

Abstract
The SARS-CoV-2 spike protein has been shown to disrupt blood–brain barrier (BBB) function, but its pathogenic mechanism of action is unknown. Whether angiotensin converting enzyme 2 (ACE2), the viral binding site for SARS-CoV-2, contributes to the spike protein-induced barrier disruption also remains unclear. Here, a 3D-BBB microfluidic model was used to interrogate mechanisms by which the spike protein may facilitate barrier dysfunction. The spike protein upregulated the expression of ACE2 in response to laminar shear stress. Moreover, interrogating the role of ACE2 showed that knock-down affected endothelial barrier properties. These results identify a possible role of ACE2 in barrier homeostasis. Analysis of RhoA, a key molecule in regulating endothelial cytoskeleton and tight junction complex dynamics, reveals that the spike protein triggers RhoA activation. Inhibition of RhoA with C3 transferase rescues its effect on tight junction disassembly. Overall, these results indicate a possible means by which the engagement of SARS-CoV-2 with ACE2 facilitates disruption of the BBB via RhoA activation. Understanding how SARS-CoV-2 dysregulates the BBB may lead to strategies to prevent the neurological deficits seen in COVID-19 patients.
Graphic Abstract
Supplementary Information
The online version contains supplementary material available at 10.1007/s11481-021-10029-0.
Keywords: Blood–brain barrier, SARS-CoV-2, RhoA, Mechanotransduction, Fluid shear stress
Introduction
The emergence of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the virus that causes coronavirus disease 2019 (COVID-19), has caused significant morbidity and mortality at a global scale (Nalbandian et al. 2021). Although widely considered a respiratory disease, studies have reported detrimental effects of the virus on multiple organ systems including the central nervous system (CNS) (Chaves Filho et al. 2021; Nalbandian et al. 2021; Qin et al. 2021). Delirium has become a common symptom among older adults with COVID-19 infection, presenting in 20–30% of patients with rates as high as 70% among severe cases (O'Hanlon and Inouye 2020; Chaves Filho et al. 2021). Delirium is characterized by sudden onset of dysphoria and disturbed attention in patients with no history of neurological complications (Acharya et al. 2015; Chaves Filho et al. 2021). Since previous studies have correlated delirium with breakdown of the blood–brain barrier (BBB) (Acharya et al. 2015), these clinical outcomes suggest that the SARS-CoV-2 is either directly or indirectly disrupting the BBB.
Cellular infection by SARS-CoV-2 is mediated by its viral spike protein binding to angiotensin-converting enzyme 2 (ACE2), which is expressed throughout the endothelium in the body including vascular beds of the kidneys, lungs, and heart (Xia and Lazartigues 2008; Chaves Filho et al. 2021). Studies have also confirmed the presence of ACE2 in cerebral vasculature, supporting clinical evidence of neuroinvasion (Buzhdygan et al. 2020; Reynolds and Mahajan 2021). ACE2 is a cell surface carboxypeptidase that converts angiotensin II (Ang-II) to angiotensin fragment 1–7(Ang 1–7) as part of the renin-angiotensin system (RAS), promoting vasodilation and decreased hypertension (Santos 2014). Several studies have posited that SARS-Cov-2 exacerbates injury in lungs and other tissues by inhibiting or downregulating ACE2 (Chaudhry et al. 2020; Ni et al. 2020). However, studies have also found that comorbidities of COVID-19 including hypertension and diabetes are associated with increased ACE2 expression in lung tissue (Pinto et al. 2020). Furthermore, a previous study found that application of shear stress to vascular endothelial cells, which is known to augment vascular integrity, transiently increased ACE2 expression (Song et al. 2020). Therefore, clarification is needed to understand the role of ACE2 expression in mediating spike-mediated breakdown of the BBB.
