

Summary
The 2019 coronavirus disease (COVID-19) became a worldwide pandemic with currently no approved effective antiviral drug. Flux balance analysis (FBA) is an efficient method to analyze metabolic networks. Here, FBA was applied on human lung cells infected with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) to reposition metabolic drugs and drug combinations against the virus replication within the host tissue. Making use of expression datasets of infected lung tissue, genome-scale COVID-19-specific metabolic models were reconstructed. Then, host-specific essential genes and gene pairs were determined through in silico knockouts that permit reducing the viral biomass production without affecting the host biomass. Key pathways that are associated with COVID-19 severity in lung tissue are related to oxidative stress, ferroptosis, and pyrimidine metabolism. By in silico screening of Food and Drug Administration (FDA)-approved drugs on the putative disease-specific essential genes and gene pairs, 85 drugs and 52 drug combinations were predicted as promising candidates for COVID-19 (https://github.com/sysbiolux/DCcov).
Subject areas: Virology, Pharmaceutical science, Bioinformatics
Graphical abstract
Highlights
-
•
Metabolic modeling of COVID-19 utilized public RNA-Seq of SARS-CoV-2-infected lung
-
•
In silico knockout identified 23 human essential genes for SARS-CoV-2 replication
-
•
Drug repositioning predicted single drugs targeting essential gene pairs
-
•
Among others, pyrimidine metabolism and ferroptosis are candidate druggable pathways
Virology; Pharmaceutical science; Bioinformatics
Introduction
Constraint-based modeling (CBM) approaches have successfully been applied in fundamental research (Blätke and Bräutigam, 2019; Marinos et al., 2020; Swainston et al., 2016) especially in cancer research (Agren et al., 2012; Larsson et al., 2020; Pacheco et al., 2019; Yizhak et al., 2015), as well as in microbial engineering (Choon et al., 2014; Reed and Palsson, 2003) among other research fields. CBM uses data- and prior knowledge-driven constraints to identify feasible metabolic flux distributions for a given condition (Schellenberger et al., 2011). Many communities and collaborative works contributed to reconstructing organism-specific generic metabolic networks which serve as starting points for CBM. Examples of such generic models are Recon 2 (Thiele et al., 2013), Recon 2.2 (Swainston et al., 2016), Recon3D (Brunk et al., 2018), Human1 (Robinson et al., 2020), and HMR (Agren et al., 2012). Other types of metabolic models are context-specific models that are built from tissue- or disease-specific data. Usually, the context-specific models are draft reconstructions built from the expression data of this condition by building algorithms such as FASTCORE (Vlassis et al., 2014), rFASTCORMICS (Pacheco et al., 2019), INIT (Agren et al., 2012), and RegrEX (Robaina Estévez and Nikoloski, 2015)/or manually curated such as for E. coli (Reed and Palsson, 2003), hepatocyte (Gille et al., 2010), and Zea mays (Saha et al., 2011). These models are often used as scaffolds for the integration of omics data or more interesting to simulate the metabolic phenotypes of organisms, tissues, or cell lines.
Within the CBM methods, flux balance analysis (FBA) is a linear programming-based approach that maximizes or minimizes an objective function, often a growth rate, to identify the optimal flux distribution(s) (Heirendt et al., 2019; Orth et al., 2010). In silico knockout studies are common in FBA through gene or reaction deletion. This deletion may be single, double, or multiple (Perumal et al., 2011). The goal of single reaction deletion is finding the most critical reactions in respect to the objective function through brute force removal of each reaction individually and calculating the ratio of the objective rates between mutated and wild-type models. Gene deletion studies are taking advantage of Boolean representations of the gene-reaction links known as gene–protein-reaction (GPR) rules (Reed et al., 2003). Gene deletion helps in defining essential genes whose deletion impacts the flux through the objective function (Heirendt et al., 2019). Essential genes are often used as targets for drug repositioning.
The 2019 coronavirus disease (COVID-19) is caused by a betacoronavirus strain called severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). COVID-19 was declared as a global pandemic on 11 March 2020 by WHO (Adhanom, 2020). Human-to-human infection can be transmitted by droplets (Huang et al., 2020) or aerosols (Morawska and Cao, 2020) by both symptomatic and asymptomatic patients (Kronbichler et al., 2020). The virus strain might have originated from the betacoronaviruses in bats and pangolins (Andersen et al., 2020). SARS-CoV-2 can cause upper and lower respiratory infections, increasing its transmissibility and severity. SARS-CoV-2 utilizes the human protein angiotensin I-converting enzyme 2 (ACE2) for cell entry with its spike protein. ACE2 is expressed on lung epithelial cells and other organs. The role of ACE2 is converting angiotensin II (AT-II) to angiotensin-(1,7) (AT-1,7) to negate the inflammatory effect of AT-II (Sparks et al., 2014). Thus, SARS-CoV-2 infection decreases the concentration of cellular unbound ACE2 molecules to facilitate the cell entry, causing an increase of AT-II which eventually increases the oxidative stress ion superoxide (Zimmerman et al., 2004). People with increased COVID-19 risk are patients with cancer (Fung and Babik, 2020), chronic kidney disease (Hirsch et al., 2020), obesity (Petrilli et al., 2020), type 2 diabetes mellitus (Fadini et al., 2020), immunocompromised (Tschopp et al., 2020), cardiac diseases (Yang et al., 2020), chronic obstructive pulmonary disease (COPD) (Yang et al., 2020), and sickle cell disease (Panepinto et al., 2020).
Acute respiratory distress syndrome (ARDS) is one of the severe symptoms of COVID-19, which may be attributed to alveolar epithelial cell injury (Li and Ma, 2020). This ARDS may become unresponsive to invasive mechanical ventilation and increase lung injury (McGuinness et al., 2020). Severe COVID-19 courses are also associated with acute injury to heart, kidney, and cerebrovascular diseases (Wu et al., 2020). In addition to the previous symptoms, long-term effects for COVID-19 survivors (or long-haulers) have been emerging. These long-term effects include new-onset diabetes, increasing severe complications in pre-existing diabetes (Rubino et al., 2020), fatigue, dyspnea, psychological distress (Halpin et al., 2021), and myocardial inflammation (Puntmann et al., 2020).
Metabolic modeling and in particular FBA were often used to understand the effects of microbes on human cells. Notably, a human alveolar model was used to assess the metabolic interaction between the host and Mycobacterium tuberculosis (Bordbar et al., 2010). Lately, a similar approach was applied in the viral genomes to model the impact of the Chikungunya, Dengue, and Zika viruses on the macrophage (Aller et al., 2018).
Only a few studies employed FBA on COVID-19 so far. Renz et al. used the viral genome information available at that time, to generate a SARS-CoV-2 specific viral biomass objective function (VBOF) (Renz et al., 2020). This VBOF generation from the genome information consisted of six steps on nucleotide, and amino acid investment, adenosine triphosphate (ATP) requirements, pyrophosphate liberation, total viral molar mass, and final construction of the VBOF (Renz et al., 2020). Then, the VBOF was added to a human alveolar macrophage model (iAB-AMØ1410) (Bordbar et al., 2010) to build a SARS-CoV-2-infected macrophage model. They identified guanylate kinase (GUK1) as an essential gene through in silico knockout that allows decreasing the viral biomass without affecting the human biomass maintenance. Several worldwide collaborative works in computational modeling of COVID-19 were established. Cheng et al. built context-specific models for different infected cell lines using multiple expression data (Cheng et al., 2021). They also predicted drug combinations to remdesivir (Cheng et al., 2021). Ostaszewski et al. built the COVID-19 Disease Map to understand the mechanistic interactions between SARS-CoV-2 and human tissues (Ostaszewski et al., 2020). In another collaborative study, Gysi et al. applied network analysis for drug repositioning using three different ranking approaches: network proximity, diffusion, and deep learning-based (Morselli Gysi et al., 2021). With the rising SARS-CoV-2 variants globally, variant-specific metabolic models were built and found GUK1 as a shared essential target (Renz et al., 2021). Previous methods for metabolic modeling of SARS-CoV-2, either focused on generating the VBOF from the viral genome (Renz et al., 2020) or on multiple cell line modeling and drug combination prediction with remdesivir (Cheng et al., 2021). To further support the search for an effective treatment for COVID-19, we employ here FBA to find candidate drugs and drug combinations that target viral-specific essential genes in SARS-CoV-2-infected lung cells through context-specific models built from expression data and the VBOF from (Renz et al., 2020) by the rFASTCORMICS workflow (Pacheco et al., 2019) (see Figure 1). We also highlight key pathways of these essential genes that might contribute to COVID-19 severity.
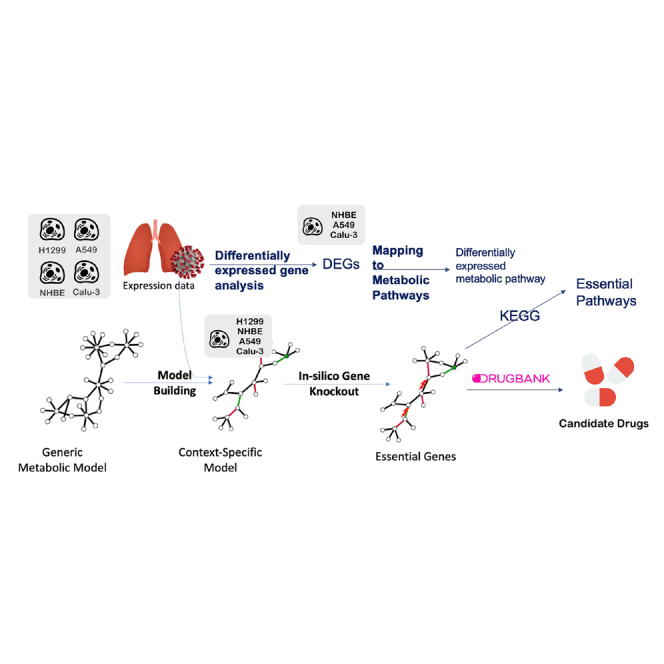
Figure 1.

