

Abstract
Background
Advances in SARS-CoV-2 sequencing have enabled identification of new variants, tracking of its evolution, and monitoring of its spread. We aimed to use whole genome sequencing to describe the molecular epidemiology of the SARS-CoV-2 outbreak and to inform the implementation of effective public health interventions for control in Zimbabwe.
Methods
We performed a retrospective study of nasopharyngeal samples collected from nine laboratories in Zimbabwe between March 20 and Oct 16, 2020. Samples were taken as a result of quarantine procedures for international arrivals or to test for infection in people who were symptomatic or close contacts of positive cases. Samples that had a cycle threshold of less than 30 in the diagnostic PCR test were processed for sequencing. We began our analysis in July, 2020 (120 days since the first case), with a follow-up in October, 2020 (at 210 days since the first case). The phylogenetic relationship of the genome sequences within Zimbabwe and global samples was established using maximum likelihood and Bayesian methods.
Findings
Of 92 299 nasopharyngeal samples collected during the study period, 8099 were PCR-positive and 328 were available for sequencing, with 156 passing sequence quality control. 83 (53%) of 156 were from female participants. At least 26 independent introductions of SARS-CoV-2 into Zimbabwe in the first 210 days were associated with 12 global lineages. 151 (97%) of 156 had the Asp614Gly mutation in the spike protein. Most cases, 93 (60%), were imported from outside Zimbabwe. Community transmission was reported 6 days after the onset of the outbreak.
Interpretation
Initial public health interventions delayed onset of SARS-CoV-2 community transmission after the introduction of the virus from international and regional migration in Zimbabwe. Global whole genome sequence data are essential to reveal major routes of spread and guide intervention strategies.
Funding
WHO, Africa CDC, Biotechnology and Biological Sciences Research Council, Medical Research Council, National Institute for Health Research, and Genome Research Limited.
Introduction
SARS-CoV-2 is a novel coronavirus and the causative agent of COVID-19.1 It is an enveloped, non-segmented, positive sense RNA virus with a diameter of about 65–125 nm. It has four main structural proteins, the most important being the spike glycoprotein.
The virus has spread to more than 220 countries, leading to more than 242 million confirmed infections, and more than 4·9 million deaths as of Oct 21, 2021, with daunting health and socioeconomic challenges.2 Low-income and middle-income countries have faced particular challenges in controlling SARS-CoV-2 infections and reducing mortality as a result of typically weak public health systems and large populations of susceptible people including those with tuberculosis, HIV, diabetes, hypertension, anaemia, or malnutrition.3
The first COVID-19 case in Africa was recorded in Egypt on Feb 14, 2020,4 and on Feb 27, 2020, the sub-Saharan African region recorded its first case in Nigeria. Further cases in sub-Saharan Africa were recorded in South Africa on March 5 and Zimbabwe on March 20, 2020.5 The initial cases identified in Africa were found to have been mostly introduced from Europe, the Middle East, and the USA.4, 6, 7 There is an urgent need to understand the epidemiology of SARS-CoV-2 in Africa, particularly pertaining to transmission and mutation capacity.8 The SARS-CoV-2 mutation rate is estimated to be approximately 2·5 single nucleotide polymorphisms (SNPs) per month.9, 10 Despite having inadequate health systems in some African countries, the number of reported cases in many African countries, specifically Zimbabwe, increased more slowly than predicted in the initial wave of infection.11
Research in context.
Evidence before this study
A narrative review of all the available literature showed that there was limited evidence of the epidemiology and genetic characteristics of SARS-CoV-2 in Africa. We searched PubMed Central, Embase, LILACS, Google Scholar, and the Cochrane Library for published literature from Jan 1, 2019, to Dec 31, 2020, using the search terms, “SARS Cov 2” OR “COVID-19” AND “Genomic Epidemiology” AND “Global” AND “Africa”. Full text articles and abstracts of SARS-CoV-2 genome epidemiology studies conducted between the search dates were included in the review. Earlier SARS-CoV-2 genomic studies were for diagnostic purposes and assessment of transmission. These earlier studies were limited to Asia, Europe, and the USA, with few reported from Africa and Zimbabwe.
Added value of this study
Genome surveillance of SARS-CoV-2 for Zimbabwe revealed that the majority were of lineage B, 97%, with the remaining being of lineage A. International and regional migration was the predominant driver of SARS-CoV-2 transmission in Zimbabwe during the early phases of the pandemic, compared with local migration. Lineage B.1.1.111 was first identified in Zimbabwe and subsequently in two other countries in the region. This finding highlights the importance of strengthening pathogen genomic surveillance in countries with high migration levels to facilitate early detection of new variants. Potential changes in transmissibility and outcome of infection of new strains compared with the ancestor lineage are key to determining appropriate interventions.
Implications of the all the available evidence
As with other previously reported pandemic influenza diseases, COVID-19 was predominantly transmitted through global and local migration. The use of lockdown measures to control transmission coupled with increasing globalisation of economies will affect economic activities, with low-income and middle-income countries worst affected due to less developed and diversified economies. Public health interventions are urgently needed to contain the pandemic before low-income countries like Zimbabwe are further impoverished. The inclusion of genomic evidence will further contextualise the pandemic regionally and globally, helping in the identification of new outbreaks through phylogenetic and phylogeographical analyses.
