

Abstract
Nearly 60 years have passed since the initial discovery by Hubel and Wiesel that changes in neuronal activity can elicit developmental rewiring of the central nervous system (CNS). Over this time, we have gained a more comprehensive picture of how both spontaneous neural activity and sensory experience-induced changes in neuronal activity guide CNS circuit development. Here, we review activity-dependent synaptic pruning in the mammalian CNS, which we define as the removal of a subset of synaptic components, while others are maintained, in response to changes in neural activity in the developing nervous system. We discuss the mounting evidence that immune and cell death molecules are important mechanistic links by which changes in neural activity guide the pruning of specific synapses, emphasizing the role of glial cells in this process. Finally, we discuss how these developmental pruning programs may go awry in neurodevelopmental disorders of the human CNS with a focus on autism spectrum disorders (ASD) and schizophrenia (SZ). Together, we intend to give an overview of how the field of activity-dependent pruning has evolved, has led to exciting new questions, and has guided the identification of new, therapeutically-relevant mechanisms by which circuit development can go awry in neurodevelopmental disorders.
Introduction
The developing nervous system undergoes remarkable remodeling to achieve the highly precise wiring diagram characteristic of mature neural circuits. This precision is achieved through pruning – an extensive process in which a large subset of axons, dendrites, and synapses that initially form in abundance are eliminated over the course of development throughout the nervous system. While it is important to recognize the vast literature on large scale pruning of axons and dendrites in vertebrate and invertebrate peripheral and central nervous systems (CNS)1–4, this review will focus on developmental pruning of synapses in the mammalian CNS. We define synapse pruning as a developmental process in which elements that comprise a bona fide structural synapse (presynaptic terminals and postsynaptic membranes) are eliminated, which may also include the removal of small segments of axonal and dendritic branches. Concomitant with elimination, remaining synapses are maintained, strengthened, and elaborated. We will review literature demonstrating roles for neural activity in developmental synapse pruning and literature that has guided a new understanding of molecular mechanisms that may link changes in activity with the physical removal of some synapses and maintenance and strengthening of others. These mechanisms include emerging new roles for immune signaling and cell death molecules in synapse removal, which can be neuron-intrinsic but can also include neuron-extrinsic, glia-driven mechanisms. Finally, we will highlight how lessons we have learned by studying synapse pruning in the healthy, developing nervous system have informed mechanisms of neurodevelopmental disorders with a focus on autism spectrum disorders (ASD) and schizophrenia (SZ).
Activity-dependent synaptic pruning: a historical perspective
Neuronal activity plays a central role in developmental synapse pruning, which involves weakening and elimination of synaptic connections and strengthening and maintenance of others5–7. In mammals, activity-dependent pruning occurs throughout the CNS at different developmental periods, depending on the brain region and neuron subtype. In this section, we will focus on model circuits that have been extensively studied in the context of activity-dependent synaptic pruning, given the stereotyped nature of the pruning and the experimental tractability of each circuit (Fig.1). Either through modulating spontaneous neural activity or sensory-evoked activity, studies of these circuits have taught us guiding principles by which neuronal activity drives synaptic pruning during development. They have also led to the identification of molecules that link changes in activity with the physical elimination of synapses, which we outline in the section below as well as in following sections.
Figure 1. Model circuits for studying activity-dependent synaptic pruning.

The classic models for studying synaptic pruning in the mammalian CNS include developmental remodeling at the retinogeniculate circuit (a), cerebellum (b), and cortical synapses (c). Within the retinogeniculate circuit (a), retinal ganglion cell (RGC) axonal arbors initially form synapses with relay neurons in overlapping territories within the lateral geniculate nucleus (LGN) (far left panel in a). Prior to eye opening (Birth-P10), spontaneous neuronal activity in the retina results in pruning of RGC synapses (dotted red and blue lines) and eye-specific segregation so that each eye occupies a discrete territory prior to eye opening (middle panel in a)8–19,233. Relay neuron synapses undergo further remodeling after eye opening in response to light exposure from P20-P30 when relay neuron spines are pruned (far right panel, green)60–62. In the cerebellum (b), Purkinje cell somas are initially innervated by multiple climbing fiber inputs (far left panel in b). In the first stage of pruning (2nd panel in b), the weaker climbing fiber inputs at the soma are pruned, while the strongest somatic input translocates to the Purkinje dendrites. In the second stage of pruning (3rd panel in b), remaining climbing fiber inputs to the soma are pruned, while the single climbing fiber input at the Purkinje dendritic arbor is maintained22,23. In the cortex (c), changes in sensory experience such as monocular deprivation or whisker manipulation (right panel in c) results in pruning of thalamocortical presynaptic terminals (red, dotted lines) and cortical dendritic spines (blue) in response to the changes in neuronal activity43–52,59,63–65.
Spontaneous neural activity-driven synapse pruning
Among the first studies to identify roles for neural activity in synaptic pruning in the CNS were those in the retinogeniculate circuit, examining synapses from retinal ganglion cells (RGCs) onto relay neurons within the lateral geniculate nucleus (LGN) of the thalamus (Fig. 1a). Initially, right and left eye RGC inputs form exuberant synapses onto relay neurons within overlapping territories of the LGN. Before eye opening (<P10 in mice), waves of spontaneous activity in the retina drive the segregation of RGC inputs into eye-specific territories in the LGN (i.e. eye-specific segregation). This process involves elimination of a subset of presynaptic inputs and strengthening and maintenance of others8–20. Appropriate refinement of eye-specific territories requires that spontaneous activity between neurons of the same retina is synchronized, while activity between the two retinas must be asynchronous17. If spontaneous activity is blocked in both eyes during eye-specific segregation or synchronized through optogenetic stimulation, RGC inputs fail to prune into eye-specific territories11,19,21. If spontaneous activity is blocked or increased in only one eye, synaptic territory from the less active RGCs is reduced and territory from the active RGCs is expanded11,20. If asynchronous activity between the two eyes is driven by optogenetics earlier in development, eye-specific segregation is achieved earlier21. This circuit, thus, supports a model in which spontaneous neuronal activity drives a Hebbian competition between synapses. The relative timing of activity between the two eyes and within a given eye is critical to dictate the elimination of some synapses and strengthening and maintenance of others.
Spontaneous neural activity also regulates pruning in the developing cerebellum (Fig. 1b). Initially, multiple climbing fibers from the inferior olivary nucleus synapse on Purkinje cell somas. By ~P11 in mice, many somatic climbing fiber synapses are eliminated and a ‘winning’ climbing fiber translocates and synapses on the Purkinje cell dendrites22,23. In a second stage (~P12-P17), any remaining somatic climbing fiber synapses are pruned away. Similar to the retinogeniculate circuit, Hebbian-type plasticity is required for climbing fiber pruning. This includes strengthening of a single climbing fiber input, while other inputs weaken and are eliminated24. This selective strengthening requires postsynaptic activity as the climbing fiber input most synchronous to the Purkinje neuron burst output becomes the “winner”25–29. Increased GABAergic innervation onto the Purkinje cell soma from cerebellar basket cells further drives the relative weakening of the “losing” climbing fiber inputs during the first stage of pruning30. During the later stage of pruning, signaling downstream of metabotropic glutamatergic receptors (mGluR1) in Purkinje cell dendrites drives the pruning of any remaining somatic climbing fiber synapses31–36.This mGluR1 signaling stimulates the expression of membrane tethered semaphorin Sema7a37,38 and the release of brain-derived neurotropic factor (BDNF)39 from Purkinje neurons onto remaining somatic climbing fiber synapses to facilitate their removal.
In addition to signals that lead to the elimination of ‘losing’ climbing fibers, other molecules stabilize the ‘winning’ climbing fibers. For example another semaphorin, Sema3A, is secreted from Purkinje neurons to promote the stabilization and maturation of ‘winning’ climbing fibers throughout all stages of pruning37. Acting in parallel to Sema3A is progranulin, which is derived from Purkinje cells and binds climbing fibers via Sort1 to promote climbing fibers stabilization and strengthening40. Finally, it is important to consider the influence of glia. Wrapping of climbing fiber synapses by specialized cerebellar astrocytes called Bergmann glia is required to strengthen the ‘winning’ synapses and prevent exuberant climbing fiber innervation along Purkinje cell dendrites during later pruning stages41. There are additional influences of microglia to promote weakening of a subset of climbing fiber synapses, which is discussed under immune-mediated synaptic pruning42,30. Together, the retinogeniculate and the cerebellar climbing fiber circuits demonstrate how differential activity in neurons drives a competitive pruning process. Cerebellar climbing fiber pruning has provided further insight into activity-dependent molecules that drive this competitive process.
Experience-driven changes in neural activity and synapse pruning
The effects of neural activity on synapse pruning have also been studied through the modulation of sensory experience. First demonstrated by Hubel and Weisel43 in the cat visual cortex, sensory deprivation caused by suturing one eye closed during development (i.e. monocular deprivation) results in the weakening of synapses corresponding to the sutured eye and strengthening of synapses corresponding to the open eye. This process is termed ocular dominance plasticity. Studies in rodents have since revealed a decrease in thalamocortical arborization and a reduction in postsynaptic dendritic spines corresponding to the deprived eye, while thalamocortical projections from the open eye increase their arborization and synaptic territory (Fig. 1c)44–52. Linking changes in activity with structural plasticity is evidence that long-term depression (LTD) also occurs during monocular deprivation53,54, which has previously been linked to spine shrinkage and pruning in the hippocampus55–57. Also, GABAergic innervation of cortical pyramidal neurons within the visual cortex by parvalbumin (PV)+ interneurons is required to detect spatiotemporal differences in thalamocortical inputs during monocular deprivation and is necessary for subsequent spine pruning58,59. Downstream of GABAergic transmission during monocular deprivation is the activation of the proteolytic enzyme tissue-type plasminogen activator (tPA), which is proposed to remodel the extracellular matrix to accommodate spine remodeling59. Similar to the visual cortex, visual experience also affects a later stage of synapse remodeling (P20-P30) in the retinogeniculate circuit when RGC synapses in the LGN consolidate and strengthen, a process that requires visual experience60. While some of this remodeling involves clustering of presynaptic terminals along a RGC arbor61, it also involves pruning of dendritic spines in the LGN62.
Experience-dependent synaptic pruning has also been well-documented in the somatosensory system. For example, whisker deprivation leads to dampened neural activity in the barrel cortex and the elimination of thalamocortical presynaptic inputs onto layer IV cortical neurons in developing rodents63–65. This also occurs in adult rodents, albeit less robustly63–65. However, there are also reports of increased spine density within the barrel cortex following whisker deprivation66,67, possibly due to an increase in cortico-cortical synapses. It is notable that synapse pruning occurs with increased activity in the somatosensory cortex, too. For example, stimulating whisker activity through environmental enrichment accelerates thalamocortical synapse maturation and pruning68. Data suggests that spines compete for synapse-stabilizing cadherin/catenin complexes when activity is increased during whisker stimulation. Upstream of the barrel cortex in this circuit is the ventral posteromedial nucleus (VPM) of the thalamus, which also undergoes experience-dependent pruning. Sensory information originating from the whisker pad is conveyed to the VPM through synapses originating from the brainstem principal trigeminal nucleus (PrV). As active whisking begins (~P14 in mice), a subset of whisker-specific inputs in the VPM are strengthened while others are pruned away69. Evidence suggests that astrocytic calcium signaling induces purinergic signaling, which then impinges on P2Y1 receptors on VPM neurons to facilitate the pruning of a subset of synapses over development70.
Together, these studies emphasize the importance of activity for driving the selective elimination and stabilization of synaptic sites. While the model circuits described above established fundamental principles of activity driven synapse pruning in mammalian circuits, there are still major gaps in our knowledge. To what extent does pruning occur throughout the entire CNS? Are all neurons and circuits pruned or is pruning limited to a subset of circuits or neurons? If so, why? A major impediment to this progress is that the developmental timing of this pruning is different across CNS regions. In addition, measuring the timing and neuron subtype undergoing pruning throughout the CNS is a major undertaking and work in one species may not necessarily align with another species. In many complex circuits, including the cortex, it is also often difficult to discern the cellular source of presynaptic inputs for each synapse, which is needed to selectively modulate activity during pruning. Instead, it has been more tractable to focus on these classic model circuits where the pre- and post-synaptic partners are clearly defined. These model circuits have further paved the way to identifying exciting new molecular mechanisms that are activity-dependent and regulate synaptic pruning (discussed below).
Immune-mediated synaptic pruning
Besides the mechanisms described above, molecules canonically involved in immune system function have also been identified as key regulators of developmental, activity-dependent synaptic pruning. In some cases, these immune molecules and cells regulate pruning in a fashion very similar to how they function in the immune system71. We give a historical perspective of these findings and highlight the most recent work outlining key roles for these molecules in shaping developing neural circuits below.
Neuron-intrinsic immune signaling regulating synaptic pruning
Excitement for immune-mediated synaptic pruning began with the discovery of activity-dependent regulation of class I major histocompatibility complex antigens (MHC class I) in the LGN72. Later, it was shown that that MHC class I molecules were neuronally expressed73. Further, mice lacking all cell surface MHC class I expression (β2M−/− or TAP 1−/− mice), mice lacking the MHC receptor component CD3ζ, and mice deficient in 2 of the 50 MHC class I genes (KbDb−/− mice) all had deficits in retinogeniculate eye-specific segregation73,74. Mice deficient in MHC class I genes or the MHC receptor PirB also had enhanced ocular dominance plasticity, diminished LTD73,75,76, and elevated spine density in the visual cortex76. While these results demonstrate a role for MHC molecules and receptors in pruning, it is important to consider that they may also affect initial synapse formation, as suggested by a study showing elevated synaptogenesis in cultured β2M−/− neurons77. It also remains unclear how MHC class I-receptor signaling leads to pruning78. In the immune system, these molecules are key factors involved in antigen presentation, which activates neighboring adaptive immune cells to initiate destruction of foreign antigens, such as viruses. The T cell receptors that bind MHC I complexes are not expressed in the CNS, but the coreceptors (CD3ζ and CD3ε) are present. The natural killer cell receptors for MHC class I molecules, PirB and Ly49, are expressed in the CNS, but exactly how MHC Class I interacts with these receptors to regulate pruning is not known. MHC Class I-mediated pruning could represent a separable phenomenon from its function in the immune system, but MHC Class I presentation could also interact with other immune molecules, like neuronal pentraxins and complement proteins (discussed below).
