

Abstract
The main protease (Mpro) of SARS-CoV-2 is a validated antiviral drug target. Several Mpro inhibitors have been reported with potent enzymatic inhibition and cellular antiviral activity, including GC376, boceprevir, calpain inhibitors II and XII, each containing a reactive warhead that covalently modifies the catalytic Cys145. Coupling structure-based drug design with the one-pot Ugi four-component reaction, we discovered one of the most potent non-covalent inhibitors 23R (Jun8-76-3A) that is structurally distinct from the canonical Mpro inhibitor GC376. Significantly, 23R is highly selective compared with covalent inhibitors such as GC376, especially towards host proteases. The co-crystal structure of SARS-CoV-2 Mpro with 23R revealed a previously unexplored binding site located in between the S2 and S4 pockets. Overall, this study discovered 23R, one of the most potent and selective non-covalent SARS-CoV-2 Mpro inhibitors reported to date, and a novel binding pocket in Mpro that can be explored for inhibitor design.
Keywords: SARS-CoV-2, COVID-19, main protease, 3CL protease, antiviral
Graphical Abstract
INTRODUCTION
The COVID-19 pandemic had a significant impact on global economy and public health, and there is an urgent need for therapeutic interventions. The viral polymerase inhibitor remdesivir gained FDA approval on Oct 22nd 2020. The combination therapy of remdesivir with a Janus kinase (JAK) inhibitor baricitinib also received the FDA emergency use authorization.1 Among the other drug targets being pursued at preclinical and clinical stages,2 the viral main protease (Mpro), also called 3-chymotrypsin-like protease (3CLpro), is one of the most extensively explored high profile antiviral drug targets.3 Mpro is a cysteine protease encoded in the viral polyprotein as non-structural protein 5 (Nsp5) that cleaves the viral polyproteins pp1a and pp1ab at more than 11 sites. Despite its multiple proteolytic sites, Mpro was shown to have a high substrate specificity of glutamine at the P1 position.4 As such, the majority of the reported Mpro inhibitors were designed to contain a 2-pyrrolidone at the P1 substitution as a mimetic of the glutamine in the substrate.5 Most advanced Mpro inhibitors including PF-07304814,6 GC376,7, 8 6j,9 MI-09 and MI-3010 all belong to this category (Figure 1). PF-07304814, an α-hydroxyl ketone prodrug, is being developed by Pfizer, which has optimal pharmacokinetic properties and recently entered human clinical trials.6 GC376 has in vivo antiviral efficacy in treating cats infected with lethal feline infectious peritonitis virus.11, 12 Recently, the GC376 analog 6j was shown to protect mice from MERS-CoV infection.9 MI-09 and MI-30 were shown to protect mice from lethal SARS-CoV-2 infection.10 These promising results highlight the translational potential of Mpro inhibitors as potent SARS-CoV-2 antivirals and validate Mpro as an antiviral drug target for coronaviruses.
Figure 1.
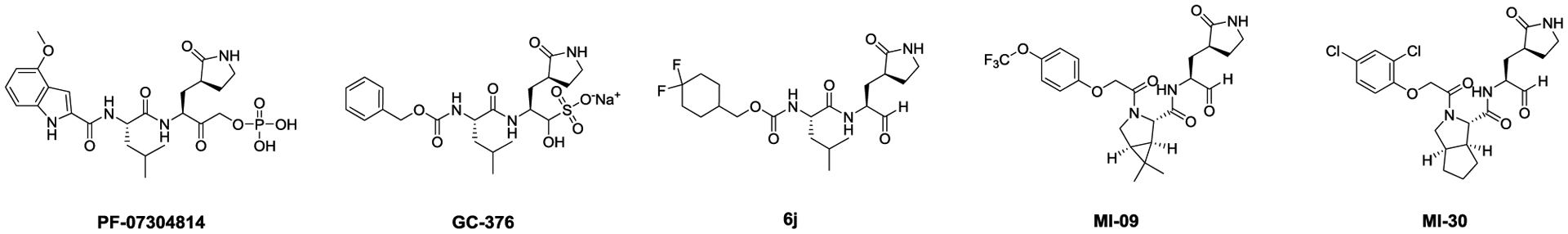
Promising SARS-CoV-2 Mpro inhibitors reported in the literature with translational potential.
Drug discovery is a lengthy process involving iterative cycles of design, synthesis, and pharmacological characterization. In the event of COVID-19 pandemic, an expedited approach with a fast turnover of this development cycle is highly desired. Using SARS-CoV-2 Mpro as a drug target, we report herein a fast-track drug discovery approach by coupling structure-based drug design and the Ugi four-component reaction (Ugi-4CR) methodology. The design was based on the superimposed structures of SARS-CoV or SARS-CoV-2 Mpro in complex with inhibitors including GC376, calpain inhibitor XII, and ML188 (R).7 8 13 The lead compound 23R from this study is the most potent non-covalent SARS-CoV-2 Mpro inhibitor reported to date in terms of enzymatic inhibition and cellular antiviral activity. The target selectivity of the designed inhibitors was profiled against a panel of viral proteases and host proteases, and the non-covalent inhibitor 23R was found to be highly selective compared to the covalent inhibitor GC376. An X-ray crystal structure of SARS-CoV-2 Mpro in complex with 23R was solved, revealing a drug-induced conformational change and a previously unexplored binding site in between the S2 and S4 pockets. Overall, this study led to the discovery of the non-covalent Mpro inhibitor 23R with potent enzymatic inhibition and in vitro cellular antiviral activity with a novel mechanism of action.
RESULTS AND DISCUSSION
Rational design of non-covalent SARS-CoV-2 Mpro inhibitors
Among the non-canonical SARS-CoV-2 Mpro inhibitors we recently discovered, calpain inhibitor XII has an unexpected binding mode showing an inverted conformation in the active site (Figure 2A).8 Instead of projecting the norvaline and leucine side chains into the S1 and S2 pockets as one would expect from its chemical structure, the pyridinyl substitution snuggly fits in the S1 pocket and forms a hydrogen bond with the H163 imidazole (Figure 2A). This hydrogen bond is essential, as replacing the pyridine with benzene led to an analog UAWJ257 with a significant loss of enzymatic inhibition.8 Examining the X-ray crystal structures of SARS-CoV and SARS-CoV-2 Mpro in the PDB database revealed another compound ML188 (R),13 which shares a similar binding mode with calpain inhibitor XII. ML188 (R) is a non-covalent SARS-CoV Mpro inhibitor derived from a high-throughput screening hit.13 The pyridinyl from ML188 (R) similarly fits in the S1 pocket and forms a hydrogen bond with the H163 side chain imidazole (Figure 2B). In addition, the furyl oxygen and its amide oxygen both form a hydrogen bond with the G143 main chain amide amine. ML188 (R) was reported to inhibit the SARS-CoV Mpro with an IC50 value of 1.5 ± 0.3 μM and the SARS-CoV viral replication in Vero E6 cells with an EC50 value of 12.9 μM.13 Several follow up studies have been conducted to optimize the enzymatic inhibition and cellular antiviral activity of this series of compounds, however, no significant improvement has been made.14, 15
Figure 2.
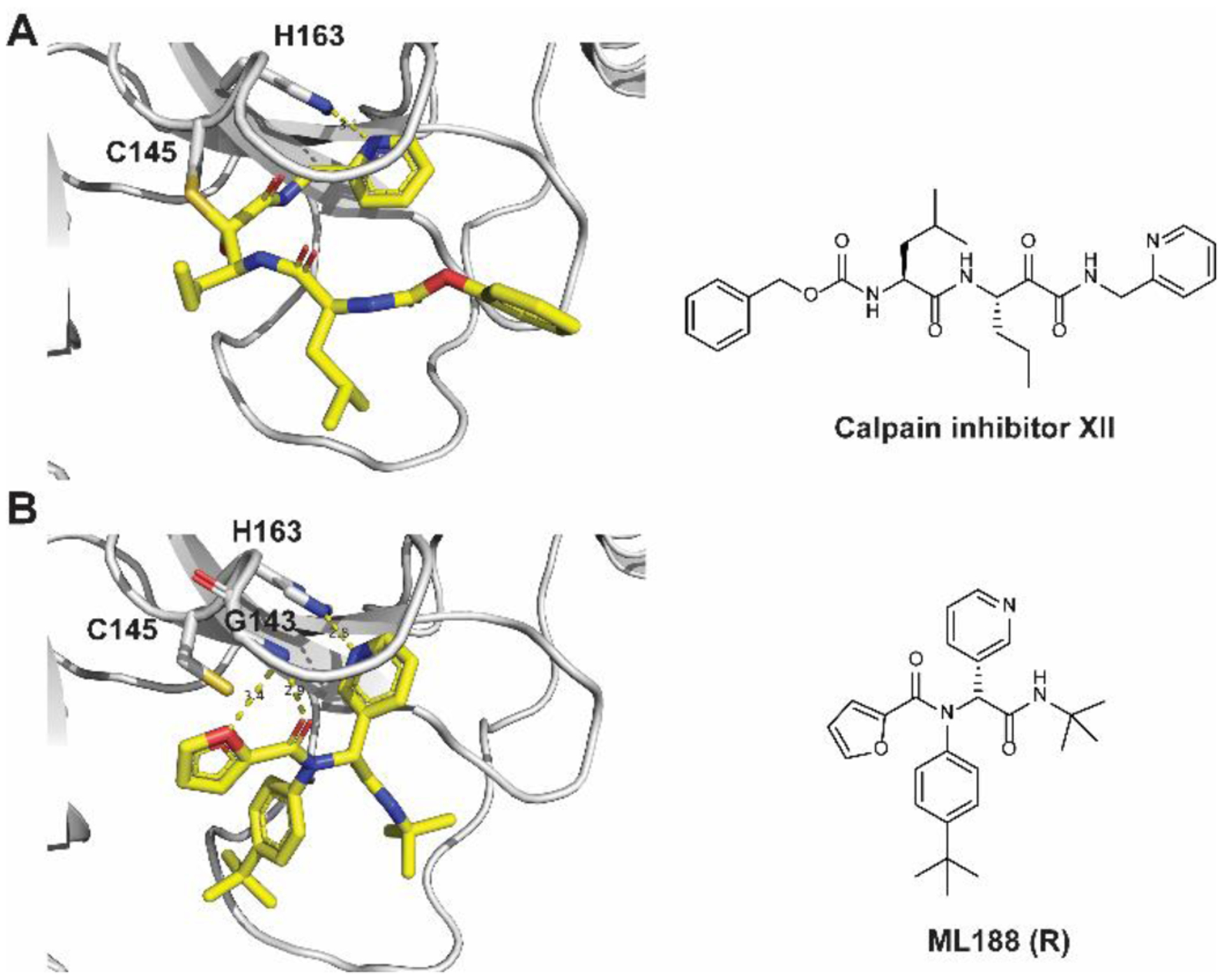
Structure of Mpro with inhibitors. (A) X-ray crystal structure of SARS-CoV-2 Mpro in complex with calpain inhibitor XII (PDB: 6XFN). (B) X-ray crystal structure of SARS-Co-V Mpro in complex with ML188 (R) (PDB: 3V3M). Hydrogen bonds are shown in dashed lines.
The similar binding mode of ML188 (R) with calpain inhibitor XII, coupled with the convenient synthesis through the one pot Ugi-4CR, inspired us to design non-covalent SARS-CoV-2 Mpro inhibitors based on the ML188 (R) scaffold. Specifically, we leverage our understanding of the Mpro inhibition mechanism based on the X-ray crystal structures of SARS-CoV-2 Mpro with multiple inhibitors to guide the lead optimization (Figure 3A to D).7, 8 Overlaying the X-ray crystal structures of SARS-CoV Mpro + ML188 (R) (PDB: 3V3M) and the SARS-CoV Mpro H41A mutant + the peptide substrate (PDB: 2Q6G) revealed that the furyl, 4-tert-butylphenyl, pyridinyl, and tert-butyl of ML188 (R) fit in the S1’, S2, S1, and S3 pockets respectively (Figure 3A, D). Therefore, the furyl, 4-tert-butylphenyl, pyridinyl, and tert-butyl substitutions in ML188 (R) were defined as P1’, P2, P1, and P3, respectively. Next, overlaying the structure of SARS-CoV Mpro + ML188 (R) (PDB: 3V3M) and SARS-CoV-2 Mpro + GC376 (PDB: 6WTT) suggested that the tert-butyl at the P3 substitution of ML188 (R) can be extended to fit in the S4 pocket (Figure 3B, D). Previous structure-activity relationship studies of GC376 indicate that P4 substitution is important, while P3 substitution does not contribute significantly to the binding affinity, as it is solvent exposed.3, 8, 9, 16 Similarly, the overlaying structures of SARS-CoV Mpro + ML188 (R) (PDB: 3V3M) and SARS-CoV-2 Mpro + UAWJ247 (PDB: 6XBH) suggested that the 4-tert-butyl at the P2 substitution of ML188 (R) can be replaced by phenyl to occupy the extra space in the S2 pocket (Figure 3C, D). Overall, binding site analysis suggests that extending the P2 and P3 substitutions of ML188 (R) might lead to better shape complementarity with the SARS-CoV-2 Mpro (Figure 3E). In practice, we adopted a stepwise optimization procedure in which the P3 and P2 substitutions were optimized individually in step 1, and then the optimal P2/P3 substitutions were combined in step 2 (Figure 3E).
Figure 3.
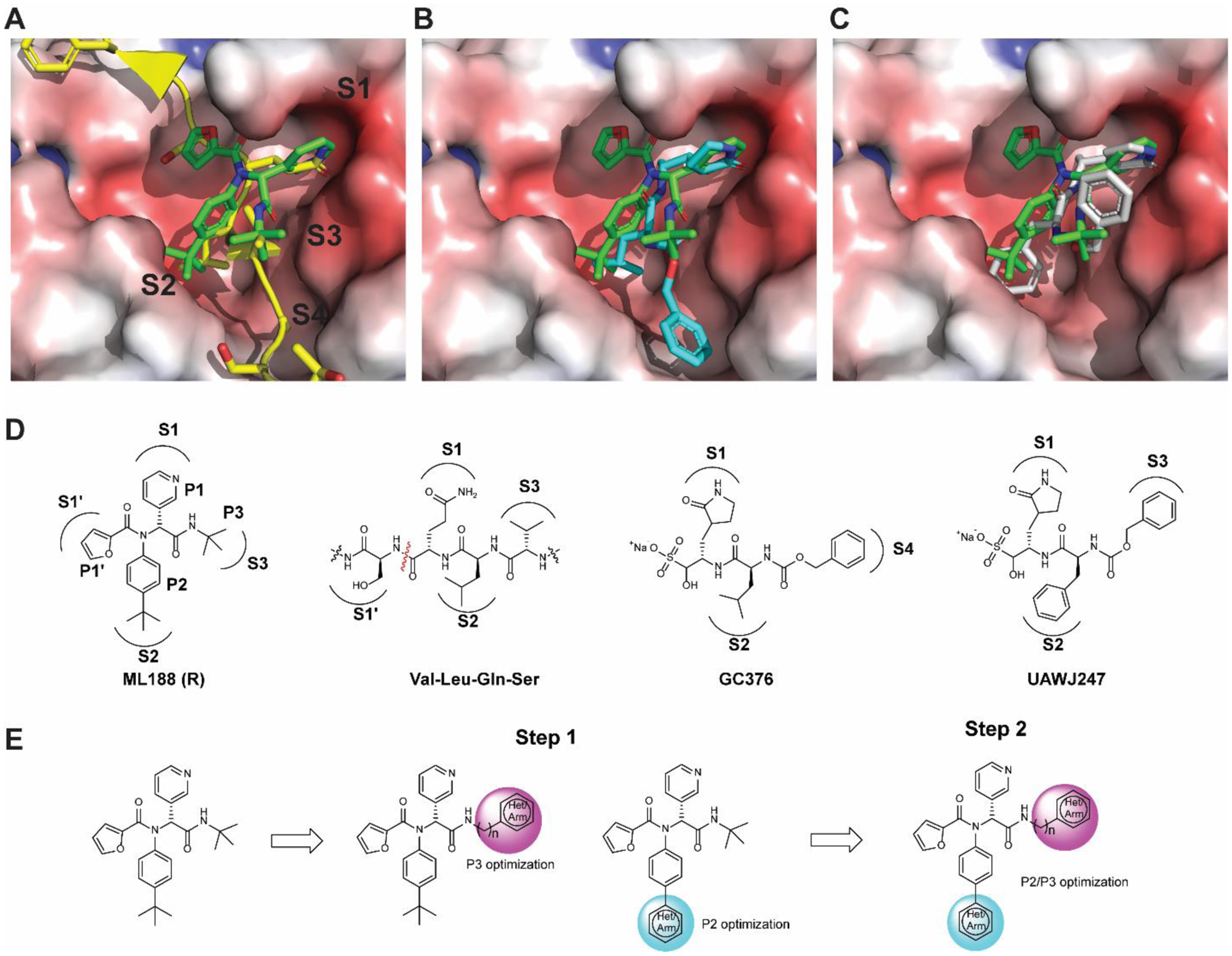
Design rationale for the non-covalent SARS-CoV-2 Mpro inhibitors. (A) Superimposed X-ray crystal structures of SARS-CoV Mpro + ML188 (R) (PDB: 3V3M, green) and SARS-CoV Mpro H41A mutant + peptide substrate (PDB: 2Q6G, yellow with backbone shown as ribbon representation). (B) Superimposed X-ray crystal structures of SARS-CoV Mpro + ML188 (R) (PDB: 3V3M) and SARS-CoV-2 Mpro + GC376 (PDB: 6WTT). (C) Superimposed X-ray crystal structures of SARS-CoV Mpro + ML188 (R) (PDB: 3V3M) and SARS-CoV-2 Mpro + UAWJ247 (PDB: 6XBH). (D) Chemical structures of ML188 (R), peptide substrate VLQS, GC376, and UAWJ247. (E) Stepwise optimization of ML188 (R) towards potent non-covalent SARS-CoV-2 Mpro inhibitor.
Guided by the design rationale elucidated above, a focused library of ML188 analogs were designed and synthesized (Figure 4). As the P1’ furyl and P1 pyridinyl both form a critical hydrogen bond with the Mpro (Figure 4A, B), the P1’ and P1 substitutions were kept with minimal variations for the design of non-covalent inhibitors (Figure 4C). All designed compounds were synthesized using the one pot Ugi four-component reaction and tested as enantiomer/diastereomer mixtures (Figure 4C). To circumvent the need of relying on expensive chiral HPLC column for the separation of enantiomers, we strategically introduced the chiral isocyanide so that the diastereomer product mixture can be separated by convenient silica gel column or reverse phase HPLC column purification.17
Figure 4.
