were then used to identify “myeloid-marker CpGs” (myld-CpGs) and subsequently deconvolute and predict the myeloid-cell composition of AMs by using the Houseman method (25).
Differential Methylation
Differentially methylated position (DMP) identification was performed by using the meffil R pipeline (R Foundation for Statistical Computing) (26), which implements epigenome-wide association study (EWAS) analysis. After covariate adjustment, quantile–quantile plots were generated and inspected for EWAS quality control and P-value inflation. A robust genome-wide significance threshold of P < 9 × 10−8, as detailed by Mansell and colleagues (27), was employed to identify significant DMPs. Differentially methylated regions (DMRs) were identified by using DMRcate (Bioconductor) (28) after covariate adjustment as outlined above. IPF DMRs contained >3 CpGs when ranked on the basis of the minimum-smoothed false discovery rate (P < 0.05).
Genomic and Epigenomic Feature Enrichment
Genomic feature distribution and annotation were identified by using HOMER (Hypergeometric Optimization of Motif EnRichment) software (Benner Laboratory, University of California San Diego) (29). Enrichment across epigenomic features from reference monocytes and macrophages (BLUEPRINT Consortium) were conducted by using eFORGE 2.0 (Altius Institute for Biomedical Sciences) (30) and EpiAnnotator (R package) (31). Additional H3K4me1 chromatin immunoprecipitation sequencing and DNase sequencing data were accessed through the International Human Epigenomics Consortium portal (https://epigenomesportal.ca/ihec/grid.html) and the ChIP-Atlas (Kyoto University and Database Center for Life Science) (32), respectively. HOMER software was used to conduct motif enrichment in DMRs.
Three-Dimensional Chromosomal Interactions
Promoter capture HiC (pcHiC) (33) was used to investigate relationships between differential DNAm and the three-dimensional (3D) chromosomal architecture. pcHiC data were overlapped with DMPs and DMRs by using bedtools (Aaron R. Quinlan and Neil Kindlon), and unique “baits” of “other ends” that overlapped DMPs and DMRs with more than five interactions in monocytes and macrophages were subsequently used to identify genes linked through a 3D chromosomal architecture to differential DNAm.
Functional Enrichment
Gene Ontology processes and Kyoto Encyclopedia of Genes and Genomes pathway enrichment of DMR-associated genes were conducted by using the goregion function as implemented in DMRcate. Additional protein–protein interaction networks were identified by using NetworkAnalyst 3.0 (McGill University) (34).
Epigenetic Clock Analysis
The minfi R package (35) was used to normalize and extract EPIC array data and was submitted for advanced analysis by using the DNAm age calculator (https://dnamage.genetics.ucla.edu) (36). Age-adjusted epigenetic age acceleration across the major blood-cell type–derived Hannum and colleagues clock (37) and pan–tissue-derived Horvath clock (36) was calculated for each donor (n = 44 total), and the findings were subsequently compared between donors with IPF and healthy donors and analyzed for myeloid-cell composition.
IPF scRNA-Seq and Other Published Data Sets
We accessed IPF lung scRNA-Seq (GSE136861) (5) by using BBrowser (BioTuring) (38). Additional Uniform Manifold Approximation and Projection for Dimension Reduction projections of gene expression across immune-cell types were generated via the IPF Cell Atlas (www.ipfcellatlas.com), and the corresponding Kaminski–Rosas data set (5) was used. Bulk RNA-Seq data and additional WGBS data from macrophage subtypes were accessed via the BLUEPRINT data analysis portal (39).
Quantitative Real-Time PCR
Gene expression was performed as outlined previously (20, 21). TaqMan probes used in this study were purchased from Thermo Fisher Scientific: LPCAT1 (lysophosphatidylcholine acyltransferase 1) (Hs00227357_m1), SLC12A7 (Hs00986431_m1), SLC6A3 (Hs00997374_m1), ARID5B (AT-rich interaction domain 5B) (Hs01382781_m1), PFKFB3 (6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3) (Hs00998698_m1), and LPCAT2 (Hs01044164_m1).
Statistical Analysis
Differences in donor data were determined by using Mann-Whitey U or chi-square tests for quantitative and categorical data, respectively. Enrichment of the overlap between DMPs or DMRs and pcHiC data were determined by using a chi-square test with Yates correction. P values for epigenetic clock residuals, quantitative real-time PCR, and RNA-sequencing data were determined by using a Mann-Whitney U test. Finally, Spearman rank analysis was used to identify correlations among differential DNAm, clinical variables, and gene expression. All analyses were performed by using GraphPad Prism version 8.4.2
Results
The DNA Methylation Profile of AMs Is Distinct from That of Peripheral Monocytes or Cultured Macrophages
Given recent work identifying AMs as being monocyte-derived and having characteristics spanning the M1–M2 spectrum of activation, we sought to determine whether these changes are also reflected at the epigenetic level by comparing the DNA methylome in AMs and other myeloid cells.
First, we enriched CD206+ AMs, obtained through BAL, from healthy donors (n = 14) and donors with IPF (n = 30) and assayed DNAm by using Illumina MethylationEPIC (850k) arrays, which interrogate >850,000 CpGs across the genome with an enrichment for functional loci (promoters and enhancers; Table 1) (40). These data were then merged with WGBS BLUEPRINT data sets from representative myeloid-cell types, including CD14+CD16− “classical” monocytes, CD14+CD16+ monocytes categorized as “other”, and in vitro–derived M0, M1, and M2 macrophages (24) (Figure 1A). We then identified the top 500 CpGs with a DNAm profile that best discriminated each monocyte and macrophage subtype (see Methods) and characterized these as myld-CpGs (Table E1).
Figure 1.