Other neuronal immune-related molecules called neuronal pentraxins also regulate synapse pruning. Pentraxins are cyclic multimeric proteins involved in calcium-dependent ligand binding and innate immune response79. Neurons express the neuronal pentraxin receptor (NPR) and also neuronal pentraxin 1 (NP1) and neuronal pentraxin 2 (NP2/Narp). These neuronal pentraxins share 20–30% homology with short pentraxins in the peripheral immune system, but they have different tertiary structure and little evidence supports roles for them in innate immunity80–82. NP1/NP2 complexes regulate AMPA receptor clustering and synapse plasticity83 and knockdown of NP1 in cultured rat neurons results in increased numbers of synapses84. Mice lacking NP1 and NP2 also have deficits in eye-specific segregation in the retinogeniculate circuit, implicating these molecules in pruning of presynaptic terminals85. In peripheral immunity, other pentraxin molecules can activate the classical complement cascade through C1q and play a role in phagocytosis86,87. One possibility is that neuronal pentraxins and MHC class I molecules could be working together with complement to mediate synapse pruning via microglia (discussed in the following section). Indeed, very recent work in adult mice has shown that NPs co-immunoprecipitate with C1q in cortex-derived synaptosomes and that these molecules are co-localized in vivo at cortical synapses88, but it remains unknown if these interactions occur during developmental synapse pruning.
Microglial-dependent immune signaling and synapse pruning
Microglia are resident macrophages of the CNS. Similar to other tissue resident macrophages, microglia are efficient phagocytes and key regulators of innate immune signaling in the CNS. The first evidence that microglia were involved in synaptic pruning was provided by work in the developing hippocampus, visual cortex, and retinogeniculate circuit51,89,90. These studies revealed that microglia engulfed synaptic components. In the visual cortex and retinogeniculate circuit, developmental synapse pruning by microglia is regulated by activity as microglia engulf synapses from less active neurons51,90,91. Microglial engulfment of synapses has also now been observed in other developing brain regions such as the auditory brainstem, auditory cortex, primary and secondary somatosensory cortices, and nucleus accumbens65,92–95.
Despite mounting evidence that microglial engulfment mechanisms underlie activity-dependent synaptic pruning, it remains to be fully deciphered whether microglia more passively “clean up” synaptic debris or play a more active role in initiating synapse pruning. Arguing for the latter, live and static imaging in rodents, and now Xenopus, have demonstrated that microglia “trogocytose” (trogo-: nibble) intact presynaptic membranes but not postsynaptic structures96,97, which is consistent with other work showing selective phagocytosis of presynaptic membranes65,90. Further arguing for a more active role, multiple groups have shown that disrupting microglial engulfment results in sustained increases in numbers of structural and functional synapses65,89,90,94,98–100. For example, mice lacking the fractalkine receptor CX3CR1 (Cx3cr1−/−), a G protein-coupled chemokine receptor (GPCR) highly enriched in microglia in the healthy CNS101, have delayed microglial infiltration into the hippocampus concomitant with delays in synapse maturation and a transient elevation in spine density89. It was also shown that adolescent to early adult Cx3cr1−/− mice had increased excitatory synaptic connectivity in the hippocampus and defects in sociability99. More recently, CX3CR1 signaling was shown to directly impact microglial synapse engulfment and activity-dependent pruning of presynaptic terminals, but not postsynaptic structures, in the somatosensory cortex following neonatal whisker lesioning65. Following whisker removal, microglia engulf and remove thalamocortical presynaptic inputs in the neonate somatosensory cortex and this process was blocked in Cx3cr1−/− mice and in mice lacking the canonical ligand fractalkine (CX3CL1). It was further shown that whisker removal elicited an increase in a metalloprotease ADAM10 that cleaves CX3CL1 into a secreted form. Pharmacological inhibition of ADAM10 phenocopied the synapse engulfment and pruning defects in Cx3cr1−/− and Cx3cl1−/− mice. Together, these results suggest a mechanism by which CX3CL1 is cleaved by ADAM10 in an activity-dependent manner to bind microglial CX3CR1 and induce pruning of presynaptic terminals (Fig. 2b). In the immune system, the canonical function for CX3CR1 is to regulate recruitment of myeloid cells102. However, in the barrel cortex following whisker ablation, microglial recruitment is unaffected and only engulfment is blocked in Cx3cr1−/− and Cx3cl1−/− mice. Interestingly, CX3CR1 is required for phagocytosis of necrotic fibers after acute skeletal muscle injury by macrophages in vivo and phagocytosis of apoptotic cells by bone marrow-derived macrophages in vitro103,104. Still, the downstream signaling of this GPCR, which leads to regulation of engulfment, is an open question.
Figure 2. Summary of glial cell engulfment mechanisms regulating synaptic pruning.

Multiple glial engulfment pathways have been identified to regulate synaptic pruning (a). A mechanism by which microglia prune synapses is through CX3CR1-CX3CL1-ADAM10 signaling (b). Neuron-expressed CX3CL1 is cleaved from the membrane by the protease ADAM10 which initiates signaling to its microglial receptor CX3CR1. CX3CR1 signaling instructs microglial synapse engulfment by a, yet-to-be identified mechanism65. Another mechanism involved in microglial synapse pruning is the complement signaling cascade. C1q induces the formation of the C3 convertase through complement factors C2 and C4. C3 convertase then cleaves and activates C3, which then directs microglia to engulf synapses via CR3 (c)90,98. In contrast, CD47 binding to its receptor SIRPα on microglia inhibits engulfment of another subset of synapses108. Similarly, the complement inhibitor SRPX2 binds C1q at the membrane in order to prevent C1q-mediated synapse engulfment and pruning109. Astrocytes also play roles in synapse pruning through the phagocytic receptors MEGF10 and MerTK. MerTK may directly bind externalized PtdSer at the synaptic membrane (d)120. Further, astrocyte-microglia crosstalk has been implicated in the pruning process (e). For example, astrocyte production of TGFβ directs C1q production by RGCs to influence microglial pruning100. Also, astrocytic IL33 expression signals to its microglial receptor IL1RL1 and instructs microglial synapse engulfment and pruning via a, yet-to-be-identified, downstream mechanism124. Cluster of differentiation 47 (CD47), signal regulatory protein α (SIPRα), sushi repeat containing protein X-linked 2 (SRPX2), transforming growth factor β (TGFβ), multiple epidermal growth factor-like domains protein 10 (MEGF10), Mer proto-oncognene tyrosine kinase (MerTK), phosphatidylserine (PtdSer), a disintegrin and metalloproteinase domain-containing protein 10 (ADAM10), C-X3-C motif chemokine ligand 1 (CX3CL1), C-X3-C motif chemokine receptor 1 (CX3CR1), interleukin 33 (IL33), interleukin 1 receptor like 1 (IL1RL1), complement receptor 3 (CR3), complement component 3 (C3), complement component 1q (C1q), complement component 4 (C4), complement component 2 (C2).
Besides CX3CR1, the classical complement cascade also regulates microglial synapse engulfment and pruning (Fig. 2c). In the innate immune system, the initiating molecule C1q induces the formation of the C3 convertase through complement factors C2 and C4. This C3 convertase then cleaves and activates C3, which then binds pathogens and cellular debris to induce cell lysis or clearance by phagocytic cells (reviewed in105,106). Similarly, C1q and C3 proteins localize to retinogeniculate synapses during developmental pruning and microglia then engulf and remove presynaptic terminals via complement receptor 3 (CR3), a C3 receptor90,98. Importantly, C1q−/−, C3−/−, and CR3−/− mice have defects in eye-specific segregation, impaired removal of structural presynaptic terminals, and failure to eliminate functional synapses in the retinogeniculate circuit98. In other brain regions, C1q−/− mice have increased numbers of axonal boutons and elevated cortical seizure activity107, and CR3-dependent phagocytosis has been implicated in the removal of neuronal dopamine D1 receptors in adolescent male mice94. It remains to be determined if data in the nucleus accumbens is a reflection of removal of postsynaptic receptors from the postsynaptic membrane or reflective of a physical elimination of structurally intact synapses. One of the biggest questions emerging from these complement-dependent pruning studies is how does complement, a secreted protein, lead to elimination of some synapses, but not others. A few recent breakthroughs suggest that molecular inhibitors of microglial synaptic engulfment are involved. First, it was shown that a canonical “don’t eat me” immune system molecule, CD47, is expressed by RGCs. CD47 and its cognate receptor SIRPα on microglia were required for activity-dependent retinogeniculate synaptic pruning (Fig. 2b)108. Another study identified an endogenous C1q inhibitor, SRPX2, is expressed by neurons109. SRPX2−/− mice have increased C3 deposition, elevated engulfment of presynaptic terminals by microglia, and abnormal eye-specific segregation in the retinogeniculate circuit (Fig. 2c)109. In addition to complement inhibition, there is evidence that changes in sialyation and/or externalization of phosphatidyl-serine residues on synapses in the retinogeniculate circuit and hippocampus target complement binding to a specific subset of synapses (discussed later)110–112.
An emerging concept from these microglia-mediated pruning studies is that mechanisms are region-specific and context-dependent. For example, early pruning in the developing retinogeniculate circuit is dependent on complement, but not CX3CR1113. CX3CR1 is also dispensable for climbing fiber pruning in the cerebellum114. Conversely, developmental pruning of hippocampal synapses and pruning of barrel cortex synapses are CX3CR1-dependent, but not CR3-dependent65,96,115. In the visual cortex, microglia engulf synaptic material during visual deprivation51,116 but CX3CR1 and complement do not regulate ocular dominance plasticity113,117. Instead, deficiency in the microglial purinergic receptor P2RY12 results in reduced microglial synapse engulfment and defects in ocular dominance plasticity91. One possibility is that microglia influence neuronal activity via P2RY12 which has recently been shown in other brain regions to modulate neural excitability118,119. This P2RY12-dependent modulation of neuronal activity by microglia, in turn, stimulates engulfment of synapses by a yet-to-be identified mechanism. Recent work has further suggested other non-phagocytic mechanisms by which microglia regulate pruning. In the cerebellum, microglia impact climbing fiber pruning by promoting GABAergic innervation of cerebellar Purkinje cell somas, which influences weakening of a subset of climbing fiber synapses42,30. Also, during a later stage of experience-dependent pruning in the retinogeniculate circuit, microglia-derived TNF family cytokine TWEAK acts via neuronal Fn14 to facilitate strengthening of synapses along thalamic relay neuron dendritic spines62. Precisely how different mechanisms drive pruning in specific circuits at specific times remains a key open question, which we discuss extensively later in this review.
Microglia-astrocyte immune crosstalk in synapse pruning
In addition to microglia, studies have also implicated astrocytes in developmental synapse pruning. Like microglia, astrocytes engulf retinogeniculate synapses in an activity-dependent manner120. Mice lacking the astrocyte-enriched engulfment receptor MEGF10 or the TAM receptor MerTK, typically used by phagocytes to engulf and clear apoptotic cells, have reduced astrocytic synapse engulfment and fail to properly prune retinogeniculate synapses (Fig. 2d)120. While MEGF10 is highly enriched in astrocytes in the mammalian CNS, both astrocytes and microglia express MerTK. In contrast to astrocytes, microglia in MerTK−/− mice engulfed more retinogeniculate inputs, but not at sufficient levels to rescue synaptic pruning deficits120. These data raise the question as to which cell type is most important for pruning. Immunofluorescence microscopy shows more engulfed material per cell in microglia, but more total engulfed material in the larger population of astrocytes120. It could be that astrocytes carry out the bulk of the pruning of retinogeniculate axons and synapses and microglia are involved in more local pruning and refinement. Interestingly, in the adult hippocampus, astrocytes were recently shown to more readily engulf synaptic material121. However, it is important to note that the kinetics of phagocytosis and degradation of phagocytosed material in vitro are faster in microglia compared to astrocytes122. Therefore, in vivo engulfment of synaptic material may be more easily detected at static imaging points in astrocytes compared to microglia, provided the kinetics of phagocytosis is similar between in vitro and in vivo contexts.
Besides engulfment, astrocyte secreted factors have also been shown to regulate pruning of synapses. For example, the astrocyte-secreted molecule Hevin is necessary for the elimination of exuberant synapses in the developing visual cortex123. It is intriguing to consider that this molecule is signaling to microglia. Indeed, astrocyte-microglia crosstalk has been shown to regulate microglial engulfment and pruning in other contexts (Fig. 2e). For example, astrocyte-derived transforming growth factor beta (TGF-β) induces C1q expression, which is necessary for complement-mediated retinogeniculate synapse pruning by microglia100. In sensorimotor circuits, astrocyte-derived IL-33 binds its receptor IL1RL1/ST2 on microglia to stimulate synapse engulfment and pruning124. Conversely, microglia may also direct astrocytic synapse engulfment. This has been suggested by data in the hippocampus where microglia were recently shown to regulate synapse pruning via triggering receptor expressed on myeloid (TREM2)112,125,126, in addition to CX3CR1. One of these studies suggested that TREM2 deficiency in microglia results in disrupted signaling of microglia to astrocytes, which subsequently results in failure of astrocytes to engulf synaptic material during hippocampal development126. However, this TREM2-deficient signal derived from microglia to stimulate astrocyte engulfment is unknown.
These studies have identified a diverse array of immune signaling mechanisms required for developmental synaptic pruning. Emphasizing the importance, many of these mechanisms are evolutionarily conserved (Box 1). However, there are still fundamental questions that remain. First, many studies only look at one timepoint once pruning is over. This raises the possibility that some of these molecules could regulate initial synaptogenesis, which would result in a phenotype that is identical to a pruning defect at older ages. Assessing synapse density pre and post pruning in mutant animals is key. Second, given the diversity of molecular players identified across different circuits, immune signals during synapse pruning are circuit-specific rather than a “one-size-fits-all” mechanism. How this circuit specificity is achieved is unclear and could be a result of molecular and functional diversity of neurons and/or glia. Differing gene expression and/or levels of activity could elicit different signaling cascades in neurons, which could then elicit diversity in glial cell responses. Third, while it is clear that immune molecules are changing expression and/or localization in response to activity, it is unclear how. One possibility is through the newly identified mechanism by which JAK2-STAT1 signaling modulates activity-dependent axonal and synaptic pruning. Recent work has provided evidence in the developing retinogeniculate and callosal projections in mice that the protein tyrosine kinase JAK2 is activated in inactive neurons and synapses, but only when neighboring neurons and synapses remain activated127. This leads to the pruning of inactive synapses via the downstream transcriptional regulator STAT1. JAK2-STAT1 is a canonical signaling cascade in the immune system and driver of cytokine signaling. Precisely which molecules are transcriptionally regulated by JAK2-STAT1 signaling to induce the selective elimination of inactive synapses is unclear, but it is intriguing to consider that many immune molecules mentioned above are impacted.
Box 1. Evolutionarily conserved pruning mechanisms: Immune and cell death signaling in invertebrate systems.
