

Summary
Identification of selective deubiquitinase (DUB) inhibitors is critical for probe development to further understand and explore DUB biological function. Here, we detail the optimization and deployment of an in vitro fluorogenic ubiquitin-rhodamine assay to conduct high-throughput screening of a small molecule library against a panel of DUBs. In screening the compound library against multiple DUBs in parallel, we describe an approach for identifying selective DUB inhibitors and provide a roadmap for enabling selective DUB inhibitor discovery.
For complete details on the use and execution of this protocol, please refer to Varca et al. (2021).
Subject areas: High Throughput Screening, Molecular/Chemical Probes, Protein Biochemistry, Protein expression and purification
Graphical abstract
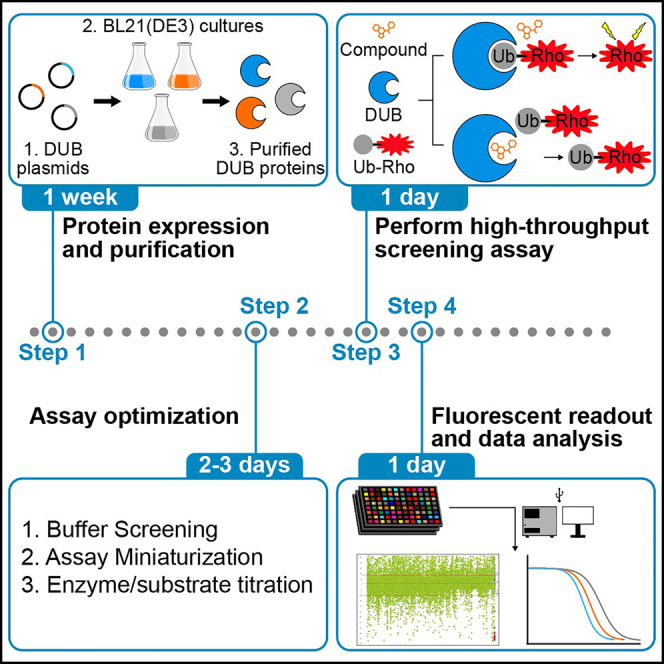
Highlights
-
•
Expression and purification of deubiquitinase (DUB) enzymes
-
•
Buffer screening, miniaturization, and deubiquitinase enzymatic assay optimization
-
•
Protocol for high-throughput deubiquitinase inhibitor discovery
Identification of selective deubiquitinase (DUB) inhibitors is critical for probe development to further understand and explore DUB biological function. Here, we detail the optimization and deployment of an in vitro fluorogenic ubiquitin-rhodamine assay to conduct high-throughput screening of a small molecule library against a panel of DUBs. In screening the compound library against multiple DUBs in parallel, we describe an approach for identifying selective DUB inhibitors and provide a roadmap for enabling selective DUB inhibitor discovery.
Before you begin
The protocol below describes the specific steps of a high-throughput screening campaign to identify small molecule DUB inhibitors using recombinant DUB enzymes and a fluorogenic DUB substrate, Ubiquitin-Rhodamine110 (Ub-Rho). All DUBs should be expressed and purified with concentrations determined. Compound stock solutions source plates should be plated out at appropriate concentrations.
General expression and purification of DUB enzymes
Timing: ∼5 days
Note: This generalizable protocol was used for the expression and purification of deubiquitinases utilized in the high-throughput screen. The protocol details the purification of both 6xHis-tagged and GST-tagged DUBs. Other affinity tags can be utilized for protein purification. Some DUBs are commercially available as purified recombinant proteins or have plasmids that can be purchased for expression and purification. We selected DUBs for screening based on multiple criteria. The first was ease of generating a large enough quantity of enzyme to be able to run the primary and dose response screens. Secondly, we sought to include multiple members of the largest DUB family (the USP subfamily) as well as representatives from the two other most well-studied cysteine proteases DUB families (UCHL and OTU) to assess compound selectivity within and amongst various DUB families. In addition, we selected DUBs associated with interesting biology or were lacking chemical probes to further explore biological function.
-
1.
Constructs encoding each DUB in the panel tested were cloned into either pET28 expression vectors with an N-terminal 6xHis tag or pGEX6P1 expression vector with an N-terminal GST tag in NEB® 10-beta Competent E. coli (High Efficiency) cells.
Note: For one of the DUBs included in the screen, OTUD3, the plasmid was obtained from Addgene and purification was carried out as previously described (Mevissen et al., 2013), which follows the general procedure for GST-tagged purification outlined below.
-
2.
Plasmids containing the desired constructs were isolated by QIAprep Spin Miniprep Kit following the manufacturer’s instructions (see QIAprep Miniprep Handbook).
-
3.
Transform BL21(DE3) Competent E. Coli cells (New England BioLabs Inc.) with the desired deubiquitinase plasmid and grow colonies at 37°C for 16–18h on LB plates with 100 μg/mL ampicillin.
Alternatives: Some constructs were obtained in plasmids with kanamycin resistance, for these constructs use LB plates supplemented with 50 μg/mL kanamycin.
Pause point: LB plates with colonies can be stored at 4°C for ∼1 week.
CRITICAL: All the following cultures need to contain antibiotics (either 100 μg/mL ampicillin or 50 μg/mL kanamycin)
-
4.
Inoculate a single colony into 5 mL of LB medium (10 g tryptone, 10 g sodium chloride, 5 g yeast extract for 1 L) with 100 μg/mL ampicillin or 50 μg/mL kanamycin and grow the cell cultures at 37°C and 1.12 × g orbital rotation for 16–18 h.
-
5.
Dilute the 5 mL culture into 1 L of LB medium and grow until OD600 is between 0.8 and 1.0.
-
6.
Remove cultures from the 37°C incubator, add IPTG to 100 mg/L to induce protein expression, and incubate at 16°C and 1.12 × g orbital rotation for 18–24 h.
-
7.
Pellet cells by centrifugation at 4,540 × g at 4°C for 20 min and decant supernatant.
