

Abstract
Background:
Homologous recombination deficiency (HRD) score is related to chemotherapy response in some cancers, but its role in endometrial cancer in not known. We determined frequency and clinical significance of alterations in the HR pathway in endometrial cancer.
Methods:
253 endometrioid endometrial adenocarcinoma (EEA) samples from two independent cohorts (discovery and replication) were tested for HRD score using the Myriad HRD assay, microsatellite instability (MSI) and tumor mutation burden (TMB) using a next generation sequencing assay. HRD scores were also generated on endometrial cancer cell lines and in vivo response to olaparib was assessed.
Results:
ROC curves were employed to determine optimal cutoffs of HRD in relation to survival impact in endometrial cancer and a cutoff of HRD ≥ 4 was suggested for DFS using discovery cohort. Patients from two independent cohorts with HRD score ≥ 4 trended toward worse survival as compared to those with HRD score <4. Both cohorts were further separated into four groups according to molecular subtypes (TMB positive; MSI positive; HRD positive; all others). When grouped by molecular subtype, there was a significant difference between groups using an HRD ≥4 cutoff in the initial (p= 0.0024) and replication (p = 0.042) cohorts. The Hec1a model (HRD score = 19) was highly sensitive to olaparib in in vitro and in vivo experiments.
Conclusions:
High HRD score was associated with worse DFS in our patient cohort. These findings suggest that HRD score may have clinical utility in patients with advanced or recurrent endometrial cancer.
Keywords: HRD, uterine cancer, platinum, PARP inhibitors, BRCA1/2
INTRODUCTION
The Cancer Genome Atlas (TCGA) analysis of endometrial carcinoma samples provided an in depth view of the genomic characteristics of this disease[1]. PTEN is the most commonly mutated gene in endometrioid endometrial carcinomas[1, 2]. PTEN loss of function and ARID1A deficiency can impair DNA damage [3], [4]. Homologous recombination (HR) is one of the mechanisms by which DNA double strand breaks, as well as inter-strand crosslinks, are detected and repaired. The clinical implications of HR deficiency (HRD) in breast and ovarian cancer due to BRCA mutation are well-established; new pathways for HR deficiency continue to emerge.
Genomic instability secondary to HRD can be caused by a multitude of genetic aberrations and has many important contributors besides BRCA deficiency. Three independent DNA-based metrics have been developed to detect major contributors to genomic instability due to HRD: loss of heterozygosity (LOH)[5], telomeric allelic imbalance (TAI)[6], and large-scale state transition (LST) scores[7]. All three metrics are associated with platinum sensitivity in breast and ovarian cancer, and correlate with mutations in BRCA1 and BRCA2 as well as other HR pathway genes [8]. While each has clinical relevance separately, the sum of the three metrics (LOH+TAI+LST), known as the HRD score, is a more robust predictor of HR deficiency than each score alone in breast cancer patients [8, 9]. HRD score is a better predictor of platinum sensitivity than clinical variables or BRCA mutation status[9].
However, there is limited knowledge about the clinical significance of HR deficiency in endometrial cancer. Here, we examined the clinical significance of HR pathway alterations in endometrial cancer, and assessed the effects of HRD on tumor growth and response to DNA damaging drugs in vivo. Our data indicate that high HRD score is associated with worse disease free survival and HRD score predicts sensitivity to PARP inhibitors (PARPi) in vivo.
MATERIALS AND METHODS
Participants and tumor samples
Analysis of data from TCGA
Level 1 TCGA exome sequencing data for 499 uterine corpus endometrial carcinoma (UCEC) tumor-normal pairs were downloaded from the Cancer Genomics Hub. VarScan2 was used to make somatic mutation calls, including both single nucleotide polymorphisms and insertion-deletions. ANNOVAR was used to annotate the mutation points and extract the exonic alterations, including synonymous single nucleotide variants (SNV), non-synonymous SNV, stop gain SNV, non-frameshift insertions, non-frameshift deletions, frameshift insertions and frameshift deletions. Samples with non-synonymous SNV, stop gain SNV or insertions-deletions in a gene were defined as having mutations in that specific gene. For samples with more than one mutation site, insertions-deletions were considered to be the most severe category, followed by stop gain SNV, non-synonymous SNV, and synonymous SNV; each sample was assigned to the most severe category matched.
Analysis of data from uterine cancer samples from discovery and replication cohorts
253 EEA and non-EEA samples from two independent discovery and replication cohorts were tested for HRD score using the Myriad HRD assay, microsatellite instability (MSI) and tumor mutation burden (TMB) using a next generation sequencing assay. After the study was approved by the local IRB, 137 FFPE tumor samples (discovery cohort) were obtained from the MD Anderson Cancer Center (MDACC). Demographic, clinical data and response to treatment for recurrent disease was recorded. 116 samples from uterine cancer patients were included for the replication study in an independent replication cohort from the Inova Fairfax Hospital Institution and MDACC (Supplementary Table 1 and Table 2).
