

Abstract
Supplemental oxygen administration is frequently used in premature infants and adults with pulmonary insufficiency. NADPH quinone oxidoreductase (NQO1) protects cells from oxidative injury by decreasing reactive oxygen species (ROS). In this investigation, we tested the hypothesis that overexpression of NQO1 in BEAS-2B cells will mitigate cell injury and oxidative DNA damage caused by hyperoxia and that A-1221C single nucleotide polymorphism (SNP) in the NQO1 promoter would display altered susceptibility to hyperoxia-mediated toxicity. Using stable transfected BEAS-2B cells, we demonstrated that hyperoxia decreased cell viability in control cells (Ctr), but this effect was differentially mitigated in cells overexpressing NQO1 under the regulation of the CMV viral promoter, the wild-type NQO1 promoter (NQO1-NQO1), or the NQO1 promoter carrying the SNP. Interestingly, hyperoxia decreased the formation of bulky oxidative DNA adducts or 8-hydroxy-2′-deoxyguanosine (8-OHdG) in Ctr cells. qPCR studies showed that mRNA levels of CYP1A1 and NQO1 were inversely related to DNA adduct formation, suggesting the protective role of these enzymes against oxidative DNA injury. In SiRNA experiments entailing the NQO1-NQO1 promoter, hyperoxia caused decreased cell viability, and this effect was potentiated in cells treated with CYP1A1 siRNA. We also found that hyperoxia caused a marked induction of DNA repair genes DDB2 and XPC in Ctr cells, supporting the idea that hyperoxia in part caused attenuation of bulky oxidative DNA lesions by enhancing nucleotide excision repair (NER) pathways. In summary, our data support a protective role for human NQO1 against oxygen-mediated toxicity and oxidative DNA lesions in human pulmonary cells, and protection against toxicity was partially lost in SNP cells. Moreover, we also demonstrate a novel protective role for CYP1A1 in the attenuation of oxidative cells and DNA injury. Future studies on the mechanisms of attenuation of oxidative injury by NQO1 should help in developing novel approaches for the prevention/treatment of ARDS in humans.
1. Introduction
Supplemental oxygen is an integral part of medical management of pediatric and adult patients with pulmonary insufficiency [1–3]. In premature infants and adults, exposure to hyperoxia contributes to the development of bronchopulmonary dysplasia (BPD) [4, 5], and in adults, it could exacerbate acute respiratory distress syndrome (ARDS) [6–8]. ARDS is a life-threatening illness that affects up to 10% of patients in intensive care units worldwide [9] and could develop following pneumonia, nonpulmonary sepsis, trauma, or aspiration [9]. Despite significant medical advances, mortality due to ARDS is high (35-46%) [8, 9], and recent studies have shown that ARDS is one of the major causes of death due to the COVID-19 infection [10]. The molecular mechanisms of oxygen-mediated lung injury are not completely understood, but reactive oxygen species (ROS) likely play an important role [11]. Hyperoxia (>95% FiO2) for 72 hours in rodents results in lung inflammation and injury, eventually leading to cell death [4, 12]. ROS generated in hyperoxic conditions lead to profound cell damage through direct DNA damage, lipid peroxidation, protein oxidation, and alteration of transcription factors [4, 12]. Recent studies from our laboratory have shown a protective effect of cytochrome P450 (CYP) 1A enzymes against hyperoxic lung injury in vivo [13–20].
NADPH quinone oxidoreductase 1 (NQO1) is a phase II enzyme whose activity in the cell is to catalyze the two-electron reduction of quinone compounds, which prevents the generation of ROS and, thus, protects cells from oxidative damage [21]. Das et al. showed that mice deficient in the genes for Nqo1 and Nqo2 are more susceptible to lung injury than wild-type mice [22]. A number of single nucleotide polymorphisms (SNPs) have been reported for NQO1 [23–28]. Although associations between genetic variants in NQO1 and ALI/ARDS have been reported [22–28], little is known regarding the mechanisms by which these genetic variants contribute to ARDS.
Prior reports have demonstrated that the A/C single nucleotide polymorphism (SNP) at -1221 of the NQO1 promoter resulted in attenuation of in vitro transcription of luciferase reporter expression following exposure to hyperoxic conditions [29]. Individuals in a cohort of trauma patients who were genotyped for the A-1221C SNP were found to have a significantly decreased incidence of acute lung injury (ALI), implying a protective role for A-1221C in ARDS patients [29].
The overall objective of this investigation was to study the role of human NQO1 and A-1221C SNP in hyperoxia-mediated cellular injury and oxidative DNA damage. Specifically, we tested the hypothesis that overexpression of NQO1 in BEAS-2B cells will mitigate cell injury and oxidative DNA damage caused by hyperoxia and that the presence of A-1221C SNP in the NQO1 promoter would display altered susceptibility to hyperoxia-mediated toxicity.
2. Materials and Methods
2.1. Cell Culture
BEAS-2B adenovirus 12-SV40-transformed, normal human bronchial epithelial cells (ATCC) were maintained in RPMI 1640 medium supplemented with 10% FBS and penicillin-streptomycin at 37°C in room air containing 5% CO2. The hyperoxia condition used was 80% O2 plus 5% CO2.
2.2. Construction of Plasmids
A 2.4 kb of human NQO1 promoter was obtained from the genomic DNA of BEAS-2B cells by the LA Taq PCR Kit (Takara) using primer pair GGCTTCTCAGACCACTCCTG and ACTAGGCTCTCGGTGAGCTG and subcloned into the pGL4.13 luciferase expression plasmid (Promega) between the SacI and XhoI sites. A-1221C mutation (rs689455) at the NQO1 promoter region of the pGL4-NQO1 plasmid was introduced by site-directed mutagenesis PCR using primer pair AGGTCGGGAGTTGGAAAC and CAGGTGATCCTACCGCCT. These two plasmids were named pGL4-NQO1 and pGL4-SNPNQO1.
To obtain the NQO1 expression plasmid pCD-NQO1, total RNA was extracted from BEAS-2B cells and subjected to reverse transcription using the SuperScript III First-Strand Synthesis System (Invitrogen). The open reading frame and the 3′-UTR of human NQO1 were obtained as one piece by the subsequent PCR (Takara) using primer pair CAGCTCACCGAGAGCCTAGT and AAAAACCACCAGTGCCAGTC and then subcloned between the NheI and XhoI sites of the pcDNA3.1(+) mammalian expression plasmid (Invitrogen). It was named pCMV-NQO1. The CMV promoter in pCD-NQO1 was replaced by the 2.4 kb wild-type or SNP-human NQO1 promoter, which was excised from pGL4-NQO1 and pGL4-SNPNQO1. The two new plasmids were named pNQO1-NQO1 and pSNPNQO1 (or pSNP). The correct sequence of each plasmid was verified by DNA sequencing.
2.3. Stable Expression of NQO1 in BEAS-2B Cells
pcDNA3.1, pCMV-NQO1, pNQO1-NQO1, or pSNP was transfected into BEAS-2B cells using SuperFect (Qiagen) and maintained in 100 μg/ml Geneticin (Invitrogen). Clones were screened by immunofluorescence staining with the A180 NQO1 antibody (Santa Cruz Biotechnology) and verified by qPCR. These 4 stable transfected BEAS-2B cell lines were named Ctr-, CMV-NQO1-, NQO1-NQO1-, and SNP-BEAS-2B cells, respectively.
2.4. NQO1 Assay
This method was adapted from Tsvetkov et al. in 2005 [30]. Cells were lysed in 25 mM Tris, pH 7.5/1 mM EDTA/0.1 mM dithiothreitol (DTT). Cell lysate (30-50 μg) was mixed in 200 μl of reaction buffer (25 mM Tris-HCl (pH 7.5), 0.01% Tween 20, 0.7 mg/ml BSA (pH 7.4), 40 μM menadione, 5 μM flavin adenine dinucleotide (FAD), and 200 μM nicotinamide adenine dinucleotide (NADH)) in a 96-well plate. Absorbance at 340 nm (A340nm) was measured repeatedly during the decay of NADH. Statistical difference between each group was calculated with Tukey's multiple comparison test in repeated measures ANOVA using GraphPad Prism 5.