Overview of the pipeline of essential gene prediction for SARS-CoV-2-infected lung cells
The viral biomass function (VBOF) was added to the generic metabolic models (Recon2.04 and Recon3D_01) (related to STAR Methods A.2.1) to build the infected generic models. Consistent versions of both the control and the infected generic models were obtained. Mock and infected lung expression data were used to build the context-specific models using rFASTCORMICS and the consistent control models as an input for the mock-specific models and the consistent infected models for infected-specific models, respectively. The objective functions were adjusted (related to STAR Methods A.2.2). Then, essential genes and gene pairs were identified by in silico gene knockout, before mapping to DrugBank V5 for drug repositioning to drugs and drug combinations.
Results
The main goal of the present study is to understand metabolic changes induced by COVID-19 in several lung cell lines, at various severity of infection, and at different time points after the infection (see Tables 1 and 2 for the metadata of the two RNA-Seq studies). We then used single and double knockouts to identify vulnerabilities that are specific to infected cells that are predicted by our network models to reduce the viral proliferation, while only moderately affecting the growth of host and control cells (see Figure 1). To further prioritize essential genes, we considered their essentiality scores across cell lines and in time and the effect of a knockout of these genes in the healthy tissues. The final aim is to identify conserved essential genes across infected models that do not provoke severe side effects when the gene is knocked out in the healthy counterpart model. To further identify vulnerabilities in the networks that could be exploited as drug targets, in silico inactivation of reactions was simulated. We further investigated the pathways harboring the predicted essential genes and reactions, to gain insight into how the virus adapts to the metabolism of lung cells. Finally, we proposed drug and drug combinations that target the predicted essential genes and synergistic essential gene pairs.
Table 1.
Severity study metadata (GEO: GSE147507)
| Condition | Cell line | Multiplicity of infection | ACE2 vector | Abbreviation | Number of samples infected/Mock |
|---|---|---|---|---|---|
| Series 1 | NHBE | 2 | No | NBHE_2 | 3/3 |
| Series 2 | A549 | 0.02 | No | A549_0.02 | 3/3 |
| Series 5 | A549 | 2 | No | A549_2 | 3/3 |
| Series 6 | A549 | 0.2 | Yes | A549_0.2_ACE2 | 3/3 |
| Series 7 | Calu-3 | 2 | No | Calu3_2 | 3/3 |
| Series 16 | A549 | 2 | Yes | A549_2_ACE2 | 3/3 |
Expression data from three lung cell lines infected with SARS-CoV-2 at three different viral loads and for some samples transfected with a vector expressing ACE2 with their controls.
Table 2.
Time-series study metadata (GEO: GSE148729)
| Condition | Cell line | Time point in hrs | Number of samples (infected/mock) |
|---|---|---|---|
| Calu3_4h | Calu-3 | 4 | 4/4 |
| Calu3_8h | Calu-3 | 8 | 2/0 |
| Calu3_12h | Calu-3 | 12 | 4/2 |
| Calu3_24h | Calu-3 | 24 | 2/2 |
| H1299_4h | H1299 | 4 | 2/2 |
| H1299_12h | H1299 | 12 | 2/0 |
| 1299_24h | H1299 | 24 | 2/0 |
| H1299_36h | H1299 | 36 | 2/2 |
Time series expression data with five time-points (4, 8, 12, 24, and 36 hrs) with infected and mock samples for two lung cell lines.
Metabolic pathway analysis of differentially expressed genes indicates COVID-19-based rewiring of core metabolism
Infection by the SARS-CoV-2 virus provokes alterations in the metabolism of the host cells. To elucidate these induced metabolic changes, we took advantage of two available expression datasets (Severity Study; Time-series Study; see STAR Methods for details). Principal component analysis (PCA) of the severity study samples shows a clear cluster separation according to the cell type (see Figure S1). Besides determining the differentially expressed genes (DEGs), we built genome-scale metabolic models applying rFASTCORMICS (related to STAR Methods A.2.1). The context-specific model reconstruction process resulted in 50 models (28 infected and 22 mock) with a median of 3646 metabolites (2465-5088) and 2456,5 reactions (1790-3474). To determine the key dysregulated pathways, we mapped the DEGs on the RECON3D_01 model and displayed the pathway alterations in the mostly dysregulated conditions (Figures 2 and S2 that shows a representation of all pathways without filtering on the number of reactions, nor the reactions per pathway). A549_0.02 condition didn't show any differentially expressed metabolic genes, thus it was discarded from the DEGs metabolic pathways. Meanwhile, the Normal Human Bronchial Epithelial cell line with MOI of 2 (NHBE_2) condition pathways were filtered (related to STAR Methods A.1.2). Among the most down-regulated pathways in the A549 cell lines with transfection (A549_2_ACE2 and A549_0.2_ACE2) in comparison to no ACE2 vector (A549_2) were chondroitin sulfate degradation, phosphatidylinositol phosphate metabolism, and phenylalanine metabolism, whereas glutathione metabolism was upregulated. For the A549 cell line with high viral load (A549_2_ACE2 and A549_2) in comparison to low (A549_0.2_ACE2), a downregulation of fatty acid synthesis, androgen and estrogen synthesis and metabolism, chondroitin synthesis, and pyruvate metabolism were also detected. Across all conditions including Calu3_2, we found a moderate downregulation of several pathways (glycerophospholipid metabolism, glycosphingolipid metabolism, sphingolipid metabolism). Other regulated pathways in Calu3_2 are the downregulated chondroitin sulfate degradation, nucleotide interconversion, and the upregulated cholesterol metabolism. The two additional DEG analyses on the ACE2 transfection showed some dysregulated pathways, such as the metabolism of folate, cholesterol, butanoate, arginine, proline, and D-alanine. Only cholesterol metabolism was shared between ACE2 transfection and Calu3_2 pathways (Figure S2).
Figure 2.

Reactions per pathway Heatmap for pathway analysis of differentially expressed genes in the severity study
Differentially expressed genes (DEGs) were computed with DESeq2 (absolute log2 fold change >1, adjusted p-value < 0.05). The Down- and up-regulated were mapped to the pathways (subSystems) of Recon3D_01. The number of up and down-regulated reactions was then summed up to identify the top altered pathways in the infected lung cell lines in the severity study (related to STAR Methods A.1.2). The color code “Reactions per Pathway [%]” represents the number of enriched metabolic reactions in a pathway divided by the overall number of reactions in this pathway. The transfection of ACE2 at an MOI of two in the A549 cell lines caused the downregulation of many pathways, which was not seen when the MOI was decreased by a factor of ten.
In silico single-gene deletion predicts common potential drug targets across infected cell lines with reduced side effects on control cells
The DEGs and the performed pathway analysis indicate a rewiring of the metabolism induced by SARS-CoV-2. The next step was then to verify if these alterations caused the appearance of infected cell-specific essential genes that could specifically be targeted by repositioned drugs. Therefore, for every condition of both lung studies, in silico single-gene deletion was performed on the respective reconstructed metabolic models. Twenty-three unique genes were predicted to be essential in the infected models (Tables S1 and S4). To assess if a drug targeting the candidate essential gene will kill the infected cell or reduce the viral proliferation, we computed an essentiality score for each essential gene, which sums up the number of models in which this gene is predicted to be essential. Essential genes that are only found in one or a few conditions might be cell line or experiment (e.g., medium) specific and hence might not have general biological relevance. Single-gene deletion of each predicted essential gene was then performed on the counterpart control model (related to STAR Methods A.2.2) to predict the effect of the gene knockout on the healthy tissue. This allowed obtaining a safety score and hence estimating the potential toxicity of each of the considered drug targets.
The obtained essentiality and safety scores are plotted for visual inspection (see Figure 3). Cardiolipin synthase 1 (CRLS1) and sphingomyelin synthase 1 (SGMS1) scored highest for essentiality, but were among the lowest for safety, thus indicating that targeting any of these genes might be effective against the virus but also reduces the growth of healthy cells, suggesting high toxicity of respective drugs. On the other hand, GUK1 gene showed a moderate essentiality score, but a higher safety score expecting fewer side effects. No gene could be identified that has high efficiency and safety. Also, nine of the 23 essential genes belong to the solute carrier (SLC) transporter gene family. Transporters are known key regulators of metabolic flux (Pacheco et al., 2015, 2019), hence modulating their expression might contribute to diverting metabolic fluxes to pathways for viral survival and proliferation. Of the 23 essential genes, 10 genes were shared between the two investigated lung studies, and many essential gene sets are shared between the investigated conditions (CRLS1, GUK1, SGMS1 in the severity study and CRLS1, ISYNA1, SGMS1, SLC27A4 in the time-series study), suggesting the existence of a consistent metabolic rewiring of the host metabolism rather than random alterations.
Figure 3.

Scatterplot and tripartite network of essential genes, and their predicted drugs and pathways, determined by in silico single-gene deletions on the infected lung models
(A) Scatterplot of essentiality and safety scores of the essential genes. Essentiality and safety scores correspond to the number of infected and healthy models, respectively, in which each gene is predicted to be essential. The y axis indicates the number of infected cell lines for which the gene is predicted to be essential, whereas the x axis indicates the number of control cell lines that are predicted to remain unharmed by the silencing of the target genes.
(B) Tripartite network of the drug-gene-pathway interactions of the essential genes: A network of the single repositioned drugs and their essential genes, predicted by in silico gene deletion, was built. The relationships between the essential genes and their pathways were mapped using Recon3D_01 subSystem. Genes, and their connected drugs, that don't have pathways in Recon3D_01 subSystems were discarded.