Following confirmation of the first case on March 20, 2020, Zimbabwe introduced stringent control measures on March 23, which included controlling travel, transportation, and public gatherings. The first death was confirmed on March 24. Due to the measures imposed in other countries, there was an influx of Zimbabwean residents returning home and a 21-day quarantine was imposed starting March 30. Zimbabwe took the initiative to sequence SARS-CoV-2 samples from individuals collected between March and October, 2020. In this study, we aimed to use the sequencing results to understand early domestic transmission of the virus in Zimbabwe, add context to the regional and global SARS-CoV-2 genome sequence data, and to evaluate the role of rapid whole genome sequencing for outbreak analysis in Zimbabwe.
Methods
Study design and data sources
We performed a retrospective study using routinely collected nasopharyngeal samples from Zimbabwe. Details of study design are provided in appendix 1 (p 2). COVID-19 testing information was obtained from the Ministry of Health and Child Care of Zimbabwe, Epidemiology and Disease Control Unit. All samples were from people present in Zimbabwe during the period of study and were sampled due to quarantine procedures, or to test for infection in people who were symptomatic and close contacts. Epidemiological data for each COVID-19 case (ie, geographical origin, case classification [imported or domestic], date of isolation, and date of death, if applicable) was extracted from surveillance data submitted to the Epidemiology and Disease Control Unit. All data were de-identified and ethical approval was waived by the Medical Research Council of Zimbabwe as routinely collected surveillance data were used. No specific consent was required from the patients whose data were used in this analysis as the National Microbiology Reference Laboratory has authority to handle patient data for public health monitoring under section 46 (notifiable diseases) of the Zimbabwe Public Health Act.
Procedures
In the selection of samples for genomic evaluation, we used a conveniently selected sample of all those that were test-positive from eight of ten provinces (appendix 1 pp 6–8) diagnosed during the study period that had a cycle threshold of less than 30 in the diagnostic PCR test12 were processed for sequencing. Demographic data (ie, age, gender, place of origin, and history of travel) associated with sequenced samples were analysed.
Genomic analysis
In the determination of SARS-CoV-2 genome sequences and sequence analysis, RNA samples for whole genome sequencing were processed and complementary DNA and multiplex PCR reactions were prepared following the ARTIC nCoV-2019 sequencing protocol version 2. Libraries were prepared for sequencing on the Illumina platform and sequenced as described previously and summarised in appendix 1 (p 3).13, 14 SARS-CoV-2 sequence data generated in this study (appendix 1 pp 8–13) or global sequences generated previously (appendix 2) are freely available in the global initiative on sharing avian influenza data (GISAID) as reported previously.5 The raw reads were demultiplexed using bcl2fastq (version 2.20, Illumina) allowing for zero mismatches in the dual barcodes to produce FASTQ files. The reads were used to generate a consensus sequence as described in appendix 1 (p 3). A consensus sequence was defined as passing quality control if greater than 50% of the genome was covered by confident calls and if there was no evidence of contamination in the negative control. This proportion is regarded as the minimum amount of data to be phylogenetically useful. A confident call was defined as having at least ten times depth of coverage. If the coverage fell below these thresholds, the bases were represented with an N character (to symbolise that the nucleotide base at this position is unknown). Low quality variants were also masked with Ns. Lineages were assigned to each genome with Pangolin15 with manual refinement. Global sequences from GISAID (downloaded on June 4, 2021) closely related to those from Zimbabwe were included in the phylogenetic analysis. All sequences were aligned against the reference Wuhan Hu-1 (GenBank accession number MN908947.3), and both maximum likelihood trees and Bayesian divergence times were estimated. Ancestral genomes were inferred over nodes of interest by empirical Bayesian reconstruction as described in appendix 1 (p 4).
Role of the funding source
The funders of the study had no role in study design, data collection, data analysis, data interpretation, or writing of the report.
Results
Of the 92 299 nasopharyngeal samples collected during the study period, 8099 were PCR positive and 328 were available for sequencing, with 156 analysed after passing sequence quality control (figure 1 ). Samples were from eight provinces (including the metropolitan Harare and Bulawayo provinces). Of these, 83 (53%) were from female participants and 73 (47%) were from male participants, ranging in age from 7 months to 10 years (five [3%]), 11–20 years (seven [4%]), 21–30 years (45 [29%]), 31–40 years (48 [31%]), 41–50 years (26 [17%]), and older than 50 years (25 [16%]). 62 (40%) samples showed evidence of local transmission, 93 (60%) were imported cases, and there was no information available for one case (1%). Among the imported cases, 67 (72%) of 93 were travellers from South Africa. Most cases were of the lineage B.1.1 (51 [33%]), B.1.1.111 (35 [22%]), or B.1 (27 [17%]).
Figure 1.