HRD score calculation
For each patient, one 5-micron slide and five 10-micron slides cut from FFPE primary endometrial tumor specimens were sent to Myriad Genetics, Inc (Salt Lake City, UT). A HE slide was reviewed by the pathologist to facilitate enrichment of tumor-derived DNA. DNA extraction was performed on 10-micron sections in the area of highest tumor cell density[9]. DNA was analyzed using one of two next-generation sequencing-based assays to generate a genome-wide single nucleotide polymorphism (SNP) profile from which the three components of the HRD score are calculated. A detailed description of the assays is available in Patel, et al[10]. In addition to the SNP probes, both panels also included probes targeting coding regions of 44 genes (AKT1, ATM, ATR, BAP1, BARD1, BRCA1, BRCA2, BRIP1, CDK12, CHEK1, CHEK2, CTNNB1, ERCC4, FAM175A, FANCA, FANCE, FANCI, FANCL, KRAS, MLH1, MRE11A, MSH2, MSH6, MUTYH, NBN, PALB2, PIK3CA, PPP2R2A, PTEN, RAD50, RAD51, RAD51B, RAD51C, RAD51D, RAD52, RAD54B, RAD54L, RPA1, TP53, TP53BP1, UBE2T, UIMC1, XRCC2, XRCC3).
MSI assay and Tumor mutation burden assay
A detailed description of these assays is available in the supplementary document.
In vitro and in vivo studies:
HRD scores were also generated on uterine cancer cell lines using Myriad HRD assay as described above and their in vivo response to olaparib was assessed (see details in the supplementary documents).
Statistical analysis
Continuous variables were compared with the two-sample t test (between two groups) or with one-way analysis of variance (ANOVA; for all groups) if normally distributed (as determined by the Kolmogrov-Smirnov test), and the Mann-Whitney test was used if distributions were non-parametric. A p-value of less than 0.05 from a two-tailed statistical test was considered statistically significant. All statistical tests were two-sided [11]. All p-values appearing on Kaplan-Meier curves are two-sided p-values from log-rank tests. HRD scores are grouped into non-continuous categories.
An initial HRD score cutoff was determined by examining the distribution of HRD scores in tumors with: somatic TP53 mutations; oncogene mutations; microsatellite instability (MSI); and tumor mutation burden (TMB). In addition, survival ROC curves and sensitivity and specificity analyses were employed to examine additional potential HRD cutoffs. Youden’s J index (sensitivity + specificity − 1) was used to determine a cutoff while giving equal weight to sensitivity and specificity[12].
RESULTS
Homologous recombination pathway alterations in endometrial cancer
Analysis of TCGA data
To determine the frequency and clinical significance of alterations in HR pathway genes in endometrial cancer, TCGA data for 499 patients with endometrial cancer were queried (Table 1). In the TCGA cohort, mutations in BRCA1 or 2 were present in 57 of the 373 EEA samples (15%). PTEN mutations were present in 286 of 373 EEA tumors (77%); concomitant BRCA1 or 2 mutation was present in 52 samples (14%; Figure 1a). In the subset of 57 patients who had BRCA mutations, 38 (67%) had stage I disease and the remainder had stage II disease or higher (Table 2).
Table 1:
Characteristics of the TCGA patient cohort (n = 499).
| EEA | nonEEA | ||
|---|---|---|---|
| Age >62 | No | 188 | 33 |
| Yes | 185 | 93 | |
| Clinical stage | Stage I | 267 | 47 |
| Stage II | 31 | 14 | |
| Stage III | 63 | 50 | |
| Stage IV | 12 | 15 | |
| Grade | G1 | 92 | 1 |
| G2 | 104 | 2 | |
| G3 | 177 | 123 |
EEA: endometrial endometriod adenocaricinoma; Non-EEA: serous-like and serous and mixed histology.
Figure 1. Clinical significance of BRCA1, BRCA2, PTEN and HRD distribution in the TCGA cohort.

A, Mutations in BRCA1, BRCA2 and PTEN in the TCGA endometrioid endometrial cancer (EEA) cohort (N=373). B, Kaplan-Meier of overall survival in the entire (including serous and mixed histology) TCGA cohort (N=498, one patient was excluded due to lack of survival data). C, Kaplan-Meier of overall survival in the TCGA EEC cohort (N=372, one patient was excluded due to lack of survival data). D. Kaplan-Meier of overall survival in the TCGA –Non EEA cohort (N=126). E. HRD score and distribution in EEA (N=271) and Non-EEA cohorts (N=64).
Table 2.