In silico single-gene deletion predicts potential drug targets for different stages and disease severity levels
Variability in the metabolism of cell lines, viral load, and time of infection gives rise to the appearance of context-specific essential genes. Core essential genes in infected cells are optimal drug targets as likely to be efficient for a majority of patients. Essential genes that are specific to the time of the disease or severity level are also of interest, as it allows modulating the specific treatment. It might be reasonable to provide drugs with strong adverse effects to more severe cases and to opt for lighter treatments for mild affections. To identify core and context-specific essential genes, we performed in silico gene knockouts on all reconstructed models and compared the sets of essential genes across all the conditions and between the severity and time-series studies. And more specifically, we focused on the effect of the transfection of the ACE2 vector, the viral load in the severity study, and the time after infection in the time-series study. ACE2 is crucial for SARS-CoV-2 cell entry by binding with its spike protein, but ACE2 also has many cellular functions crucial to the host cells, such as in the angiotensin–renin system. By comparing the essential genes in the absence (A549_2) and the presence of the ACE2 vector (A549_2_ACE2 & A549_0.2_ACE2), we could identify one set of genes (ISYNA1, SLC3A2, SLC7A11) that are essential for the virus in the absence of the ACE2 vector. By comparing the essential genes in the A549 cell line in the severity study with a high multiplicity of infection (MOI) (A549_2 & A549_2_ACE2) against low MOI (A549_0.02 & A549_0.2_ACE2), we identified essential genes for high viral load (CMPK1, CTH, PTDSS1, SLC2A13, SLC3A1, SLC5A3, SLC7A9) in A549_2_ACE2 and one essential gene, DTYMK, in A549_2, where the gene set (AGXT, DHFR, SLC27A4, TYMS) were unique for low viral load in A549_0.2_ACE2.
For the time-series study, a list of core essential genes (7–10 genes) was common to every time point and for each cell line (see Table S2). Besides the core essential genes, there were time point-specific essential genes that were essential only at very specific time-points owing to the inactivation of alternative pathways (Table S2). The Calu-3 cell line has eight core essential genes (CRLS1, GUK1, ISYNA1, PEPD, SGMS1, SLC27A4, SLC3A2, SLC7A11) and three-time point-specific genes (see Table S2). Out of the eight core essential genes, five (SLC27A4, CRLS1, GUK1, PEPD, SGMS1) were also in the six essential genes of the Calu3_2 condition in the severity study. Six core essential genes (CRLS1, ISYNA1, PLD2, SGMS1, SLC27A4, SLC7A6) and two time-specific genes were predicted for the H1299 cell line. Jaccard similarity of the essential genes between different conditions shows cell type-specific essential genes in the time-series study (see Figure S4). Clustering of the reconstructed models by core reactions using Jaccard similarity (see Figure S3) shows four clusters by cell type (A549, H1299, NHBE, Calu-3), even for cell lines between the two studies (Calu-3). This cell line-specific clustering is more apparent in Recon 3D than in Recon2. Moreover, the infection state (Mock, infected) forms sub-clusters within each of the four main clusters.
Essential genes and reactions are predicted to be harbored in 8 unique pathways among which is methionine and cysteine metabolism
To obtain a comprehensive picture of viral essentiality, we apply pathway analysis for core and context-specific essential genes to identify pathways that are major players in the determination of the severity as well as the stage of the infections. For both studies, the predicted essential genes were enriched in 13 unique pathways, of which eight were shared between both studies (Figure 4B) (fatty acid oxidation, glycerophospholipid metabolism, inositol phosphate metabolism, methionine, and cysteine metabolism, nucleotide interconversion, sphingolipid metabolism, starch, and sucrose metabolism, extracellular transport). Glycerophospholipid metabolism was enriched in all conditions across both studies. Nucleotide interconversion and extracellular transport were highly enriched in the severity and in the time-series study, respectively. Also, two pathways were shared with the DEGs pathways (glycerophospholipid metabolism, sphingolipid metabolism). The essential gene ISYNA1, encoding a synthase in the inositol phosphate metabolism pathway, was specific to cell lines without ACE2 vectors. No unique change in the set of essential genes' pathways was found in the function of the viral load in both conditions (A549_2 & A549_2_ACE2).
Figure 4.

Pathway analysis of the essential genes in the two lung studies
Identification of the pathways harboring the identified essential genes using Enrichr and Recon3D_01 subSystem, and hence the most critical pathways for the viral survival and proliferation across cell type, severity level, and time after infection. On the x axis from the left to the right, are the conditions of both the severity study and the time-series lung studies, respectively (related to STAR Methods B). Conditions in the severity study were named as cell line + ACE2 vector (if exists) + MOI. Conditions in the second lung cell line were named as cell line + time point.
(A) Enrichr enrichment: The color code in (A) "Enrichment [%]" represents the number of enriched genes in this pathway divided by the overall number of genes in this pathway.
(B) Metabolic pathway analysis: The color code in (B) "Reactions per Pathway [%]" represents the number of enriched metabolic reactions in a pathway divided by the overall number of reactions in this pathway.
To also explore pathways harboring essential genes that are not directly linked to metabolism or that are not captured by the metabolic genes and pathways in Recon3D_01, and Enrichr pathway analysis was performed (Kuleshov et al., 2016). Among others, ferroptosis, selenocompound metabolism, cysteine, and methionine metabolism, mTOR signaling pathway, and ether lipid metabolism were enriched for the essential genes (Figure 4A). Ferroptosis was the only Enrichr derived pathway that was associated with non-ACE2 vector samples owing to context-specific essential genes (SLC3A2, SLC7A11). Protein digestion and absorption were also enriched in some conditions with high viral load, whereas glycerophospholipid metabolism was highly enriched in both lung studies. Finally, pyrimidine metabolism was enriched in most severity study conditions; meanwhile, sphingolipid metabolism was enriched in all time-series study conditions. Only folate metabolism was shared between the essential genes' metabolic pathways and the dysregulated pathways of ACE2 transfection (Figure S2).
Prediction of candidates for repositioning of drugs and drug combinations targeting essential genes and synergetic gene pairs
Out of the 23 predicted essential genes, eight genes are druggable by 45 unique drugs (Table S3) from DrugBank (Wishart et al., 2018). Six antiviral drugs (acyclovir, valaciclovir, lamivudine, sofosbuvir, methotrexate, trifluridine) were identified in these 45 drugs. These drugs cover many modes of actions such as immunosuppressive, antiviral, folic acid antagonists, antirheumatic, and hypolipidemic actions besides some known nutraceutical cofactors such as lactose and folic acid (see Figure S5). The mode of actions were downloaded from the Drug Repurposing Hub (Corsello et al., 2017), while side effects were extracted from the MedDRA database (downloaded on 26th May 2020) (Mozzicato, 2009) with selecting only side effects containing the pattern “toxic.” The tripartite network of individual repositioned drugs (Figure 5B) shows a multi-target effect of four drugs (pralatrexate, pemetrexed, methotrexate, gemcitabine). Gemcitabine affects the nucleotide interconversion pathway through both CPMK1 and TYMS essential genes. Meanwhile, pralatrexate, pemetrexed, and methotrexate affect both nucleotide interconversion and folate metabolism pathways through thymidylate synthetase (TYMS) and dihydrofolate reductase (DHFR) essential genes, respectively.
Figure 5.

Tripartite network of the drug-gene-pathway interactions of the synergetic gene pairs determined by double gene deletion
Determination for the individual repositioned drugs for the synergistic gene pairs, and also mapping the relationships of the genes to pathways determined by Recon3D_01 subSystem.
(A) Tripartite network of the reduced list of double gene deletion drug pairs: Candidate gene pairs causing synergistic lethality were determined by double gene knockout (DKO) (related to STAR Methods A.2.3). Gene pairs for which both genes are targeted by the same drug were excluded from the candidate list for drug combinations and added to the list of single drugs. Genes, and their associated drugs, that are not present in Recon3D_01 were discarded.
(B) Tripartite network of single drugs targeting two genes that reduce biomass when knocked-out together: The gene pairs were determined by DKO (related to STAR Methods A.2.3). Only targets present in Recon3D_01 were included in the analysis.
Double gene deletion produced 598 unique gene pairs across the two lung studies. Out of these 598 gene pairs, 56 gene pairs are druggable by 3411 unique drugs or drug pairs. We found 47 single drugs with two paired targets (Table S4), owing to multiple identified targets per drug. As these 3411 drug pairs could target more than one gene-pair, safety scores and essentiality scores were calculated using the average of these scores. To prioritize among the 3411 drug pairs, we filtered by keeping drug pairs with more than two essentiality scores, and more than one in either the number of gene pairs or safety scores. This reduced drug pair list has 52 drug pairs that consist of 37 individual drugs (Table S5). The top-ranked drug pairs in the number of gene pairs are (azathioprine-pemetrexed and mercaptopurine-pemetrexed) affecting five essential gene pairs, while (imexon–valaciclovir and imexon–acyclovir) are top-ranked in the essentiality score of 10. The pathway analysis of the druggable essential gene pairs (see Figure 5) shows that most of the single drugs with two paired, targets the extracellular transport pathway. Meanwhile, the reduced drug pairs cover more diverse pathways. These pathways include new pathways in addition to the single druggable pathways such as purine catabolism, purine synthesis, nucleotide salvage pathway, and NAD metabolism. The aforementioned azathioprine–pemetrexed drug pair targets seven metabolic pathways such as transporter pathway and purine synthesis and catabolism. Also, among the 47 predicted single drugs with two paired targets, four drugs are affecting more than one gene-pair (gemcitabine, trifluridine, mercaptopurine, tegafur–uracil).
Discussion
In the present study, we analyzed changes in transcriptomic data of lung cell lines infected with COVID-19 at various viral loads and at different time points after infection. The main focus was on the alteration of expression of metabolic genes that could be evidence of a metabolic rewiring induced by the virus. Then, in silico single and gene double knockouts were performed to identify potential infected cell-specific essential genes that arise from this metabolic rewiring and that could be used as potential drug targets. To extend the list of targets and identify critical pathways for the growth or survival of the virus, reactions were inactivated in silico and the resulting impact on the viral biomass production was estimated. In addition, we explored pathways enriched for predicted essential genes and reactions to obtain a better picture of the occurring metabolic rewiring. Furthermore, we predicted a set of 85 repositionable single drugs (45 drugs on single targets in Table S3 and 47 drugs on gene pairs in Table S4 with 7 drugs shared between the 2 drug lists), and 52 drug combinations that could be explored as a treatment against COVID-19. Finally, we compared our results against two recent studies that cover the candidate metabolic pathways' alternation in COVID-19 infection (Bernardes et al., 2020; Thomas et al., 2020).