2.5. qPCR
Total RNA was extracted from the cell lysates using the Qiagen RNeasy Kit. The mRNA level was quantified with the BioRad iScript Reverse Transcription Supermix and the iQ SYBR Green Supermix RT-qPCR method, while the primers for CYP1B1 and the reference gene OAZ1were obtained following the method of Dinu et al. in 2016 [31]. Primers for AHR, CYP1A1, and NQO1 were obtained following the method of Shivanna et al. in 2011 [32]. Other primers included the following: NME1, tcattgcgatcaaaccagat and caacgtagtgttccttgaga; PCNA, aggcactcaaggacctcatca and gagtccatgctctgcaggttt; ERCC1, ggcgacgtaattcccgacta and agttcttccccaggctctgc; OGG1, gatgttgttgttggaggaa and aagaggtggctcagaaat; XPC, taaatagcaaatctcctttcc and acacctactacctctcaa; PARP1, cacttgctgcttgttgaa and gaacgacctgatctggaa; DDB2, gcattctgagattccaaagc and tgtagcctggatgtgtct; XAB2, cccccaaaatatgccaagacct and tgctcgtccgacagcacctc; and NEIL2, gcactcaggactgaaccga and ctgtctgctatacactgctgga.
2.6. Cell Viability Assays
Cell viability was determined by the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) Proliferation Assay Kit from ATCC and the live protease assay using the ApoTox-Glo Triplex Assay Kit from Promega, according to the manufacturers' instructions and the method of Dinu et al. in 2016 [31].
2.7. ApoTox-Glo Triplex Assay
Cytotoxicity and cell viability of cells in 96-well black-walled plates were determined using the ApoTox-Glo Triplex Assay (Promega) according to the manufacturers' instructions and the method of Dinu et al. in 2016 [31]. Cell viability (live cell protease activity) and dead cell level (dead cell protease activity) were determined by fluorescence absorption at 505 nm and 520 nm, respectively. Caspase 3/7 assays were determined by bioluminescence as reported earlier [31].
2.8. Knockdown of CYP1A1 in Ctr and NQO1-NQO1 Cells
Ctr and NQO1-NQO1 cells were transfected with human CYP1A1 siRNA (Thermo Fisher Scientific #4392420, Assay ID s3800) or negative control siRNA (Thermo Fisher Scientific #4390843) using the Lipofectamine RNAiMAX Transfection Reagent (Invitrogen) according to the manufacturers' instructions. The cells were subjected to hyperoxia treatment 24 h after the transfection.
2.9. Detection of Oxidative DNA Lesions by the 32P-Postlabeling Assay
BEAS-2B human cells were grown in culture and transfected with pcDNA3.1, pCMV-NQO1, pNQO1-NQO1, or pSNP. Cells were exposed to 80% oxygen or room air for 48 hours. DNA was extracted from the cells and subjected to enzymatic digestion and enrichment of the oxidative products (pNp-cAP) from the DNA digest. Dinucleotide adducts were labeled with [32P]-orthophosphate from [γ-32]-ATP mediated by polynucleotide kinase and then separated by two-dimensional thin-layer chromatography per the previously described method [16, 33–36]. The labeled nucleotides were chromatographed on polyethyleneimine-impregnated cellulose thin-layer chromatography (TLC) plates and imaged by the InstantImager (Packard Instruments, Merien, Connecticut). Levels of total 8,5-cyclo-20-deoxyadenosine (cA) oxidative DNA adducts as well as the individual dinucleotides adenine cA (AcA), guanine cA (GcA), cytosine (GcA), and thymine cA (TcA) were analyzed as reported previously [25, 35]. 32P-labeled DNA adducts were quantified by InstantImager [35, 36]. The oxidative dinucleotide adducts of cells on TLC maps were identified by comparing with those from genomic DNA obtained from endotracheal aspirate of an ARDS patient who was subjected to supplemental oxygen and mechanical ventilation. The ARDS patient sample was obtained from Ben Taub General Hospital, Houston, TX, as part of an ongoing IRB-approved study at Baylor College of Medicine.
2.10. Detection of 8-Hydroxy-2′-Deoxyguanosine (8-OHdG) by LC-MS/MS
Total DNA was isolated from cells using proteinase K digestion followed by phenol/chloroform extraction and ethanol precipitation. After undergoing a series of digestion with micrococcal endonuclease, spleen phosphodiesterase, nuclease P1, and calf intestinal phosphatase, 0.2 μg DNA in 50 μl of a 1 : 1 methanol/water mixture was subjected to LC-MS/MS analysis [37, 38].
2.11. Statistical Analyses
All data were analyzed by comparing mean ± SE of at least 3 independent experiments. Mean values among different groups were compared using Student's t-test, unless specified, and P < 0.05 was considered significant.
3. Results
3.1. Effect of Hyperoxia on NQO1, CYP1A1, CYP1B1, and AHR Gene Expression in Control and NQO1 Overexpressing Cells
Stable cell lines transfected with pcDNA3.1 (Ctr), pCMV-NQO1 (CMV-NQO1), pNQO1NQO1 (NQO1-NQO1), and pSNPNQO1 (SNP) were cultured under room air (RA) or 80% oxygen (O2) for 48 h. qPCR, using OAZ1 as the reference gene, indicated that NQO1 mRNA level was significantly induced by hyperoxia in Ctr cells (Figure 1(a)). In cells stably transfected with NQO1-containing cDNA plasmids, hyperoxia augmented the NQO1 expression by 77%, 118%, and 66% in CMV-NQO1, NQO1-NQO1, and SNP cells, respectively, compared to room air conditions (Figure 1(a)). NQO1 mRNA level was significantly higher in each of the NQO1 overexpressed cells compared to Ctr cells even in room air (Figure 1(a)), with SNP cells showing greater NQO1 expression compared to NQO1-NQO1 cells. The extent of NQO1 induction from baseline levels in SNP cells by hyperoxia appeared to be less than that of NQO1-NQO1 cells (Figure 1(a)).
Figure 1.
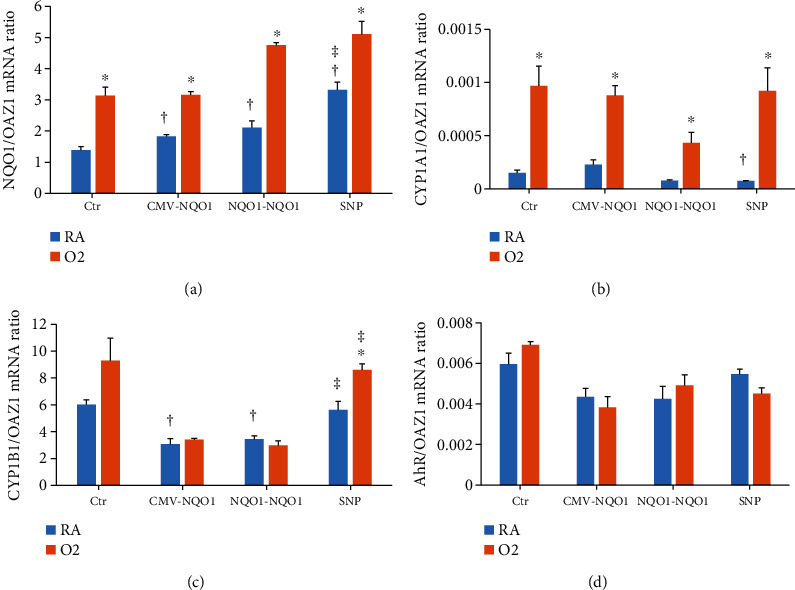
Overexpression of NQO1 in NQO1-stable transfected cells. BEAS-2B cells stably transfected with pcDNA3.1 (Ctr), pCD-NQO1 (CMV-NQO1), pWT-NQONQO1 (NQO1-NQO1), and pmut-NQONQO1 (SNP) were incubated under room air (RA) or 80% O2 (O2) conditions for 48 h and subjected to qPCR using total RNA extracted from these cells. Gene expression of NQO1 (a), CYP1A1 (b), CYP1B1 (c), and AHR (d) were determined. ∗Statistically significant difference between room air and hyperoxia. †Statistically significant difference with Ctr. ‡Statistically significant difference between the NQO1-NQO1 and SNP-NQO1 promoter (n = 3; P < 0.05).