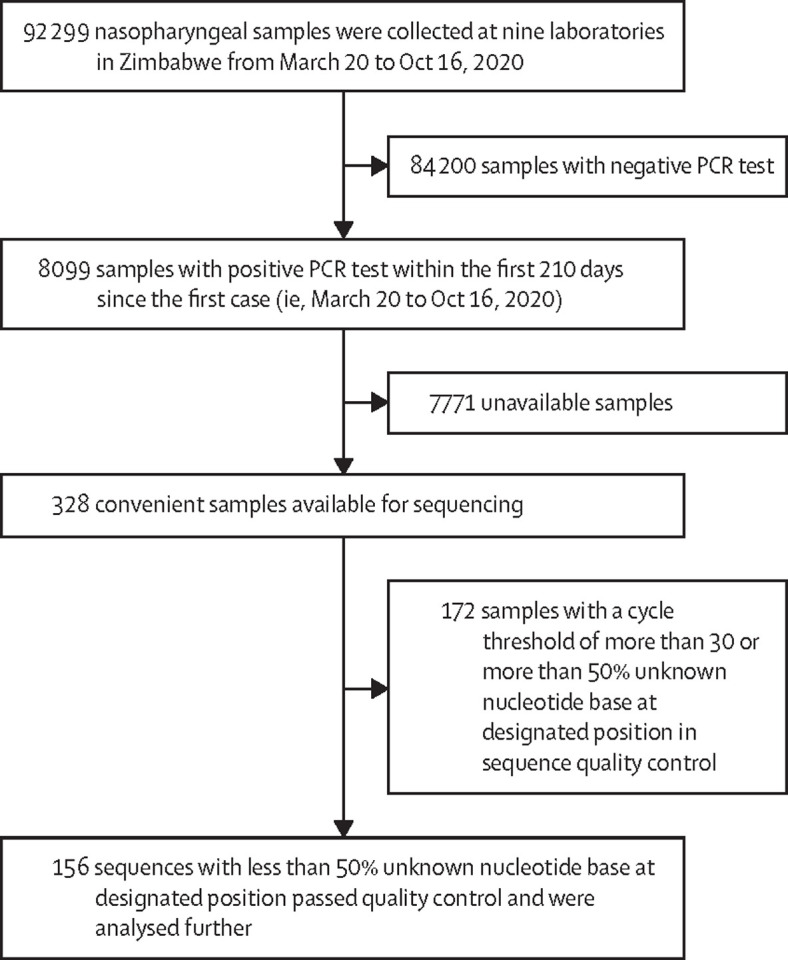
Study profile
Most of the initial COVID-19 cases in Zimbabwe between March 20 and June 30, 2020, were Zimbabwean residents returning from neighbouring Botswana, Mozambique, and South Africa, the majority of whom were aged 25–45 years (Survellance data, Department of Epidemiology and Disease Control, Ministry of Health and Child Care, Zimbabwe, personal communication). The country recorded a case fatality rate of 2·9% (231 of 8099) with those in the 31–40 year age group being most affected. The epidemiological curve plotted (figure 2A, B ) was based on 8099 reported positive cases identified from the 92 299 national samples made available between March 20 (first case) and Oct 16, 2020. From the time the first case was recorded until 120 days later (ie, until July 18), more travel-associated cases than locally transmitted cases were detected. Most travel-related cases were asymptomatic (appendix 1 p 8). The number of cases with no reported history of travel in the previous 2 weeks remained less than 50 during this period. People travelling from South Africa accounted for the greatest proportion (72 [72%] of 100) of travel-associated cases during the first 120 days. In late July, there was a rapid increase in the number of reported cases with no travel history, confirming the start of community transmission.
Figure 2.
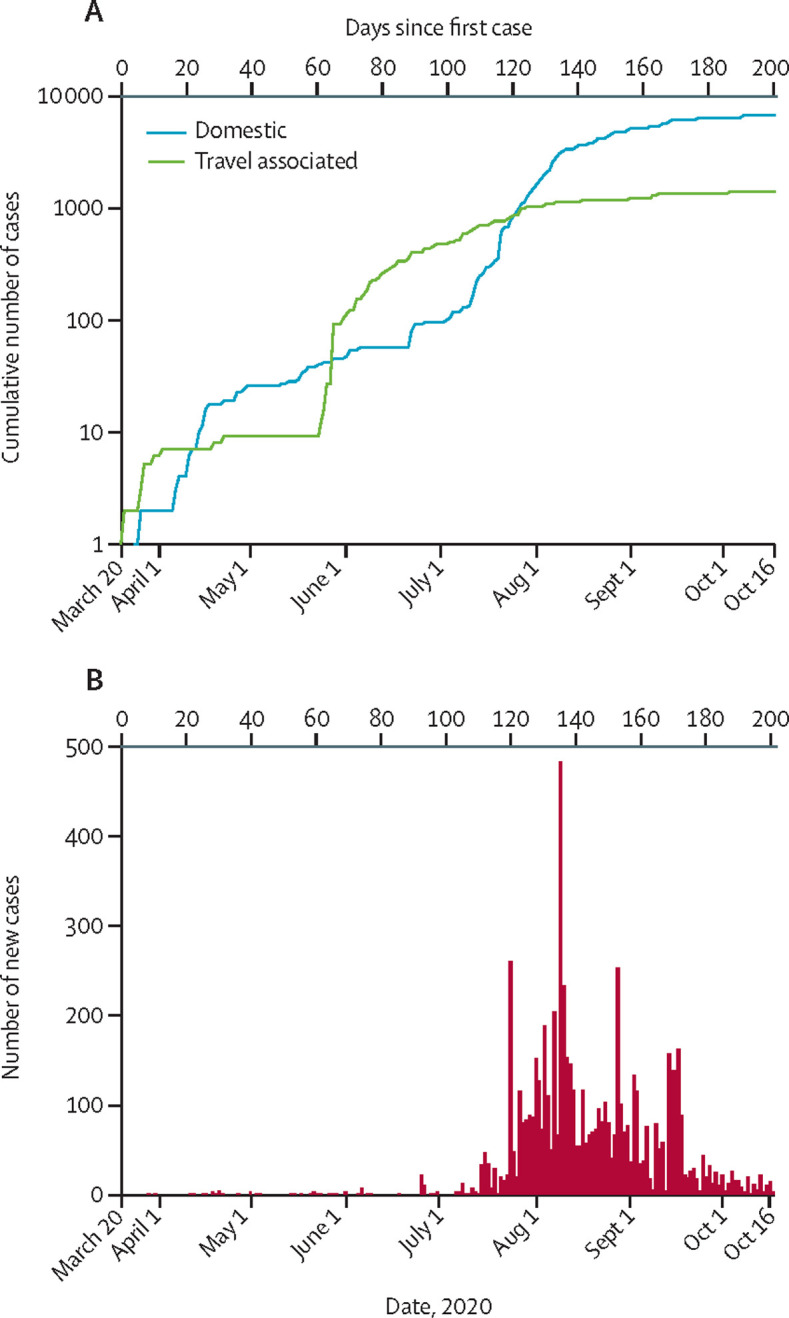
Epidemic curve of SARS-CoV-2 confirmed cases as of Oct 16, 2020
(A) Cumulative number of positive tests for SARS-CoV-2 in individuals reporting travel in the previous 2 weeks (probable imported cases) or domestic cases by day. (B) Number of reported positive tests for SARS-CoV-2 by day.
To place the genomes from Zimbabwe SARS-CoV-2 samples into a global context, closely related genomes from abroad were analysed together, to produce a maximum likelihood phylogenetic tree of 472 genomes (figure 3 ). The 156 genomes from Zimbabwe could be placed within 12 previously defined global lineages (table ). Four samples (3%), were from high-order lineage A, with the remaining genomes (n=152, 97%) derived from lineage B. The most common lineage, B.1.1, was first observed in April, 2020; table). Lineage B.1.1.111 was first observed in Zimbabwe, followed by a further eight sequences in Zambia and one in Kenya in June.
Figure 3.

Phylogenetic relationship of SARS-CoV-2 genomes from Zimbabwe and closely related genomes from global cases
Maximum likelihood phylogenetic tree of 156 SARS-CoV-2 genomes from Zimbabwe together with their closest 316 genomes from global samples. Lineages connecting Zimbabwe clusters (red) and global lineages (black) are indicated.