Distribution of BRCA mutations by stage in the TCGA endometrioid cohort (EEA tumors and nonEEA: serous-like+mixed tumors)
| Stage | EEA (%) | nonEEA(%) |
|---|---|---|
| I | 38 (66.7) | 3 (42.9) |
| II | 6 (10.5) | 3 (42.9) |
| III | 11 (19.3) | 1 (14.3) |
| IV | 2 (3.5) | 0 (0) |
| Total | 57 (100) | 7 (100) |
When BRCA and PTEN somatic mutations were examined in relation to overall survival in the TCGA cohort, patients classified by BRCA and PTEN somatic mutation status (wild-type; BRCA mutation only; PTEN mutation only; BRCA and PTEN mutations) showed no significant differences in survival (p=0.13, Figure 1b). When BRCA and PTEN somatic mutations were examined in relation to overall survival in TCGA cohorts of EEA tumors (N=372) and Non-EEA tumors (n=126), there was no significant difference in survival (p=0.37; Figure 1c; p=0.28; Figure 1d respectively).
To address the possible role of HR pathway alterations in endometrial cancer, we first identified a HRD score for 271 EEA samples, 12 mixed serous and endometrioid cancers and 52 serous endometrial cancers from TCGA data set. The median HRD score for EEA samples was 3, with a range of 0 to 83 (Figure 1E). The data set is skewed right and has a mean HRD score of 6.9. The median HRD score for the mixed and serous samples was 15.5 and 28.5 respectively, with a range of 1 to 55 (Figure 1E).
Analysis of two independent endometrial cancer cohorts (discovery and replication)
To further investigate the possible role of HR deficiency in endometrial cancer, we first utilized a discovery cohort of 137 patients enriched for endometrioid histology (Table 3). HRD score was determined for 112 samples and MSI status was determined for 125 samples; the remainder of the samples failed the analysis. TMB status was determined for all 137 samples. All three molecular tests were determined on 103 samples. The median HRD score was 3, with a range of 0 to 54 (Figure 2A). The data set is skewed right and has a mean HRD score of 5.45.
Table 3:
Characteristics of the discovery cohort (n=137).
| Characteristics | Number of patients (%) | |
|---|---|---|
| Age | ≤60 | 68 (50) |
| >60 | 68 (50) | |
| Stage | I | 74 (54) |
| II | 15 (11) | |
| III | 37 (27) | |
| IV | 11 (8) | |
| Grade | I | 2 (1) |
| II | 100 (73) | |
| III | 35 (26) | |
| Treatment | ||
| Chemotherapy | 65 (47.4) | |
| Stage | I | 14 (21.5) |
| II | 7 (10.8) | |
| III | 33 (50.7) | |
| IV | 11 (16.9) | |
| Radiation | 71 (51.8) | |
| Stage | I | 60 (84.5) |
| II | 8 (11.3) | |
| III | 3 (4.2) | |
| IV | 0 | |
| 1 (0.7) |
Figure 2. Clinical significance of HRD in the discovery cohort.
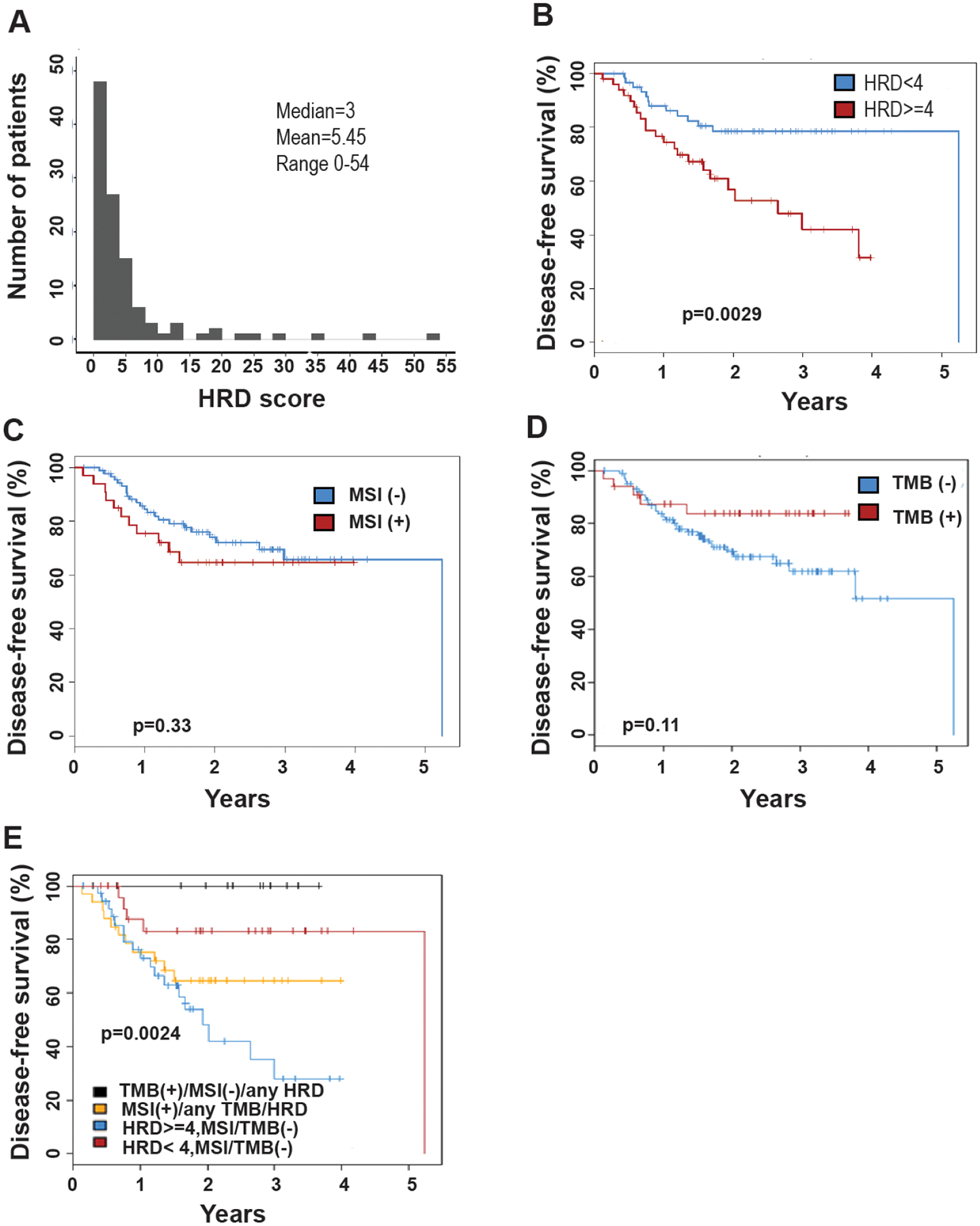
A, Histogram of HRD scores in the MD Anderson patient cohort (n=112). B, Kaplan-Meier plot of disease free survival, HRD score ≥4 vs HRD score <4. C, Kaplan-Meier plot of disease free survival, MSI-positive vs MSI -negative tumors. D, Kaplan-Meier plot of disease free survival, TMB-positive vs TMB-negative tumors. E, Kaplan-Meier plot of disease free survival in relation to HRD score ≥4 vs HRD score <4, MSI and TMB status for the entire MD Anderson cohort.