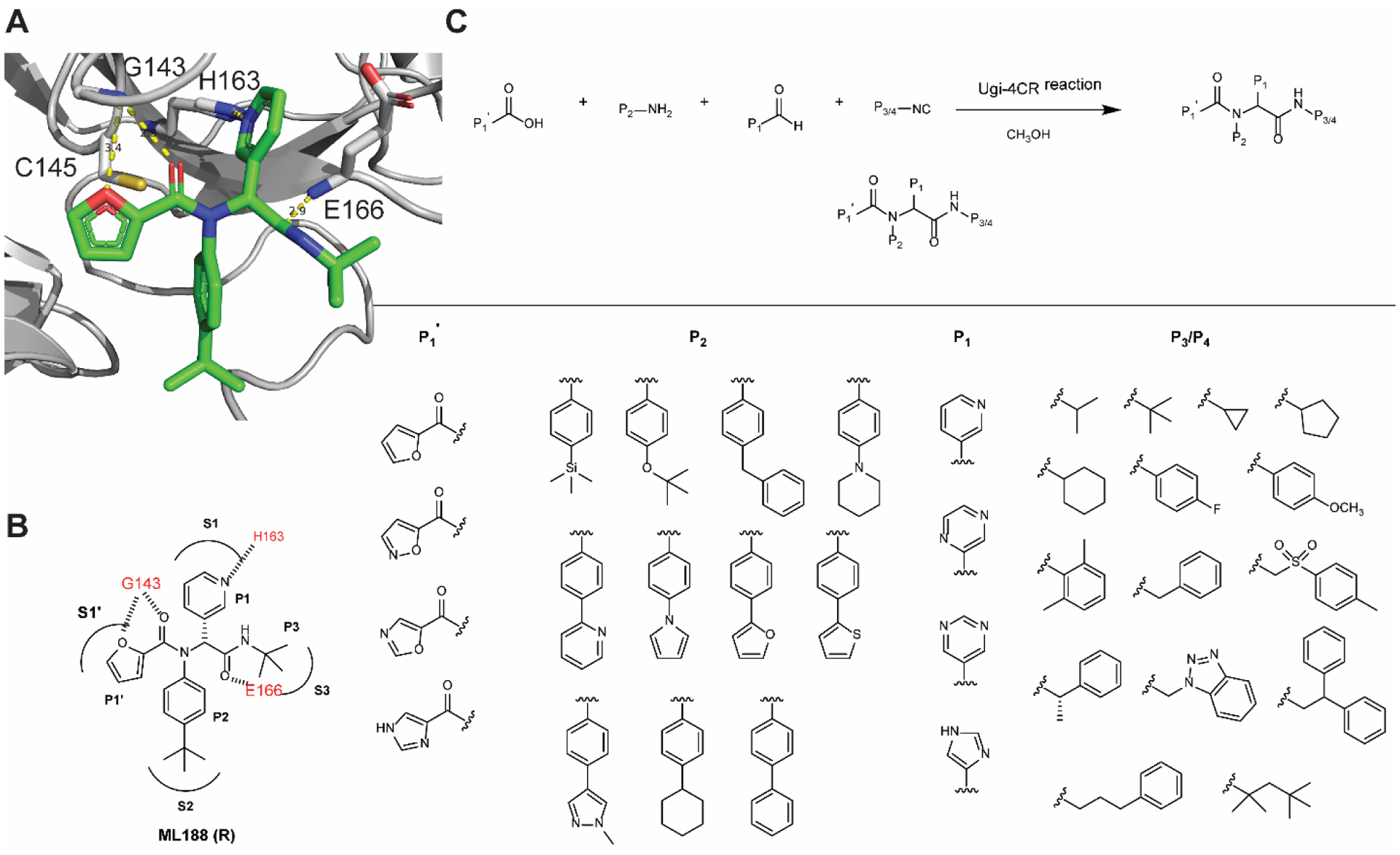
Design and synthesis of a focused library of non-covalent SARS-CoV-2 Mpro inhibitors. (A) X-ray crystal structure of SARS-CoV Mpro + ML188 (R) (PDB: 3V3M). (B) Binding interactions of ML188 (R) with SARS-CoV Mpro. (C) Synthesis of ML188 analogs using the Ugi four-component reaction.
Structure-activity relationship studies of non-covalent SARS-CoV-2 Mpro inhibitors
In total, 39 compounds were synthesized (Figure 5 A to E) and all compounds were initially tested as a mixture of enantiomers or diastereomers in the FRET-based enzymatic assay against SARS-CoV-2 Mpro at 20 μM (Figure 5F). Compounds showing more than 50% inhibition at 20 μM were further titrated to determine the IC50 values. Next, compounds with IC50 values lower than 5 μM were selected for cellular cytotoxicity profiling in Vero E6 cells, the cell line which was used for the SARS-CoV-2 antiviral assay. The purpose was to prioritize lead candidates for the in vitro cellular antiviral assay with infectious SARS-CoV-2. As shown in Figure 5, the majority of the designed compounds showed more than 50% inhibition when tested at 20 μM. Specifically, Figure 5A lists compounds with P4 variations. As a reference, ML188 (1) (racemic mixture) inhibits SARS-CoV-2 Mpro with an IC50 value of 10.96 ± 1.58 μM. It was found that compounds 2, 3, 5, 6, 7, 8, 10, and 13 had improved enzymatic inhibition compared to ML188 (1). These results suggest that: a) isopropyl (2), cyclopropyl (3), cyclopentyl (5), cyclohexyl (6), and phenyl (7 and 8) are the more favorable substitutions at the P3 position than tert-butyl; b) compound 13 with the (S)-α-methylbenzyl substitution at the P3 position had improved potency, which suggests that extending the substitutions to the S4 pocket might improve the enzymatic inhibition (Figure 3B). Given the advantage of convenient separation of diastereomers over enantiomers, we therefore decided to fix the P3/P4 substitution as α-methylbenzyl substitution during the P2 optimization (Figure 5B). All compounds in Figure 5B were designed to have extended substitutions at the 4-position of benzyl to occupy the extra space in the S2 pocket (Figure 3C). Consistent with the design hypothesis, several compounds including 14, 17, 18, 19, 20, 21, and 23 had significantly improved enzymatic inhibition (IC50 < 3 μM) compared to compound 13. Replacing the tert-butyl in compound 13 with the bulkier trimethylsilyl led to compound 14 with a 2.9-fold increase in Mpro inhibition. Cyclohexyl (17), thienyl (19), pyrrolyl (20), pyridinyl (21), and phenyl (23) were found to be the most favorable substitutions at the S2 pocket. Compound 16 with piperidyl substitution had similar potency as compound 13, while compound 15 with O-tert butyl was less active. Further extending the substitution to benzyl led to compound 22 that was inactive, suggesting biphenyl might be the longest substitution that can be accommodated at the S2 pocket.
Figure 5.
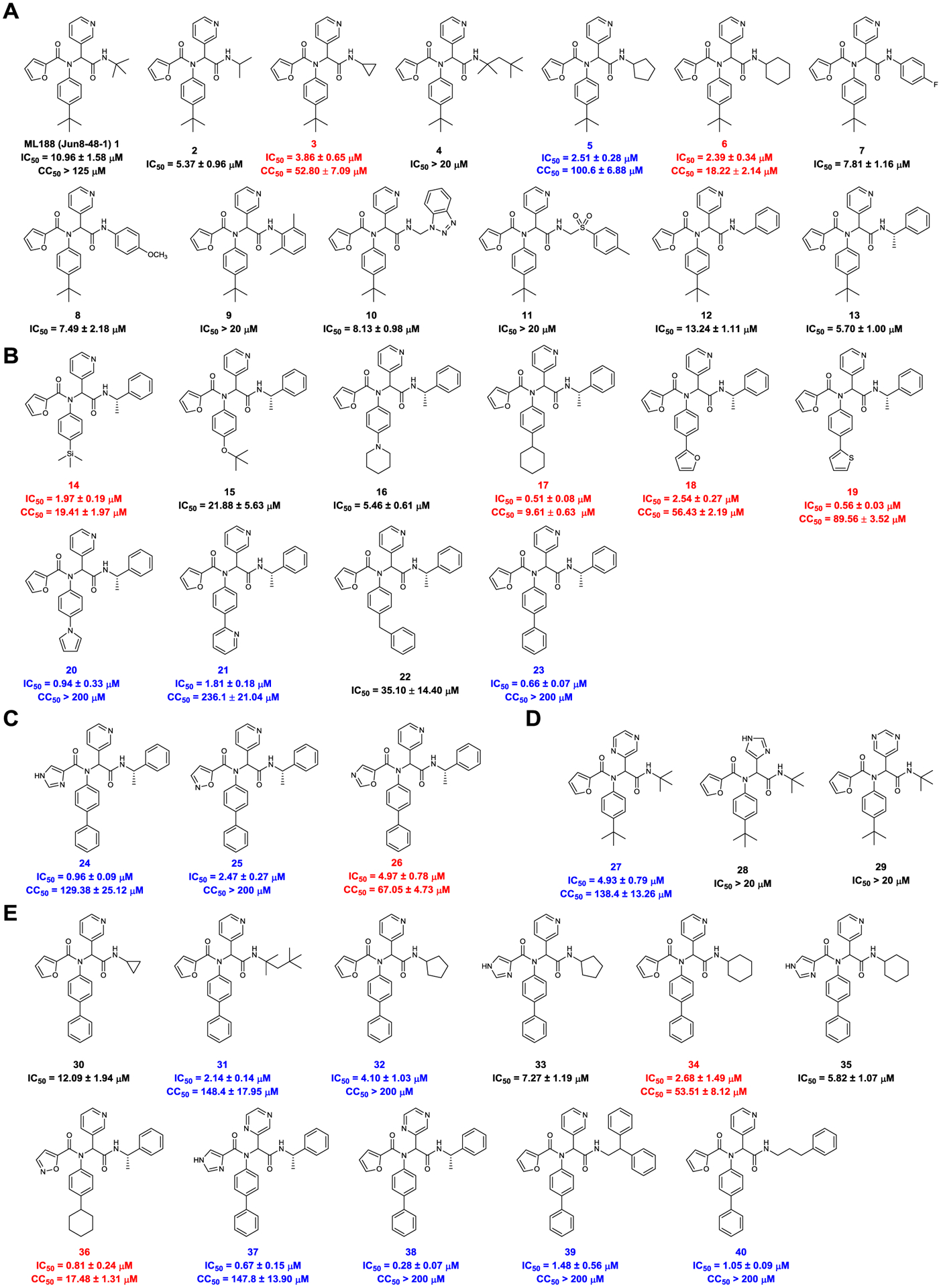
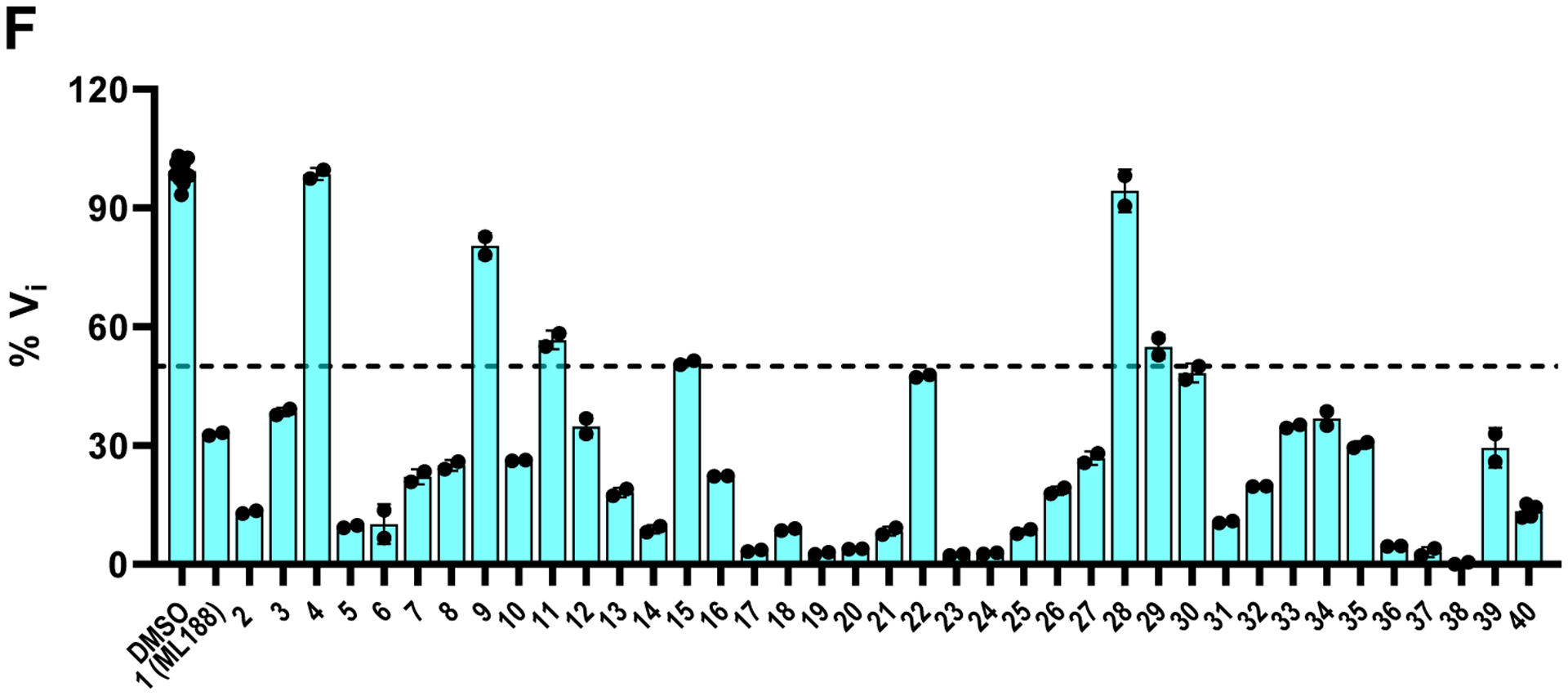
Structures of non-covalent and covalent SARS-CoV-2 Mpro inhibitors and the enzymatic inhibition against SARS-CoV-2 Mpro. (A) Non-covalent analogs with P3/P4 modifications. (B) Non-covalent analogs with P2 modifications. (C) Non-covalent analogs with P1’ modifications. (D) Non-covalent analogs with P1 modifications. (E) Non-covalent analogs with combined P1’, P1, P2, and P3/P4 modifications. Compounds with potent enzymatic inhibition (IC50 < 5 μM) but moderate to high cellular cytotoxicity (CC50 < 100 μM) were labeled in red. Compounds with both potent enzymatic inhibition (IC50 < 5 μM) and low cellular cytotoxicity (CC50 > 100 μM) were labeled in blue. (F) Percentage enzymatic activity of SARS-CoV-2 Mpro in the presence of the designed compounds at 20 μM compound concentration.
The P1’ and P1 substitutions (Figure 5C, D) were chosen to retain the critical hydrogen bonds in ML188 (Figure 4A). It was found that imidazole (24) was tolerated at the P1’ position (IC50 = 0.96 ± 0.09 μM), followed by isoxazole (25) (IC50 = 2.47 ± 0.27 μM), and oxazole (26) (IC50 = 4.97 ± 0.78 μM). Pyrazine (27) was tolerated at the P1 position (IC50 = 4.93 ± 0.79 μM); however, imidazole (28) and pyrimidine (29) were not preferred (IC50 > 20 μM).
Next, the above identified favorable P1’, P2, P1, and P3/P4 substitutions were combined and the designed compounds were shown in Figure 5E. Compounds 36, 37, and 38 were the most potent leads with IC50 values of 0.81 ± 0.24, 0.67 ± 0.15, and 0.28 ± 0.07 μM, respectively. Compound 39 and 40 were also highly active with IC50 values of 1.48 ± 0.56 and 1.05 ± 0.09 μM, respectively.
Among the active compounds with IC50 value lower than 5 μM, compounds 3, 6, 14, 17, 18, 19, 26, 34, and 36 had moderate to high cellular cytotoxicity in Vero E6 cells (Figure 5A to E red), while compounds 5, 20, 21, 23, 24, 25, 27, 31, 32, 37, 38, 39, and 40 were well tolerated and the CC50 values were greater than 100 μM (Figure 5A to E blue).
Cellular antiviral activity of non-covalent SARS-CoV-2 Mpro inhibitors
Next, compounds with potent enzymatic inhibition (IC50 ≤ 1 μM) and low cellular cytotoxicity (CC50 > 100 μM) were prioritized for the cellular antiviral assay with infectious SARS-CoV-2 in Vero E6 cells using the immunofluorescence assay as the primary assay (Figure 6A). ML188 (1) was included as a control. It was found that ML188 (1) was inactive in the antiviral assay (EC50 > 20 μM), probably due to its incomplete inhibition of the Mpro in the cellular content. Gratifyingly, compounds 20, 23, 37, 38, and 40 all had potent cellular antiviral activity with EC50 values ranging from 0.82 to 4.54 μM. Compound 24 was less active (EC50 = 13.06 ± 2.30 μM), possibly due to the poor cellular membrane permeability.
Figure 6.
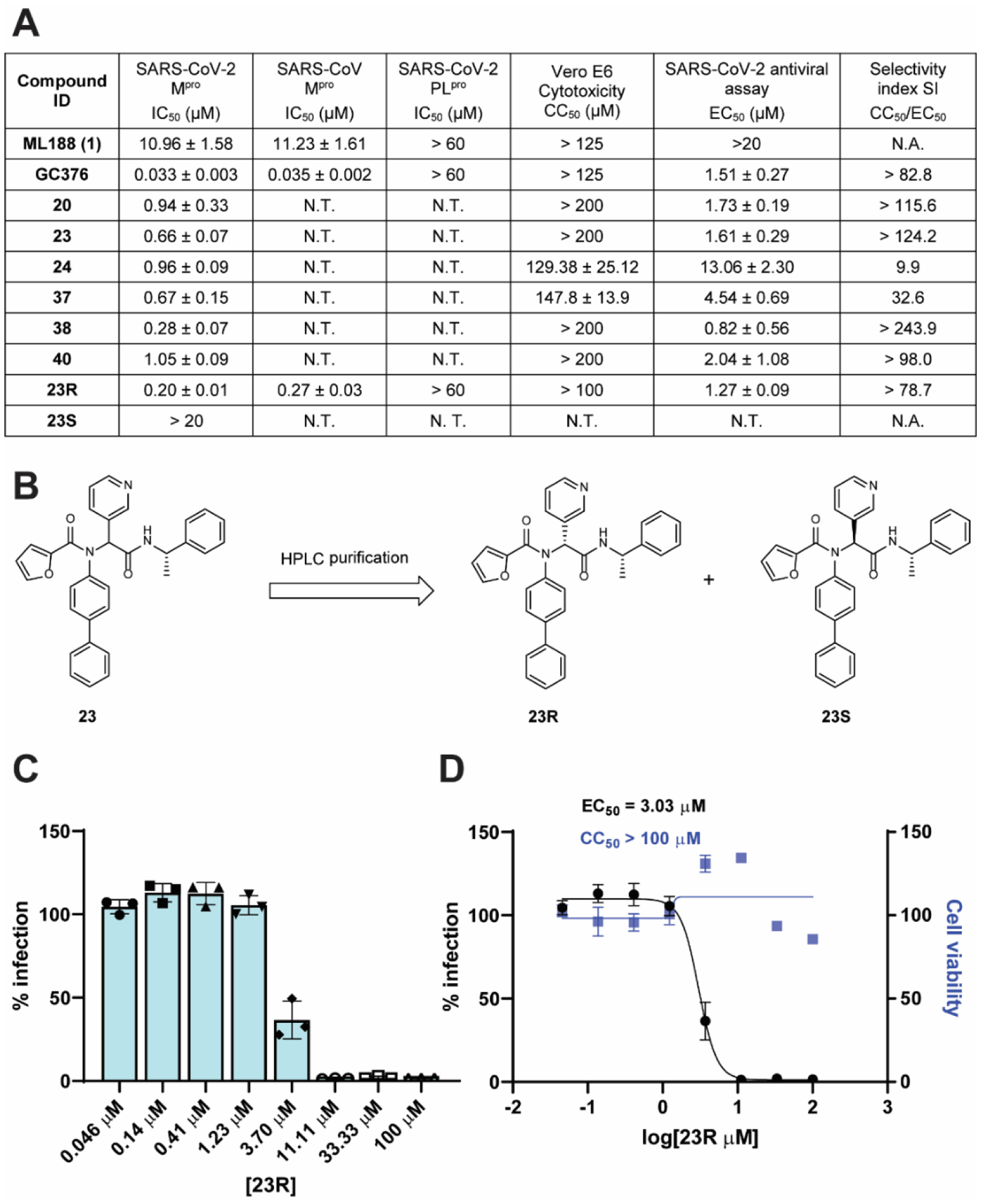
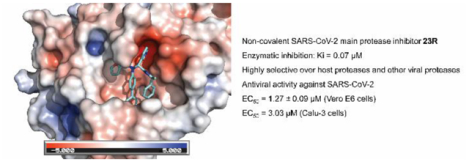
Enzymatic inhibition, cellular antiviral activity, and selectivity index of non-covalent Mpro inhibitors. (A) Antiviral activity and selectivity index of non-covalent SARS-CoV-2 Mpro inhibitors. (Selection criteria IC50 < 1 μM, CC50 > 100 μM). Antiviral assay was performed using the SARS-CoV-2 USA-WA1/2020 isolate in Vero E6 cells with an MOI of 0.05. (B) Chemical structures of the two diastereomers 23R and 23S. The absolute stereochemistry of compound 23R was determined in the co-crystal structure of this diastereomer with SARS-CoV-2 Mpro (PDB: 7KX5). (C and D) Antiviral activity of 23R against SARS-CoV-2 in Calu-3 cells. (C) Raw data of the percentage of immunofluorescence positive cells with different concentrations of 23R. (D) Antiviral potency and cytotoxicity plots. N.T. = not tested. N.A. = not applicable.