Two of the major immune signaling pruning pathways, MHC class I and complement, appear to be vertebrate-specific synaptic pruning pathways. However, many of the other molecular players identified in activity-dependent synaptic pruning were first identified during axonal or dendrite pruning in invertebrates. For example, glial cell-mediated engulfment of neuronal compartments during pruning is highly conserved. Indeed, in Drosophila glial cells engulf axons and sensory endings during developmental pruning238. This process is regulated by Draper239–241, the homologue of the mammalian MEGF10 which regulates astrocyte-mediated pruning120. The homologue in C. Elegans CED-1 performs similar functions in cholinergic synapse pruning242. There is also evidence that secreted immune molecules involved in mammalian synaptic pruning regulate pruning in invertebrates. For example, in Drosophila the chemokine-like protein Orion, which has some similarity to mammalian fractalkine (CX3CL1), was recently shown to regulate glial recruitment and engulfment of axons during mushroom body pruning243. In addition, as astrocyte-derived TGF-β signaling has been implicated in driving complement-mediated pruning in mammals100, TGF-β/Myoglianin drives axonal pruning in developing Drosophila244. Unlike mammals, TGF-β appears to be acting neuron-intrinsically to promote pruning in Drosophila.
Cell death signaling during pruning is also evolutionarily conserved. Caspase was first identified in C. elegans whereby the homologue ced-3 regulated cell survival245. It is now appreciated that this signaling can be compartmentalized to regulate cellular remodeling without inducing cell death246. In the C. elegans nervous system, caspase CED-3 cleaves and activates the actin severing protein gesolin/GLSN-1 to promote disassembly of presynaptic machinery during synapse remodeling247. In addition, in GABAergic neurons, local calcium increases activates calcineurin, which, in turn, activates the caspase CED-3 to promote dismantling of presynaptic machinery248. Downstream of caspase activation and similar to the mammalian system, it has recently been shown in C. elegans that local PtdSer exposure regulates subsequent AMsh glial cell engulfment of sensory endings249.
Together, these data point to evolutionary conserved mechanisms driving synaptic pruning by glia and immune and cell death molecules. Better understanding how changes in activity could regulate invertebrate pruning could open up powerful new systems to better understand activity-dependent synapse pruning across species.
Cell death molecules at the synapse
Besides immune signaling, evolutionarily conserved molecules (Box 1) canonically involved in cell death signaling have been implicated in activity-dependent synaptic pruning. The mechanisms described below are recognized as primary drivers of programmed cell death/apoptosis and clearance of apoptotic debris by phagocytes. However, it is becoming increasingly appreciated that these mechanisms can be subcellularly localized and temporally restricted to avoid cell death and, instead, drive pruning. In some cases, they may also be operating upstream of immune molecules to localize them to synapses prior to pruning.
Activation of caspases and phosphatases
Caspase-3 is a protease activated during mitochondria-mediated apoptosis. It cleaves hundreds of different protein substrates that mediate DNA fragmentation, phosphatidyl-serine (PtdSer) exposure, and membrane blebbing128. In vitro studies have revealed that mitochondrial capase-3 activation can be compartmentalized in dendritic spines, leading to LTD via AKT1 cleavage and AMPAR internalization129,130, and subsequent spine pruning131. Additionally, caspase-3 knockout mice have defects in hippocampal spine pruning in vivo131. Compared to apoptosis, LTD and pruning involve lower levels and durations of caspase-3 activation, possibly due to proteasomal degradation of caspase-3 in the dendrites to prevent precocious cell death130,131. Besides caspase-3, caspase-2 has also been implicated in dendritic spine pruning132. Similar to caspase-3 knockout mice129,130, deficiency in caspase-2 results in defects in LTD and increased numbers of dendritic spines in hippocampal neurons both in vitro and in vivo. Finally, another molecule involved in cell death133, which has been implicated in pruning, is the calcium-dependent protein phosphatase calcineurin. In mammalian systems, calcineurin has been shown to be regulated by myocyte enhancer factor 2 (MEF2) to initiate degradation of PSD-95 in an activity-dependent manner, leading to dendritic spine elimination134–136.
Phosphatidyl-serine exposure
Downstream of caspases, phosphatidylserine (PtdSer) residues normally restricted to the inner leaflet of the plasma membrane become externalized on the cell surface by scramblases, which are activated by caspases. During apoptosis, TAM receptor complexes on phagocytes bind externalized PtdSer, leading to engulfment and clearance of apoptotic cell bodies137. Similarly, recent studies have indicated that PtdSer is exposed on a subset of largely presynaptic terminals during developmental synaptic pruning in the retinogeniculate circuit and hippocampus, which are subsequently cleared by phagocytes111,112,138. It is unclear whether TAM receptors, such as MerTK, bind PtdSer during synapse pruning. However, other immune molecules required for synaptic pruning have been shown to bind PtdSer, including C1q139, GPR56138, and TREM2140. Annexin-V treatment, which binds externalized PtdSer, or deletion of TREM2 in vitro leads to reduced microglial engulfment and inhibition of hippocampal synapse pruning112. In addition, deletion of C1q in vivo leads to accumulation of exposed PtdSer, primarily at presynaptic structures, and reduced microglia-mediated pruning of presynaptic terminals in the retinogeniculate circuit112,138. Although an earlier study provided ex vivo data to support that C1q-tagged synapses were enriched for activated caspase-3 and exposed PtdSer compared to untagged synapses111, this later work in the retinogeniculate circuit showed that stimulation of microglia-mediated synaptic pruning by PtdSer exposure was not downstream of caspase-3 activation112. Published in parallel, another group showed that the microglial adhesion G-protein coupled receptor, GPR56 binds directly to PtdSer138. Similar to C1q-deficient mice, PtdSer exposure and pruning of presynaptic terminals is blocked when GPR56 is conditionally ablated in microglia. Thus, data points towards GPR56, TREM2, and complement proteins working in concert to regulate microglial pruning of synapses via binding to PtdSer. It remains to be determined whether these pathways regulate each other or whether they are working in parallel to perform a similar function.
Potential role for mitochondria
While data are suggestive that molecules that normally regulate cell death are compartmentalized at the synapse to regulate synapse pruning, it remains less clear how they are initially activated at some synapses and not others. One potential mechanism is through JAK-STAT signaling (described above), which is activated in inactive synapses leading to their removal127. In addition to regulating immune molecules, JAK-STAT signaling is also known to regulate the expression of caspases during cell death in other contexts (reviewed in141). Another possibility is that mitochondria may be an important link between changes in activity at a given synapse, compartmentalized cell death signaling, and the physical removal of the synapse. Indeed, elevated mitochondrial calcium is required for LTD-dependent elimination of spines in cultured hippocampal neurons129. Also, reducing the amount of mitochondria in dendrites leads to a reduction in dendritic spine density in cultured neurons while an increase in mitochondrial content supports synaptogenesis and plasticity142. Further, live imaging in adult mouse cortex and hippocampal slices has revealed that presynaptic terminals with more mitochondria are more stable and decreased presynaptic mitochondria motility is associated with increased synaptic strength143,144. It remains critical to understand how these mitochondrial changes may be relevant to synapse pruning during development in vivo as this may be an important link by which activity drives the selective elimination of some synapses and maintenance of others.
Together, these data demonstrate that calcium-dependent cell death pathways involving caspases and calcineurin are used to facilitate synapse pruning. Externalized PtdSer, which may be downstream of local caspase activation, provides a substrate to initiate immune-mediated synapse pruning by glial cells (Fig. 3). Upstream JAK-STAT signaling and/or mitochondria may be key.
Figure 3. Local “death of a synapse.”.
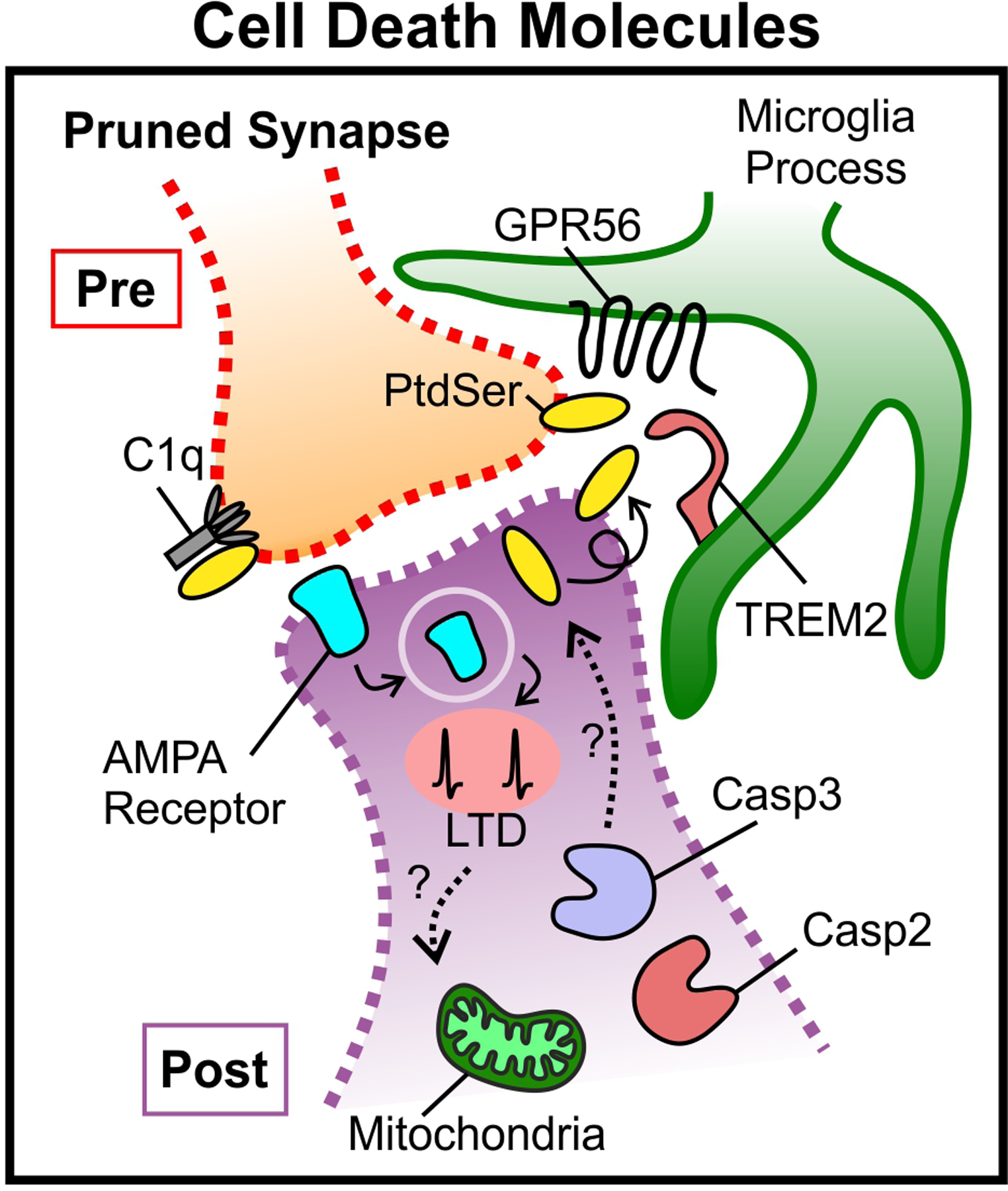
During synaptic pruning, mitochondrial molecules in the dendrite activate Caspase 3 which drives AMPA receptor internalization from the postsynaptic membrane and LTD129,130. Caspase 2 also impacts AMPAR internalization although though a separate pathway132. Activity-dependent changes at the synapse, for example via LTD, may also influence mitochondrial processes and compartmentalized cell death molecules in order to regulate pruning (dotted lines). In some, but not all cases, caspase signaling leads to externalization of to the outer membrane at synapses. This exposed PtdSer can subsequently bind molecules such as C1q, GPR56, and TREM2 to induce pruning112,125,138. α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic-acid (AMPA), phosphatidylserine (PtdSer), G protein coupled receptor 56 (GPR56), triggering receptor expressed on myeloid cells (TREM2), complement component 1q (C1q).
Disease-associated genes, environment, and synaptic pruning: implications for neurodevelopmental disorders
In humans, synaptic pruning primarily occurs during two developmental periods: the first two years after birth and adolescence (Fig. 4)1. Studies suggest defects in synaptic pruning during both developmental periods underlie several neurodevelopmental disorders145–153. Here, we focus on autism spectrum disorders (ASD) and schizophrenia (SZ) to highlight how, in addition to the molecules described above, many disease-associated genes contribute to changes in neural circuit structure and function through disruptions in synaptic pruning (summarized in Table 1).
Figure 4. Synaptic pruning and neurodevelopmental disorders.

A summary of published findings depicting development changes in synapse density in the brains of neurotypical individuals compared to patients with autism spectrum disorders (ASD)155,156 or schizophrenia (SZ)198,199,200,201,202–204,205 (based on figure from153). The developmental periods in early life and adolescence when synaptic pruning primarily occurs are highlighted in yellow1. Key molecules implicated in aberrant pruning during these two periods in ASD (mTOR182,183,155,95, TSC155,184, PTEN234,235,236, FMRP161,172,173,174,175,179,136,169–171,177,178, eIF4E192,193,194) and SZ (C4213,214,215,216, MHC class I211,78,212) are also indicated. Environmental risk factors which may, in concert, affect pruning are also indicated218–223,219, such as maternal infection223,225,226,227 and diet228,229 in utero and pollution, infection, and stress later in development. Genetic factors, which likely interact with environment factors237, are present throughout life (signified by the double helix). Mammalian target of rapamycin (mTOR), tuberous sclerosis (TSC), phosphatase and tensin homolog (PTEN), fragile X mental retardation protein (FMRP), eukaryotic translation initiation factor 4E (eIF4E), major histocompatibility complex class I (MHC I), complement 4 (C4).
Table 1.