Unlike serous ovarian and serous endometrial cancer samples, the median HRD score for EEA samples was found to be 3. There was insufficient evidence of HR deficiency because BRCA1 or 2 mutations were predominantly monoallelic and the number of tumors with biallelic BRCA1 or 2 loss was insufficient to define an HRD score threshold. Therefore, we used ROC curves to determine optimal cutoffs of HRD score in relation to survival impact in EEA samples. The HRD score cutoffs and corresponding sensitivity and specificity are illustrated in supplementary Table 3. When giving equal weight to sensitivity and specificity, a cutoff of HRD score ≥ 4 was suggested using discovery cohort. When using an HRD score of 4 in this discovery cohort, there was statistically significant difference in survival between groups (p=0.0029, Figure 2B).
When testing for MSI, there was no statistically significant difference in survival between groups (p=0.33; Figure 2C). When testing for TMB, there was no significant difference in survival between patients with TMB positive tumors and TMB negative tumors (p=0.11; Figure 2D).
The cohort was then separated into four groups according to molecular subtype when the threshold of 4 was used to define HRD score. There was statistical significance in survival durations between the four groups (p = 0.0024) (Figure 2E). To further evaluate the possible role of HRD score on survival in endometrial cancer in the replication cohort (Figure 3A, Supplementary Table 1 and Table 2), patients with an HRD score ≥ 4 cut-off had significantly worse survival (p = 0.0011, Figure 3B). We also tested TMB and MSI in this replication cohort. There was no significant difference in disease free survival between the groups (p>0.05) (Figure 3C–3D). When grouped by molecular subtype, there was a significant difference between groups (TMB positive MSI positive; HRD score positive; All others) using an HRD score ≥ 4 cutoff (p = 0.042; Figure 3E) Overall, the replication study supports that high HRD score was associated with worse survival in endometrial cancer patients, and patients with HRD score ≥ 4 had worse DFS as compared to patients with HRD score <4.
Figure 3. Clinical significance of HRD in the replication cohort.

A, Histogram of HRD scores in the replication cohort (n=102). B, Kaplan-Meier plot of disease free survival, HRD score ≥4 vs HRD score <4. C, Kaplan-Meier plot of disease free survival in relation to MSI-positive vs MSI -negative tumors. D, Kaplan-Meier plot of disease free survival in relation to TMB-positive vs TMB-negative tumors. E, Kaplan-Meier plot of disease free survival in relation to HRD score≥4, MSI and TMB status for the entire replication cohort.
Mutation status was also assessed for both the discovery and replication cohorts. In the discovery cohort, 14/137 (10%) of all individuals had at least one somatic BRCA mutation, none of which had loss of heterozygosity (LOH). The majority of BRCA mutations were observed in tumors which tested positive for MSI, or had high TMB, and in MSI positive tumors the mutations tended to be deletions in simple repeats consistent with the tumors MSI positive status (Supplementary Table 4). The lack of biallelic loss, presence of hypermutation phenotypes, and low HRD scores in these BRCA mutant tumors would suggest that these tumors are not BRCA (or HR) deficient. 97 individuals (71%) had a PTEN mutation and 17 patients (12.4%) had a TP53 mutation.