Table.
Summary of lineages circulating in Zimbabwe, South Africa, and globally in the first 210 days of known cases
|
Zimbabwe (n=156) |
South Africa (n=3015) |
Global (n=299 405) |
||||
|---|---|---|---|---|---|---|
| Number of samples | Percentage | Number of samples | Percentage | Number of samples | Percentage | |
| B.1.1 | 51 | 32·7% | 301 | 10·0% | 32 163 | 10·7% |
| B.1.1.111 | 35 | 22·4% | 0 | 0 | 27 | <0·1% |
| B.1 | 27 | 17·3% | 379 | 12·6% | 57 071 | 19·1% |
| B.1.446 | 16 | 10·3% | 0 | 0 | 237 | <0·1% |
| B.1.1.459 | 8 | 5·1% | 32 | 1·1% | 32 | <0·1% |
| B.1.1.57 | 5 | 3·2% | 62 | 2·1% | 67 | <0·1% |
| A | 4 | 2·6% | 4 | 0·1% | 2153 | 0·1% |
| B.1.381 | 4 | 2·6% | 69 | 2·3% | 69 | <0·1% |
| B.1.1.200 | 3 | 1·9% | 0 | 0 | 126 | <0·1% |
| B.1.1.306 | 1 | 0·6% | 0 | 0 | 1383 | 0·5% |
| B.1.1.62 | 1 | 0·6% | 25 | 0·8% | 26 | <0·1% |
| B.39 | 1 | 0·6% | 2 | 0·1% | 412 | 0·1% |
Data submitted to GISAID16 before Oct 16, 2020 (using PANGO lineages from GISAID June 4, 2021). GISAID=global initiative on sharing avian influenza data.
A maximum likelihood tree based on variation in the nucleotide sequence revealed a population structure for Zimbabwean cases of COVID-19 that grouped into 13 clusters. Each comprised multiple genomes that emerged from nodes distributed throughout the tree; 15 genomes were present as separate lineages (appendix 1 p 6). If each of these clusters represent separate introductions, we can infer that at least 28 independent introductions took place for which there was good supporting evidence, as given by the existence of close genomes on GISAID from other countries separating the Zimbabwean clusters in the tree. However, in the case of four clusters or single genome lineages, the tree topology suggested that the genomes arose by divergence from just two introductions into Zimbabwe. In each case, two lineages shared a common ancestor with no intermediary nodes formed with genomes from other countries. By this more conservative estimation, the minimum number of introductions into Zimbabwe that our data support was 26. By reconstructing the ancestral locations on the Bayesian time tree, the number of transitions (from abroad to Zimbabwe) was estimated to be 29 (appendix 1 p 6). The 13 clusters were each composed of between two and 30 genomes with 0–3 SNPs between members of the cluster. Seven clusters contained more than one genome that had an identical genome sequence to at least one genome in the same cluster.