In the present study, a three-dimensional model of the BBB (Partyka et al. 2017) was utilized to examine the role of ACE2 and SARS-CoV-2 spike protein on BBB homeostasis. The model employs human cerebral microvascular endothelial cells (HCMEC/d3) (Weksler et al. 2005) in combination with fluid shear stress in a collagen/HA hydrogel matrix in order to mimic the human BBB. This platform enables the real time assessment of BBB function in response to spike protein such as permeability and transendothelial electrical resistance (TEER) measurements. Here, evidence suggests that barrier breach in response to SARS-CoV-2 spike protein is RhoA-mediated and inhibition of RhoA can mitigate BBB hyperpermeability.
Methods
In Vitro BBB Models
Three-dimensional models of the BBB (3D BBB) were fabricated as described previously (Partyka et al. 2017). Hydrogels composed of 5 mg/mL type I collagen, 1 mg/mL HA(> 1MDa), 1 mg/mL Matrigel were created within microfluidic devices. Prior to polymerization, 180-µm needles were removed to create voids, and HCMEC/D3 (P22-P23) (provided by the laboratory of Dr. Robert Nagele) were injected at a density of 10 million per mL (10 µL per channel). Following cell seeding, channels were either exposed to flow using a linear syringe pump (Kent Scientific) or incubated in static conditions in a 6-well plate. For GTPase activity assays, a larger diameter (1-mm) model was used to generate sufficient protein quantities (Bouhrira et al. 2019). For ACE2 protein expression assays, the same hydrogel formulation was polymerized on treated PDMS coated plates, and HCMEC/D3 were seeded on the gel at a density of 4 k/cm2. Monolayers were incubated for 3 days in static culture to ensure confluency, then exposed to fluid shear stress. Fluid shear stress was applied using a 40-mm 1-degree cone plate on a rheometer (TA Instruments) for 24-h on a Peltier plate set to 37 °C. The media was supplemented with HEPES buffer to a final concentration of 10 mM to maintain physiological pH.
Permeability Assessment
After exposure to experimental conditions, channels were perfused with 4-kDa dextran-FITC at a flow rate of 5 µL/min using a syringe pump for 10 min. This flow rate was selected to assure fully developed flow throughout the majority of the channel and to maintain consistency with previous work (Partyka et al. 2017). Images were taken at 30-s intervals for 10 min, and the diffusion coefficients were established using the following equation from previous work (Adamson et al. 1994).
| 1 |
Sample numbers of at least n = 3 were used to determine the mean and standard deviation for each condition.
Transendothelial Electrical Resistance (TEER) Measurements
TEER measurements by impedance spectroscopy were obtained using a stingray DS1M12 USB oscilloscope adapter (USB Instruments) that measured current across a reference resistor for a range of frequencies. Impendence, defined as Z = V/I, was measured at 15-Hz, where capacitance of the electrodes dominates and 15.6-kHz, where resistance of the culture media dominates (Benson et al. 2013); the difference between these two values yielded the TEER measurement. These values were normalized using the impedance of an acellular hydrogel within the microfluidic device.
SARS-CoV-2 Subunit S1 Exposure
Following 4 days of perfusion, vessels were exposed to EGM-2 supplemented with 50 nM recombinant SARS-CoV-2 subunit S1 (RayBiotech) for three hours. The S1 subunit of the spike protein is used because it contains the receptor binding domain that interacts with ACE2, in contrast with the S2 subunit that mediates membrane fusion of the viral particles to the host cells. In some cases Rho Inhibitor I (C3 Transferase) (Cytoskeleton) was added in addition to the S1 protein. TEER measurements were taken at 0-h, 1.5-h, 3-h with dextran permeability measurements taken at 0-h and 3-h time points.
Immunocytochemistry
Vessels were fixed in 4% paraformaldehyde (Alfa Aesar) for 30 min at room temperature. Following fixation, the hydrogel was removed from the device and placed in 0.1% Triton X-100 to permeabilize the cell membrane. Gels were blocked in 5% normal donkey serum and 3% BSA for 30 min at room temperature followed by incubating overnight with primary antibodies for either angiotensin converting enzyme II (ACE2) (Santa Cruz) or zonula occludin-1 (ZO-1)(CST), a tight-junction scaffolding protein (Dejana 2004). Following primary incubation, gels were washed with PBS then incubated with the appropriate secondary antibody conjugated to Alexa 555 (CST) or Dylight 650 (Thermo Scientific). Gels were counter-stained with DAPI to label nuclei and FITC-phalloidin for actin. All gels were imaged using a Nikon A-1 confocal scanning microscope.