Deleterious TP53 mutations were observed in 17/137 tumors from the discovery cohort and 9/116 tumors from the replication cohort. In both cohorts, there was significant association between TP53 mutation status and DFS (p = 0.031 and 0.03 respectively). TP53 mutations were frequently observed in tumors with MSI and/or high TMB scores. To determine whether DFS in TP53 mutation-positive tumors was related to MSI and/or TMB status, TP53 mutation positive tumors from both cohorts were combined (n =25) and the mutants classified as MSI+ and/or TMB+, or negative for both biomarkers. One individual was excluded due to unknown MSI/TMB status. A significant difference in DFS was observed between patients with the TP53 mutant tumors and patients with MSI/TMB positive tumors and TP53 mutations (p-value = 0.0091; Supplementary Figure 1).
HRD score predicts drug sensitivity of endometrial cancer cell lines
First, HRD scores were determined for 12 endometrial cancer cell lines (Figure 4A), and ranged from 2 to 24. Supplementary Table 5 demonstrates the BRCA1, BRCA2 and PTEN mutation status as obtained from the Cancer Cell Line Encyclopedia. The three cell lines (Hec1a, Hec1b, KLE) with highest HRD scores have wild-type BRCA1, BRCA2 and PTEN (Figure 4A–4C). To determine the sensitivity of the selected HRD-high or low cell lines to cisplatin, paclitaxel and olaparib, MTT and colony formation assays were performed with each drug. The three cell lines with the highest HRD scores (Hec1a, Hec1b and KLE, HRD>19) were more sensitive to all three agents tested than were the cell lines with relative low HRD score (Ishikawa, AN3CA, Hec265)(p<0.001, Figure 4D–4E). Olaparib was selected due to its known efficacy in the setting of HRD due to BRCA mutation. IC50 values ranged from 10.7 μM to 223.43 μM after olaparib treatment for 7 days (Figure 4E). Hec1a was the most sensitive cell line with IC50 of 11.8 μM, and Ishikawa was the most resistant with IC50 of 209.5 μM.
Figure 4: HRD predicts chemo-sensitivity of endometrial cancer cell lines.
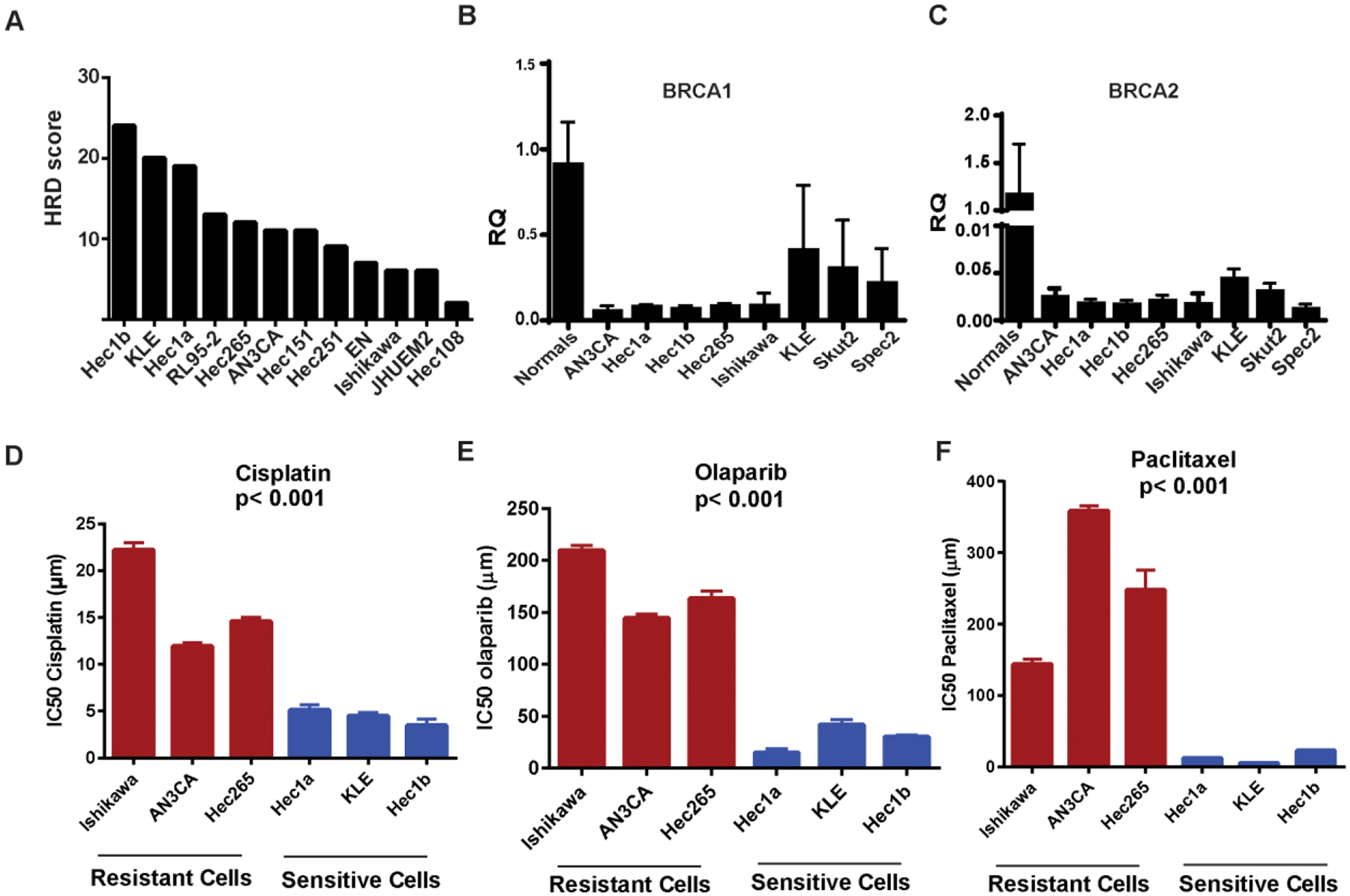
A, HRD scores of endometrial cancer cell lines. B and C, Expression of BRCA1 and 2 in endometrial cancer cell lines. D, Effect of cisplatin treatment on endometrial cancer cell lines. E, Effect of olaparib treatment on endometrial cancer cell lines. F, Effect of paclitaxel treatment on endometrial cancer cell lines. The three cell lines with the highest HRD scores (Hec1a, Hec1b and KLE) were significantly more sensitive to all three agents tested (cisplatin, paclitaxel and olaparib) than were the cell lines with low (Ishikawa) or intermediate HRD score (AN3CA, Hec265)(p<0.001).
Effect of olaparib on high HRD score orthotopic model