Two large clusters within lineage B.1.1 were present on extended branches of the phylogenetic tree, composed of genomes that were distinct by 4–5 SNPs from all other sequences in the GISAID database. One of these clusters met all the requirements for a new lineage according to the PANGO classification (ie, well supported monophyletic group, introduced and circulating in a new region, with defining SNPs). These defining features led to the designation of lineage B.1.1.111. The branch leading to this cluster was estimated to contain five SNPs, including the replacements Phe35Ser in open reading frame (ORF)10, Lys1895Asn in ORF1A, and Lys2557Arg in ORF1B. The other cluster consists of eight sequences from lineage B.1.1.459 (appendix 1 p 6). Using a Bayesian relaxed clock model, we estimated the time to the most recent common ancestor for each of these clusters to a few weeks before their sampling: beginning of April, 2020, for the B.1.1.111 cluster and mid-May for B.1.1.459 (appendix 1 p 6). These introductions into Zimbabwe might have been from geographical locations where the sequence of SARS-CoV-2 was not reported. The results could be caused by sequence divergence from an earlier case within the country not included in our analysis. Due to the sparse sampling due to incomplete surveillance, it is not possible to pinpoint the direction of transmission or to establish better phylogeographical estimates for this dataset. By superposing the ancestral shifts from abroad to Zimbabwe on the Bayesian time estimates of tree nodes, we inferred that most transmissions occurred between mid-February and mid-March, 2020 (appendix 1 p 6).
We established the distribution and frequency of the Asp614Gly variant in SARS-CoV-2 genomes from Zimbabwe. 151 (97%) of 156 samples contained the Asp614Gly mutation. Four of the five samples with the ancestral 614Gly were from a single cluster of identical genomes from lineage A, which was associated with travel from Dubai (United Arab Emirates); the other was from lineage B.39 and associated with travel from the USA. All five were from cases presenting in March or April, 2020.
Genomic epidemiology identified intercontinental transmission and local transmission in the first 210 days of the epidemic. National surveillance in Zimbabwe identified the first three cases of COVID-19 in the second half of March, 2020, in three individuals with a history of international travel within the previous 2 weeks. These individuals had arrived from the UK, the USA, and Dubai and corresponded to samples ZW-25, ZW-29 and ZW-70, respectively (appendix 1 p 6). The genome sequence of samples ZW-25 and ZW-29 were identical, suggesting a recent common source or direct transmission. These genomes together with a few others were classified as B.1.446, although other sequences in the same phylogenetic cluster did not have enough defining SNPs for a more specific PANGO classification. This mismatch between the PANGO classification and the phylogeny is due to the presence of ambiguous bases in the genomic DNA, in particular N (ie, unknown nucleotide base at designated position. Pangolin thus conservatively classified them as B.1, a very common lineage in the GISAID database (figure 4 ).16 After visual inspection of the alignments and tree clustering, a correction of their classification was made.
Figure 4.

SNP differences between genomes from the cluster containing the B.1.446 lineage and the ancestral sequence
The sequences classified by Pangolin as B.1.446 are in red, while the others were classified as B.1. The plot was created with the software snipit.
Genome ZW-70 from a traveller arriving from Dubai was identical to three other genomes: ZW-168, ZW-169, and ZW-214. Dubai is an important hub for flights from many locations in the world including Asia. Local epidemiological investigation indicated that these were infections resulting from cohabitation and might represent the earliest cases of local transmission. This interpretation was supported by whole genome sequencing data. Genomes ZW-168, ZW-169, and ZW-214 all belonged to lineage A, which is most widely distributed in China.
An additional eight genomes were associated with arrivals from the UK and three with arrivals from the USA in late March or April, 2020. International travel was suspended following the arrival of these cases. Nine of these 11 genomes belonged to the B.1 lineage, while one, from a case imported from the USA, belonged to the B.39 lineage. Although in the same B.1 lineage, eight of these travel-associated cases were from different sublineages and, with one exception, had no identical genomes from non-travel associated COVID-19 cases in our dataset. The one exception was sample ZW-25BY, which was from a case recorded in Bulawayo in April, which was identical to samples ZW-391 and ZW-EC, also from Bulawayo, with no travel history recorded. This finding represents potential local transmission. Furthermore, they are phylogenetically very close to samples assigned to lineage B.1.446, and by visual inspection of the genomes, it was confirmed that they could not be classified with confidence by the software due to singletons or ambiguous bases (figure 4).
An increase in cases associated with Zimbabwean residents returning from neighbouring countries, especially South Africa, during May, 2020, was reported by local surveillance. As SARS-CoV-2 was reported to be spreading rapidly in South Africa at the time, a 2-week quarantine in accommodation designated by the Ministry of Health and Child Care was introduced. Although most returnees were asymptomatic, they were tested and on multiple occasions were positive for SARS-CoV-2. 26 samples from quarantined individuals housed in six quarantine accommodation settings were sequenced. In several quarantine accommodation settings, genomes from multiple samples from different individuals resident in the same quarantine location were identical or differed by only a single nucleotide in the genome. For example, in quarantine location G (appendix 1 p 8), two clusters were identified that separately consisted of ten and 14 samples (May, 2020). All of these were of the B.1.1 lineage with genomes that were indistinguishable from one another, consistent with direct transmission. Since returnees from multiple locations were quarantined together, the most probable explanation is that transmission occurred in the quarantine facility rather than from a common outside source. Later spread was primarily linked to returnees from South Africa and potential transmission in quarantine facilities.