At protein level, NQO1 overexpression was detected by the NQO1 assay (Figure 2(a)). NADH decay was measured by A340nm in 50 μg lysate protein from Ctr cells as well as CMV-NQO1, NQO1-NQO1, and SNP cells in the presence of menadione (substrate) and FAD (coenzyme), a reaction catalyzed by the NQO1 enzyme in the lysate. The decay of NADH appeared to be significantly faster in CMV-NQO1, NQO1-NQO1, and SNP cells when comparing with Ctr cells, which was represented by slightly but statistically significant higher Kdecay value and shorter half-life (Figure 2(a)). This result indicated that CMV-NQO1, NQO1-NQO1, and SNP cells expressed higher NQO1 activities than Ctr cells.
Figure 2.
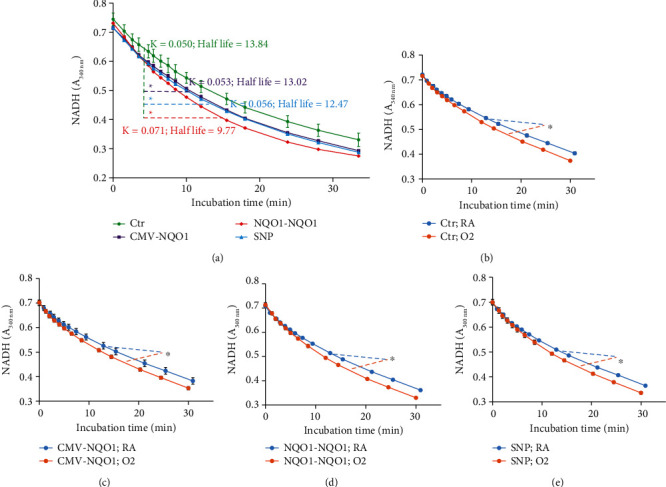
NADH decay curve indicated enhanced NQO1 enzyme activity in cells stably transfected with NQO1 cDNA (a), or by hyperoxia (b–e). (a) 50 μg lysate from each of the stably transfected BEAS-2B cell lines Ctr, NQO1-NQO1, and SNP was subjected to the NQO1 assay. (b–e) Four cell lines were incubated under room air (RA) or 80% O2 (O2) conditions for 48 h. 30 μg lysate was subjected to the NQO1 assay. One way ANOVA indicated statistically significant difference between specified curves. Kdecay value and half-life were the curve fitting results using the “one phase decay” model in GraphPad Prism 5. ∗Statistically significant difference with Ctr cells (a) or between RA and O2 (b–e) (n = 3; P < 0.05).
The NQO1 assay also showed that hyperoxia (80% O2, 48 h) significantly induced the NQO1 enzyme in CMV-NQO1, NQO1-NQO1, and SNP cells when comparing with Ctr cells (Figures 2(b)–2(e)). Western blot analyses detected NQO1 protein in each of the constructs (Supplemental Figure 1), and hyperoxia induced NQO1 expression in each of the cells, albeit the basal expression of NQO1 was not different among the constructs.
At mRNA level, hyperoxia elicited marked induction of CYP1A1 (~10-fold) in Ctr cells and CMV-NQO1 cells (~5-fold) (Figure 1(b)). Hyperoxia also caused induction of CYP1A1 in NQO1-NQO1 and SNP cells (Figure 1(b)), with the extent of induction being greater in the SNP cells (~20-fold). Hyperoxia induced CYP1B1 gene expression in SNP cells but not in the other cell lines (Figure 1(c)). CYP1B1 expression in room air in the CMV-NQO1 and NQO1-NQO1 was lower than Ctr cells. On the other hand, CYP1B1 expression in SNP cells was higher than that of NQO1-NQO1 in both room air and hyperoxic conditions (Figure 1(c)). The expression of AHR gene was not altered by hyperoxia in any of the cells (Figure 1(d)).
3.2. Overexpression of NQO1 Altered Hyperoxic Cytotoxicity
Hyperoxia significantly decreased cell viability. The A590nm of the Ctr cells decreased by 41% in the MTT assay (Figure 3(a)). Overexpression of NQO1 resulted in improvement in cell viability (16-33%) in the 3 NQO1-overexpressed cell lines. The live cell protease assay (Figure 3(b)) exhibited a comparable result, in which hyperoxia decreased cell viability, and it was rescued in part by overexpression of NQO1, with CMV-NQO1 cells showing an increase in cell viability after hyperoxia. The dead cell protease activities represented the number of dead cells in the wells. In NQO1-NQO1 (and CMV-NQO1) cells, cell death was 45-50% lower compared to room air controls, and as in NQO1-NQO1 cells, cell death in hyperoxic cells was lower than that in the Ctr group (Figure 3(c)). Cell death was also decreased in SNP cells by hyperoxia, but the number of dead cells were higher in SNP cells exposed to hyperoxia compared to those of NQO1-NQO1 (Figure 3(c)). Interestingly, there was an increase of caspase 3/7 activities in the live cells (Figure 3(d)) overexpressing NQO1. This result suggested that overexpression of NQO1 might redirect the hyperoxia-stressed cells into an apoptotic pathway rather than necrotic death. This redirection was decreased in cells harboring the A-1221C SNP on the NQO1 promoter because SNP cells appeared to not be different from Ctr cells (Figure 3(d)). In all these experiments, an equal number of cells were plated from all cell lines.
Figure 3.
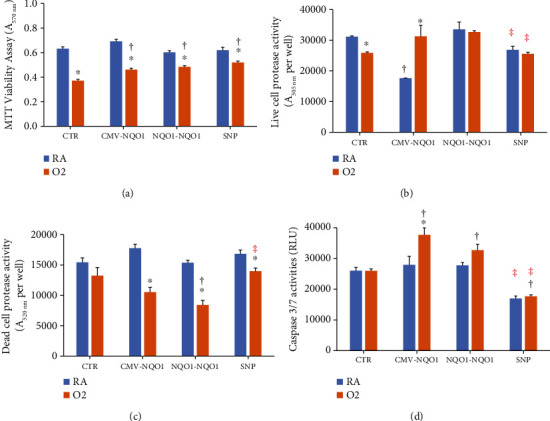
NQO1 overexpression protected cells from hyperoxic toxicity. BEAS-2B cells stably transfected with pcDNA3.1 (Ctr), pCMV-NQO1 (CMV-NQO1), pWT-NQONQO1 (NQO1-NQO1), and pSNP-NQONQO1 (SNP) were incubated under room air (RA) or 80% O2 (O2) condition for 48 h and the MTT cell viability assay (a), the live cell protease activity assay (b), the dead cell protease activity assay (c), and the caspase 3/7 activity assay (d) were determined using the Promega ApoTox-Glo Triplex Assay. ∗Statistically significant differences between the RA and O2 groups. †Statistically significant difference with Ctr. ‡Statistically significant difference between the WT- and SNP-NQO1 promoter (n = 3; P < 0.05). Hyperoxia decreased cell viability, which was attenuated by overexpression of NQO1 (a and b).
3.3. Effect of Hyperoxia on Oxidative DNA Adduct Formation
Previous studies have shown that hyperoxia increases oxidative DNA adduct formation [34]. Levels of total 8,5-cyclo-2-deoxyadenosine (cA) oxidative DNA adducts as well as the individual dinucleotides adenine cA (AcA) and guanine cA (GcA) were determined by Veith et al. in 2018 [34] and by Zhou and Moorthy in 2015 [35] (Figure 3(a)). The DNA in Figure 4(a) was obtained from an endotracheal aspirate sample from an ARDS patient as described under Materials and Methods. The cA adducts are formed by hydroxyl radical attack on 2′-deoxyadenosine, which then binds covalently with the adjacent nucleotide [33, 35]. The location of these adducts on the thin-layer chromatography (TLC) plates was based on cochromatography and rechromatography using structurally characterized adducts [36]. Total pulmonary adducts were quantified in Figure 4(b), which included the aggregate values of the AcA, CcA, GcA, and TcA adducts. The individual dinucleotide adducts were also analyzed as well. Our main finding was that in all cells, the formation of the DNA adducts AcA, CcA, GcA, and TcA was mostly decreased in the hyperoxia groups. The hyperoxia-mediated decrease in total adduct levels was significant in Ctr cells and CMV-NQO1 cells but not significant in NQO1-NQO1 or SNP cells (Figure 4(b)).