Summary of synaptic pruning studies in neurodevelopmental disorders discussed in this review
| Species | Evidence for changes in pruning | Brain Region | Reference |
|---|---|---|---|
| Human | |||
| ASD | Increased spine density Failure to eliminate spines over development |
Temporal cortex | Tang, Neuron, 2014, Ref #155 |
| Increased spine density | Temporal cortex, frontal cortex, parietal cortex | Hutsler, Brain Research, 2010, Ref #156 | |
| Transcriptional profiling reveals changes in immune/glial mRNA related to pruning | Combined frontal/temporal cortex | Voineagu, Nature, 2011, Ref #157 | |
| Hypomethylation and overexpression of complement genes involved in pruning | Prefrontal cortex, cingulate cortex | Nardone, Transl Psychiatry, 2014, Ref #158 | |
| Genetic association to synaptic plasticity genes | N/A | De Rubeis, Nature, 2014, Ref #160 | |
| Fragile-X | Increased immature spine morphology | Cingulate cortex, temporal cortex | Hinton, Am J Med Genet, 1991, Ref #172 |
| Increased immature spine morphology | Cortex | Rudelli, Acta Neuropathol, 1985, Ref #173 | |
| Schiophrenia | Reduced gray matter volume | Cortex | Zipursky, Arch Gen Psychiatry, 1992, Ref #198 |
| Progressive loss of grey matter voume | Cerebral cortex, frontal cortex, thalamus | Andreasen, Biologial Psychiatry, 2011, Ref #199 | |
| Reduced spine density Normal spine density |
Dorsolateral prefrontal cortex Visual cortex |
Glantz, Arch Gen Psychiatry, 2000, Ref #200 | |
| Normal spine density | Dorsolateral prefrontal cortex | Kolluri, Am J Psychiatry, 2005, Ref #201 | |
| Reduced synaptophysin immunoreactivity | Prefrontal cortex | Glantz, Arch Gen Psychiatry, 1997, Ref #202 | |
| Reduced synaptic vesicle proteins | Frontal cortex, parietal cortex, cingulate cortex, hippocampus, thalamus | Davidsson, Schizophr Research, 1999, Ref #203 | |
| Reduced VGluT1 boutons | Prefrontal cortex | Bitanihirwe, BMC Psychiatry, 2009, Ref #204 | |
| Reduced SV2A (PET) | Frontal cortex, cingulate cortex | Onwordi, Nature, 2020, Ref #205 | |
| Genetic association to synaptic gene loci | N/A | Fromer, Nature, 2014, Ref #206; SWG of PGC, Nature, 2014, Ref #207 |
|
| Genetic association to MHC gene locus | N/A | Stefansson, Nature, 2009, Ref #211 | |
| Genetic association to C4 allelic variation | N/A | Sekar, Nature, 2016, Ref #213 | |
| Human Cell Models | |||
| C4 variant microglia-neuron co-cultures | Increased microglial engulfment | N/A | Sellgren, Nature Neuroscience, 2019, Ref #216 |
| Mouse Models | |||
| Tsc2+/− | Failure to eliminate spines over development Increased levels of PSD95 and Synaptophysin |
Temporal cortex | Tang, Neuron, 2014, Ref #155 |
| Atg7 flox/flox; CamKIIa-Cre | Failure to eliminate spines over development | Temporal cortex | Tang, Neuron, 2014, Ref #155 |
| Atg7 flox/flox; CamKIIa-Cre; Tsc2+/− | Failure to eliminate spines over development | Temporal cortex | Tang, Neuron, 2014, Ref #155 |
| Nse-Cre; Pten flox/flox | Macrocephaly Increased spine density |
Hippocampus, cortex | Kwon, Neuron, 2006, Ref #183 |
| L7-Cre; Tsc1 flox/flox | Increased spine density | Cerebellum | Tsai, Nature, 2012, Ref #184 |
| Fmr1-KO* | Increased spine density Reduced microglial engulfment of PSD95 |
Hippocampus | Jawaid, Glia, 2018, Ref #175 |
| Impaired ocular dominance plasticity | Visual Cortex | Dolen, Neuron, 2007, Ref #176 | |
| Failure to eliminate spines over development | Somatosensory Cortex | Patel, J. Neuroscience, 2014, Ref #179 | |
| Impaired synapse elimination | Hippocampus | Pfeiffer, Neuron, 2010, Ref #169 | |
| Impaired activity-dependent spine elimination | Somatosensory Cortex | Pan, PNAS, 2010, Ref #178 | |
| Lyz2-Cre; Atg7 flox/flox | Increased spine density Higher PSD-95, Shank3 protein levels Reduced degradation of synptophysin, PSD-95 by microglia |
Somatosensory Cortex | Kim, Molecular Psychiatry, 2016, Ref #95 |
| Cx3cr1-CreER/+; R26(Eif4e/Eif4e) | Increased spine density Altered microglial gene expression Impaired microglial phagocytosis |
Prelimbic cortex, hippocampus | Xu, Nature Communications, 2020, Ref #192 |
| eIF4E transgenic mice | Increased spine density | Prelimbic cortex | Santini, Nature, 2013, Ref #194 |
| Setd1a+/− | Reduced spine density | Prelimbic cortex | Mukai, Neuron, 2019, Ref #208 Nagahama, Cell Reports, 2020, Ref #209 |
| DISC1 | |||
| C4 KO | Impaired eye-specific segregation Reduction in synaptic C3 labeling |
Thalamus | Sekar, Nature, 2016, Ref #213 |
| Human C4 overexpression (in utero electroporation) | Increased microglial engulfment of PSD-95 Increased spine elimination |
Prefrontal cortex | Comer, PLoS Biol, 2020, Ref #214 |
| Human C4 overexpression (transgenic mice) | Reduced synapse density Increased microglial synapse engulfment |
Thalamus, prefrontal cortex | Yilmaz, Nature Neuroscience, 2020, Ref #215 |
| Maternal Immune Activation** | Increased synapse density Reduced Cx3cr1 expression |
Hippocampus | Fernandez de Cossio, Brain, behavior, and Immunity, 2017, Ref #224 |
| Increased synapse density Reduced microglia synaptic engulfment |
Hippocampus | Andoh, Cell Reports, 2019, Ref #225 | |
| Altered microglial gene expression Increased spine density Increased microglia-spine interations Hyperramified microglia |
Prefrontal cortex | Ikezu, Molecular Psychiatry, 2020, Ref #226 | |
| Maternal n-3 PUFA deficiency | Decreased synapse density Increased microglial engulfement |
Hippocampus | Madore, Nature Communications, 2020, Ref #229 |
Note: Some studies also report normal or decreased spine densities depending on age, brain region, and method of analysis. See Review by He, Neuroscience, 2013, Ref #174
Note: Some studies also report normal or decreased spine densities depending on age, brain region, and infection paradigm. See Review by Pekala, Dev Neurobiol, 2020, Ref #227
Genes associated with autism spectrum disorders (ASD)
Individuals with ASD are typically diagnosed within the first five years of life coincident with the first wave of synaptic pruning1,154. Analyses of postmortem ASD tissue have shown increased synapse density compared to neurotypical controls in the frontal, temporal, and parietal lobes, suggesting an under-pruning phenotype155,156. Transcriptomic analyses of postmortem ASD brains further show significant changes in genes associated with pruning157 and hypomethylation of immune genes involved in synaptic pruning including C1Q, C3, and C3R158. Genetic analyses also point to the involvement of molecules that regulate synapse formation and synaptic plasticity159,160.
In addition to these genetic associations, recent studies in mice harboring mutations or deletions in syndromic ASD-linked genes further support activity-dependent synaptic pruning deficits. One example is Fragile X Mental Retardation Protein (FMRP). Mutations in FMRP cause Fragile-X Syndrome (FXS), a disorder with high incidence of ASD. FMRP is a known regulator of mRNA translation through eIF4E161, is regulated by neuronal activity162,163, and regulates activity-dependent synaptic plasticity164,165. FMRP also works in concert with another ASD-linked gene, MEF2166–168, which can no longer carry out its normal role in activity-dependent spine elimination in Fmr1−/− mice136,169–171. In addition, increased spines have been reported in the neocortex in FXS172,173 and there are increased spines in the hippocampus and cortex and impaired ocular dominance plasticity in mice deficient in FMRP (Fmr1−/− mice)174–176. Linking immune mechanisms discussed previously with FMRP regulation of pruning, data suggests an impairment in microglia-mediated engulfment of postsynaptic structures in the hippocampus of Fmr1−/− mice175. However, it is important to note that assessment of synaptic pruning is complicated by evidence that there are defects in synapse stabilization and synaptogenesis in Fmr1−/− mice177,178. Also, other studies find the opposite or no change in spines in Fmr1−/− mice, likely due to differences in age, region, and methodologies174. Still, supportive of aberrant synaptic pruning, a time-course of paired electrophysiological recordings of L5 pyramidal neurons demonstrated that excess cell-to-cell connections in Fmr1−/− mice were, indeed, due to impaired pruning179. Given that activity-dependent pruning is dependent on the strengthening and stabilization of ‘winning’ synapses, it is possible that defects in spine stabilization or spine dynamics contribute to these changes in pruning. Indeed, enhanced spine dynamics has been noted in mice harboring genetic mutations linked to syndromic ASD: 15q11–13 duplication and neuroligin-3 R451C point mutation180.
Other syndromic ASD-linked genes that regulate synaptic pruning are molecules belonging to the mTOR signaling pathway, another set of mRNAs that are regulated by FMRP181. For example, deletion of phosphatase and tensin homolog (PTEN) in neurons, a gene that accounts for ~1–4% of ASD cases182 and an upstream inhibitor of mTOR, leads to increased spine density and altered social behavior in mice183. Other negative regulators of mTOR, tuberous sclerosis 1 (TSC1) and 2 (TSC2), cause tuberous sclerosis (TSC), which is another disorder with a high incidence of ASD. In mice deficient in Tsc1 (Tsc1−/−) or heterozygous for Tsc2 (Tsc+/−), spine density is increased in the cerebellum and cortex155,184. These synaptic changes occur concomitant with overactive mTOR signaling in both ASD tissue and in Tsc+/− neurons154. Tang et al. further showed that overactive mTOR signaling in ASD and in Tsc2+/− mice likely exerts its effects on spines and behavior by modulating neuronal autophagy155, a cellular degradative process for proteins and organelles. The exact mechanism by which autophagy leads to synaptic pruning remains unknown. However, there is evidence that mitochondria, well-known targets of autophagy, are depleted at presynaptic sites in Tsc1/2 deficient neurons185, which could impact synaptic pruning. Autophagy can also be induced presynaptically by neuronal activity186,187 and autophagy can regulate synaptic transmission and plasticity by degrading synaptic vesicles and postsynaptic proteins188–191. Thus, autophagy via mTOR signaling may play a similar role in activity-dependent pruning and disruptions may contribute to aberrant pruning in ASD.
These studies demonstrate clear links between alterations in mRNA translation and autophagy in neurons to alterations in synapse numbers in ASD. Yet, all of these molecules are ubiquitously expressed by numerous cell types throughout the CNS and the contribution of non-neuronal cell types to synaptic alterations in ASD must be considered. In fact, a recent study showed that increasing protein synthesis specifically in microglia by overexpressing eIF4E, which is regulated by FMRP, results in increased spine density and ASD-like behaviors192. This phenotype largely recapitulates phenotypes observed in mice with global overexpression of eIF4E193,194. A separate study found that selectively disrupting a core autophagy gene (Atg7) in Lyz2+ myeloid cells, which may include microglia, also impairs synaptic pruning95. Thus, disruptions in mRNA translation and autophagy in multiple cell types may contribute to synaptic alterations in ASD.
Genes associated with schizophrenia (SZ)
SZ is diagnosed later than ASD, typically between late adolescence and mid to late 20’s195. However, even before the full onset of SZ, cognitive deficits and structural changes can be detected in “high-risk” individuals196. The emergence of symptoms towards the end of the second wave of pruning led to the hypothesis that aberrant synaptic pruning during adolescence contributes to SZ197. In contrast to ASD, data from SZ patients and animal models suggest an over-pruning defect. For instance, studies in SZ patients indicate progressive loss of gray matter volume198, especially during the initial phase of the disease199. Examination of postmortem tissue has also revealed a layer-specific loss of spines200,201 and presynaptic proteins202–204, particularly in layer III of the prefrontal cortex. Similarly, new PET imaging with a ligand for the synaptic marker SV2A has revealed reduced density of presynaptic terminals in SZ brains205.
Synaptic pruning defects in SZ are also supported by genetics. Genetic variations overrepresented in SZ predominantly affect synaptic genes160,206,207. In several cases, reduced spine density has been observed in transgenic mouse models lacking or harboring mutations in SZ risk genes208–210, including recent studies of Setd1a+/− mice. However, it is unknown if these changes are due to over-pruning or reduced spine formation. The most significant genetic associations for SZ are within the extended MHC locus, which spans ~8Mb and contains numerous immune-related genes211. Given the role of MHC molecules in synaptic pruning, it is intriguing to speculate that defects in synaptic pruning due to genetic variations in MHC molecules can result in SZ78,212. This MHC locus gained even more attention recently with work showing that allelic variations in another gene within this locus, complement C4, confer genetic risk for SZ213. C4 is downstream of C1q in the classical complement cascade, which is necessary to form the C3 convertase to cleave and activate C390,98 (Fig 2c). C4 exists as two isoforms: C4A and C4B. Sekar et al. showed allelic variations that increase C4A, but not C4B, are correlated with heightened SZ risk213. In addition, synaptic pruning in the retinogeniculate circuit was defective in C4 knockout mice. Supporting this initial work, AAV-mediated overexpression of C4 leads to microglial synapse engulfment and hypoconnectivity in the mouse prefrontal cortex, which is accompanied by defects in social interactions214. Most recently, human C4A, the isoform that confers higher SZ risk, was shown to more efficiently bind synapses compared to C4B215. Also, C3 deposition and microglial synapse engulfment in microglia-neuron co-cultures was higher in cells derived from SZ patients harboring C4A allelic variants216. Finally, overexpressing human C4A in vivo in mice resulted in increased synapse engulfment by microglia, enhanced synaptic pruning in the cortex, and social and cognitive behaviors related to SZ215. Intriguingly, co-expression analysis of SZ brains show that C4A has a selective, inverse correlation with synaptic SZ risk genes217. Together, these studies provide evidence linking SZ-risk genes, particularly C4, with over-pruning in SZ.
ASD and SZ: Environmental influences leading to pruning and behavioral defects
In addition to genetics, there are clear indications that environmental factors play roles in the etiology and progression of ASD, SZ, and related disorders. For example, epidemiological evidence suggests that environmental factors, which affect the immune system, such as maternal infections, maternal obstetric complications, maternal nutrition, and pollution, increase disease risk218–223. Thus, a “two-hit” hypothesis has been proposed in which risk genes impact susceptibility, but also require a secondary environmental influence to initiate abnormal developmental programs, including synaptic pruning219.
