

Abstract
Abstract
The global spread of COVID-19 has raised the importance of pharmaceutical drug development as intractable and hot research. Developing new drug molecules to overcome any disease is a costly and lengthy process, but the process continues uninterrupted. The critical point to consider the drug design is to use the available data resources and to find new and novel leads. Once the drug target is identified, several interdisciplinary areas work together with artificial intelligence (AI) and machine learning (ML) methods to get enriched drugs. These AI and ML methods are applied in every step of the computer-aided drug design, and integrating these AI and ML methods results in a high success rate of hit compounds. In addition, this AI and ML integration with high-dimension data and its powerful capacity have taken a step forward. Clinical trials output prediction through the AI/ML integrated models could further decrease the clinical trials cost by also improving the success rate. Through this review, we discuss the backend of AI and ML methods in supporting the computer-aided drug design, along with its challenge and opportunity for the pharmaceutical industry.
Graphic abstract
 From the available information or data, the AI and ML based prediction for the high throughput virtual screening. After this integration of AI and ML, the success rate of hit identification has gained a momentum with huge success by providing novel drugs.
From the available information or data, the AI and ML based prediction for the high throughput virtual screening. After this integration of AI and ML, the success rate of hit identification has gained a momentum with huge success by providing novel drugs.
Keywords: Artificial intelligence, Machine learning, Deep learning, Pharmaceutical industry, Imaging
Introduction
The ground-breaking development in biomedical research has shown an increase in the availability of biomedical data [1]. But researchers have raised whether the transmission and transfer of biomedical data are done correctly to extract practical knowledge [2]. Especially in the pharmaceutical sector, there have been several pieces of research, research outcomes, clinical data, and ethnic population-wise data, and other experimental data are available [3]. In this case, the pharmaceutical sector with drug discovery is a process that is very costly, time-consuming, and subject to many formalities. The average cost for getting a new drug by the various phase of drug development can range from $1 to $2 billion and consumes up to 15 years [4]. Upon considering the research question, the available data on this domain can be used to develop new drugs, which can be more accurate, timely, and cost-effective [5]. Researchers worldwide are continuously developing innovative methods and algorithms to obtain suitable molecules with a short time and cost-effectiveness. Significantly, the introduction of artificial intelligence (AI), deep learning (DL), machine learning (ML), and computational chemistry towards drug discovery has shown a significant impact on its success rate. These methods alone or jointly combine to form new strategies that incorporate a wide range of efficient algorithms that enhance the predictions [6].
Computation power and algorithms for developing new leads with therapeutic importance in the modern drug designing process play a vital role. This current technological era is showing the technical update regularly [7]. Nowadays, several types of research offer deep atomic insights, which tend to establish the cause of disease, function, or inhibitions. Based on that, the algorithms are also updated to respond to the atom’s realistic mechanistic action that is core important for the drug designing process. For the Computer-Aided Drug Designing (CADD), the designing process initiates with two methods. One approach is structure-based drug design (SBDD), and the other one is ligand-based drug design (LBDD) [8]. But both ways heavily rely on the backend algorithms, scoring functions, and force fields for ranking and evaluating the energy contribution of lead molecules in the molecular systems. As of now, there have been many programmes or software applications that run with various algorithms, interpreting the results with predefined scoring functions for both SBDD and LBDD methods. But predicting the exact parameterization and obtaining the accurate energy levels and transferable force fields are challenging tasks for filtering the possible drug molecules [9]. To solve those issues, the parameterization process with the input of quantum physics of significant dimensions with a small number of parameters and a simple, functional analytic form is also introduced. In this way, the CADD and molecular modelling approaches enhance the efficiency of predicting the lead molecules by lowering the error components [10]. The specialty of CADD and molecular modelling drives many small molecules or whole small molecules databases in a limited time and can show realistic interactions between the hit molecules and macromolecules. Macromolecules are composed of polymeric units of amino acids or nucleic acids and the predefined algorithms. The force fields are programmed to adjust these atoms in these macromolecules. There are several programmes, software applications, and web servers available, but the user must choose the backend algorithm and force fields according to the macromolecules of their requirements [11].
The introduction of modern AI methods offers highly reliable computational methods in pharmaceutics and biomedical science. AI simulates human intelligence to machine models to rehabilitate or imitate human performance [12, 13]. Specifically, the MI approach can correct complex chemical problems in the drug identification process. This field of interest is not limited to certain areas, as every domain applies the automation traits in link with human minds for thinking, learning, and problem-solving efficiency [14]. The integration of AI and ML into biomedical applications is shown in Fig. 1. It clearly shows the AI and ML with computational advances and statistical methods are incorporated into biomedical applications to mimic human thinking, reasoning, and implementation [15].
Fig. 1.
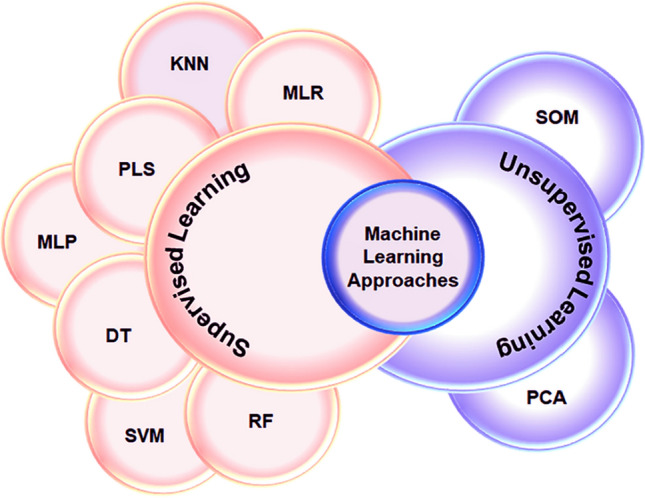
Classified machine learning approaches into supervised and unsupervised learnings into respective categories. Here, MLR: multiple linear regression; PLS: partial least squares; DT: decision trees; RF: random forest, KNN: K-nearest neighbours, MLP: multilayer perceptron; SVM: support vector machines; SOM: self-organizing maps; PCA: principal component analysis
Artificial neural networks
Artificial neural networks (ANNs) are favourably recognized computer models developed based on the Homo sapiens brain and its networking trends [16]. The simplest case shows a fully connected network or feed-forward that shapes the computation chart with three layers (input layer, hidden layer, and output layer) [17]. The layer-wise single computing unit called neurons works as a nonlinear transformation to the input data. This information is propagated in layer-wise mode and receives the output of the preceding layer as shown in Fig. 2. Molecular modelling and drug design predominantly rely on ANNs. It resolves the complexity associated with statistical models used in HTVS (high-throughput virtual screening), QSAR (quantitative structure–activity relationship), and pharmacokinetics and pharmacodynamics studies [18]. For the numerical values determining the output, ANNs perform excellently in interpreting nonlinear relationships and predict the process of success in the drug finding process [19].
Fig. 2.
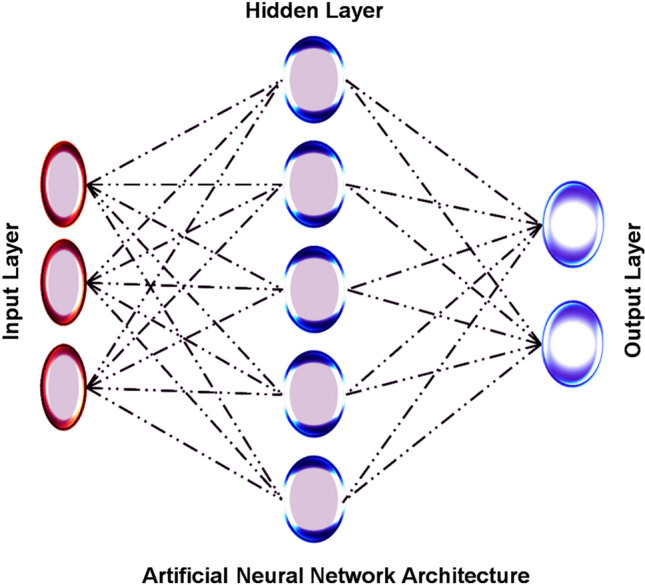
Basic model of artificial neural network architecture proposed where the input layer is modelled for network inputs, output layer is modelled for network outputs, and in between the hidden layer is modelled for the feed-forward and back-propagation functions
Application of artificial neural networks in drug discovery
As discussed above, ANNs have high reliability towards improving efficiency and target-based drug discovery. It has a massive ability towards complex investigation and nonlinear relationships, and so this ANN is alternatively called “Digitalized Model Brains” [20]. Neural network (NN) applications are highly accountable for STEM (Science, Technology, Engineering and Medicine). Especially in the molecular modelling and pharmaceutical sciences, the ANNs’ application sets the trend by providing high reliability of results [21]. Notably, the ANNs used to scrutinize the extensive database of small molecules (HTVS), property prediction (ADME/T), QSAR, pharmacophore analysis, pose validation, formulation and development of leads are shown in Fig. 3.
Fig. 3.
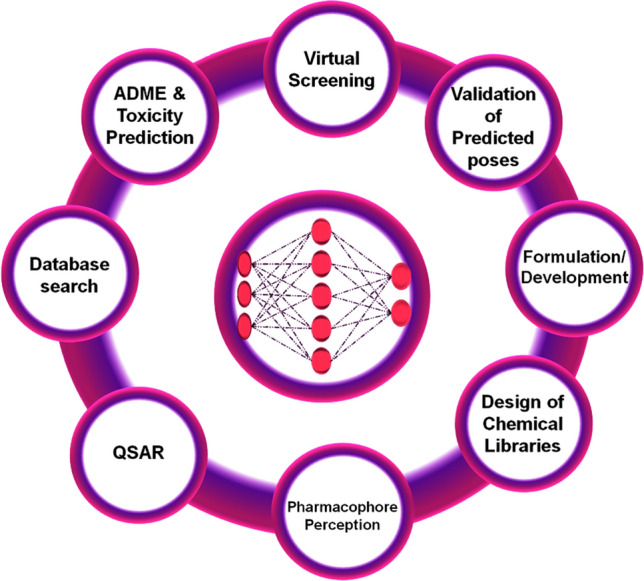
ANN applications are widespread in the molecular modelling, especially in increasing the efficiency of drug discovery
Deep learning methods
DL method represents the neural network by possessing multiple hidden layers, and it is highly used for its flexibility to learn arbitrarily complex functions, as shown in Fig. 4. It can learn as much as possible with adequate data and the computational time investment, providing highly reliable outputs. The multiple layers hidden in DL patterns offer flexible access to learn arbitrarily complex modules, which directly provide suitable neurons and trained sets. The DL applies a backpropagation algorithm and a gradient-based optimization method that allows neuronal network training, resulting in end-to-end differentiation [22]. In addition to that, the feed-forward networks are interlinked with layers, conventional and graph convolutional architecture that proceeded towards the development of various domains and data types. The updated data reading tendency and advanced products in algorithms and computational hardware are driving the DL methodology. The neural networks are highly recognized among deep networks due to their difficulty training and understanding small sets [23]. From an algorithmic point of view, DL networks with more layers have often suffered from disappearance gradients and prevent models from learning efficiently. The novel method initialization schemes, neural activation function, and gradient-based optimization methods significantly improve efficiently trained deep networks [24]. The recurrent neural networks (RNNs) are described as recurrent units mainly used to capture the temporal dependence in sequence-level data information. Conventional neural networks (CNNs) are notably emerging for image processing by catching local and spatial relationships with learning filters [25, 26]. Graph neural networks (GNNs) are mostly operated with unordered data like analysing the social networks, but this is a well-suited model for representing the small molecules [27]. The representation and difference between these types of neural networks are represented in Fig. 5.
Fig. 4.