Hec1a was selected for in vivo (Supplementary Figure 2A – 5D) due to its high HRD score, wild-type BRCA status, and high sensitivity to olaparib. In the group receiving control siRNA, olaparib resulted in a significant reduction in tumor weight (p<0.001, Supplementary Figure 2A). In the group receiving BRCA1 siRNA, olaparib also resulted in a significant reduction in tumor weight (Supplementary Figure 2A). The same trend was observed in the number of tumor nodules (Supplementary Figure 2B). There was no significant change in mouse weight in any of the treatment groups (Supplementary Figure 2C).
In the group treated with control siRNA, the addition of olaparib to control siRNA resulted in significant reduction in Ki67 counts (Supplementary Figure 2E). In the group treated with control siRNA, the addition of olaparib to control siRNA resulted in significant increase in apoptosis as determined by cleaved caspase 3 (Supplementary Figure 2F). In the group treated with BRCA1 siRNA, addition of olaparib to BRCA1 siRNA resulted in significantly increased apoptosis.
Histone H2AX is phosphorylated in response to DNA double strand breaks and is an early marker for HR function [13–15]. Immunofluorescence staining for γ-H2AX was performed to detect possible differences in the number of double strand breaks between groups (Supplementary Figure 2G). Olaparib significantly decreased the number of γ-H2AX per cell. We determined the efficacy of BRCA1 silencing in the four treatment groups (Supplementary Figure 2H, top panel) using tumor samples obtained at the time of necropsy. In the groups treated with BRCA1 siRNA, there was fairly uniform silencing as shown by decreased protein expression. Differences within groups are likely due to tumor heterogeneity. We also determined RAD51 expression in all four groups using tumor samples obtained at the time of necropsy (Supplementary Figure 2H, middle panel). In the two groups treated with olaparib, there is decreased expression of RAD51 as compared to the untreated groups. When the left middle panel (siRNA only) is compared to the right middle panel (siRNA plus olaparib), there is decreased expression of RAD51 in the groups treated with olaparib.
DISCUSION
Our work demonstrates that a subset of patients with endometrial cancer who had high HRD scores have a trend toward poor DFS in comparison to patients with low HRD scores, which is in contrast to published platinum treated breast and ovarian studies where in both cases platinum treated patients with HRD high tumors showed improved outcome. One possible explanation is that the HRD high endometrioid tumors are more aggressive than their HRD low counterparts. While the biology supporting the use of PARP inhibitors in endometrial cancer is less robust than that in breast or ovarian cancer, our in vivo work and HRD analysis in patient samples suggests that in the setting of HR deficiency, PARP inhibitors may be beneficial, regardless of BRCA1 function.