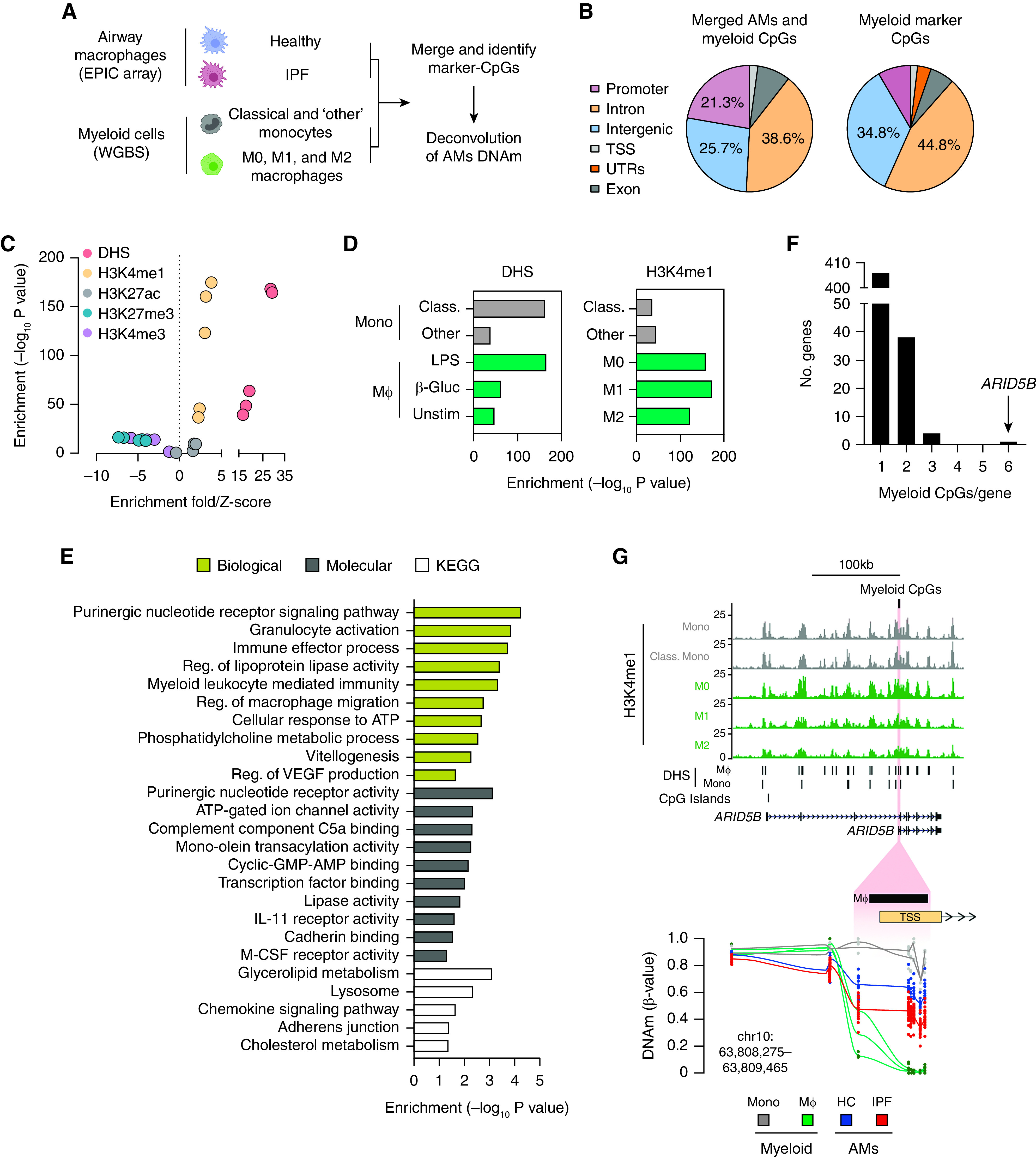
(A) Outline of an approach to investigating the relationship between airway MΦ (AMs) and other myeloid-cell types (i.e., Mono and MΦ) by using DNA methylation (DNAm) as determined through the use of EPIC arrays and WGBS, respectively. Cell-type depictions were generated by using https://www.biorender.com/. (B) Genomic feature distribution for merged AM and myeloid-cell DNAm data sets and the n = 500 myeloid-marker cytosine–guanine dinucleotides (CpGs) to be used in deconvolution analysis. (C and D) Enrichment of myeloid-marker CpGs across histone modifications and DNase hypersensitivity sites (DHS) identified in myeloid cells. (E) Gene Ontology processes and KEGG pathway enrichment analysis for myeloid-marker CpGs. (F) Distribution of myeloid-marker CpGs per gene. (G) Genome track depicting epigenomic features (H3K4me1 chromatin immunoprecipitation sequencing, DHS [BLUEPRINT data] [24]) of myeloid cells across the ARID5B (AT-rich interaction domain 5B) loci. The location of n = 6 myeloid-marker CpGs that cluster at the ARID5B variant 2 TSS is highlighted in red and magnified further below to show DNAm profiles (β values) across each myeloid-cell type and across AMs. Lines indicate the average DNAm across all of the CpGs assayed in the magnified region. β-Gluc = β-glucan; chr = chromosome; Class. = classical; HC = healthy control; IPF = idiopathic pulmonary fibrosis; KEGG = Kyoto Encyclopedia of Genes and Genomes; Mono = monocytes; MΦ = macrophages; Reg. = regulator; TSS = transcription start site; Unstim = unstimulated; UTR = untranslated region; WGBS = whole-genome bisulfite sequencing.
We found that myld-CpGs reside predominately in intronic and intergenic regions (Figure 1B) that are enriched for other epigenetic features in myeloid cells, including histone modifications indicative of poised enhancers (H3K4me1 without H3K27ac) and open chromatin (DNase-I hypersensitivity sites [DHS]; Figures 1C and 1D). Functional enrichment analysis additionally indicated that myld-CpGs encompass a diverse range of receptor signaling, immune-cell activation, and chemokine- and metabolism-related processes and pathways (Figure 1E). Although annotated to n = 449 genes, we found that ARID5B, a transcriptional cofactor that has been shown to regulate glucose metabolism (41), contained the most myld-CpGs (Figure 1F, Table E1)
We focused further on ARID5B, and mining scRNA-Seq data sets from healthy and diseased lungs (IPF/chronic obstructive pulmonary disease) established that ARID5B is expressed across immune cells, including monocytes and macrophages (Figure E1A). At the ARID5B locus, we found that the myld-CpGs are clustered at the promoter region of a shorter transcript, variant 2, and that they overlap with DHS and H3K4me1 enrichment (Figure 1G). We then confirmed the expression of the shorter ARID5B transcript in AMs and in the M1 macrophage subset (Figures E1B and E1C). Finally, closer inspection revealed dramatic changes in DNAm toward the shorter ARID5B variant promoter region, with AMs exhibiting a DNAm profile that was intermediate (average [avg.], 50.8%) compared with that of other myeloid cells (monocytes: avg., 85.4%; macrophages: avg., 0.6%; Table E1). Taken together, these results indicate that the DNAm profile of human AMs is distinct from that of peripheral monocytes/cultured macrophages, and we identify ARID5B DNAm status as a marker of AM development.
Changes in the AM Methylome Define IPF AMs
Next, to determine whether the methylome was distinct in each cell type or disease state, we clustered DNAm profiles for genes with more than two myld-CpGs. Interestingly, our analysis indicated that epigenetic heterogeneity is a feature of IPF, as DNAm profiles across myld-CpGs were distinct when comparing healthy and IPF AMs (Figure E1D). Furthermore, the DNAm of AMs overlapped significantly with that of other myeloid cells, potentially indicating monocytic origin.
To clarify this further, we performed deconvolution analysis of AM DNAm data sets with myld-CpGs (see Methods). Deconvolution revealed that although healthy and IPF AMs were predicted to be largely a composite of other monocytes and M0/M2 macrophages at the epigenetic level, clustering identified the separation of healthy and IPF AMs (Figure 2A), driven largely by differences in the minor classical monocyte and M1 macrophage fractions (Figure 2B). However, further investigation revealed that the specific differences in subsets was related to donor age (Figure 2C).
Figure 2.

(A) Heat map depicting predicted myeloid-cell composition of airway macrophages (AMs) (columns) after deconvolution of DNA methylation (DNAm) profiles with myeloid-marker CpGs generated from reference monocyte and macrophage methylomes (rows). (B) Differences in myeloid-cell composition were evident for AMs derived from healthy donors and donors with idiopathic pulmonary fibrosis (IPF) for “classical” (Class.) and “M1 macrophages,” respectively. P values were determined by using one-way ANOVA with Tukey’s correction for multiple testing. (C) Spearman rank correlation between donor age and the composition of AM DNAm attributed to Class. monocytes (mono) and M1 macrophages. (D) Epigenetic clock analysis indicating a correlation between chronological age and epigenetic age as determined by using the Hannum and colleagues (37) “clock” (top). By comparing residuals from two age-adjusted epigenetic clocks (Hannum and colleagues [37] and Horvath [36]), it was determined that IPF AMs exhibited no epigenetic age acceleration compared with AMs from HC subjects. (E) Spearman rank correlation between epigenetic age acceleration and the composition of AM DNAm attributed to Class. mono and M1 macrophages. CpG = cytosine–guanine dinucleotide; HC = healthy control; IPF = idiopathic pulmonary fibrosis.