Given the potent antiviral activity and a high selectivity index of these potent lead compounds, we then selected the non-covalent inhibitor 23 for further characterization. The two diastereomers of 23 were separated by reverse phase HPLC (Figure 6B). Both diastereomers were tested in the FRET-based enzymatic assay. GC376 was included as a positive control. It was found that 23R is the active diastereomer with an IC50 value of 0.20 ± 0.01 μM, while the 23S diastereomer was not active (IC50 > 20 μM) (Figure 6A). The stereochemistry of 23R was determined by the co-crystal structure with SARS-CoV-2 Mpro as described in the following section. Compared with the parent compound ML188 (1), the optimized lead 23R had more than a 54-fold increase in enzymatic inhibition against SARS-CoV-2 Mpro. Compound 23R also showed comparable potency against SARS-CoV Mpro with an IC50 value of 0.27 ± 0.03 μM. Neither ML188 (1) nor 23R inhibited the SARS-CoV-2 papain-like protease (PLpro) (IC50 > 20 μM) (Figure 6A), suggesting the inhibition of SARS-CoV-2 Mpro by 23R is specific.
Next, the antiviral activity of 23R was tested against SARS-CoV-2 (USA-WA1/2020 isolate) in Vero E6 cells using the immunofluorescence assay. It was found that compound 23R had an EC50 value of 1.27 μM (Figure 6A), which was similar to the antiviral potency of the covalent inhibitor GC376 (EC50 = 1.51 μM). Compound 23R was also not cytotoxic to Vero E6 cells at up to 100 μM. In contrast, the parent compound ML188 (1) had no detectable antiviral activity when tested at up to 20 μM. To further confirm the antiviral activity of compound 23R, we performed secondary antiviral assay in the human lung epithelial Calu-3 cell line, which endogenously expresses TMPRSS2 and is widely used as a physiological relevant cell line for SARS-CoV-2 infection. It was found that compound 23R inhibited SARS-CoV-2 (USA-WA1/2020 isolate) replication in Calu-3 cells with an EC50 value of 3.03 μM and it was not cytotoxic at up to 100 μM (Figure 6C, D).
Profiling the target selectivity of 23R against other viral cysteine proteases and host proteases
One of the major challenges facing cysteine protease inhibitors is the target selectivity.18, 19 It was recently reported that GC376 inhibits host cathepsins B and L in addition to SARS-CoV-2 Mpro,20 and GC376 analogs compound_1 and zyy16 that contained the aldehyde and cyanohydrin warheads, respectively, inhibit both calpain 1 and cathepsin K.21 These results raised the concern of the selectivity of this class of covalent inhibitors. To profile the target selectivity of the lead compounds developed in this study, we selected compound 23R as a representative example of the non-covalent inhibitor. ML188 (1) and GC376 were included as controls. The compounds were tested against a panel of viral cysteine proteases including SARS-CoV Mpro, MERS-CoV Mpro, HCoV-OC43 Mpro, EV-A71 3Cpro, EV-D68 3Cpro, EV-A71 2Apro, EV-D68 2Apro, and SARS-CoV-2 PLpro, as well as host cysteine proteases calpain 1, cathepsin K, cathepsin L, and serine protease trypsin. It was found that GC376 inhibited all the Mpro and 3Cpro tested including SARS-CoV-2 Mpro, SARS-CoV Mpro, MERS-CoV Mpro, HCoV-OC43 Mpro, EV-A71 3Cpro and EV-D68 3Cpro (IC50 ≤ 0.16 μM), but not the unrelated EV-A71 2Apro, EV-D68 2Apro and SARS-CoV-2 PLpro (IC50 > 50 μM) (Figure 7A and Table S1). In addition, GC376 also inhibited host cysteine proteases calpain 1, cathepsin K and cathepsin L (IC50 ≤ 0.074 μM), but not the serine protease trypsin (IC50 > 50 μM) (Figure 7B and Table S1). In contrast, the non-covalent inhibitor 23R developed in this study only selectively inhibited SARS-CoV-2 Mpro and SARS-CoV Mpro with high potency (IC50 ≤ 0.27 μM), but not other viral proteases tested, which was similar to ML188 (1) (Figure 7A and Table S1). Compound 23R showed weak inhibition against calpain 1 and cathepsin L with IC50 values of 6.00 μM and 10.5 μM, respectively, corresponding to selectivity indexes of 40 and 70 regards to SARS-CoV-2 Mpro inhibition (Figure 7B and Table S1).
Figure 7.
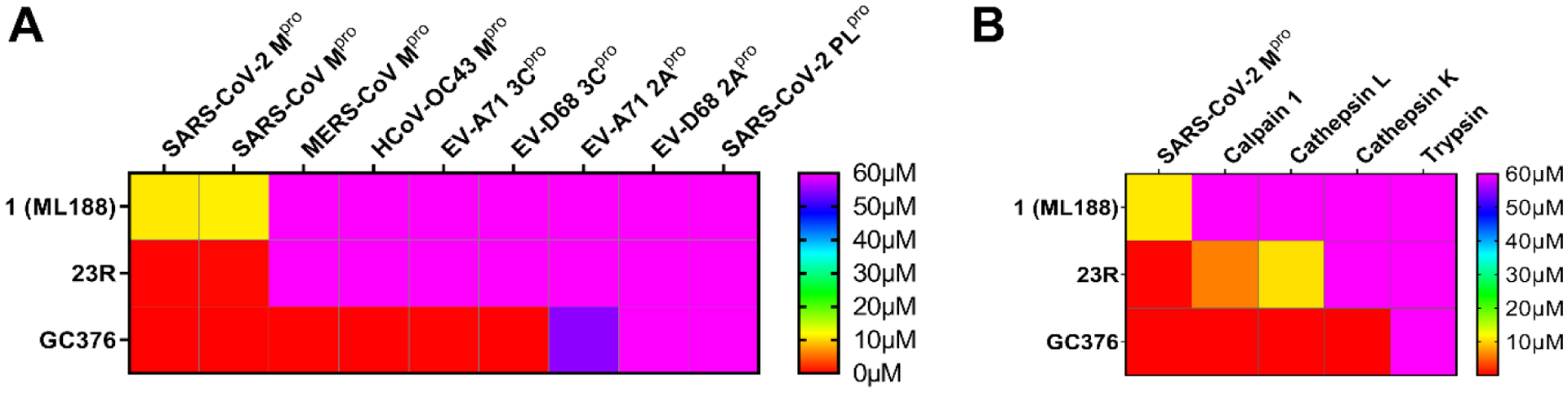
Selectivity of non-covalent and covalent SARS-CoV-2 Mpro inhibitors shown in color map. (A) Selectivity against viral cysteine proteases. (B) Selectivity against host proteases. The values plotted were the IC50 values from the FRET-based enzymatic assay.
Mechanism of action of non-covalent inhibitor 23R in inhibiting SARS-CoV-2 Mpro
The mechanism of action was characterized using the native mass spectrometry, the thermal shift-binding assay, and the enzymatic kinetic studies (Figure 8). In the native mass spectrometry binding assay, compound 23R showed dose-dependent binding to SARS-CoV-2 Mpro, similar to the positive control GC376, with a binding stoichiometry of one drug per monomer (Figure 8A). Similarly, compound 23R showed dose-dependent stabilization of the SARS-CoV-2 Mpro in the thermal shift binding assay with an apparent Kd value of 9.43 μM, a 9.3-fold decrease compared to ML188 (1) (Figure 8B). In the enzymatic kinetic studies, 23R was shown to be a non-covalent inhibitor with a Ki value of 0.07 μM (Figure 8C, D top and middle panels). In comparison, the Ki for the parent compound ML188 (1) is 2.29 μM. The Lineweaver-Burk or double-reciprocal plot with different compound concentrations yielded an intercept at the Y-axis, suggesting that 23R is a competitive inhibitor similar to ML188 (1) (Figure 8C, D bottom panel). Overall, the enzymatic kinetic studies confirmed that compound 23R is a non-covalent inhibitor of SARS-CoV-2 Mpro.
Figure 8.
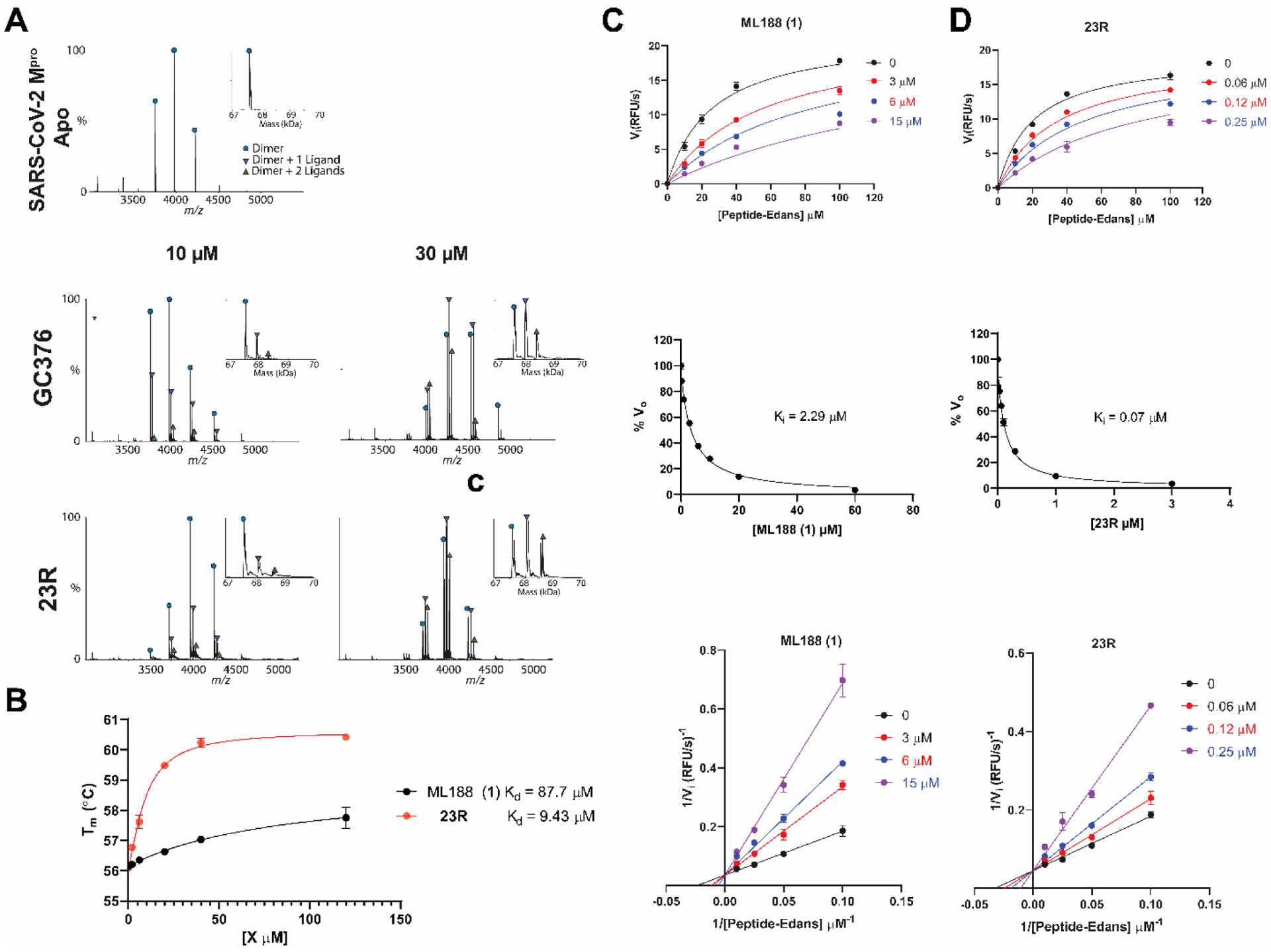
Characterization of the mechanism of action of 23R to SARS-CoV-2 Mpro using native mass-spectrometry, thermal shift assay, and enzyme kinetic studies. (A) Binding of 23R to SARS-CoV-2 Mpro using native mass spectrometry. Native mass spectra with the inset deconvolved spectra revealing ligand binding with 10 μM or 30 μM GC376 added (middle panel), and 10 μM and 30 μM 23R (bottom panel) with 4 mM DTT added. The peaks are annotated with the blue circle as the dimer, green down triangle as the dimer with one ligand bound, and the purple up triangle as the dimer with two ligands bound. (B) Dose-dependent melting temperature (Tm) shift in thermal shift assay. 3 μM SARS-CoV-2 Mpro protein was incubated with various concentrations of ML188 or 23R in the presence of 4 mM DTT. Measured Tm was plotted against compound concentration with one-site binding function in Prism 8. (C and D) Enzymatic kinetic assay with ML188 and compound 23R. Kinetic parameters in the presence of various concentrations of ML188 or 23R were globally fitted with Michaelis-Menten function in prism 8 (top panels); double reciprocal plots were shown in the bottom panels. The middle panels show the Morrison plots of compound ML188 and 23R with 20 μM FRET substrate was used.
X-ray crystal structure of SARS-CoV-2 Mpro with 23R
Using X-ray crystallography, we successfully determined the binding pose of 23R with SARS-CoV-2 Mpro at 2.6 Å resolution (Figure 9A, Table S2). Electron density reveals the body of 23R extends throughout the substrate binding channel, with side chains occupying the S1’, S1, S2, and a previously unexplored sub-pocket in between S2 and S4. The binding pose is similar to the previously solved structure of SARS-CoV Mpro with ML188 (R) (PDB: 3V3M)13, consistent with the similarities between the two compounds and between the two proteins (Figure 9B). The furyl moiety of 23R binds to a portion of the P1’ site, which normally accommodates small hydrophobic residues. While the furylamide carbonyl group of 23R does not insert into the oxyanion hole, it does form a bifurcated hydrogen bond with the apical residue of this oxyanion hole, Gly143. However, the furan ring oxygen is likely a weaker hydrogen bond acceptor than the amide oxygen and it lies outside of the plane of Gly143’s amide NH. Directly attached to the furylamide moiety is a P2 biphenyl group and a P1 pyridinyl ring. The P2 biphenyl group projects directly into the S2 pocket, which prefers hydrophobic residues such as leucine and phenylalanine. As expected, the P1 pyridinyl ring occupies the S1 pocket, which is known for its strict preference for glutamine. While most Mpro inhibitors bear a pyrrolidinone glutamine mimetic at the P1 position, we determined that more hydrophobic residues can also bind to the S1 site, and that hydrogen bond formation with His163 is critical for inhibition.8 In this instance, the pyridinyl ring of 23R is nearly superimposable with the same moiety from calpain inhibitor XII (Figure 9C) forming a close (2.9 Å) hydrogen bond with His163. An amide bond connecting the pyridinyl ring to the α-methylbenzyl moiety forms a hydrogen bond with the main chain of Glu166. The benzyl ring of the α- methylbenzyl moiety is partially positioned both in the S2 and S4 pockets, a novel binding pose that has not been observed with existing Mpro inhibitors. Normally, a substituent at this position would be expected to flip away from the enzyme core towards the solvent-exposed S3 pocket, which explains why P3 substitutions have little to no influence on the enzymatic inhibition.4 However, the chirality and hydrophobic nature of the benzyl ring in 23R causes it to project towards the core near the S2 pocket, forcing Gln189 to rotate outwards (Figure 9D). This conformation is reinforced by pi-stacking interactions with the first phenyl of the biphenyl substituent. Notably, the binding pose of 23R features continuous intramolecular pi-stacking, where the phenyl is sandwiched by furan and benzyl groups, potentially contributing to its potent inhibition of Mpro. Meanwhile, the S4 pocket remains largely unoccupied by 23R, leaving room for further improvement. In summary, the X-ray crystal structure of SARS-CoV-2 Mpro in complex with 23R revealed two interesting structural features: 1) The P2 biphenyl is probably the longest substitution that can be accommodated in the S2 pocket, which is consistent with our design hypothesis. 2) The benzyl group from the terminal α-methylbenzyl fits in a pocket in between the S2 and S4 pockets, and this is a ligand-induced binding site that has not been previously explored. Although this is unexpected from the design perspective, this novel binding mode suggests that the new binding pocket in between S2 and S4 that can be explored for inhibitor design.
Figure 9.
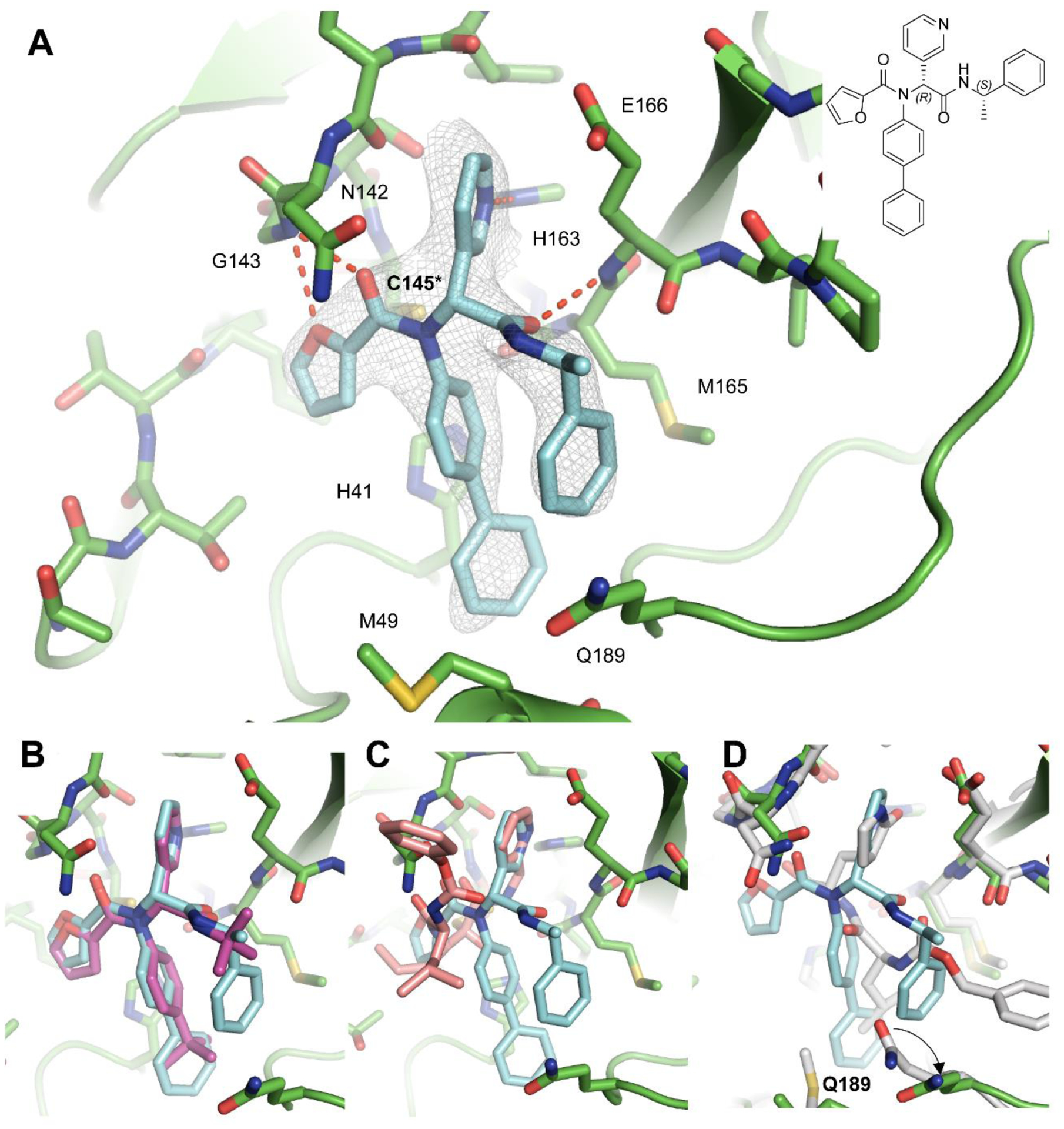
X-ray crystal structure of SARS-CoV-2 Mpro in complex with 23R. (A) X-ray crystal structure of SARS-CoV-2 Mpro in complex with 23R (PDB: 7KX5). (B) Superimposed structures of SARS-CoV-2 Mpro + 23R (PDB: 7KX5) and SARS-CoV Mpro + ML188 (R) (PDB: 3V3M). (C) Superimposed structures of SARS-CoV-2 Mpro + 23R (PDB: 7KX5) and SARS-CoV-2 Mpro + calpain inhibitor XII (PDB: 6XFN). (D) Superimposed structures of SARS-CoV-2 Mpro + 23R (PDB: 7KX5) and SARS-CoV-2 Mpro + GC376 (PDB: 6WTT).