The analysis provided here describes the molecular characteristics of a cohort of patients with advanced stage or high-intermediate risk endometrial cancer. Of particular interest, the three cell lines in our study with the highest HRD scores (Hec1a, Hec1b and KLE) have mutations in TP53. In endometrioid endometrial cancer, mutations in TP53 are more common with increasing grade[16]. The reported rate of TP53 mutations in endometrioid endometrial cancer is 11.4% in the TCGA analysis. In contrast, the rate of TP53 mutations in serous uterine carcinoma is approximately 90%[1], which is comparable to that of high grade serous ovarian cancer [17]. Likewise, approximately 25% of the high grade endometrioid samples in the TCGA analysis had mutational profiles similar to that of uterine serous carcinoma[1], which has a more aggressive clinical course[18]. Interpretation of mutation profiles in EEA is complicated by the high frequency of MSI positive and high TMB tumors as these tumors have high rates of somatic mutations, which tend to be monoallelic leaving one functional copy of the gene intact. The group of patients in our cohort with EEA tumors and HRD scores ≥ 10 appears to resemble uterine serous carcinoma, both biologically and clinically. Indeed, preclinical studies have shown that administration of PARP inhibitors can sensitize TP53 mutated breast cancer cells[19]. Clinical trials are already underway examining the role of PARP inhibitors in endometrial cancer (NCT02208375 and others). Furthermore, there are several specific subsets of patients with endometrial cancer that may benefit from HRD testing and use of the HRD score to triage into further therapy. First are the patients with endometrioid histology who have stage III or IV disease and/or grade 3 histology. Second, uterine serous tumors have a clinical course, mutational profile, and recommended treatment similar to that of high grade serous ovarian cancer, making this tumor type an ideal candidate for further study in this area[1, 17, 18].
In our patient cohort, BRCA1 and BRCA2 mutations were observed at a rate of 10% in the initial cohort and 15% in the validation cohort. However, the mutations were generally present in only one allele, indicating BRCA was likely functional. In addition, the majority (7/11) of these patients had HRD scores less than five, suggesting that HR deficiency was not present. Our initial analysis using TCGA data suggested that classifying by BRCA and PTEN mutation status did not have a significant impact on survival. Furthermore, our in vivo model demonstrates that whether or not BRCA1 is silenced, mice who received olaparib treatment had significantly less tumor growth than controls. The HRD score has been shown to be superior to both clinical variables and BRCA mutation status in identifying likely responders to platinum chemotherapy in breast cancer [5, 9], and there is ample evidence that mutations in genes other than BRCA1 and BRCA2 can produce clinically significant HR deficiency [20–22].
There are some limitations for this study, which include p53 mutation rate variation in the discovery and replication cohorts, mainly due to the tumor heterogeneity and small sample size, and lack of prospective validation of HRD markers in uterine cancer patients on PARP inhibitor trials. This is the first examination of the HRD score in a cohort of endometrial cancer patient samples, and of the HRD score in an in vivo model of endometrial cancer. Given that these tumors with high HRD score in vitro show good sensitivity to both PARP inhibitor and platinum, it is possible that patients with high -HRD score tumors may derive benefit from PARP inhibitor therapy. It is also possible that the threshold defined by survival is likely not optimal for drug sensitivity, which explains the disconnect between the tumor analysis and the in vivo dosing studies where some cell lines with HRD scores ≥4 show little sensitivity to PARP inhibitor or /platinum therapy. Moreover, mutations in genes other than BRCA1 and BRCA2 might produce clinically significant HR deficiency. A larger panel of genes related to genomic instability could be tested in this disease for future study. Whether HRD score could predict the response to PARP inhibitors warrants further validation on uterine cancer patients using matched tumor and blood samples.
Supplementary Material
Highlights.
Patients from two independent cohorts with HRD score ≥ 4 trended toward worse survival as compared to those with HRD score <4.
When grouped by molecular subtype, there was a significant difference between groups
Using an HRD ≥4 cutoff in the initial (p= 0.0024) and replication (p = 0.042) cohorts.
The Hec1a model (HRD score = 19) was highly sensitive to olaparib in in vitro and in vivo experiments.
These findings suggest that HRD score may have clinical utility in patients with advanced or recurrent endometrial cancer.
Acknowledgment
We thank Myriad Genetics, Inc. for performing the HRD score testing in uterine cancer patients.
Portions of this work were supported by the National Institutes of Health (P50 CA098258, P50 CA217685, CA 209904), the Frank McGraw Memorial Chair in Cancer Research, the American Cancer Society, and the Institutional Core Grant (CA16672) to MDACC. JMH was supported by a T32 Training Grant (T32CA101642) from the NIH/NCI. SW was supported by the Andrew Sabin Family Fellowship and an NRG Scholar Investigator Award.
Conflict of interest statement:
AKS: Consulting (Merck, Kiyatec); research funding (M-Trap); shareholder (BioPath). KMT, SG, PT and CN are the employees of Myriad Genetics, Inc. WH: Research funding (Geistlich Pharma AG).