COVID-19-induced dysregulated pathways may not be essential
To unravel the metabolic rewiring induced by COVID-19 on the host lung cell after ACE2 or a mock transfection at different viral loads, we computed the metabolic differentially expressed genes (DEGs) in the severity study. Among the metabolic pathways with a different activation pattern after the transfection with the ACE2 vector, are chondroitin sulfate degradation and synthesis that have a link to oxidative stress. Chondroitin is a glycosaminoglycan (GAG) with antioxidant and neuroprotective effects against oxidative stress through the upregulation of phosphoinositide 3-kinases (PI3K)/Akt signaling and heme oxygenase-1 (HMOX1) (Cañas et al., 2007). Chondroitin sulfate degradation was downregulated after the transfection with ACE2 vector, while chondroitin synthesis was downregulated with a high viral load. This dysregulation is consistent with the hypothesis of chondroitin accumulation in the infected cells to balance the oxidative stress induced by the virus. The phosphoinositol phosphate pathway, which includes PI3K, was also downregulated after transfection with the ACE2 vector, further supporting the protective role of chondroitin in COVID-19 infection. Although this hypothesis was not tested in vitro, in an in vivo study on the Vero cell line, chondroitin sulfate showed weak inhibition of SARS-CoV-2 cell entry in comparison to other types of GAGs such as heparin and enoxaparin (Tandon et al., 2020). Finally, HMOX1 has been found to bind to SARS-CoV-2 open reading frame 3 A (ORF3a) (Gordon et al., 2020). The ineffectiveness of chondroitin sulfate as an antiviral agent in SARS-CoV-2 was expected owing to its unspecific mode of action. Reducing oxidative stress may alleviate the symptoms but may not kill the virus nor does it reduce its ability to replicate itself. The assessment of ACE2 transfection alone (see Figure S2) didn't show that shared dysregulation after infection with SARS-CoV-2. Meanwhile, ACE2 transfection increased the viral reads to ∼54% of the total mapped reads (Blanco-Melo et al., 2020). Also, ACE2 transfection decreases the interferons IFN-I and IFN-III through the inactivation of the kinase TBK1 (Blanco-Melo et al., 2020).
Pyrimidine metabolism as a candidate druggable essential pathway
To be more effective, drug candidates have to target genes, reactions, or pathways that are key and specific to viral metabolism. Hence, in silico single and double gene knockouts were performed to identify genes essential to the viral biomass production but whose knockout has little or no effect on the host biomass production. Among the 23 predicted essential genes for viral biomass, two genes of the phospholipid metabolism (CRLS1 and SGMS1) showed the highest essentiality score. GUK1, which was the main essential gene identified in (Renz et al., 2020), is also among the top predicted targets and displays moderate essentiality and safety score. Also, pyrimidine biosynthesis was among the pathways with consistent flux changes in the metabolic modeling of multiple cell lines (Cheng et al., 2021). Furthermore, three essential genes (DTYMK, CMPK1, and TYMS) are part of pyrimidine metabolism (through pyrimidine deoxyribonucleotides de novo biosynthesis) that were enriched in all conditions in the severity study, but for the last time point of the time-series study in the Calu-3 cell line. In two separate in vivo studies on the Vero cell line, inhibition of de novo pyrimidine biosynthesis pathway through dihydroorotate dehydrogenase (DHODH) inhibitors, showed broad-spectrum antiviral activity, stopped, or halted SARS-CoV-2 replication, respectively (Luban et al., 2020; Xiong et al., 2020). Although DHODH inhibitor PTC299 showed a little cytotoxic effect on SARS-CoV2, they were proven to have an immunomodulatory effect on IL-6, IL-7A, IL-17F, and VEGF (Luban et al., 2020). Alterations in the expression of genes of the pyrimidine metabolism were significantly higher in the A549, suggesting a response that could be specific to this cell line. Additionally, using pyrimidine biosynthesis inhibitors on SARS-CoV-2-infected mouse models, reduced individually viral infectivity, and reduced lung inflammation when used in combinations (Schultz et al., 2021).
Ferroptosis as a candidate prognostic and target pathway for COVID-19
To further understand why a gene is essential for viral biomass production, we examined the pathways harboring the essential genes, where eight out of 13 pathways were found between the two lung studies. Pathways harboring essential genes were also enriched for DEGs. Exploring the ferroptosis-specific database FerrDb shows that three out of the 23 essential genes are related to ferroptosis. SLC7A11 and SLC3A2 are classified as suppressors; meanwhile, SLC7A5 is a marker. Also, FerrDb, classified only one of our predicted drugs, sulfasalazine, as an inducer of ferroptosis. Ferroptosis is an iron-dependent programmed cell death that can be inhibited by Selenium. In a cross-sectional study, selenium level was found higher in tissue samples from COVID-19 survivors in comparison to non-survivors (Moghaddam et al., 2020). Similarly, the analysis of blood single-cell expression data found that four genes governing ferroptosis were upregulated in infected patients than recovered (Huang et al., 2021). Also in a population retrospective analysis, the selenium concentration in the hair in the population of Chinese cities outside Hubei province was correlated with the COVID-19 cure rate in Chinese cities (Zhang et al., 2020a). Even though the last retrospective study using city population-level data instead of patient-level data, might be less reliable, these studies suggest a role of ferroptosis in the survival of patients with COVID.
Comparison to a metabolomics study shows altered polyunsaturated fatty acids
We further compared our enriched pathways from the DEGs and essential genes (Figure 2) with a recent metabolomics study (Thomas et al., 2020). In this study, serum metabolites were compared among SARS-CoV-2 positive and negative patients, also altered IL-6 levels measures as an indication of COVID-19 severity. The study found several altered pathways and dysregulation, notably of nitrogen and tryptophan metabolism associated with increased severity. Also, some metabolite levels were increased in patients with COVID-19 such as kynurenines, methionine sulfoxide, cystine, and free polyunsaturated fatty acids (PUFAs). Up-regulated fatty acid oxidation in DEGs and glycerophospholipid metabolism in essential gene pathways is consistent with the increased levels of PUFAs (Thomas et al., 2020). The increased PUFAs are biomarkers for ferroptosis which was predicted in the condition without ACE2 vector. The recent evidence for the role of selenium in COVID-19 and the significant presence of PUFAs as a biomarker in severe COVID-19 cases might be a further indication of the role of ferroptosis regarding COVID-19 severity. Moreover, the SARS-CoV-2 spike protein was discovered to have a binding pocket for free fatty acids (Toelzer et al., 2020). This seems to allow the PUFA linoleic acid to have a synergistic effect with the antiviral remdesivir against SARS-CoV-2 in vitro (Toelzer et al., 2020). The use of 5-aminosalicylate or sulfasalazine, a 5-aminosalicylate prodrug, has recently been shown to increase COVID-19 severity in patients with inflammatory bowel disease (IBD) in a retrospective study (n = 525) (Brenner et al., 2020). As sulfasalazine was found as a ferroptosis inducer in FerrDb (Sehm et al., 2016), this could strengthen the evidence of a role of ferroptosis in COVID-19 severity. It also illustrates the need for a careful assessment of the toxicity of the predicted drugs in follow-up in vitro and in vivo studies as well as a patient or group of patient-tailored approaches.
Methionine, cysteine, and pyrimidine metabolism are enriched in a multi-omics study
To discover which lung essential pathways might be shared with other infected organs, we also compared our identified pathways with a multi-omics study on three cell types: megakaryocytes, erythroid cells, and plasmablasts (Bernardes et al., 2020). In this longitudinal study of COVID-19 severity, cell-/tissue-specific metabolic models were reconstructed from single-cell/bulk RNA-seq, respectively (Bernardes et al., 2020). The goal of the metabolic reconstruction in this study was to find cell-specific metabolic pathways associated with different disease progression and recovery time points (Bernardes et al., 2020). The essentiality of genes and reactions in these pathways across the three cell types are unknown as single-gene or reaction deletions were not applied. Many identified metabolic pathways in this multi-omics study across megakaryocytes, erythroid cells, and plasmablasts were also shared with our essential pathways on the lung such as pyrimidine metabolism and cysteine and methionine metabolism. Also, our lung essential metabolic pathways such as inositol phosphate metabolism and sphingolipid metabolism have been identified as both erythroid cells- and plasmablasts-specific. Meanwhile, the lung-essential fatty acid oxidation and non-essential pyruvate metabolism have been identified as megakaryocytes-specific. Interestingly, a high upregulation of pyruvate kinase M in PI3K/Akt signaling was found in critical patients in megakaryocytes (Bernardes et al., 2020), that participate in the dysregulated chondroitin sulfate metabolism (Cañas et al., 2007). Furthermore, serum sphingosine-1-phosphate, a metabolite in the lung-essential sphingolipid metabolism, was found to significantly decrease with COVID-19 severity in a small study (n = 111) (Marfia et al., 2020). Moreover, clofazimine, an inhibitor to the acid sphingomyelinase in the sphingolipid metabolism pathway, was found to have antiviral activity in the golden Syrian hamster model against MERS & SARS-CoV-2 (Yuan et al., 2021). Taken together, the shared metabolic pathways between the different studies such as pyrimidine metabolism and methionine and cysteine metabolism across different tissues might represent core viral-specific pathways that could harbor efficient drug targets that would eliminate or slow down the virus regardless of the infected tissue.
Some candidate drugs have antiviral, immunomodulatory, and angiotensin I-converting enzyme inhibitor actions
To prioritize drug and drug combinations and as many conditions in the time-series study were lacking mock samples, we relied for the present work rather on the essentiality score of each gene identified in terms of reducing the viral proliferation rather than the predicted toxicity on control tissue models (safety score). In total, SARS-COV-2-specific essential genes and gene pairs were predicted by rFASTCORMICS-based lung models that can be targeted by 85 single repositionable drugs and 52 drug combinations. The safety of the drugs was assessed by simulation knockouts on the biomass of the counterpart mock sample. This strategy allows estimating which drugs might be potential candidates for not having too drastic side effects. Although the drug candidates are all FDA-approved drugs, some treatments are associated with severe side effects, and combining two drugs can have additional unexpected side effects. Hence, further tests would be required on other tissues and using other optimization functions as well as in vitro and in vivo validations before considering any predictions as potential drug candidates.