CONCLUSION
The viral Mpro is a high profile antiviral drug target and several Mpro inhibitors are now in animal model studies and human clinical trial.6 Among the known Mpro inhibitors, the majority are covalent inhibitors such as GC376 analogs that contain a pyrrolidone in the P1 position as a glutamine mimetic. Several structurally distinct compounds including ebselen, disulfiram, carmofur, PX-12, tideglusib, and shiknonin were claimed as Mpro inhibitors,22, 23 but were later proved to be promiscuous non-specific cysteine protease inhibitors.24, 25 In addition, non-covalent inhibitors such as ML188 (R) were developed and validated as SARS-CoV Mpro inhibitors.5, 13 Several follow up studies have been conducted to optimize the enzymatic potency of this series of compounds against SARS-CoV Mpro and the SARS-CoV-2 Mpro. However, no significant improvement has been made and ML188 (R) remains the only non-covalent inhibitor with moderate antiviral activity against SARS-CoV (EC50 = 12.9 ± 0.7 μM).14, 15 Nevertheless, given the sequence and structure similarities between SARS-CoV and SARS-CoV-2 Mpro, and the similar binding mode of ML188 (R) and calpain inhibitor XII, we hypothesize that ML188 (R) is a promising scaffold for structure-based drug design. In this study, we developed an expedited drug discovery approach for the design of non-covalent SARS-CoV-2 Mpro inhibitors. The design approach couples the superimposed X-ray crystal structures with the one-pot Ugi 4CR synthetic methodology. We were able to improve the enzymatic inhibition potency of ML188 (1) by 54-fold from a focused library of 39 non-covalent inhibitors. This is a significant advantage compared to covalent inhibitors such as GC376, which involves at least a five-step synthesis. In addition, by introducing the chiral isocyanide, the diastereomer product can be conveniently separated by either silica gel column or reverse phase HPLC column, bypassing the need for an expensive chiral HPLC column. Target selectivity profiling showed that the non-covalent inhibitor 23R only selectively inhibits SARS-CoV-2 and SARS-CoV Mpro, but not other viral proteases and host proteases including calpain 1, cathepsins L and K, and trypsin. In contrast, the covalent Mpro inhibitor GC376 is not selective and inhibits host cysteine proteases, which might result in potential side effects. Furthermore, the X-ray crystal structure of SARS-CoV-2 Mpro in complex with 23R reveals a ligand-induced binding pocket in between S2 and S4 sites that can be explored for drug design. Overall, using the expedited drug discovery approach, this study revealed a promising non-covalent Mpro inhibitor 23R with a well characterized mechanism of action and potent cellular antiviral activity for further development.
EXPERIMENTAL SECTION
MATERIALS AND METHODS
Cell lines and viruses.
VERO E6 cells (ATCC, CRL-1586) were cultured in Dulbecco’s modified Eagle’s medium (DMEM), supplemented with 5% heat inactivated FBS in a 37°C incubator with 5% CO2. SARS-CoV-2, isolate USA-WA1/2020 (NR-52281), was obtained through BEI Resources and propagated once on VERO E6 cells before it was used for this study. Studies involving the SARS-CoV-2 were performed at the UTHSCSA biosafety level-3 laboratory by personnel wearing powered air purifying respirators.
Protein expression and purification.
SARS CoV-2 main protease (Mpro or 3CL) gene from strain BetaCoV/Wuhan/WIV04/2019 and SARS-CoV main protease from strain CDC#200301157 in the pET29a(+) vector with E. coli codon optimization were ordered from GenScript (Piscataway, NJ). The Mpro gene was then subcloned into pE-SUMO vector as described previously.7, 8 The expression and purification of SARS-CoV and SARS-CoV-2 Mpro with unmodified N- and C-termini was detailed in our previous publication.8
The expression and purification of SARS CoV-2 papain-like protease (PLpro) was also described in our previous publications.7, 8, 24
The expression and purification of EV-A71 2Apro and 3Cpro, EV-D68 2Apro and 3Cpro was described in our previous publications.24, 26
Cathepsin K and Cathepsin L were purchased from EMD Millipore (catalog #. 219461 and 219402 respectively).
Calpain 1 and trypsin were purchased from Sigma-Aldrich (catalog #. C6108 and T6763 respectively).
Peptide synthesis.
The SARS-CoV-2 Mpro FRET substrate Dabcyl-KTSAVLQ/SGFRKME(Edans) was synthesized as described before.7 The SARS-CoV-2 PLpro, EV-A71 2Apro and 3Cpro, and EV-D68 2Apro and 3Cpro FRET substrates were listed in our previous publication24 and were synthesized by solid-phase synthesis through iterative cycles of coupling and deprotection using the previously optimized procedure.27
General chemical methods.
All chemicals were purchased from commercial vendors and used without further purification unless otherwise noted. 1H and 13C NMR spectra were recorded on a Bruker-400 or −500 NMR spectrometer. Chemical shifts are reported in parts per million referenced with respect to residual solvent (CD3OD) 3.31 ppm, (DMSO-d6) 2.50 ppm, and (CDCl3) 7.26 ppm or from internal standard tetramethylsilane (TMS) 0.00 ppm. The following abbreviations were used in reporting spectra: s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; dd, doublet of doublets; ddd, doublet of doublet of doublets. All reactions were carried out under N2 atmosphere, unless otherwise stated. HPLC-grade solvents were used for all reactions. Flash column chromatography was performed using silica gel (230–400 mesh, Merck). High resolution mass spectra were obtained using an OrbitrapTM for all the compounds, obtained in an Ion Cyclotron Resonance (ICR) spectrometer. The purity was assessed by using Shimadzu UPLC with Shimdazu C18-AQ column (4.6×150 mm P/N #227–30767-05) at a flow rate of 1 mL/min; λ = 254 and 220 nm; mobile phase A, 0.1% trifluoroacetic acid in H2O, and mobile phase B, 0.1% trifluoroacetic acid in 90% CH3CN and 10% H2O. The gradients are 0–2 mins 10% B, 2–15mins 10%−100% B, 15–18mins, 100% B, 18.1–20mins 10% B. All compounds submitted for testing were confirmed to be > 95.0% purity by HPLC traces. All final products were characterized by proton and carbon NMR, HPLC and HRMS.
General Procedure for Ugi-4CR Reaction:
![]()
Ug-4CR reaction was performed according to the reported procedure with modifications.13 Amine (1.0 equiv) and aldehyde (1.0 equiv) were mixed in methanol (10 ml) and stirred at room temperature for 30 minutes. Then carboxylic acid (1.0 equiv) and isocyanide (1.0 equiv) were added sequentially and the resulting mixture was stirred at room temperature overnight. After that, the solvent was removed under reduced pressure and the crude product was purified with flash silica gel chromatography (methanol in dichloromethane 1–5% or acetone in hexane 30–80%).
General procedures for the synthesis of compounds 24, 28, 33, 35, and 37 by TFA deprotection:
To a solution of N-Trityl-protected Ugi-4CR compound in dichloromethane (5 mL) was added TFA (1 mL). The mixture was stirred at room temperature for 2 h, and the solvent was removed under reduced pressure. The crude mixture was diluted in dichloromethane and purified by silica gel flash column chromatography (ammonia 10 % methanol in dichloromethane 10 – 15 %) to give the final product.
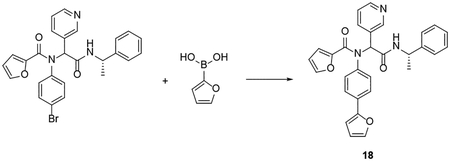
Procedure for the synthesis of compound 18 by Suzuki -Miyamura Cross-coupling:
To solution of 2-[N-(4-bromophenyl)-1-(furan-2-yl)formamido]-N-[(1S)-1-phenylethyl]-2-(pyridin-3-yl)acetamide (1 mmol) and furan-2-boronic acid (1 mmol) in 1,4-dioxane in a microwave reaction vial was added an aqueous solution of K2CO3 (4 mmol). The resulting solution was purged with N2 for 5 min. The catalyst, Pd(PPh3)4 (0.1 mmol), was added in one portion. The vial was capped and heated to 140 °C for 30 minutes with microwave irradiation. After cooling down to room temperature, the reaction solution was diluted with dichloromethane and extracted with water, followed by brine. The organic layer was dried over MgSO4, filtrated, and concentrated under reduced pressure. The crude product was purified by silica gel flash column chromatography (Methanol in dichloromethane 1–5%) to give the final product.
Procedure for the synthesis of 4-(thiophen-2-yl)aniline by Suzuki -Miyamura Cross-coupling:
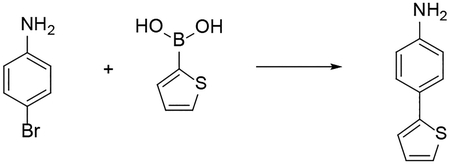
The starting material 4-(thiophen-2-yl)aniline used for the synthesis of compound 19 was prepared using the following procedure. To suspension of thiophene-2-boronic acid (1.0 mmol), sodium carbonate (1.0 mmol) in toluene/methanol (4:1, 40 mL) was added 4-bromoaniline (1.0 mmol). The resulting solution was purged with N2 for 10 min. The catalyst, Pd(PPh3)4 (0.1 mmol), was added in one portion. The reaction mixture was stirred for 16 hr. at 100 °C. After cooling to room temperature, the reaction mixture was diluted with ethyl acetate (100 mL) and washed with water (100 mL). The organic layer was dried over MgSO4, filtrated, and concentrated under reduced pressure. The crude product was purified by silica gel flash column chromatography (Ethyl acetate in Hexane 20 – 40 %) to give the final product.
4-(thiophen-2-yl)aniline. White solid. 31% yield. 1H NMR (500 MHz, CDCl3) δ 7.43 – 7.38 (m, 2H), 7.17 – 7.12 (m, 2H), 7.01 (dd, J = 5.0, 3.6 Hz, 1H), 6.69 – 6.63 (m, 2H), 3.68 (s, 2H). 13C NMR (126 MHz, CDCl3) δ 146.10, 145.11, 127.93, 127.25, 125.24, 123.17, 121.36, 115.39.
The isocyanide used for the synthesis of compound 40 was prepared used the method below:
Procedure for synthesis of N-(3-phenylpropyl)formamide:
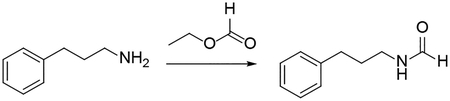
3-phenyl propylamine (2 mmol) in a microwave reaction vial was mixed with ethyl formate (6 mL). The vial was capped and heated to 70 °C overnight. After reaction residual ethyl formate was evaporated under reduced pressure. The crude product was purified by silica gel flash column chromatography (methanol in dichloromethane 1 – 3 %) to give the product N-(3-phenylpropyl)formamide. Oil, 98% yield. 1H NMR (500 MHz, CDCl3) δ 8.15 – 7.96 (m, 1H), 7.33 – 7.24 (m, 2H), 7.24 – 7.12 (m, 3H), 6.18 – 5.73 (m, 1H), 3.35 – 3.17 (m, 2H), 2.69 – 2.60 (m, 2H), 1.89 – 1.78 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 170.33, 164.88, 161.44, 141.51, 141.27, 140.65, 128.67, 128.55, 128.52, 128.42, 128.41, 126.32, 126.13, 126.07, 41.15, 39.39, 37.84, 33.36, 33.22, 32.60, 32.54, 31.17, 31.16, 23.31.
Procedure for the synthesis of compound 40:
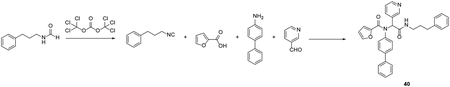
To solution of N-(3-phenylpropyl)formamide (2 mmol) in dichloromethane (50 mL) was added triethylamine (4.8 mmol). The solution was cooled to approximately – 10 °C using an ethanol ice bath. Triphosgene (0.4 mmol) was added to the stirring mixture. The resulting mixture was stirred at – 10 °C for 10 minutes. In separate 250 ml round-bottomed flask, 4-aminobiphneyl (2 mmol) and 3-pyridinecarboxaldehyde (2 mmol) were mixed in methanol (100 ml) and stirred at room temperature for 30 minutes. Then carboxylic acid (2 mmol) and the resulting isocyanide solution (1.0 equiv) were added sequentially and the resulting mixture was stirred at room temperature overnight. After that, the solvent was removed under reduced pressure and the crude product was purified with flash silica gel chromatography (Methanol in dichloromethane 1–5%) to give the final product.
N-tert-butyl-2-[N-(4-tert-butylphenyl)-1-(furan-2-yl)formamido]-2-(pyridin-3-yl)acetamide (1). Light yellow solid, 81% yield. 1H NMR (400 MHz, DMSO-d6) δ 8.38 – 8.24 (m, 2H), 7.86 (s, 1H), 7.69 – 7.66 (m, 1H), 7.37 (dt, J = 7.9, 2.0 Hz, 1H), 7.26 – 7.17 (m, 2H), 7.26 – 7.02 (br s, 1H), 7.14 – 7.04 (m, 2H), 6.31 (dd, J = 3.6, 1.7 Hz, 1H), 6.15 (s, 1H), 5.24 (d, J = 3.5 Hz, 1H), 1.22 (s, 9H), 1.19 (s, 9H). 13C NMR (101 MHz, DMSO-d6) δ 168.1, 158.1, 151.1, 150.9, 148.5, 146.4, 145.2, 137.3, 136.5, 131.2, 130.8, 125.2, 122.7, 115.8, 111.2, 62.3, 50.4, 34.3, 31.0, 28.3. C26H31N3O3, HRMS calculated for m/z [M+H]+: 434.24437, found 434.2438. HPLC purity: 97%.
2-[N-(4-tert-butylphenyl)-1-(furan-2-yl)formamido]-N-(propan-2-yl)-2-(pyridin-3-yl)acetamide (2). White solid. 82% yield. 1H NMR (400 MHz, DMSO-d6) δ 8.37 – 8.28 (m, 2H), 8.10 (d, J = 7.5 Hz, 1H), 7.76 – 7.61 (m, 1H), 7.37 (dt, J = 7.9, 2.0 Hz, 1H), 7.30 – 7.17 (m, 2H), 7.24 – 7.00 (br s, 1H), 7.17 – 7.07 (m, 2H), 6.30 (dd, J = 3.6, 1.7 Hz, 1H), 6.15 (s, 1H), 5.26 (d, J = 3.6 Hz, 1H), 3.99 – 3.76 (m, 1H), 1.20 (s, 9H), 1.07 (d, J = 6.6 Hz, 3H), 0.95 (d, J = 6.5 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 167.6, 158.2, 151.1, 151.0, 148.6, 146.3, 145.2, 137.4, 136.5, 131.0, 130.7, 125.2, 122.8, 115.8, 111.2, 62.1, 40.8, 34.3, 31.0, 22.0, 22.0. C25H29N3O3, HRMS calculated for m/z [M+H]+: 420.2287 (calculated), 420.2282 (found). HPLC purity: 98%.
2-[N-(4-tert-butylphenyl)-1-(furan-2-yl)formamido]-N-cyclopropyl-2-(pyridin-3-yl)acetamide (3). White solid. 80% yield. 1H NMR (400 MHz, DMSO-d6) δ 8.34 – 8.28 (m, 3H), 7.67 (dd, J = 1.7, 0.7 Hz, 1H), 7.37 (dt, J = 8.0, 1.9 Hz, 1H), 7.26 – 7.19 (m, 2H), 7.25 – 6.94 (br s, 1H), 7.16 – 7.09 (m, 2H), 6.31 (dd, J = 3.6, 1.7 Hz, 1H), 6.09 (s, 1H), 5.28 (dd, J = 3.6, 0.8 Hz, 1H), 2.71 – 2.60 (m, 1H), 1.20 (s, 9H), 0.67 – 0.53 (m, 2H), 0.40 – 0.25 (m, 2H).13C NMR (101 MHz, DMSO-d6) δ 169.8, 158.2, 151.1, 148.7, 146.3, 145.2, 137.4, 136.4, 130.7, 125.3, 122.8, 115.4, 111.2, 62.0, 34.3, 31.0, 22.0, 5.1. C25H27N3O3, HRMS calculated for m/z [M+H]+: 418.2131 (calculated), 418.2125 (found). HPLC purity: 96%.
2-[N-(4-tert-butylphenyl)-1-(furan-2-yl)formamido]-2-(pyridin-3-yl)-N-(2,4,4-trimethylpentan-2-yl)acetamide (4). Light yellow solid. 60% yield. 1H NMR (500 MHz, DMSO-d6) δ 8.36 – 8.28 (m, 2H), 7.79 – 7.59 (m, 2H), 7.43 – 7.35 (m, 1H), 7.32–6.97 (br s, 1H), 7.30 – 7.18 (m, 2H), 7.12 (dd, J = 8.0, 4.8 Hz, 1H), 6.36 – 6.26 (m, 1H), 6.18 (s, 1H), 5.25 (d, J = 3.6 Hz, 1H), 1.67 (dd, J = 75.5, 14.6 Hz, 2H), 1.35 – 1.23 (m, 7H), 1.21 (s, 9H), 0.86 (s, 9H). 13C NMR (101 MHz, DMSO-d6) δ 167.6, 158.0, 151.1, 150.9, 148.5, 146.3, 145.1, 137.4, 136.6, 131.0, 130.8, 125.2, 122.5, 115.7, 111.2, 62.4, 54.3, 50.6, 34.2, 31.1, 31.1, 31.1, 31.0, 28.9, 28.3. C30H39N3O3, HRMS calculated for m/z [M+H]+: 490.3069 (calculated), 490.3064 (found). HPLC purity: 97%.
2-[N-(4-tert-butylphenyl)-1-(furan-2-yl)formamido]-N-cyclopentyl-2-(pyridin-3-yl)acetamide (5). White solid. 85% yield. 1H NMR (400 MHz, DMSO-d6) δ 8.36 – 8.28 (m, 2H), 8.18 (d, J = 7.0 Hz, 1H), 7.70 – 7.65 (m, 1H), 7.37 (dt, J = 7.9, 2.0 Hz, 1H), 7.28 – 7.18 (m, 2H), 7.25 – 6.94 (br s, 1H) 7.18 – 7.06 (m, 2H), 6.30 (dd, J = 3.6, 1.7 Hz, 1H), 6.16 (s, 1H), 5.26 (d, J = 3.6 Hz, 1H), 4.02 (h, J = 6.9 Hz, 1H), 1.86 – 1.67 (m, 2H), 1.65 – 1.31 (m, 5H), 1.30 – 1.21 (m, 1H), 1.20 (s, 9H). 13C NMR (101 MHz, DMSO-d6) δ 168.1, 158.2, 151.1, 151.0, 148.6, 146.3, 145.2, 137.3, 136.5, 131.0, 130.7, 125.2, 122.8, 115.8, 111.2, 62.0, 50.6, 34.3, 32.0, 31.8, 31.0, 23.5, 23.4. C27H31N3O3, HRMS calculated for m/z [M+H]+: 446.2443 (calculated), 446.2438 (found). HPLC purity: 100%.