SNW: Consulting (AstraZeneca, Clovis Oncology, Tesaro, Roche/Genentech, Novartis, Takeda, Merck, Pfizer, Circulogene); Research Funding (ArQule, AstraZeneca, Clovis Oncology, Tesaro, Roche/Genentech, Bayer, Cotinga Pharmaceuticals, Inc., Novartis).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Cancer Genome Atlas Research N, Kandoth C, Schultz N, Cherniack AD, Akbani R, Liu Y, et al. Integrated genomic characterization of endometrial carcinoma. Nature. 2013;497:67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Mutter GL, Lin MC, Fitzgerald JT, Kum JB, Baak JP, Lees JA, et al. Altered PTEN expression as a diagnostic marker for the earliest endometrial precancers. J Natl Cancer Inst. 2000;92:924–30. [DOI] [PubMed] [Google Scholar]
- [3].Ming M, He YY. PTEN in DNA damage repair. Cancer Lett. 2012;319:125–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Shen J, Peng Y, Wei L, Zhang W, Yang L, Lan L, et al. ARID1A Deficiency Impairs the DNA Damage Checkpoint and Sensitizes Cells to PARP Inhibitors. Cancer Discov. 2015;5:752–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Abkevich V, Timms KM, Hennessy BT, Potter J, Carey MS, Meyer LA, et al. Patterns of genomic loss of heterozygosity predict homologous recombination repair defects in epithelial ovarian cancer. Br J Cancer. 2012;107:1776–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Birkbak NJ, Wang ZC, Kim JY, Eklund AC, Li Q, Tian R, et al. Telomeric allelic imbalance indicates defective DNA repair and sensitivity to DNA-damaging agents. Cancer Discov. 2012;2:366–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Popova T, Manie E, Rieunier G, Caux-Moncoutier V, Tirapo C, Dubois T, et al. Ploidy and large-scale genomic instability consistently identify basal-like breast carcinomas with BRCA1/2 inactivation. Cancer Res. 2012;72:5454–62. [DOI] [PubMed] [Google Scholar]
- [8].Timms KM, Abkevich V, Hughes E, Neff C, Reid J, Morris B, et al. Association of BRCA1/2 defects with genomic scores predictive of DNA damage repair deficiency among breast cancer subtypes. Breast Cancer Res. 2014;16:475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Telli ML, Timms KM, Reid JE, Hennessy B, Mills GB, Jensen KC, et al. Homologous Recombination Deficiency (HRD) Score Predicts Response to Platinum-Containing Neoadjuvant Chemotherapy in Patients with Triple Negative Breast Cancer. Clin Cancer Res. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Patel JN, Braicu I, Timms KM, Solimeno C, Tshiaba P, Reid J, et al. Characterisation of homologous recombination deficiency in paired primary and recurrent high-grade serous ovarian cancer. Br J Cancer. 2018;119:1060–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Pradeep S, Huang J, Mora EM, Nick AM, Cho MS, Wu SY, et al. Erythropoietin Stimulates Tumor Growth via EphB4. Cancer Cell. 2015;28:610–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Youden WJ. Index for rating diagnostic tests. Cancer. 1950;3:32–5. [DOI] [PubMed] [Google Scholar]
- [13].Pilch DR, Sedelnikova OA, Redon C, Celeste A, Nussenzweig A, Bonner WM. Characteristics of gamma-H2AX foci at DNA double-strand breaks sites. Biochem Cell Biol. 2003;81:123–9. [DOI] [PubMed] [Google Scholar]
- [14].Celeste A, Petersen S, Romanienko PJ, Fernandez-Capetillo O, Chen HT, Sedelnikova OA, et al. Genomic instability in mice lacking histone H2AX. Science. 2002;296:922–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Maser RS, Monsen KJ, Nelms BE, Petrini JH. hMre11 and hRad50 nuclear foci are induced during the normal cellular response to DNA double-strand breaks. Mol Cell Biol. 1997;17:6087–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Lax SF, Kendall B, Tashiro H, Slebos RJ, Hedrick L. The frequency of p53, K-ras mutations, and microsatellite instability differs in uterine endometrioid and serous carcinoma: evidence of distinct molecular genetic pathways. Cancer. 2000;88:814–24. [PubMed] [Google Scholar]
- [17].Cancer Genome Atlas Research N. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Moore KN, Fader AN. Uterine papillary serous carcinoma. Clin Obstet Gynecol. 2011;54:278–91. [DOI] [PubMed] [Google Scholar]
- [19].Munoz-Gamez JA, Martin-Oliva D, Aguilar-Quesada R, Canuelo A, Nunez MI, Valenzuela MT, et al. PARP inhibition sensitizes p53-deficient breast cancer cells to doxorubicin-induced apoptosis. Biochem J. 2005;386:119–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Casadei S, Norquist BM, Walsh T, Stray S, Mandell JB, Lee MK, et al. Contribution of inherited mutations in the BRCA2-interacting protein PALB2 to familial breast cancer. Cancer Res. 2011;71:2222–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Jenner ZB, Sood AK, Coleman RL. Evaluation of rucaparib and companion diagnostics in the PARP inhibitor landscape for recurrent ovarian cancer therapy. Future Oncol. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Liu FW, Tewari KS. New Targeted Agents in Gynecologic Cancers: Synthetic Lethality, Homologous Recombination Deficiency, and PARP Inhibitors. Curr Treat Options Oncol. 2016;17:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.