Because IPF and aging are linked and many age-related diseases exhibit “accelerated” changes to the epigenome, we next used DNAm clock analyses to clarify the contribution of aging toward the predicted myeloid-cell composition of AMs. Epigenetic clocks use changes in DNAm to estimate the sample donor age. Specifically, the clocks are constructed by using elastic net regression to parsimoniously reflect the aging-related DNA methylome changes with a very small set of CpGs (e.g., Horvath [36], 355 CpGs; Hannum and colleagues [37], 71 CpGs) (42). The clocks provide a strongly validated age estimate in years and, subsequently, have led to the observation of a predicted age that is older than the actual chronological age, or positively “accelerated” aging, with a number of specific diseases (43–45) as well as morbidity and mortality (46). Age-adjusted clock results were adjusted for chronological age by forming a residual and were then compared by using t tests (see Methods). We found that although a strong correlation between chronological and epigenetic age was present for the blood- and tissue-derived Hannum and colleagues (37) and Horvath (36) clocks, respectively (Figures 2D and E1E), no differences in age-adjusted DNAm clock age acceleration was observed between healthy AMs and IPF AMs (Figure 2D). Furthermore, although both the Horvath (36) and Hannum and colleagues (37) clocks may underestimate the age of older individuals (>60 yr) because of nonlinear effects potentially driven by signal saturation (47), we also found no positive age acceleration in IPF across other epigenetic clocks that are less likely to be influenced these effects (Figure E1F). This does not exclude the role of aging-related changes, but there is no supportive evidence for an acceleration beyond chronological age of the robust aging-related changes captured by DNAm clocks at the power resolution of this purified cell-type study (42). Importantly, there was no relationship between the predicted myeloid-cell composition and epigenetic age acceleration (Figures 2E and E1G), implying that myeloid-cell composition was a feature of IPF AMs and not of the more generalized DNAm clock changes. Taken together, these data indicate that epigenetic heterogeneity is present in AMs and is a characteristic of IPF.
Identification of DMPs in IPF
We next sought to determine whether AM DNAm profiles are impacted during IPF and to identify the impact of myeloid-cell composition in these analyses. Initial principal component analysis indicated a separation of donors by disease group (Figure E2A), with myeloid-cell composition and donor age being comparable drivers of variance within the data set (Figure E2D).
We then undertook analysis to identify DMPs in IPF, employing a robust genome-wide significance threshold P value of <9 ×10−8 that controls for the false-positive rate of the DNAm EPIC arrays (27) and observed a dramatic impact when adjusting for myeloid-cell composition in addition to other study covariates (Figure 3A and Methods). This was equally evident when investigating the direction of DNAm change in IPF, with all myeloid-adjusted DMPs identified (n = 11) losing DNAm compared with healthy control subjects (Figures 3B, E2C, and E2D and Table E2).
Figure 3.

(A) Quantile–quantile plots depicting the impact of adjustment (adjust.) for myeloid-cell composition in addition to other study covariates on identification of differentially methylated positions (DMPs) in idiopathic pulmonary fibrosis (IPF). Those DMPs reaching the epigenome-wide association study significance threshold of P < 9 × 10−8 are highlighted in red. (B) Volcano plot depicting impact of myeloid-cell adjust. and direction of DNA methylation (DNAm) changes of DMPs in IPF. The dashed line represents the epigenome-wide association study P-value threshold. (C) Genome track depicting epigenomic features (H3K4me1 chromatin immunoprecipitation sequencing, DHS [BLUEPRINT data] [24]) of myeloid cells across the LPCAT1 (lysophosphatidylcholine acyltransferase 1) loci. Regions interacting with the LPCAT1 promoter in a 3D fashion as determined by pcHiC are indicated in gray. Interactions with frequency threshold >5 in myeloid cells are highlighted in purple. The location of n = 3 intronic IPF DMPs are highlighted in red and magnified further below to show methylation profiles (β values) of healthy and IPF airway MΦ (AMs) across the respective cytosine–guanine dinucleotides (CpGs) (*). Lines indicate average methylation across all of the CpGs assayed in the magnified region. (D) Relationship between DNAm of IPF DMPs and gene expression for LPCAT1 across HC (blue) and IPF (red) donor AMs. (E and F) Relationship between gene expression (E) and methylation of an IPF-associated DMP (F) for LPCAT1 and FVC. 3D = three-dimensional; chr = chromosome; Class. = classical; DHS = DNase-I hypersensitivity; HC = healthy control; Mono = monocytes; MΦ = macrophages; pcHiC = promoter capture HiC; POLE = DNA polymerase epsilon, catalytic subunit; Rel. = relative.
IPF DMPs were either intronic (n = 9) or intergenic (n = 2) and occurred in regions enriched for open chromatin in myeloid cells (DHS; Figure E2E). We found n = 3 IPF DMPs clustered at LPCAT1, an enzyme that mediates the conversion of lysophosphatidylcholine to phosphatidylcholine (28) (Figure 3B). Mining of IPF scRNA-Seq data indicates that LPCAT1 is expressed across monocytes and macrophages in the lung, and we confirmed these findings in our study AMs (Figures E2F and E2G).
Although LPCAT1 DMPs are intronic, distal regions can influence gene expression through 3D interactions. To investigate this further, we used pcHiC data to investigate the relationship between IPF DMPs and 3D interactions in myeloid cells (33). Remarkably, although the LPCAT1 promoter interacted with other genes/regions specifically in monocytes (Figure E2H), the strongest interaction occurred with the region containing the IPF DMPs (Figure 3C). We identified a correlation between IPF DMP methylation and gene expression that occurred only for LPCAT1 (Figure 3D) and not any other interacting genes (Figure E2I).
Finally, we investigated the relationship between LPCAT1 and the clinical features of IPF (Table 1) and found that although no relationship was present for gene expression (Figure 3E), there was a strong correlation between methylation and the FVC, a measure of disease severity and progression in IPF (48) (Figure 3F). Taken together, these data suggest AMs’ similarity to monocytes on the epigenetic and higher-order 3D-interaction level and point to a function of the DNAm of AMs in IPF pathogenesis.
Identification of DMRs in IPF
Given the clustering of IPF DMPs, we next conducted an analysis to identify DMRs containing changes across more than three CpGs with a false discovery rate–adjusted P value of <0.05 in IPF (49). Similar to what we observed in the DMP analysis, we saw a reduction in the total number of DMRs identified after adjusting for myeloid-cell composition (Figure 4A). However, we found n = 49 myeloid-adjusted DMRs that included regions that were both gaining and losing DNAm compared with healthy control DMRs (Table E3). We also found n = 2 DMRs that encompassed the previously identified DMPs of LPCAT1 and POLE (DNA polymerase epsilon, catalytic subunit) (Figure 3B).
Figure 4.