Mechanistic evidence for environmental influences impacting developmental synaptic pruning and subsequent ASD or SZ-like behaviors largely stems from animal models. For example, an immune challenge in pregnant rodents (i.e. maternal immune activation/MIA), which is known to induce ASD and SZ-like behaviors in offspring223, increases neuronal spine density and reduces expression of the microglial pruning gene Cx3cr1 in the hippocampi of offspring224. Two other studies found decreased microglial synapse engulfment in a similar model225 and synaptic and behavioral deficits of MIA were prevented by postnatal depletion of microglia226. Thus, microglia may be the conduit by which early immune activation results in synaptic pruning defects and behavioral changes (reviewed in227). Other prenatal environmental risk factors for ASD and SZ such as insufficient maternal dietary omega-3 fatty acids (n-3 PUFAs)228 also result in increased microglial engulfment of synaptic material, decreased hippocampal synapses, and altered spatial working memory in the offspring229. Connecting with immune-mediated pruning, in n-3 PUFA-deficient offspring, C1q, C3, and CR3 are elevated in the hippocampus and spine and memory defects are dependent on C3. With dietary changes, it was shown that n-3PUFAs deficiency affects brain lipids, which then stimulates 12/15-LOX/12-HETE signaling in microglia to induce changes in CR3 expression229. Another possibility is that these environmental stressors first impact the peripheral immune system, including the microbiome, which then impacts immune signaling in the brain. This has been shown in rodent MIA in which the autism-like behaviors and cortical circuit abnormalities in offspring manifest as a result of gut microbiome-dependent development of T helper 17 cells (TH17) and their release of IL17a230–232. Thus, it could be that, for those with genetic susceptibility, this environmental influence becomes the “second hit” to induce changes in developmental pruning. This may affect pruning in early life in the case of ASD. It could also “prime” the system for later disruptions in pruning during SZ.
In summary, multiple lines of evidence point towards impaired synaptic pruning as an important mechanism in ASD, SZ, and related disorders (summarized in Table 1). As discussed previously, many studies look at only one endpoint and compare synapse density. It is important to consider that besides the physical elimination of synapses, these changes in synapse densities could be a consequence of exuberant synaptogenesis or increased turnover of synaptic structures. In ASD, most evidence points towards elevated synaptic connectivity and an under-pruning phenotype. However, depending on the mutation, neuron type, and brain region affected, over or under-pruning could have similar functional outcomes. In SZ, over-pruning prevails across multiple studies. Animal models demonstrate that many genetic risk factors that underlie syndromic forms of these disorders impact pruning. In SZ, there is the clearest link to disruptions in MHC and complement-mediated pruning. Further linking to immune-mediated pruning is work showing that environmental stressors may work cooperatively to impact the immune system, pruning, and risk of developing ASD, SZ, and related disorders (Fig. 4).
Perspectives and outstanding questions
Synaptic pruning in the CNS is a developmental program necessary for establishing appropriate brain wiring and function. In many mammalian circuits, spontaneous and experience-driven changes in neuronal activity drive an activity-dependent Hebbian competition between synaptic inputs for synaptic territory, which is further shaped by local LTD and GABAergic innervation at individual synapses. Neuronal and glial immune signaling mechanisms and cell death pathways spatially restricted to synapses are activated downstream of changes in neural activity and lead to synaptic pruning. Yet, whether and how activity directly regulates these molecules and cells is unknown. One possibility is JAK2-STAT1 signaling. Canonically, JAK2-STAT1 is involved in innate immune and cell death transcriptional programs. During pruning, it was very recently shown to be activated specifically in less active neurons and localized to less active synapses, which were subsequently eliminated138. It remains unknown how this transcriptional program is elicited by changes in activity or is compartmentalized, leading to pruning of some synapses but not others. Another possibility is that local changes in calcium due to, for example, LTD results in perturbations of local mitochondria. These early mitochondrial changes can then elicit recruitment and/or activation of cell death signaling molecules, which then stimulates immune signaling and glial cells for synapse removal. It also remains unknown whether pruning occurs in all neurons and circuits. Also, for those circuits that do prune, it remains a mystery why different mechanisms are used for pruning depending on the brain region and circuit. The extent of pruning could prove challenging to measure given the differences across species, and the timing of pruning will be different depending on the circuit. Understanding why different pruning mechanisms are elicited for different types of neurons should be more tractable. This could result from regional molecular and functional heterogeneity of neurons and glia throughout the mammalian CNS, which is becoming increasingly more appreciated with single-cell RNAseq and spatial transcriptomic studies. Finally, the lessons learned regarding the mechanistic underpinnings of synaptic pruning are now informing disease. This is reflected by a number of studies, which point to synaptic pruning as a common feature disrupted across multiple neurodevelopmental disorders, including ASD and SZ. However, in many of these studies, as well as studies of the fundamental biology underlying activity-dependent pruning, only one timepoint is assessed. It is possible that synaptic density changes could also result from defects in synaptogenesis. Also, while pruning defects could be reflective of increases or decreases in elimination of the “losing” synapses, these phenotypes could also be a consequence of impaired stability or maintenance of the “winning” synapses.
The field of activity-dependent synaptic pruning has made tremendous progress since the initial studies by Hubel and Wiesel. A picture is emerging by which activity is eliciting immune and cell death signaling that, ultimately, leads to the clearance of some synapses and maintenance and strengthening of others. We are now at a point to address these critical questions in the field. This will result in fundamental new insight into how CNS circuits develop and how defects in pruning can drive changes in connectivity and behavior in a myriad of neurodevelopmental and neuropsychiatric disorders.
Acknowledgments:
We thank P. Feinberg for his careful review of this manuscript. This work was supported by NIMH-R01MH113743 (DPS), NINDS-R01NS117533 (DPS), NINDS- F31NS117053 (GG).
Glossary
- Synapse pruning
A developmental process in which elements that comprise a bona fide structural synapse (presynaptic terminal and postsynaptic membranes) are eliminated, which may also include some pruning back small branches of axonal arbors and dendrites while remaining synapses are maintained and strengthened.
- Spontaneous neural activity
Neuronal activity that is not driven by an external stimulus
- Experience-driven neural activity
Neuronal activity driven by external changes affecting sensory experience
- Long-term depression
A process by which changes in neuronal activity, such as sustained low-frequency stimulation, induces a reduction in synaptic strength.
- Eye-specific segregation
A process involving synaptic pruning by which spontaneous retinal activity drives presynaptic inputs from retinal ganglion cells (RGCs) to segregate and synapse in discrete, non-overlapping territories within the lateral geniculate nucleus (LGN) during postnatal development.
- Monocular deprivation
The loss of sensory input to one eye, typically performed by suturing one eye closed for a defined period of time.
- Ocular dominance plasticity
A process by which disruption of visual input to one eye (see Monocular deprivation) results in strengthening of synaptic inputs from the open eye and weakening and elimination of synapses corresponding to the sutured, deprived eye.
- Critical window
A limited period during development in which a cellular developmental process, such as synaptic pruning, is responsive to intrinsic or extrinsic (environmental) factors, including changes in neuronal activity.
- Engulfment
The internalization or phagocytosis of material into a cell for degradation.
- Trogocytosis
Partial phagocytosis of material (“trogo” – “nibble”) while leaving the remaining membrane intact.
- Opsonization
The process by which molecules, such as complement, adhere to material destined for removal by phagocytes or cell lysis.
- Apoptosis
A well-controlled process of programmed cell death which may naturally occur in development and involves membrane blebbing, cell shrinkage, and DNA fragmentation.
Footnotes
Competing interests: None
References
- 1.Riccomagno MM & Kolodkin AL Sculpting neural circuits by axon and dendrite pruning. Annual review of cell and developmental biology 31, 779–805, doi: 10.1146/annurev-cellbio-100913-013038 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schuldiner O & Yaron A Mechanisms of developmental neurite pruning. Cell Mol Life Sci 72, 101–119, doi: 10.1007/s00018-014-1729-6 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Luo L & O’Leary DD Axon retraction and degeneration in development and disease. Annual review of neuroscience 28, 127–156, doi: 10.1146/annurev.neuro.28.061604.135632 (2005). [DOI] [PubMed] [Google Scholar]
- 4.Sanes JR & Lichtman JW Development of the vertebrate neuromuscular junction. Annual review of neuroscience 22, 389–442 (1999). [DOI] [PubMed] [Google Scholar]
- 5.Katz LC & Shatz CJ Synaptic activity and the construction of cortical circuits. Science 274, 1133–1138, doi: 10.1126/science.274.5290.1133 (1996). [DOI] [PubMed] [Google Scholar]
- 6.Hua JY & Smith SJ Neural activity and the dynamics of central nervous system development. Nature neuroscience 7, 327–332, doi: 10.1038/nn1218 (2004). [DOI] [PubMed] [Google Scholar]
- 7.Kano M & Hashimoto K Synapse elimination in the central nervous system. Current opinion in neurobiology 19, 154–161, doi: 10.1016/j.conb.2009.05.002 (2009). [DOI] [PubMed] [Google Scholar]
- 8.Shatz C & Stryker M Prenatal Tetrodotoxin Infusion Blocks Segregation of Retinogeniculate Afferents. Science 242, 87–89 (1988). [DOI] [PubMed] [Google Scholar]
- 9.Cang J et al. Development of precise maps in visual cortex requires patterned spontaneous activity in the retina. Neuron 48, 797–809, doi: 10.1016/j.neuron.2005.09.015 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Muir-Robinson G, Hwang BJ & Feller MB Retinogeniculate axons undergo eye-specific segregation in the absence of eye-specific layers. J Neurosci 22, 5259–5264, doi:20026563 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Penn A, Riquelme P, Feller MB & Shatz C Competition in retinogeniculate patterning driven by spontaneous activity. Science 279, 2108–2112 (1998). [DOI] [PubMed] [Google Scholar]
- 12.Torborg CL & Feller MB Spontaneous patterned retinal activity and the refinement of retinal projections. Prog Neurobiol 76, 213–235, doi: 10.1016/j.pneurobio.2005.09.002 (2005). [DOI] [PubMed] [Google Scholar]
- 13.Butts DA, Kanold PO & Shatz CJ A burst-based “Hebbian” learning rule at retinogeniculate synapses links retinal waves to activity-dependent refinement. PLoS Biol 5, e61, doi: 10.1371/journal.pbio.0050061 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huberman AD et al. Eye-specific retinogeniculate segregation independent of normal neuronal activity. Science 300, 994–998, doi: 10.1126/science.1080694 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McLaughlin T, Torborg CL, Feller MB & O’Leary DD Retinotopic map refinement requires spontaneous retinal waves during a brief critical period of development. Neuron 40, 1147–1160, doi: 10.1016/s0896-6273(03)00790-6 (2003). [DOI] [PubMed] [Google Scholar]
- 16.Grubb MS, Rossi FM, Changeux JP & Thompson ID Abnormal functional organization in the dorsal lateral geniculate nucleus of mice lacking the beta 2 subunit of the nicotinic acetylcholine receptor. Neuron 40, 1161–1172, doi: 10.1016/s0896-6273(03)00789-x (2003). [DOI] [PubMed] [Google Scholar]
- 17.Burbridge TJ et al. Visual circuit development requires patterned activity mediated by retinal acetylcholine receptors. Neuron 84, 1049–1064, doi: 10.1016/j.neuron.2014.10.051 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ziburkus J & Guido W Loss of binocular responses and reduced retinal convergence during the period of retinogeniculate axon segregation. Journal of neurophysiology 96, 2775–2784, doi: 10.1152/jn.01321.2004 (2006). [DOI] [PubMed] [Google Scholar]
- 19.Rossi FM et al. Requirement of the nicotinic acetylcholine receptor beta 2 subunit for the anatomical and functional development of the visual system. Proceedings of the National Academy of Sciences of the United States of America 98, 6453–6458, doi: 10.1073/pnas.101120998 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stellwagen D & Shatz C An Instructive Role for Retinal Waves in the Development of Retinogeniculate Connectivity. Neuron 33, 357–367 (2002). [DOI] [PubMed] [Google Scholar]
- 21.Zhang J, Ackman JB, Xu HP & Crair MC Visual map development depends on the temporal pattern of binocular activity in mice. Nature neuroscience 15, 298–307, doi: 10.1038/nn.3007 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kano M & Watanabe T Developmental synapse remodeling in the cerebellum and visual thalamus. F1000Res 8, doi: 10.12688/f1000research.18903.1 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Watanabe M & Kano M Climbing fiber synapse elimination in cerebellar Purkinje cells. The European journal of neuroscience 34, 1697–1710, doi: 10.1111/j.1460-9568.2011.07894.x (2011). [DOI] [PubMed] [Google Scholar]
- 24.Andjus PR, Zhu L, Cesa R, Carulli D & Strata P A change in the pattern of activity affects the developmental regression of the Purkinje cell polyinnervation by climbing fibers in the rat cerebellum. Neuroscience 121, 563–572, doi: 10.1016/s0306-4522(03)00556-6 (2003). [DOI] [PubMed] [Google Scholar]
- 25.Hashimoto K et al. Postsynaptic P/Q-type Ca2+ channel in Purkinje cell mediates synaptic competition and elimination in developing cerebellum. Proc Natl Acad Sci U S A 108, 9987–9992, doi: 10.1073/pnas.1101488108 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miyazaki T et al. Cav2.1 in cerebellar Purkinje cells regulates competitive excitatory synaptic wiring, cell survival, and cerebellar biochemical compartmentalization. J Neurosci 32, 1311–1328, doi: 10.1523/JNEUROSCI.2755-11.2012 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mikuni T et al. Arc/Arg3.1 is a postsynaptic mediator of activity-dependent synapse elimination in the developing cerebellum. Neuron 78, 1024–1035, doi: 10.1016/j.neuron.2013.04.036 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lorenzetto E et al. Genetic perturbation of postsynaptic activity regulates synapse elimination in developing cerebellum. Proceedings of the National Academy of Sciences of the United States of America 106, 16475–16480, doi: 10.1073/pnas.0907298106 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kawamura Y et al. Spike timing-dependent selective strengthening of single climbing fibre inputs to Purkinje cells during cerebellar development. Nature communications 4, 2732, doi: 10.1038/ncomms3732 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nakayama H et al. GABAergic inhibition regulates developmental synapse elimination in the cerebellum. Neuron 74, 384–396, doi: 10.1016/j.neuron.2012.02.032 (2012). [DOI] [PubMed] [Google Scholar]