-
8.
Resuspend cell pellet in 50–100 mL lysis buffer (25 mM Tris, pH 8, 10 mM β-mercaptoethanol, 1 M NaCl) and stir at 4°C for ∼30 min.
-
9.Add phenylmethylsulfonyl fluoride (PMSF) to 10 μg/mL then lyse the homogenized solution by sonication on ice (Fisher Scientific Model 505 Sonic Dismembrator). Alternate 10 s of sonication at 70% amplitude with 5 s rest periods twelve times for a total of 120 s (2 min) sonication.
-
a.After first sonication, mix lysate with a spatula to check for complete lysis (no cellular globs should be observed). If needed, repeat sonication step above to complete lysis.
-
a.
Note: If proper lysis has occurred, the solution should have a thinner consistency and be more translucent than the starting solution.
-
10.
Centrifuge the lysate at 30,000 × g for 40 min at 4°C.
-
11.For every 1 L of culture, equilibrate 0.5–1 mL of Ni-NTA Agarose (50% slurry in storage solution, QIAGEN) in lysis buffer.Note: Some constructs had a GST-tag instead of a 6xHis-tag. For purification of GST-tagged DUBs, Pierce™ Glutathione Superflow Agarose was utilized instead of Ni-NTA Agarose. 2–5 mL of Pierce™ Glutathione Superflow Agarose (stored in 70% ethanol, Thermo Scientific) was equilibrated in lysis buffer.
-
a.Wash the resin 2–3 times with sterile water, 2–3 times with lysis buffer, then resuspend in lysis buffer.
-
a.
-
12.
Add cell lysate to the resin (either the Ni-NTA or Agarose resin depending on the tag present on the expressed DUB) in a gravity flow column and incubate at 4°C for 2–4 h with gentle agitation.
-
13.
Drain the column via gravity flow until the liquid level is just above the top of the resin, usually around 2–5 min, and collect the eluate (loading eluate)
-
14.Flow 50 mL of wash buffer (25 mM Tris, pH 8, 10 mM β-mercaptoethanol, 1 M NaCl, 25 mM imidazole, pH 8) and collect the eluate (wash eluate)
-
a.Repeat wash step 14 10x collecting all of the wash eluate
-
a.
Note: For GST-tag purification, the wash buffer used was the same as the lysis buffer (25 mM Tris, pH 8, 10 mM β-mercaptoethanol, 1 M NaCl)
-
15.Elute desired protein by adding 10 mL of elution buffer (25 mM Tris, pH 8, 10 mM β-mercaptoethanol, 1 M NaCl, 300 mM imidazole, pH 8), stirring gently, letting rest for 2 min and then eluting via gravity flow.
-
a.Repeat elution step 15 for a total of 5 times, combining eluent collected from all elutions.
-
a.
Note: For GST-tag purification, a different elution buffer was used (25 mM Tris pH 8, 10 mM β-mercaptoethanol, 200 mM NaCl, GST 3C protease at 1 mg/mL). The elution buffer was added to the resin and incubated at 4°C 16–18 h. Eluent was then collected via gravity filtration the following day.
Note: For the 6xHis tagged DUBs the tags were not removed. For the GST-tagged DUBs, the elution buffer contained GST 3C protease which removed the GST-tag from the DUB as part of the purification step.
Optional: Reserve ∼15–20 μL of each elution from steps 9–15 above (loading eluate, 25 mM imidazole wash eluate, 300 mM imidazole eluate, etc.) as well as a small sample of the resin after the final elution to run an SDS-PAGE and total protein stain analysis to identify fractions containing the DUB of interest. In instances of low or no yield, the SDS-PAGE gel and protein stain analysis can be used to determine at which step in the purification process the desired DUB was eluted off the Ni-NTA or Agarose resin. See Troubleshooting problem 1.
-
16.
Use a centrifugal filter unit (Amicon® Ultra-15 Centrifugal Filter Unit, Millipore Sigma) to concentrate the DUB containing fraction(s) from step 15 to <1 mL.
-
17.Purify protein sample via fast protein liquid chromatography (FPLC, BioRad) using a Superdex 200 (GE healthcare) size exclusion column.
-
a.Prepare FPLC by washing sample loop with size exclusion column buffer (25 mM HEPES, pH 7.5, 200 mM NaCl, 1 mM DTT) twice.Optional: Centrifuge protein sample for 10 minutes at 16,162 × g to pellet any protein that may have precipitated out of solution.
-
b.Inject protein sample into sample loop and run size exclusion method (isocratic flow of size exclusion buffer from step 17a, 0.4 mL/min, detection at 280 nm) on FPLC and collect fractions containing desired protein.
-
a.
-
18.
Run SDS-PAGE gel with fractions from FPLC followed by Coomassie staining to determine which fractions contain the pure DUB of interest. Combine fractions and use a centrifugal filter unit (Amicon® Ultra-15 Centrifugal Filter Unit, Millipore) to concentrate the DUB containing fraction(s) to ∼1 mL
-
19.
Determine protein concentration by Tyrosine/Tryptophan fluorescence on a NanoDrop instrument at 280 nm absorbance along with the calculated extinction coefficient of the protein construct.
Note: Protein extinction coefficients can be calculated from online tools such as ProtParam (https://web.expasy.org/protparam/)
-
20.
To confirm DUB activity, purified DUBs can be assessed by a ubiquitin-rhodamine110 (Ub-Rho) cleavage assay.
Note: The Ub-Rho cleavage assay involves mixing a solution of purified DUB at varying concentrations in assay buffer and a solution of Ub-Rho at varying concentrations in assay buffer and monitoring fluorescence over time. Active DUB will result in increased fluorescence over time. A generalized protocol for this assay can be found in the enzyme and substrate titrations section below.
Note: While many DUBs have demonstrated activity against Ub-Rho, there are some DUBs that are less active or do not readily cleave Ub-Rho. Some of these DUBs have been shown to cleave alternative substrates that can be deployed in high-throughput screens. See Troubleshooting problem 2.