To investigate SARS-CoV-2 sequences associated with the first wave of the pandemic from July to October, 2020, we sequenced an additional 56 genomes from infections in this period. During this period, most infections were thought to result from domestic transmission rather than additional introductions from returning travellers. Consistent with this hypothesis, the vast majority of genomes were from the same lineages as those observed in the period in which transmission was mainly associated with imported cases in the first 120 days (appendix 1 p 6). The only exceptions to this pattern was a single lineage B.1.1.306 sample in July and four samples of lineage B.1.1.200 in October, which had not been sampled in the first 120 days.
Discussion
Epidemiological analysis identified two phases of the SARS-CoV-2 epidemic in Zimbabwe. Most cases in the first 90 days were associated with intercontinental or intracontinental travel. This finding was confirmed by the epidemiological data linking most confirmed cases with a history of travel outside of Zimbabwe, from international locations in which there were rising cases and local transmission. A smaller number of cases were imported from Asia, Europe, and the USA, the early epicentres of the SARS-CoV-2 epidemic.
For the first 120 days after the first case of COVID-19 was identified in Zimbabwe, the total number of detected cases remained less than 50, suggesting that public health interventions were effective in limiting local transmission to a minimum. The rise in cases was slower than initially predicted and mirrored what was seen in many African countries.11 This slow rise in cases could be attributed to the measures the country instituted, which were shown to minimise and contain the outbreak in other countries, including France.17, 18, 19, 20 Evidence suggests that quarantine of travel-associated cases and early suspension of tourism-associated travel might have been effective.
After July, 2020, a rapid increase in the number of cases was associated with probable domestic transmission, consisting predominantly of SARS-CoV-2 lineages present in the first 120 days that were imported into the country by travellers. Notably, no samples of lineage A were detected after March, 2020. The greatest number of cases centred around cities with close proximity to land borders or international airports and densely populated areas—namely Bulawayo, Gweru, and Harare. These factors might explain how the COVID-19 epidemic curve showed propagated transmission.
The case fatality rate of COVID-19 in Zimbabwe was 2·9%, which was similar to that reported elsewhere in Asia, Europe, and the USA. However, low access to testing in the community and health-care settings might have underestimated the true magnitude of the case fatality rate. This under-reporting was difficult to quantify.21, 22, 23 Compared with reports from other countries, younger men aged 31–40 years were the most likely to be infected with SARS-CoV-2 in Zimbabwe during the first 120 days of the epidemic, but individuals older than 60 years were more likely to die from COVID-19, as observed in other regions.24, 25 The WHO report for the period of January to July, 2020, indicated that 64% of those infected were aged 25–64 years. However, older members of the population with pre-existing medical conditions were also at increased risk of dying after being diagnosed with COVID-19.26, 27 In the UK, girls and women were significantly over-represented in the first wave of infection.28 The greater proportion of boys and men infected in Zimbabwe might reflect their prevalence in the mobile work force who move to other countries in search of employment.
14 of the 156 SARS-CoV-2 cases sequenced reported that the individual infected had travelled from outside the African region (ie, Dubai, the UK, and the USA). The sequences of SARS-CoV-2 genomes associated with travellers returning from neighbouring southern African countries were distinct from those associated with travellers from these three places, and recorded earlier in the epidemic. Among those returning from southern Africa, most were from lineage B.1.1, which was also the most common lineage among 2016 sequences from 18 African countries reported by the WHO Regional Office for Africa. Lineages B.1.1.448 and C.1, which together accounted for 26% of the samples reported in South Africa before November, 2020, were not found in our analysis of samples from Zimbabwe.
There has been considerable interest in the emergence of a variant lineage of SARS-CoV-2 with a non-synonymous mutation resulting in substitution of 614Gly in which a glycine residue was replaced by an aspartate residue at position 614 (Asp614Gly) of the spike protein. The spike protein is thought to be important for entry of the virus into host cells and Asp614Gly has been implicated in increased cell entry.29, 30 Consistent with this idea, the 614Gly variant increased in frequency in the UK relative to the 614Asp ancestral variant, suggesting a selective advantage for 614Gly. The 614Gly genotype was the most frequent variant in early spread events in Zimbabwe, consistent with the reported trend for increased frequency of this genotype in many locations globally.29 The 614Asp genotype was found in just five samples from a single household-linked cluster of infections that occurred very early in the epidemic and were not found after this point. The dominance of the 614Gly genotype in Zimbabwe probably reflects the high frequency of the 614Gly genotype in countries from which the virus was introduced. Although in the present work we cannot establish an increase in 614Gly infectivity, the notable absence of 614Asp, despite evidence of at least two introductions and household transmission, supports the fitness advantage of the 614Gly genotype.
We analysed data from most provinces in Zimbabwe; however, as convenience sampling was used, it is not clear whether this population is representative of the whole population. Also, as in all studies investigating COVID-19, the case burden will be underestimated as cases are only tested at prespecified intervals (in quarantine facilities) or for symptomatic cases meaning asymptomatic individuals are missed, thus underestimating case burden.