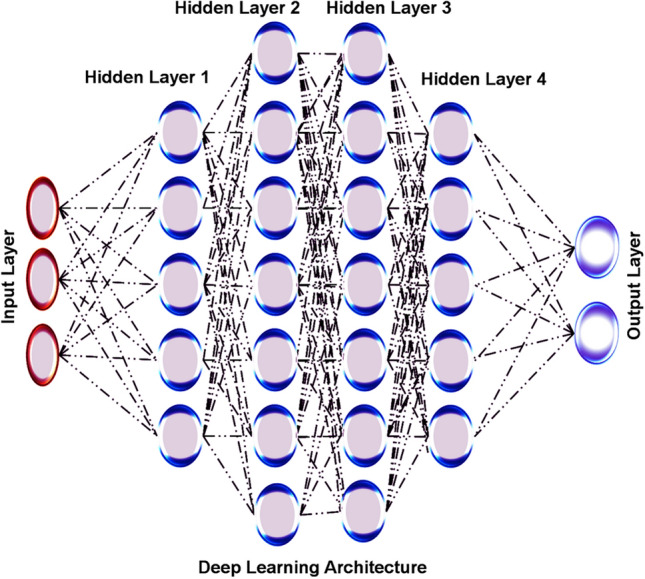
Deep learning architecture showing the input layer, and output layer in the external, while multiple hidden layers in the middle layer
Fig. 5.

Other types of neural networks showing the input, output, and hidden layer architecture
Machine learning method
ML is the basic paradigm that involves multiple method-based domains and several algorithms to recognize the pattern within the data. Every automation-based method uses DL and ML but holds the difference, as shown in Fig. 6. ML is further classified into several types, and DL is the subfield of ML which engages artificial networks that interlinks computing elements [28]. This is equivalent to human biological neurons and imitates the transmission of electronic impulses. This model is the well-established mathematical model that shows underlying patterns available in the data and information and applies to learn methods for predicting future data. The ML strength was due to solving complex mathematical problems and is used in various fields of modern biology. In terms of generalized ML methods, it has been applied to accurately predict the unseen data set in choosing the method for performance in complicated situations. Several models are used to train a single dataset to avoid brute force sensitivity and optimize specifically by understanding the viewpoint in various model architectures. In classification, ML methods are defined by supervised or unsupervised models, as shown in Fig. 1. In supervised models, the mathematical model relationship of variables found in the dataset is known as input and output variables. It is too difficult to comprehend the linear regression between the known bi-variables by supervised learning models. At the same time, the unsupervised learning models find the hidden patterns within input data that build the clusters based on intrinsic structure and predict the relationship between the data points [29]. Reinforcement learning is another major category of ML method that processes mainly towards the application in dealing complex environments that learn the optimal series of actions in response to opposed environmental information to the output similar to the supervised learning [30, 31]. In AutoDock, the ML methods are well prepared and trained for those specific types of protein/DNA, active site residues, small molecule, or drugs instead of showing scoring functions that include the Drug score [32, 33]. The ML deals with multiple applications that offer too much success in various stages of HTVS of typical drug discovery in understanding the novel drugs and lead components. The binding site features of receptor information are mainly analysed by ML methods, in association with receptor homology and disease information [34, 35].
Fig. 6.

Difference between machine learning and deep learning algorithm based on feature extraction
Machine learning for target identification
The typical drug discovery requires identifying target proteins with casual aspects of pathophysiology and a plausible framework. Misunderstanding of target protein information may lead to modulation in the disease information, and in this sense, target selection is a mandatory step [36, 37]. Evidence of successful drug response will be considered and subsequently, lead efficiency in the randomized clinical trial tends to the identification of prominent drug targets. The ML algorithm predicts the unseen biological happenings, events, and problems [38]. Costa et al. [39] developed a computational model for predicting the morbidity and druggable genes on a genome-wide scale. That model has been widespread in reducing the laborious experimental procedures and identifying the putative molecular drug targets linked with disease mechanisms. Here, this classifier is modelled to uncover the biological rationale from a data-driven view. The main classification features are mRNA expression, gene essentiality, occurrence of mutations, and protein–protein network interactions. The meta-classifier analysis results initiated from 65% of known morbid gene recovery and 78% of unknown druggable gene recovery [40, 41]. The decision tree (DT) and uncover rules inspect the parameters that include the membrane localization and regulation of multiple transcription factors to identify biological traits [42]. This can also exhibit understanding and designing the Biosystems principles by applying the reverse engineering methods [43, 44]. Volk et al. [45] applied the ML methods to model the challenges at DNA, protein, specific pathway levels and process them for the genome and cellular communities. Jeon et al. [46] stated and developed a Support Vector Machine (SVM) method to analyse the genomic variety and systematic data set to distinguish the protein based on homologs or likelihood for drug binding in breast cancer cases, pancreatic cancer, and ovarian cancers. Momoshina et al. [47] applied the same concept of identifying the drug target in the complicated disease by using the biomarker discovery approach in muscle tissue to detect the druggable targets considering the molecular basis of human ageing. In this approach, the SVM model is contracted with linear kernel and deep feature selection to find the gene of expression linked with ageing. This model also evaluates the gene expression samples from Genotype-Tissue Expression (GTEx) project and has obtained a 0.80 accuracy level [48].
Machine learning for imaging analysis
ML approaches are used in investigatory screens and automated or robotic image acquisition and investigations. For example, the potent inhibitors against the β2 adrenoceptor target and radiological binding assay through novel molecules are screened based on ability to interfere with radiolabels ligand with binding affinity [49]. This acute effect of small molecules may cause the alteration in the surface plasma resonance (SPR) detected in the receptors. This tendency allows the selection of small molecule inhibitors processed into the lead optimization stage [50]. But this process is laborious and a long approach, and therefore, alternative methods such as phenotypic screening are highly focused. At this stage, ML-based analytics are applied to identify complex phenotypes that have tended to increase the efficiency of the small molecule [51, 52]. Another technique, namely advanced imaging, is a mechanism applicable to finding the phenotypes and perturbation of small molecules, and this method is known to enhance prediction [53]. More broadly, imaging can be composed of two camps: 1. typically called phenotypic screening, which targets the predefined phenotypes of intracellular signalling molecules associated with the disease mechanism; 2. the various subcellular structures with antibodies, infective or chemical agents, and fluorescent dyes categorize their responses.
Machine learning for high-throughput screening
Identification and experimental validation of novel drug targets are costly and time-consuming. Significantly, virtual screening or HTVS is essential for the drug discovery process and integrated with new methods for technological updates. Hence, AI/ML methods are used to understand promising drug targets’ priorities, which are carried forward for the subsequent experiments [54]. Valentini et al. (2014) developed the combination of functionally different gene networks with kernel-based methods to rank the order of genes [55]. Later, Ferrero et al. [56] worked with target–disease association data available in public databases for predicting novel drug targets. Arabfard et al. [57] have predicted and ranked about 3000 targets associated with an ageing gene with three positive unlabelled methods such as Naïve Bayes, Spy, and Rocchio—SVM, categorized by ranking the human genes according to this implication ageing. Elaborate discussions are on the correlation between the drug targets and disease in drug discovery [58]. The subsequent imperial process begins with finding a small apt molecule to disturb the disease mechanism. Generally, a suitable drug candidate is designed and exposed to pharma companies or deposited in large compound libraries. Vapnik et al. [59] has developed the SVM method, and this SVM model is integrated with cheminformatics by Burbidge et al. [60]. The SVM classification resembles other sample methods as linear discrimination, and in this hyperplane is the important feature to classify the dependency of boundary conditions [61]. Particularly, while the ranking is essential, the length between the instance and hyperplane is maximized and only a small subset of the training instance defines the boundary [62]. Experimental error data or noisy data can also be considered for allotting some instances positioned on the wrong side of the hyperplane. The Kernel trick is the SVM method’s unique feature applied in various applications, which extends the classification with both linear and nonlinear hyperplanes associated with Mercer’s theorem [63]. This feature allows the calculation of distances in high-dimensional nonlinear spaces that do not require the explicit distance transformation [64]. A variety of decomposition algorithms are also used in SVMs to analyse the large dataset, extended to its regression (SVR). Syngenta and Willett used SVM methods and were markedly inferior to the use of BKD to test about 35,991 compounds in the NCI (National Cancer Institute) AIDS data set associated with UNITY fingerprint as descriptors [65, 66]. After that, 125,657 compounds were studied for pesticide activity and the polynomial kernel of degree five outperformed binary kernel discrimination (BKD). Franke et al. (2005) has scrutinized 2.7 million small molecules from the COBRA ligand database based on 94 reference compounds having Cox-2 antagonist molecules as the training set. For this screening, several compounds were studied and yielded only three molecules with potent activity. Notably, one compound has shown stronger inhibitory profiles than the celecoxib and rofecoxib [67]. Lepp et al. [68] have screened 21 data sets related to depression from MDDR and classified 30,000 ligand molecules per data set with SVM and descriptors of atom counts. In the ML-based screening, the true negatives are well predicted and classified as positive recall ranging from 44 to 89% [69]. Jorissen and Gilson [70] have reported the screening of small molecules against four receptors, namely 1AAR, CoX-2, PDE5, and CDK2, from the 50 known compounds as a training set for each drug target and reported 1892 small molecules from the NCI database and 25,175 compounds from the Maybridge database [64, 71]. Using the SVM approach, 2D descriptors are predicted through the DRAGON software (http://www.disat.unimib.it/chm/), and through this, they have got more than 10% of the screened database than other fusion methods using fingerprints [72]. Saeh et al. (2005) used SVM methods for G protein-coupled receptor (GPCR) and found 1573 compounds as top-ranked from the list of 129,994 compounds, which also yield the success rate 69 times higher in comparison with random choice-based selections [73]. The application of support vector regression (SVR) in virtual screening is reported by Byvatov et al. (2005). By constructing the model based on 331 domain receptor inhibitors for screening the SPECS database (https://www.specs.net/) and inter-bio-screen database, which have more than 225,000 molecules yield 11 hit compounds with strong binding selectivity against the D3 receptor [74]. Recent reports on SVM support develop a variety of kernels that are applied for the data with various structures. The same process is also applied in the HTVS process. It represents the molecules as information data rather than vector descriptors and focuses kernels representing the pharmacophores, graphs, and trees [75]. The model workflow of ML-integrated virtual screening is provided in Fig. 7. Eitrich et al. [76] suggested the kernel function to demonstrate the various costs linked with misclassified sections of both active and inactive molecules; different slack variables can consider that for various classes [77]. Prediction of the scoring function is the core component of molecular docking used to validate the binding affinities of hit compounds with respective drug targets. Due to the stronger nonlinear mapping ability, the ML-driven scoring function shows the best results by extracting properties like chemical and geometric features and physical force fields [78]. These scoring methods are tagged as the data-driven black box model for the binding affinity prediction that forms the interactions between the protein–ligand complex from the experimental data to eliminate the errors in physical function related to molecular docking methods [79]. ML tools such as SVM and random forest (RF) significantly improve the performance of a scoring function [80]. This method uses a nonlinear relationship of individual energy values obtained through the docking program for the lead molecules and experimental binding affinity data instead of linear additive assumption energy. Hence, it shows improved screening power and scoring power. Wang and Zhang reported ΔvinaRF parameterization correction with a combination of RF and AutoDock scoring function with Glide XP Score for good performance [81]. Jimenez et al. (2018) have developed the 3D graph-based CNN model to predict protein–ligand interaction results in precise prediction of binding affinities that highly match the experimental data [82, 83]. A DL method predicts the binding affinity by extracting the feature from the protein–ligand interaction image, similar to the knowledge-based scoring function [84]. Therefore, it was crucial to depict basic features such as atomic information representing its types, distance, charges, and amino acids.
Fig. 7.