2-[N-(4-tert-butylphenyl)-1-(furan-2-yl)formamido]-N-cyclohexyl-2-(pyridin-3-yl)acetamide (6). White solid. 81% yield. 1H NMR (500 MHz, DMSO-d6) δ 8.36 – 8.30 (m, 2H), 8.09 (d, J = 7.7 Hz, 1H), 7.67 (dd, J = 1.7, 0.7 Hz, 1H), 7.38 (dt, J = 7.9, 2.0 Hz, 1H), 7.28 – 7.02 (br s, 1H), 7.25 – 7.19 (m, 2H), 7.17 – 7.10 (m, 2H), 6.31 (dd, J = 3.6, 1.7 Hz, 1H), 6.18 (s, 1H), 5.28 (d, J = 3.6 Hz, 1H), 3.63 – 3.52 (m, 1H), 1.86 – 1.40 (m, 4H), 1.32 – 1.14 (m, 13H), 1.14 – 0.92 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 167.6, 157.9, 151.1, 151.0, 148.6, 146.3, 145.2, 137.3, 136.5, 131.0, 130.7, 125.2, 122.7, 116.2, 111.2, 62.0, 47.9, 34.2, 32.0, 31.0, 25.1, 24.5, 24.3. C28H33N3O3, HRMS calculated for m/z [M+H]+: 460.2600 (calculated), 460.2595 (found). HPLC purity: 96%.
2-[N-(4-tert-butylphenyl)-1-(furan-2-yl)formamido]-N-(4-fluorophenyl)-2-(pyridin-3-yl)acetamide (7). Brown solid. 86% yield. 1H NMR (400 MHz, MeOD-d4) δ 8.43 (d, J = 2.3 Hz, 1H), 8.34 (dd, J = 4.9, 1.6 Hz, 1H), 7.63 (dt, J = 8.0, 2.0 Hz, 1H), 7.59 – 7.53 (m, 2H), 7.52 (d, J = 1.8 Hz, 1H), 7.45 – 6.60 (br s, 2H), 7.30 (s, 2H), 7.25 – 7.17 (m, 1H), 7.08 – 6.98 (m, 2H), 6.37 (s, 1H), 6.25 (dd, J = 3.6, 1.7 Hz, 1H), 5.46 (d, J = 3.6 Hz, 1H), 1.26 (s, 9H). 13C NMR (101 MHz, MeOD-d4) δ 169.5, 161.4, 153.8, 152.2, 149.9, 147.4, 146.7, 140.3, 139.4, 137.4, 134.5, 132.2, 131.9, 127.1, 125.5, 123.1, 123.0, 118.4, 116.4, 116.2, 112.3, 65.5, 35.5, 31.6. C28H26FN3O3, HRMS calculated for m/z [M+H]+: 472.2036 (calculated), 472.2031 (found). HPLC purity: 98%.
2-[N-(4-tert-butylphenyl)-1-(furan-2-yl)formamido]-N-(4-methoxyphenyl)-2-(pyridin-3-yl)acetamide (8). Brown solid. 79% yield. 1H NMR (400 MHz, DMSO-d6) δ 10.22 (s, 1H), 8.42 (d, J = 2.3 Hz, 1H), 8.34 (dd, J = 4.8, 1.6 Hz, 1H), 7.72 – 7.67 (m, 1H), 7.54 – 7.46 (m, 2H), 7.43 (dt, J = 8.0, 2.0 Hz, 1H), 7.35 – 7.05 (br s, 2H), 7.27 – 7.21 (m, 2H), 7.17 – 7.10 (m, 1H), 6.92 – 6.83 (m, 2H), 6.32 (dd, J = 3.6, 1.7 Hz, 1H), 6.30 (s, 1H), 5.29 (d, J = 3.6 Hz, 1H), 3.71 (s, 3H), 1.20 (s, 9H). 13C NMR (101 MHz, DMSO-d6) δ 167.3, 158.3, 155.3, 151.3, 151.2, 149.0, 146.2, 145.4, 137.5, 136.3, 131.9, 130.8, 130.2, 125.4, 122.9, 120.7, 116.0, 113.9, 111.8, 62.7, 55.1, 34.3, 31.0. C29H29N3O4, HRMS calculated for m/z [M+H]+: 484.2236 (calculated), 484.2231 (found). HPLC purity: 96%.
2-[N-(4-tert-butylphenyl)-1-(furan-2-yl)formamido]-N-(2,6-dimethylphenyl)-2-(pyridin-3-yl)acetamide (9). Light yellow solid. 75% yield. 1H NMR (400 MHz, DMSO-d6) δ 9.66 (s, 1H), 8.48 (s, 1H), 8.37 (d, J = 4.9 Hz, 1H), 7.70 (dd, J = 1.7, 0.7 Hz, 1H), 7.50 (dt, J = 8.0, 2.0 Hz, 1H), 7.28 – 7.08 (br s, 2H), 7.28 – 7.21 (m, 2H), 7.21 – 7.13 (m, 1H), 7.10 – 6.99 (m, 3H), 6.35 (s, 1H), 6.32 (dd, J = 3.6, 1.7 Hz, 1H), 5.28 (dd, J = 3.6, 0.8 Hz, 1H), 2.07 (s, 6H), 1.21 (s, 9H). 13C NMR (101 MHz, DMSO-d6) δ 167.7, 158.3, 151.4, 151.2, 149.0, 146.2, 145.3, 137.7, 136.4, 135.4, 134.6, 130.8, 130.2, 127.6, 126.5, 125.4, 122.7, 115.9, 111.2, 62.6, 34.3, 31.0, 17.9. C30H31N3O3, HRMS calculated for m/z [M+H]+: 482.2443 (calculated), 482.2438 (found). HPLC purity: 98%.
N-[(1H-1,2,3-benzotriazol-1-yl)methyl]-2-[N-(4-tert-butylphenyl)-1-(furan-2-yl)formamido]-2-(pyridin-3-yl)acetamide (10). Yellow solid. 22% yield. 1H NMR (500 MHz, CDCl3) δ 8.80 (t, J = 6.7 Hz, 1H), 8.50 – 8.46 (m, 1H), 8.40 – 8.36 (m, 1H), 8.10 (d, J = 8.3 Hz, 1H), 8.02 (d, J = 8.6 Hz, 1H), 7.63 – 7.55 (m, 1H), 7.52 – 7.38 (m, 3H), 7.24 (s, 1H), 7.12 – 6.83 (m, 3H), 6.38 (s, 1H), 6.33 – 6.20 (m, 3H), 5.47 – 5.40 (m, 2H), 1.37 (s, 9H). 13C NMR (126 MHz, CDCl3) δ 169.8, 159.8, 152.7, 151.6, 149.7, 146.0, 145.9, 145.3, 138.3, 136.1, 132.5, 130.1, 129.6, 127.9, 126.2, 124.4, 122.9, 119.6, 117.5, 111.3, 111.0, 63.0, 51.4, 34.7, 31.3. C29H28N6O3, HRMS calculated for m/z [M+H]+: 509.2301 (calculated), 509.2296 (found). HPLC purity: 96%.
2-[N-(4-tert-butylphenyl)-1-(furan-2-yl)formamido]-N-[(4-methylbenzenesulfonyl)methyl]-2-(pyridin-3-yl)acetamide (11). Yellow solid. 49% yield. 1H NMR (400 MHz, DMSO-d6) δ 9.28 – 9.20 (m, 1H), 8.37 (dd, J = 4.3, 2.2 Hz, 1H), 8.23 (s, 1H), 7.69 (dd, J = 1.8, 0.7 Hz, 1H), 7.56 – 7.49 (m, 2H), 7.30 – 7.26 (m, 2H), 7.24 – 7.17 (m, 2H), 7.15 – 7.05 (m, 2H), 6.96 (s, 2H), 6.31 (dd, J = 3.6, 1.7 Hz, 1H), 6.23 (s, 1H), 5.26 (d, J = 3.6 Hz, 1H), 4.93 (dd, J = 14.1, 7.5 Hz, 1H), 4.61 (dd, J = 14.1, 5.6 Hz, 1H), 2.34 (s, 3H), 1.20 (s, 9H). 13C NMR (101 MHz, DMSO-d6) δ 169.3, 158.1, 151.4, 151.2, 148.9, 146.1, 145.4, 144.4, 137.7, 136.0, 134.4, 130.7, 129.5, 128.4, 125.3, 122.6, 116.0, 111.8, 61.6, 60.1, 34.3, 30.9, 21.0. C30H31N3O5S, HRMS calculated for m/z [M+H]+: 546.2062 (calculated), 546.2057 (found). HPLC purity: 96%.
N-benzyl-2-[N-(4-tert-butylphenyl)-1-(furan-2-yl)formamido]-2-(pyridin-3-yl)acetamide (12). Light yellow solid. 49% yield. 1H NMR (400 MHz, DMSO-d6) δ 8.74 (t, J = 5.9 Hz, 1H), 8.37 – 8.31 (m, 2H), 7.72 – 7.66 (m, 1H), 7.39 (dt, J = 8.0, 1.9 Hz, 1H), 7.33 – 7.18 (m, 7H), 7.21 – 7.05 (br s, 1H), 7.17 – 7.08 (m, 2H), 6.31 (dd, J = 3.6, 1.7 Hz, 1H), 6.22 (s, 1H), 5.28 (d, J =4.21 Hz, 1H), 4.42 – 4.27 (m, 2H), 1.21 (s, 9H). 13C NMR (101 MHz, DMSO-d6) 13C NMR (101 MHz, DMSO) δ 168.9, 158.2, 151.3, 151.1, 148.8, 146.3, 145.3, 139.1, 137.6, 136.5, 130.7, 128.1, 127.1, 126.7, 125.3, 122.7, 115.9, 111.2, 62.4, 42.3, 34.3, 31.0. C29H29N3O3, HRMS calculated for m/z [M+H]+: 468.2287 (calculated), 468.2282 (found). HPLC purity: 98%.
2-[N-(4-tert-butylphenyl)-1-(furan-2-yl)formamido]-N-[(1S)-1-phenylethyl]-2-(pyridin-3-yl)acetamide (13). Light yellow solid. 84% yield. 1H NMR (400 MHz, DMSO-D6) δ 8.68 (dd, J = 7.8, 2.7 Hz, 1H), 8.41 – 8.23 (m, 2H), 7.67 (ddd, J = 5.7, 1.7, 0.7 Hz, 1H), 7.46 – 7.24 (m, 3H), 7.28 – 6.94 (br s, 1H), 7.23 – 7.11 (m, 5H), 7.11 – 7.01 (m, 2H), 6.34 – 6.24 (m, 2H), 5.28 (dd, J = 3.6, 0.8 Hz, 0.5H), 5.22 (dd, J = 3.6, 0.8 Hz, 0.5H), 5.04 – 4.91 (m, 1H), 1.37 (d, J = 7.0 Hz, 1.5H), 1.25 (d, J = 7.0 Hz, 1.5H), 1.19 (d, J = 6.0 Hz, 9H). 13C NMR (101 MHz, DMSO) δ 168.1, 167.9, 158.2, 158.1, 151.3, 151.2, 151.0, 148.7, 146.3, 146.2, 145.2, 145.2, 144.3, 144.0, 137.5, 137.4, 136.4, 136.3, 130.8, 130.4, 128.1, 128.0, 126.6, 126.5, 126.1, 125.7, 125.2, 122.8, 122.5, 115.8, 115.8, 111.2, 111.2, 62.0, 48.3, 48.2, 34.2, 31.0, 30.9, 22.2, 22.1. C30H31N3O3, HRMS calculated for m/z [M+H]+: 482.2443 (calculated), 482.2438 (found). HPLC purity: 97%.
2-[1-(furan-2-yl)-N-[4-(trimethylsilyl)phenyl]formamido]-N-[(1S)-1-phenylethyl]-2-(pyridinyl)acetamide (14). Light yellow solid. 50% yield. 1H NMR (500 MHz, CDCl3) δ 8.50 – 8.41 (m, 2H), 7.47 (dd, J = 49.6, 8.2 Hz, 1H), 7.39 – 7.16 (m, 8H), 7.10 – 6.81 (m, 4H), 6.25 (d, J = 13.4 Hz, 1H), 6.18 – 6.13 (m, 1H), 5.50 – 5.45 (m, 0.5H), 5.44 – 5.39 (m, 0.5H), 5.22 – 5.12 (m, 1H), 1.54 (d, J = 6.9 Hz, 1.5H), 1.48 (d, J = 7.0 Hz, 1.5H), 0.23 (s, 9H). 13C NMR (126 MHz, CDCl3) δ 168.0, 167.7, 159.8, 159.7, 151.5, 151.4, 149.8, 149.7, 146.2, 146.2, 145.1, 145.1, 143.1, 142.1, 142.0, 139.7, 139.5, 138.4, 138.1, 134.1, 134.1, 130.2, 130.1, 130.0, 129.8, 128.7, 128.6, 127.4, 127.3, 126.3, 126.1, 122.9, 122.9, 117.4, 117.4, 111.3, 111.3, 63.4, 62.9, 49.5, 49.5, 22.0, 21.9, 0.1. C29H31N3O3Si, HRMS calculated for m/z [M+H]+: 498.2212 (calculated), 498.2220 (found). HPLC purity: 96%.
2-{N-[4-(tert-butoxy)phenyl]-1-(furan-2-yl)formamido}-N-[(1S)-1-phenylethyl]-2-(pyridin-3-yl)acetamide (15). Yellow solid. 75% yield. 1H NMR (500 MHz, DMSO-d6) δ 8.72 (t, J = 8.17 Hz, 1H), 8.31 (dd, J = 55, 2.4 Hz, 1H), 8.33 (ddd, J = 13.5, 4.8, 1.6 Hz, 1H), 7.69 (ddd, J = 7.0, 1.7, 0.8 Hz, 1H), 7.50–7.00 (br s, 1H), 7.42–7.06 (m, 3H), 7.43 – 7.32 (m, 1H), 7.31 – 7.12 (m, 3H), 6.76 (s, 3H), 6.34 (ddd, J = 8.2, 3.6, 1.7 Hz, 1H), 6.27 (d, J = 13.2 Hz, 1H), 5.46 (d, J = 3.6 Hz, 0.5H), 5.41 (d, J = 3.6 Hz, 0.5H), 5.05 – 4.94 (m, 1H), 1.38 (d, J = 7.1 Hz, 1.5H), 1.25 (d, J = 7.0 Hz, 1.5H), 1.19 (d, J = 9.5 Hz, 9H). 13C NMR (126 MHz, DMSO-d6) δ 168.3, 168.1, 166.3, 158.2, 158.1, 154.7, 154.7, 151.4, 151.4, 148.7, 146.4, 146.3, 145.3, 145.3, 144.3, 144.0, 137.6, 137.6, 134.1, 134.1, 132.0, 130.7, 130.3, 128.2, 128.1, 126.7, 126.6, 126.2, 125.7, 123.7, 122.9, 122.6, 116.1, 116.0, 111.2, 111.2, 78.3, 62.0, 48.4, 48.2, 28.4, 22.3, 22.1. C30H31N3O4, HRMS calculated for m/z [M+H]+: 498.2392 (calculated), 498.2387 (found). HPLC purity: 97%.
2-[1-(furan-2-yl)-N-[4-(piperidin-1-yl)phenyl]formamido]-N-[(1S)-1-phenylethyl]-2-(pyridin-3-yl)acetamide (16). Light brown solid. 78% yield. 1H NMR (400 MHz, DMSO-d6) δ 8.65 (dd, J = 7.8, 3.6 Hz, 1H), 8.40 – 8.19 (m, 2H), 7.70 (ddd, J = 6.3, 1.7, 0.7 Hz, 1H), 7.64–7.01 (br s, 1H), 7.48 – 7.29 (m, 3H), 7.28 – 7.03 (m, 4H), 6.68 (s, 3H), 6.35 – 6.28 (m, 1H), 6.25 (d, J = 7.8 Hz, 1H), 5.30 – 5.23 (m, 0.5H), 5.22 – 5.13 (m, 0.5H), 4.98 (h, J = 7.1 Hz, 1H), 3.11 – 3.02 (m, 4H), 1.53 (s, 6H), 1.37 (d, J = 7.0 Hz, 1.5H), 1.24 (d, J = 7.0 Hz, 1.5H). 13C NMR (101 MHz, DMSO-d6) δ 168.3, 168.1, 158.5, 158.5, 151.3, 151.2, 150.8, 148.7, 146.4, 146.3, 145.1, 145.1, 144.3, 144.0, 137.6, 137.6, 131.7, 128.8, 128.7, 128.1, 128.0, 126.6, 126.5, 126.1, 125.7, 115.7, 115.7, 114.5, 111.3, 62.0, 54.9, 48.6, 48.3, 48.1, 30.6, 25.7, 24.9, 24.9, 24.1, 23.7, 22.3. C31H32N4O3, HRMS calculated for m/z [M+H]+: 509.2552 (calculated), 509.2547 (found). HPLC purity: 97%.
2-[N-(4-cyclohexylphenyl)-1-(furan-2-yl)formamido]-N-[(1S)-1-phenylethyl]-2-(pyridin-3-yl)acetamide (17). White solid. 83% yield. 1H NMR (400 MHz, DMSO-d6) δ 8.68 (dd, J = 7.7, 3.4 Hz, 1H), 8.32 (dd, J = 10.5, 4.7 Hz, 1H), 8.31 (d, J = 48 Hz, 1H), 7.70 – 7.64 (m, 1H), 7.44 – 7.28 (m, 3H), 7.28 – 7.11 (m, 4H), 7.10 – 6.98 (m, 4H),6.33 – 6.24 (m, 2H), 5.25 (d, J = 3.6 Hz, 0.5H), 5.20 (d, J = 3.6 Hz, 0.5H), 5.05 – 4.91 (m, 1H), 2.48 – 2.31 (m, 1H), 1.84 – 1.57 (m, 5H), 1.39–1.10 (m, 5H), 1.31 (dd, J = 41.17, 7.02 Hz, 3H) 13C NMR (101 MHz, DMSO-d6) δ 168.1, 167.9, 158.2, 158.1, 151.3, 151.2, 148.7, 147.8, 146.3, 146.2, 145.2, 145.2, 144.3, 144.0, 137.5, 136.6, 136.6, 131.1, 128.1, 128.0, 126.7, 126.6, 126.5, 126.1, 125.7, 115.8, 115.8, 111.2, 62.0, 48.3, 48.2, 43.1, 33.8, 33.8, 33.7, 26.1, 25.4, 22.2, 22.1. C32H33N3O3, HRMS calculated for m/z [M+H]+: 508.2600 (calculated), 508.2595 (found). HPLC purity: 98%.
2-[1-(furan-2-yl)-N-[4-(furan-2-yl)phenyl]formamido]-N-[(1S)-1-phenylethyl]-2-(pyridin-3-yl)acetamide (18). Light brown solid. 35% yield. 1H NMR (500 MHz, DMSO-d6) δ 8.80 (dd, J = 13.9, 7.8 Hz, 1H), 8.66 – 8.45 (m, 2H), 7.82 (dt, J = 8.1, 1.9 Hz, 1H), 7.71 (dddd, J = 24.9, 6.9, 1.8, 0.8 Hz, 2H), 7.64 – 7.46 (m, 3H), 7.45 – 7.07 (m, 7H), 6.96 (ddd, J = 5.9, 3.4, 0.8 Hz, 1H), 6.62 – 6.55 (m, 1H), 6.38 (d, J = 11.7 Hz, 1H), 6.35 (ddd, J = 8.2, 3.6, 1.7 Hz, 1H), 5.66 (dd, J = 2.8 Hz, 0.8 Hz, 0.5H), 5.62 (d, J = 3.6, 0.8 Hz, 0.5H), 5.06 – 4.94 (m, 1H), 1.37 (d, J = 7.0 Hz, 1.5H), 1.29 (d, J = 7.0 Hz, 1.5H). 13C NMR (126 MHz, DMSO-d6) δ 167.4, 167.2, 158.2, 158.2, 151.9, 148.3, 148.2, 146.2, 146.1, 145.5, 145.5, 144.1, 143.8, 143.4, 141.3, 141.1, 138.1, 138.0, 132.4, 132.0, 131.6, 131.6, 130.0, 128.2, 128.1, 126.7, 126.6, 126.1, 125.8, 124.4, 124.1, 123.5, 116.6, 116.6, 112.2, 111.4, 111.4, 107.0, 61.9, 48.5, 48.3, 22.2, 21.9. C30H25N3O4, HRMS calculated for m/z [M+H]+: 492.1923 (calculated), 492.1918 (found). HPLC purity: 98%.