Figure 4.
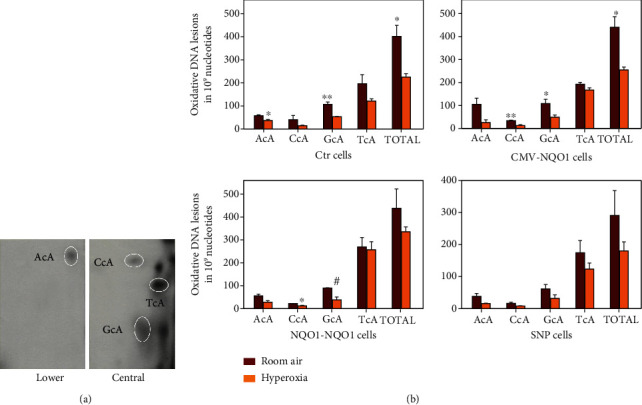
Hyperoxia decreased DNA adduct formation in 32P-postlabeling experiments, and high cellular levels of NQO1 and CYP1A1 might attenuate such effect. (a) A typical result of a 32P-postlabeling experiment showing chromatographic images labeled with AcA, GcA, CcA, and TcA. (b) Quantitation of the 32P-postlabeling experiment. Three stably transfected BEAS-2B cell lines Ctr, CMV-NQO1, NQO1-NQO1, and SNP (see Figure 2) were incubated under RA or O2 for 48 h, followed by DNA extraction and 32P-postlabeling experiments (n = 3; ∗P < 0.05; ∗∗P < 0.01; #P = 0.05).
3.4. Inverse Correlation between Oxidative DNA Adducts and CYP1A1/NQO1 Expression
In order to determine if a mechanistic relationship exists between oxidative DNA adducts and gene expression of CYP1A1 or NQO1, we conducted a regression analyses between levels of each of the cyclopurines (AcA, GcA, TcA, and CcA) and total adducts, and that of CYP1A1 (Figure 5(a)) or NQO1 (Figure 5(b)). The data were compared among these parameters after combining the data obtained from at least 3 individual experiments in all the 4 cell lines, which were either maintained in room air or exposed to hyperoxia (Figure 5). The results showed that each of the cyclopurine dinucleotides and total adducts inversely correlated with CYP1A (Figure 5(a)) or NQO1 (Figure 5(b)) gene expression.
Figure 5.
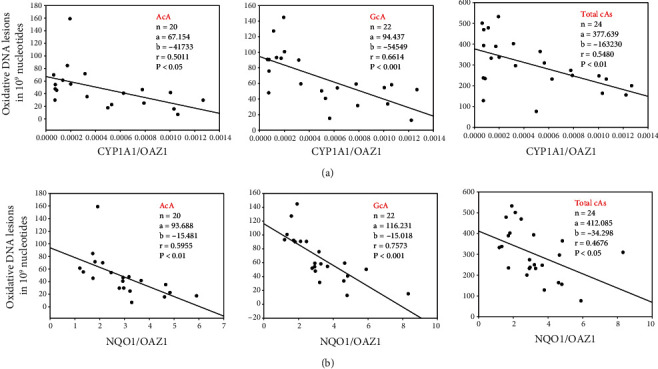
Linear correlations between levels of 8,5′-cyclopurine-2′-deoxynucleotides (oxidative DNA lesions in 109 normal nucleotides) and CYP1A1/OAZ1 (a) or NQO1/OAZ1 (b) in lung cell lines. Data of DNA adducts from all the individual samples (n = 20‐24) in room air or hyperoxic condition in each cell line were combined and plotted against the mean CYP1A1 (a) or NQO1 (b) gene expression using data from all individual samples. Significant inverse correlations were observed between levels of AcA, GcA, and Total cA (sum of AcA, CcA, GcA, and TcA) and CYP1A1/OAZ1 (a) or NQO1/OAZ1 (b).
3.5. Effect of Hyperoxia on 8-OHdG Levels
In order to determine if the oxidative DNA adduct data correlated with 8-OHdG levels, we measured 8-OHdG levels under similar experimental conditions by LC-MS/MS in the total DNA extracted from Ctr, NQO1-NQO1, and SNP cells. The 8-OHdG level in the genomic DNA from Ctr cells was significantly (34.2%) decreased by hyperoxia, and this decrease was not observed in NQO1-NQO1 cells or the SNP cells (Figure 6).
Figure 6.
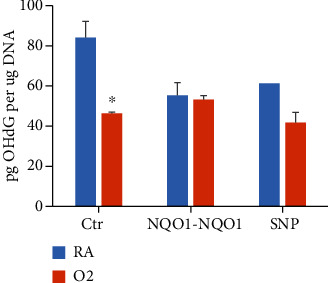
Hyperoxia or NQO1 overexpression decreased 8-OHdG formation. Three stably transfected BEAS-2B cell lines Ctr, NQO1-NQO1, and SNP were incubated under RA or O2 for 48 h. Genomic DNA was isolated from the cells and digested with micrococcal endonuclease, spleen phosphodiesterase, nuclease P1, and calf intestinal phosphatase, followed with LC-MS of 8-OHdG using 0.2 μg enzyme-treated DNA (n = 3; ∗P < 0.05).
3.6. Role of CYP1A1 in the Modulation of Cell Toxicity and 8-OHdG
In order to determine if inhibiting CYP1A1 would modulate the cell toxicity and oxidative DNA damage responses by hyperoxia, we determined cell viability (Figure 7(a)) and levels of 8-OHdG (Figure 7(b)) in Ctr and NQO1-NQO1 cells that had been treated with control or CYP1A1 siRNA. CYP1A1 mRNA knockdown by CYP1A1 siRNA was verified by qPCR (Supplemental Figure 2). As shown in Figure 7(a), hyperoxia decreased cell viability in cells treated with control or CYP1A1 siRNA. In NQO1-NQO1 cells, the decrease in cell viability by hyperoxia was higher in those that were treated with CYP1A1 siRNA (Figure 7(a)) compared to those treated with control siRNA, suggesting that CYP1A1 might protect the cell from hyperoxia in an NQO1-dependent manner.
Figure 7.
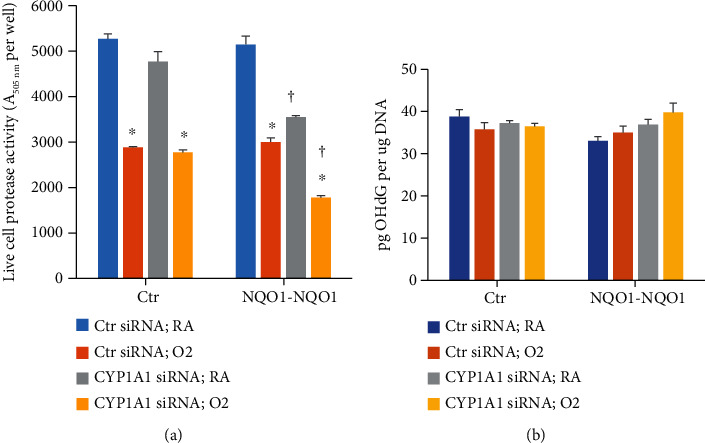
Effect of CYP1A1 silencing on cell viability (a) and 8-OHdG (b) levels. Stably transfected BEAS-2B cell lines Ctr or NQO1-NQO1 were transfected with CYP1A1 siRNA or control siRNA and incubated under RA or O2 for 48 h, followed by determination of live cell protease activity using the Promega ApoTox-Glo Triplex Assay (a) or the LS-MS/MS assay (b) (n = 3; ∗P < 0.05 by Student's t-test.
In order to study the role of CYP1A1 on oxidative DNA damage, we measured 8-OHdG levels in Ctr and NQO1 cells that were treated with control or CYP1A1 siRNA. As shown in Figure 7(b), there were no significant changes in 8-OHdG levels in Ctr cells that were treated with CYP1A1 siRNA. However, in NQO1-NQO1 cells, 8-OHdG was slightly increased by hyperoxia by about 7% and by 12% in cells that were treated with CYP1A1 siRNA compared to those that were treated with control siRNA (Figure 7(b)), although the results were not statistically significant.