Model workflow for the prediction of lead molecules using machine learning approaches for the high-throughput virtual screening
Machine learning for structure-based drug design
The computational drug discovery process initiates from the drug target identification, target evaluation, and finding the suitable drug candidates [85, 86]. Hence, target selection plays an imperial role in disease pathology, assessing the druggability of lead molecules and prioritizing candidate targets [87, 88]. However, due to the complex nature of the human disease, the target selection process needs comprehensive methods that take part in the heterogeneous data and understand the molecular-level mechanism of disease phenotypes and also help to identify the patient-specific changes [89]. Advanced methods like AI/ML have been applied to overcome these challenges. For example, the DL codes help in the prediction of retrosynthetic pathways to small molecules with the desired bioactivity and develop new chemical structures [90]. One of the ML methods is self-organizing maps (SOM)-based prediction of the drug equivalence relationship (SPiDER) that widely applies the algorithm of neural network for discretizing the input vectors into feature maps in an unsupervised fashion [91]. The predictions of drug–protein relationships are inferred based on the similarity of descriptors to the reference lead molecules in the same neuron without unambiguously allowing for the target identification [92]. This method is performed with the available set of pharmacophore descriptors and its topological information (CATS2) to identify the non-hydrogen atoms. SOM is built based on the topological feature autocorrelation molecules by using physicochemical parameters [93]. This software has been extensively applied in the de novo approach, especially to design natural products with high inhibitory potential. Several studies reported the application of SPiDER including identification of farnesoid X receptor (EC50 = 0.2), 5-lypoxygenase (EC50 = 11 mM), and peroxisome proliferator-activated gamma receptor (EC50 = 8 mM) as drug targets [94]. In addition, it also provides the structural difference between small molecules used as reference compounds. The lead molecules such as isomacroin, graveolinine, and piperlongumine are potent bioactive molecules with SPiDER as serotonin 2B receptor and transient receptor potential channel vanilloid modulators [95]. Recently, SPiDER has been applied in the de novo drug design to identify the new chemical structures fitting a defined model pharmacological space for the small molecule synthesis and their experimental validation [96]. Another method DEcRyPT is known for predicting network pharmacology; it can be performed either in single or in combination mode with SPiDER by employing random forest and CATS2 descriptors [97]. Through this method, the predictors defined by users independently analyse a different portion of the training set before aggregation of result outputs. For example, the application of β-lapachone leads the workflow confidently of the results of 5-lipoxygenase as a target [98]. Following the identification of an ideal target, a novel medicinal strategy hinges on finding befitting small molecules that corrupt the native structure of the target [99]. Contemporary biology, especially current cancer research, depends on these small molecules and innovative drug processes. We feel the necessity for molecule bearing structural resemblances to ligand with a few functional modifications to alter receptor molecule function; on the other hand, numerous desirable drug targets—might lack the domain described above for ligand binding (e.g. PARP), may get activated without the presence of ligand (e.g. EGFR), might have multiple ligands (e.g. CXCR2), or may have unknown ligand (e.g. HER2) [100]. Those above are the reasons behind the cross-reactivity of small molecules with other receptors. “Biologics” is generally regarded as a multitude of drug-targeting approaches that help overcome the limitations mentioned above. Oncolytic viruses, bi-specific antibodies, humanized monoclonal antibodies, engineered T-cells, and chimeric receptors are a few to name in oncology [101]. Through these advancements, a large number of small molecules are predicted as drug-level candidates [102]. Meanwhile, contemporary in silico methods have shown the believability of modelling protein structures, which is closer to the experimental structure [103]. Protein modelling techniques like homology modelling begin with a known protein structure that is > 40% in homology with the target sequence. It is considered most reliable compared to other methods, and homology modelled structures are usually validated by considering their stereochemical properties (e.g. Ramachandran plot). Following that, the folding protein’s binding energy in exposing to charges in functional groups is taken into consideration for modelling potential binding sites [104]. The Q-SiteFinder is one such energy-based method to predict stable conformations of binding sites [105]. Protein building blocks linked with these active sites are annotated for the functional predictions. Synthesized or computationally modelled target proteins are then extensively screened through experimental high-throughput screens and virtual screening using the lead molecules’ renowned small molecule database [106]. On execution of molecular docking methods with the predicted binding site information of a target protein, the hit compounds based on SBDD have obtained stable and strong binding free energies. Conversely, a de novo drug design can be utilized if the binding pocket is of sufficient resolution. Based on predefined criteria like pharmacodynamic, pharmacokinetic, and toxicological criteria, the hit compounds are optimized. For instance, in an attempt to find the varying levels of success, several studies have tried to perform the ligand-based screening using ANNs [107]. DeepChem by Ram Sundar and colleagues is one such utilization of multitasking deep ANN. DeepChem is an open-source tool with simple Python scripts that enable the construction, fit, and evaluation of complex models and drive ligand screening for commercial drugs. Usually, multitask models of ANNs outperform standard ANNs such as random forests by synthesizing information from distinct sources. The authors intended to alleviate the hurdles with software availability amidst drug discovery industries, and their validations illustrated that multitask ANNs are robust [108, 109]. Wu and colleagues generated a small molecule database of 700 K compounds and its binding data, integrated into DeepChem to help with the benchmarking. Unknown ligand–receptor interaction mechanisms can be identified by combining multiple ANNs, Markov state models, and one-shot learning to reduce the amount of data required in a new experimental set-up [110]. The discovery of new allosteric sites, especially in analgesia and GPCR biology as new drug targets, will let modification or fine-tuning of receptors easily by eliminating the competition for receptor occupancy by the ligand. Drug pharmacokinetics properties prediction can be made using ANNs [111]. ANNs excelled in 13 out of 15 assay-based classification tasks to determine drug-like molecules and ADME parameters compared to other ML methods and RF in a competition [112]. DeepTox is a multitask ANN by Mayer et al. (2016) that calculates the chemical descriptors and trains the ANN for determining the nuclear toxicity by normalizing chemical structures. This ANN was implemented in the Tox21 data set to predict silico toxicity of 12,000 small molecules through 12 high-throughput toxicity assays [113]. Apart from optimization and virtual screening of small molecules, ML-based approaches can be utilized to generate innovative chemical entities for enhancing de novo drug design [114]. Kadurin and colleagues reported that variational autoencoders combined with generalized adversarial networks (GANs) can be used for the computational design of selective antitumor agents. In de novo drug design, GANs are very intriguing because they train two ANNs simultaneously through various objective functions (the generator and the discriminator). To generate the best molecular structure, GAN must compete in the zero-sum game. Using variation autoencoders for finding the chemical structures from available databases in the latent space is an important task to be carried out before converting the molecules into SMILES format strings with latent vectors [115]. In this method, the 3D structure of the drug target is mandatory to predict the binding of lead molecules with the active site. Structure-based methods employed in virtual screening are the most acceptable methods for predicting high success rate candidates with prominent interactions and energy [116]. The ML methods are implemented in structure-based methods to improve the robustness and functional scoring accuracy. ML-based algorithms such as RF, SVM, and NN were used to develop streamflow (SF) for the best prediction compared to other approaches [117]. ML-based SF shows superior prediction in all respects. However, the prediction of SF varies depending on the types of targets. Advanced ML approaches target SFs to improve the efficiency of the available methods for the targets such as GPCRs, cytochrome p450 aromatase, and histone methyltransferases [118]. Moreover, ML methods are applied to the post-docking methods for improving the accuracy of molecular docking methods and scoring function [119].
Machine learning in druggability prediction
Several ML models are utilized to evaluate the druggability pockets of the drug targets by using different features. Surface Cavity Recognition and Evaluation (SCREEN) is one of the known ML web servers built based on the RF classifier to analyse the structural, geometric, and physicochemical properties of drug interactions and non-drug-binding cavities in the drug targets [120]. The protein active site surface, allocation space, and shape geometry of the active site cavities are essential in the classification process [118–123]. Previous studies have applied the SVM method to predict the active sites in the target proteins based on their physicochemical properties available in the protein sequences [124]. The DT-based meta-classifier has been used to predict non-druggable and druggable genes based on the network topology, tissue expression profile, and subcellular localization [125]. Dezso and Ceccarelli have developed an RF model to analyse the druggability prediction of cancer targets by comparing the similarity of approved drug targets [126].
Artificial intelligence models for de novo design
Various software and methodologies have been introduced in de novo drug designing to generate the novel potent molecules without the information from the reference compounds. Unfortunately, these de novo methods are not widespread application in the drug design methods compared to the other structure-based screening methods. In this method, the compounds which are difficult to synthesize are generated. Variational autoencoder has two neuronal networks: the encoder and the decoder network [127]. The encoder network actively involves translating compounds’ chemical structure from its SMILE notation into a real-value continuous vector. Most of the dominant molecule’s back translation dominates and light conformational changes exist with a more negligible probability. In another study, the performance of the variations autoencoder was compared with the adversarial autoencoder [128]. These adversarial autoencoders have a generative model for the production of novel chemical structures [129]. Prediction of novel structures in combination with in silico model shows more active compounds against dopamine receptor type 2. Similarly, Kadurin et al. used a generative adversarial network (GAN) and suggested the compounds with potent and effective anticancer properties [130]. In addition, recursive neural networks (RNNs) are intensively applied in de novo drug design. It emerged in natural language processing, and through this method, sequential information has been provided as input. Since SMILES notation strings encode the chemical structure format in a letter sequence, the RNN is applied to develop the chemical structures. RNNs are trained with a massive data set of chemical compounds taken from the collection, like ChEMBL or a large set of commercially available compounds to train the neuronal network for the SMILES string [131]. This approach has also been used in generating new and novel peptides either in sequence or in structural forms. Reinforcement learning is used to bias the generated compounds towards predicting their important core properties. On the other hand, transfer learning is applied as a different strategy for generating potent novel compounds with the expected biological activity. In addition, the various types of architecture models are implemented in ML methods capable of producing potent novel structures. The new chemical features can be explored by this approach with similar properties of training set drug molecules [132].
Prediction of protein folding from sequence
Most of the pathological conditions are associated with protein dysfunctions. Hence, understanding the structural aspects of proteins plays an imperial role in the SBDD approach. It can be used to identify the potentially active molecules towards the drug target proteins. Predicting and measuring the 3D structure of all the protein targets via in vitro approaches will be a cost-effective and time-consuming process [133]. Hence, the development of appropriate algorithms for predicting the model structure of a protein is highly appreciable at present [134]. Though the protein sequence is available, it is still an unresolvable problem to expect accurate de novo prediction of corresponding 3D structures [135]. Due to the presence of advanced features, DL techniques are applied for the prediction of secondary structure, topology information, backbone torsion angle, and residue contacts. DL methods were also used to combine the one-dimensional and two-dimensional structures, and CNN was used to predict the residue-wise contacts. The advanced feature of DL may accurately study the association between the sequence and structure of the protein via feature extraction approach; currently, it is still a goal to precisely predict the 3D structure and DL shows a highlight of the future development. Homology modelling and de novo methods are traditionally used to predict the 3D structure of target proteins and have become more accurate and sophisticated [136]. However, the recent valuation of protein structure with AI tools is developed and performed to predict the 3D structure of the protein. For instance, AI tool AlphaFold is used for the 3D structure of target protein, which accurately predicts about 25 structures from 45 structures.
Prediction of protein–protein interactions
Evaluation of protein–protein interactions (PPIs) is functionally essential for several biological processes and diseases. STRING is one of the widely used PPI databases comprising 1.4 billion PPIs obtained by computational and experimental approaches. The interface of PPI is predicted by protein–protein binding sites composed of many amino acids [137]. It can be raised as a new class of drug targets entirely different from the traditional drug targets, including GPCR, nuclear receptor, ion channels, and kinases. For instance, about 1756 non-peptide inhibitors from 18 families of PPIs are reported as the protein–protein blockers in the protein–protein database (iPPI-DB) [138]. It extends the target space and promotes the development of potent small molecules. Compared to the available traditional methods, PPIs reduced the adverse effects, because it has efficiently increased its biological activity. Compound DCAC50 is the potent inhibitor of ion channels; it can block copper (Cu2+) ion transport in cells by binding with the Cu2+ transfer interfaces and effectively inhibits the specific cell proliferation of cancer cells without affecting the normal cells [139]. For example, eFindSite is a web server that predicts the interface of protein–protein interaction based on the templates. AI methods such as SVM and NBS models are used to predict the residue-based and sequence-based features. Based on this principle, various protein–protein docking (ZDOCK, SymmDock) can be used to predict the interface of PPIs [140]. Among these methods, the prediction of conformational changes when two proteins bind is a more challenging issue. DL methods effectively extract the most relevant sequence features for the prediction of the PPI interface, which shows improvements compared to other ML methods like SVM. Due to the large buried area of the interface, the binding site or local regions should predict in the interface. With the contribution of a large amount of energy, the hot spots of the protein may be the druggable sites at the interface [141]. Bai et al. used other unique properties such as fragment docking and direct coupling analysis (FD-DC) to predict druggability available in PPI sites. In this method, they developed a fragment-based docking methodology initially called iFitDock to seek the hot spot in the PPI interface. Then, the predicted host spot is categorized with clusters to form the enzyme binding site. Finally, the scoring function based on the algorithm provides the scoring values based on the conservative evolutionary levels to find the active site information which is not available or identified in previous [142].