(A) Volcano plot depicting impact of myeloid-cell adjust. and direction of DNAm changes of differentially methylated regions (DMRs) in idiopathic pulmonary fibrosis (IPF). (B) Genomic feature distribution for all EPIC array CpGs and those encompassed by IPF DMRs. (C and D) Enrichment of IPF DMRs across histone modifications and DHS in Mono and MΦ. (E) DNA motif enrichment in IPF DMRs. adjust. = adjustment; β-Gluc = β-glucan; Class. = classical; CpG = cytosine–guanine dinucleotide; DHS = DNase-I hypersensitivity sites; DNAm = DNA methylation; FDR = false discovery rate; HC = healthy control; LPCAT1 = lysophosphatidylcholine acyltransferase 1; Mono = monocytes; MΦ = macrophages; PFKFB3 = 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3; POLE = DNA polymerase epsilon, catalytic subunit; TSS = transcription start site; Unstim = unstimulated; UTR = untranslated region.
IPF DMRs were distributed across various genomic features, including promoters, introns, and exons (Figure 4B); occurred in regions enriched for open chromatin in myeloid cells (Figures 4C and 4D); and were more likely to be linked in a 3D fashion to distal genes and regions (Figures E3A–E3C). We additionally found motifs matching transcription factors previously implicated in macrophage polarization to be enriched in IPF DMRs (e.g., KLF4, FOXO1; Figure 4E) and the subsequent cell-type expression profiles of transcription factor–encoding genes across lung immune cells (Figure E3D).
Although mining of scRNA-Seq data revealed that only some DMR-associated genes were expressed/detected in the monocyte and macrophage subsets identified in the IPF lung (Figure E3E), donors with IPF and healthy donors remarkably clustered separately on the basis of their respective DMR-associated gene expression profiles (Figure E3F).
We then undertook functional enrichment analysis and found enrichment across various processes and pathways pertinent to macrophage biology (e.g., extravasations) and IPF pathogenesis (e.g., platelet activation, response to wound healing; Figure E4A).
To gain a better insight into the biological implications of changes in DNAm, we undertook additional protein–protein interaction analysis and found that DMR-associated genes form the central hubs of large interconnection networks (Figures 5A and E4B). We refined our analysis further and undertook functional enrichment of networks by DNAm status and found that hub genes gaining DNAm in IPF predominately encompass metabolic processes, whereas those losing DNAm play a role in processes and pathways pertinent to macrophage biology and fibrogenesis (e.g., phagocytosis, cell proliferation, and TGF-β signaling; Figure 5B).
Figure 5.

(A) Network depicting protein–protein interactions of differentially methylated region (DMR)-associated genes. (B) Gene Ontology processes and KEGG pathway enrichment analysis for DMR-associated genes gaining or losing DNA methylation (DNAm) in idiopathic pulmonary fibrosis (IPF). (C) Genome track depicting epigenomic features (H3K4me1 chromatin immunoprecipitation sequencing, DHS [BLUEPRINT data] [24]) of myeloid cells across the PFKFB3 (6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3) loci. The location of the IPF DMR is highlighted in red and magnified further below to show methylation profiles (β values) at the associated n = 3 cytosine–guanine dinucleotide (CpGs) (*) across healthy and IPF airway MΦ (AMs). Lines indicate average methylation across all of the CpGs assayed in the magnified region. (D) Relationship between DNAm of IPF DMPs and gene expression for PFKFB3 across HC (blue) and IPF (red) donor AMs. (E and F) Relationship between gene expression (E) and methylation of DMR-associated CpG (F) for PFKFB3 and FVC. chr = chromosome; Class. = classical; DHS = DNase-I hypersensitivity; DMP = differentially methylated position; ER = endoplasmic reticulum; HC = healthy control; KEGG = Kyoto Encyclopedia of Genes and Genomes; LPCAT1 = lysophosphatidylcholine acyltransferase 1; Mono = monocytes; MΦ = macrophages; POLE = DNA polymerase epsilon, catalytic subunit; pos. reg. = positive regulator; rel. = relative.
Given that work from our laboratory has identified an altered state of AM metabolism in IPF (20, 21), we focused on PFKFB3, a potent driver of glycolysis (50), and found that the associated IPF DMR was located in an intergenic region upstream of the PFKFB3 promoter and that it overlapped with H3K4me1 and DHS enrichment (Figure 5C). Remarkably, a complete loss of methylation for CpGs at the PFKFB3 transcription start site was observed for study AMs and across macrophage subsets (Figure E4C and E4D).
We then found that PFKFB3 was differentially expressed between healthy conditions and IPF conditions not only in our study AMs but also across macrophages in scRNA-Seq data from the IPF lung and macrophage subsets (Figures E4E–E4G). We subsequently identified a correlation in AMs between DNAm and gene expression for two of the three CpGs encompassing the PFKFB3 DMR (Figure 5D) and identified relationships between the PFKFB3 gene expression and methylation and the severity of IPF as determined by using the FVC (Figures 5E and 5F). Taken together, these data strongly suggest that changes in the epigenome underpin the distinct metabolic phenotype observed in AMs isolated from IPF lungs as well as their contribution toward disease pathogenesis.
Discussion
AMs are key regulators of the lung environment and are implicated in the pathogenesis of lung fibrosis. By comparing AMs with reference myeloid cells, we determined that epigenetic heterogeneity is present in AMs and is furthermore a characteristic of IPF (Figure 2). Although identified computationally, our findings mirror scRNA-Seq studies of the IPF lung in which AMs exhibit transcriptional heterogeneity (5–7). Differences in myeloid-cell composition also suggest that the IPF lung influences monocyte-to-macrophage developmental trajectories. Interestingly, transcriptomic signatures reflective of blood monocytes are already altered in association with IPF severity (51–53), potentially indicating that the effects of IPF extend across tissue compartments rather than being isolated to the lung.
In the absence of single-cell data, computational deconvolution of epigenetic data is essential to deciphering disease effects within samples consisting of mixed cell populations (54). Even though we had enriched AMs on the basis of cell surface expression of CD206, we identified epigenetic heterogeneity and found a tremendous impact of myeloid-cell composition in identification of DMPs and DMRs in IPF. Our work has implications for previous studies of DNAm in IPF that have largely assayed whole-lung tissue in bulk without accounting for cell-type heterogeneity (55, 56). Furthermore, our study employed a genome-wide approach, providing better insights into the influence of IPF on the wider epigenome than previously conducted gene-specific studies in this disease area (57).
Advanced age is a key risk factor for IPF (58), and numerous studies have shown that molecular changes commonly associated with aging such as telomere shortening (59, 60) and augmented markers of senescence (61, 62) are found in patients with IPF. Although the identification of epigenetic heterogeneity in AMs was important for deciphering DNAm changes in IPF, we additionally found that donor age correlated with this predicted heterogeneity. To address the potential interaction of heterogeneity and age, we conducted epigenetic clock analysis, as these signatures are actively being explored for possible novel, age-related disease insights (63). However, we found no differences in age acceleration between healthy AMs and IPF AMs or in the relationship to the predicted myeloid-cell composition. These findings are in contrast to those for many other age-related diseases (42) and indicate that although IPF predominately occurs in later decades, the DNAm changes detected are likely specific to IPF rather than representing epigenome changes that occur as a consequence of otherwise “healthy aging” (64). Although these analyses and other adjustments for donor age in differential analysis revealed the influence of IPF on AM DNAm, IPF and age remain inexplicably linked. Future studies of epigenetics in IPF should therefore strive to include healthy age- and sex-matched control subjects.