Note: For these experiments Ub-Rho was purchased from R&D Systems, however, Ub-Rho can be synthesized based on previously established protocols (Hassiepen et al., 2007).
Note: Purified DUB stocks should be aliquoted to appropriately sized aliquots for desired experiments and flash frozen using liquid N2 to minimize freeze-thaws.
Design of experiment (DOE) operation
Note: The DOE approach was deployed to test for optimal buffer. This involves first deciding on relevant buffer components to vary, taking from literature sources or baseline buffer conditions. Once all the factors are chosen, the experimental design is created in JMP statistical software (developed by SAS Institute), which provides the optimal combination of factors to achieve ∼2% coverage of all potential combinations, allowing adequate modeling of each component’s effect on the assay.
-
21.Starting buffer components (50 mM HEPES pH 7.6, 0.5 mM EDTA pH 8, 11 μM ovalbumin, and 5 mM DTT) based on previously reported assay conditions (Lamberto et al., 2017) and variable conditions for each component were entered into the JMP software.
-
a.Variable conditions for each component were selected follows: HEPES and Tris buffers were prepared at three pH values (7, 7.5, and 8) and NaCl was varied at 0, 25, and 50 mM. Bovine serum albumin was varied at 0 and 1% and detergents CHAPS, Tween-20, PF127, NP-40, and Triton-X, were varied at 0×, 0.25×, and 0.5× their critical micelle concentration (CMC) values. Reducing reagents DTT and TCEP were varied at 0 and 1 mM and EDTA was varied at 0 and 1 mM. See Table 1 for summary of conditions assessed.
-
a.
Note: The output from the JMP design was used to create a dispense list to deliver the buffer components to the assay plate using a Formulatrix TEMPEST liquid handler that is capable of dispensing up to 12 reagents in varying volumes and combinations. Two 384 well plates were prepared of the various buffer conditions, one for screening with enzyme and the other for screening without enzyme.
-
22.
8 μL of the variable DOE buffers as determined by the JMP software were dispensed to 384 well plates.
-
23.
Prepare a 10x solution of DUB in DOE assay buffer (50 mM HEPES pH 7.6, 0.5 mM EDTA pH 8, and 5 mM DTT).
-
24.
Prepare a 10x solution of Ub-Rho in DOE assay buffer (50 mM HEPES pH 7.6, 0.5 mM EDTA pH 8, and 5 mM DTT).
-
25.
1 μL of the 10x DUB solution was added to the 384 well plate containing varied buffers as selected by JMP software from step 22.
-
26.
1 μL of the 10x Ub-Rho solution was then added to the plates.
-
27.
Fluorescence was recorded using a PHERAstar plate reader (reading at excitation and emission wavelengths of 485 nm and 535 nm respectively) every 5 min for the first 70 min and every 10 min until 100 min.
Note: The DOE was performed for two DUBs, USP7 and UCHL1, following the protocol above. Based on the DOE performed for USP7 and UCHL1, we selected a single buffer composition that was used in the subsequent titration experiments as well as the primary and dose-response screens to maximize fluorescence signal.
Alternatives: A lower throughput method for buffer testing can be deployed by preparing a smaller selection of buffers manually and running the assay in those buffers as described. DOE results for DUBs suggests that a more basic pH (pH 8) and presence of reducing agent are the most important factors for DUB activity observed while many of the other buffer components had minimal effect on observed signal. See Troubleshooting problems 3 and 4.
Table 1.
Variable conditions for design of experiment (DOE) buffer testing
| Variables | Conditions |
|---|---|
| Buffer | HEPES, Tris |
| pH | 7, 7.5, 8 |
| NaCl concentration | 0, 25, 50 mM |
| BSA concentration | 0, 1% |
| EDTA concentration | 0, 1 mM |
| Detergent | CHAPS, Tween20, PF127, NP-40, Triton-X |
| Detergent concentration | 0×, 0.25×, 0.5× critical micellar concentration (CMC) |
| Reducing agent | DTT, TCEP |
| Reducing agent concentration | 0, 1 mM |
Enzyme and substrate titrations
Note: Once buffer conditions were selected from the DOE, a titration of DUB and substrate was carried out for each enzyme to minimize reagent consumption while still producing fluorescence values with greater than a 5-fold signal-to-noise ratio. Initial titrations were carried out with USP7 and UCHL1 using the following protocol.
-
28.
Prepare 3 mL of a 2x DUB solution in assay buffer (50 mM Tris pH 8, 50 mM NaCl, 0.002% Tween-20, and 5 mM DTT).
-
29.
Perform a series of 2-fold dilutions to prepare a range of concentrations of the DUB solution from step 28 from 500 nM to 7.8 nM
-
30.
Add 5 μL of assay buffer (no DUB) to columns 1–3 on a 384 well plate
-
31.Add 5 μL of the lowest concentration DUB solution prepared in step 29 to columns 4–6 on the 384 well plate.
-
a.Add the next lowest concentrated DUB solution to the next 3 columns and repeat until the entire plate is filled.
-
a.
-
32.
Prepare 2 mL of a 2x Ub-Rho solution in assay buffer (50 mM Tris pH 8, 50 mM NaCl, 0.002% Tween-20, and 5 mM DTT).
-
33.
Perform a series of 2-fold dilutions to prepare a range of concentrations of the Ub-Rho solution from step 32 from 2000 nM to 31.25 nM.
Note: DUB and Ub-Rho concentrations can be altered to assess different ranges of enzyme and substrate. Depending on how active the DUB is, multiple titrations may need to be performed to assess the appropriate concentrations for screening.
-
34.
Add 5 μL of different Ub-Rho solutions to rows on the 384 well plate from step 31 such that the first 2 rows received 0 nM Ub-Rho (assay buffer only) and every subsequent 2 rows received the next highest concentration of Ub-Rho solutions prepared.
-
35.
Centrifuge the plate (16 × g, 5 s) and incubate at 20°C–25°C.