- 31.Kano M et al. Impaired synapse elimination during cerebellar development in PKC gamma mutant mice. Cell 83, 1223–1231, doi: 10.1016/0092-8674(95)90147-7 (1995). [DOI] [PubMed] [Google Scholar]
- 32.Ichikawa R et al. Territories of heterologous inputs onto Purkinje cell dendrites are segregated by mGluR1-dependent parallel fiber synapse elimination. Proc Natl Acad Sci U S A 113, 2282–2287, doi: 10.1073/pnas.1511513113 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ichise T et al. mGluR1 in cerebellar Purkinje cells essential for long-term depression, synapse elimination, and motor coordination. Science 288, 1832–1835, doi: 10.1126/science.288.5472.1832 (2000). [DOI] [PubMed] [Google Scholar]
- 34.Kano M et al. Persistent multiple climbing fiber innervation of cerebellar Purkinje cells in mice lacking mGluR1. Neuron 18, 71–79, doi: 10.1016/s0896-6273(01)80047-7 (1997). [DOI] [PubMed] [Google Scholar]
- 35.Kano M, Hashimoto K & Tabata T Type-1 metabotropic glutamate receptor in cerebellar Purkinje cells: a key molecule responsible for long-term depression, endocannabinoid signalling and synapse elimination. Philos Trans R Soc Lond B Biol Sci 363, 2173–2186, doi: 10.1098/rstb.2008.2270 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kano M et al. Phospholipase cbeta4 is specifically involved in climbing fiber synapse elimination in the developing cerebellum. Proc Natl Acad Sci U S A 95, 15724–15729, doi: 10.1073/pnas.95.26.15724 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Uesaka N et al. Retrograde semaphorin signaling regulates synapse elimination in the developing mouse brain. Science 344, 1020–1023, doi: 10.1126/science.1252514 (2014). [DOI] [PubMed] [Google Scholar]
- 38.Uesaka N et al. Retrograde signaling for climbing fiber synapse elimination. Cerebellum 14, 4–7, doi: 10.1007/s12311-014-0615-y (2015). [DOI] [PubMed] [Google Scholar]
- 39.Choo M et al. Retrograde BDNF to TrkB signaling promotes synapse elimination in the developing cerebellum. Nat Commun 8, 195, doi: 10.1038/s41467-017-00260-w (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Uesaka N et al. Retrograde Signaling from Progranulin to Sort1 Counteracts Synapse Elimination in the Developing Cerebellum. Neuron 97, 796–805 e795, doi: 10.1016/j.neuron.2018.01.018 (2018). [DOI] [PubMed] [Google Scholar]
- 41.Miyazaki T et al. Glutamate transporter GLAST controls synaptic wrapping by Bergmann glia and ensures proper wiring of Purkinje cells. Proceedings of the National Academy of Sciences of the United States of America 114, 7438–7443, doi: 10.1073/pnas.1617330114 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nakayama H et al. Microglia permit climbing fiber elimination by promoting GABAergic inhibition in the developing cerebellum. Nat Commun 9, 2830, doi: 10.1038/s41467-018-05100-z (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wiesel TN & Hubel DH Single-Cell Responses in Striate Cortex of Kittens Deprived of Vision in One Eye. Journal of neurophysiology 26, 1003–1017 (1963). [DOI] [PubMed] [Google Scholar]
- 44.Antonini A, Fagiolini M & Stryker MP Anatomical correlates of functional plasticity in mouse visual cortex. The Journal of neuroscience : the official journal of the Society for Neuroscience 19, 4388–4406 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Antonini A & Stryker MP Plasticity of geniculocortical afferents following brief or prolonged monocular occlusion in the cat. The Journal of comparative neurology 369, 64–82, doi: (1996). [DOI] [PubMed] [Google Scholar]
- 46.Antonini A & Stryker MP Effect of sensory disuse on geniculate afferents to cat visual cortex. Vis Neurosci 15, 401–409, doi: 10.1017/s0952523898153105 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhou Y, Lai B & Gan WB Monocular deprivation induces dendritic spine elimination in the developing mouse visual cortex. Sci Rep 7, 4977, doi: 10.1038/s41598-017-05337-6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sun YJ, Espinosa JS, Hoseini MS & Stryker MP Experience-dependent structural plasticity at pre- and postsynaptic sites of layer 2/3 cells in developing visual cortex. Proc Natl Acad Sci U S A 116, 21812–21820, doi: 10.1073/pnas.1914661116 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Huh CYL et al. Long-term Monocular Deprivation during Juvenile Critical Period Disrupts Binocular Integration in Mouse Visual Thalamus. The Journal of neuroscience : the official journal of the Society for Neuroscience 40, 585–604, doi: 10.1523/JNEUROSCI.1626-19.2019 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yu H, Majewska AK & Sur M Rapid experience-dependent plasticity of synapse function and structure in ferret visual cortex in vivo. Proceedings of the National Academy of Sciences of the United States of America 108, 21235–21240, doi: 10.1073/pnas.1108270109 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tremblay ME, Lowery RL & Majewska AK Microglial interactions with synapses are modulated by visual experience. PLoS Biol 8, e1000527, doi: 10.1371/journal.pbio.1000527 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhou Y et al. REM sleep promotes experience-dependent dendritic spine elimination in the mouse cortex. Nature communications 11, 4819, doi: 10.1038/s41467-020-18592-5 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rittenhouse CD, Shouval HZ, Paradiso MA & Bear MF Monocular deprivation induces homosynaptic long-term depression in visual cortex. Nature 397, 347–350, doi: 10.1038/16922 (1999). [DOI] [PubMed] [Google Scholar]
- 54.Sidorov MS, Kaplan ES, Osterweil EK, Lindemann L & Bear MF Metabotropic glutamate receptor signaling is required for NMDA receptor-dependent ocular dominance plasticity and LTD in visual cortex. Proc Natl Acad Sci U S A 112, 12852–12857, doi: 10.1073/pnas.1512878112 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhou Q, Homma KJ & Poo MM Shrinkage of dendritic spines associated with long-term depression of hippocampal synapses. Neuron 44, 749–757, doi: 10.1016/j.neuron.2004.11.011 (2004). [DOI] [PubMed] [Google Scholar]
- 56.Shinoda Y, Tanaka T, Tominaga-Yoshino K & Ogura A Persistent synapse loss induced by repetitive LTD in developing rat hippocampal neurons. PLoS One 5, e10390, doi: 10.1371/journal.pone.0010390 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wiegert JS & Oertner TG Long-term depression triggers the selective elimination of weakly integrated synapses. Proc Natl Acad Sci U S A 110, E4510–4519, doi: 10.1073/pnas.1315926110 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hensch TK et al. Local GABA circuit control of experience-dependent plasticity in developing visual cortex. Science 282, 1504–1508, doi: 10.1126/science.282.5393.1504 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mataga N, Mizuguchi Y & Hensch TK Experience-dependent pruning of dendritic spines in visual cortex by tissue plasminogen activator. Neuron 44, 1031–1041, doi: 10.1016/j.neuron.2004.11.028 (2004). [DOI] [PubMed] [Google Scholar]
- 60.Hooks BM & Chen C Vision triggers an experience-dependent sensitive period at the retinogeniculate synapse. The Journal of neuroscience : the official journal of the Society for Neuroscience 28, 4807–4817, doi: 10.1523/JNEUROSCI.4667-07.2008 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hong YK et al. Refinement of the retinogeniculate synapse by bouton clustering. Neuron 84, 332–339, doi: 10.1016/j.neuron.2014.08.059 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cheadle L et al. Sensory Experience Engages Microglia to Shape Neural Connectivity through a Non-Phagocytic Mechanism. Neuron 108, 451–468 e459, doi: 10.1016/j.neuron.2020.08.002 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wimmer VC, Broser PJ, Kuner T & Bruno RM Experience-induced plasticity of thalamocortical axons in both juveniles and adults. J Comp Neurol 518, 4629–4648, doi: 10.1002/cne.22483 (2010). [DOI] [PubMed] [Google Scholar]
- 64.Sadaka Y, Weinfeld E, Lev DL & White EL Changes in mouse barrel synapses consequent to sensory deprivation from birth. The Journal of comparative neurology 457, 75–86, doi: 10.1002/cne.10518 (2003). [DOI] [PubMed] [Google Scholar]
- 65.Gunner G et al. Sensory lesioning induces microglial synapse elimination via ADAM10 and fractalkine signaling. Nature neuroscience 22, 1075–1088, doi: 10.1038/s41593-019-0419-y (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chen CC, Bajnath A & Brumberg JC The impact of development and sensory deprivation on dendritic protrusions in the mouse barrel cortex. Cereb Cortex 25, 1638–1653, doi: 10.1093/cercor/bht415 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zuo Y, Yang G, Kwon E & Gan WB Long-term sensory deprivation prevents dendritic spine loss in primary somatosensory cortex. Nature 436, 261–265, doi: 10.1038/nature03715 (2005). [DOI] [PubMed] [Google Scholar]
- 68.Bian WJ, Miao WY, He SJ, Qiu Z & Yu X Coordinated Spine Pruning and Maturation Mediated by Inter-Spine Competition for Cadherin/Catenin Complexes. Cell 162, 808–822, doi: 10.1016/j.cell.2015.07.018 (2015). [DOI] [PubMed] [Google Scholar]
- 69.Midorikawa M & Miyata M Distinct functional developments of surviving and eliminated presynaptic terminals. Proceedings of the National Academy of Sciences of the United States of America 118, doi: 10.1073/pnas.2022423118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yang J et al. Astrocytes contribute to synapse elimination via type 2 inositol 1,4,5-trisphosphate receptor-dependent release of ATP. eLife 5, e15043, doi: 10.7554/eLife.15043 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Morimoto K & Nakajima K Role of the Immune System in the Development of the Central Nervous System. Front Neurosci 13, 916, doi: 10.3389/fnins.2019.00916 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Corriveau RA, Huh GS & Shatz CJ Regulation of class I MHC gene expression in the developing and mature CNS by neural activity. Neuron 21, 505–520, doi: 10.1016/s0896-6273(00)80562-0 (1998). [DOI] [PubMed] [Google Scholar]
- 73.Lee H et al. Synapse elimination and learning rules co-regulated by MHC class I H2-Db. Nature 509, 195–200, doi: 10.1038/nature13154 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Huh GS et al. Functional requirement for class I MHC in CNS development and plasticity. Science 290, 2155–2159, doi: 10.1126/science.290.5499.2155 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Syken J, Grandpre T, Kanold PO & Shatz CJ PirB restricts ocular-dominance plasticity in visual cortex. Science 313, 1795–1800, doi: 10.1126/science.1128232 (2006). [DOI] [PubMed] [Google Scholar]
- 76.Adelson JD et al. Developmental Sculpting of Intracortical Circuits by MHC Class I H2-Db and H2-Kb. Cereb Cortex 26, 1453–1463, doi: 10.1093/cercor/bhu243 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Glynn MW et al. MHCI negatively regulates synapse density during the establishment of cortical connections. Nature neuroscience 14, 442–451, doi: 10.1038/nn.2764 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.McAllister AK Major histocompatibility complex I in brain development and schizophrenia. Biological psychiatry 75, 262–268, doi: 10.1016/j.biopsych.2013.10.003 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Du Clos TW Pentraxins: structure, function, and role in inflammation. ISRN Inflamm 2013, 379040, doi: 10.1155/2013/379040 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Schlimgen AK, Helms JA, Vogel H & Perin MS Neuronal pentraxin, a secreted protein with homology to acute phase proteins of the immune system. Neuron 14, 519–526, doi: 10.1016/0896-6273(95)90308-9 (1995). [DOI] [PubMed] [Google Scholar]
- 81.Tsui CC et al. Narp, a novel member of the pentraxin family, promotes neurite outgrowth and is dynamically regulated by neuronal activity. J Neurosci 16, 2463–2478 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Dodds DC, Omeis IA, Cushman SJ, Helms JA & Perin MS Neuronal pentraxin receptor, a novel putative integral membrane pentraxin that interacts with neuronal pentraxin 1 and 2 and taipoxin-associated calcium-binding protein 49. J Biol Chem 272, 21488–21494, doi: 10.1074/jbc.272.34.21488 (1997). [DOI] [PubMed] [Google Scholar]
- 83.Xu D et al. Narp and NP1 form heterocomplexes that function in developmental and activity-dependent synaptic plasticity. Neuron 39, 513–528, doi: 10.1016/s0896-6273(03)00463-x (2003). [DOI] [PubMed] [Google Scholar]
- 84.Figueiro-Silva J et al. Neuronal pentraxin 1 negatively regulates excitatory synapse density and synaptic plasticity. J Neurosci 35, 5504–5521, doi: 10.1523/JNEUROSCI.2548-14.2015 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bjartmar L et al. Neuronal pentraxins mediate synaptic refinement in the developing visual system. J Neurosci 26, 6269–6281, doi: 10.1523/JNEUROSCI.4212-05.2006 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ma YJ & Garred P Pentraxins in Complement Activation and Regulation. Front Immunol 9, 3046, doi: 10.3389/fimmu.2018.03046 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lu J, Mold C, Du Clos TW & Sun PD Pentraxins and Fc Receptor-Mediated Immune Responses. Front Immunol 9, 2607, doi: 10.3389/fimmu.2018.02607 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kovacs RA et al. Identification of Neuronal Pentraxins as Synaptic Binding Partners of C1q and the Involvement of NP1 in Synaptic Pruning in Adult Mice. Front Immunol 11, 599771, doi: 10.3389/fimmu.2020.599771 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Paolicelli RC et al. Synaptic pruning by microglia is necessary for normal brain development. Science 333, 1456–1458, doi: 10.1126/science.1202529 (2011). [DOI] [PubMed] [Google Scholar]
- 90.Schafer DP et al. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 74, 691–705, doi: 10.1016/j.neuron.2012.03.026 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sipe GO et al. Microglial P2Y12 is necessary for synaptic plasticity in mouse visual cortex. Nat Commun 7, 10905, doi: 10.1038/ncomms10905 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Milinkeviciute G et al. Microglia Regulate Pruning of Specialized Synapses in the Auditory Brainstem. Front Neural Circuits 13, 55, doi: 10.3389/fncir.2019.00055 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Mallya AP, Wang HD, Lee HNR & Deutch AY Microglial Pruning of Synapses in the Prefrontal Cortex During Adolescence. Cereb Cortex 29, 1634–1643, doi: 10.1093/cercor/bhy061 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kopec AM, Smith CJ, Ayre NR, Sweat SC & Bilbo SD Microglial dopamine receptor elimination defines sex-specific nucleus accumbens development and social behavior in adolescent rats. Nature communications 9, 3769, doi: 10.1038/s41467-018-06118-z (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kim HJ et al. Deficient autophagy in microglia impairs synaptic pruning and causes social behavioral defects. Molecular psychiatry, doi: 10.1038/mp.2016.103 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Weinhard L et al. Microglia remodel synapses by presynaptic trogocytosis and spine head filopodia induction. Nat Commun 9, 1228, doi: 10.1038/s41467-018-03566-5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lim TK & Ruthazer ES Microglial trogocytosis and the complement system regulate axonal pruning in vivo. eLife 10, doi: 10.7554/eLife.62167 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Stevens B et al. The classical complement cascade mediates CNS synapse elimination. Cell 131, 1164–1178, doi: 10.1016/j.cell.2007.10.036 (2007). [DOI] [PubMed] [Google Scholar]