Among the 85 predicted single drugs, five are broad-spectrum antivirals (lamivudine, methotrexate, sofosbuvir, valaciclovir, zalcitabine) (Coronavirus Data Download - Targeting COVID-19 Portal, 2020). Also, five drugs in the candidate drug combinations have broad-spectrum antivirals (ezetimibe, lamivudine, methotrexate, sofosbuvir, valaciclovir). In a small clinical trial (n = 62), the combination of sofosbuvir-daclatasvir decreased the COVID-19 mortality rate (6%) in comparison to ribavirin (33%) (Eslami et al., 2020). Of the predicted drugs, two drugs (acyclovir, valaciclovir) target the GUK1 gene, which shows relative essentiality and safety. Acetylcysteine, another predicted drug by our workflow, is mucolytic and antioxidant in high doses through regenerating glutathione. Acetylcysteine alone or with bromelain was able in vitro to fragment the recombinant spike and envelope SARS-CoV-2 proteins (Akhter et al., 2020). Moreover, gemcitabine has been shown to have antiviral activity against SARS-CoV-2 in the Vero-E6 cell line (Zhang et al., 2020b). Methotrexate shows antiviral activity against SARS-CoV-2 in Vero-E and Calu-3 cell lines (Stegmann et al., 2020). This antiviral activity was better than the only authorized antiviral for emergency use for COVID-19 remdesivir. Till 29 July 2021, out of the 85 single drugs, nine drugs are being tested currently in clinical trials (acetylcysteine, liothyronine, melphalan, methotrexate, moexipril, quinapril, ramipril, rosuvastatin, sofosbuvir, trandolapril) according to DrugBank COVID-19 Clinical Trial Summary (Wishart et al., 2018).
Among the single drugs targeting gene pairs, nine were drugs belonging to angiotensin-converting enzyme inhibitors (ACEIs) such as ramipril, affecting the gene-pair SLC15A1-SLC15A2 through targeting the extracellular transport pathway. Interestingly, in a prospective study of COVID-19 (n = 19,486), patients taking ACEIs have a reduced risk of COVID-19, with differences according to ethnicity (Hippisley-Cox et al., 2020). Meanwhile, ACEIs did not reduce the risk of receiving ICU care (Hippisley-Cox et al., 2020). Furthermore, statins, lipid-lowering drugs that were enriched among the predicted drugs were debated for their efficacy in reducing COVID-19 severity at the onset of the pandemic and their usefulness for COVID-19 is still unclear (Subir et al., 2020). Also, a retrospective study (n = 13,981) has shown an association between statins and reduced COVID-19 mortality from 9.4% in patients not taking statins to 5.2% with statins (Zhang et al., 2020a). Owing to the relative number of the different statin recipients, this study couldn't rank the different statin types. But, a recent in vitro study of different statins showed an antiviral effect on SARS-CoV-2 (Moeller et al., 2020), where rosuvastatin was ranked second in the antiviral activity (Moeller et al., 2020). In the candidate drug combinations, immunomodulators appear such as mercaptopurine, azathioprine, pemetrexed, and methotrexate. Also, three predicted nucleoside analogs (azathioprine, mercaptopurine, gemcitabine), were among 16 nucleoside analogs and 122 drugs validated in vitro against Calu-3 (Schultz et al., 2021).
In conclusion, unlike drug repositioning using expression reversal or drug docking that lack targets' identification or genome-scale multi-targeting, respectively, constraint-based metabolic modeling is a powerful in silico tool for drug repositioning with genome-scale information and producing known targets. These powerful advantages come from gene essentiality prediction. In this work, context-specific models from expression data from infected lung cell lines were built, then constrained by both viral and host biomass. In silico gene deletion identified 23 single essential genes and 598 essential gene pairs. Drug repositioning using approved drugs in DrugBank V5 identified 85 single drugs and 52 drug combinations, of which 47 single drugs are targeting both genes in the gene-pair. Pathway analysis of the essential genes identifies ferroptosis as a candidate biomarker pathway of COVID-19 severity. Gemcitabine was predicted to target two single essential genes in the nucleotide interconversion pathway and three gene pairs in drugs identified by both single and double gene deletion, respectively. Finally, we predicted the GUK1 gene as both relatively safe and essential against SARS-CoV2 as reported by a previous in silico modeling.
Limitations of study
Although this study predicts some interesting drug candidates and drug combinations, the work is limited by the modeled lung cell lines (A549, Calu-3, H1299, NHBE). Another limitation to this work is that the identified drug and drug pairs are based on targets identified by network effects on the host metabolome (as the virus is only modeled through its biomass function) rather than direct docking on the viral proteome. Thus, further in vitro single- and double-gene deletion studies are needed to determine the essentiality of the identified single genes and gene pairs. These could, for example, involve some selected drug and drug combinations on the different cell lines at various concentrations in order to obtain drug response curves and landscapes, respectively, to identify IC-50 and synergy scores for the drug combinations. These experiments would be more beneficial for genes with predicted essentiality across different cell lines such as (CRLS1, SGMS1, SLC27A4) which were essential in the four lung cell lines.
STAR★Methods
Key resources table
Resource availability
Lead contact
Further information and requests for code or datasets should be directed to and will be fulfilled by the lead contact, Prof. Thomas Sauter (Thomas.Sauter@uni.lu).
Materials availability
This study did not generate new unique reagents.
Methods details
A. SARS-CoV-2 essentiality analysis in lung
A.1 Differentially expressed genes analysis
A.1.1 Data preprocessing
At the onset of the pandemic, two datasets were available, focusing mainly on the effects of the virus on lung tissues. These two bulk RNA seq datasets GSE147507 (Blanco-Melo et al., 2020) and GSE148729 (Emanuel et al., 2020) of human cell lines hosting SARS-CoV-2, as well as of mock samples, were downloaded from the NCBI Gene Expression Omnibus (GEO) (Clough and Barrett, 2016) data repository on April 23, and May 15, 2020, respectively. The GSE147507 dataset, which focuses on the expression changes at various severity levels of Infection (severity study), contains 36 samples originating from healthy epithelial, A549, and Calu-3 cells infected by SARS-CoV-2 at three different viral loads, as well as control samples with a mock infection. As the level of plasma ACE2 is a potential predictor of COVID-19 severity (Kragstrup et al., 2021), the comparison between conditions with different levels of ACE2 and viral load would allow identifying essential genes, and hence drug targets for different stages and severity levels of COVID-19. Using drugs associated with severe effects would not be beneficial for milder forms of COVID-19, but could be crucial for the cure of more severe forms. The A549 cell line was found to express ACE2 at a lower level than Calu-3, which doesn't allow the cell entry of SARS-CoV-2 (Blanco-Melo et al., 2020). For this reason, the A549 cell line was transfected with a vector expressing ACE2 (Table 1). Conditions with two replicates only or subjected to drug perturbations were not considered for the analysis. Raw counts were converted to the Reads Per Kilobase of transcript (RPKM) using an in-house Python script.
For the GSE148729 dataset, which monitors how expression changes at different points after the infection (time-series study), the normalized Fragments Per Kilobase of transcript per Million (FPKM) values of Calu-3 and H1299 cell lines infected by SARS-CoV-2, as well as controls were retrieved from GEO.
A.1.2 Differentially expressed genes analysis
The differential gene expression analysis was only applied to the severity study, as the time-series has either missing mock samples or two samples in some conditions. First, a principal component analysis was first performed using FactoMineR (Lê et al., 2008) to identify and if necessary, remove outliers by visual inspection. Then, genes with low expression values were filtered out using edgeR's filterByExpr function (Version 3.30.3). This function keeps genes based on a minimum count-per-million in at least k samples, determined by the lowest sample size between all conditions (Robinson et al., 2009). DESeq2 identifies the significant DEGs using the Wald test and adjusted for multiple testing by Benjamini and Hochberg yielding adjusted p-values. DESeq2 (Version 1.28.1) (Love et al., 2014) via R (Version 4.0.1) was run on the preprocessed data to identify differentially expressed genes (DEG) between the infected and the mock samples applying an adjusted p-value threshold of 0.05, and an absolute log fold change threshold of 1. To assess if the gene expression changes observed after ACE2 transfection were caused by the transfection itself, rather than the overexpression of ACE2, a DEG analysis was performed to compare two conditions (A549_2_ACE2 against A549_2), using first the mock samples only (ACE2_Mock), then only the infected samples (ACE2_Infected). DEGs were mapped to the genes of the generic model Recon3D_01 (Brunk et al., 2018) via the GPR rules to retrieve differentially expressed reactions (DERs) as well as their associated pathways. For each pathway with at least three reactions, the ratio of up-and down-regulated reactions over all reactions was computed. To improve the readability of the plot, only pathways with more than 5% of DERs were depicted.
A.2 Essentiality analysis
A.2.1 Condition-specific model building
To further elucidate, the metabolic alteration provoked by the virus, metabolic models for the infected samples and mock samples were built. Therefore, the VBOF from the infected alveolar macrophage model iAB_AM O 1410_SARS-CoV-2 (BioModels: MODEL2003020001) (Renz et al., 2020) was added to both the generic reconstructions Recon2.04 (Thiele et al., 2013) and Recon3D_01 (Brunk et al., 2018) using the addReaction function of the COBRA Toolbox v.3.0 (Heirendt et al., 2019). The identifiers of metabolites included in the biomass had first to be modified to match the ones of the generic reconstruction. Then, FASTCC (Vlassis et al., 2014) was run to remove blocked reactions. For each condition, the RPKM values and the modified consistent generic reconstruction were used as input for the rFASTCORMICS (Pacheco et al., 2019) to get condition-specific models. COBRA Toolbox v.3.0 and FASTCC were used via MATLAB (R2019a).
A.2.2: Single gene knockout
The metabolic models were then used to identify viral-specific vulnerabilities, using a single gene deletion approach on the mock and infected models. For the infected model, to ensure that both host and viral biomass's objective functions can carry simultaneously a flux in the infected models, the objective coefficients were set to 100 and 1, respectively, and the upper bound of the host biomass was fixed to 10% of its maximal flux. This setting constrains the model to guarantee cell homeostasis and protein turnover in the host model while diverting all non-essential resources for viral reproduction.
model.c(viral biomass) =1
model.c(host biomass) = 100
model.ub(host biomass) = 10% of max flux determined by FBA
In silico single-gene knockouts (SKO) were performed on the infected models using a corrected version of the singleGeneDeletion function of the COBRA Toolbox v.3.0 (Heirendt et al., 2019) to assess the impact of the knockout of each gene on the viral biomasses. The 0.2 threshold was used as a cutoff for gene growth rate Ratio (grRatio) to identify essential genes.