2-[1-(furan-2-yl)-N-[4-(thiophen-2-yl)phenyl]formamido]-N-[(1S)-1-phenylethyl]-2-(pyridin-3-yl)acetamide (19). Light yellow solid. 81% yield. 1H NMR (500 MHz, DMSO-d6) δ 8.79 – 8.71 (m, 1H), 8.48 – 8.23 (m, 2H), 7.74 – 7.64 (m, 1H), 7.58 – 7.45 (m, 4H), 7.44 – 7.31 (m, 3H), 7.30 – 7.14 (m, 4H), 7.14 – 7.05 (m, 3H), 6.40 – 6.28 (m, 2H), 5.63 – 5.69 (m, 0.5H), 5.58 – 5.54 (m, 0.5H), 5.07 – 4.96 (m, 1H), 1.39 (d, J = 7.1 Hz, 1.5H), 1.26 (d, J = 7.0 Hz, 1.5H). 13C NMR (126 MHz, DMSO-d6) δ 168.1, 167.9, 158.2, 158.1, 151.4, 151.3, 148.9, 146.3, 146.2, 145.4, 145.3, 144.2, 143.9, 142.0, 138.2, 138.2, 137.6, 137.6, 133.4, 133.3, 133.3, 131.9, 130.6, 130.2, 128.6, 128.5, 128.2, 128.1, 126.6, 126.6, 126.2, 126.1, 126.1, 125.7, 125.1, 124.4, 124.4, 123.0, 122.8, 116.3, 116.3, 111.3, 111.3, 62.02, 62.00, 48.3, 48.2, 22.3, 22.1. C30H25N3O3S, HRMS calculated for m/z [M+H]+: 508.1694 (calculated), 508.1689 (found). HPLC purity: 96%.
2-[1-(furan-2-yl)-N-[4-(1H-pyrrol-1-yl)phenyl]formamido]-N-[(1S)-1-phenylethyl]-2-(pyridin-3-yl)acetamide (20). Yellow solid. 80% yield. 1H NMR (400 MHz, DMSO-d6) δ 8.76 (dd, J = 7.8, 4.0 Hz, 1H), 8.37 (d, J = 51.9 Hz 1H), 8.34 (dd, J = 12.0, 4.4 Hz, 1H), 7.72 – 7.66 (m, 1H), 7.57 – 7.30 (m, 7H), 7.30 – 7.12 (m, 4H), 7.11 – 7.05 (m, 2 H), 6.38 – 6.30 (m, 2H), 6.23 (dt, J = 5.2, 2.2 Hz, 2H), 5.58 (d, J = 3.6 Hz, 0.5H), 5.53 (d, J = 3.6 Hz, 0.5H), 5.01 (h, J = 7.3 Hz, 1H), 1.39 (d, J = 7.0 Hz, 1.5H), 1.26 (d, J = 7.0 Hz, 1.5H). 13C NMR (101 MHz, DMSO-d6) δ 168.3, 168.1, 158.3, 158.2, 151.5, 151.4, 148.9, 146.3, 146.3, 145.5, 145.4, 144.3, 144.0, 139.1, 139.1, 137.7, 137.6, 135.8, 135.7, 132.6, 128.2, 128.1, 126.7, 126.7, 126.2, 125.8, 118.7, 118.3, 116.4, 116.4, 111.4, 110.9, 110.9, 61.9, 54.9, 48.4, 48.3, 22.3, 22.2. C30H26N4O3, HRMS calculated for m/z [M+H]+: 491.2083 (calculated), 491.2078 (found). HPLC purity: 100%.
2-[1-(furan-2-yl)-N-[4-(pyridin-2-yl)phenyl]formamido]-N-[(1S)-1-phenylethyl]-2-(pyridin-3-yl)acetamide (21). Yellow solid. 88% yield. 1H NMR (500 MHz, DMSO-d6) δ 8.77 (dd, J = 7.8, 5.8 Hz, 1H), 8.65 – 8.59 (m, 1H), 8.46–8.27 (m, 2H), 7.97 – 7.88 (m, 3H), 7.88 – 7.78 (m, 1H), 7.68 (ddd, J = 7.6, 1.7, 0.8 Hz, 1H), 7.57 – 7.00 (m, 10H), 6.36 (d, J = 14.4 Hz, 1H), 6.32 (ddd, J = 9.0, 3.6, 1.7 Hz, 1H), 5.59 (d, J = 3.6, 0.8 Hz, 0.5H), 5.54 (d, J = 3.6, 0.8 Hz, 0.5H), 5.09 – 4.94 (m, 1H), 1.39 (d, J = 7.0 Hz, 1.5H), 1.27 (d, J = 7.0 Hz, 1.5H). 13C NMR (126 MHz, DMSO-d6) δ 168.2, 168.0, 158.2, 158.1, 154.6, 151.4, 151.3, 149.5, 149.5, 149.0, 148.9, 146.3, 146.3, 145.4, 145.4, 144.3, 144.0, 139.9, 139.9, 138.0, 137.7, 137.7, 137.3, 137.3, 131.6, 130.6, 130.3, 128.2, 128.1, 126.7, 126.6, 126.5, 126.2, 125.8, 123.1, 122.9, 122.8, 120.3, 120.3, 116.4, 116.4, 111.43, 111.41, 62.1, 48.4, 48.3, 22.3, 22.1. C31H26N4O3, HRMS calculated for m/z [M+H]+: 503.2083 (calculated), 503.2078 (found). HPLC purity: 98%.
2-[N-(4-benzylphenyl)-1-(furan-2-yl)formamido]-N-[(1S)-1-phenylethyl]-2-(pyridin-3-yl)acetamide (22). White solid. 80% yield. 1H NMR (400 MHz, DMSO-d6) δ 8.71 (dd, J = 7.8, 4.2 Hz, 1H), 8.44 – 8.23 (m, 2H), 7.66 (ddd, J = 5.5, 1.7, 0.7 Hz, 1H), 7.45 – 7.31 (m, 3H), 7.31 – 7.11 (m, 7H), 7.10 – 6.96 (m, 6H), 6.39 – 6.18 (m, 2H), 5.43 – 5.38 (m, 0.5H), 5.36 – 5.30 (m, 0.5H), 5.06 – 4.89 (m, 1H), 3.86 (d, J = 6.6 Hz, 2H), 1.37 (d, J = 7.0 Hz, 1.5H), 1.25 (d, J = 7.0 Hz, 1.5H). 13C NMR (101 MHz, DMSO-d6) δ 168.1, 167.9, 158.2, 158.1, 151.4, 151.3, 148.8, 146.3, 146.3, 145.2, 145.2, 144.2, 143.9, 141.1, 140.9, 137.5, 137.1, 137.0, 131.3, 130.7, 130.3, 129.2, 128.4, 128.4, 128.3, 128.3, 128.2, 128.0, 126.6, 126.6, 126.1, 125.9, 125.9, 125.7, 122.9, 122.6, 116.0, 116.0, 111.1, 62.0, 48.3, 48.2, 40.2, 22.3, 22.1. C33H29N3O3, HRMS calculated for m/z [M+H]+: 516.2287 (calculated), 516.2282 (found). HPLC purity: 98%.
2-(N-{[1,1’-biphenyl]-4-yl}-1-(furan-2-yl)formamido)-N-[(1S)-1-phenylethyl]-2-(pyridin-3-yl)acetamide (23). White solid. 89% yield. 1H NMR (400 MHz, DMSO-d6) δ 8.75 (dd, J = 7.8, 5.0 Hz, 1H), 8.53 – 8.21 (m, 2H), 7.72 – 7.66 (m, 1H), 7.66 – 7.58 (m, 2H), 7.58 – 7.24 (m, 10H), 7.24 – 7.05 (m, 4H), 6.47 – 6.23 (m, 2H), 5.63 – 5.53 (m, 0.5H), 5.52 – 5.43 (m, 0.5H), 5.10 – 4.92 (m, 1H), 1.39 (d, J = 7.0 Hz, 1.5H), 1.27 (d, J = 7.0 Hz, 1.5H). 13C NMR (101 MHz, DMSO-d6) δ 168.1, 167.9, 158.2, 158.1, 151.3, 151.3, 148.9, 146.3, 146.2, 145.3, 145.3, 144.2, 143.9, 139.4, 138.55, 138.50, 138.4, 137.6, 131.8, 128.95, 128.93, 128.2, 128.0, 127.8, 126.6, 126.6, 126.5, 126.4, 126.1, 125.7, 116.2, 116.2, 111.3, 62.1, 48.4, 48.3, 22.2, 22.1. C32H27N3O3, HRMS calculated for m/z [M+H]+: 502.2130 (calculated), 502.2132 (found). HPLC purity: 97%.
(2R)-2-(N-{[1,1’-biphenyl]-4-yl}-1-(furan-2-yl)formamido)-N-[(1S)-1-phenylethyl]-2-(pyridin- 3-yl)acetamide (23R, Jun8-76-3A). White solid, 45% yield. 1H NMR (500 MHz, DMSO-d6) δ 8.74 (d, J = 7.9 Hz, 1H), 8.43 (d, J = 2.3 Hz, 1H), 8.35 (dd, J = 4.8, 1.6 Hz, 1H), 7.71 – 7.66 (m, 1H), 7.66 – 7.61 (m, 2H), 7.60 – 7.52 (m, 2H), 7.50 (dt, J = 8.0, 2.0 Hz, 1H), 7.46 – 7.32 (m, 8H), 7.30 – 7.15 (m, 3H), 6.36 (s, 1H), 6.32 (dd, J = 3.6, 1.7 Hz, 1H), 5.50 (d, J = 3.6 Hz, 1H), 5.02 (p, J = 7.1 Hz, 1H), 1.27 (d, J = 7.0 Hz, 3H). 13C NMR (126 MHz, DMSO-d6) δ 168.1, 158.1, 151.3, 148.9, 146.2, 145.3, 143.9, 139.4, 138.5, 138.5, 137.6, 131.8, 130.7, 128.9, 128.2, 127.8, 126.6, 126.5, 126.4, 126.1, 123.0, 116.2, 111.3, 62.1, 48.2, 22.1. C32H27N3O3, HRMS calculated for m/z [M+H]+: 502.2130 (calculated), 502.2132 (found). HPLC purity: 100%.
(2S)-2-(N-{[1,1’-biphenyl]-4-yl}-1-(furan-2-yl)formamido)-N-[(1S)-1-phenylethyl]-2-(pyridin-3- yl)acetamide (23S). White solid, 45% yield. 1H NMR (500 MHz, DMSO-d6) δ 8.75 (d, J = 7.6 Hz, 1H), 8.38 – 8.25 (m, 2H), 7.73 – 7.66 (m, 1H), 7.66 – 7.59 (m, 2H), 7.53 (d, J = 8.0 Hz, 2H), 7.47 – 7.31 (m, 4H), 7.30 – 7.13 (m, 5H), 7.12 – 7.06 (m, 3H), 6.36 – 6.32 (m, 2H), 5.54 (d, J = 3.6 Hz, 1H), 5.00 (p, J = 7.1 Hz, 1H), 1.38 (d, J = 7.0 Hz, 3H). 13C NMR (126 MHz, DMSO) δ 168.0, 158.2, 151.4, 148.9, 146.3, 145.4, 144.3, 139.4, 138.5, 138.5, 137.6, 131.8, 130.3, 128.9, 128.1, 127.8, 126.6, 126.5, 126.5, 125.8, 122.7, 116.3, 111.4, 62.1, 48.4, 22.3. C32H27N3O3, HRMS calculated for m/z [M+H]+ : 502.2130 (calculated), 502.2132 (found). HPLC purity: 97%.
2-(N-{[1,1’-biphenyl]-4-yl}-1-(1H-imidazol-4-yl)formamido)-N-[(1S)-1-phenylethyl]-2-(pyridin-3-yl)acetamide (24). White solid. 38% yield. 1H NMR (500 MHz, DMSO-d6) δ 12.76 (s, 1H), 8.74 (d, J = 7.9 Hz, 1H), 8.44 (d, J = 2.4 Hz, 1H), 8.36 (dd, J = 4.8, 1.7 Hz, 1H), 7.73 – 7.32 (br s, 1H), 7.68 – 7.60 (m, 3H), 7.60 – 7.49 (m, 3H), 7.48 – 7.23 (m, 9H), 7.19 (ddd, J = 7.8, 4.8, 0.8 Hz, 1H), 6.40 (s, 1H), 5.53 (s, 1H), 5.03 (p, J = 7.2 Hz, 1H), 1.26 (d, J = 7.0 Hz, 3H). 13C NMR (126 MHz, DMSO-d6) δ 168.3, 151.3, 148.8, 144.0, 139.6, 138.6, 137.6, 137.0, 132.1, 130.9, 128.9, 128.2, 127.8, 126.6, 126.5, 126.1, 123.0, 62.0, 48.2, 22.2. C31H27N5O2, HRMS calculated for m/z [M+H]+: 502.2242 (calculated), 502.2238 (found). HPLC purity: 96%.
2-(N-{[1,1’-biphenyl]-4-yl}-1-(1,2-oxazol-5-yl)formamido)-N-[(1S)-1-phenylethyl]-2-(pyridin-3-yl)acetamide (25). Light yellow solid. 82% yield. 1H NMR (400 MHz, DMSO-d6) δ 8.83 (dd, J = 7.7, 5.7 Hz, 1H), 8.56 – 8.26 (m, 3H), 7.65 – 7.45 (m, 5H), 7.44 – 7.06 (m, 11H), 6.35 (d, J = 11.2 Hz, 1H), 5.88 – 5.77 (m, 1H), 5.13 – 4.94 (m, 1H), 1.40 (d, J = 7.0 Hz, 1.5H), 1.28 (d, J = 7.0 Hz, 1.5H). 13C NMR (101 MHz, DMSO-d6) δ 167.5, 167.3, 161.7, 161.6, 157.0, 157.0, 151.4, 151.3, 150.7, 149.1, 144.1, 143.8, 139.7, 138.4, 137.6, 137.4, 137.3, 131.4, 130.1, 129.7, 128.9, 128.9, 128.2, 128.1, 127.9, 126.7, 126.7, 126.56, 126.54, 126.1, 125.8, 123.1, 122.8, 107.11, 107.10, 62.3, 48.5, 48.4, 22.3, 22.1. C31H26N4O3, HRMS calculated for m/z [M+H]+: 503.2083 (calculated), 503.2078 (found). HPLC purity: 98%.
2-(N-{[1,1’-biphenyl]-4-yl}-1-(1,3-oxazol-5-yl)formamido)-N-[(1S)-1-phenylethyl]-2-(pyridin-3-yl)acetamide (26). Yellow solid. 85% yield. 1H NMR (400 MHz, DMSO-d6) δ 8.80 (dd, J = 7.8, 4.7 Hz, 1H), 8.57 – 8.23 (m, 3H), 7.69 – 7.49 (m, 5H), 7.48 – 7.32 (m, 6H), 7.31 – 7.07 (m, 5H), 6.34 (d, J = 9.3 Hz, 1H), 5.87 (d, J = 16.3 Hz, 1H), 5.02 (h, J = 7.6 Hz, 1H), 1.40 (d, J = 7.0 Hz, 1.5H), 1.27 (d, J = 7.0 Hz, 1.5H). 13C NMR (101 MHz, DMSO-d6) δ 167.8, 167.6, 156.9, 156.8, 153.69, 153.65, 151.4, 151.3, 149.1, 144.2, 144.24, 144.21, 143.9, 140.1, 138.4, 137.6, 137.6, 137.5, 137.4, 131.8, 130.4, 130.3, 129.9, 128.9, 128.9, 128.2, 128.1, 127.9, 126.8, 126.7, 126.65, 126.64, 126.1, 125.7, 123.1, 122.8, 62.1, 48.4, 48.3, 22.2, 22.1. C31H26N4O3, HRMS calculated for m/z [M+H]+: 503.2083 (calculated), 503.2078 (found). HPLC purity: 97%.
N-tert-butyl-2-[N-(4-tert-butylphenyl)-1-(furan-2-yl)formamido]-2-(pyrazin-2-yl)acetamide (27). Light yellow solid. 86% yield. 1H NMR (500 MHz, DMSO-d6) δ 8.53 (dd, J = 2.5, 1.5 Hz, 1H), 8.47 (d, J = 1.5 Hz, 1H), 8.43 (d, J = 2.6 Hz, 1H), 8.00 (s, 1H), 7.68 (dd, J = 1.7, 0.7 Hz, 1H), 7.35 – 7.16 (m, 4H), 6.33 (dd, J = 3.6, 1.7 Hz, 1H), 6.28 (s, 1H), 5.39 (d, J = 3.6 Hz, 1H), 1.22 (s, 9H), 1.14 (s, 9H). 13C NMR (126 MHz, DMSO-d6) δ 166.0, 158.3, 152.0, 151.0, 146.2, 145.3, 145.2, 143.6, 143.0, 136.9, 130.2, 125.2, 116.0, 111.2, 65.1, 50.5, 34.3, 31.0, 28.1. C25H30N4O3, HRMS calculated for m/z [M+H]+: 435.2396 (calculated), 435.2391 (found). HPLC purity: 96%.
N-tert-butyl-2-[N-(4-tert-butylphenyl)-1-(furan-2-yl)formamido]-2-(1H-imidazol-4-yl)acetamide (28). Light yellow solid. 60% yield. 1H NMR (400 MHz, DMSO-d6) δ 8.91 (d, J = 1.3 Hz, 1H), 8.02 (s, 1H), 7.73 – 7.66 (m, 1H), 7.36 – 7.29 (m, 2H), 7.23 (dd, J = 1.3, 0.7 Hz, 3H), 6.34 (dd, J = 3.6, 1.7 Hz, 1H), 6.27 (s, 1H), 5.34 (d, J = 3.6 Hz, 1H), 1.24 (s, 9H), 1.23 (s, 9H). 13C NMR (101 MHz, DMSO-d6) δ 165.4, 158.2, 151.4, 146.0, 145.5, 136.4, 134.5, 129.9, 128.3, 125.5, 119.6, 116.2, 111.4, 56.2, 50.7, 34.4, 31.0, 28.2. C24H30N4O3, HRMS calculated for m/z [M+H]+: 423.2396 (calculated), 423.2391 (found). HPLC purity: 97%.
N-tert-butyl-2-[N-(4-tert-butylphenyl)-1-(furan-2-yl)formamido]-2-(pyrimidin-5-yl)acetamide (29). Yellow solid. 78% yield. 1H NMR (400 MHz, DMSO-d6) δ 8.93 (s, 1H), 8.46 (s, 2H), 7.95 (s, 1H), 7.68 (dd, J = 1.7, 0.8 Hz, 1H), 7.33–7.12 (br s, 2H)7.27 (d, J = 8.0 Hz, 2H), 6.32 (dd, J = 3.6, 1.7 Hz, 1H), 6.16 (s, 1H), 5.32 (d, J = 3.6 Hz, 1H), 1.22 (s, 9H), 1.21 (s, 9H). 13C NMR (101 MHz, DMSO-d6) δ 167.1, 158.1, 157.8, 157.2, 151.3, 146.2, 145.3, 136.4, 130.7, 129.6, 125.5, 116.0, 111.3, 60.5, 50.5, 34.3, 30.9, 28.2. C25H30N4O3, HRMS calculated for m/z [M+H]+: 435.2396 (calculated), 435.2391 (found). HPLC purity: 98%.
2-(N-{[1,1’-biphenyl]-4-yl}-1-(furan-2-yl)formamido)-N-cyclopropyl-2-(pyridin-3-yl)acetamide (30). Light yellow solid. 89% yield. 1H NMR (500 MHz, DMSO-d6) δ 8.46 – 8.26 (m, 3H), 7.69 (dd, J = 1.7, 0.8 Hz, 1H), 7.66 – 7.62 (m, 2H), 7.62 – 7.50 (m, 2H), 7.49–7.14 (br s, 1H), 7.48 – 7.31 (m, 5H), 7.17 (dd, J = 7.9, 4.8 Hz, 1H), 6.34 (dd, J = 3.6, 1.7 Hz, 1H), 6.18 (s, 1H), 5.55 (dd, J = 3.6, 0.8 Hz, 1H), 2.74 – 2.65 (m, 1H), 0.73 – 0.55 (m, 2H), 0.46 – 0.26 (m, 2H). 13C NMR (126 MHz, DMSO-d6) δ 169.9, 158.2, 151.1, 148.9, 146.3, 145.4, 139.5, 138.5, 138.5, 137.5, 131.7, 130.6, 128.9, 127.8, 126.5, 126.5, 123.0, 116.3, 111.3, 62.0, 22.5, 5.6, 5.5. C27H23N3O3, HRMS calculated for m/z [M+H]+: 438.1817 (calculated), 438.1812 (found). HPLC purity: 98%.