Quantum chemistry with artificial intelligence and machine learning
Understanding the drug binding mechanism and the interaction with target proteins is a primary step in the drug designing pipelines. Quantum mechanics (QM) or integrated quantum mechanics/molecular mechanics (QM/MM) hybrid methods are used to predict protein–ligand interaction in the drug discovery process [143]. These methods are highly ruminated quantum effects for the simulation of the system at the atomistic level and offer higher accuracy levels than the other available classical methods [144]. In classical MM methods, the simple energy function is considered based on the atomic coordinates involved. Recently, the implementation of AI methods to QM calculation offers much better accuracy due to the inclusion of QM charges and favourable time–cost than the classical MM method [145]. In AI, methods are highly trained to deliver the actual energies available in atom coordinates and the MM methods speed. Generally, AI methods are used to predict electrical properties with atomic simulation. In contrast, DL methods are applied extensively for the energy prediction of small molecules, hence replacing it with accurate computationally demanding quantum chemistry calculation via ML methods. For example, quantum chemistry-derived density functional theory (DFT)-based potential energies are calculated even for large database holding 2 million elpasolite crystals, and the accuracy of the ML model is enhanced for increasing the sample data size and reached 0.1 eV/atom for DFT calculation trained on 10,000 molecules [146]. ML approach with QM properties has made drug development into atomist potential by integrating Kernel ridge regression (KRR), Gaussian process regression (GPR), and neural network (NN). Considering the reliability of low numerical complexity and high accuracy of ML algorithms makes it comfortable, attractive, and an alternative for ab initio and DFT calculations [147]. With the remarkable ability to understand the complexity in the data, the ML integrated methods are most efficient for calculating the force fields and empirical QM. However, the predicted ML models relied on the trained quality and quantity of the data. Neural networks effectively and efficiently model baseline data with high flexibility, and reliable, cost-effective, and a large amount of baseline data are used to train them for accurate prediction. Recently, Smith et al. (2018) developed an ANAKIN-ME (Accurate NeurAl networK engINe for Molecular Energies) or ANI to evaluate the extensive data set of 22 million minor molecule conformations that yield potential capability for predicting energies in large systems, which are much different from the training sets. The performance, applicability of the system shown up to 70 atoms. ANI-1 is exceptional for predicting energy with external molecular size with RMSE versus DFT energy at room temperature. Koohy et al. reports the extensive organic system, trained into fragmented small molecules with the addition of DFT for the larger systems [148].
Ligand-based virtual screening
Ligand-based virtual screening (LBVS) methods are considerably applied when the 3D structure information of the protein drug target is unknown or lacking [149, 150]. It stands opposite to the available docking method by predicting the binding orientation of ligand, excluded volumes, and charge space requirement of the binding pocket of the drug target [151, 152]. The basic principle of LBVS is that structurally similar lead molecules with known active compounds have similar reference activity. In the LBVS, known compounds information is required, and based on it, active molecules set is predicted from the test molecules set in the functional assays, even without the information of the target protein structure [153]. In such a case, LBVS methods are effectively used to find the potent lead molecules by assessing the similar structures of active molecules. Recently, ML has been implemented in LBVS methods to find more potent lead molecules and boost the predictive ability of LBVS models [154]. The crucial goal of ML methods is to advance the prediction of the active molecules against a specific protein target using the trained data set on input that discriminates lead molecules from a huge non-drug compound database and prioritizes the excellent lead molecules activity with the statistical probabilities. To overcome these issues, researchers have applied the SVM models, Bayesian architecture, and ANNs. For example, Stokes et al. have successfully identified several new antibiotics using graph convolutional networks (GCNs), performed by ML models in predicting molecular properties [155]. Researchers have executed a high-throughput screening on a large data scale in implementing the GCN model and have identified a promising new antibiotic, namely halicin. The potency of the DL methods in drug discovery approaches has come up with extraordinary techniques for finding the new lead molecule with DL and Spark-H2O platform Python for LBVS [156]. In the ligand-based methods, QSAR models are applied to understand the statistical values of small molecule physicochemical properties represented by molecular descriptors, along with its biological activity. These QSAR models play a crucial role in small molecule optimization. Also, they offer primary theoretical evaluation of essential characteristic features related to inhibitory activity, binding selectivity, and toxicity of the lead molecules. The QSAR approach drastically decreases the count of lead molecules to be tested in vitro and in vivo experiments [157]. This method can be assessed based on a regression or classification model that depends on the computational strategy. Implementing the AI/ML approach in the QSAR model has been extensively used in recent years. The ML tools such as RF, SVM, Naïve Bayesian, and ANN are the often used algorithms in QSAR [158, 159]. In the implementation of NN with QSAR models, single assay data with molecular descriptors are utilized for training an NN and record activities of training labels [160]. In 2012, a state-of-the-art method was developed with multiple DNNs to predict the accuracy by 15% over the baseline RF method. Since it was implemented in the QSAR model, the RF-based QSAR method was often used in drug discovery approaches [161]. Recently, Zakharov et al. developed a QSAR model combined with multitasking DNNs and consensus modelling to model the large-scale QSAR prediction for improved accuracy and better prediction over the concept of QSAR models [162]. ML approaches with ensemble integration are combined with several available basic models to overcome the weakness of each learning model and aimed to improve the performance task of QSAR. Various ensemble models like data sampling ensembles, representation ensembles, and methods ensembles are integrated into QSAR. Kwon et al. developed a model with a combination of all the three ensemble models with end-to-end NN-based individual classifiers to achieve better performance than individual models [163].
Artificial intelligence and machine learning for ADME/T properties
Predicting poor physiochemical properties of a lead molecule in the primary drug discovery stage pipeline will significantly reduce the risk of failure. For that, the model representation is provided in Fig. 8. Several DL-based methods have been developed and implemented with classical models. The study carried out by Duvenaud et al. used CNN-ANN algorithms for the solubility prediction of a lead molecule by extracting information from a molecular graph with an effective predictive performance with high interpretability [164]. In this method, the back-tracking method can obtain molecular fragments of solubility like the hydrophilic R-OH group. Followed by Devenaud, Coley et al. developed a tensor-based convolutional embedding of attributes molecular graph method for predicting the solubility of molecules in an aqueous environment, which outperformed Duvenaud’s model. Coley’s model applies the deep atom-level information to predict the solubility of lead molecules in an aqueous medium. Since a suitable correlation matrix is obtained between oral drug absorption and the Caco-2 permeability coefficient, the prediction of this Caco-2 permeability coefficient plays a crucial role in evaluating the pharmacokinetic properties of lead molecules [165]. The study carried out by Wang et al. included 1272 ligand molecules with Caco-2 permeability data and boosted the models with SVM regression, partial least squares (PLS), and multiple linear regression (MLR) algorithms to develop the prediction models with 30 descriptors. These boosting models showed better results with the good predictive ability (R2 = 0.81, RMSE = 0.31) for the test set molecules, and this model strictly follows the principles of organization for economic cooperation and development (OECD) about QSAR/QSPR, which assures the rationality and reliability of the model [166].
Fig. 8.

Development of ML- and DL-based toxicity predictions from the source of clinical data information
In early times, pharmaceutical companies were spending approximately more than 5 billion dollars per year. Later in the 2000s, it was estimated to be 30 billion USD dollars. The R&D investment rose about 875 billion USD; in 2010, it was estimated around 96 billion USD. In the last decades, pharmaceutical companies have applied some rules based on Lipinski’s rule, drug-likeness, and lead-like filters to avoid the undesirable ADME/T profiles [167]. The bioavailability of the lead molecule is a crucial pharmacokinetic parameter. Hence, the prediction of bioavailability can direct the medicinal chemist for the optimization of test molecules. ML techniques and implementation in ADME studies focus on building predictive models that extract the effective training data patterns and predict the PK values of new lead molecules [168]. Several types of ML models were used in the ADME process and are highly helpful to establish the relationship between the molecular descriptors such as PLS, MR, and DT matrix. Followed by absorption, drug distribution is the important prediction where the drug circulation of blood to intestinal fluid and intercellular fluid was predicted; the steady state of drug distribution of the drug is the ratio of its dose in vivo to its plasma concentration. Predicting the steady-state distribution in the tissues is an important criterion for evaluating the drug distribution mechanism [169]. Hence, the prediction drug metabolism site with high accuracy can lead to the optimization of drugs for obtaining the stability mechanism of the molecules. Large data sets are predicted with ML methods. It is also used to predict the site of metabolism and the enzymes involved in metabolisms, such as cytochrome P450, UDP-glucuronosyltransferase (UGTs), and aldehyde oxidase. For instance, the development of the neural network method XenoSite provides the possibility of the site of metabolism with an overall accuracy of 87%. Excretion is the crucial step for the consumed drugs, and their metabolites should be excreted from the body. Drug metabolites are known for their solubility in water and can be excreted easily, while most of the drug can be directly eliminated without metabolism. Lambardo et al. apply the PLS model to predict human clearance, which shows good discrimination results with an accuracy of prediction of 84%. Based on the mechanism of elimination, the predicted PLS model was used to predict the drug excretion [170].
During the drug development process, most of the lead molecules can fail in pre-clinical and clinical toxicity. Therefore, toxicity prediction is application-wise mandatory in drug optimization and decreases the risk of error. Traditionally, liver and kidney toxicity studies have been practised to predict the drug toxicity profile by rule-based expert knowledge and structural alert [171]. Hence, in recent years, DL methods have been implemented to automatically handle the various chemical characteristic features and their merits and obtain strong performance in toxicity prediction. For instance, a molecular graph encoding convolutional neural network (MGE-CNN), with the acute oral toxicity prediction model, obtains more accurate results than previously reported SVM model-based results. Their study mapped all the toxicological properties of fingerprints in atom-level information with structural alerts defined by the ToxAlters [172]. In another study, Mayr et al. have developed a multitask DNN model (DeepTox) for toxicity prediction, and this model provides better outperformance than others. The ADME/T properties of drug molecules show some relevance, and multitasking NNs can improve the performance [173]. The study was carried out with ML-driven approaches that accurately predict some important physicochemical properties like water solubility, lipophilicity, etc. An improved model of ML algorithm results in the better prediction of molecular properties with limited progress. The other classification models such as DT, K-nearest neighbour (KNN), SVM, NN, and RF have been widely used to predict ADME/T properties of the lead molecules, though they need further developments in this area. The recent advancement of AI- and ML-based ADME/T prediction imputes heterogeneous drug discovery data like cell-based assay and biochemical activities [174]. Prediction of lipophilicity is an important physicochemical property in the drug discovery process because lipophilicity stands in modulating several key pharmacokinetic properties. Lipophilicity of lead molecules explicitly affects the membrane permeability of lead molecules and impacts ADME behaviours [172–177]. Traditionally, octanol–water partition coefficient/pH-dependent distribution coefficient (log D) and alternative method liposome/water partitioning and immobilized artificial membrane (IAM) methods are used as a standard gold method for predicting quantitative characterization lipophilicity. However, the conventional computational approaches such as group contribution method (GC), equation state, quantum chemistry-driven methods like molecular simulation, and linear/nonlinear QSAR are used as highly adopted methods to predict the highly correlated log P/log D with molecular descriptors [178]. Riniker et al. developed novel molecular dynamics feature representing MDFP + to predict the log P. They found more information-rich fingerprints than rigorous MD calculation [179, 180]. Hence, the AI-based methods are utilized and predict log P. However, the protocols may differ from each other based on the accuracy and efficiency of all trained datasets, which limits the applicability domain compared to other physics-based methods [181].
Challenges and scope
In recent years, AI has often been used in the pharmaceutical and biomedical industries. These industries heavily adopt several AI-based tools with more efficient and automated processes that integrate predictive and data-driven decisions (Table 1). It provides a significant amount of data that is utilized for successive trained models. The data accession model from various databases highly suffered due to extra cost to a company. Also, the data should be reliable and in large quantity to ensure the pharmaceutical analysis [182]. In addition, the lack of skilled persons to operate the AI-based platforms is the other challenge that prevents full-fledged adoption of AI in the pharma and biomedical industries. The limited budget for a small organization and the replacement of humans in pharma and other large-scale companies with AI and ML tools leads to job loss. In such cases, the data generated by AL may be incredulity due to the black box phenomenon. Many companies widely adopt AI platforms, and it is projected that revenues of US$2.199 billion will be generated by 2022 in the pharmaceutical sector. Hence, pharma and biomedical companies and other organizations need clarity about the potential of AI- and ML-based tools to find the appropriate solution to problems once implemented with a clear understanding of the problems [183]. The specific task of drug development and clinical trials and sales will take more time, and all these can be narrowed by AI and ML programmes. Hence, it needs programmers with deep knowledge of thinking abilities of AI- and ML-based techniques and skilled data scientists and also a transparent company business target that can be adapted to use the full potential of AI and ML platforms [184].