A.2.3 Double gene knockout
To identify potential targets for drug combinations, double gene knockouts (DKO) for all gene-pair combinations were simulated using the doubleGeneDeletion function on the infected models. From the analysis, we obtained two lists of synergistic gene-pairs: non-essential gene-pairs that allow reducing the growth rate below the specified thresholds when simultaneously knocked out and pairs of essential and non-essential genes that induced a stronger reduction of the growth than the knockout of the essential gene alone. Both non-essential and essential gene-pairs were concatenated as DKO outputs for further drug repositioning.
A.2.4 Essentiality and safety scoring
To test the knockout impact of SARS-CoV-2- infected host-specific predicted essential genes and gene-pairs on the healthy counterpart tissue, SKO, and DKO of these genes were performed on the healthy models. Genes or gene-pairs that cause a reduction of biomass only in the infected models are considered safe, whereas those that cause also a reduction of biomass in the healthy models are regarded as potentially toxic. The essentiality score of an essential gene is the sum of infected models that show this gene as essential. The safety score of an essential gene is the sum of healthy models that show this gene as safe. A healthy model, SKO and DKO of a gene was applied only if the gene is determined as essential in its respective infected model. A scatterplot of essentiality scores against safety scores was plotted (see Figure 3).
B. Gene enrichment of the potential targets
The identifiers of the essential genes and synergetic genes were translated into HGNC gene symbols using GSEApy (Version 0.9.17, https://github.com/zqfang/GSEApy/) Python package (3.7.4) and then uploaded to Enrichr API (Kuleshov et al., 2016) to identify enrichment of these genes in KEGG pathways (KEGG2019 human (Kanehisa and Goto, 2000) database with 0.05 p-value cutoff). Fisher exact test (default hypothesis test) was performed for calculating the p-value based on the assumption of the binomial distribution of the input gene set. All enrichment results based on only one gene were discarded. Then, the enrichment percentage was calculated (related to STAR Methods A.1.2). Metabolic pathway analysis was also applied using the Recon3D_01 subSystem as background instead of KEGG pathways on the essential genes (related to STAR Methods A.1.2) without further filtering of the pathways. Comparison of pathways of Calu-3 and NHBE cell lines were excluded from the analysis of the effect of viral load and ACE2 vector in the severity study because these cell lines didn't have ACE2 vector conditions.
C. Drug repositioning of the essential genes
To identify drugs targeting the predicted viral-specific essential genes, drug-target interactions were downloaded from DrugBank V5 (Wishart et al., 2018) on April 23, 2020. Drugs that were withdrawn, nutraceutical, or experimental were discarded from the analysis. Drugs that are described as having any effect on the potential targets were selected as candidate drugs (Table S3) and drug combinations (Tables S4 and S5). To determine which drugs, have a multi-target effect, tripartite networks of the drug-gene-pathway interactions were constructed for the single and double knockout drugs using Recon3D_01 subSystems as pathways (Figures 3B and 5). The tripartite networks were constructed using the sankeyNetwork function in networkD3 (Allaire et al., 2017, p. 3) (version 0.4) package in R.
D. Relationship with ferroptosis
As SKO targets were enriched for many pathways related to ferroptosis, the potential targets and SKO drugs were searched in a curated database (FerrDb) (Zhou and Bao, 2020) for ferroptosis genes, and related drugs. FerrDb classifies genes into driver, suppressor, and marker, while it classifies drugs into inducer and inhibitor. These classes were also used to identify the role of the potential targets and SKO drugs in the ferroptosis pathway.
Acknowledgments
The experiments presented in this paper were carried out using the HPC facilities of the University of Luxembourg (Varrette et al., 2014).
Author contributions
A.K. carried out the experiments. M.P.P. and T.S. conceived and planned the experiment. A.K., M.P.P., and T.S. analyzed the data. All the authors wrote, read, and revised the manuscript.
Declaration of interests
The authors declare no competing interests.
Published: November 19, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.103331.
Supplemental information
Data and code availability
This paper analyses existing, publicly available data. The source of the data is listed in the key resources table.
The models and code generated during this study are available at GitHub (https://github.com/sysbiolux/DCcov).
References
- Adhanom T. World Health Organization; 2020. WHO Director-General’s Opening Remarks at the Media Briefing on COVID-19.https://www.who.int/dg/speeches/detail/who-director-general-s-opening-remarks-at-the-media-briefing-on-covid-19-11-march-2020 [Google Scholar]
- Agren R., Bordel S., Mardinoglu A., Pornputtapong N., Nookaew I., Nielsen J. Reconstruction of genome-scale Active metabolic networks for 69 human cell types and 16 cancer types using INIT. PLoS Comput. Biol. 2012;8:e1002518. doi: 10.1371/journal.pcbi.1002518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akhter J., Pillai K., Badar S., Mekkawy A., Valle S.J., Morris D.L. In vitro study of BromAc on SARS-CoV-2 spike and envelope protein shows synergy and disintegration at modest concentrations. bioRxiv. 2020 doi: 10.1101/2020.09.07.286906. [DOI] [Google Scholar]
- Allaire J.J., Ellis P., Gandrud C., Kuo K., Lewis B.W., Owen J., Russell K., Rogers J., Sese C., Yetman C.J. 2017. https://cran.r-project.org/web/packages/networkD3/index.html
- Aller S., Scott A., Sarkar-Tyson M., Soyer O.S. Integrated human-virus metabolic stoichiometric modelling predicts host-based antiviral targets against Chikungunya, Dengue and Zika viruses. J. R. Soc. Interf. 2018;15:20180125. doi: 10.1098/rsif.2018.0125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen K.G., Rambaut A., Lipkin W.I., Holmes E.C., Garry R.F. The proximal origin of SARS-CoV-2. Nat. Med. 2020;26:450–452. doi: 10.1038/s41591-020-0820-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardes J.P., Mishra N., Tran F., Bahmer T., Best L., Blase J.I., Bordoni D., Franzenburg J., Geisen U., Josephs-Spaulding J., et al. Longitudinal multi-omics analysis identifies responses of megakaryocytes, erythroid cells and plasmablasts as hallmarks of severe COVID-19 trajectories. medRxiv. 2020 doi: 10.1101/2020.09.11.20187369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco-Melo D., Nilsson-Payant B.E., Liu W.-C., Uhl S., Hoagland D., Møller R., Jordan T.X., Oishi K., Panis M., Sachs D., et al. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell. 2020;181:1036–1045.e9. doi: 10.1016/j.cell.2020.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blätke M.-A., Bräutigam A. Evolution of C4 photosynthesis predicted by constraint-based modelling. eLife. 2019;8:e49305. doi: 10.7554/eLife.49305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordbar A., Lewis N.E., Schellenberger J., Palsson B.Ø., Jamshidi N. Insight into human alveolar macrophage and M. tuberculosis interactions via metabolic reconstructions. Mol. Syst. Biol. 2010;6:422. doi: 10.1038/msb.2010.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner E.J., Ungaro R.C., Gearry R.B., Kaplan G.G., Kissous-Hunt M., Lewis J.D., Ng S.C., Rahier J.-F., Reinisch W., Ruemmele F.M., et al. Corticosteroids, but not TNF antagonists, are associated with adverse COVID-19 outcomes in patients with inflammatory bowel diseases: results from an international registry. Gastroenterology. 2020;159:481–491.e3. doi: 10.1053/j.gastro.2020.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunk E., Sahoo S., Zielinski D.C., Altunkaya A., Dräger A., Mih N., Gatto F., Nilsson A., Preciat Gonzalez G.A., Aurich M.K., et al. Recon3D enables a three-dimensional view of gene variation in human metabolism. Nat. Biotechnol. 2018;36:272–281. doi: 10.1038/nbt.4072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cañas N., Valero T., Villarroya M., Montell E., Vergés J., García A.G., López M.G. Chondroitin sulfate protects SH-SY5Y cells from oxidative stress by inducing heme oxygenase-1 via phosphatidylinositol 3-kinase/Akt. J. Pharmacol. Exp. Ther. 2007;323:946–953. doi: 10.1124/jpet.107.123505. [DOI] [PubMed] [Google Scholar]
- Cheng K., Riva L., Sinha S., Pal L.R., Nair N.U., Martin-Sancho L., Chanda S.K., Ruppin E. Genome-scale metabolic modeling reveals SARS-CoV-2-induced host metabolic reprogramming and identifies metabolic antiviral targets. bioRxiv. 2021 doi: 10.1101/2021.01.27.428543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choon Y.W., Mohamad M.S., Deris S., Illias R.M., Chong C.K., Chai L.E., Omatu S., Corchado J.M. Differential bees flux balance analysis with OptKnock for in silico microbial strains optimization. PLoS One. 2014;9:e102744. doi: 10.1371/journal.pone.0102744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clough E., Barrett T. The gene expression Omnibus database. Methods Mol. Biol. 2016;1418:93–110. doi: 10.1007/978-1-4939-3578-9_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coronavirus data download - targeting COVID-19 portal 2020. https://ghddi-ailab.github.io/Targeting2019-nCoV/CoV_Experiment_Data/#broad-spectrum-antiviral-agents