3.7. Modulation of DNA Repair Genes by Hyperoxia
We determined the effect of hyperoxia on a number of DNA repair genes to test the hypothesis that hyperoxia would induce DNA repair enzymes, which could play a protective role against oxygen toxicity. We also wanted to determine if DNA repair genes could in part explain the decrease in oxidative DNA damage caused by hyperoxia and also if NQO1 overexpression would modulate DNA repair mechanisms. Exposure to hyperoxic conditions significantly increased the expression of damage-specific DNA binding protein 2 (DDB2) and xeroderma pigmentosum, complementation group C (XPC) (Figures 8(a) and 8(f)) in Ctr, CMV-NQO1, NQO1-NQO1, and SNP-transfected cells. In hyperoxic cells, significantly less XPC expression was detected in cells transfected with NQO1-NQO1 promoter constructs compared to control cells, whereas hyperoxic cells transfected with SNP-containing promoter constructs had levels of XPC expression that were not significantly different from control-transfected cells (Figure 8(f)). Nei-like DNA glycosylase 2 (NEIL2) expression (Figure 8(b)) did not show any increase with hyperoxia; however, the expression was decreased under normoxic conditions in CMV-NQO1 and NQO1-NQO1 cells compared to Ctr cells.
Figure 8.
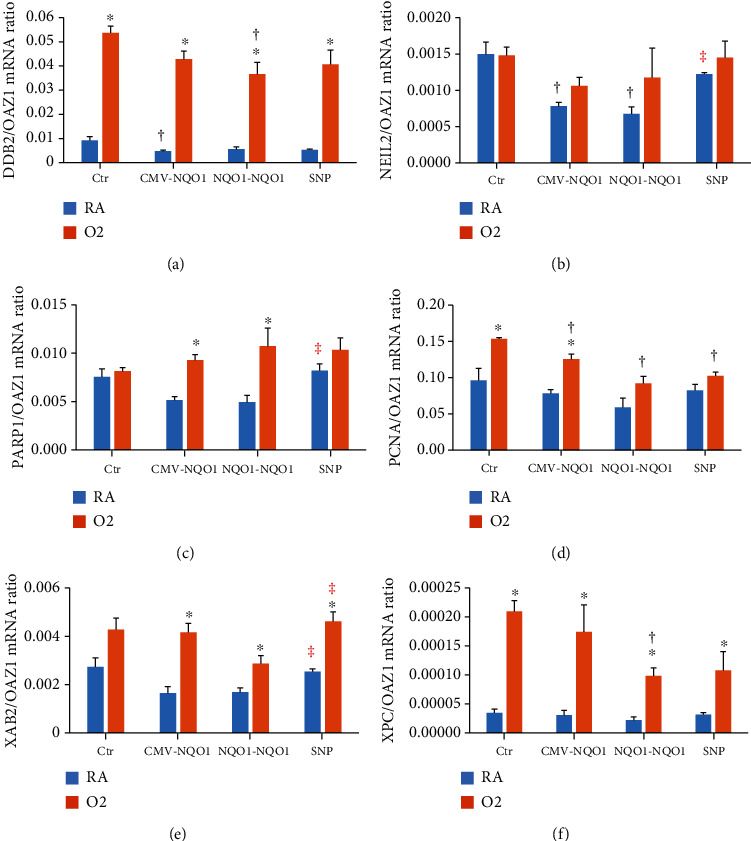
Effect of hyperoxia on DNA repair genes. Four stably transfected BEAS-2B cell lines Ctr, CMV-NQO1, NQO1-NQO1, and SNP were incubated in RA or O2 for 48 h and subjected to qPCR. ∗Statistically significant difference between RA and O2 groups. †Statistically significant difference compared to Ctr. ‡Statistically significant difference between NQO1-NQO1 and SNP (n = 3; P < 0.05).
Poly(ADP-ribose polymerase 1 (PARP1) expression was increased in CMV-NQO1 and NQO1-NQO1 cells upon exposure to hyperoxia (Figure 8(c)). In cells with SNP, PARP1 expression was higher than NQO1-NQO1 cells under normoxia (Figure 8(c)). Hyperoxia caused induction of proliferating cell nuclear antigen (PCNA) under hyperoxic conditions in Ctr and CMV-NQO1 but not in NQO1-NQO1 or SNP-containing cells (Figure 8(d)). Under hyperoxia, the expression of PCNA was lower in CMV-NQO1, NQO1-NQO1, and SNP-containing cells compared to Ctr cells. Additionally, there were significantly increased levels of XPA binding protein 2 (XAB2) mRNA levels in hyperoxia-exposed CMV-NQO1, NQO1-NQO1, and SNP cells with levels being relatively higher in the latter compared to NQO1-NQO1 cells (Figure 8(e)). In cells carrying the SNP, the expression was increased in normoxia as well as compared to NQO1-NQO1 cells (Figure 8(e)).
4. Discussion
The overall goal of this study was to determine the role of human NQO1 in hyperoxia-mediated cellular injury and oxidative DNA damage. Specifically, we tested the hypothesis that overexpression of NQO1 in BEAS-2B cells will mitigate cell injury and oxidative DNA damage caused by hyperoxia and that A-1221C SNP in the NQO1 promoter would display altered susceptibility to hyperoxia-mediated toxicity.
Our results showing increased hyperoxia-mediated NQO1 expression in Ctr cells and in cells overexpressing NQO1 in CMV-NQO1 and NQO1-NQO1 cells (Figure 1(a)) were in agreement with earlier studies showing induction of NQO1 by hyperoxia [29, 39]. Our observation that SNP cells showed lesser extent induction of NQO1 expression by hyperoxia compared to NQO1-NQO1 cells was probably due to the regulatory elements in the SNP construct that were masked, leading to reduced induction of the gene (Figure 1(a)). However, we did see increased NQO1 expression per se in the SNP cells exposed to hyperoxia, and additional work needs to be done to explain this discrepancy. The induction of the CYP1A1 gene by hyperoxia (Figure 1(b)) was in agreement with earlier reports of induction of the CYP1A1 enzyme in vitro [40] and in vivo [13–17]. The suppression of induction of CYP1A1 in NQO1-NQO1 cells was probably due to the metabolism of ROS-mediated AHR ligands [41] that contributed to CYP1A1 enhancement by hyperoxia [34]. The restoration of CYP1A1 induction in the SNP cells by hyperoxia (Figure 1(b)) could have been due to an increase in ROS levels in these cells, which in turn may have resulted in increased formation of endogenous ligands that contributed to CYP1A1 induction by hyperoxia.
The suppression of CYP1B1 gene expression (Figure 1(c)) in CMV-NQO1 and NQO1-NQO1 cells in room air conditions could be explained by the metabolism of ROS-mediated endogenous AHR ligands that were responsible for CYP1B1 induction probably by CYP1A1. The fact that CYP1B1 expression was restored in SNP cells in room air and was induced in these cells by hyperoxia lends credence to the theory that endogenous AHR ligands contributed to CYP1B1 induction.
The fact that the decay of NADH was significantly faster in CMV-NQO1, NQO1-NQO1, and SNP cells compared to Ctr cells (Figure 2(a)) suggested that CMV-NQO1, NQO1-NQO1, and SNP cells expressed higher NQO1 activities than Ctr cells. Given that NQO1 is an antioxidant enzyme, we first sought to evaluate the role of oxygen toxicity in human lung cells that had been transfected with the WT- (NQO1-NQO1) and SNP-containing NQO1 promoter/gene construct compared to controls. Cells that had not been transfected with the NQO1 constructs displayed decreased cell viability, decreased live cell protease, and increased cell death under hyperoxic conditions (Figures 3(a)–3(c)), suggesting that oxidative stress contributed to cell injury. In the live cell and dead cell protease assays (Figures 3(b) and 3(c)), cells transfected with the constitutively active CMV promotor/NQO1gene construct demonstrated enhanced ratio of live/dead cell protease activities under hyperoxic conditions compared to room air, which implied that the overexpression of CMV-NQO1 might prevent the disruption of the cell membrane and keep the proteases inside the cells. In cells transfected with SNP A-1221C, the live cell protease activity was lesser in both room air and hyperoxic conditions compared to the NQO1-NQO1 group (Figure 3(b)), probably due to a partial loss of protection to cell membrane integrity by NQO1 due to the SNP.