Our whole genome sequence analysis provides a baseline view of the SARS-CoV-2 lineages introduced into Zimbabwe, which will further inform analysis of later domestic spread of the virus. As the national response to the SARS-CoV-2 pandemic continues, the capability to test, isolate, and track the evolution of SARS-CoV-2 is crucial to assess ongoing mutation of the virus and inform public health measures to control the spread of this deadly disease.
Data sharing
Individual participant data that underlie the results reported in this Article (text, tables, figures, and supplementary material), after de-identification, will be made available immediately after publication without an end date, to anyone who wishes to access the data for any purpose. Data will also be available on the GISAID website.
Declaration of interests
We declare no competing interests.
Acknowledgments
Acknowledgments
This work was supported by a combination of routine work of scientists at the National Microbiology Reference Laboratory, Biomedical Research and Training Institute, Beatrice Road Infectious Diseases Hospital, National Virology Laboratory, and National TB Reference Laboratory (Zimbabwe). The authors gratefully acknowledge the support of the Biotechnology and Biological Sciences Research Council (BBSRC); this research was funded in part by the BBSRC Institute Strategic Programme Microbes in the Food Chain (BB/R012504/1) and its constituent projects (BBS/E/F/000PR10348, BBS/E/F/000PR10349, BBS/E/F/000PR10351, and BBS/E/F/000PR10352) and by the Quadram Institute Bioscience BBSRC funded Core Capability Grant (project number BB/CCG1860/1). The COVID-19 Genomics UK Consortium is supported by funding from the Medical Research Council, part of UK Research & Innovation, the National Institutes of Health Research and Genome Research Limited, operating as the Wellcome Sanger Institute. The authors thank all local clinical and laboratory staff for their contribution and dedication to the SARS-CoV-2 surveillance network. Special thanks go to Elizabeth Tabitha Abbew, for all her expert advice and proofreading of the Article. The authors acknowledge the Epidemiology and Diseases Control Department of the Ministry of Health and Child Care who provided full access to all the COVID-19 datasets used in this study. We thank members of the COVID-19 Genomics UK Consortium for their contributions in generating some of the data used in this study. We thank the laboratories who submitted their data to the GISAID; full details are listed in the appendix 1 (p 14).
Contributors
AJP, RAK, JOG, and SM-Z were responsible for leadership and supervision. AJP, RAK, and JOG were responsible for funding acquisition. TM, GT, SM-Z, GM, and MG-M were responsible for metadata curation. TM, RAK, AJP, and GM were responsible for project administration. TM, GT, AVG, FTT, MG-M, AT, HG, and the SARS-CoV-2 Research Group, Zimbabwe, were responsible for sample acquisition and logistics. DB, AJP, and LOM were responsible for sequencing and bioinformatics analysis. TM, FTT, LOM, AJP, RAK, IP, PM, AG, SD, IC, JJ, RM, KM, IM, DM, and MH were responsible for data analysis. TM, RAK, and LOM verified the data. TM, RAK, LM, LOM, JC, BVC, FTT, JC, MG-M, GM, and SM-Z were responsible for writing of the manuscript. All authors had full access to all the data in the study and had final responsibility for the decision to submit for publication.
Supplementary Materials
References
- 1.Huang C, Wang Y, Li X, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395:497–506. doi: 10.1016/S0140-6736(20)30183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Khandelwal SK. Debating the process, impact, and handling of social and health determinants of the COVID-19 pandemic. Indian J Soc Psychiatry. 2020;36:64. [Google Scholar]
- 3.Kaseje N. Why Sub-Saharan Africa needs a unique response to COVID-19. World Economic Forum. 2020. https://www.weforum.org/agenda/2020/03/why-sub-saharan-africa-needs-a-unique-response-to-covid-19/
- 4.Lone SA, Ahmad A. COVID-19 pandemic—an African perspective. Emerg Microbes Infect. 2020;9:1300–1308. doi: 10.1080/22221751.2020.1775132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Msomi N, Mlisana K, de Oliveira T, et al. A genomics network established to respond rapidly to public health threats in South Africa. Lancet Microbe. 2020;1:e229–e230. doi: 10.1016/S2666-5247(20)30116-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hâncean M-G, Perc M, Lerner J. Early spread of COVID-19 in Romania: imported cases from Italy and human-to-human transmission networks. R Soc Open Sci. 2020;7 doi: 10.1098/rsos.200780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mehtar S, Preiser W, Lakhe NA, et al. Limiting the spread of COVID-19 in Africa: one size mitigation strategies do not fit all countries. Lancet Glob Health. 2020;8:e881–e883. doi: 10.1016/S2214-109X(20)30212-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kalk A, Schultz A. SARS-CoV-2 epidemic in African countries-are we losing perspective? Lancet Infect Dis. 2020;20 doi: 10.1016/S1473-3099(20)30563-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Duchene S, Featherstone L, Haritopoulou-Sinanidou M, Rambaut A, Lemey P, Baele G. Temporal signal and the phylodynamic threshold of SARS-CoV-2. Virus Evol. 2020;6 doi: 10.1093/ve/veaa061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Meredith LW, Hamilton WL, Warne B, et al. Rapid implementation of SARS-CoV-2 sequencing to investigate cases of health-care associated COVID-19: a prospective genomic surveillance study. Lancet Infect Dis. 2020;20:1263–1271. doi: 10.1016/S1473-3099(20)30562-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nkengasong JN, Mankoula W. Looming threat of COVID-19 infection in Africa: act collectively, and fast. Lancet. 