Table1.
Various ML/DL tools in the drug discovery process with respective descriptions
| Description | Tool | Technique | Websites |
|---|---|---|---|
| Library of high-quality AI algorithm for drug discovery | DeepChem | Python | https://github.com/deepchem/deepchem |
| Molecular properties prediction | Neural graph fingerprints | CNN to generate molecular fingerprints | https://github.com/HIPS/neural-fingerprint |
| Conv_qsar_fast | Tensor-based CNN | https://github.com/connorcoley/conv_qsar_fast | |
| InnerOuterRNN | Two kinds of RNN | https://github.com/Chemoinformatics/InnerOuterRNN | |
| Molecular activity prediction | DeepNeuralNet-QSAR | Multitask DNN | https://github.com/Merck/DeepNeuralNet-QSAR |
| de novo molecule design with desired properties | ORGANIC (Sanchez-Lengeling 2017) | Generative model | https://github.com/aspuru-guzik-group/ORGANIC |
| REINVENT | Generative model using RNN and reinforcement learning | https://github.com/MarcusOlivecrona/REINVENT | |
| JunctionTree VAE (Jin et al. 2018) | Generative model based on JunctionTree VAE | https://github.com/wengong-jin/icml18-jtnn/tree/master/molvae | |
| Synthetic complexity of the molecule | SCScore | Score evaluation | https://github.com/connorcoley/scscore |
| Combining the RF with AutoDock scoring function | DeltaVina | Rescoring approach | https://github.com/chengwang88/deltavina |
AI- and ML-based platforms also make a significant contribution to the drug development process to find the correct dosage form and its optimization, helping to aid quick decision-making for faster manufacturing with good-quality products [185]. It can help to predict the customers using AI, and lead to improvements in predicting the ability [186]. In future, multiple research opportunities will emerge related to different customer purchase behaviours and marketing. In health care domains, AI- and ML-based techniques will be helpful to ensure the adoption in daily clinical practice [187]. The utilization of AI and ML systems increases the efficiency of handling large-scale clinical data and increases their efforts to care for patients [188]. The research finding should use AI and ML technologies for future investigation in smart logistics in industrial firms for effective implementation of technologies in various research areas like mechanical engineering, statistics, or mathematics in future projects. It also pays to establish the safety-cum efficacy of the clinical trials and ensure proper positioning via comprehensive market analysis and prediction. Hence, AI and ML tools will become vital approaches in the pharma and biomedical industries in future.
Concluding remarks and future perspectives
ML and DL methods of AI can be widely used in the pharmaceutical field to understand the chemical structure and activity relationship of lead molecules from many pharmaceutical data. In recent days, AI-based tools have been used in computer-assisted drug development because of their excellent data mining capability. However, this method has some issues, for example, a large amount of data in the data mining technology directly influence the performance of both deep learning and machine learning methods. Since the successful formation of deep learning methods has a potential approach to overcoming these problems, another major disadvantage is that understanding the mechanism of deep learning models remains unclear. In addition, neural models of formation are involved in adjusting different parameters, but only a few practical guidelines have optimized these models. The applications of AI-based techniques have mainly been increased in recent years. The large data sets form the drug discovery process, such as de novo design and identification of lead molecules. With advances in different fields, it can expect the trend towards the more automated drug discovery process with the help of computers and produce more accurate results than other methods. Therefore, further research in essential and missing areas, new ideas in biological research fields, and a drug discovery pipeline could provide several findings in the drug design process.
Acknowledgements
The authors CS and SKS thankfully acknowledge the Tamil Nadu State Council for Higher Education (TANSCHE) for the research grant (Au/S.o. (P&D): TANSCHE Projects: 117/2021). The authors acknowledge Dr. John David Raja, Department of English, Thiyagarajar College, Madurai for language and Grammatical error Check.
Declarations
Conflict of interest
The authors declare that there is no conflict of interest.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Chandrabose Selvaraj, Email: selnikraj@bioclues.org.
Sanjeev Kumar Singh, Email: skysanjeev@gmail.com.
References
- 1.Sanal MG, Paul K, Kumar S, et al. Artificial intelligence and deep learning: the future of medicine and medical practice. J Assoc Physicians India. 2019;67:71–73. [PubMed] [Google Scholar]
- 2.Sousa MJ, Pesqueira AM, Lemos C, et al. Decision-making based on big data analytics for people management in healthcare organizations. J Med Syst. 2019;43:290. doi: 10.1007/s10916-019-1419-x. [DOI] [PubMed] [Google Scholar]
- 3.Vamathevan J, Clark D, Czodrowski P, et al. Applications of machine learning in drug discovery and development. Nat Rev Drug Discov. 2019;18:463–477. doi: 10.1038/s41573-019-0024-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mohs RC, Greig NH. Drug discovery and development: role of basic biological research. Alzheimers Dement (N Y) 2017;3:651–657. doi: 10.1016/j.trci.2017.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Paul D, Sanap G, Shenoy S, et al. Artificial intelligence in drug discovery and development. Drug Discov Today. 2021;26:80–93. doi: 10.1016/j.drudis.2020.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mak KK, Pichika MR. Artificial intelligence in drug development: present status and future prospects. Drug Discov Today. 2019;24:773–780. doi: 10.1016/j.drudis.2018.11.014. [DOI] [PubMed] [Google Scholar]
- 7.Chan HCS, Shan H, Dahoun T, et al. Advancing drug discovery via artificial intelligence. Trends Pharmacol Sci. 2019;40:592–604. doi: 10.1016/j.tips.2019.06.004. [DOI] [PubMed] [Google Scholar]
- 8.Vanommeslaeghe K, Guvench O, MacKerell AD., Jr Molecular mechanics. Curr Pharm Des. 2014;20:3281–3292. doi: 10.2174/13816128113199990600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bryce RA, Hillier IH. Quantum chemical approaches: semiempirical molecular orbital and hybrid quantum mechanical/molecular mechanical techniques. Curr Pharm Des. 2014;20:3293–3302. doi: 10.2174/13816128113199990601. [DOI] [PubMed] [Google Scholar]
- 10.Nagarajan N, Yapp EKY, Le NQK, et al. Application of computational biology and artificial intelligence technologies in cancer precision drug discovery. Biomed Res Int. 2019;2019:8427042. doi: 10.1155/2019/8427042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Souza PCT, Thallmair S, Conflitti P, et al. Protein-ligand binding with the coarse-grained Martini model. Nat Commun. 2020;11:3714. doi: 10.1038/s41467-020-17437-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ahuja AS. The impact of artificial intelligence in medicine on the future role of the physician. PeerJ. 2019;7:e7702. doi: 10.7717/peerj.7702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Davenport T, Kalakota R. The potential for artificial intelligence in healthcare. Future Healthc J. 2019;6:94–98. doi: 10.7861/futurehosp.6-2-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carpenter KA, Cohen DS, Jarrell JT, et al. Deep learning and virtual drug screening. Future Med Chem. 2018;10:2557–2567. doi: 10.4155/fmc-2018-0314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aguiar-Pulido V, Gestal M, Cruz-Monteagudo M, et al. Evolutionary computation and QSAR research. Curr Comput Aided Drug Des. 2013;9:206–225. doi: 10.2174/1573409911309020006. [DOI] [PubMed] [Google Scholar]
- 16.Zador AM. A critique of pure learning and what artificial neural networks can learn from animal brains. Nat Commun. 2019;10:3770. doi: 10.1038/s41467-019-11786-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alzahab NA, Apollonio L, Di Iorio A, et al. Hybrid deep learning (hDL)-based brain-computer interface (BCI) systems: a systematic review. Brain Sci. 2021 doi: 10.3390/brainsci11010075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ekins S, Mestres J, Testa B. In silico pharmacology for drug discovery: methods for virtual ligand screening and profiling. Br J Pharmacol. 2007;152:9–20. doi: 10.1038/sj.bjp.0707305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ahmed Z, Mohamed K, Zeeshan S, et al. Artificial intelligence with multi-functional machine learning platform development for better healthcare and precision medicine. Database (Oxford) 2020 doi: 10.1093/database/baaa010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Henry J, Wlodkowic D. Towards high-throughput chemobehavioural phenomics in neuropsychiatric drug discovery. Mar Drugs. 2019 doi: 10.3390/md17060340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pesapane F, Tantrige P, Patella F, et al. Myths and facts about artificial intelligence: why machine- and deep-learning will not replace interventional radiologists. Med Oncol. 2020;37:40. doi: 10.1007/s12032-020-01368-8. [DOI] [PubMed] [Google Scholar]
- 22.Sakellaropoulos T, Vougas K, Narang S, et al. A deep learning framework for predicting response to therapy in cancer. Cell Rep. 2019;29(3367–3373):e3364. doi: 10.1016/j.celrep.2019.11.017. [DOI] [PubMed] [Google Scholar]
- 23.Hodas NO, Stinis P. Doing the impossible: why neural networks can be trained at all. Front Psychol. 2018;9:1185. doi: 10.3389/fpsyg.2018.01185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Poggio T, Banburski A, Liao Q. Theoretical issues in deep networks. Proc Natl Acad Sci U S A. 2020;117:30039–30045. doi: 10.1073/pnas.1907369117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Del Fiol G, Michelson M, Iorio A, et al. A deep learning method to automatically identify reports of scientifically rigorous clinical research from the biomedical literature: comparative analytic study. J Med Internet Res. 2018;20:e10281. doi: 10.2196/10281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yamashita R, Nishio M, Do RKG, et al. Convolutional neural networks: an overview and application in radiology. Insights Imag. 2018;9:611–629. doi: 10.1007/s13244-018-0639-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Trabelsi A, Chaabane M, Ben-Hur A. Comprehensive evaluation of deep learning architectures for prediction of DNA/RNA sequence binding specificities. Bioinformatics. 2019;35:i269–i277. doi: 10.1093/bioinformatics/btz339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ben-Bassat I, Chor B, Orenstein Y. A deep neural network approach for learning intrinsic protein-RNA binding preferences. Bioinformatics. 2018;34:i638–i646. doi: 10.1093/bioinformatics/bty600. [DOI] [PubMed] [Google Scholar]
- 29.Graupe D, Vern B. On the inter-relations between artificial and physiological neural networks. Neurol Res. 2001;23:482–488. doi: 10.1179/016164101101198875. [DOI] [PubMed] [Google Scholar]
- 30.Lee D, Yoon SN. Application of artificial intelligence-based technologies in the healthcare industry: opportunities and challenges. Int J Environ Res Public Health. 2021 doi: 10.3390/ijerph18010271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Agatonovic-Kustrin S, Beresford R. Basic concepts of artificial neural network (ANN) modeling and its application in pharmaceutical research. J Pharm Biomed Anal. 2000;22:717–727. doi: 10.1016/s0731-7085(99)00272-1. [DOI] [PubMed] [Google Scholar]
- 32.Narhi M, Salmela L, Toivonen J, et al. Machine learning analysis of extreme events in optical fibre modulation instability. Nat Commun. 2018;9:4923. doi: 10.1038/s41467-018-07355-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ravindranath PA, Forli S, Goodsell DS, et al. AutoDockFR: advances in protein-ligand docking with explicitly specified binding site flexibility. PLoS Comput Biol. 2015;11:e1004586. doi: 10.1371/journal.pcbi.1004586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lavecchia A, Di Giovanni C. Virtual screening strategies in drug discovery: a critical review. Curr Med Chem. 2013;20:2839–2860. doi: 10.2174/09298673113209990001. [DOI] [PubMed] [Google Scholar]