-
36.
Read the plate on a PHERAstar plate reader (excitation/emission 485 nm/535 nm) every 20 min for 100 min.
-
37.
DUB and substrate concentrations for screening were selected based on the conditions that produced a signal to noise ratio greater than 5 while using lesser amounts of DUB and substrate. The signal to noise ratio was calculated by average signal of the assay wells at a given concentration divided by the average for the 0 nM substrate wells at that DUB concentration.
Note: DUB and substrate conditions could be, and in some cases were, selected to increase the signal-to-noise ratio beyond 5. A signal-to-noise ratio of 5 represented the minimal size for consideration of a set of conditions to be deployed in the high-throughput screens. If signal is low at all tested concentrations of enzyme and substrate that could indicate either an issue with the protein, buffer preparation, or enzyme/substrate solution preparation. See Troubleshooting problems 3 and 4.
Assay miniaturization
Note: Once enzyme and substrate concentrations were established based on the previous step, we performed a miniaturization to 1536 well plates with 5 μL total reaction volume to assess if we could further reduce reagent consumption while retaining good signal to noise ratios. If desired, the assays can be run in 384 well plates, in which case this step can be skipped.
-
38.
Prepare a 2x DUB solution in assay buffer (50 mM Tris pH 8, 50 mM NaCl, 0.002% Tween-20, and 5 mM DTT).
-
39.
Prepare a 2x Ub-Rho solution in assay buffer (50 mM Tris pH 8, 50 mM NaCl, 0.002% Tween-20, and 5 mM DTT).
Note: Two different DUB and Ub-Rho solutions were prepared for each DUB such that final assay concentrations match with the two best conditions selected from the enzyme/substrate titration experiment for specific DUBs.
-
40.Add 2.5 μL of DUB solution to rows A-F on a 1536 well plate.
-
a.Columns 1–3 received assay buffer only (no DUB), columns 4–9 received DUB solutions with columns 4–6 and 7–9 receiving different concentrations of DUB.
-
a.
-
41.Add 2.5 μL of Ub-Rho solutions to rows A-F on the 1536 well plate from step 40.
-
a.Rows A-C received assay buffer only (no Ub-Rho), rows D-I received Ub-Rho solutions with rows D-F and G-I receiving different concentrations of Ub-Rho.
-
a.
-
42.
Read the plate on a PHERAstar plate reader (excitation/emission 485 nm/535 nm) at 0 min and 60 min.
Note: Miniaturization of the assay produced robust signal even at reduced reaction volumes for both USP7 and UCHL1. Based on this finding, all future DUB titrations were performed in 1536 well plates using the 5 μL reaction volumes and subsequent primary screening efforts were also carried out in this format.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| NEB® 10-beta Competent E. coli (High Efficiency) | New England Biolabs Inc | Cat#C3019H |
| BL21(DE3) Competent E. coli | New England Biolabs Inc | Cat#C2527H |
| Chemicals, peptides, and recombinant proteins | ||
| Isopropyl β-D-1-thiogalactopyranoside (IPTG) | UBP Bio | Cat#P1010-100 |
| β-Mercaptoethanol | Sigma-Aldrich | Cat#M3148-25ML |
| Phenylmethylsulfonyl fluoride (PMSF) | RPI | Cat#P20270-25.0 |
| Ampicillin sodium salt | Fisher Scientific | Cat#BP1760-25 |
| Kanamycin sulfate, ultrapure | Thermo Scientific | Cat# J1792414 |
| Ubiquitin-Rhodamine 110 (Ub-Rho) | R&D Systems | Cat#U-555-050 |
| Deposited data | ||
| Primary and dose response confirmation screening data | PubChem | https://pubchem.ncbi.nlm.nih.gov/bioassay/1645869 |
| Recombinant DNA | ||
| pOPINK-OTUD3 (OTU+UBA, aa 52-275) | (Mevissen et al., 2013) | Addgene plasmid# 61411 |
| pET28aLIC-USP7 (His-tagged, aa 207-532) | (Varca et al., 2021) | N/A |
| pET28aLIC-USP8 (His-tagged, aa 742-1110) | (Varca et al., 2021) | N/A |
| pET28PP-USP10 (His-tagged, aa 376-798) | (Varca et al., 2021) | N/A |
| pET28b-USP17 (His-tagged, aa 1-530) | (Varca et al., 2021) | N/A |
| pET28a USP25 (His-tagged, aa 207-532) | (Varca et al., 2021) | N/A |
| pET28aLIC-USP28 (His-tagged, aa 207-532) | (Varca et al., 2021) | N/A |
| pET28PP-USP30 (His-tagged, aa 207-532) | (Varca et al., 2021) | N/A |
| pGEX6P1-UCHL1 (GST-tagged, aa 1-223) | (Varca et al., 2021) | N/A |
| Software and algorithms | ||
| Prism 8 | GraphPad |
https://www.graphpad.com/scientific-software/prism/; RRID: SCR_002798 |
| JMP | SAS Institute | RRID:SCR_014242 |
| Other | ||
| Ni-NTA Agarose | QIAGEN | Cat#30210 |
| Pierce™ Glutathione Superflow Agarose | Thermo Scientific | Cat#25237 |
| QIAprep Spin Miniprep Kit | QIAGEN | Cat#27104 |
| Amicon® Ultra-15 Centrifugal Filter Unit | Millipore Sigma | Cat#UFC903024 |
| Fisher Scientific Model 505 Sonic Dismembrator | Fisher Scientific | Cat#FB505 |
| NanoDrop 2000 | Thermo Scientific | Cat#ND-2000 |
| 384-Well plates | Greiner | Cat#784201 |
| 1536-Well plates | Greiner | Cat#782076 |
| Echo | Labcyte | Cat#ECHO555 |
| Multidrop™ Combi Reagent Dispenser | Thermo Fisher Scientific | Cat#5840300 |
| PHERAstar | BMG Labtech | Cat#PHERAstarFSX |
| Tecan Freedom EVO 200 | Tecan | Cat#Freedom EVO 200 |
| TEMPEST liquid handler | Formulatrix | Cat#TEMPEST |
Materials and equipment
Compound transfer from source plates was done via acoustic transfer using an Echo Liquid Handler. Dispensing of assay buffer and enzyme/substrate solutions can be done either using a BioRaptr™ Microfluidic Dispenser or Multidrop™ Combi Reagent Dispenser. Results can be measured on any plate reader equipped to monitor fluorescence.