On the other hand, both CMV and NQO1-NQO1 cells showed significantly decreased dead cell protease activities under hyperoxic conditions, which was probably due to protection of cell membrane integrity by NQO1 overexpression in these cells (Figure 3(c)). Figure 3(d) shows the increase of caspase 3/7 activities by hyperoxia in CMV-NQO1 and NQO1-NQO1 cells. This increase suggested that part of the hyperoxia-damaged cells might have entered an apoptotic pathway. This would also explain why the CMV and NQO1-NQO1 cells exhibited increased live cell protease activities compared to Ctr cells under hyperoxic conditions (Figure 3(b)).
To further characterize the toxic effect of high levels of oxygen exposure on cells transfected with the various NQO1 promoter/gene constructs, we investigated the effect of hyperoxia on oxidative DNA lesions by 32P-postlabeling. Our observations (Figure 4(b)) showing decreased levels of AcA, GcA, and total adducts in all cells were surprising, as we would expect increased oxidative DNA damage by hyperoxia due to increased oxidative stress.
Firstly, we found an inverse correlation between oxidative DNA adducts and CYP1A1 and NQO1 gene expression (Figures 5(a) and 5(b)). This observation supports the hypothesis that these enzymes are protective against oxidative DNA damage. Our numerous studies in animal models [13–19, 42] have clearly shown the role of both CYP1A1 and NQO1 in the protection against oxidative injury. Our recent study [19] showing the increased susceptibility to hyperoxic lung injury of mice lacking the gene for nrf2, and the rescue of this phenotype by the CYP1A1 inducer β-napthoflavone, lends further credence to the hypothesis that both Nrf2-regulated enzymes (e.g., NQ01) and CYP1A enzymes play a beneficial role in oxygen injury. While CYP1A1 might protect the cells from oxidative stress by metabolizing toxic lipid hydroperoxides [16–20], it is possible that NQO1 in the current study might have protected cells from oxidative stress by metabolizing quinones and semiquniones [21, 22]. The innovative aspect of our current study is that our results show a decrease in the extent of induction of CYP1A1 by hyperoxia in NQO1-NQO1 cells, suggesting a role for NQO1 in the regulation of CYP1A1 expression.
Our results showing the attenuation of 8-OHdG by hyperoxia (Figure 6) in Ctr cells but not in NQO1-NQO1 or SNP cells were in agreement with our studies on bulky oxidative lesions (Figure 4). Although studies reported in the literature show increased levels of OHdG in rat alveolar type II cells exposed to hyperoxia [43], Jin et al. [44] showed that human 8-oxoguanine DNA glycolyase increases resistance to hyperoxic toxicity in alveolar epithelial A549 cells. In our studies, it is possible that hyperoxia in BEAS-2B cells caused a decrease in OHdG levels in part by inducing DNA repair.
Because hyperoxia-mediated induction of DNA repair pathways [45] could in part play a role in the attenuation of oxidative DNA lesions by hyperoxia in Ctr cells (Figures 4 and 6), we determined the effect of hyperoxia on base excision repair (BER) as well as nucleotide excision repair pathways. We studied NEIL2, PARP1, and PCNA as representative of the BER pathway and DDB2, XAB2, and XPC as representative of the NER pathway [46]. While 8-OHdG is repaired by BER [44], the oxidative DNA adducts are repaired by NER mechanisms [36, 47]. Our observations showing a marked induction of DDB2 and XPC by hyperoxia in Ctr cells (Figure 8) supported the idea that hyperoxia in part caused attenuation of bulky oxidative DNA lesions by enhancing NER pathways. The decrease of 8-OHdG by hyperoxia in Ctr cells (Figure 6), but not in NQO1-NQO1 cells or SNP cells, was probably due to significant induction of the proliferating cell nuclear antigen (PCNA), which repairs DNA via BER in Ctr but not in NQO1-NQO1 or SNP cells (Figure 8(d)). Also, the induction of XPC, a NER enzyme, was induced by hyperoxia to a much higher degree in Ctr than NQO1-NQO1 or SNP cells (Figure 8(f)). Thus, we observed a significant modulation of both BER and NER genes by hyperoxia in CMV-NQO1, NQO1-NQO1, and SNP cells.
We did not see a striking difference of DNA repair gene expression among the NQO1-NQO1 and SNP cells, suggesting that the SNP A-1221C did not play a major role in the regulation of DNA repair pathways. Our finding that the protection against hyperoxic toxicity in SNP cells was partially lost in spite of these cells having high NQO1 mRNA (Figure 1(a)) could have been due to the fact that this SNP produced a gene product that had lower NQO1 activity. Previous reports have implicated NQO1 promotor SNPs, specifically the A-1221C SNP, as having a potential protective effect on the severity of acute lung injury in patients suffering from ALI/ARDS [29]. That we did not observe a similar protective effect could have been due to the fact that the current study was in the human BEAS-2B cell line that was exposed to hyperoxia (80% O2 and 5% CO2) for 48 h, and that mechanisms independent of NQO1 may have contributed to the protective effects in humans expressing the SNP A 1221C variant. Future successful creation of in vivo knock-in mouse models that carry the wild-type NQO1 or the A-1221C SNP will help us delineate the mechanistic role of A-1221C SNP in oxygen toxicity in relation to ARDS.
In summary, our data support a protective role for human NQO1 against oxygen-mediated toxicity and oxidative DNA lesions in human pulmonary cells, and this protection is partially lost in cells carrying the A-1221C SNP. Moreover, we also demonstrate a novel protective role for CYP1A1 in the attenuation of oxidative cell and DNA injury. Future studies on the mechanisms of attenuation of oxidative injury by NQO1 should help in developing novel approaches for the prevention/treatment of ARDS in humans.
Acknowledgments
This work was supported in part by USPHS grants 5R01ES009132, R01HL129794, 1R01ES029382, and 1P42 ES0327725 and grants from the Cancer Prevention and Research Institute of Texas (CPRIT) to BM (RP190279) and KL (R01144775-01A1). The metabolomics core was supported by the CPRIT Core Facility Support Award RP170005 “Proteomic and Metabolomic Core Facility,” the NCI Cancer Center Support Grant P30CA125123, intramural funds from the Dan L. Duncan Cancer Center, Baylor College of Medicine, the American Cancer Society (ACS) Award 127430-RSG-15-105-01-CNE (NP), NIH/NCI R01CA220297 (NP), NIH/NCI U01CA214263 (NP), and NIH/NCI R01CA216426 (NP).
Data Availability
The data used to support the findings of this study are available from the corresponding author upon request.
Conflicts of Interest
The authors declare that there is no conflict of interest regarding the publication of this paper.
Authors' Contributions
Rebecca Burke and Chun Chu contributed equally to this work.
Supplementary Materials
Supplementary Materials S1 Hyperoxia increased NQO1 protein expression. BEAS-2B cells stably transfected with pcDNA3.1 (Ctr), pCD-NQO1 (CMV-NQO1), pWT-NQO1-NQO1 (NQO1-NQO1), and pmut-NQO1-NQO1 (SNP) were incubated under room air (RA) or 80% O2 conditions for 48 h and subjected to western blotting using 20 μg total protein of cell lysates per well and 1 : 1000 dilution of A-180 NQO1 antibody (Santa Cruz Biotechnology). (A) Colorimetry image of a representative blot. (B) Densitometry analysis of 2 replicate blots. The result indicated that hyperoxia slightly induced NQO1 protein expression in all 4 cell lines. However, it did not show increased basal NQO1 expression in any of the three NQO1-overexpressed cell lines. S2. CYP1A1 siRNA downregulated CYP1A1 mRNA in both Ctr cells and NQO1-NQO1 cells. Cells were transfected with control siRNA or CYP1A1 siRNA and cultured in RA condition for 48 h. Total RNA were extracted and subjected to qPCR of CYP1A1 and the reference gene OAZ1. The Ct value of CYP1A1 was not detected in one group of sample, probably “Ct value > 39.” (n = 1‐3).