- Corsello S.M., Bittker J.A., Liu Z., Gould J., McCarren P., Hirschman J.E., Johnston S.E., Vrcic A., Wong B., Khan M., et al. The drug repurposing hub: A next-generation drug library and information resource. Nat. Med. 2017;23:405–408. doi: 10.1038/nm.4306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emanuel W., Kirstin M., Vedran F., Asija D., Theresa G.L., Roberto A., Filippos K., David K., Salah A., Christopher B., et al. Bulk and single-cell gene expression profiling of SARS-CoV-2 infected human cell lines identifies molecular targets for therapeutic intervention. bioRxiv. 2020 doi: 10.1101/2020.05.05.079194. [DOI] [Google Scholar]
- Eslami G., Mousaviasl S., Radmanesh E., Jelvay S., Bitaraf S., Simmons B., Wentzel H., Hill A., Sadeghi A., Freeman J., et al. The impact of sofosbuvir/daclatasvir or ribavirin in patients with severe COVID-19. J. Antimicrob. Chemother. 2020;75:3366–3372. doi: 10.1093/jac/dkaa331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fadini G.P., Morieri M.L., Longato E., Avogaro A. Prevalence and impact of diabetes among people infected with SARS-CoV-2. J. Endocrinol. Invest. 2020;43:867–869. doi: 10.1007/s40618-020-01236-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fung M., Babik J.M. COVID-19 in immunocompromised hosts: what we know so far. Clin. Infect. Dis. 2020;72:340–350. doi: 10.1093/cid/ciaa863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gille C., Bölling C., Hoppe A., Bulik S., Hoffmann S., Hübner K., Karlstädt A., Ganeshan R., König M., Rother K., et al. HepatoNet1: a comprehensive metabolic reconstruction of the human hepatocyte for the analysis of liver physiology. Mol. Syst. Biol. 2010;6:411. doi: 10.1038/msb.2010.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon D.E., Jang G.M., Bouhaddou M., Xu J., Obernier K., White K.M., O’Meara M.J., Rezelj V.V., Guo J.Z., Swaney D.L., et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature. 2020;583:459–468. doi: 10.1038/s41586-020-2286-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halpin S.J., McIvor C., Whyatt G., Adams A., Harvey O., McLean L., Walshaw C., Kemp S., Corrado J., Singh R., et al. Postdischarge symptoms and rehabilitation needs in survivors of COVID-19 infection: a cross-sectional evaluation. J. Med. Virol. 2021;93:1013–1022. doi: 10.1002/jmv.26368. [DOI] [PubMed] [Google Scholar]
- Heirendt L., Arreckx S., Pfau T., Mendoza S.N., Richelle A., Heinken A., Haraldsdóttir H.S., Wachowiak J., Keating S.M., Vlasov V., et al. Creation and analysis of biochemical constraint-based models using the COBRA toolbox v.3.0. Nat. Protoc. 2019;14:639–702. doi: 10.1038/s41596-018-0098-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hippisley-Cox J., Young D., Coupland C., Channon K.M., Tan P.S., Harrison D.A., Rowan K., Aveyard P., Pavord I.D., Watkinson P.J. Risk of severe COVID-19 disease with ACE inhibitors and angiotensin receptor blockers: cohort study including 8.3 million people. Heart. 2020;106:1503–1511. doi: 10.1136/heartjnl-2020-317393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch J.S., Ng J.H., Ross D.W., Sharma P., Shah H.H., Barnett R.L., Hazzan A.D., Fishbane S., Jhaveri K.D., Abate M., et al. Acute kidney injury in patients hospitalized with COVID-19. Kidney Int. 2020;98:209–218. doi: 10.1016/j.kint.2020.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C., Wang Y., Li X., Ren L., Zhao J., Hu Y., Zhang L., Fan G., Xu J., Gu X., et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395:497–506. doi: 10.1016/S0140-6736(20)30183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L., Shi Y., Gong B., Jiang L., Zhang Z., Liu X., Yang J., He Y., Jiang Z., Zhong L., et al. Dynamic blood single-cell immune responses in patients with COVID-19. Signal Transduct. Target. Ther. 2021;6:110. doi: 10.1038/s41392-021-00526-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M., Goto S. KEGG: kyoto Encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kragstrup T.W., Singh H.S., Grundberg I., Nielsen A.L.-L., Rivellese F., Mehta A., Goldberg M.B., Filbin M.R., Qvist P., Bibby B.M. Plasma ACE2 predicts outcome of COVID-19 in hospitalized patients. PLoS One. 2021;16:e0252799. doi: 10.1371/journal.pone.0252799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kronbichler A., Kresse D., Yoon S., Lee K.H., Effenberger M., Shin J.I. Asymptomatic patients as a source of COVID-19 infections: a systematic review and meta-analysis. Int. J. Infect. Dis. 2020;98:180–186. doi: 10.1016/j.ijid.2020.06.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuleshov M.V., Jones M.R., Rouillard A.D., Fernandez N.F., Duan Q., Wang Z., Koplev S., Jenkins S.L., Jagodnik K.M., Lachmann A., et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016;44:W90–W97. doi: 10.1093/nar/gkw377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson I., Uhlén M., Zhang C., Mardinoglu A. Genome-scale metabolic modeling of glioblastoma reveals promising targets for drug development. Front. Genet. 2020;11:381. doi: 10.3389/fgene.2020.00381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lê S., Josse J., Husson F. FactoMineR: an R package for multivariate analysis. J. Stat. Softw. 2008;25:1–18. doi: 10.18637/jss.v025.i01. [DOI] [Google Scholar]
- Li X., Ma X. Acute respiratory failure in COVID-19: is it “typical” ARDS? Crit. Care. 2020;24:198. doi: 10.1186/s13054-020-02911-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luban J., Sattler R., Mühlberger E., Graci J.D., Cao L., Weetall M., Trotta C., Colacino J.M., Bavari S., Strambio-De-Castillia C., et al. The DHODH inhibitor PTC299 arrests SARS-CoV-2 replication and suppresses induction of inflammatory cytokines. bioRxiv. 2020 doi: 10.1101/2020.08.05.238394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marfia G., Navone S., Guarnaccia L., Campanella R., Mondoni M., Locatelli M., Barassi A., Fontana L., Palumbo F., Garzia E., et al. Social Science Research Network; 2020. Serum Sphingosine-1-Phosphate as Novel Prognostic and Predictive Biomarker for COVID-19 Severity and Morbidity and its Implications in Clinical Management (SSRN Scholarly Paper No. ID 3668364) [DOI] [Google Scholar]
- Marinos G., Kaleta C., Waschina S. Defining the nutritional input for genome-scale metabolic models: a roadmap. PLoS One. 2020;15:e0236890. doi: 10.1371/journal.pone.0236890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuinness G., Zhan C., Rosenberg N., Azour L., Wickstrom M., Mason D.M., Thomas K.M., Moore W.H. High incidence of barotrauma in patients with COVID-19 infection on invasive mechanical ventilation. Radiology. 2020:202352. doi: 10.1148/radiol.2020202352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moeller R., Zapatero-Belinchon F.J., Lasswitz L., Kirui J., Brogden G., Gunesch A.P., Pietschmann T., Wichmann D., Kluge S., Gerold G. Effect of statins on SARS-CoV-2 infection. medRxiv. 2020 doi: 10.1101/2020.07.13.20152272. [DOI] [Google Scholar]
- Moghaddam A., Heller R.A., Sun Q., Seelig J., Cherkezov A., Seibert L., Hackler J., Seemann P., Diegmann J., Pilz M., et al. Selenium deficiency is associated with mortality risk from COVID-19. Nutrients. 2020;12:2098. doi: 10.3390/nu12072098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morawska L., Cao J. Airborne transmission of SARS-CoV-2: the world should face the reality. Environ. Int. 2020;139:105730. doi: 10.1016/j.envint.2020.105730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morselli Gysi D., do Valle Í., Zitnik M., Ameli A., Gan X., Varol O., Ghiassian S.D., Patten J.J., Davey R.A., Loscalzo J., Barabási A.-L. Network medicine framework for identifying drug-repurposing opportunities for COVID-19. Proc Natl Acad Sci USA. 2021;118 doi: 10.1073/pnas.2025581118. e2025581118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mozzicato P. MedDRA. Pharm. Med. 2009;23:65–75. doi: 10.1007/BF03256752. [DOI] [Google Scholar]
- Pacheco M.P., John E., Kaoma T., Heinäniemi M., Nicot N., Vallar L., Bueb J.L., Sinkkonen L., Sauter T. Integrated metabolic modelling reveals cell-type specific epigenetic control points of the macrophage metabolic network. BMC Genomics. 2015;16:809. doi: 10.1186/s12864-015-1984-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orth J.D., Thiele I., Palsson B.Ø. What is flux balance analysis? Nat. Biotechnol. 2010;28:245–248. doi: 10.1038/nbt.1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostaszewski M., Mazein A., Gillespie M.E., Kuperstein I., Niarakis A., Hermjakob H., Pico A.R., Willighagen E.L., Evelo C.T., Hasenauer J., et al. COVID-19 disease map, building a computational repository of SARS-CoV-2 virus-host interaction mechanisms. Sci. Data. 2020;7:136. doi: 10.1038/s41597-020-0477-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacheco M.P., Bintener T., Ternes D., Kulms D., Haan S., Letellier E., Sauter T. Identifying and targeting cancer-specific metabolism with network-based drug target prediction. EBioMedicine. 2019;43:98–106. doi: 10.1016/j.ebiom.2019.04.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panepinto J.A., Brandow A., Mucalo L., Yusuf F., Singh A., Taylor B., Woods K., Payne A.B., Peacock G., Schieve L.A. Coronavirus disease among persons with sickle cell disease, United States, March 20–may 21, 2020. Emerg. Infect. Dis. 2020;26:2473–2476. doi: 10.3201/eid2610.202792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perumal D., Samal A., Sakharkar K.R., Sakharkar M.K. Targeting multiple targets in Pseudomonas aeruginosa PAO1 using flux balance analysis of a reconstructed genome-scale metabolic network. J. Drug Target. 2011;19:1–13. doi: 10.3109/10611861003649753. [DOI] [PubMed] [Google Scholar]