- 35.You J, McLeod RD, Hu P. Predicting drug-target interaction network using deep learning model. Comput Biol Chem. 2019;80:90–101. doi: 10.1016/j.compbiolchem.2019.03.016. [DOI] [PubMed] [Google Scholar]
- 36.Schenone M, Dancik V, Wagner BK, et al. Target identification and mechanism of action in chemical biology and drug discovery. Nat Chem Biol. 2013;9:232–240. doi: 10.1038/nchembio.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Singh RK, Lee JK, Selvaraj C, et al. Protein engineering approaches in the post-genomic era. Curr Protein Pept Sci. 2018;19:5–15. doi: 10.2174/1389203718666161117114243. [DOI] [PubMed] [Google Scholar]
- 38.Lima AN, Philot EA, Trossini GH, et al. Use of machine learning approaches for novel drug discovery. Expert Opin Drug Discov. 2016;11:225–239. doi: 10.1517/17460441.2016.1146250. [DOI] [PubMed] [Google Scholar]
- 39.Costa PR, Acencio ML, Lemke N. A machine learning approach for genome-wide prediction of morbid and druggable human genes based on systems-level data. BMC Genom. 2010;11(Suppl 5):S9. doi: 10.1186/1471-2164-11-S5-S9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Qi Y, Bar-Joseph Z, Klein-Seetharaman J. Evaluation of different biological data and computational classification methods for use in protein interaction prediction. Proteins. 2006;63:490–500. doi: 10.1002/prot.20865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang M, Su Q, Lu Y, et al. Application of machine learning approaches for protein-protein interactions prediction. Med Chem. 2017;13:506–514. doi: 10.2174/1573406413666170522150940. [DOI] [PubMed] [Google Scholar]
- 42.Sakkiah S, Selvaraj C, Gong P, et al. Development of estrogen receptor beta binding prediction model using large sets of chemicals. Oncotarget. 2017;8:92989–93000. doi: 10.18632/oncotarget.21723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Doane AS, Elemento O. Regulatory elements in molecular networks. Wiley Interdiscip Rev Syst Biol Med. 2017 doi: 10.1002/wsbm.1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu ZP. Reverse engineering of genome-wide gene regulatory networks from gene expression data. Curr Genom. 2015;16:3–22. doi: 10.2174/1389202915666141110210634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Volk MJ, Lourentzou I, Mishra S, et al. Biosystems design by machine learning. ACS Synth Biol. 2020;9:1514–1533. doi: 10.1021/acssynbio.0c00129. [DOI] [PubMed] [Google Scholar]
- 46.Jeon J, Nim S, Teyra J, et al. A systematic approach to identify novel cancer drug targets using machine learning, inhibitor design and high-throughput screening. Genome Med. 2014;6:57. doi: 10.1186/s13073-014-0057-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mamoshina P, Volosnikova M, Ozerov IV, et al. Machine learning on human muscle transcriptomic data for biomarker discovery and tissue-specific drug target identification. Front Genet. 2018;9:242. doi: 10.3389/fgene.2018.00242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Consortium GT The genotype-tissue expression (GTEx) project. Nat Genet. 2013;45:580–585. doi: 10.1038/ng.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ljosa V, Caie PD, Ter Horst R, et al. Comparison of methods for image-based profiling of cellular morphological responses to small-molecule treatment. J Biomol Screen. 2013;18:1321–1329. doi: 10.1177/1087057113503553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Aristotelous T, Ahn S, Shukla AK, et al. Discovery of beta2 adrenergic receptor ligands using biosensor fragment screening of tagged wild-type receptor. ACS Med Chem Lett. 2013;4:1005–1010. doi: 10.1021/ml400312j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Swinney DC, Lee JA. Recent advances in phenotypic drug discovery. F1000Research. 2020 doi: 10.12688/f1000research.25813.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lee JA, Berg EL. Neoclassic drug discovery: the case for lead generation using phenotypic and functional approaches. J Biomol Screen. 2013;18:1143–1155. doi: 10.1177/1087057113506118. [DOI] [PubMed] [Google Scholar]
- 53.Scheeder C, Heigwer F, Boutros M. Machine learning and image-based profiling in drug discovery. Curr Opin Syst Biol. 2018;10:43–52. doi: 10.1016/j.coisb.2018.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhavoronkov A, Vanhaelen Q, Oprea TI. Will artificial intelligence for drug discovery impact clinical pharmacology? Clin Pharmacol Ther. 2020;107:780–785. doi: 10.1002/cpt.1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Valentini G, Paccanaro A, Caniza H, et al. An extensive analysis of disease-gene associations using network integration and fast kernel-based gene prioritization methods. Artif Intell Med. 2014;61:63–78. doi: 10.1016/j.artmed.2014.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ferrero E, Dunham I, Sanseau P. In silico prediction of novel therapeutic targets using gene-disease association data. J Transl Med. 2017;15:182. doi: 10.1186/s12967-017-1285-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Arabfard M, Ohadi M, Rezaei Tabar V, et al. Genome-wide prediction and prioritization of human aging genes by data fusion: a machine learning approach. BMC Genom. 2019;20:832. doi: 10.1186/s12864-019-6140-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Selvaraj C, Vierra M, Dinesh DC, et al. Structural insights of macromolecules involved in bacteria-induced apoptosis in the pathogenesis of human diseases. Adv Protein Chem Struct Biol. 2021;126:1–38. doi: 10.1016/bs.apcsb.2021.02.001. [DOI] [PubMed] [Google Scholar]
- 59.Vapnik VN. An overview of statistical learning theory. IEEE Trans Neural Netw. 1999;10:988–999. doi: 10.1109/72.788640. [DOI] [PubMed] [Google Scholar]
- 60.Burbidge R, Trotter M, Buxton B, et al. Drug design by machine learning: support vector machines for pharmaceutical data analysis. Comput Chem. 2001;26:5–14. doi: 10.1016/s0097-8485(01)00094-8. [DOI] [PubMed] [Google Scholar]
- 61.Abd-Alrazaq A, Alajlani M, Alhuwail D, et al. Artificial intelligence in the fight against COVID-19: scoping review. J Med Internet Res. 2020;22:e20756. doi: 10.2196/20756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Maltarollo VG, Kronenberger T, Espinoza GZ, et al. Advances with support vector machines for novel drug discovery. Expert Opin Drug Discov. 2019;14:23–33. doi: 10.1080/17460441.2019.1549033. [DOI] [PubMed] [Google Scholar]
- 63.Li J, Weng Z, Xu H, et al. Support vector machines (SVM) classification of prostate cancer Gleason score in central gland using multiparametric magnetic resonance images: a cross-validated study. Eur J Radiol. 2018;98:61–67. doi: 10.1016/j.ejrad.2017.11.001. [DOI] [PubMed] [Google Scholar]
- 64.Tao Q, Chu D, Wang J. Recursive support vector machines for dimensionality reduction. IEEE Trans Neural Netw. 2008;19:189–193. doi: 10.1109/TNN.2007.908267. [DOI] [PubMed] [Google Scholar]
- 65.Wilton DJ, Harrison RF, Willett P, et al. Virtual screening using binary kernel discrimination: analysis of pesticide data. J Chem Inf Model. 2006;46:471–477. doi: 10.1021/ci050397w. [DOI] [PubMed] [Google Scholar]
- 66.Geppert H, Horvath T, Gartner T, et al. Support-vector-machine-based ranking significantly improves the effectiveness of similarity searching using 2D fingerprints and multiple reference compounds. J Chem Inf Model. 2008;48:742–746. doi: 10.1021/ci700461s. [DOI] [PubMed] [Google Scholar]
- 67.Franke L, Byvatov E, Werz O, et al. Extraction and visualization of potential pharmacophore points using support vector machines: application to ligand-based virtual screening for COX-2 inhibitors. J Med Chem. 2005;48:6997–7004. doi: 10.1021/jm050619h. [DOI] [PubMed] [Google Scholar]
- 68.Lepp Z, Kinoshita T, Chuman H (2006) Screening for new antidepressant leads of multiple activities by support vector machines. J chem inform model 46:158–67. 10.1021/ci050301y [DOI] [PubMed]
- 69.Chicco D, Jurman G. The advantages of the Matthews correlation coefficient (MCC) over F1 score and accuracy in binary classification evaluation. BMC Genom. 2020;21:6. doi: 10.1186/s12864-019-6413-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jorissen RN, Gilson MK (2005) Virtual screening of molecular databases using a support vector machine. J chem inform model 45:549–61. 10.1021/ci049641u [DOI] [PubMed]
- 71.Aversa A, Duca Y, Condorelli RA, et al. Androgen deficiency and phosphodiesterase type 5 expression changes in aging male: therapeutic implications. Front Endocrinol (Lausanne) 2019;10:225. doi: 10.3389/fendo.2019.00225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lo YC, Rensi SE, Torng W, et al. Machine learning in chemoinformatics and drug discovery. Drug Discov Today. 2018;23:1538–1546. doi: 10.1016/j.drudis.2018.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Huang S, Cai N, Pacheco PP, et al. Applications of support vector machine (SVM) learning in cancer genomics. Cancer Genom Proteom. 2018;15:41–51. doi: 10.21873/cgp.20063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lionta E, Spyrou G, Vassilatis DK, et al. Structure-based virtual screening for drug discovery: principles, applications and recent advances. Curr Top Med Chem. 2014;14:1923–1938. doi: 10.2174/1568026614666140929124445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mahe P, Ralaivola L, Stoven V, et al. The pharmacophore kernel for virtual screening with support vector machines. J Chem Inf Model. 2006;46:2003–2014. doi: 10.1021/ci060138m. [DOI] [PubMed] [Google Scholar]
- 76.Eitrich T, Kless A, Druska C, Meyer W, Grotendorst J (2007) Classification of highly unbalanced CYP450 data of drugs using cost sensitive machine learning techniques. J chem inform model 47:92–103. 10.1021/ci6002619 [DOI] [PubMed]
- 77.Ben-Hur A, Ong CS, Sonnenburg S, et al. Support vector machines and kernels for computational biology. PLoS Comput Biol. 2008;4:e1000173. doi: 10.1371/journal.pcbi.1000173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ballester PJ, Mitchell JB. A machine learning approach to predicting protein-ligand binding affinity with applications to molecular docking. Bioinformatics. 2010;26:1169–1175. doi: 10.1093/bioinformatics/btq112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Nguyen DD, Wei GW. AGL-score: algebraic graph learning score for protein-ligand binding scoring, ranking, docking, and screening. J Chem Inf Model. 2019;59:3291–3304. doi: 10.1021/acs.jcim.9b00334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Guedes IA, Pereira FSS, Dardenne LE. Empirical scoring functions for structure-based virtual screening: applications, critical aspects, and challenges. Front Pharmacol. 2018;9:1089. doi: 10.3389/fphar.2018.01089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Brown BP, Mendenhall J, Geanes AR, et al. General purpose structure-based drug discovery neural network score functions with human-interpretable pharmacophore maps. J Chem Inf Model. 2021;61:603–620. doi: 10.1021/acs.jcim.0c01001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Li H, Leung KS, Wong MH, et al. Improving AutoDock Vina using random forest: the growing accuracy of binding affinity prediction by the effective exploitation of larger data sets. Mol Inform. 2015;34:115–126. doi: 10.1002/minf.201400132. [DOI] [PubMed] [Google Scholar]
- 83.Jimenez J, Skalic M, Martinez-Rosell G, et al. KDEEP: protein-ligand absolute binding affinity prediction via 3D-convolutional neural networks. J Chem Inf Model. 2018;58:287–296. doi: 10.1021/acs.jcim.7b00650. [DOI] [PubMed] [Google Scholar]
- 84.Kumar S, Kim MH. SMPLIP-score: predicting ligand binding affinity from simple and interpretable on-the-fly interaction fingerprint pattern descriptors. J Cheminform. 2021;13:28. doi: 10.1186/s13321-021-00507-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sharma K, Patidar K, Ali MA, et al. Structure-based virtual screening for the identification of high affinity compounds as potent VEGFR2 inhibitors for the treatment of renal cell carcinoma. Curr Top Med Chem. 2018;18:2174–2185. doi: 10.2174/1568026619666181130142237. [DOI] [PubMed] [Google Scholar]