- 99.Zhan Y et al. Deficient neuron-microglia signaling results in impaired functional brain connectivity and social behavior. Nat Neurosci 17, 400–406, doi: 10.1038/nn.3641 (2014). [DOI] [PubMed] [Google Scholar]
- 100.Bialas AR & Stevens B TGF-beta signaling regulates neuronal C1q expression and developmental synaptic refinement. Nature neuroscience 16, 1773–1782, doi: 10.1038/nn.3560 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 101.Jung S et al. Analysis of fractalkine receptor CX(3)CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol Cell Biol 20, 4106–4114 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lee M, Lee Y, Song J, Lee J & Chang SY Tissue-specific Role of CX3CR1 Expressing Immune Cells and Their Relationships with Human Disease. Immune Netw 18, e5, doi: 10.4110/in.2018.18.e5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Zhao W, Lu H, Wang X, Ransohoff RM & Zhou L CX3CR1 deficiency delays acute skeletal muscle injury repair by impairing macrophage functions. FASEB J 30, 380–393, doi: 10.1096/fj.14-270090 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Blomster LV et al. CX(3)CR1 deficiency exacerbates neuronal loss and impairs early regenerative responses in the target-ablated olfactory epithelium. Molecular and cellular neurosciences 48, 236–245, doi: 10.1016/j.mcn.2011.08.004 (2011). [DOI] [PubMed] [Google Scholar]
- 105.Ingram G, Hakobyan S, Robertson NP & Morgan BP Complement in multiple sclerosis: its role in disease and potential as a biomarker. Clinical and experimental immunology 155, 128–139, doi: 10.1111/j.1365-2249.2008.03830.x (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Barrington R, Zhang M, Fischer M & Carroll MC The role of complement in inflammation and adaptive immunity. Immunol Rev 180, 5–15, doi: 10.1034/j.1600-065x.2001.1800101.x (2001). [DOI] [PubMed] [Google Scholar]
- 107.Chu Y et al. Enhanced synaptic connectivity and epilepsy in C1q knockout mice. Proc Natl Acad Sci U S A 107, 7975–7980, doi: 10.1073/pnas.0913449107 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lehrman EK et al. CD47 Protects Synapses from Excess Microglia-Mediated Pruning during Development. Neuron 100, 120–134 e126, doi: 10.1016/j.neuron.2018.09.017 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Cong Q, Soteros BM, Wollet M, Kim JH & Sia GM The endogenous neuronal complement inhibitor SRPX2 protects against complement-mediated synapse elimination during development. Nat Neurosci 23, 1067–1078, doi: 10.1038/s41593-020-0672-0 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Linnartz B, Kopatz J, Tenner AJ & Neumann H Sialic acid on the neuronal glycocalyx prevents complement C1 binding and complement receptor-3-mediated removal by microglia. The Journal of neuroscience : the official journal of the Society for Neuroscience 32, 946–952, doi: 10.1523/JNEUROSCI.3830-11.2012 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Gyorffy BA et al. Local apoptotic-like mechanisms underlie complement-mediated synaptic pruning. Proceedings of the National Academy of Sciences of the United States of America 115, 6303–6308, doi: 10.1073/pnas.1722613115 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Scott-Hewitt N et al. Local externalization of phosphatidylserine mediates developmental synaptic pruning by microglia. EMBO J 39, e105380, doi: 10.15252/embj.2020105380 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Schecter RW et al. Experience-Dependent Synaptic Plasticity in V1 Occurs without Microglial CX3CR1. J Neurosci 37, 10541–10553, doi: 10.1523/JNEUROSCI.2679-16.2017 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kaiser N, Patz C, Brachtendorf S, Eilers J & Bechmann I Undisturbed climbing fiber pruning in the cerebellar cortex of CX3 CR1-deficient mice. Glia 68, 2316–2329, doi: 10.1002/glia.23842 (2020). [DOI] [PubMed] [Google Scholar]
- 115.Shi Q et al. Complement C3-Deficient Mice Fail to Display Age-Related Hippocampal Decline. The Journal of neuroscience : the official journal of the Society for Neuroscience 35, 13029–13042, doi: 10.1523/JNEUROSCI.1698-15.2015 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Lowery RL, Tremblay ME, Hopkins BE & Majewska AK The microglial fractalkine receptor is not required for activity-dependent plasticity in the mouse visual system. Glia 65, 1744–1761, doi: 10.1002/glia.23192 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Welsh CA, Stephany CE, Sapp RW & Stevens B Ocular Dominance Plasticity in Binocular Primary Visual Cortex Does Not Require C1q. J Neurosci 40, 769–783, doi: 10.1523/JNEUROSCI.1011-19.2019 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Badimon A et al. Negative feedback control of neuronal activity by microglia. Nature 586, 417–423, doi: 10.1038/s41586-020-2777-8 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Peng J et al. Microglial P2Y12 receptor regulates ventral hippocampal CA1 neuronal excitability and innate fear in mice. Mol Brain 12, 71, doi: 10.1186/s13041-019-0492-x (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Chung WS et al. Astrocytes mediate synapse elimination through MEGF10 and MERTK pathways. Nature 504, 394–400, doi: 10.1038/nature12776 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Lee JH et al. Astrocytes phagocytose adult hippocampal synapses for circuit homeostasis. Nature, doi: 10.1038/s41586-020-03060-3 (2020). [DOI] [PubMed] [Google Scholar]
- 122.Byun YG & Chung WS A Novel In Vitro Live-imaging Assay of Astrocyte-mediated Phagocytosis Using pH Indicator-conjugated Synaptosomes. Journal of visualized experiments : JoVE, doi: 10.3791/56647 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Risher WC et al. Astrocytes refine cortical connectivity at dendritic spines. eLife 3, doi: 10.7554/eLife.04047 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Vainchtein ID et al. Astrocyte-derived interleukin-33 promotes microglial synapse engulfment and neural circuit development. Science 359, 1269–1273, doi: 10.1126/science.aal3589 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Filipello F et al. The Microglial Innate Immune Receptor TREM2 Is Required for Synapse Elimination and Normal Brain Connectivity. Immunity 48, 979–991 e978, doi: 10.1016/j.immuni.2018.04.016 (2018). [DOI] [PubMed] [Google Scholar]
- 126.Jay TR et al. TREM2 is required for microglial instruction of astrocytic synaptic engulfment in neurodevelopment. Glia 67, 1873–1892, doi: 10.1002/glia.23664 (2019). [DOI] [PubMed] [Google Scholar]
- 127.Yasuda M, Nagappan-Chettiar S, Johnson-Venkatesh EM & Umemori H An activity-dependent determinant of synapse elimination in the mammalian brain. Neuron 109, 1333–1349 e1336, doi: 10.1016/j.neuron.2021.03.006 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Nagata S Apoptosis and Clearance of Apoptotic Cells. Annu Rev Immunol 36, 489–517, doi: 10.1146/annurev-immunol-042617-053010 (2018). [DOI] [PubMed] [Google Scholar]
- 129.Li Z et al. Caspase-3 activation via mitochondria is required for long-term depression and AMPA receptor internalization. Cell 141, 859–871, doi: 10.1016/j.cell.2010.03.053 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Jiao S & Li Z Nonapoptotic function of BAD and BAX in long-term depression of synaptic transmission. Neuron 70, 758–772, doi: 10.1016/j.neuron.2011.04.004 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Erturk A, Wang Y & Sheng M Local pruning of dendrites and spines by caspase-3-dependent and proteasome-limited mechanisms. The Journal of neuroscience : the official journal of the Society for Neuroscience 34, 1672–1688, doi: 10.1523/JNEUROSCI.3121-13.2014 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Xu ZX et al. Caspase-2 promotes AMPA receptor internalization and cognitive flexibility via mTORC2-AKT-GSK3beta signaling. Nature communications 10, 3622, doi: 10.1038/s41467-019-11575-1 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Creamer TP Calcineurin. Cell Commun Signal 18, 137, doi: 10.1186/s12964-020-00636-4 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Flavell SW et al. Activity-dependent regulation of MEF2 transcription factors suppresses excitatory synapse number. Science 311, 1008–1012, doi: 10.1126/science.1122511 (2006). [DOI] [PubMed] [Google Scholar]
- 135.Tian X, Kai L, Hockberger PE, Wokosin DL & Surmeier DJ MEF-2 regulates activity-dependent spine loss in striatopallidal medium spiny neurons. Molecular and cellular neurosciences 44, 94–108, doi: 10.1016/j.mcn.2010.01.012 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Wilkerson JR et al. A role for dendritic mGluR5-mediated local translation of Arc/Arg3.1 in MEF2-dependent synapse elimination. Cell reports 7, 1589–1600, doi: 10.1016/j.celrep.2014.04.035 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Zagorska A, Traves PG, Lew ED, Dransfield I & Lemke G Diversification of TAM receptor tyrosine kinase function. Nat Immunol 15, 920–928, doi: 10.1038/ni.2986 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Li T et al. A splicing isoform of GPR56 mediates microglial synaptic refinement via phosphatidylserine binding. EMBO J 39, e104136, doi: 10.15252/embj.2019104136 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Paidassi H et al. C1q binds phosphatidylserine and likely acts as a multiligand-bridging molecule in apoptotic cell recognition. J Immunol 180, 2329–2338, doi: 10.4049/jimmunol.180.4.2329 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Lemke G & Burstyn-Cohen T TAM receptors and the clearance of apoptotic cells. Ann N Y Acad Sci 1209, 23–29, doi: 10.1111/j.1749-6632.2010.05744.x (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Owen KL, Brockwell NK & Parker BS JAK-STAT Signaling: A Double-Edged Sword of Immune Regulation and Cancer Progression. Cancers (Basel) 11, doi: 10.3390/cancers11122002 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Li Z, Okamoto K, Hayashi Y & Sheng M The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell 119, 873–887, doi: 10.1016/j.cell.2004.11.003 (2004). [DOI] [PubMed] [Google Scholar]
- 143.Lees RM, Johnson JD & Ashby MC Presynaptic Boutons That Contain Mitochondria Are More Stable. Front Synaptic Neurosci 11, 37, doi: 10.3389/fnsyn.2019.00037 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Sun T, Qiao H, Pan PY, Chen Y & Sheng ZH Motile axonal mitochondria contribute to the variability of presynaptic strength. Cell reports 4, 413–419, doi: 10.1016/j.celrep.2013.06.040 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Koyama R & Ikegaya Y Microglia in the pathogenesis of autism spectrum disorders. Neuroscience research 100, 1–5, doi: 10.1016/j.neures.2015.06.005 (2015). [DOI] [PubMed] [Google Scholar]
- 146.Pardo CA, Vargas DL & Zimmerman AW Immunity, neuroglia and neuroinflammation in autism. Int Rev Psychiatry 17, 485–495, doi: 10.1080/02646830500381930 (2005). [DOI] [PubMed] [Google Scholar]
- 147.Vargas DL, Nascimbene C, Krishnan C, Zimmerman AW & Pardo CA Neuroglial activation and neuroinflammation in the brain of patients with autism. Ann Neurol 57, 67–81, doi: 10.1002/ana.20315 (2005). [DOI] [PubMed] [Google Scholar]
- 148.Belmonte MK et al. Autism and abnormal development of brain connectivity. The Journal of neuroscience : the official journal of the Society for Neuroscience 24, 9228–9231, doi: 10.1523/JNEUROSCI.3340-04.2004 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Peca J & Feng G Cellular and synaptic network defects in autism. Current opinion in neurobiology 22, 866–872, doi: 10.1016/j.conb.2012.02.015 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Thomas MS, Davis R, Karmiloff-Smith A, Knowland VC & Charman T The over-pruning hypothesis of autism. Developmental science 19, 284–305, doi: 10.1111/desc.12303 (2016). [DOI] [PubMed] [Google Scholar]
- 151.Waites CL & Garner CC Presynaptic function in health and disease. Trends in neurosciences 34, 326–337, doi: 10.1016/j.tins.2011.03.004 (2011). [DOI] [PubMed] [Google Scholar]
- 152.Melom JE & Littleton JT Synapse development in health and disease. Curr Opin Genet Dev 21, 256–261, doi: 10.1016/j.gde.2011.01.002 (2011). [DOI] [PubMed] [Google Scholar]
- 153.Penzes P, Cahill ME, Jones KA, VanLeeuwen JE & Woolfrey KM Dendritic spine pathology in neuropsychiatric disorders. Nature neuroscience 14, 285–293, doi: 10.1038/nn.2741 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Maenner MJ et al. Prevalence of Autism Spectrum Disorder Among Children Aged 8 Years - Autism and Developmental Disabilities Monitoring Network, 11 Sites, United States, 2016. MMWR Surveill Summ 69, 1–12, doi: 10.15585/mmwr.ss6904a1 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Tang G et al. Loss of mTOR-dependent macroautophagy causes autistic-like synaptic pruning deficits. Neuron 83, 1131–1143, doi: 10.1016/j.neuron.2014.07.040 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Hutsler JJ & Zhang H Increased dendritic spine densities on cortical projection neurons in autism spectrum disorders. Brain research 1309, 83–94, doi: 10.1016/j.brainres.2009.09.120 (2010). [DOI] [PubMed] [Google Scholar]
- 157.Voineagu I et al. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature 474, 380–384, doi: 10.1038/nature10110 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Nardone S et al. DNA methylation analysis of the autistic brain reveals multiple dysregulated biological pathways. Transl Psychiatry 4, e433, doi: 10.1038/tp.2014.70 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Bourgeron T From the genetic architecture to synaptic plasticity in autism spectrum disorder. Nature reviews. Neuroscience 16, 551–563, doi: 10.1038/nrn3992 (2015). [DOI] [PubMed] [Google Scholar]
- 160.De Rubeis S et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 515, 209–215, doi: 10.1038/nature13772 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Darnell JC & Richter JD Cytoplasmic RNA-binding proteins and the control of complex brain function. Cold Spring Harbor perspectives in biology 4, a012344, doi: 10.1101/cshperspect.a012344 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Antar LN, Afroz R, Dictenberg JB, Carroll RC & Bassell GJ Metabotropic glutamate receptor activation regulates fragile x mental retardation protein and FMR1 mRNA localization differentially in dendrites and at synapses. The Journal of neuroscience : the official journal of the Society for Neuroscience 24, 2648–2655, doi: 10.1523/JNEUROSCI.0099-04.2004 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Weiler IJ et al. Fragile X mental retardation protein is translated near synapses in response to neurotransmitter activation. Proceedings of the National Academy of Sciences of the United States of America 94, 5395–5400, doi: 10.1073/pnas.94.10.5395 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Huber KM, Gallagher SM, Warren ST & Bear MF Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proceedings of the National Academy of Sciences of the United States of America 99, 7746–7750, doi: 10.1073/pnas.122205699 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Li J, Pelletier MR, Perez Velazquez JL & Carlen PL Reduced cortical synaptic plasticity and GluR1 expression associated with fragile X mental retardation protein deficiency. Molecular and cellular neurosciences 19, 138–151, doi: 10.1006/mcne.2001.1085 (2002). [DOI] [PubMed] [Google Scholar]