In mice, recruited monocyte-derived AMs, as opposed to fetal-derived tissue-resident AMs, have been implicated in the pathogenesis of pulmonary fibrosis (2). However, the origins of AMs during IPF have not been defined. By attempting to clarify the epigenetic events related to macrophage development in the lung, we found that DNAm patterns that discriminate by myeloid-cell type occur largely in intronic and intergenic regions. This supports previous work indicating that epigenomic changes during immune-cell lineage commitment occur within noncoding regions (65). However, we identified intergenic DNAm within ARID5B at the promoter for a shorter transcript, variant 2, as a mark of monocyte-to-macrophage development. ARID5B is a chromatin modifier that acts as a transcriptional coactivator by removing repressive histone modifications (66). In addition, ARID5B has been linked to adipogenesis (67) and metabolism in hepatocytes (68) and natural killer cells, where altered DNAm, particularly of the short transcript variant 2, characterized a HMCV+-adaptive natural-killer-cell subtype (69). More relevant to this study was work using a multiomics approach that identified ARID5B’s association with atherosclerosis in CD14+ blood monocytes and implicated 3D interactions in linking intronic ARID5B DNAm (and other regulatory regions) with the ARID5B promoter (70). Thus, our data indicate that the DNAm profile of mature AMs is distinct from that of peripheral monocytes and identify ARID5B DNAm as a marker of AM development.
Recently, AM metabolic function has been implicated in the pathogenesis of IPF. Increased expression of the glucose transporter GLUT1 has been described in IPF AMs (71), as has accumulation of dysmorphic mitochondria and reduced oxidative phosphorylation–related gene expression (72). Indeed, work from our laboratory has indicated the crucial role of AM metabolic rewiring in IPF (20, 21). Here, our data demonstrate that aberrant AM metabolism during IPF may be in part under epigenetic control. We found that IPF DMRs encompassed hub genes of networks associated with lipid, iron, and glycolytic metabolic processes, suggesting that epigenetic control likely occurs at key genes in a discrete manner rather than across pathways or processes at large.
Indicative of the impact of changes in DNAm was the DMR located at PFKFB3, an enzyme responsible for the synthesis and degradation of fructose 2,6-bisphosphate, a key regulator of glycolysis. Recent work has identified an important role of PFKFB3 in controlling macrophage plasticity and activation during liver fibrosis (73), in maintaining cell viability under hypoxic and inflammatory conditions (74), and in promoting macrophage antiviral responses (75). Furthermore, 3 of 11 EWAS DMPs in IPF clustered within an intronic region of LPCAT1, an evolutionarily conserved enzyme that is involved in phospholipid metabolism and performs a key role in surfactant production in alveolar type 2 cells (76) and in the inflation of the lungs upon birth (77). Recent work has also implicated LPCAT1 in aberrant metabolism and plasma membrane remodeling in cancer (78).
In IPF, reduction of LPCAT1 gene expression was shown to characterize subsets of IPF-specific airway epithelial cells (79). Interestingly, despite not reaching EWAS significance, we observed changes in DNAm in IPF AMs at the related paralog LPCAT2 (Figure E2J). Although no difference in LPCAT2 gene expression was observed in IPF AMs, the expression of LPCAT2 is higher than that of LPCAT1 across macrophage subtypes (Figures E2K–E2M). These data suggest that epigenetic regulation of phospholipid metabolism, specifically in AMs, may occur in an LPCAT1-dependent manner and further support the role of discrete changes in DNAm contributing to altered lipid mechanisms and other metabolic defects in IPF (71, 72, 80).
Our work has several limitations that should be considered. First, similar to work in blood monocytes (70), our work investigated whether integrating 3D interactions could help elucidate the potential impact of changes in DNAm on gene expression (33). Remarkably, we found that the LPCAT1 promoter “self-interacted” with regions containing IPF DMPs and that over half of all DMRs were linked in a 3D fashion to other genomic regions in myeloid cells. Although these data suggest that AMs share a similarity with DNAm in their having a higher-order chromatin structure similar to that of monocytes and macrophages, bias from the EPIC array design needs to be taken into consideration. Future studies should therefore aim to determine the 3D interactions of AMs from healthy donors and donors with IPF empirically.
Second, given that the composition of AMs in IPF was more “M1-like” and that the progressive remodeling of the IPF lung results in an inflammatory hypoxic environment, these results raise the question of whether developmental and epigenetic changes identified in IPF AMs are a cause or consequence of the fibrotic milieu of the IPF lung. Future studies should seek to enrich individual pulmonary cell types (as well as their circulating precursors) to assess how the duration of exposure in the IPF lung alters the epigenome by comparing more transient cell types (i.e., immune cells) to those unable to “escape” the hypoxic/fibrotic milieu (i.e., stromal/epithelial cells). Furthermore, although our work identifies epigenomic changes during the monocyte-to-macrophage transition in the lung, these data are inferred from computational deconvolution of bulk populations. Future studies of IPF should seek to generate matched transcriptome and epigenomic data sets across the blood and lung to comprehensively address the molecular events and influence of IPF on AM developmental trajectories.
Finally, we did not investigate avenues to recapitulate or reverse the changes in DNAm observed in our study. In addition to more traditional chemical-based methods (e.g., DNAm inhibitors), advances in epigenome editing techniques that can effectively recapitulate diseased epigenomes in vitro (81) should be employed in future studies to allow functional characterization of epigenetic changes occurring in monocyte-to-macrophage transitions and IPF pathogenesis (82).
In conclusion, our study has identified a role of the aberrant epigenetic regulation of AMs, independent of aging alone, which appears to be involved in IPF pathogenesis. Our study provides a foundation for further investigations to clarify the role of epigenetics during monocyte-to-macrophage development in healthy and diseased airways. Furthermore, our data highlight the possibility that therapeutic agents targeting epigenetic modification may have a role in the treatment of IPF.
Acknowledgments
Acknowledgment
The authors thank the Imperial College South Kensington flow cytometry facility, particularly Ms. Jane Srivastava and Dr. Jessica Rowley, for support. The authors also thank Dr. Mark Kristiansen and Gaganjit Kaur Madhan at University College London Genomics for processing EPIC arrays. They thank Dr. Michael Scherer at the Department of Genetics/Epigenetics, Saarland University, Germany, for advice with RnBeads analysis and for providing additional code.
Footnotes
Supported by a National Heart and Lung Institute Pilot Award (P.M.); a Joan Bending, Evelyn Bending, Mervyn Stephens, and Olive Stephens Memorial Fellowship from Asthma UK (AUK-SNF-2017-381; A.J.B.); a National Institute for Health Research Clinician Scientist Fellowship from the Health Services and Delivery Research Programme (CS-2013-13-017; T.M.M.); British Lung Foundation Chair in Respiratory Research (C17-3; T.M.M.); and a Wellcome Trust Senior Fellowship in Basic Biomedical Science (107059/Z/15/Z; C.M.L.).
Author Contributions: P.M., A.J.B. C.M.L., and T.M.M. designed the study. R.J.H., P.L.M., and T.M.M. obtained consent from patients and carried out bronchoscopies. P.P.O., P.G., S.K., G.J.A., E.C., and P.M. processed samples and conducted gene expression analysis. P.M., C.G.B., Z.B., and S.B. analyzed and interpreted DNA methylation data. All authors were involved in the interpretation of the results and drafting and/or revising the manuscript, approved the final version to be published, and vouch for the content of the final manuscript.
Data Availability: All EPIC methylation array data has been deposited on the Gene Expression Omnibus (GSE159655).