2-(N-{[1,1’-biphenyl]-4-yl}-1-(furan-2-yl)formamido)-2-(pyridin-3-yl)-N-(2,4,4-trimethylpentan-2-yl)acetamide (31). Light yellow solid. 62% yield. 1H NMR (400 MHz, DMSO-d6) δ 8.48 – 8.22 (m, 2H), 7.75 – 7.66 (m, 2H), 7.66 – 7.60 (m, 2H), 7.60 – 7.49 (m, 2H), 7.51–7.09 (br s, 1H), 7.49 – 7.30 (m, 5H), 7.15 (ddd, J = 7.9, 4.8, 0.8 Hz, 1H), 6.32 (dd, J = 3.6, 1.7 Hz, 1H), 6.25 (s, 1H), 5.49 (dd, J = 3.6, 0.8 Hz, 1H), 1.68 (dd, J = 75.4, 14.5 Hz, 2H), 1.29 (d, J = 16.4 Hz, 6H), 0.87 (s, 9H). 13C NMR (101 MHz, DMSO-d6) δ 167.7, 158.0, 151.3, 148.6, 146.3, 145.2, 139.4, 138.6, 138.6, 137.6, 131.9, 130.9, 128.9, 127.8, 126.5, 126.4, 122.8, 116.1, 111.3, 62.4, 50.7, 31.2, 31.1, 29.0, 28.4. C32H35N3O3, HRMS calculated for m/z [M+H]+: 510.2756 (calculated), 510.2751 (found). HPLC purity: 100%.
2-(N-{[1,1’-biphenyl]-4-yl}-1-(furan-2-yl)formamido)-N-cyclopentyl-2-(pyridin-3-yl)acetamide (32). White solid. 78% yield. 1H NMR (400 MHz, DMSO-d6) δ 8.35 (d, J = 20.6 Hz, 2H), 8.24 (d, J = 7.0 Hz, 1H), 7.68 (dd, J = 1.7, 0.7 Hz, 1H), 7.66 – 7.60 (m, 2H), 7.55 (d, J = 8.1 Hz, 2H), 7.51–7.12 (br s, 1H), 7.50 – 7.29 (m, 5H), 7.16 (dd, J = 7.9, 4.7 Hz, 1H), 6.33 (dd, J = 3.6, 1.7 Hz, 1H), 6.24 (s, 1H), 5.53 (d, J = 3.6 Hz, 1H), 4.06 (h, J = 6.6 Hz, 1H), 1.90 – 1.70 (m, 2H), 1.68 – 1.37 (m, 5H), 1.33 – 1.19 (m, 1H). 13C NMR (101 MHz, DMSO-d6) δ 168.2, 158.2, 151.2, 148.8, 146.3, 145.3, 139.4, 138.5, 137.5, 131.7, 130.9, 128.9, 127.8, 126.5, 126.4, 123.0, 116.2, 111.3, 62.0, 50.7, 32.1, 31.8, 23.5, 23.4. C29H27N3O3, HRMS calculated for m/z [M+H]+: 466.2130 (calculated), 466.2125 (found). HPLC purity: 96%.
2-(N-{[1,1’-biphenyl]-4-yl}-1-(1H-imidazol-4-yl)formamido)-N-cyclopentyl-2-(pyridin-3-yl)acetamide (33). White solid. 44% yield. 1H NMR (500 MHz, DMSO-d6) δ 8.38 (d, J = 2.3 Hz, 1H), 8.33 (dd, J = 4.8, 1.7 Hz, 1H), 8.24 (d, J = 7.1 Hz, 1H), 7.82–7.32 (br s, 1H), 7.73 – 7.55 (m, 5H), 7.53 – 7.29 (m, 5H), 7.23 – 7.12 (m, 1H), 6.24 (s, 1H), 5.54 (s, 1H), 4.12 – 4.00 (m, 1H) 1.90 – 1.70 (m, 2H), 1.67 – 1.40 (m, 5H), 1.32 – 1.19 (m, 1H). 13C NMR (126 MHz, DMSO-d6) δ 168.4, 151.2, 148.8, 139.6, 138.7, 138.6, 137.5, 132.1, 131.1, 129.0, 127.9, 126.6, 123.0, 62.0, 50.7, 32.1, 31.9, 23.5, 23.4. C28H27N5O2, HRMS calculated for m/z [M+H]+: 466.2242 (calculated), 466.2238 (found). HPLC purity: 98%.
2-(N-{[1,1’-biphenyl]-4-yl}-1-(furan-2-yl)formamido)-N-cyclohexyl-2-(pyridin-3-yl)acetamide (34). White solid. 83% yield. 1H NMR (500 MHz, CDCl3/ MeOD-d4) δ 8.43 (s, 1H), 8.38 – 8.31 (m, 1H), 7.55 – 7.48 (m, 3H), 7.48 – 7.27 (m, 6H), 7.26 – 6.95 (br s, 1H), 7.16 – 7.05 (m, 2H), 6.22 (s, 1H), 6.16 – 6.08 (m, 1H), 5.55 (d, J = 3.6 Hz, 1H), 3.79 (s, 1H), 3.77 – 3.69 (m, 1H), 2.04 – 1.46 (m, 5H), 1.43 – 0.9 (m, 5H). 13C NMR (126 MHz, CDCl3/ MeOD-d4) δ 168.1, 168.0, 159.8, 150.9, 148.9, 145.9, 145.1, 141.5, 139.3, 138.5, 137.9, 131.3, 130.8, 128.8, 127.8, 127.4, 126.8, 123.3, 117.5, 111.3, 62.7, 62.6, 32.5, 32.4, 25.3, 24.8, 24.7. C30H29N3O3, HRMS calculated for m/z [M+H]+: 480.2287 (calculated), 480.2282 (found). HPLC purity: 97%.
2-(N-{[1,1’-biphenyl]-4-yl}-1-(1H-imidazol-4-yl)formamido)-N-cyclohexyl-2-(pyridin-3-yl)acetamide (35). White solid. 42% yield. 1H NMR (500 MHz, DMSO-d6) δ 8.40 (d, J = 2.3 Hz, 1H), 8.33 (dd, J = 4.8, 1.7 Hz, 1H), 8.17 (d, J = 7.7 Hz, 1H), 7.89 (s, 1H), 7.69 – 7.63 (m, 2H), 7.59 (d, J = 7.2 Hz, 2H), 7.54–6.99 (br s, 1 H), 7.51 – 7.32 (m, 5H), 7.17 (ddd, J = 7.9, 4.8, 0.8 Hz, 1H), 6.26 (s, 1H), 5.56 (s, 1H), 3.67–3.57 (m, 1H), 1.86 – 1.48 (m, 5H), 1.35 – 1.16 (m, 3H), 1.15 – 0.96 (m, 2H). 13C NMR (126 MHz, DMSO-d6) δ 167.8, 159.8, 151.2, 148.8, 139.8, 138.5, 138.4, 137.6, 136.8, 132.0, 130.9, 129.0, 127.9, 126.7, 126.6, 123.0, 62.1, 48.0, 32.1, 25.1, 24.5, 24.4. C29H29N5O2, HRMS calculated for m/z [M+H]+: 480.2399 (calculated), 480.2394 (found). HPLC purity: 96%.
2-[N-(4-cyclohexylphenyl)-1-(1,2-oxazol-5-yl)formamido]-N-[(1S)-1-phenylethyl]-2-(pyridin-3-yl)acetamide (36). Yellow solid. 76% yield. 1H NMR (400 MHz, DMSO-d6) δ 8.69 (dd, J = 7.8, 3.1 Hz, 1H), 8.32 (ddd, J = 10.3, 4.8, 1.6 Hz, 1H), 8.31 (dd, J = 46.3, 2.2 Hz, 1H), 7.74 – 7.60 (m, 1H), 7.46 – 7.29 (m, 3H), 7.28 – 6.96 (m, 7H), 6.35 – 6.19 (m, 2H), 5.26 – 5.22 (m, 0.5H), 5.21 – 5.15 (m, 0.5H), 5.08 – 4.87 (m, 1H), 2.48 – 2.34 (m, 1H), 1.87 – 1.59 (m, 5H), 1.47 – 1.07 (m, 5H), 1.31 (dd, J = 47.0 7.0 Hz, 3 H) 13C NMR (101 MHz, DMSO-d6) δ 168.2, 167.9, 158.2, 158.1, 151.3, 151.3, 148.8, 147.8, 146.3, 146.2, 145.3, 145.2, 144.3, 144.0, 137.5, 136.6, 136.6, 131.1, 130.7, 130.4, 128.2, 128.0, 126.7, 126.6, 126.6, 126.1, 125.7, 122.8, 122.5, 115.8, 115.8, 111.2, 62.0, 54.9, 48.3, 48.2, 43.1, 33.8, 33.8, 33.7, 26.1, 25.4, 22.3, 22.1. C31H32N4O3, HRMS calculated for m/z [M+H]+: 509.2552 (calculated), 509.2547 (found). HPLC purity: 97%.
2-(N-{[1,1’-biphenyl]-4-yl}-1-(1H-imidazol-4-yl)formamido)-N-[(1S)-1-phenylethyl]-2-(pyrazin-2-yl)acetamide (37). Yellow solid. 42% yield. 1H NMR (500 MHz, DMSO-d6) δ 8.89 – 8.37 (m, 5H), 7.70 – 7.61 (m, 3H), 7.57 (dd, J = 16.7, 8.1 Hz, 2H), 7.52 – 7.41 (m, 3H), 7.40 – 7.29 (m, 4H), 7.29 – 7.16 (m, 3H), 6.61 – 6.47 (m, 1H), 5.07 – 4.93 (m, 1H), 1.33 (d, J = 7.0 Hz, 1.5H), 1.30 (d, J = 7.0 Hz, 1.5H). 13C NMR (126 MHz, DMSO-d6) δ 166.7, 166.5, 151.4, 151.3, 146.1, 146.0, 144.2, 144.1, 143.8, 143.7, 143.4, 138.7, 131.6, 131.5, 129.0, 128.2, 128.1, 127.9, 126.6, 126.6, 126.1, 126.0, 64.5, 48.4, 48.3, 22.2, 22.2. C30H26N6O2, HRMS calculated for m/z [M+H]+: 503.2195 (calculated), 503.2190 (found). HPLC purity: 99%.
2-(N-{[1,1’-biphenyl]-4-yl}-1-(furan-2-yl)formamido)-N-[(1S)-1-phenylethyl]-2-(pyrazin-2-yl)acetamide (38). Yellow solid. 83% yield. 1H NMR (500 MHz, DMSO-d6) δ 8.79 (dd, J = 24.5, 7.8 Hz, 1H), 8.68 – 8.34 (m, 3H), 7.72 – 7.67 (m, 1H), 7.66 – 7.61 (m, 2H), 7.60 – 7.51 (m, 2H), 7.49–7.26 (br s, 1H), 7.48 – 7.41 (m, 2H), 7.39 – 7.30 (m, 4H), 7.29 – 7.16 (m, 3H), 6.56 – 6.46 (m, 1H), 6.40 – 6.31 (m, 1H), 5.66 – 5.64 (m, 0.5H), 5.62 – 5.59 (m, 0.5H), 5.10 – 4.90 (m, 1H), 1.39 – 1.24 (m, 3H). 13C NMR (126 MHz, DMSO-d6) δ 166.5, 166.3, 158.3, 158.3, 151.2, 151.1, 146.2, 146.1, 146.0, 145.5, 145.4, 144.1, 144.0, 143.8, 143.7, 143.4, 139.6, 139.5, 138.8, 138.8, 138.6, 138.6, 131.2, 131.1, 129.0, 128.2, 128.1, 127.9, 126.7, 126.6, 126.1, 126.0, 116.6, 116.5, 111.4, 111.4, 64.5, 64.4, 48.5, 48.4, 22.2. C31H26N4O3, HRMS calculated for m/z [M+H]+: 503.2083 (calculated), 503.2078 (found). HPLC purity: 100%.
2-(N-{[1,1’-biphenyl]-4-yl}-1-(furan-2-yl)formamido)-N-(2,2-diphenylethyl)-2-(pyrazin-2-yl)acetamide (39). Yellow solid. 65% yield. 1H NMR (500 MHz, DMSO-d6) δ 8.39 – 8.19 (m, 3H), 7.69 (dd, J = 1.7, 0.7 Hz, 1H), 7.67 – 7.60 (m, 2H), 7.58 – 7.50 (m, 2H), 7.46 – 7.42 (m, 2H), 7.38 – 7.33 (m, 1H), 7.32 – 7.10 (m, 12H), 7.05 (dt, J = 7.9, 2.0 Hz, 1H), 6.96 (dd, J = 8.0, 4.8 Hz, 1H), 6.33 (dd, J = 3.6, 1.7 Hz, 1H), 6.21 (s, 1H), 5.52 (dd, J = 3.6, 0.8 Hz, 1H), 4.23 (t, J = 7.9 Hz, 1H), 4.06 – 3.90 (m, 1H), 3.73 – 3.57 (m, 1H). 13C NMR (126 MHz, DMSO-d6) δ 168.7, 158.2, 151.4, 148.5, 146.3, 145.3, 142.7, 142.6, 139.4, 138.5, 138.5, 137.4, 131.6, 130.5, 129.0, 128.4, 128.3, 127.9, 127.8, 127.8, 126.5, 126.5, 126.3, 126.3, 122.7, 116.3, 111.3, 62.1, 50.0, 43.6. C38H31N3O3, HRMS calculated for m/z [M+H]+: 578.2443 (calculated), 578.2438 (found). HPLC purity: 96%.
2-(N-{[1,1’-biphenyl]-4-yl}-1-(furan-2-yl)formamido)-N-(3-phenylpropyl)-2-(pyridin-3-yl)acetamide (40). Light yellow solid. 76% yield. 1H NMR (500 MHz, DMSO-d6) δ 8.55 (d, J = 2.2 Hz, 1H), 8.49 (dd, J = 5.2, 1.6 Hz, 1H), 8.32 (t, J = 5.5 Hz, 1H), 7.89 – 7.79 (m, 1H), 7.64 (d, J = 1.5 Hz, 1H), 7.63 – 7.51 (m, 4H), 7.49 – 7.42 (m, 1H), 7.42 – 7.21 (m, 5H), 7.21 – 7.13 (m, 2H), 7.12 – 7.06 (m, 3H), 6.30 (dd, J = 3.6, 1.7 Hz, 1H), 6.19 (s, 1H), 5.58 (d, J = 3.5 Hz, 1H), 3.08 (q, J = 6.6 Hz, 2H), 2.48 – 2.39 (m, 2H), 1.69 – 1.55 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 167.7, 158.3, 146.1, 145.5, 141.6, 139.7, 138.8, 138.5, 132.8, 131.2, 128.9, 128.2, 128.2, 127.9, 126.8, 126.6, 125.7, 124.5, 116.6, 111.4, 62.6, 38.6, 32.4, 30.7. C33H29N3O3, HRMS calculated for m/z [M+H]+: 516.2287 (calculated), 516.2282 (found). HPLC purity: 98%.
Native Mass Spectrometry.
Prior to analysis, the protein was buffer exchanged into 0.2 M ammonium acetate (pH 6.8) and diluted to 10 μM. DTT was dissolved in water and prepared at a 400 mM stock. Each ligand was dissolved in ethanol and diluted to 10X stock concentrations. The final mixture was prepared by adding 4 μL protein, 0.5 μL DTT stock, and 0.5 μL ligand stock for final concentration of 4 mM DTT and 70 μM protein. Final ligand concentrations were 10 μM and 30 μM. The mixtures were then incubated for 10 minutes at room temperature prior to analysis. Each sample was mixed and analyzed in triplicate.
Native mass spectrometry (MS) was performed using a Q-Exactive HF quadrupole-Orbitrap mass spectrometer with the Ultra-High Mass Range research modifications (Thermo Fisher Scientific). Samples were ionized using nano-electrospray ionization in positive ion mode using 1.0 kV capillary voltage at a 150 °C capillary temperature. The samples were all analyzed with a 1,000–25,000 m/z range, the resolution set to 30,000, and a trapping gas pressure set to 3. Between 10 and 50 V of source fragmentation was applied to all samples to aid in desolvation. Data were deconvolved and analyzed with UniDec.28
Enzymatic assays.
The main protease (Mpro) enzymatic assays were carried out in Mpro reaction buffer containing 20 mM HEPES pH 6.5, 120 mM NaCl, 0.4 mM EDTA, 20% glycerol and 4 mM DTT, and the SARS-CoV-2 papain-like protease (PLpro) enzymatic assays were carried out in PLPro reaction buffer containing 50 mM HEPES, pH7.5, 0.01% triton-100 and 5 mM DTT. The percentage of inhibition and enzymatic IC50 values were calculated as previously described.7, 8 Briefly, the assay was performed in 96-well plates with 100 μL of 100 nM Mpro protein or 200 nM PLPro protein in their respective reaction buffers. Then 1 μL testing compound at various concentrations was added to each well and incubated at 30 °C for 30 min. The enzymatic reaction was initiated by adding 1 μL of 1 mM corresponding FRET substrate (the final substrate concentration is 10 μM). The reaction was monitored in a Cytation 5 image reader with filters for excitation at 360/40 nm and emission at 460/40 nm at 30 °C for 1 hr. The initial velocity of the enzymatic reaction with and without testing compounds was calculated by linear regression for the first 15 min of the kinetic progress curve.
For the Morrison plot, 10 μL 100 nM SARS-CoV-2 Mpro protein was added to 190 μl of Mpro reaction buffer containing testing compound and the FRET substrate, and the reaction was monitored for 2 hr. The final FRET substrate concentration in this assay is 20 μM. Detailed curve fitting and KI determination was described previously.7, 8
For Michaelis-Menten and Lineweaver-Burk plots, assay was carried as follows: 50 μL of 50 μM Mpro protein was added to 50 μL reaction buffer containing testing compound and various concentrations of FRET substrate to initiate the enzyme reaction. The initial velocity of the enzymatic reaction with and without testing compounds was calculated by linear regression for the first 15 min of the kinetic progress curve, the plotted against substrate concentrations in Prism 8 with Michaelis-Menten equation.
Cathepsin K and cathepsin L enzymatic assay was carried out as follows: cathepsin K or cathepsin L was activated by incubating in reaction buffer [20 mM sodium acetate, 1 mM EDTA, and 5 mM DTT (pH 5.5)] for 30 min at 30°C. Upon activation, the assay was assembled in 96-well plates with 100 μL of cathepsin K at 200 pM or cathepsin L at 300 pM in reaction buffer. Then, 1 μL of testing compound at various concentrations was added to each well and incubated at 30°C for 30 min. The enzymatic reaction was initiated by adding 1 μL of FRET substrate Z-Phe-Arg-AMC (BACHEM, catalog #. 4003379.0050) (the final substrate concentration is about 5 μM for cathepsin K and 1 μM for cathepsin L). The reaction was monitored in a Cytation 5 image reader with filters for excitation at 360/40 nm and emission at 460/40 nm at 30°C for 1 hour. The IC50 values were calculated as described in the previous section.
Calpain 1 enzymatic assay was carried out as follows: 1 μL of 1 μg/μl calpain 1 protein was added to 100 μL calpain I reaction buffer [50 mM HEPES, 50 mM NaCl, and 10 mM DTT and 5 mM CaCl2 (pH 7.5)]; the enzymatic reaction was iniated by adding 1 μL of 4 mM N-succinyl-Leu-Leu-Val-Tyr-7-amido-4-methylcoumarin substrate (Sigma-Aldrich catalog #. S6510). The reaction was monitored a Cytation 5 image reader with filters for excitation at 360/40 nm and emission at 460/40 nm at 30°C for 1 hour. The IC50 values were calculated as described in the previous section.
Trypsin enzymatic assay was carried out in 50 μL volume containing 100 nM trypsin in, 50 mM HEPES (pH7.6) reaction buffer, 100 μM Bz-Arg-AMC·HCl (BACHEM, catalog no. 4002540.0050) and serial concentrations of test compounds in duplicates. The reaction was monitored a Cytation 5 image reader with filters for excitation at 360/40 nm and emission at 460/40 nm at 30°C for 1 hour. The IC50 values were calculated as described in the previous section.