- Petrilli C.M., Jones S.A., Yang J., Rajagopalan H., O’Donnell L.F., Chernyak Y., Tobin K., Cerfolio R.J., Francois F., Horwitz L.I. Factors associated with hospitalization and critical illness among 4,103 patients with COVID-19 disease in New York City. medRxiv. 2020 doi: 10.1101/2020.04.08.20057794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puntmann V.O., Carerj M.L., Wieters I., Fahim M., Arendt C., Hoffmann J., Shchendrygina A., Escher F., Vasa-Nicotera M., Zeiher A.M., et al. Outcomes of cardiovascular magnetic resonance imaging in patients recently recovered from coronavirus disease 2019 (COVID-19) JAMA Cardiol. 2020;5:1265–1273. doi: 10.1001/jamacardio.2020.3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed J.L., Palsson B.Ø. Thirteen years of building constraint-based in silico models of Escherichia coli. J. Bacteriol. 2003;185:2692–2699. doi: 10.1128/JB.185.9.2692-2699.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed J.L., Vo T.D., Schilling C.H., Palsson B.O. An expanded genome-scale model of Escherichia coli K-12 (iJR904 GSM/GPR) Genome Biol. 2003;4:R54. doi: 10.1186/gb-2003-4-9-r54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renz A., Widerspick L., Dräger A. Genome-scale metabolic model of infection with SARS-CoV-2 mutants confirms guanylate kinase as robust potential antiviral target. Genes. 2021;12:796. doi: 10.3390/genes12060796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renz A., Widerspick L., Dräger A. FBA reveals guanylate kinase as a potential target for antiviral therapies against SARS-CoV-2. Zenodo. 2020 doi: 10.5281/zenodo.3752641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robaina Estévez S., Nikoloski Z. Context-specific metabolic model extraction based on regularized least squares optimization. PLoS One. 2015;10:e0131875. doi: 10.1371/journal.pone.0131875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson J.L., Kocabaş P., Wang H., Cholley P.-E., Cook D., Nilsson A., Anton M., Ferreira R., Domenzain I., Billa V., et al. An atlas of human metabolism. Sci. Signal. 2020;13:eaaz1482. doi: 10.1126/scisignal.aaz1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson M.D., McCarthy D.J., Smyth G.K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2009;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubino F., Amiel S.A., Zimmet P., Alberti G., Bornstein S., Eckel R.H., Mingrone G., Boehm B., Cooper M.E., Chai Z., et al. New-onset diabetes in covid-19. N. Engl. J. Med. 2020;383:789–791. doi: 10.1056/NEJMc2018688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha R., Suthers P.F., Maranas C.D. Zea mays iRS1563: a comprehensive genome-scale metabolic reconstruction of maize metabolism. PLoS One. 2011;6:e21784. doi: 10.1371/journal.pone.0021784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schellenberger J., Que R., Fleming R.M.T., Thiele I., Orth J.D., Feist A.M., Zielinski D.C., Bordbar A., Lewis N.E., Rahmanian S., et al. Quantitative prediction of cellular metabolism with constraint-based models: the COBRA Toolbox v2.0. Nat. Protoc. 2011;6:1290–1307. doi: 10.1038/nprot.2011.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz D.C., Johnson R.M., Ayyanathan K., Miller J., Whig K., Kamalia B., Dittmar M., Weston S., Hammond H.L., Dillen C., et al. Pyrimidine biosynthesis inhibitors synergize with nucleoside analogs to block SARS-CoV-2 infection. bioRxiv. 2021 doi: 10.1101/2021.06.24.449811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sehm T., Fan Z., Ghoochani A., Rauh M., Engelhorn T., Minakaki G., Dörfler A., Klucken J., Buchfelder M., Eyüpoglu I.Y., Savaskan N. Sulfasalazine impacts on ferroptotic cell death and alleviates the tumor microenvironment and glioma-induced brain edema. Oncotarget. 2016;7:36021–36033. doi: 10.18632/oncotarget.8651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparks M.A., Crowley S.D., Gurley S.B., Mirotsou M., Coffman T.M. Classical renin-angiotensin system in kidney physiology. Compr. Physiol. 2014;4:1201–1228. doi: 10.1002/cphy.c130040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stegmann K.M., Dickmanns A., Gerber S., Nikolova V., Klemke L., Manzini V., Schlösser D., Bierwirth C., Freund J., Sitte M., et al. The folate antagonist methotrexate diminishes replication of the coronavirus SARS-CoV-2 and enhances the antiviral efficacy of remdesivir in cell culture models. bioRxiv. 2020 doi: 10.1101/2020.07.18.210013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subir R., Jagat J M., Kalyan K G. Pros and cons for use of statins in people with coronavirus disease-19 (COVID-19) Diabetes Metab. Syndr. Clin. Res. Rev. 2020;14:1225–1229. doi: 10.1016/j.dsx.2020.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swainston N., Smallbone K., Hefzi H., Dobson P.D., Brewer J., Hanscho M., Zielinski D.C., Ang K.S., Gardiner N.J., Gutierrez J.M., et al. Recon 2.2: from reconstruction to model of human metabolism. Metabolomics. 2016;12:109. doi: 10.1007/s11306-016-1051-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tandon R., Sharp J.S., Zhang F., Pomin V.H., Ashpole N.M., Mitra D., Jin W., Liu H., Sharma P., Linhardt R.J. Effective inhibition of SARS-CoV-2 entry by heparin and enoxaparin derivatives. bioRxiv. 2020 doi: 10.1101/2020.06.08.140236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiele I., Swainston N., Fleming R.M.T., Hoppe A., Sahoo S., Aurich M.K., Haraldsdottir H., Mo M.L., Rolfsson O., Stobbe M.D., et al. A community-driven global reconstruction of human metabolism. Nat. Biotechnol. 2013;31:419–425. doi: 10.1038/nbt.2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas T., Stefanoni D., Reisz J.A., Nemkov T., Bertolone L., Francis R.O., Hudson K.E., Zimring J.C., Hansen K.C., Hod E.A., et al. COVID-19 infection alters kynurenine and fatty acid metabolism, correlating with IL-6 levels and renal status. JCI Insight. 2020;5:e140327. doi: 10.1172/jci.insight.140327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toelzer C., Gupta K., Yadav S.K.N., Borucu U., Davidson A.D., Kavanagh Williamson M., Shoemark D.K., Garzoni F., Staufer O., Milligan R., et al. Free fatty acid binding pocket in the locked structure of SARS-CoV-2 spike protein. Science. 2020;370:725–730. doi: 10.1126/science.abd3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tschopp J., L’Huillier A., Mombelli M., Mueller N., Khanna N., Garzoni C., Meloni D., Papadimitriou-Olivgeris M., Neofytos D., Hirsch H., et al. First experience of SARS-CoV-2 infections in solid organ transplant recipients in the Swiss Transplant Cohort Study. Am. J. Transpl. 2020;20:2876–2882. doi: 10.1111/ajt.16062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varrette S., Bouvry P., Cartiaux H., Georgatos F. 2014 International Conference on High Performance Computing Simulation (HPCS). Presented at the 2014 International Conference on High Performance Computing Simulation (HPCS) 2014. Management of an academic HPC cluster: the UL experience; pp. 959–967. [DOI] [Google Scholar]
- Vlassis N., Pacheco M.P., Sauter T. Fast reconstruction of compact context-specific metabolic network models. PLoS Comput. Biol. 2014;10:e1003424. doi: 10.1371/journal.pcbi.1003424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart D.S., Feunang Y.D., Guo A.C., Lo E.J., Marcu A., Grant J.R., Sajed T., Johnson D., Li C., Sayeeda Z., et al. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res. 2018;46:D1074–D1082. doi: 10.1093/nar/gkx1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu T., Zuo Z., Kang S., Jiang L., Luo X., Xia Z., Liu J., Xiao X., Ye M., Deng M. Multi-organ dysfunction in patients with COVID-19: a systematic review and meta-analysis. Aging Dis. 2020;11:874–894. doi: 10.14336/AD.2020.0520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong R., Zhang L., Li S., Sun Y., Ding M., Wang Y., Zhao Y., Wu Y., Shang W., Jiang X., et al. Novel and potent inhibitors targeting DHODH are broad-spectrum antivirals against RNA viruses including newly-emerged coronavirus SARS-CoV-2. Protein Cell. 2020;11:723–739. doi: 10.1007/s13238-020-00768-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J., Zheng Y., Gou X., Pu K., Chen Z., Guo Q., Ji R., Wang H., Wang Y., Zhou Y. Prevalence of comorbidities and its effects in patients infected with SARS-CoV-2: a systematic review and meta-analysis. Int. J. Infect. Dis. 2020;94:91–95. doi: 10.1016/j.ijid.2020.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yizhak K., Chaneton B., Gottlieb E., Ruppin E. Modeling cancer metabolism on a genome scale. Mol. Syst. Biol. 2015;11:817. doi: 10.15252/msb.20145307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan S., Yin X., Meng X., Chan J.F.-W., Ye Z.-W., Riva L., Pache L., Chan C.C.-Y., Lai P.-M., Chan C.C.-S., et al. Clofazimine broadly inhibits coronaviruses including SARS-CoV-2. Nature. 2021;593:418–423. doi: 10.1038/s41586-021-03431-4. [DOI] [PubMed] [Google Scholar]
- Zhang J., Taylor E.W., Bennett K., Saad R., Rayman M.P. Association between regional selenium status and reported outcome of COVID-19 cases in China. Am. J. Clin. Nutr. 2020;111:1297–1299. doi: 10.1093/ajcn/nqaa095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.-N., Zhang Q.-Y., Li X.-D., Xiong J., Xiao S.-Q., Wang Z., Zhang Z.-R., Deng C.-L., Yang X.-L., Wei H.-P., et al. Gemcitabine, lycorine and oxysophoridine inhibit novel coronavirus (SARS-CoV-2) in cell culture. Emerg. Microbes Infect. 2020;9:1170–1173. doi: 10.1080/22221751.2020.1772676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou N., Bao J. FerrDb: a manually curated resource for regulators and markers of ferroptosis and ferroptosis-disease associations. Database. 2020;2020 doi: 10.1093/database/baaa021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerman M.C., Lazartigues E., Sharma R.V., Davisson R.L. Hypertension caused by angiotensin II infusion involves increased superoxide production in the central nervous system. Circ. Res. 2004;95:210–216. doi: 10.1161/01.RES.0000135483.12297.e4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This paper analyses existing, publicly available data. The source of the data is listed in the key resources table.
The models and code generated during this study are available at GitHub (https://github.com/sysbiolux/DCcov).