This article has an online supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1164/rccm.202101-0004OC on July 19, 2021
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1. Byrne AJ, Maher TM, Lloyd CM. Pulmonary macrophages: a new therapeutic pathway in fibrosing lung disease? Trends Mol Med. 2016;22:303–316. doi: 10.1016/j.molmed.2016.02.004. [DOI] [PubMed] [Google Scholar]
- 2. Misharin AV, Morales-Nebreda L, Reyfman PA, Cuda CM, Walter JM, McQuattie-Pimentel AC, et al. Monocyte-derived alveolar macrophages drive lung fibrosis and persist in the lung over the life span. J Exp Med. 2017;214:2387–2404. doi: 10.1084/jem.20162152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Byrne AJ, Powell JE, O’Sullivan BJ, Ogger PP, Hoffland A, Cook J, et al. Dynamics of human monocytes and airway macrophages during healthy aging and after transplant. J Exp Med. 2020;217:e20191236. doi: 10.1084/jem.20191236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lavin Y, Winter D, Blecher-Gonen R, David E, Keren-Shaul H, Merad M, et al. Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell. 2014;159:1312–1326. doi: 10.1016/j.cell.2014.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Adams TS, Schupp JC, Poli S, Ayaub EA, Neumark N, Ahangari F, et al. Single-cell RNA-seq reveals ectopic and aberrant lung-resident cell populations in idiopathic pulmonary fibrosis. Sci Adv. 2020;6:eaba1983. doi: 10.1126/sciadv.aba1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Habermann AC, Gutierrez AJ, Bui LT, Yahn SL, Winters NI, Calvi CL, et al. Single-cell RNA sequencing reveals profibrotic roles of distinct epithelial and mesenchymal lineages in pulmonary fibrosis. Sci Adv. 2020;6:eaba1972. doi: 10.1126/sciadv.aba1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Reyfman PA, Walter JM, Joshi N, Anekalla KR, McQuattie-Pimentel AC, Chiu S, et al. Single-cell transcriptomic analysis of human lung provides insights into the pathobiology of pulmonary fibrosis. Am J Respir Crit Care Med. 2019;199:1517–1536. doi: 10.1164/rccm.201712-2410OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wallner S, Schröder C, Leitão E, Berulava T, Haak C, Beißer D, et al. Epigenetic dynamics of monocyte-to-macrophage differentiation. Epigenetics Chromatin. 2016;9:33. doi: 10.1186/s13072-016-0079-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tirado-Magallanes R, Rebbani K, Lim R, Pradhan S, Benoukraf T. Whole genome DNA methylation: beyond genes silencing. Oncotarget. 2017;8:5629–5637. doi: 10.18632/oncotarget.13562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Schübeler D. Function and information content of DNA methylation. Nature. 2015;517:321–326. doi: 10.1038/nature14192. [DOI] [PubMed] [Google Scholar]
- 11. Chen Y, Armstrong DA, Salas LA, Hazlett HF, Nymon AB, Dessaint JA, et al. Genome-wide DNA methylation profiling shows a distinct epigenetic signature associated with lung macrophages in cystic fibrosis. Clin Epigenetics. 2018;10:152. doi: 10.1186/s13148-018-0580-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fricker M, Gibson PG. Macrophage dysfunction in the pathogenesis and treatment of asthma. Eur Respir J. 2017;50:1700196. doi: 10.1183/13993003.00196-2017. [DOI] [PubMed] [Google Scholar]
- 13. He L-X, Tang Z-H, Huang Q-S, Li W-H. DNA methylation: a potential biomarker of chronic obstructive pulmonary disease. Front Cell Dev Biol. 2020;8:585. doi: 10.3389/fcell.2020.00585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Armstrong DA, Chen Y, Dessaint JA, Aridgides DS, Channon JY, Mellinger DL, et al. DNA methylation changes in regional lung macrophages are associated with metabolic differences. Immunohorizons. 2019;3:274–281. doi: 10.4049/immunohorizons.1900042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Martinez FJ, Collard HR, Pardo A, Raghu G, Richeldi L, Selman M, et al. Idiopathic pulmonary fibrosis. Nat Rev Dis Primers. 2017;3:17074. doi: 10.1038/nrdp.2017.74. [DOI] [PubMed] [Google Scholar]
- 16. Chambers RC, Mercer PF. Mechanisms of alveolar epithelial injury, repair, and fibrosis. Ann Am Thorac Soc. 2015;12:S16–S20. doi: 10.1513/AnnalsATS.201410-448MG. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Allen RJ, Guillen-Guio B, Oldham JM, Ma SF, Dressen A, Paynton ML, et al. Genome-wide association study of susceptibility to idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2020;201:564–574. doi: 10.1164/rccm.201905-1017OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Molyneaux PL, Willis-Owen SAG, Cox MJ, James P, Cowman S, Loebinger M, et al. Host–microbial interactions in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2017;195:1640–1650. doi: 10.1164/rccm.201607-1408OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Vancheri C, Failla M, Crimi N, Raghu G. Idiopathic pulmonary fibrosis: a disease with similarities and links to cancer biology. Eur Respir J. 2010;35:496–504. doi: 10.1183/09031936.00077309. [DOI] [PubMed] [Google Scholar]
- 20. Allden SJ, Ogger PP, Ghai P, McErlean P, Hewitt R, Toshner R, et al. The transferrin receptor CD71 delineates functionally distinct airway macrophage subsets during idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2019;200:209–219. doi: 10.1164/rccm.201809-1775OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ogger PP, Albers GJ, Hewitt RJ, O’Sullivan BJ, Powell JE, Calamita E, et al. Itaconate controls the severity of pulmonary fibrosis. Sci Immunol. 2020;5:eabc1884. doi: 10.1126/sciimmunol.abc1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Müller F, Scherer M, Assenov Y, Lutsik P, Walter J, Lengauer T, et al. RnBeads 2.0: comprehensive analysis of DNA methylation data. Genome Biol. 2019;20:55. doi: 10.1186/s13059-019-1664-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pidsley R, Y Wong CC, Volta M, Lunnon K, Mill J, Schalkwyk LC. A data-driven approach to preprocessing Illumina 450K methylation array data. BMC Genomics. 2013;14:293. doi: 10.1186/1471-2164-14-293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Stunnenberg HG, Hirst M. International Human Epigenome Consortium. The International Human Epigenome Consortium: a blueprint for scientific collaboration and discovery. Cell. 2016;167:1145–1149. doi: 10.1016/j.cell.2016.11.007. [DOI] [PubMed] [Google Scholar]