ACE2 Knockdown
SMARTpool siRNA (Horizon Discovery) was used to knockdown ACE2 expression in HCMEC/D3 cells. Cells were plated in a 6-well plate at 250 k per well. When cells reached 60–70% confluency, siRNA was added according to the manufacturer’s protocol. Knockdown efficiency was verified using western blotting after the cells were cultured in a well plate for 96 h, since this period matched the timing of permeability testing in the 3D-BBB model.
ELISA-based Quantification of Small GTPase Activity
Commercial ELISA kits were purchased from Cytoskeleton (G-LISA) to quantify RhoA activation (Cytoskeleton). Cell lysates were prepared using Cytoskeleton’s protocols. Following exposure to flow with and without S1 peptide (Ray Biotech), the hydrogel was removed from the device and washed in ice cold 1 × PBS for 30 s. 80 µL of ice-cold cell lysis buffer with 1 × protease inhibitor was injected into vessels and collected in a microcentrifuge tube. The cell lysate solution was spun at 10,000 g for 1 min at 4 °C to pellet cell debris. Prior to measuring GTPase activity, samples were thawed in a room temperature water bath and equilibrated to 0.5 mg/mL.
ACE2 Expression
Quantification of ACE2 expression was performed using lysates extracted from cell monolayers cultured on hydrogels exposed to 24 h of fluid shear stress applied by a cone and plate rheometer. 50 mM SARS-CoV-1 subunit S1 was added during the final 3 h of flow. Cell lysates were separated using a tris–glycine gel (Invitrogen) in tris–glycine running buffer (Invitrogen). Separated protein was transferred to 0.45-μm PVDF membrane (Invitrogen). The membrane was probed over night with ACE2 (Santa Cruz Biotech) (1:250) and beta-Actin (1:400) (Santa Cruz Biotech) antibodies and visualized with anti-mouse HRP conjugated secondary antibodies (1:4000) (Azure).
Statistical Analysis
The open-source statistics package, R, was used to perform all statistical calculations. Data sets were tested for normality with Shapiro–Wilk tests prior to testing for significance. One and two-way ANOVA tests followed by Tukey HSD post-hoc comparisons were used to evaluate significant differences between multiple conditions. All two sample comparisons were made using 2-tailed Student’s t-tests. Each statistical test used sample numbers greater than or equal to 3 unless otherwise noted and p < 0.05 was considered significant. All error bars indicate standard deviation of the mean.
Results
Fluid Shear Stress and SARS-CoV-2 Alter ACE2 Expression
Previous results have demonstrated that ACE2, the main viral binding target of the SARS-CoV-2 spike protein, is expressed by endothelial cells in the brain (Buzhdygan et al. 2020). In order to demonstrate that the enzyme is also expressed by HCMEC/D3 cells in our 3D BBB model, microvessels were cultured for 96 h under constant perfusion with 0.7 dyn/cm2 shear stress to establish barrier function. During the final 3 h, vessels were exposed to three different conditions: (i) flow cessation (static conditions) (ii) no change in flow (continuation of flow with a shear stress of 0.7 dyn/cm2) or (iii) continuation of flow with the addition of 50 nM SARS-CoV-2 spike S1 subunit. ACE2 staining was present in the vessels in all of the conditions (Fig. 1A−C). These images show the effect of shear stress and S1 spike protein on the morphology of the endothelial cells lining the in vitro vessel and localization of ZO-1 to the cell–cell junctions. After 4 days of perfusion, vessels exposed to 3 h of static conditions (Fig. 1A) exhibited a substantial amount of perinuclear ZO-1 staining, with irregular localization to the junctions. In contrast, the vessel exposed to 0.7 dyn/cm2, indicated clear localization of ZO-1 to the cell–cell junctions without any gaps in the endothelial monolayer (Fig. 1B). Addition of the spike protein disrupted the monolayer and localization of ZO-1 at the cell–cell junctions in a manner similar to static culture (Fig. 1C). Due to the limited number of cells in the 3D microvessel model, we utilized a 2D rheological model for quantification of ACE2 protein levels, which maintained the extracellular matrix properties of the 3D configuration (Fig. 1D). Endothelial cells were exposed to 2.1 dyn/cm2 of shear stress in a 2D geometrical configuration for 24-h, after three days of culturing to allow the monolayer to reach confluency. The relative quantity of ACE2 was normalized to the static condition. Exposure to shear stress significantly decreased the level of ACE2 expression in the monolayers compared to static conditions by approximately 60%. However, addition of the spike protein under flow resulted in the upregulated expression of ACE2, so that there was no significant difference with static conditions (Fig. 1E). Exposing static cultures to the spike protein indicated no significant increase in ACE2 expression compared to controls (Supplemental Fig. 1). Therefore, the spike protein does not increase ACE2 expression in endothelial cells cultured in static culture, but it does so in the presence of fluid shear stress.