- 86.Patidar K, Deshmukh A, Bandaru S, et al. Virtual screening approaches in identification of bioactive compounds Akin to delphinidin as potential HER2 inhibitors for the treatment of breast cancer. Asian Pac J Cancer Prev. 2016;17:2291–2295. doi: 10.7314/apjcp.2016.17.4.2291. [DOI] [PubMed] [Google Scholar]
- 87.Sliwoski G, Kothiwale S, Meiler J, et al. Computational methods in drug discovery. Pharmacol Rev. 2014;66:334–395. doi: 10.1124/pr.112.007336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Reddy KK, Singh SK. Combined ligand and structure-based approaches on HIV-1 integrase strand transfer inhibitors. Chem Biol Interact. 2014;218:71–81. doi: 10.1016/j.cbi.2014.04.011. [DOI] [PubMed] [Google Scholar]
- 89.Subramanian I, Verma S, Kumar S, et al. Multi-omics Data Integration, Interpretation, and Its Application. Bioinform Biol Insights. 2020;14:1177932219899051. doi: 10.1177/1177932219899051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Cova T, Pais A. Deep learning for deep chemistry: optimizing the prediction of chemical patterns. Front Chem. 2019;7:809. doi: 10.3389/fchem.2019.00809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Brereton RG. Self organising maps for visualising and modelling. Chem Cent J. 2012;6(Suppl 2):S1. doi: 10.1186/1752-153X-6-S2-S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Cherkasov A, Muratov EN, Fourches D, et al. QSAR modeling: where have you been? Where are you going to? J Med Chem. 2014;57:4977–5010. doi: 10.1021/jm4004285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Palyulin VA, Radchenko EV, Zefirov NS. Molecular field topology analysis method in QSAR studies of organic compounds. J Chem Inf Comput Sci. 2000;40:659–667. doi: 10.1021/ci980114i. [DOI] [PubMed] [Google Scholar]
- 94.Mouchlis VD, Afantitis A, Serra A, et al. Advances in de novo drug design: from conventional to machine learning methods. Int J Mol Sci. 2021 doi: 10.3390/ijms22041676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Gurevich EV, Gurevich VV. Therapeutic potential of small molecules and engineered proteins. Handb Exp Pharmacol. 2014;219:1–12. doi: 10.1007/978-3-642-41199-1_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Yang X, Wang Y, Byrne R, et al. Concepts of artificial intelligence for computer-assisted drug discovery. Chem Rev. 2019;119:10520–10594. doi: 10.1021/acs.chemrev.8b00728. [DOI] [PubMed] [Google Scholar]
- 97.Lind AP, Anderson PC. Predicting drug activity against cancer cells by random forest models based on minimal genomic information and chemical properties. PLoS ONE. 2019;14:e0219774. doi: 10.1371/journal.pone.0219774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Rodrigues T, Werner M, Roth J, et al. Machine intelligence decrypts beta-lapachone as an allosteric 5-lipoxygenase inhibitor. Chem Sci. 2018;9:6899–6903. doi: 10.1039/c8sc02634c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Garscha U, Voelker S, Pace S, et al. BRP-187: a potent inhibitor of leukotriene biosynthesis that acts through impeding the dynamic 5-lipoxygenase/5-lipoxygenase-activating protein (FLAP) complex assembly. Biochem Pharmacol. 2016;119:17–26. doi: 10.1016/j.bcp.2016.08.023. [DOI] [PubMed] [Google Scholar]
- 100.Park EJ, Myint PK, Ito A, et al. Integrin-ligand interactions in inflammation, cancer, and metabolic disease: insights into the multifaceted roles of an emerging ligand irisin. Front Cell Dev Biol. 2020;8:588066. doi: 10.3389/fcell.2020.588066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Freedman JD, Hagel J, Scott EM, et al. Oncolytic adenovirus expressing bispecific antibody targets T-cell cytotoxicity in cancer biopsies. EMBO Mol Med. 2017;9:1067–1087. doi: 10.15252/emmm.201707567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Smak P, Chandrabose S, Tvaroska I, et al. Pan-selectin inhibitors as potential therapeutics for COVID-19 treatment: in silico screening study. Glycobiology. 2021 doi: 10.1093/glycob/cwab021.10.1093/glycob/cwab021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Batool M, Ahmad B, Choi S. A structure-based drug discovery paradigm. Int J Mol Sci. 2019 doi: 10.3390/ijms20112783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Rai DK, Rieder E. Homology modeling and analysis of structure predictions of the bovine rhinitis B virus RNA dependent RNA polymerase (RdRp) Int J Mol Sci. 2012;13:8998–9013. doi: 10.3390/ijms13078998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Laurie AT, Jackson RM. Q-SiteFinder: an energy-based method for the prediction of protein-ligand binding sites. Bioinformatics. 2005;21:1908–1916. doi: 10.1093/bioinformatics/bti315. [DOI] [PubMed] [Google Scholar]
- 106.Cheng T, Li Q, Zhou Z, et al. Structure-based virtual screening for drug discovery: a problem-centric review. AAPS J. 2012;14:133–141. doi: 10.1208/s12248-012-9322-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Dos Santos RN, Ferreira LG, Andricopulo AD. Practices in molecular docking and structure-based virtual screening. Methods Mol Biol. 2018;1762:31–50. doi: 10.1007/978-1-4939-7756-7_3. [DOI] [PubMed] [Google Scholar]
- 108.Esteva A, Robicquet A, Ramsundar B, et al. A guide to deep learning in healthcare. Nat Med. 2019;25:24–29. doi: 10.1038/s41591-018-0316-z. [DOI] [PubMed] [Google Scholar]
- 109.Ramsundar B, Liu B, Wu Z, et al. Is Multitask deep learning practical for pharma? J Chem Inf Model. 2017;57:2068–2076. doi: 10.1021/acs.jcim.7b00146. [DOI] [PubMed] [Google Scholar]
- 110.Akinc A, Zumbuehl A, Goldberg M, et al. A combinatorial library of lipid-like materials for delivery of RNAi therapeutics. Nat Biotechnol. 2008;26:561–569. doi: 10.1038/nbt1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hauser AS, Attwood MM, Rask-Andersen M, et al. Trends in GPCR drug discovery: new agents, targets and indications. Nat Rev Drug Discov. 2017;16:829–842. doi: 10.1038/nrd.2017.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Riddick G, Song H, Ahn S, et al. Predicting in vitro drug sensitivity using random forests. Bioinformatics. 2011;27:220–224. doi: 10.1093/bioinformatics/btq628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Mayr A, Klambauer G, Unterthiner T, Hochreiter S. DeepTox: toxicity prediction using deep learning. Front Environ Sci. 2016;3:1–15. doi: 10.3389/fenvs.2015.00080. [DOI] [Google Scholar]
- 114.Idakwo G, Thangapandian S, Jt L, et al. Deep learning-based structure-activity relationship modeling for multi-category toxicity classification: a case study of 10K Tox21 chemicals with high-throughput cell-based androgen receptor bioassay data. Front Physiol. 2019;10:1044. doi: 10.3389/fphys.2019.01044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Bjerrum EJ, Sattarov B. Improving chemical autoencoder latent space and molecular de novo generation diversity with heteroencoders. Biomolecules. 2018 doi: 10.3390/biom8040131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Pradiba D, Aarthy M, Shunmugapriya V, Singh SK, Vasanthi M (2018) Structural insights into the binding mode of flavonols with the active site of matrix metalloproteinase-9 through molecular docking and molecular dynamic simulations studies. J Biomol Struct Dynamics 36:3718-3739. 10.1080/07391102.2017.1397058 [DOI] [PubMed]
- 117.Ashtawy HM, Mahapatra NR. Boosted neural networks scoring functions for accurate ligand docking and ranking. J Bioinform Comput Biol. 2018;16:1850004. doi: 10.1142/S021972001850004X. [DOI] [PubMed] [Google Scholar]
- 118.Seo S, Choi J, Ahn SK, et al. Prediction of GPCR-ligand binding using machine learning algorithms. Comput Math Methods Med. 2018;2018:6565241. doi: 10.1155/2018/6565241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Pinzi L, Rastelli G. Molecular docking: shifting paradigms in drug discovery. Int J Mol Sci. 2019 doi: 10.3390/ijms20184331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Bruno A, Costantino G, Sartori L, et al. The in silico drug discovery toolbox: applications in lead discovery and optimization. Curr Med Chem. 2019;26:3838–3873. doi: 10.2174/0929867324666171107101035. [DOI] [PubMed] [Google Scholar]
- 121.Nayarisseri A, Khandelwal R, Madhavi M, et al. Shape-based machine learning models for the potential novel COVID-19 protease inhibitors assisted by molecular dynamics simulation. Curr Top Med Chem. 2020;20:2146–2167. doi: 10.2174/1568026620666200704135327. [DOI] [PubMed] [Google Scholar]
- 122.Omer A, Suryanarayanan V, Selvaraj C, et al. Explicit drug re-positioning: predicting novel drug-target interactions of the shelved molecules with QM/MM based approaches. Adv Protein Chem Struct Biol. 2015;100:89–112. doi: 10.1016/bs.apcsb.2015.07.001. [DOI] [PubMed] [Google Scholar]
- 123.Selvaraj C, Omer A, Singh P, et al. Molecular insights of protein contour recognition with ligand pharmacophoric sites through combinatorial library design and MD simulation in validating HTLV-1 PR inhibitors. Mol Biosyst. 2015;11:178–189. doi: 10.1039/c4mb00486h. [DOI] [PubMed] [Google Scholar]
- 124.Li Q, Lai L. Prediction of potential drug targets based on simple sequence properties. BMC Bioinform. 2007;8:353. doi: 10.1186/1471-2105-8-353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Gao M, Zhou H, Skolnick J. DESTINI: a deep-learning approach to contact-driven protein structure prediction. Sci Rep. 2019;9:3514. doi: 10.1038/s41598-019-40314-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Liu W, Meng X, Xu Q, et al. Quantitative prediction of mouse class I MHC peptide binding affinity using support vector machine regression (SVR) models. BMC Bioinform. 2006;7:182. doi: 10.1186/1471-2105-7-182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Lin E, Lin CH, Lane HY. Relevant applications of generative adversarial networks in drug design and discovery: molecular de novo design, dimensionality reduction, and de novo peptide and protein design. Molecules. 2020 doi: 10.3390/molecules25143250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Grisoni F, Moret M, Lingwood R, et al. Bidirectional molecule generation with recurrent neural networks. J Chem Inf Model. 2020;60:1175–1183. doi: 10.1021/acs.jcim.9b00943. [DOI] [PubMed] [Google Scholar]
- 129.Pogany P, Arad N, Genway S, et al. De novo molecule design by translating from reduced graphs to SMILES. J Chem Inf Model. 2019;59:1136–1146. doi: 10.1021/acs.jcim.8b00626. [DOI] [PubMed] [Google Scholar]
- 130.Kell DB, Samanta S, Swainston N. Deep learning and generative methods in cheminformatics and chemical biology: navigating small molecule space intelligently. Biochem J. 2020;477:4559–4580. doi: 10.1042/BCJ20200781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Prykhodko O, Johansson SV, Kotsias PC, et al. A de novo molecular generation method using latent vector based generative adversarial network. J Cheminform. 2019;11:74. doi: 10.1186/s13321-019-0397-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Schneider P, Walters WP, Plowright AT, et al. Rethinking drug design in the artificial intelligence era. Nat Rev Drug Discov. 2020;19:353–364. doi: 10.1038/s41573-019-0050-3. [DOI] [PubMed] [Google Scholar]
- 133.Aparoy P, Reddy KK, Reddanna P. Structure and ligand based drug design strategies in the development of novel 5- LOX inhibitors. Curr Med Chem. 2012;19:3763–3778. doi: 10.2174/092986712801661112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Schmidt T, Bergner A, Schwede T. Modelling three-dimensional protein structures for applications in drug design. Drug Discov Today. 2014;19:890–897. doi: 10.1016/j.drudis.2013.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Fang C, Shang Y, Xu D. Prediction of protein backbone torsion angles using deep residual inception neural networks. IEEE/ACM Trans Comput Biol Bioinform. 2018 doi: 10.1109/TCBB.2018.2814586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Baker D, Sali A. Protein structure prediction and structural genomics. Science. 2001;294:93–96. doi: 10.1126/science.1065659. [DOI] [PubMed] [Google Scholar]