- 25. Houseman EA, Kile ML, Christiani DC, Ince TA, Kelsey KT, Marsit CJ. Reference-free deconvolution of DNA methylation data and mediation by cell composition effects. BMC Bioinformatics. 2016;17:259. doi: 10.1186/s12859-016-1140-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Min JL, Hemani G, Davey Smith G, Relton C, Suderman M. Meffil: efficient normalization and analysis of very large DNA methylation datasets. Bioinformatics. 2018;34:3983–3989. doi: 10.1093/bioinformatics/bty476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mansell G, Gorrie-Stone TJ, Bao Y, Kumari M, Schalkwyk LS, Mill J, et al. Guidance for DNA methylation studies: statistical insights from the Illumina EPIC array. BMC Genomics. 2019;20:366. doi: 10.1186/s12864-019-5761-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Du Y, Wang Q, Zhang X, Wang X, Qin C, Sheng Z, et al. Lysophosphatidylcholine acyltransferase 1 upregulation and concomitant phospholipid alterations in clear cell renal cell carcinoma. J Exp Clin Cancer Res. 2017;36:66. doi: 10.1186/s13046-017-0525-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Breeze CE, Reynolds AP, van Dongen J, Dunham I, Lazar J, Neph S, et al. eFORGE v2.0: updated analysis of cell type-specific signal in epigenomic data. Bioinformatics. 2019;35:4767–4769. doi: 10.1093/bioinformatics/btz456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pageaud Y, Plass C, Assenov Y. Enrichment analysis with EpiAnnotator. Bioinformatics. 2018;34:1781–1783. doi: 10.1093/bioinformatics/bty007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Oki S, Ohta T, Shioi G, Hatanaka H, Ogasawara O, Okuda Y, et al. ChIP-Atlas: a data-mining suite powered by full integration of public ChIP-seq data. EMBO Rep. 2018;19:e46255. doi: 10.15252/embr.201846255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Javierre BM, Burren OS, Wilder SP, Kreuzhuber R, Hill SM, Sewitz S, et al. BLUEPRINT Consortium. Lineage-specific genome architecture links enhancers and non-coding disease variants to target gene promoters. Cell. 2016;167:1369–1384, e19. doi: 10.1016/j.cell.2016.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhou G, Soufan O, Ewald J, Hancock REW, Basu N, Xia J. NetworkAnalyst 3.0: a visual analytics platform for comprehensive gene expression profiling and meta-analysis. Nucleic Acids Res. 2019;47:W234–W241. doi: 10.1093/nar/gkz240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Aryee MJ, Jaffe AE, Corrada-Bravo H, Ladd-Acosta C, Feinberg AP, Hansen KD, et al. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics. 2014;30:1363–1369. doi: 10.1093/bioinformatics/btu049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Horvath S. DNA methylation age of human tissues and cell types. Genome Biol. 2013;14:R115. doi: 10.1186/gb-2013-14-10-r115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hannum G, Guinney J, Zhao L, Zhang L, Hughes G, Sadda S, et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol Cell. 2013;49:359–367. doi: 10.1016/j.molcel.2012.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Le T, Phan T, Pham M, Tran D, Lam L, Nguyen T, et al. BBrowser: making single-cell data easily accessible [preprint] bioRxiv. 2020 https://www.biorxiv.org/content/10.1101/2020.12.11.414136v1
- 39. Fernández JM, de la Torre V, Richardson D, Royo R, Puiggròs M, Moncunill V, et al. BLUEPRINT Consortium. The BLUEPRINT data analysis portal. Cell Syst. 2016;3:491–495, e5. doi: 10.1016/j.cels.2016.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pidsley R, Zotenko E, Peters TJ, Lawrence MG, Risbridger GP, Molloy P, et al. Critical evaluation of the Illumina MethylationEPIC BeadChip microarray for whole-genome DNA methylation profiling. Genome Biol. 2016;17:208. doi: 10.1186/s13059-016-1066-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Okazaki Y, Murray J, Ehsani A, Clark J, Whitson RH, Hirose L, et al. Increased glucose metabolism in Arid5b−/− skeletal muscle is associated with the down-regulation of TBC1 domain family member 1 (TBC1D1) Biol Res. 2020;53:45. doi: 10.1186/s40659-020-00313-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Horvath S, Raj K. DNA methylation-based biomarkers and the epigenetic clock theory of ageing. Nat Rev Genet. 2018;19:371–384. doi: 10.1038/s41576-018-0004-3. [DOI] [PubMed] [Google Scholar]
- 43. Horvath S, Levine AJ. HIV-1 infection accelerates age according to the epigenetic clock. J Infect Dis. 2015;212:1563–1573. doi: 10.1093/infdis/jiv277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Horvath S, Ritz BR. Increased epigenetic age and granulocyte counts in the blood of Parkinson’s disease patients. Aging (Albany NY) 2015;7:1130–1142. doi: 10.18632/aging.100859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Horvath S, Garagnani P, Bacalini MG, Pirazzini C, Salvioli S, Gentilini D, et al. Accelerated epigenetic aging in Down syndrome. Aging Cell. 2015;14:491–495. doi: 10.1111/acel.12325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Marioni RE, Shah S, McRae AF, Chen BH, Colicino E, Harris SE, et al. DNA methylation age of blood predicts all-cause mortality in later life. Genome Biol. 2015;16:25. doi: 10.1186/s13059-015-0584-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. El Khoury LY, Gorrie-Stone T, Smart M, Hughes A, Bao Y, Andrayas A, et al. Systematic underestimation of the epigenetic clock and age acceleration in older subjects. Genome Biol. 2019;20:283. doi: 10.1186/s13059-019-1810-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Richeldi L, Ryerson CJ, Lee JS, Wolters PJ, Koth LL, Ley B, et al. Relative versus absolute change in forced vital capacity in idiopathic pulmonary fibrosis. Thorax. 2012;67:407–411. doi: 10.1136/thoraxjnl-2011-201184. [DOI] [PubMed] [Google Scholar]
- 49. Peters TJ, Buckley MJ, Statham AL, Pidsley R, Samaras K, V Lord R, et al. De novo identification of differentially methylated regions in the human genome. Epigenetics Chromatin. 2015;8:6. doi: 10.1186/1756-8935-8-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Finucane OM, Sugrue J, Rubio-Araiz A, Guillot-Sestier MV, Lynch MA. The NLRP3 inflammasome modulates glycolysis by increasing PFKFB3 in an IL-1β-dependent manner in macrophages. Sci Rep. 2019;9:4034. doi: 10.1038/s41598-019-40619-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Herazo-Maya JD, Sun J, Molyneaux PL, Li Q, Villalba JA, Tzouvelekis A, et al. Validation of a 52-gene risk profile for outcome prediction in patients with idiopathic pulmonary fibrosis: an international, multicentre, cohort study. Lancet Respir Med. 2017;5:857–868. doi: 10.1016/S2213-2600(17)30349-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Herazo-Maya JD, Noth I, Duncan SR, Kim S, Ma S-F, Tseng GC, et al. Peripheral blood mononuclear cell gene expression profiles predict poor outcome in idiopathic pulmonary fibrosis. Sci Transl Med. 2013;5:205ra136. doi: 10.1126/scitranslmed.3005964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Scott MKD, Quinn K, Li Q, Carroll R, Warsinske H, Vallania F, et al. Increased monocyte count as a cellular biomarker for poor outcomes in fibrotic diseases: a retrospective, multicentre cohort study. Lancet Respir Med. 2019;7:497–508. doi: 10.1016/S2213-2600(18)30508-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Titus AJ, Gallimore RM, Salas LA, Christensen BC. Cell-type deconvolution from DNA methylation: a review of recent applications. Hum Mol Genet. 2017;26:R216–R224. doi: 10.1093/hmg/ddx275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Yang IV, Pedersen BS, Rabinovich E, Hennessy CE, Davidson EJ, Murphy E, et al. Relationship of DNA methylation and gene expression in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2014;190:1263–1272. doi: 10.1164/rccm.201408-1452OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sanders YY, Ambalavanan N, Halloran B, Zhang X, Liu H, Crossman DK, et al. Altered DNA methylation profile in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2012;186:525–535. doi: 10.1164/rccm.201201-0077OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Yang IV, Schwartz DA. Epigenetics of idiopathic pulmonary fibrosis. Transl Res. 2015;165:48–60. doi: 10.1016/j.trsl.2014.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Selman M, López-Otín C, Pardo A. Age-driven developmental drift in the pathogenesis of idiopathic pulmonary fibrosis. Eur Respir J. 2016;48:538–552. doi: 10.1183/13993003.00398-2016. [DOI] [PubMed] [Google Scholar]