Fig. 1.

A−C Fluorescent images of 3D vessels stained with DAPI (blue), Phalloidin (green), ZO-1( red) (isolated in ii) and ACE2 (magenta)(isolated in iii) for 3 conditions: Static (A), Flow (B), (C) Flow + S1 subunit exposure for 3 h. D Schematic of rheometer setup used to apply fluid shear stress to 2D cultures for protein expression assays. E Western blot of cells exposure to static, flow, or flow + S1 protein (3 h) (i), and ratio of ACE2 to beta-actin normalized to a flow sample (ii). * Indicates p < 0.05 compared to flow. n = 3 for all experiments
ACE2 Expression Contributes to Shear Stress-mediated BBB Formation
Previous studies have shown that SARS-CoV-2 spike protein promotes endothelial dysfunction when bound to ACE2 (Kumar et al. 2020). Therefore, ACE2 knockdown was conducted to determine whether its expression has any observable effect on barrier integrity. siRNA was used to achieve 40–50% reduction in protein expression in the HCMEC/D3 cells (Fig. 2Ai). Knockdown efficiency was measured 5 days after transfection to match the timing of the permeability tests in the 3D model (Fig. 2Aii). Analysis of permeability containing the knockdown for ACE2 revealed significantly higher permeability compared to the scrambled controls (Fig. 2B). The higher permeability may be caused by disassembly of the tight junction complex and subsequently increased paracellular transport. Immunofluorescence images confirmed altered morphology in the presentation of ZO-1 in cells with attenuated ACE2 expression. Specifically, ZO-1 expression lacked the typical membrane-like expression patterned and instead showed a more diffused and displaced cytosolic expression (Fig. 2C). These results indicate that ACE2 expression contributes to the integrity of the tight junction complex in response to fluid shear stress.
Fig. 2.