References
- 1.Northway W. H., Jr., Rosan R. C. Radiographic features of pulmonary oxygen toxicity in the newborn: bronchopulmonary dysplasia. Radiology . 1968;91:49–58. doi: 10.1148/91.1.49. [DOI] [PubMed] [Google Scholar]
- 2.Tsai S. H., Anderson W. R., Strickland M. B., Pliego M. Bronchopulmonary dysplasia associated with oxygen therapy in infants with respiratory distress syndrome. Radiology . 1972;105:107–112. doi: 10.1148/105.1.107. [DOI] [PubMed] [Google Scholar]
- 3.Matthay M. A., Zemans R. L., Zimmerman G. A., et al. Acute respiratory distress syndrome. Nature Reviews Disease Primers. . 2019;5:p. 18. doi: 10.1038/s41572-019-0069-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Warner B. B., Stuart L. A., Papes R. A., Wispe J. R. Functional and pathological effects of prolonged hyperoxia in neonatal mice. American Journal of Physiology . 1998;275(1):L110–L117. doi: 10.1152/ajplung.1998.275.1.L110. [DOI] [PubMed] [Google Scholar]
- 5.Vento M., Moro M., Escrig R., et al. Preterm resuscitation with low oxygen causes less oxidative stress, inflammation, and chronic lung disease. Pediatrics . 2009;124(3):e439–e449. doi: 10.1542/peds.2009-0434. [DOI] [PubMed] [Google Scholar]
- 6.Ranieri V. M., Rubenfeld G. D., Thompson B. T. Acute respiratory distress syndrome: the Berlin Definition. Journal of the American Medical Association. . 2012;307:2526–2533. doi: 10.1001/jama.2012.5669. [DOI] [PubMed] [Google Scholar]
- 7.Agarwal R., Aggarwal A. N., Gupta D., Behera D., Jindal S. K. Etiology and outcomes of pulmonary and extrapulmonary acute lung injury/ARDS in a respiratory ICU in North India. Chest . 2006;130:724–729. doi: 10.1378/chest.130.3.724. [DOI] [PubMed] [Google Scholar]
- 8.Cochi S. E., Kempker J. A., Annangi S., Kramer M. R., Martin G. S. Mortality trends of acute respiratory distress syndrome in the United States from 1999 to 2013. Annals of the American Thoracic Society. . 2016;13:1742–1751. doi: 10.1513/AnnalsATS.201512-841OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bellani G., Laffey J. G., Pham T., et al. Epidemiology, patterns of care, and mortality for patients with acute respiratory distress syndrome in intensive care units in 50 countries. Journal of the American Medical Association. . 2016;315:788–800. doi: 10.1001/jama.2016.0291. [DOI] [PubMed] [Google Scholar]
- 10.Ramanathan K., Antognini D., Combes A., et al. Planning and provision of ECMO services for severe ARDS during the COVID-19 pandemic and other outbreaks of emerging infectious diseases. Lancet Respiratory Medicine . 2020;8(5):518–526. doi: 10.1016/S2213-2600(20)30121-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Villar J., Pérez-Méndez L., López J., et al. An early PEEP/FiO2Trial identifies different degrees of lung injury in patients with acute respiratory distress syndrome. American Journal of Respiratory and Critical Care Medicine. . 2007;176(8):795–804. doi: 10.1164/rccm.200610-1534OC. [DOI] [PubMed] [Google Scholar]
- 12.O'Reilly M., Thebaud B. Animal models of bronchopulmonary dysplasia. The term rat models. American Journal of Physiology-Lung Cellular and Molecular Physiology . 2014;307 doi: 10.1152/ajplung.00160.2014. [DOI] [PubMed] [Google Scholar]
- 13.Sinha A., Muthiah K., Jiang W., Couroucli X., Barrios R., Moorthy B. Attenuation of hyperoxic lung injury by the CYP1A inducer beta-naphthoflavone. Toxicological Sciences. . 2005;87(1):204–212. doi: 10.1093/toxsci/kfi226. [DOI] [PubMed] [Google Scholar]
- 14.Moorthy B., Parker K. M., Smith C. V., Bend J. R., Welty S. E. Potentiation of oxygen-induced lung injury in rats by the mechanism-based cytochrome P-450 inhibitor, 1-aminobenzotriazole. Journal of Pharmacology and Experimental Therapeutics . 2000;292 [PubMed] [Google Scholar]
- 15.Jiang W., Welty S. E., Couroucli X. I. Disruption of the Ah receptor gene alters the susceptibility of mice to oxygen-mediated regulation of pulmonary and hepatic cytochromes P4501A expression and exacerbates hyperoxic lung injury. Journal of Pharmacology and Experimental Therapeutics . 2004;310(2):512–519. doi: 10.1124/jpet.103.059766. [DOI] [PubMed] [Google Scholar]
- 16.Lingappan K., Jiang W., Wang L., et al. Mice deficient in the gene for cytochrome P450 (CYP)1A1 are more susceptible than wild-type to hyperoxic lung injury: evidence for protective role of CYP1A1 against oxidative stress. Toxicological Sciences . 2014;141(1):68–77. doi: 10.1093/toxsci/kfu106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang L., Lingappan K., Jiang W., et al. Disruption of cytochrome P4501A2 in mice leads to increased susceptibility to hyperoxic lung injury. Free Radical Biology and Medicine . 2015;82:147–159. doi: 10.1016/j.freeradbiomed.2015.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stading R., Chu C., Couroucli X., Lingappan K., Moorthy B. Molecular role of cytochrome P4501A enzymes in oxidative stress. Current Opinion in Toxicology . 2020;20-21:77–84. doi: 10.1016/j.cotox.2020.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stading R., Couroucli X., Lingappan K., Moorthy B. The role of cytochrome P450 (CYP) enzymes in hyperoxic lung injury. Expert Opinion on Drug Metabolism & Toxicology . 2021;17(2):171–178. doi: 10.1080/17425255.2021.1853705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Callaway D. A., Jiang W., Wang L., Lingappan K., Moorthy B. Oxygen-mediated lung injury in mice lacking the gene for NRF2: Rescue with the cytochrome P4501A-inducer, beta-naphthoflavone (BNF), and differential sex- specific effects. Free Radical Biology and Medicine . 2020;160:208–218. doi: 10.1016/j.freeradbiomed.2020.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Oh E. T., Park H. J. Implications of NQO1 in cancer therapy. BMB Reports . 2015;48(11):609–617. doi: 10.5483/BMBRep.2015.48.11.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Das A., Kole L., Wang L., Barrios R., Moorthy B., Jaiswal A. K. BALT development and augmentation of hyperoxic lung injury in mice deficient in NQO1 and NQO2. Free Radical Biology and Medicine . 2006;40(10):1843–1856. doi: 10.1016/j.freeradbiomed.2006.01.025. [DOI] [PubMed] [Google Scholar]
- 23.Larson R. A., Wang Y., Banerjee M., et al. Prevalence of the inactivating 609C→T polymorphism in the NAD(P)H:Quinone oxidoreductase (NQO1) gene in patients with primary and therapy-related myeloid leukemia. Blood . 1999;94(2):803–807. doi: 10.1182/blood.V94.2.803. [DOI] [PubMed] [Google Scholar]
- 24.Traver R. D., Horikoshi T., Danenberg K. D. NAD(P)H:quinone oxidoreductase gene expression in human colon carcinoma cells: characterization of a mutation which modulates DT-diaphorase activity and mitomycin sensitivity. Cancer Research . 1992;52(4):797–802. [PubMed] [Google Scholar]
- 25.Hong C.-C., Ambrosone C. B., Ahn J., et al. Genetic variability in iron-related oxidative stress pathways (Nrf2, NQ01, NOS3, andHO-1), iron intake, and risk of postmenopausal breast cancer. Cancer Epidemiology, Biomarkers & Prevention. . 2007;16(9):1784–1794. doi: 10.1158/1055-9965.EPI-07-0247. [DOI] [PubMed] [Google Scholar]
- 26.Palming J., Sjoholm K., Jernaas M., et al. The expression of NAD(P)H:quinone oxidoreductase 1 is high in human adipose tissue, reduced by weight loss, and correlates with adiposity, insulin sensitivity, and markers of liver dysfunction. Journal of Clinical Endocrinology and Metabolism. . 2007;92(6):2346–2352. doi: 10.1210/jc.2006-2476. [DOI] [PubMed] [Google Scholar]