2020;395:841–842. doi: 10.1016/S0140-6736(20)30464-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Corman VM, Landt O, Kaiser M, et al. Detection of 2019 novel coronavirus (2019-nCoV) by real-time RT-PCR. Euro Surveill. 2020;25 doi: 10.2807/1560-7917.ES.2020.25.3.2000045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Quick J. NCoV-2019 Sequencing Protocol V2. https://www.protocols.io/view/ncov-2019-sequencing-protocol-v2-bdp7i5rn?version_warning=no
- 14.Baker DJ, Kay GL, Aydin A, et al. CoronaHiT: large scale multiplexing of SARS-CoV-2 genomes using Nanopore sequencing. bioRxiv. 2020 doi: 10.1101/2020.06.24.162156. published online June 24. (preprint). [DOI] [Google Scholar]
- 15.Rambaut A, Holmes EC, Hill V, et al. A dynamic nomenclature proposal for SARS-CoV-2 to assist genomic epidemiology. Nat Microbiol. 2020;5:1403–1407. doi: 10.1038/s41564-020-0770-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shu Y, McCauley J. GISAID: global initiative on sharing all influenza data—from vision to reality. Euro Surveill. 2017;22 doi: 10.2807/1560-7917.ES.2017.22.13.30494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cauchemez S, Kiem CT, Paireau J, Rolland P, Fontanet A. Lockdown impact on COVID-19 epidemics in regions across metropolitan France. Lancet. 2020;396:1068–1069. doi: 10.1016/S0140-6736(20)32034-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.La Regina M, Tanzini M, Fineschi V, et al. Responding to COVID-19: the experience from Italy and recommendations for management and prevention. Int J Qual Health Care. 2021;33 doi: 10.1093/intqhc/mzaa057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shekhar R, Sheikh AB, Upadhyay S, Atencio J, Kapuria D. Early experience with COVID-19 patients at academic hospital in Southwestern United States. Infect Dis. 2020;52:596–599. doi: 10.1080/23744235.2020.1774645. [DOI] [PubMed] [Google Scholar]
- 20.Xu W, Wu J, Cao L. COVID-19 pandemic in China: context, experience and lessons. Health Policy Technol. 2020;9:639–648. doi: 10.1016/j.hlpt.2020.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Niforatos JD, Melnick ER, Faust JS. Covid-19 fatality is likely overestimated. BMJ. 2020;368 doi: 10.1136/bmj.m1113. [DOI] [PubMed] [Google Scholar]
- 22.Pachetti M, Marini B, Giudici F, et al. Impact of lockdown on Covid-19 case fatality rate and viral mutations spread in 7 countries in Europe and North America. J Transl Med. 2020;18:338. doi: 10.1186/s12967-020-02501-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Samaddar A, Gadepalli R, Nag VL, Misra S. The enigma of low COVID-19 fatality rate in India. Front Genet. 2020;11:854. doi: 10.3389/fgene.2020.00854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dhama K, Patel SK, Kumar R, et al. Geriatric population during the COVID-19 Pandemic: problems, considerations, exigencies, and beyond. Front Public Health. 2020;8 doi: 10.3389/fpubh.2020.574198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brandén M, Aradhya S, Kolk M, et al. Residential context and COVID-19 mortality among adults aged 70 years and older in Stockholm: a population-based, observational study using individual-level data. Lancet Healthy Longev. 2020;1:e80–e88. doi: 10.1016/S2666-7568(20)30016-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sanyaolu A, Okorie C, Marinkovic A, et al. Comorbidity and its impact on patients with COVID-19. SN Compr Clin Med. 2020;2:1–8. doi: 10.1007/s42399-020-00363-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schultze A, Walker AJ, MacKenna B, et al. Risk of COVID-19-related death among patients with chronic obstructive pulmonary disease or asthma prescribed inhaled corticosteroids: an observational cohort study using the OpenSAFELY platform. Lancet Respir Med. 2020;8:1106–1120. doi: 10.1016/S2213-2600(20)30415-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Page AJ, Mather AE, Le Viet T, et al. Large scale sequencing of SARS-CoV-2 genomes from one region allows detailed epidemiology and enables local outbreak management. Microb Genom. 2021;7 doi: 10.1099/mgen.0.000589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Korber B, Fischer WM, Gnanakaran S, et al. Tracking changes in SARS-CoV-2 spike: evidence that D614G increases infectivity of the COVID-19 virus. Cell. 2020;182:812–827. doi: 10.1016/j.cell.2020.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yurkovetskiy L, Wang X, Pascal KE, et al. Structural and functional analysis of the D614G SARS-CoV-2 spike protein variant. Cell. 2020;183:739–751. doi: 10.1016/j.cell.2020.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Individual participant data that underlie the results reported in this Article (text, tables, figures, and supplementary material), after de-identification, will be made available immediately after publication without an end date, to anyone who wishes to access the data for any purpose. Data will also be available on the GISAID website.