A Western blot of cells treated with DsiRNA targeting ACE2 (i), and relative intensity of ACE2 in DsiRNA and control cells normalized to beta-actin (ii). B Permeability coefficients measured in channels seeded with ACE2-Knockdown (KD) HCMEC/D3 cells. *indicates p < 0.05 compared to control. n = 3 for all experiments. C−D Fluorescent images of vessels stained with DAPI (blue), Phalloidin (green), ZO-1( magenta) (isolated in ii)for 3 conditions: siRNA control (C), and siRNA ACE2-KD (D)
SARS-CoV-2 Spike Protein Disrupts the Barrier through RhoA
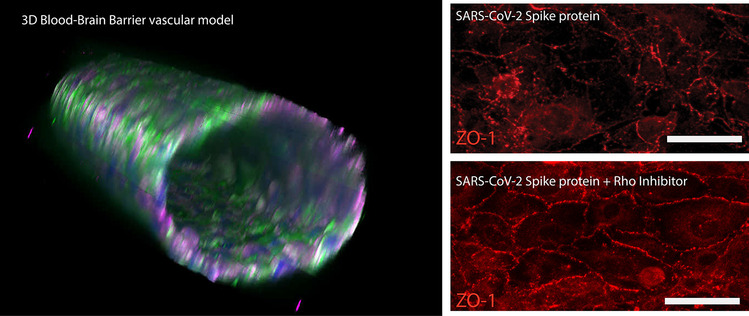
Multiple studies have demonstrated that activation of RhoA disrupts vascular integrity (Yamamoto et al. 2008; Mikelis et al. 2015). Therefore, we evaluated whether the spike protein could trigger signaling events that activate RhoA. Using ELISAs that measure the active form of the RhoA-GTPase, endothelial cells were incubated with and without the spike S1 subunit. Figure 3A shows that 24 h post-perfusion with the spike protein significantly increased RhoA activity compared to no S1 flow only controls (Fig. 3A). Consequently, measurements of dextran permeability (Fig. 3B) and TEER (Fig. 3C) show that inhibition of RhoA with C3 transferase while in the presence of S1 mitigated barrier breach. A 3-h period was used for the evaluation of permeability and both 1.5-h and 3-h time points were used for the TEER measurements. Immunofluorescence verified that RhoA inhibition increased compartmentalization of ZO-1 to the cell–cell junctions in the presence of the spike protein (Fig. 3D). Together these results show that RhoA is activated by the SARS-CoV-2 spike protein S1 subunit leading to a compromise in barrier integrity.
Fig. 3.

A Relative intensity (RQ) of RhoA activation in channels exposed to flow measured with ELISA. B Permeability measurement of vessels exposed to experimental conditions at 0-h and 3-h time points. *Indicates p < 0.05 compared to all conditions. C TEER values of vessels exposed to experimental conditions at 0-h,1.5 h and 3-h time points. * Indicates p < 0.05 compared to all 0-h and 1.5-h time points and ** indicates p < 0.05 compared all other conditions. n = 3 for all experiments. D Fluorescent images of vessels treated with C3 Transferase and S1 spike protein stained with DAPI (blue), Phalloidin (green), ACE2 (magenta), and ZO-1 ( red) (isolated in ii)
Discussion
The results presented here provide preliminary insight into the mechanisms underlying the effect of the SARS-CoV-2 spike protein on barrier integrity and identify RhoA as a key mediator of spike-induced barrier disruption. We first examined the main cellular binding target for the spike protein, ACE2. Application of fluid shear stress, which is needed for barrier formation and constitutes a crucial physiological parameter for the maturity of endothelial cells, results in lower levels of ACE2 expression compared to static conditions. However, when brain endothelial cells are exposed to the S1 subunit of the spike protein in the presence of fluid shear stress, expression of ACE2 is upregulated to static levels. This result contrasts with a previous finding that fluid shear stress transiently increases ACE2 expression, though that specific study was performed with non-brain endothelial cells on tissue culture plastic (Song et al. 2020). It is notable that the spike protein only increased ACE2 expression in endothelial cells exposed to fluid shear stress, given the uncertainty regarding whether ACE2 levels increase or decrease during COVID-19 infection (Chaudhry et al. 2020; Pinto et al. 2020). Further studies are required to elucidate the molecular mechanisms underlying the interaction between the spike protein and fluid shear stress and their combined effects on ACE2 expression. Our results also showed for the first time a possible link between homeostatic barrier maintenance and the ACE2 protein. Indeed, the tightness of the barrier appears to be greatly impacted in experiments in which the expression of the ACE2 gene is knocked down (Fig. 2). This result may help to reconcile observations of the protective effects of ACE2-containing exosomes in the aftermath of ischemia/reoxygenation injury (Zhang et al. 2018). Thus, our analysis merits a closer look at a direct role for ACE2 in maintaining BBB tightness.