- 137.Rao VS, Srinivas K, Sujini GN, et al. Protein-protein interaction detection: methods and analysis. Int J Proteomics. 2014;2014:147648. doi: 10.1155/2014/147648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Legrain P, Selig L. Genome-wide protein interaction maps using two-hybrid systems. FEBS Lett. 2000;480:32–36. doi: 10.1016/s0014-5793(00)01774-9. [DOI] [PubMed] [Google Scholar]
- 139.Noh S, Lee SR, Jeong YJ, et al. The direct modulatory activity of zinc toward ion channels. Integr Med Res. 2015;4:142–146. doi: 10.1016/j.imr.2015.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Ding Z, Kihara D. Computational methods for predicting protein-protein interactions using various protein features. Curr Protoc Protein Sci. 2018;93:e62. doi: 10.1002/cpps.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Gao W, Coley CW. The synthesizability of molecules proposed by generative models. J Chem Inf Model. 2020;60:5714–5723. doi: 10.1021/acs.jcim.0c00174. [DOI] [PubMed] [Google Scholar]
- 142.Wang S, Sun S, Li Z, et al. Accurate de novo prediction of protein contact map by ultra-deep learning model. PLoS Comput Biol. 2017;13:e1005324. doi: 10.1371/journal.pcbi.1005324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Selvaraj C, Dinesh DC, Panwar U, et al. High-throughput screening and quantum mechanics for identifying potent inhibitors against Mac1 Domain of SARS-CoV-2 Nsp3. IEEE/ACM Trans Comput Biol Bioinform. 2021;18:1262–1270. doi: 10.1109/TCBB.2020.3037136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Aminpour M, Montemagno C, Tuszynski JA. An overview of molecular modeling for drug discovery with specific illustrative examples of applications. Molecules. 2019 doi: 10.3390/molecules24091693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Kubar T, Elstner M. A hybrid approach to simulation of electron transfer in complex molecular systems. J R Soc Interface. 2013;10:20130415. doi: 10.1098/rsif.2013.0415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Tkatchenko A. Machine learning for chemical discovery. Nat Commun. 2020;11:4125. doi: 10.1038/s41467-020-17844-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Huang L, Massa L, Karle J. Kernel energy method: the interaction energy of the collagen triple helix. J Chem Theory Comput. 2007;3:1337–1341. doi: 10.1021/ct7000649. [DOI] [PubMed] [Google Scholar]
- 148.Smith JS, Zubatyuk R, Nebgen B, et al. The ANI-1ccx and ANI-1x data sets, coupled-cluster and density functional theory properties for molecules. Sci Data. 2020;7:134. doi: 10.1038/s41597-020-0473-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Singh SK, Dessalew N, Bharatam PV. 3D-QSAR CoMFA study on oxindole derivatives as cyclin dependent kinase 1 (CDK1) and cyclin dependent kinase 2 (CDK2) inhibitors. Med Chem. 2007;3:75–84. doi: 10.2174/157340607779317517. [DOI] [PubMed] [Google Scholar]
- 150.Selvaraj C, Selvaraj G, Mohamed Ismail R, et al. Interrogation of Bacillus anthracis SrtA active site loop forming open/close lid conformations through extensive MD simulations for understanding binding selectivity of SrtA inhibitors. Saudi J Biol Sci. 2021;28:3650–3659. doi: 10.1016/j.sjbs.2021.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Reddy KK, Singh SK, Dessalew N, et al. Pharmacophore modelling and atom-based 3D-QSAR studies on N-methyl pyrimidones as HIV-1 integrase inhibitors. J Enzyme Inhib Med Chem. 2012;27:339–347. doi: 10.3109/14756366.2011.590803. [DOI] [PubMed] [Google Scholar]
- 152.Suryanarayanan V, Singh SK, Tripathi SK, et al. A three-dimensional chemical phase pharmacophore mapping, QSAR modelling and electronic feature analysis of benzofuran salicylic acid derivatives as LYP inhibitors. SAR QSAR Environ Res. 2013;24:1025–1040. doi: 10.1080/1062936X.2013.821421. [DOI] [PubMed] [Google Scholar]
- 153.Ferreira LG, Dos Santos RN, Oliva G, et al. Molecular docking and structure-based drug design strategies. Molecules. 2015;20:13384–13421. doi: 10.3390/molecules200713384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Carpenter KA, Huang X. Machine learning-based virtual screening and its applications to Alzheimer's drug discovery: a review. Curr Pharm Des. 2018;24:3347–3358. doi: 10.2174/1381612824666180607124038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Vatansever S, Schlessinger A, Wacker D, et al. Artificial intelligence and machine learning-aided drug discovery in central nervous system diseases: state-of-the-arts and future directions. Med Res Rev. 2020 doi: 10.1002/med.21764.10.1002/med.21764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Vink G, Nebel JC, Wren SP. In silico design of bioisosteric modifications of drugs for the treatment of diabetes. Future Med Chem. 2021 doi: 10.4155/fmc-2020-0374.10.4155/fmc-2020-0374. [DOI] [PubMed] [Google Scholar]
- 157.Wang T, Yuan XS, Wu MB, et al. The advancement of multidimensional QSAR for novel drug discovery—where are we headed? Expert Opin Drug Discov. 2017;12:769–784. doi: 10.1080/17460441.2017.1336157. [DOI] [PubMed] [Google Scholar]
- 158.Dobchev D, Karelson M. Have artificial neural networks met expectations in drug discovery as implemented in QSAR framework? Expert Opin Drug Discov. 2016;11:627–639. doi: 10.1080/17460441.2016.1186876. [DOI] [PubMed] [Google Scholar]
- 159.Hong H, Rua D, Sakkiah S, et al. Consensus modeling for prediction of estrogenic activity of ingredients commonly used in sunscreen products. Int J Environ Res Public Health. 2016 doi: 10.3390/ijerph13100958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Baskin II, Palyulin VA, Zefirov NS. Neural networks in building QSAR models. Methods Mol Biol. 2008;458:137–158. [PubMed] [Google Scholar]
- 161.Meftahi N, Walker ML, Enciso M, et al. Predicting the enthalpy and gibbs energy of sublimation by QSPR modeling. Sci Rep. 2018;8:9779. doi: 10.1038/s41598-018-28105-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Ponzoni I, Sebastian-Perez V, Requena-Triguero C, et al. Hybridizing feature selection and feature learning approaches in QSAR modeling for drug discovery. Sci Rep. 2017;7:2403. doi: 10.1038/s41598-017-02114-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Goodarzi M, Dejaegher B, Vander Heyden Y. Feature selection methods in QSAR studies. J AOAC Int. 2012;95:636–651. doi: 10.5740/jaoacint.sge_goodarzi. [DOI] [PubMed] [Google Scholar]
- 164.Hefti FF. Requirements for a lead compound to become a clinical candidate. BMC Neurosci. 2008;9(Suppl 3):S7. doi: 10.1186/1471-2202-9-S3-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Meanwell NA. Improving drug candidates by design: a focus on physicochemical properties as a means of improving compound disposition and safety. Chem Res Toxicol. 2011;24:1420–1456. doi: 10.1021/tx200211v. [DOI] [PubMed] [Google Scholar]
- 166.Wang NN, Dong J, Deng YH, et al. ADME properties evaluation in drug discovery: prediction of Caco-2 cell permeability using a combination of NSGA-II and boosting. J Chem Inf Model. 2016;56:763–773. doi: 10.1021/acs.jcim.5b00642. [DOI] [PubMed] [Google Scholar]
- 167.Hou TJ, Zhang W, Xia K, et al. ADME evaluation in drug discovery. 5. Correlation of Caco-2 permeation with simple molecular properties. J Chem Inf Comput Sci. 2004;44:1585–1600. doi: 10.1021/ci049884m. [DOI] [PubMed] [Google Scholar]
- 168.Yamashita F, Wanchana S, Hashida M. Quantitative structure/property relationship analysis of Caco-2 permeability using a genetic algorithm-based partial least squares method. J Pharm Sci. 2002;91:2230–2239. doi: 10.1002/jps.10214. [DOI] [PubMed] [Google Scholar]
- 169.Castillo-Garit JA, Marrero-Ponce Y, Torrens F, et al. Estimation of ADME properties in drug discovery: predicting Caco-2 cell permeability using atom-based stochastic and non-stochastic linear indices. J Pharm Sci. 2008;97:1946–1976. doi: 10.1002/jps.21122. [DOI] [PubMed] [Google Scholar]
- 170.Pham-The H, Cabrera-Perez MA, Nam NH, et al. In silico assessment of ADME properties: advances in Caco-2 cell monolayer permeability modeling. Curr Top Med Chem. 2018;18:2209–2229. doi: 10.2174/1568026619666181130140350. [DOI] [PubMed] [Google Scholar]
- 171.Milanetti E, Raimondo D, Tramontano A. Prediction of the permeability of neutral drugs inferred from their solvation properties. Bioinformatics. 2016;32:1163–1169. doi: 10.1093/bioinformatics/btv725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Xu Y, Pei J, Lai L. Deep learning based regression and multiclass models for acute oral toxicity prediction with automatic chemical feature extraction. J Chem Inf Model. 2017;57:2672–2685. doi: 10.1021/acs.jcim.7b00244. [DOI] [PubMed] [Google Scholar]
- 173.Li X, Xu Y, Lai L, et al. Prediction of human cytochrome P450 inhibition using a multitask deep autoencoder neural network. Mol Pharm. 2018;15:4336–4345. doi: 10.1021/acs.molpharmaceut.8b00110. [DOI] [PubMed] [Google Scholar]
- 174.Koromina M, Pandi MT, Patrinos GP. Rethinking drug repositioning and development with artificial intelligence, machine learning, and omics. OMICS. 2019;23:539–548. doi: 10.1089/omi.2019.0151. [DOI] [PubMed] [Google Scholar]
- 175.Damiati SA. Digital pharmaceutical sciences. AAPS PharmSciTech. 2020;21:206. doi: 10.1208/s12249-020-01747-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Daina A, Michielin O, Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep. 2017;7:42717. doi: 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Temml V, Kutil Z. Structure-based molecular modeling in SAR analysis and lead optimization. Comput Struct Biotechnol J. 2021;19:1431–1444. doi: 10.1016/j.csbj.2021.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Avdeef A, Box KJ, Comer JE, et al. pH-metric logP 10. Determination of liposomal membrane-water partition coefficients of ionizable drugs. Pharm Res. 1998;15:209–215. doi: 10.1023/a:1011954332221. [DOI] [PubMed] [Google Scholar]
- 179.Taskinen J, Yliruusi J. Prediction of physicochemical properties based on neural network modelling. Adv Drug Deliv Rev. 2003;55:1163–1183. doi: 10.1016/s0169-409x(03)00117-0. [DOI] [PubMed] [Google Scholar]
- 180.Selvaraj C, Sakkiah S, Tong W, et al. Molecular dynamics simulations and applications in computational toxicology and nanotoxicology. Food Chem Toxicol. 2018;112:495–506. doi: 10.1016/j.fct.2017.08.028. [DOI] [PubMed] [Google Scholar]
- 181.Haghighatlari M, Li J, Heidar-Zadeh F, et al. Learning to make chemical predictions: the interplay of feature representation, data, and machine learning methods. Chem. 2020;6:1527–1542. doi: 10.1016/j.chempr.2020.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Esmaeilzadeh P. Use of AI-based tools for healthcare purposes: a survey study from consumers' perspectives. BMC Med Inform Decis Mak. 2020;20:170. doi: 10.1186/s12911-020-01191-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Thakur A, Mishra AP, Panda B, et al. Application of artificial intelligence in pharmaceutical and biomedical studies. Curr Pharm Des. 2020;26:3569–3578. doi: 10.2174/1381612826666200515131245. [DOI] [PubMed] [Google Scholar]
- 184.Ahsan MA, Liu Y, Feng C, et al. Bioinformatics resources facilitate understanding and harnessing clinical research of SARS-CoV-2. Brief Bioinform. 2021;22:714–725. doi: 10.1093/bib/bbaa416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Mansouri K, Kleinstreuer N, Abdelaziz AM, et al. CoMPARA: collaborative modeling project for androgen receptor activity. Environ Health Perspect. 2020;128:27002. doi: 10.1289/EHP5580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Henstock PV. Artificial intelligence for pharma: time for internal investment. Trends Pharmacol Sci. 2019;40:543–546. doi: 10.1016/j.tips.2019.05.003. [DOI] [PubMed] [Google Scholar]
- 187.Lamberti MJ, Wilkinson M, Donzanti BA, et al. A study on the application and use of artificial intelligence to support drug development. Clin Ther. 2019;41:1414–1426. doi: 10.1016/j.clinthera.2019.05.018. [DOI] [PubMed] [Google Scholar]
- 188.Ranjan J. Data mining in pharma sector: benefits. Int J Health Care Qual Assur. 2009;22:82–92. doi: 10.1108/09526860910927970. [DOI] [PubMed] [Google Scholar]