Assay Buffer for DUB HTS
| Reagent | Stock concentration | Final concentration | Volume |
|---|---|---|---|
| Tris pH 8 | 1 M | 50 mM | 50 mL |
| NaCl | 5 M | 50 mM | 10 mL |
| Tween20 | 10% | 0.002% | 200 μL |
| DTT | 1 M | 5 mM | 5 mL |
| H2O | – | – | 934.8 mL |
| Total | – | – | 1000 mL |
Assay buffer was prepared fresh for each experiment. DTT as a 1 M stock solution was stored at 4°C, other reagents were stored at 20°C–25°C.
STOP buffer for DUB HTS
| Reagent | Stock concentration | Final concentration | Volume |
|---|---|---|---|
| Trifluoroacetic acid (TFA) | 10% | 0.2% | 8.4 mL |
| Assay buffer | – | – | 131.6 mL |
| Total | – | – | 140 mL |
STOP buffer should be prepared fresh for each experiment.
Alternatives: Other reducing agents such as TCEP can be used in place of DTT. The activity of each DUB in any alternate buffers should be confirmed prior to running any screening assays.
Step-by-step method details
Primary screen enzyme/substrate solution preparation
Prepare assay buffer, DUB, and substrate solutions for assay.
-
1.
Prepare 170 mL of a 2x DUB solution in assay buffer (50 mM Tris pH 8, 50 mM NaCl, 0.002% Tween-20, and 5 mM DTT).
-
2.
Prepare 170 mL of a 2x Ub-Rho solution in assay buffer (50 mM Tris pH 8, 50 mM NaCl, 0.002% Tween-20, and 5 mM DTT).
-
3.
Prepare 160 mL of assay buffer (50 mM Tris pH 8, 50 mM NaCl, 0.002% Tween-20, and 5 mM DTT) + 0.8% DMSO.
-
4.
Prepare 140 mL of STOP buffer (50 mM Tris pH 8, 50 mM NaCl, 0.002% Tween-20, and 5 mM DTT + 0.2% TFA).
Note: All enzyme and substrate concentrations were optimized for each enzyme prior to running all primary and confirmation screens
Primary screen plate preparation
Prepare primary screen assay plates by adding compound to each well via acoustic transfer.
-
5.
Single concentration compound source plates for primary screening were made by adding 2 μL of 10 mM stock solution to each well of a 1536 well plate.
-
6.
2 μL of 90% DMSO was then added to all wells to dilute each compound to 5 mM.
-
7.
20–50 nL of each compound were acoustically transferred from source plates to new 1536 well assay plates by Labcyte Echo acoustic transfer.
Note: Compound concentrations for primary screens varied from 20 μM to 50 μM depending on DUB. Columns 45–46 contained DMSO only as a neutral control. Columns 47–48 were left blank to be used as an active control (no DUB).
Primary screen DUB enzymatic reaction
DUB enzymatic cleavage of Ub-Rho substrate in the presence of small molecule inhibitors.
Note: Prior to running any of the full high-throughput screens, it is recommended to check all liquid handling steps first to ensure proper function of all liquid handlers. See Troubleshooting problem 3.
-
8.
2.5 μL of a 2x DUB solution in assay buffer (50 mM Tris pH 8, 50 mM NaCl, 0.002% Tween-20, 5 mM DTT) was added to columns 1–46 on a Greiner medium binding 1536 well plate (catalog number: 782076) prepared in step 7.
-
9.
2.5 μL of assay buffer was dispensed to columns 47–48.
Optional: Plates can be stored at 20°C–25°C and up to 95% humidity for up to an hour if needed.
-
10.
2.5 μL of a 2x Ub-Rho solution in assay buffer (50 mM Tris pH 8, 50 mM NaCl, 0.002% Tween-20, 5 mM DTT) was then added to all wells on the plate.
-
11.
Plates were incubated at 20°C–25°C.
Note: Incubation times varied for different DUBs based on the enzyme/substrate titration experiments. Incubation times ranged from 30 minutes to 3 hours.
Alternatives: For some DUB screens, 2.5 μL of STOP buffer was added to each well on the plate to prevent further substrate cleavage. This allowed multiple DUB screens to be run concurrently with plates being read on the plate reader at a later timepoint. See Troubleshooting problem 5.
-
12.
Plates were centrifuged at ∼16 × g for 5 s.
-
13.
Fluorescence was recorded using a BMG PHERAStar fluorescence plate reader at excitation and emission wavelengths of 485 nm and 535 nm respectively.
Note:Figure 1 shows a general workflow for the primary and dose-response screens
Figure 1.

General workflow for primary and dose-response screens
Prior to beginning the screening process, DUB and ubiquitin-rhodamine (Ub-Rho) solutions are prepared in assay buffer and compound source plates are prepared from DMSO stocks. Step 1: Compounds are added from source plates to 1536 well assay plates in single concentrations (for primary screen) or in either 4 or 8-point dose-response (for dose-response screens). Columns 45–46 contain DMSO only as a neutral control. Step 2: A 2x solution of DUB in assay buffer is then added to all wells on the plate except columns 47–48 (which receive only assay buffer). Step 3: A 2x solution of Ub-Rho is then added to all wells on the plate. Step 4: Plates are then incubated to allow the DUB enzymatic reaction to occur. Step 5: Fluorescence measurements for each plate are then recorded using a fluorescence plate reader followed by data analysis.