ELISA assays indicated that S1 spike protein significantly increased the activation of RhoA, demonstrating that the small GTPase influences barrier breakdown in response to SARS-CoV-2. The activation of RhoA has been shown to induce cell contractility and cytoskeleton restructuring, resulting in enhanced cell motility and disrupted barrier integrity (Shaw et al. 1998; Mikelis et al. 2015). This conclusion is supported by permeability and TEER testing showing that the detrimental effects of S1 spike protein on the BBB are ablated by inhibiting RhoA activation (Fig. 3). Given that RhoA activates the Rho kinase (ROCK), it is noteworthy that the therapeutic effects of ROCK inhibition have already been considered for the treatment of severe COVID-19 (Abedi et al. 2020b). In fact, preclinical studies have shown the benefits of inhibiting the Rho-ROCK pathway to improve pulmonary outcomes (Xu et al. 2019; Abedi et al. 2020a). Additionally, ROCK inhibitors have been shown to protect pulmonary tissue during severe respiratory illness (Abedi et al. 2020a, b). The effects of ROCK inhibition seems to also confer vascular protection by showing improved neurological outcomes following ischemic stroke (Shibuya et al. 2005). Perhaps a similar therapeutic approach could be developed to prevent neurological deficits associated with COVID-19.
Additional studies are required to further probe the underlying mechanisms of spike protein-mediated increase in ACE2 expression and RhoA activity. It has been shown previously that binding of angiotensin II to its receptor, angiotensin II type 1 receptor, increases RhoA activity (Kimura and Eguchi 2009). Therefore, if ACE2 is inhibited by the spike protein of SARS-CoV-2, less angiotensin II can be converted to angiotensin fragment 1–7(Ang 1–7) and can potentially accumulate to increase RhoA activity. However, in the context of these in vitro studies, it is unclear how much angiotensin II is present within the fetal bovine serum used to supplement the culture medium used in these experiments. Moreover, these studies use only the subunit of the spike protein containing the receptor binding domain, so it is unclear how interaction between ACE2 and the full virus may impact the results. Spike protein binding to ACE2 may also directly activate signaling pathways through the cytoplasmic tail of ACE2, given that previous studies have found binding sites in this region for calmodulin, an important mediator of cytoskeletal processes (Lambert et al. 2008). Therefore, future studies can be conducted in the presence of physiological levels of angiotensin II to more completely elucidate how the interplay between the SARS-CoV-2 spike protein, ACE2, and RhoA activation leads to barrier disruption. Additionally, future work in this system can also determine whether variants of the SARS-CoV-2 virus alter the RhoA activation observed here.
Supplementary Information
Below is the link to the electronic supplementary material.
Author Contributions
B.J.D. and P.A.G conceived and planned experiments. B.J.D and P.A.G conducted experiments and analyzed data. P.A.G provided reagents and supplies. B.J.D., K.A.T., A.M.A, S.H.R., and P.A.G. prepared figures and wrote the manuscript.
Funding
This work was supported by a National Science Foundation GRFP fellowship (2019287762) awarded to B.J.D, and National Science Foundation grant to P.A.G. (2034780).
Availability of Data
The datasets used and/or analyzed during the current study are available from the corresponding author upon request.