- 59. Alder JK, Chen JJ, Lancaster L, Danoff S, Su SC, Cogan JD, et al. Short telomeres are a risk factor for idiopathic pulmonary fibrosis. Proc Natl Acad Sci USA. 2008;105:13051–13056. doi: 10.1073/pnas.0804280105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kropski JA, Pritchett JM, Zoz DF, Crossno PF, Markin C, Garnett ET, et al. Extensive phenotyping of individuals at risk for familial interstitial pneumonia reveals clues to the pathogenesis of interstitial lung disease. Am J Respir Crit Care Med. 2015;191:417–426. doi: 10.1164/rccm.201406-1162OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Minagawa S, Araya J, Numata T, Nojiri S, Hara H, Yumino Y, et al. Accelerated epithelial cell senescence in IPF and the inhibitory role of SIRT6 in TGF-β-induced senescence of human bronchial epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2011;300:L391–L401. doi: 10.1152/ajplung.00097.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Disayabutr S, Kim EK, Cha SI, Green G, Naikawadi RP, Jones KD, et al. miR-34 miRNAs regulate cellular senescence in type II alveolar epithelial cells of patients with idiopathic pulmonary fibrosis. PLoS One. 2016;11:e0158367. doi: 10.1371/journal.pone.0158367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Bell CG, Lowe R, Adams PD, Baccarelli AA, Beck S, Bell JT, et al. DNA methylation aging clocks: challenges and recommendations. Genome Biol. 2019;20:249. doi: 10.1186/s13059-019-1824-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Shchukina I, Bagaitkar J, Shpynov O, Loginicheva E, Porter S, Mogilenko DA, et al. Epigenetic aging of classical monocytes from healthy individuals [preprint] bioRxiv. 2020 https://www.biorxiv.org/content/10.1101/2020.05.10.087023v1.full
- 65. Corces MR, Buenrostro JD, Wu B, Greenside PG, Chan SM, Koenig JL, et al. Lineage-specific and single-cell chromatin accessibility charts human hematopoiesis and leukemia evolution. Nat Genet. 2016;48:1193–1203. doi: 10.1038/ng.3646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Okuno Y, Inoue K, Imai Y. Novel insights into histone modifiers in adipogenesis. Adipocyte. 2013;2:285–288. doi: 10.4161/adip.25731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Claussnitzer M, Dankel SN, Kim K-H, Quon G, Meuleman W, Haugen C, et al. FTO obesity variant circuitry and adipocyte browning in humans. N Engl J Med. 2015;373:895–907. doi: 10.1056/NEJMoa1502214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Baba A, Ohtake F, Okuno Y, Yokota K, Okada M, Imai Y, et al. PKA-dependent regulation of the histone lysine demethylase complex PHF2-ARID5B. Nat Cell Biol. 2011;13:668–675. doi: 10.1038/ncb2228. [DOI] [PubMed] [Google Scholar]
- 69. Cichocki F, Wu C-Y, Zhang B, Felices M, Tesi B, Tuininga K, et al. ARID5B regulates metabolic programming in human adaptive NK cells. J Exp Med. 2018;215:2379–2395. doi: 10.1084/jem.20172168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Liu Y, Reynolds LM, Ding J, Hou L, Lohman K, Young T, et al. Blood monocyte transcriptome and epigenome analyses reveal loci associated with human atherosclerosis. Nat Commun. 2017;8:393. doi: 10.1038/s41467-017-00517-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. El-Chemaly S, Malide D, Yao J, Nathan SD, Rosas IO, Gahl WA, et al. Glucose transporter-1 distribution in fibrotic lung disease: association with [18F]-2-fluoro-2-deoxyglucose-PET scan uptake, inflammation, and neovascularization. Chest. 2013;143:1685–1691. doi: 10.1378/chest.12-1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Tsitoura E, Vasarmidi E, Bibaki E, Trachalaki A, Koutoulaki C, Papastratigakis G, et al. Accumulation of damaged mitochondria in alveolar macrophages with reduced OXPHOS related gene expression in IPF. Respir Res. 2019;20:264. doi: 10.1186/s12931-019-1196-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Leslie J, Macia MG, Luli S, Worrell JC, Reilly WJ, Paish HL, et al. c-Rel orchestrates energy-dependent epithelial and macrophage reprogramming in fibrosis. Nat Metab. 2020;2:1350–1367. doi: 10.1038/s42255-020-00306-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Tawakol A, Singh P, Mojena M, Pimentel-Santillana M, Emami H, MacNabb M, et al. HIF-1α and PFKFB3 mediate a tight relationship between proinflammatory activation and anerobic metabolism in atherosclerotic macrophages. Arterioscler Thromb Vasc Biol. 2015;35:1463–1471. doi: 10.1161/ATVBAHA.115.305551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Jiang H, Shi H, Sun M, Wang Y, Meng Q, Guo P, et al. PFKFB3-driven macrophage glycolytic metabolism is a crucial component of innate antiviral defense. J Immunol. 2016;197:2880–2890. doi: 10.4049/jimmunol.1600474. [DOI] [PubMed] [Google Scholar]
- 76. Chen X, Hyatt BA, Mucenski ML, Mason RJ, Shannon JM. Identification and characterization of a lysophosphatidylcholine acyltransferase in alveolar type II cells. Proc Natl Acad Sci USA. 2006;103:11724–11729. doi: 10.1073/pnas.0604946103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Bridges JP, Ikegami M, Brilli LL, Chen X, Mason RJ, Shannon JM. LPCAT1 regulates surfactant phospholipid synthesis and is required for transitioning to air breathing in mice. J Clin Invest. 2010;120:1736–1748. doi: 10.1172/JCI38061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Bi J, Ichu T-A, Zanca C, Yang H, Zhang W, Gu Y, et al. Oncogene amplification in growth factor signaling pathways renders cancers dependent on membrane lipid remodeling. Cell Metab. 2019;30:525–538.e8. doi: 10.1016/j.cmet.2019.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Xu Y, Mizuno T, Sridharan A, Du Y, Guo M, Tang J, et al. Single-cell RNA sequencing identifies diverse roles of epithelial cells in idiopathic pulmonary fibrosis. JCI Insight. 2016;1:e90558. doi: 10.1172/jci.insight.90558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Suryadevara V, Ramchandran R, Kamp DW, Natarajan V. Lipid mediators regulate pulmonary fibrosis: potential mechanisms and signaling pathways. Int J Mol Sci. 2020;21:4257. doi: 10.3390/ijms21124257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Lavender P, Kelly A, Hendy E, McErlean P. CRISPR-based reagents to study the influence of the epigenome on gene expression. Clin Exp Immunol. 2018;194:9–16. doi: 10.1111/cei.13190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Qu J, Zhu L, Zhou Z, Chen P, Liu S, Locy ML, et al. Reversing mechanoinductive DSP expression by CRISPR/dCas9-mediated epigenome editing. Am J Respir Crit Care Med. 2018;198:599–609. doi: 10.1164/rccm.201711-2242OC. [DOI] [PMC free article] [PubMed] [Google Scholar]