Dose response enzyme/substrate solution preparation
Prepare assay buffer, DUB, and substrate solutions for dose-response confirmation of primary screen hits.
-
14.
Prepare 170 mL of a 2x DUB solution in assay buffer (50 mM Tris pH 8, 50 mM NaCl, 0.002% Tween-20, and 5 mM DTT).
-
15.
Prepare 170 mL of a 2x Ub-Rho solution in assay buffer (50 mM Tris pH 8, 50 mM NaCl, 0.002% Tween-20, and 5 mM DTT)
-
16.
Prepare 160 mL of assay buffer (50 mM Tris pH 8, 50 mM NaCl, 0.002% Tween-20, and 5 mM DTT) + 0.8% DMSO.
-
17.
Prepare 140 mL of STOP buffer (50 mM Tris pH 8, 50 mM NaCl, 0.002% Tween-20, and 5 mM DTT + 0.2% TFA).
Note: As with the primary screen all enzyme and substrate concentrations were optimized for each enzyme prior to running all primary and confirmation screens. The same concentrations of DUB and substrate used in the primary screen were used for each enzyme in the dose-response confirmation.
Dose response screen plate preparation
Prepare assay plates by adding compound to each well via acoustic transfer.
-
18.For preparation of dose-response compound source plates, 10 mM DMSO stocks were added to an intermediary 384 well plate.
-
a.11.7 μL of 10mM compound in 100% DMSO was transferred from Matrix storage tubes to top concentration wells (row A and row I for 8-point dose response or row A, row E, row I, and row M for 4-point dose response) using a Tecan Freedom EVO-2 200 Base automated pipetting robot.
-
a.
-
19.
8 μL of 90% DMSO is dispensed to remaining wells in the 384 well polypropylene plate (row B-H and row J-P for 8-point dose response or row B-D, row F-G, row J-L, and row N-P for 4-point dose response) with a Thermo Fischer MultiDrop Combi.
-
20.A serial half-log dilution series is prepared by transferring compounds from rows with the highest concentration wells to the row directly below using the Tecan Freedom EVO-2 200 Base pipetting robot.
-
a.For 8-point dose response plates 3.7 μL is transferred from wells in row A to wells in row B, and from wells in row I to wells in row J followed by mixing with 7 cycles of aspiration and dispense.
-
b.For 4-point dose response plates 3.7 μL is transferred from wells in row A to wells in row B, from wells in row E to wells in row F, from wells in row I to wells in row J, and from wells in row M to wells in row N followed by mixing with 7 cycles of aspiration and dispense.
-
a.
-
21.This process is repeated vertically down the plate to prepare the half-log dose response series for each compound.
-
a.For the 8-point dose response plates 3.7 μL of solution from row H and P are removed after the final dilution and disposed to waste.
-
b.For the 4-point dose response plates 3.7 μL of solution from row D, row H, row L, and P are removed after the final dilution and disposed to waste.
-
a.
Note: DMSO percentage in the dose response compound source plates range from 100% at the highest concentration to 90.0% at the lowest concentration.
-
22.5 μL of compound are transferred from each well of the intermediary plate to the 1536 well source plate for screening.
-
a.Four 384 well intermediary plates are stacked in an interleaved pattern to make each 1536 well source plate.
-
a.
-
23.
Compounds were acoustically transferred from source plates to new 1536 well assay plates by Labcyte Echo acoustic transfer.
Note: For all DUBs, hits from the primary screen were prepared in compound source plates in either 8-point or 4-point half-log dose response in DMSO depending on DUB. Initial concentrations of the dose-response varied from 20 μM to 50 μM depending on DUB. Columns 45–46 contained DMSO only as a neutral control. Columns 47–48 were left blank to be used as an active control (no DUB).
Dose response DUB enzymatic reaction
DUB enzymatic cleavage of Ub-Rho substrate in the presence of primary screen hits plated out in dose-response.
-
24.
2.5 μL of a 2x DUB solution in assay buffer (50 mM Tris pH 8, 50 mM NaCl, 0.002% Tween-20, 5 mM DTT) was added to columns 1–46 on a Greiner medium binding 1536 well plate (catalog number: 782076) prepared in step 23.
-
25.
2.5 μL of assay buffer was dispensed to columns 47–48.
Optional: Plates can be stored at 20°C–25°C and up to 95% humidity.
-
26.
2.5 μL of a 2x Ub-Rho solution in assay buffer (50 mM Tris pH 8, 50 mM NaCl, 0.002% Tween-20, 5 mM DTT) was then added to all wells on the plate.
-
27.
Plates were incubated at 20°C–25°C.
Note: Incubation times varied for different DUBs based on the enzyme/substrate titration experiments. Incubation times ranged from 30 minutes to 3 hours.
Alternatives: For some DUB screens, 2.5 μL of STOP buffer was added to each well on the plate to prevent further substrate cleavage. This allowed multiple DUB screens to be run concurrently with plates being read on the plate reader at a later timepoint. See Troubleshooting problem 5.
-
28.
Plates were centrifuged at ∼16 × g for 5s.
-
29.
Fluorescence was recorded using a BMG PHERAStar fluorescence plate reader at excitation and emission wavelengths of 485 nm and 535 nm respectively.
Note:Figure 1 shows a general workflow for the primary and dose-response screens
Expected outcomes
Compounds demonstrating DUB inhibition should show reduced fluorescence signal. The DMSO neutralcontrol wells compared to the active control wells (no DUB) should show >5-fold signal to noise ratio. Z′ values were calculated for all plates in the high-throughput screen and ranged from 0.49–0.92 across the primary and dose-response screens. For assessing the IC50 values of compounds in the dose-response screen, active compounds should demonstrate a clear dose-response curve across the concentrations tested. The quantification and statistical analysis section provide additional detail on how the data was analyzed.
Quantification and statistical analysis
Z-prime factors were calculated for all plates in the primary and dose-response screens as follows.
Compute the threshold value for active controls (no DUB wells) as the mean signal of the active controls (μAC) plus three times their standard deviation (σAC).