Declarations
Conflict of Interest
The authors report no conflicts of interest.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- Abedi F, Hayes AW, Reiter R, Karimi G (2020a) Acute lung injury: The therapeutic role of Rho kinase inhibitors. Pharmacol Res 155:104736 [DOI] [PubMed]
- Abedi F, Rezaee R, Karimi G (2020b) Plausibility of therapeutic effects of Rho kinase inhibitors against Severe Acute Respiratory Syndrome Coronavirus 2 (COVID-19). Pharmacol Res 156:104808 [DOI] [PMC free article] [PubMed]
- Acharya NK, Goldwaser EL, Forsberg MM, Godsey GA, Johnson CA, Sarkar A, et al. Sevoflurane and Isoflurane induce structural changes in brain vascular endothelial cells and increase blood-brain barrier permeability: Possible link to postoperative delirium and cognitive decline. Brain Res. 2015;1620:29–41. doi: 10.1016/j.brainres.2015.04.054. [DOI] [PubMed] [Google Scholar]
- Adamson RH, Lenz JF, Curry FE. Quantitative laser scanning confocal microscopy on single capillaries: permeability measurement. Microcirculation. 1994;1(4):251–265. doi: 10.3109/10739689409146752. [DOI] [PubMed] [Google Scholar]
- Benson K, Cramer S, Galla HJ. Impedance-based cell monitoring: barrier properties and beyond. Fluids Barriers CNS. 2013;10(1):5. doi: 10.1186/2045-8118-10-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouhrira N, Deore BJ, Sazer DW, Chiaradia Z, Miller JS, Galie PA (2019) Disturbed flow disrupts the blood-brain barrier in a 3D bifurcation model. Biofabrication [DOI] [PubMed]
- Buzhdygan TP, Deore BJ, Baldwin-Leclair A, Bullock TA, Mcgary HM, Khan JA et al (2020) The SARS-CoV-2 spike protein alters barrier function in 2D static and 3D microfluidic in-vitro models of the human blood-brain barrier. Neurobiol Dis 146:105131 [DOI] [PMC free article] [PubMed]
- Chaudhry F, Lavandero S, Xie X, Sabharwal B, Zheng YY, Correa A et al (2020) Manipulation of ACE2 expression in COVID-19. Open Heart 7(2) [DOI] [PMC free article] [PubMed]
- Chaves Filho AJM, Gonçalves F, Mottin M, Andrade CH, Fonseca SNS, Macedo DS. Repurposing of Tetracyclines for COVID-19 Neurological and Neuropsychiatric Manifestations: A Valid Option to Control SARS-CoV-2-Associated Neuroinflammation?. J Neuroimmune Pharmacol. 2021;16(2):213–218. doi: 10.1007/s11481-021-09986-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dejana E. Endothelial cell-cell junctions: happy together. Nat Rev Mol Cell Biol. 2004;5(4):261–270. doi: 10.1038/nrm1357. [DOI] [PubMed] [Google Scholar]
- Kimura K, Eguchi S (2009) Angiotensin II type-1 receptor regulates RhoA and Rho-kinase/ROCK activation via multiple mechanisms. Focus on "Angiotensin II induces RhoA activation through SHP2-dependent dephosphorylation of the RhoGAP p190A in vascular smooth muscle cells". Am J Physiol Cell Physiol 297(5):C1059–1061 [DOI] [PMC free article] [PubMed]
- Kumar A, Narayan RK, Kumari C, Faiq MA, Kulandhasamy M, Kant K et al (2020) SARS-CoV-2 cell entry receptor ACE2 mediated endothelial dysfunction leads to vascular thrombosis in COVID-19 patients. Med Hypotheses 145:110320 [DOI] [PMC free article] [PubMed]
- Lambert DW, Clarke NE, Hooper NM, Turner AJ. Calmodulin interacts with angiotensin-converting enzyme-2 (ACE2) and inhibits shedding of its ectodomain. FEBS Lett. 2008;582(2):385–390. doi: 10.1016/j.febslet.2007.11.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikelis CM, Simaan M, Ando K, Fukuhara S, Sakurai A, Amornphimoltham P, et al. RhoA and ROCK mediate histamine-induced vascular leakage and anaphylactic shock. Nat Commun. 