Differential scanning fluorimetry (DSF).
The thermal shift binding assay (TSA) was carried out using a Thermal Fisher QuantStudio™ 5 Real-Time PCR System as described previously.7, 8, 29 Briefly, 3 μM SARS-CoV-2 Mpro protein in Mpro reaction buffer was incubated with various concentrations of compound ML188 or 23R at 30 °C for 30 min. 1X SYPRO orange dye was added and fluorescence of the well was monitored under a temperature gradient range from 20 °C to 90 °C with 0.05 °C/s incremental step. Measured Tm was plotted against compound concentration with one-site binding function in Prism 8.
Cytotoxicity measurement.
Evaluation of the cytotoxicity of compounds were carried out using the neutral red uptake assay.30, 31 Briefly, 80,000 cells/mL of the tested cell lines were dispensed into 96-well cell culture plates at 100 μL/well. Twenty-four hours later, the growth medium was removed and washed with 150 μL PBS buffer. 200 μL fresh serum-free medium containing serial diluted compounds was added to each well. After incubating for 5 days at 37 °C, the medium was removed and replaced with 100 μL DMEM medium containing 40 μg/mL neutral red and incubated for 2–4 h at 37 °C. The amount of neutral red taken up was determined by measuring the absorbance at 540 nm using a Multiskan FC Microplate Photometer (Fisher Scientific). The CC50 values were calculated from best-fit dose response curves with variable slope in Prism 8.
Immunofluorescence assay.
Antiviral immunofluorescence assay was carried out as previously described.8 Briefly, Vero E6 cells in 96-well plates (Corning) were infected with SARS-CoV-2 (USA-WA1/2020 isolate) at MOI of 0.05 in DMEM supplemented with 1% FBS. Immediately before the viral inoculation, the tested compounds in a three-fold dilution concentration series were also added to the wells in triplicate. The infection proceeded for 48 h without the removal of the viruses or the compounds. The cells were then fixed with 4% paraformaldehyde, permeabilized with 0.1% Triton-100, blocked with DMEM containing 10% FBS, and stained with a rabbit monoclonal antibody against SARS-CoV-2 NP (GeneTex, GTX635679) and an Alexa Fluor 488-conjugated goat anti-mouse secondary antibody (ThermoFisher Scientific). Hoechst 33342 was added in the final step to counterstain the nuclei. Fluorescence images of approximately ten thousand cells were acquired per well with a 10x objective in a Cytation 5 (BioTek). The total number of cells, as indicated by the nuclei staining, and the fraction of the infected cells, as indicated by the NP staining, were quantified with the cellular analysis module of the Gen5 software (BioTek).
Antiviral assay in Calu-3 cells.
Calu-3 cells (ATCC, HTB-55) grown in Minimal Eagles Medium supplemented with 1% non-essential amino acids, 1% penicillin/streptomycin, and 10% FBS are plated in 384 well plates. The next day, 50 nL of drug suspended in DMSO is added as an 8-pt dose response with three-fold dilutions between test concentrations in triplicate, starting at 40 μM final concentration. The negative control (DMSO, n=32) and positive control (10 μM Remdesivir, n=32) are included on each assay plate. Calu3 cells are pretreated with controls and test drugs (in triplicate) for 2 hours prior to infection. In BSL3 containment, SARS-CoV-2 (isolate USA-WA1/2020) diluted in serum free growth medium is added to plates to achieve an MOI=0.5. Cells are incubated continuously with drugs and SARS-CoV-2 for 48 hours. Cells are fixed and then immunstained with anti-dsRNA (J2) and nuclei are counterstained with Hoechst 33342 for automated microscopy. Automated image analysis quantifies the number of cells per well (toxicity) and the percentage of infected cells (dsRNA+ cells/cell number) per well. SARS-CoV-2 infection at each drug concentration was normalized to aggregated DMSO plate control wells and expressed as percentage-of-control (POC=% Infection sample/Avg % Infection DMSO cont). A non-linear regression curve fit analysis (GraphPad Prism 8) of POC Infection and cell viability versus the log10 transformed concentration values to calculate EC50 values for Infection and CC50 values for cell viability. Selectivity index (SI) was calculated as a ratio of drug’s CC50 and EC50 values (SI = CC50/IC50).
Mpro crystallization and structure determination.
23R was added to 20 mg/mL SARS-CoV-2 Mpro to a final concentration of 1.75 mM and incubated overnight at 4°C. This mixture was then diluted four-fold with protein stock buffer (20 mM Tris pH 7.5, 200 mM NaCl, 1 mM DTT) then spun down at 13,000 × g for 1 min to remove precipitate. Crystals were grown by mixing the protein-inhibitor sample with an equal volume of crystallization buffer (20% PEG 3350, 0.2 M NaF) in a vapor diffusion, hanging drop apparatus. Crystals were then transferred to a drop with crystallization buffer containing 5 mM 23R for 1 h, followed by a brief soaking in a cryoprotectant solution of 30% PEG 3350 and 15% glycerol with 2 mM 23R. Crystals were then flash frozen in liquid nitrogen for X-ray diffraction.
X-ray diffraction data for the SARS-CoV-2 Mpro structures were collected on the SBC 19-ID beamline at the Advanced Photon Source (APS) in Argonne, IL, and processed with the HKL3000 software suite. The CCP4 versions of MOLREP32 was used for molecular replacement using a previously solved SARS-CoV-2 Mpro structure, 6YB7. Structural refinement was performed using REFMAC533 and COOT.34 The crystallographic statistics is shown in Supplementary Materials table S2. The complex structure for SARS-CoV-2 Mpro with 23R has been deposited in the Protein Data Bank with the accession ID of 7KX5 (SARS-CoV-2 MPro + Jun8-76-3A).
Supplementary Material
ACKNOWLEDGEMENTS
This research was partially supported by the National Institutes of Health (NIH) (Grants AI147325 and AI157046) and the Arizona Biomedical Research Centre Young Investigator grant (ADHS18-198859) to J. W. The antiviral assay in Calu-3 cells was conducted through the NIAID preclinical service under a non-clinical evaluation agreement. J. A. T. and M. T. M. were funded by the National Institute of General Medical Sciences and National Institutes of Health (Grant R35 GM128624 to M. T. M.). We thank Michael Kemp for assistance with crystallization and X-ray diffraction data collection. We also thank the staff members of the Advanced Photon Source of Argonne National Laboratory, particularly those at the Structural Biology Center (SBC), with X-ray diffraction data collection. SBC-CAT is operated by UChicago Argonne, LLC, for the U.S. Department of Energy, Office of Biological and Environmental Research under contract DE-AC02-06CH11357. The SARS-CoV-2 experiments were supported by a COVID-19 pilot grant from UTHSCSA and NIH grant AI151638 to Y.X. SARS-Related Coronavirus 2, Isolate USA-WA1/2020 (NR-52281) was deposited by the Centers for Disease Control and Prevention and obtained through BEI Resources, NIAID, NIH.
ABBREVIATIONS
- SARS-CoV-2
severe acute respiratory syndrome coronavirus 2
- JAK
janus kinase
- Ugi-4CR
Ugi four-component reaction
- Mpro
main protease
- 3CLpro
3-chymotrypsin-like protease
- Nsp5
non-structural protein 5
- 2Apro
2A protease
- 3Cpro
3C protease
- PLpro
papain-like protease
Footnotes
Competing interests
Naoya Kitamura, Chunlong Ma, Yanmei Hu, and Jun Wang are inventors of a filed patent claiming the use of 23R and related compounds as potential COVID-19 antiviral drugs.
REFERENCES
- 1.Kalil AC; Patterson TF; Mehta AK; Tomashek KM; Wolfe CR; Ghazaryan V; Marconi VC; Ruiz-Palacios GM; Hsieh L; Kline S; Tapson V; Iovine NM; Jain MK; Sweeney DA; El Sahly HM; Branche AR; Regalado Pineda J; Lye DC; Sandkovsky U; Luetkemeyer AF; Cohen SH; Finberg RW; Jackson PEH; Taiwo B; Paules CI; Arguinchona H; Goepfert P; Ahuja N; Frank M; Oh MD; Kim ES; Tan SY; Mularski RA; Nielsen H; Ponce PO; Taylor BS; Larson L; Rouphael NG; Saklawi Y; Cantos VD; Ko ER; Engemann JJ; Amin AN; Watanabe M; Billings J; Elie MC; Davey RT; Burgess TH; Ferreira J; Green M; Makowski M; Cardoso A; de Bono S; Bonnett T; Proschan M; Deye GA; Dempsey W; Nayak SU; Dodd LE; Beigel JH; Members A-SG Baricitinib plus remdesivir for hospitalized adults with Covid-19. N. Engl. J. Med 2020, 384, 795–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gil C; Ginex T; Maestro I; Nozal V; Barrado-Gil L; Cuesta-Geijo MA; Urquiza J; Ramirez D; Alonso C; Campillo NE; Martinez A COVID-19: drug targets and potential treatments. J. Med. Chem 2020, 63, 12359–12386. [DOI] [PubMed] [Google Scholar]
- 3.Ullrich S; Nitsche C The SARS-CoV-2 main protease as drug target. Bioorg. Med. Chem. Lett 2020, 30, 127377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rut W; Groborz K; Zhang L; Sun X; Zmudzinski M; Pawlik B; Wang X; Jochmans D; Neyts J; Młynarski W; Hilgenfeld R; Drag M SARS-CoV-2 Mpro inhibitors and activity-based probes for patient-sample imaging. Nat. Chem. Biol 2020, 17, 222–228. [DOI] [PubMed] [Google Scholar]
- 5.Ghosh AK; Brindisi M; Shahabi D; Chapman ME; Mesecar AD Drug development and medicinal chemistry efforts toward SARS-coronavirus and Covid-19 therapeutics. ChemMedChem 2020, 15, 907–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boras B; Jones RM; Anson BJ; Arenson D; Aschenbrenner L; Bakowski MA; Beutler N; Binder J; Chen E; Eng H; Hammond J; Hoffman R; Kadar EP; Kania R; Kimoto E; Kirkpatrick MG; Lanyon L; Lendy EK; Lillis JR; Luthra SA; Ma C; Noell S; Obach RS; O’Brien MN; O’Connor R; Ogilvie K; Owen D; Pettersson M; Reese MR; Rogers T; Rossulek MI; Sathish JG; Steppan C; Ticehurst M; Updyke LW; Zhu Y; Wang J; Chatterjee AK; Mesecar AD; Anderson AS; Allerton C Discovery of a novel inhibitor of coronavirus 3CL protease as a clinical candidate for the potential treatment of COVID-19. bioRxiv 2020, 2020.09.12.293498, doi: 10.1101/2020.09.12.293498. [DOI] [Google Scholar]
- 7.Ma C; Sacco MD; Hurst B; Townsend JA; Hu Y; Szeto T; Zhang X; Tarbet B; Marty MT; Chen Y; Wang J Boceprevir, GC-376, and calpain inhibitors II, XII inhibit SARS-CoV-2 viral replication by targeting the viral main protease. Cell. Res 2020, 30, 678–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sacco MD; Ma C; Lagarias P; Gao A; Townsend JA; Meng X; Dube P; Zhang X; Hu Y; Kitamura N; Hurst B; Tarbet B; Marty MT; Kolocouris A; Xiang Y; Chen Y; Wang J Structure and inhibition of the SARS-CoV-2 main protease reveal strategy for developing dual inhibitors against M(pro) and cathepsin L. Sci. Adv 2020, 6, eabe0751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rathnayake AD; Zheng J; Kim Y; Perera KD; Mackin S; Meyerholz DK; Kashipathy MM; Battaile KP; Lovell S; Perlman S; Groutas WC; Chang KO 3C-like protease inhibitors block coronavirus replication in vitro and improve survival in MERS-CoV-infected mice. Sci. Transl. Med 2020, 12, eabc5332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Qiao J; Li YS; Zeng R; Liu FL; Luo RH; Huang C; Wang YF; Zhang J; Quan B; Shen C; Mao X; Liu X; Sun W; Yang W; Ni X; Wang K; Xu L; Duan ZL; Zou QC; Zhang HL; Qu W; Long YH; Li MH; Yang RC; Liu X; You J; Zhou Y; Yao R; Li WP; Liu JM; Chen P; Liu Y; Lin GF; Yang X; Zou J; Li L; Hu Y; Lu GW; Li WM; Wei YQ; Zheng YT; Lei J; Yang S SARS-CoV-2 M(pro) inhibitors with antiviral activity in a transgenic mouse model. Science 2021, 371, 1374–1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pedersen NC; Kim Y; Liu H; Galasiti Kankanamalage AC; Eckstrand C; Groutas WC; Bannasch M; Meadows JM; Chang KO Efficacy of a 3C-like protease inhibitor in treating various forms of acquired feline infectious peritonitis. J. Feline Med. Surg 2018, 20, 378–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim Y; Liu H; Galasiti Kankanamalage AC; Weerasekara S; Hua DH; Groutas WC; Chang KO; Pedersen NC Reversal of the progression of fatal coronavirus infection in cats by a broad-spectrum coronavirus protease inhibitor. PLoS Pathog 2016, 12, e1005531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jacobs J; Grum-Tokars V; Zhou Y; Turlington M; Saldanha SA; Chase P; Eggler A; Dawson ES; Baez-Santos YM; Tomar S; Mielech AM; Baker SC; Lindsley CW; Hodder P; Mesecar A; Stauffer SR Discovery, synthesis, and structure-based optimization of a series of N-(tert-butyl)-2-(N-arylamido)-2-(pyridin-3-yl) acetamides (ML188) as potent noncovalent small molecule inhibitors of the severe acute respiratory syndrome coronavirus (SARS-CoV) 3CL protease. J. Med. Chem 2013, 56, 534–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Turlington M; Chun A; Tomar S; Eggler A; Grum-Tokars V; Jacobs J; Daniels JS; Dawson E; Saldanha A; Chase P; Baez-Santos YM; Lindsley CW; Hodder P; Mesecar AD; Stauffer SR Discovery of N-(benzo[1,2,3]triazol-1-yl)-N-(benzyl)acetamido)phenyl) carboxamides as severe acute respiratory syndrome coronavirus (SARS-CoV) 3CLpro inhibitors: Identification of ML300 and noncovalent nanomolar inhibitors with an induced-fit binding. Bioorg. Med. Chem. Lett 2013, 23, 6172–6177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zaidman D; Gehrtz P; Filep M; Fearon D; Prilusky J; Duberstein S; Cohen G; Owen D; Resnick E; Strain-Damerell C; Lukacik P; Barr H; Walsh MA; von Delft F; London N An automatic pipeline for the design of irreversible derivatives identifies a potent SARS-CoV-2 Mpro inhibitor. bioRxiv 2020, 2020.09.21.299776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang LL; Lin DZ; Sun XYY; Curth U; Drosten C; Sauerhering L; Becker S; Rox K; Hilgenfeld R Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved alpha-ketoamide inhibitors. Science 2020, 368, 409–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang J; Hu Y; Foley C; Wang Y; Musharrafieh R; Xu S; Zhang Y; Ma C; Hulme C; Wang J Exploring ugi-azide four-component reaction products for broad-spectrum influenza antivirals with a high genetic barrier to drug resistance. Sci. Rep 2018, 8, 4653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Siklos M; BenAissa M; Thatcher GRJ Cysteine proteases as therapeutic targets: does selectivity matter? A systematic review of calpain and cathepsin inhibitors. Acta. Pharm. Sin. B 2015, 5, 506–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cianni L; Feldmann CW; Gilberg E; Gutschow M; Juliano L; Leitao A; Bajorath J; Montanari CA Can cysteine protease cross-class inhibitors achieve selectivity? J. Med. Chem 2019, 62, 10497–10525. [DOI] [PubMed] [Google Scholar]
- 20.Steuten K; Kim H; Widen JC; Babin BM; Onguka O; Lovell S; Bolgi O; Cerikan B; Cortese M; Muir RK; Bennett JM; Geiss-Friedlander R; Peters C; Bartenschlager R; Bogyo M Challenges for targeting SARS-CoV-2 proteases as a therapeutic strategy for COVID-19. ACS Infect. Dis 2021, doi: 10.1021/acsinfecdis.0c00815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ma Y; Li L; He S; Shang C; Sun Y; Liu N; Meek TD; Wang Y; Shang L Application of dually activated michael acceptor to the rational design of reversible covalent inhibitor for enterovirus 71 3C protease. J. Med. Chem 2019, 62, 6146–6162. [DOI] [PubMed] [Google Scholar]
- 22.Jin Z; Du X; Xu Y; Deng Y; Liu M; Zhao Y; Zhang B; Li X; Zhang L; Peng C; Duan Y; Yu J; Wang L; Yang K; Liu F; Jiang R; Yang X; You T; Liu X; Yang X; Bai F; Liu H; Liu X; Guddat LW; Xu W; Xiao G; Qin C; Shi Z; Jiang H; Rao Z; Yang H Structure of M(pro) from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [DOI] [PubMed] [Google Scholar]
- 23.Li J; Zhou X; Zhang Y; Zhong F; Lin C; McCormick PJ; Jiang F; Luo J; Zhou H; Wang Q; Fu Y; Duan J; Zhang J Crystal structure of SARS-CoV-2 main protease in complex with the natural product inhibitor shikonin illuminates a unique binding mode. Sci Bull (Beijing) 2021, 66, 661–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ma C; Hu Y; Townsend JA; Lagarias PI; Marty MT; Kolocouris A; Wang J Ebselen, disulfiram, carmofur, PX-12, tideglusib, and shikonin are nonspecific promiscuous SARS-CoV-2 main protease inhibitors. ACS Pharmacol. Transl. Sci 2020, 3, 1265–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gurard-Levin ZA; Liu C; Jekle A; Jaisinghani R; Ren S; Vandyck K; Jochmans D; Leyssen P; Neyts J; Blatt LM; Beigelman L; Symons JA; Raboisson P; Scholle MD; Deval J Evaluation of SARS-CoV-2 3C-like protease inhibitors using self-assembled monolayer desorption ionization mass spectrometry. Antiviral Res 2020, 182, 104924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Musharrafieh R; Ma C; Zhang J; Hu Y; Diesing JM; Marty MT; Wang J Validating enterovirus D68–2A(pro) as an antiviral drug target and the discovery of telaprevir as a potent D68–2A(pro) inhibitor. J. Virol 2019, 93, e02221–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cady SD; Wang J; Wu Y; DeGrado WF; Hong M Specific binding of adamantane drugs and direction of their polar amines in the pore of the influenza M2 transmembrane domain in lipid bilayers and dodecylphosphocholine micelles determined by NMR spectroscopy. J. Am. Chem. Soc 2011, 133, 4274–4284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marty MT; Baldwin AJ; Marklund EG; Hochberg GK; Benesch JL; Robinson CV Bayesian deconvolution of mass and ion mobility spectra: from binary interactions to polydisperse ensembles. Anal. Chem 2015, 87, 4370–4376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hu Y; Meng X; Zhang F; Xiang Y; Wang J The in vitro antiviral activity of lactoferrin against common human coronaviruses and SARS-CoV-2 is mediated by targeting the heparan sulfate co-receptor. Emerg. Microbes Infect 2021, 10, 317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Repetto G; del Peso A; Zurita JL Neutral red uptake assay for the estimation of cell viability/cytotoxicity. Nat. Protoc 2008, 3, 1125–1131. [DOI] [PubMed] [Google Scholar]
- 31.Ma C; Zhang J; Wang J Pharmacological characterization of the spectrum of antiviral activity and genetic barrier to drug resistance of M2-S31N channel blockers. Mol. Pharmacol 2016, 90, 188–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vagin A; Teplyakov A MOLREP: an automated program for molecular replacement. J. Appl. Crystallogr 1997, 30, 1022–1025. [Google Scholar]
- 33.Murshudov GN; Skubak P; Lebedev AA; Pannu NS; Steiner RA; Nicholls RA; Winn MD; Long F; Vagin AA REFMAC5 for the refinement of macromolecular crystal structures. Acta. Crystallogr. D 2011, 67, 355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Emsley P; Cowtan K Coot: model-building tools for molecular graphics. Acta Crystallogr. D 2004, 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.