- 27.Sarbia M., Bitzer M., Siegel D., et al. Association betweenNAD(P)H: Quinone oxidoreductase 1 (NQ01) inactivating C609T polymorphism and adenocarcinoma of the upper gastrointestinal tract. International Journal of Cancer. . 2003;107(3):381–386. doi: 10.1002/ijc.11430. [DOI] [PubMed] [Google Scholar]
- 28.Martin N. J., Collier A. C., Bowen L. D., et al. Polymorphisms in the NQO1, GSTT and GSTM genes are associated with coronary heart disease and biomarkers of oxidative stress. Mutation Research . 2009;674(1-2):93–100. doi: 10.1016/j.mrgentox.2008.09.009. [DOI] [PubMed] [Google Scholar]
- 29.Reddy A. J., Christie J. D., Aplenc R., Fuchs B., Lanken P. N., Kleeberger S. R. Association of human NAD(P)H:quinone oxidoreductase 1 (NQO1) polymorphism with development of acute lung injury. Journal of Cellular and Molecular Medicine. . 2009;13(8b):1784–1791. doi: 10.1111/j.1582-4934.2008.00581.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tsvetkov P., Asher G., Reiss V., Shaul Y., Sachs L., Lotem J. Inhibition of NAD(P)H:quinone oxidoreductase 1 activity and induction of p53 degradation by the natural phenolic compound curcumin. Proceedings of the National Academy of Sciences of the United States of America. . 2005;102(15):5535–5540. doi: 10.1073/pnas.0501828102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dinu D., Chu C., Veith A., et al. Mechanistic role of cytochrome P450 (CYP)1B1 in oxygen-mediated toxicity in pulmonary cells: a novel target for prevention of hyperoxic lung injury. Biochemical and Biophysical Research Communications. . 2016;476(4):346–351. doi: 10.1016/j.bbrc.2016.05.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shivanna B., Chu C., Welty S. E., et al. Omeprazole attenuates hyperoxic injury in H441 cells via the aryl hydrocarbon receptor. Free Radical Biology and Medicine. . 2011;51(10):1910–1917. doi: 10.1016/j.freeradbiomed.2011.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Reddy M. V., Randerath K. Nuclease P1-mediated enhancement of sensitivity of 32P-postlabeling test for structurally diverse DNA adducts. Carcinogenesis . 1986;7(9):1543–1551. doi: 10.1093/carcin/7.9.1543. [DOI] [PubMed] [Google Scholar]
- 34.Veith A. C., Aram B. B., Jiang W., et al. Mice lacking the cytochrome P450 1B1 gene are less susceptible to hyperoxic lung injury than wild type. Toxicological Sciences. . 2018;165(2):462–474. doi: 10.1093/toxsci/kfy154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhou G. D., Moorthy B. Detection of bulky endogenous oxidative DNA lesions derived from 8,5’‐Cyclo‐2’‐deoxyadenosine by32P‐Postlabeling assay. Current Protocols in Toxicology. . 2015;64(1) doi: 10.1002/0471140856.tx1717s64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Randerath K., Zhou G. D., Somers R. L., Robbins J. H., Brooks P. J. A 32P-postlabeling assay for the oxidative DNA lesion 8,5′-cyclo-2′-deoxyadenosine in mammalian tissues: evidence that four type II I-compounds are dinucleotides containing the lesion in the 3′ nucleotide. Journal of Biological Chemistry. . 2001;276(38):36051–36057. doi: 10.1074/jbc.M105472200. [DOI] [PubMed] [Google Scholar]
- 37.Vantaku V., Amara C. S., Piyarathna D. W. B., et al. DNA methylation patterns in bladder tumors of African American patients point to distinct alterations in xenobiotic metabolism. Carcinogenesis . 2019;40(11):1332–1340. doi: 10.1093/carcin/bgz128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Black C. N., Bot M., Scheffer P. G., Penninx B. W. Sociodemographic and lifestyle determinants of plasma oxidative stress markers 8-OHdG and F2-isoprostanes and associations with metabolic syndrome. Oxidative Medicine and Cellular Longevity. . 2016;2016, article 7530820 doi: 10.1155/2016/7530820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pietrofesa R., Velalopoulou A., Lehman S., et al. Novel double-hit model of radiation and hyperoxia-induced oxidative cell damage relevant to space travel. International Journal of Molecular Sciences . 2016;17(6) doi: 10.3390/ijms17060953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bhakta K. Y., Jiang W., Couroucli X. I., Fazili I. S., Muthiah K., Moorthy B. Regulation of cytochrome P4501A1 expression by hyperoxia in human lung cell lines: implications for hyperoxic lung injury. Toxicology and Applied Pharmacology. . 2008;233(2):169–178. doi: 10.1016/j.taap.2008.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kaiser H., Yu K., Pandya C., et al. Kynurenine, a tryptophan metabolite that increases with age, induces muscle atrophy and lipid peroxidation. Oxidative Medicine and Cellular Longevity. . 2019;2019, article 9894238 doi: 10.1155/2019/9894238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Maturu P., Wei-Liang Y., Androutsopoulos V. P., et al. Quercetin attenuates the hyperoxic lung injury in neonatal mice: implications for bronchopulmonary dysplasia (BPD) Food and Chemical Toxicology. . 2018;114:23–33. doi: 10.1016/j.fct.2018.02.026. [DOI] [PubMed] [Google Scholar]
- 43.Kannan S., Pang H., Foster D. C., Rao Z., Wu M. Human 8-oxoguanine DNA glycosylase increases resistance to hyperoxic cytotoxicity in lung epithelial cells and involvement with altered MAPK activity. Cell Death and Differentiation . 2006;13(2):311–323. doi: 10.1038/sj.cdd.4401736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jin L., Yang H., Fu J., Xue X., Yao L., Qiao L. Association between oxidative DNA damage and the expression of 8-oxoguanine DNA glycosylase 1 in lung epithelial cells of neonatal rats exposed to hyperoxia. Molecular Medicine Reports . 2015;11(6):4079–4086. doi: 10.3892/mmr.2015.3339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.O’Reilly M. A. DNA damage and cell cycle checkpoints in hyperoxic lung injury: braking to facilitate repair. American Journal of Physiology-Lung Cellular and Molecular Physiology. . 2001;281:L291–L305. doi: 10.1152/ajplung.2001.281.2.L291. [DOI] [PubMed] [Google Scholar]
- 46.Chatterjee N., Walker G. C. Mechanisms of DNA damage, repair, and mutagenesis. Environmental and Molecular Mutagenesis. . 2017;58(5):235–263. doi: 10.1002/em.22087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brooks P. J., Wise D. S., Berry D. A., et al. The oxidative DNA lesion 8,5′-(S)-cyclo-2′-deoxyadenosine is repaired by the nucleotide excision repair pathway and blocks gene expression in mammalian cells. Journal of Biological Chemistry. . 2000;275(29):22355–22362. doi: 10.1074/jbc.M002259200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Materials S1 Hyperoxia increased NQO1 protein expression. BEAS-2B cells stably transfected with pcDNA3.1 (Ctr), pCD-NQO1 (CMV-NQO1), pWT-NQO1-NQO1 (NQO1-NQO1), and pmut-NQO1-NQO1 (SNP) were incubated under room air (RA) or 80% O2 conditions for 48 h and subjected to western blotting using 20 μg total protein of cell lysates per well and 1 : 1000 dilution of A-180 NQO1 antibody (Santa Cruz Biotechnology). (A) Colorimetry image of a representative blot. (B) Densitometry analysis of 2 replicate blots. The result indicated that hyperoxia slightly induced NQO1 protein expression in all 4 cell lines. However, it did not show increased basal NQO1 expression in any of the three NQO1-overexpressed cell lines. S2. CYP1A1 siRNA downregulated CYP1A1 mRNA in both Ctr cells and NQO1-NQO1 cells. Cells were transfected with control siRNA or CYP1A1 siRNA and cultured in RA condition for 48 h. Total RNA were extracted and subjected to qPCR of CYP1A1 and the reference gene OAZ1. The Ct value of CYP1A1 was not detected in one group of sample, probably “Ct value > 39.” (n = 1‐3).
Data Availability Statement
The data used to support the findings of this study are available from the corresponding author upon request.