2015;6:6725. doi: 10.1038/ncomms7725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalbandian A, Sehgal K, Gupta A, Madhavan MV, Mcgroder C, Stevens JS, et al. Post-acute COVID-19 syndrome. Nat Med. 2021;27(4):601–615. doi: 10.1038/s41591-021-01283-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni W, Yang X, Yang D, Bao J, Li R, Xiao Y, et al. Role of angiotensin-converting enzyme 2 (ACE2) in COVID-19. Crit Care. 2020;24(1):422. doi: 10.1186/s13054-020-03120-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'hanlon S, Inouye SK. Delirium: a missing piece in the COVID-19 pandemic puzzle. Age Ageing. 2020;49(4):497–498. doi: 10.1093/ageing/afaa094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partyka PP, Godsey GA, Galie JR, Kosciuk MC, Acharya NK, Nagele RG, et al. Mechanical stress regulates transport in a compliant 3D model of the blood-brain barrier. Biomaterials. 2017;115:30–39. doi: 10.1016/j.biomaterials.2016.11.012. [DOI] [PubMed] [Google Scholar]
- Pinto BGG, Oliveira AER, Singh Y, Jimenez L, Goncalves ANA, Ogava RLT, et al. ACE2 Expression Is Increased in the Lungs of Patients With Comorbidities Associated With Severe COVID-19. J Infect Dis. 2020;222(4):556–563. doi: 10.1093/infdis/jiaa332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin Y, Wu J, Chen T, Li J, Zhang G, Wu D et al (2021) Long-term microstructure and cerebral blood flow changes in patients recovered from COVID-19 without neurological manifestations. J Clin Investig 131(8) [DOI] [PMC free article] [PubMed]
- Reynolds JL, Mahajan SD. SARS-COV2 Alters Blood Brain Barrier Integrity Contributing to Neuro-Inflammation. J Neuroimmune Pharmacol. 2021;16(1):4–6. doi: 10.1007/s11481-020-09975-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos RA. Angiotensin-(1–7) Hypertension. 2014;63(6):1138–1147. doi: 10.1161/HYPERTENSIONAHA.113.01274. [DOI] [PubMed] [Google Scholar]
- Shaw RJ, Henry M, Solomon F, Jacks T. RhoA-dependent phosphorylation and relocalization of ERM proteins into apical membrane/actin protrusions in fibroblasts. Mol Biol Cell. 1998;9(2):403–419. doi: 10.1091/mbc.9.2.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibuya M, Hirai S, Seto M, Satoh S-I, Ohtomo E. Effects of fasudil in acute ischemic stroke: Results of a prospective placebo-controlled double-blind trial. J Neurol Sci. 2005;238(1):31–39. doi: 10.1016/j.jns.2005.06.003. [DOI] [PubMed] [Google Scholar]
- Song J, Hu B, Qu H, Wang L, Huang X, Li M, et al. Upregulation of angiotensin converting enzyme 2 by shear stress reduced inflammation and proliferation in vascular endothelial cells. Biochem Biophys Res Commun. 2020;525(3):812–818. doi: 10.1016/j.bbrc.2020.02.151. [DOI] [PubMed] [Google Scholar]
- Weksler BB, Subileau EA, Perriere N, Charneau P, Holloway K, Leveque M, et al. Blood-brain barrier-specific properties of a human adult brain endothelial cell line. FASEB J. 2005;19(13):1872–1874. doi: 10.1096/fj.04-3458fje. [DOI] [PubMed] [Google Scholar]
- Xia H, Lazartigues E. Angiotensin-converting enzyme 2 in the brain: properties and future directions. J Neurochem. 2008;107(6):1482–1494. doi: 10.1111/j.1471-4159.2008.05723.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Shi L, Ma X, Su H, Ma G, Wu X, et al. RhoA-Rho associated kinase signaling leads to renin-angiotensin system imbalance and angiotensin converting enzyme 2 has a protective role in acute pulmonary embolism. Thromb Res. 2019;176:85–94. doi: 10.1016/j.thromres.2019.02.016. [DOI] [PubMed] [Google Scholar]
- Yamamoto M, Ramirez SH, Sato S, Kiyota T, Cerny RL, Kaibuchi K, et al. Phosphorylation of Claudin-5 and Occludin by Rho Kinase in Brain Endothelial Cells. Am J Pathol. 2008;172(2):521–533. doi: 10.2353/ajpath.2008.070076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Wang J, Ma X, Wang W, Zhao B, Chen Y, et al. ACE2-EPC-EXs protect ageing ECs against hypoxia/reoxygenation-induced injury through the miR-18a/Nox2/ROS pathway. J Cell Mol Med. 2018;22(3):1873–1882. doi: 10.1111/jcmm.13471. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author upon request.