Compute the threshold value for neutral controls (DMSO wells) as the mean signal of the positive controls (μnc) minus three times their standard deviation (σNC).
Compute the difference between the two thresholds as the ‘separation band’ of the assay, S. If the threshold computed in step 1 is less than the one computed in step 2, then this difference is positive. Otherwise, this difference will have a negative value.
Compute the absolute value of the difference between the two means as the ‘dynamic range’ of the assay, R.
Compute the Z′ as S/R
Normalization of raw data to active and neutral control wells, systematic pattern models, and curve fitting were applied to the data using Helios, a high-throughput screening data analysis program developed at Novartis (Gubler et al., 2018). The median value of DMSO control (neutral control) wells (columns 45–46 on each plate) was calculated for each plate. Individual DMSO wells with raw fluorescence values greater than 4 standard deviations from the median were masked from analysis. Fluorescence data was normalized by the following equation for each fluorescence value:
Where x is the raw fluorescence value for each well, NC is the DMSO neutral control (DMSO + DUB + substrate), and AC is the active control (no DUB). The calculation was multiplied by −1 to represent the active control median being −100% (inhibitory effect). Results were reported as Percent DUB Activity (Table 2, column 3). For measuring compound potency, a four-parameter sigmoid Hill curve model was fitted to the data (Gubler et al., 2018) and the absolute IC50 was used for potency measurements, that is the concentration of compound where the data showed 50% inhibition. If no data points achieved 50% inhibition, then the absolute IC50 was reported as >X μM (where X is the highest tested concentration) Figure 2 shows a representative IC50 curve based on the data in Table 1.
Table 2.
Example data and data processing from the dose response of a compound from the small molecule library against USP7
| Compound (μM) | Raw fluorescence | Percent DUB activity ((fluorescence – NC)/(AC – NC)) ∗100∗−1 |
|---|---|---|
| 40 | 11190 | −93.97 |
| 12.66 | 34769 | −74.58 |
| 4 | 80793 | −36.73 |
| 1.266 | 107757 | −14.55 |
| 0.4 | 112614 | −10.56 |
| 0.126 | 112856 | −10.36 |
| 0.04 | 119232 | −5.11 |
| 0.012 | 114746 | −8.80 |
| DMSO (Neutral control, NC) | 125451.5 | 0 |
| No enzyme (Active control, AC) | 3853 | −100 |
The calculation for Percent DUB activity was multiplied by −1 to represent the active control median being −100% (inhibitory effect).
Figure 2.

Inhibition curve of normalized fluorescence data
Dose-response curve of normalized fluorescence data from Table 2.
Limitations
The Ub-Rho screening assay is an in vitro screening assay that doesn’t accurately reflect the cellular conditions that DUBs experience. We have tested some of the hit compounds in cellular experiments to confirm DUB inhibition, however, it is not a given that hits will always confirm when working in cellular contexts (Varca et al., 2021). High-throughput screens, especially using a fluorescence readout, can also be subject to assay interference through a myriad of ways including PAINS compounds, compounds that are autofluorescent, or fluorescence quenchers. While the compound library was curated to avoid these PAINS compounds there is still the possibility that certain compounds demonstrate assay interference phenotypes that can confound results. This assay platform is best utilized to identify potential selective and potent starting points for optimization of DUB inhibitors against a given DUB target which require additional biophysical and cellular validation to confirm them as bona-fide DUB inhibitors.
Troubleshooting
Problem 1
No or low protein yield from protein purification.
Potential solution
Examples of issues that may arise and lead to no or low protein yields include not allowing the protein of interest enough time to bind to the resin or using an older batch of resin that may be less efficient at binding the protein tag, both of which can result in much of the protein being discarded after draining the column following the initial incubation step. Alternatively, if the elution solutions are prepared incorrectly, it’s possible that the protein of interest will not elute off of the resin at all. As there are numerous steps during which problems may occur, it is recommended to keep all eluents from each step of the protein purification process to be able to diagnose where potential issues may have arisen in isolating the desired protein. Running an SDS-PAGE gel on a sample of each eluent obtained in the protein purification process can determine which eluents contain the desired protein of interest. Seeing which eluents contain the protein, and which eluents no longer contain the protein, can help determine if there is an issue with a specific step. As a general guideline, it is recommended to use fresh resin, if possible, or if you suspect your resin may be less efficient at binding the protein tag you can increase the amount of resin used. It is also recommended to prepare buffers and solutions fresh on the day of the purification.
Problem 2
Certain DUBs are much less active against or are unable to cleave Ub-Rho.
Potential solution
It is always recommended to assess the purity of proteins prior to testing to rule out low activity based on lower purity or degraded protein due to things like multiple freeze-thaw cycles. If protein purity is high but signal is still low, alternative substrates include ubiquitin-like proteins conjugated to rhodamine or short ubiquitin chains functionalized for a fluorescence polarization or fluorescence resonance energy transfer readout(Cho et al., 2020; Orcutt et al., 2012). In general, most cysteine protease DUBs are amenable to using Ub-Rho as a reporter substrate while zinc metalloprotease DUBs typically require a di-ubiquitin substrate with preference for K63 linked ubiquitin. For example, a K63 linked di-ubiquitin TAMRA probe was used in a fluorescence polarization assay to assess small molecule inhibitors of the zinc metalloprotease DUBs Rpn11(Li et al., 2017). If cost is not an issue, higher concentrations of Ub-Rho can be used to make up for lower signal or alternative substrates can be used. Use of these alternative substrates would require re-optimization of the assay conditions and potentially the use of alternate readouts than the fluorescence readout described in this protocol.
Problem 3
Low activity observed in assay plates can result from a number of reasons, most likely issues in addition of DUB or substrate.
Potential solution
Check liquid handling equipment to ensure the proper addition of the correct volumes of enzyme and substrate to assay plates. Insufficient or inaccurate addition of either DUB or Ub-Rho can result in low signal observed as the fluorogenic rhodamine species is not generated. A test run to check out liquid handling steps is recommended followed by a visual inspection of the plate to ensure wells all look like they have received the proper amount of solution and equal amounts. In addition, the volume added to wells can be checked by aspirating the contents of individual wells with a pipette and confirming the volume added that way.
Problem 4
Preparation of the DUB and Ub-Rho solutions are an important step of the screening protocol. Insufficient mixing or poor-quality reducing agent stock can result in low DUB activity and lead to low assay signal.
Potential solution
It is crucial that fresh reducing agent is used and is accurately added to all assay buffers. The catalytic cysteine residues of many DUBs are prone to oxidation and require a reducing environment to observe DUB function. Comparison of the assay signal of compound wells to the no DUB control wells (columns 47–48) can give an initial sense of the activity of the DUBs. The results from the DOE and enzyme/substrate titrations indicates the reducing agent concentration selected for our buffer is sufficient for all DUBs included in the screen; however, the concentration of reducing agent could be increased if the reducing agent is suspected to be an issue.
Problem 5
Efficient readout of assay plates at specific timepoints can be difficult with a large number of plates required for screening the entire compound library.
Potential solution
This high-throughput screening campaign assessed a roughly 50,000 compound library against each DUB selected. In both the primary screen and dose-response screen this equated to upwards of 40 separate 1536 well plates. Timing of enzyme and substrate incubation with compound is important to keep consistent and exact for each plate. We observed that the time required for the readout of each plate was causing some plates at the end of the stack to incubate for a slightly longer period of time than the first plates being read. We found that addition of a STOP buffer to cease all enzymatic activity enabled us to stop the enzymatic reactions on each plate allowing us to better compare results from plate to plate when reading out the fluorescence values. For larger compound libraries with a larger number of plates required, or if running a similar size using 384 well plates, the use of this STOP buffer or something similar can permit the reactions on all plates to be stopped first and then read out without fear of variable incubation times between plates.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dr. Sara Buhrlage (saraj_buhrlage@dfci.harvard.edu).
Materials availability
The materials generated in this study will be distributed upon request to the lead contact. There are restrictions to availability due to a Material Transfer Agreement (MTA).
Acknowledgments
S.J.B. acknowledges generous support from the Mark Foundation ASPIRE program, NIH CA233800, and NIH CA247671.
Author contributions
A.C.V., D.C., D.A., and S.J.B. conceived of and designed the study. A.C.V. and D.C. carried out the assay optimization, high-throughput screening, and initial dose response validation, which were reviewed under the supervision of D.A. and S.J.B. The paper was written by A.C.V. and S.J.B and reviewed by all co-authors.
Declaration of interests
S.J.B. serves on the SAB of Adenoid Cystic Carcinoma Foundation. The authors have submitted patent applications related to compounds in this manuscript.
Contributor Information
Dominick Casalena, Email: dominick.casalena@novartis.com.
Sara J. Buhrlage, Email: saraj_buhrlage@dfci.harvard.edu.
Data and code availability
The primary and dose-response screening datasets generated during this study are available at PubChem (PubChem AID: 1645869). https://pubchem.ncbi.nlm.nih.gov/bioassay/1645869
References
- Cho J., Park J., Kim E.E., Song E.J. Assay systems for profiling deubiquitinating activity. Int. J. Mol. Sci. 2020;21:5638. doi: 10.3390/ijms21165638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gubler H., Clare N., Galafassi L., Geissler U., Girod M., Herr G. Helios: History and anatomy of a successful in-house enterprise high-throughput screening and profiling data analysis system. SLAS Discov. 2018;23:474–488. doi: 10.1177/2472555217752140. [DOI] [PubMed] [Google Scholar]
- Hassiepen U., Eidho U., Meder G., Bulber J.-F., Hein A., Bodendorf U., Lorthiois E., Martoglio B. A sensitive fluorescence intensity assay for deubiquitinating proteases using ubiquitin-rhodamine110-glycine as substrate. Anal. Biochem. 2007;371:201–207. doi: 10.1016/j.ab.2007.07.034. [DOI] [PubMed] [Google Scholar]
- Lamberto I., Liu X., Seo H.S., Schauer N.J., Iacob R.E., Hu W., Das D., Mikhailova T., Weisberg E.L., Engen J.R. Structure-guided development of a potent and selective non-covalent active-site inhibitor of USP7. Cell Chem. Biol. 2017;24:1490–1500.e11. doi: 10.1016/j.chembiol.2017.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J., Yakushi T., Parlati F., MacKinnon A.L., Perez C., Ma Y., Carter K.P., Colayco S., Magnuson G., Brown B. Capzimin is a potent and specific inhibitor of proteasome isopeptidase Rpn11. Nat. Chem. Biol. 2017;13:486–493. doi: 10.1038/nchembio.2326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mevissen T.E., Hospenthal M.K., Geurink P.P., Elliott P.R., Akutsu M., Arnaudo N., Ekkebus R., Kulathu Y., Wauer T., El Oualid F. OTU deubiquitinases reveal mechanisms of linkage specificity and enable ubiquitin chain restriction analysis. Cell. 2013;154:169–184. doi: 10.1016/j.cell.2013.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orcutt S.J., Wu J., Eddins M.J., Leach C.A., Strickler J.E. Bioluminescence assay platform for selective and sensitive detection of Ub/Ubl proteases. Biochim. Biophys. Acta. 2012;1823:2079–2086. doi: 10.1016/j.bbamcr.2012.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varca A.C., Casalena D., Chan W.C., Hu B., Magin R.S., Roberts R.M., Liu X., Zhu H., Seo H.S., Dhe-Paganon S. Identification and validation of selective deubiquitinase inhibitors. Cell Chem. Biol. 2021 doi: 10.1016/j.chembiol.2021.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The primary and dose-response screening datasets generated during this study are available at PubChem (PubChem AID: 1645869). https://pubchem.ncbi.nlm.nih.gov/bioassay/1645869