- 166.Le Meur N et al. MEF2C haploinsufficiency caused by either microdeletion of the 5q14.3 region or mutation is responsible for severe mental retardation with stereotypic movements, epilepsy and/or cerebral malformations. J Med Genet 47, 22–29, doi: 10.1136/jmg.2009.069732 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Nowakowska BA et al. Severe mental retardation, seizures, and hypotonia due to deletions of MEF2C. American journal of medical genetics. Part B, Neuropsychiatric genetics : the official publication of the International Society of Psychiatric Genetics 153B, 1042–1051, doi: 10.1002/ajmg.b.31071 (2010). [DOI] [PubMed] [Google Scholar]
- 168.Paciorkowski AR et al. MEF2C Haploinsufficiency features consistent hyperkinesis, variable epilepsy, and has a role in dorsal and ventral neuronal developmental pathways. Neurogenetics 14, 99–111, doi: 10.1007/s10048-013-0356-y (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Pfeiffer BE et al. Fragile X mental retardation protein is required for synapse elimination by the activity-dependent transcription factor MEF2. Neuron 66, 191–197, doi: 10.1016/j.neuron.2010.03.017 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Tsai NP, Wilkerson JR, Guo W & Huber KM FMRP-dependent Mdm2 dephosphorylation is required for MEF2-induced synapse elimination. Human molecular genetics 26, 293–304, doi: 10.1093/hmg/ddw386 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Tsai NP et al. Multiple autism-linked genes mediate synapse elimination via proteasomal degradation of a synaptic scaffold PSD-95. Cell 151, 1581–1594, doi: 10.1016/j.cell.2012.11.040 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Hinton VJ, Brown WT, Wisniewski K & Rudelli RD Analysis of neocortex in three males with the fragile X syndrome. Am J Med Genet 41, 289–294, doi: 10.1002/ajmg.1320410306 (1991). [DOI] [PubMed] [Google Scholar]
- 173.Rudelli RD et al. Adult fragile X syndrome. Clinico-neuropathologic findings. Acta Neuropathol 67, 289–295, doi: 10.1007/BF00687814 (1985). [DOI] [PubMed] [Google Scholar]
- 174.He CX & Portera-Cailliau C The trouble with spines in fragile X syndrome: density, maturity and plasticity. Neuroscience 251, 120–128, doi: 10.1016/j.neuroscience.2012.03.049 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Jawaid S et al. Alterations in CA1 hippocampal synapses in a mouse model of fragile X syndrome. Glia 66, 789–800, doi: 10.1002/glia.23284 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Dolen G et al. Correction of fragile X syndrome in mice. Neuron 56, 955–962, doi: 10.1016/j.neuron.2007.12.001 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Cruz-Martin A, Crespo M & Portera-Cailliau C Delayed stabilization of dendritic spines in fragile X mice. The Journal of neuroscience : the official journal of the Society for Neuroscience 30, 7793–7803, doi: 10.1523/JNEUROSCI.0577-10.2010 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Pan F, Aldridge GM, Greenough WT & Gan WB Dendritic spine instability and insensitivity to modulation by sensory experience in a mouse model of fragile X syndrome. Proceedings of the National Academy of Sciences of the United States of America 107, 17768–17773, doi: 10.1073/pnas.1012496107 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Patel AB, Loerwald KW, Huber KM & Gibson JR Postsynaptic FMRP promotes the pruning of cell-to-cell connections among pyramidal neurons in the L5A neocortical network. The Journal of neuroscience : the official journal of the Society for Neuroscience 34, 3413–3418, doi: 10.1523/JNEUROSCI.2921-13.2014 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Isshiki M et al. Enhanced synapse remodelling as a common phenotype in mouse models of autism. Nature communications 5, 4742, doi: 10.1038/ncomms5742 (2014). [DOI] [PubMed] [Google Scholar]
- 181.Darnell JC et al. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell 146, 247–261, doi: 10.1016/j.cell.2011.06.013 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Gabrielli AP, Manzardo AM & Butler MG GeneAnalytics Pathways and Profiling of Shared Autism and Cancer Genes. Int J Mol Sci 20, doi: 10.3390/ijms20051166 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Kwon CH et al. Pten regulates neuronal arborization and social interaction in mice. Neuron 50, 377–388, doi: 10.1016/j.neuron.2006.03.023 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Tsai PT et al. Autistic-like behaviour and cerebellar dysfunction in Purkinje cell Tsc1 mutant mice. Nature 488, 647–651, doi: 10.1038/nature11310 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Ebrahimi-Fakhari D et al. Impaired Mitochondrial Dynamics and Mitophagy in Neuronal Models of Tuberous Sclerosis Complex. Cell reports 17, 1053–1070, doi: 10.1016/j.celrep.2016.09.054 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Wang T et al. Flux of signalling endosomes undergoing axonal retrograde transport is encoded by presynaptic activity and TrkB. Nature communications 7, 12976, doi: 10.1038/ncomms12976 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Wang T et al. Control of autophagosome axonal retrograde flux by presynaptic activity unveiled using botulinum neurotoxin type a. The Journal of neuroscience : the official journal of the Society for Neuroscience 35, 6179–6194, doi: 10.1523/JNEUROSCI.3757-14.2015 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Binotti B et al. The GTPase Rab26 links synaptic vesicles to the autophagy pathway. eLife 4, e05597, doi: 10.7554/eLife.05597 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Hernandez D et al. Regulation of presynaptic neurotransmission by macroautophagy. Neuron 74, 277–284, doi: 10.1016/j.neuron.2012.02.020 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Shehata M, Matsumura H, Okubo-Suzuki R, Ohkawa N & Inokuchi K Neuronal stimulation induces autophagy in hippocampal neurons that is involved in AMPA receptor degradation after chemical long-term depression. The Journal of neuroscience : the official journal of the Society for Neuroscience 32, 10413–10422, doi: 10.1523/JNEUROSCI.4533-11.2012 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Nikoletopoulou V, Sidiropoulou K, Kallergi E, Dalezios Y & Tavernarakis N Modulation of Autophagy by BDNF Underlies Synaptic Plasticity. Cell Metab 26, 230–242 e235, doi: 10.1016/j.cmet.2017.06.005 (2017). [DOI] [PubMed] [Google Scholar]
- 192.Xu ZX et al. Elevated protein synthesis in microglia causes autism-like synaptic and behavioral aberrations. Nature communications 11, 1797, doi: 10.1038/s41467-020-15530-3 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Gkogkas CG et al. Autism-related deficits via dysregulated eIF4E-dependent translational control. Nature 493, 371–377, doi: 10.1038/nature11628 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Santini E et al. Exaggerated translation causes synaptic and behavioural aberrations associated with autism. Nature 493, 411–415, doi: 10.1038/nature11782 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.American Psychiatric Association. & American Psychiatric Association. DSM-5 Task Force. Diagnostic and statistical manual of mental disorders : DSM-5. Fifth edition. edn. [Google Scholar]
- 196.Fusar-Poli P et al. The psychosis high-risk state: a comprehensive state-of-the-art review. JAMA Psychiatry 70, 107–120, doi: 10.1001/jamapsychiatry.2013.269 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Feinberg I Schizophrenia: caused by a fault in programmed synaptic elimination during adolescence? J Psychiatr Res 17, 319–334, doi: 10.1016/0022-3956(82)90038-3 (1982). [DOI] [PubMed] [Google Scholar]
- 198.Zipursky RB, Lim KO, Sullivan EV, Brown BW & Pfefferbaum A Widespread cerebral gray matter volume deficits in schizophrenia. Arch Gen Psychiatry 49, 195–205, doi: 10.1001/archpsyc.1992.01820030027004 (1992). [DOI] [PubMed] [Google Scholar]
- 199.Andreasen NC et al. Progressive brain change in schizophrenia: a prospective longitudinal study of first-episode schizophrenia. Biological psychiatry 70, 672–679, doi: 10.1016/j.biopsych.2011.05.017 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Glantz LA & Lewis DA Decreased dendritic spine density on prefrontal cortical pyramidal neurons in schizophrenia. Arch Gen Psychiatry 57, 65–73, doi: 10.1001/archpsyc.57.1.65 (2000). [DOI] [PubMed] [Google Scholar]
- 201.Kolluri N, Sun Z, Sampson AR & Lewis DA Lamina-specific reductions in dendritic spine density in the prefrontal cortex of subjects with schizophrenia. Am J Psychiatry 162, 1200–1202, doi: 10.1176/appi.ajp.162.6.1200 (2005). [DOI] [PubMed] [Google Scholar]
- 202.Glantz LA & Lewis DA Reduction of synaptophysin immunoreactivity in the prefrontal cortex of subjects with schizophrenia. Regional and diagnostic specificity. Arch Gen Psychiatry 54, 943–952, doi: 10.1001/archpsyc.1997.01830220065010 (1997). [DOI] [PubMed] [Google Scholar]
- 203.Davidsson P et al. The synaptic-vesicle-specific proteins rab3a and synaptophysin are reduced in thalamus and related cortical brain regions in schizophrenic brains. Schizophr Res 40, 23–29, doi: 10.1016/s0920-9964(99)00037-7 (1999). [DOI] [PubMed] [Google Scholar]
- 204.Bitanihirwe BK, Lim MP, Kelley JF, Kaneko T & Woo TU Glutamatergic deficits and parvalbumin-containing inhibitory neurons in the prefrontal cortex in schizophrenia. BMC Psychiatry 9, 71, doi: 10.1186/1471-244X-9-71 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Onwordi EC et al. Synaptic density marker SV2A is reduced in schizophrenia patients and unaffected by antipsychotics in rats. Nature communications 11, 246, doi: 10.1038/s41467-019-14122-0 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Fromer M et al. De novo mutations in schizophrenia implicate synaptic networks. Nature 506, 179–184, doi: 10.1038/nature12929 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Schizophrenia Working Group of the Psychiatric Genomics, C. Biological insights from 108 schizophrenia-associated genetic loci. Nature 511, 421–427, doi: 10.1038/nature13595 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Mukai J et al. Recapitulation and Reversal of Schizophrenia-Related Phenotypes in Setd1a-Deficient Mice. Neuron 104, 471–487 e412, doi: 10.1016/j.neuron.2019.09.014 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Nagahama K et al. Setd1a Insufficiency in Mice Attenuates Excitatory Synaptic Function and Recapitulates Schizophrenia-Related Behavioral Abnormalities. Cell reports 32, 108126, doi: 10.1016/j.celrep.2020.108126 (2020). [DOI] [PubMed] [Google Scholar]
- 210.Jones CA, Watson DJ & Fone KC Animal models of schizophrenia. Br J Pharmacol 164, 1162–1194, doi: 10.1111/j.1476-5381.2011.01386.x (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Stefansson H et al. Common variants conferring risk of schizophrenia. Nature 460, 744–747, doi: 10.1038/nature08186 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Mokhtari R & Lachman HM The Major Histocompatibility Complex (MHC) in Schizophrenia: A Review. J Clin Cell Immunol 7, doi: 10.4172/2155-9899.1000479 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Sekar A et al. Schizophrenia risk from complex variation of complement component 4. Nature, doi: 10.1038/nature16549 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Comer AL et al. Increased expression of schizophrenia-associated gene C4 leads to hypoconnectivity of prefrontal cortex and reduced social interaction. PLoS Biol 18, e3000604, doi: 10.1371/journal.pbio.3000604 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Yilmaz M et al. Overexpression of schizophrenia susceptibility factor human complement C4A promotes excessive synaptic loss and behavioral changes in mice. Nature neuroscience, doi: 10.1038/s41593-020-00763-8 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Sellgren CM et al. Increased synapse elimination by microglia in schizophrenia patient-derived models of synaptic pruning. Nature neuroscience 22, 374–385, doi: 10.1038/s41593-018-0334-7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Kim M et al. Brain gene co-expression networks link complement signaling with convergent synaptic pathology in schizophrenia. Nature neuroscience, doi: 10.1038/s41593-021-00847-z (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Al-Haddad BJS et al. The fetal origins of mental illness. Am J Obstet Gynecol 221, 549–562, doi: 10.1016/j.ajog.2019.06.013 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Bayer TA, Falkai P & Maier W Genetic and non-genetic vulnerability factors in schizophrenia: the basis of the “two hit hypothesis”. J Psychiatr Res 33, 543–548, doi: 10.1016/s0022-3956(99)00039-4 (1999). [DOI] [PubMed] [Google Scholar]
- 220.Bolte S, Girdler S & Marschik PB The contribution of environmental exposure to the etiology of autism spectrum disorder. Cell Mol Life Sci 76, 1275–1297, doi: 10.1007/s00018-018-2988-4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Brown AS The environment and susceptibility to schizophrenia. Prog Neurobiol 93, 23–58, doi: 10.1016/j.pneurobio.2010.09.003 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.van Os J, Kenis G & Rutten BP The environment and schizophrenia. Nature 468, 203–212, doi: 10.1038/nature09563 (2010). [DOI] [PubMed] [Google Scholar]
- 223.Patterson PH Immune involvement in schizophrenia and autism: etiology, pathology and animal models. Behavioural brain research 204, 313–321, doi: 10.1016/j.bbr.2008.12.016 (2009). [DOI] [PubMed] [Google Scholar]
- 224.Fernandez de Cossio L, Guzman A, van der Veldt S & Luheshi GN Prenatal infection leads to ASD-like behavior and altered synaptic pruning in the mouse offspring. Brain, behavior, and immunity 63, 88–98, doi: 10.1016/j.bbi.2016.09.028 (2017). [DOI] [PubMed] [Google Scholar]
- 225.Andoh M et al. Exercise Reverses Behavioral and Synaptic Abnormalities after Maternal Inflammation. Cell reports 27, 2817–2825 e2815, doi: 10.1016/j.celrep.2019.05.015 (2019). [DOI] [PubMed] [Google Scholar]
- 226.Ikezu S et al. Inhibition of colony stimulating factor 1 receptor corrects maternal inflammation-induced microglial and synaptic dysfunction and behavioral abnormalities. Molecular psychiatry, doi: 10.1038/s41380-020-0671-2 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Pekala M, Doliwa M & Kalita K Impact of maternal immune activation on dendritic spine development. Dev Neurobiol, doi: 10.1002/dneu.22804 (2020). [DOI] [PubMed] [Google Scholar]
- 228.McNamara RK, Vannest JJ & Valentine CJ Role of perinatal long-chain omega-3 fatty acids in cortical circuit maturation: Mechanisms and implications for psychopathology. World J Psychiatry 5, 15–34, doi: 10.5498/wjp.v5.i1.15 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Madore C et al. Essential omega-3 fatty acids tune microglial phagocytosis of synaptic elements in the mouse developing brain. Nature communications 11, 6133, doi: 10.1038/s41467-020-19861-z (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Choi GB et al. The maternal interleukin-17a pathway in mice promotes autism-like phenotypes in offspring. Science 351, 933–939, doi: 10.1126/science.aad0314 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Kim S et al. Maternal gut bacteria promote neurodevelopmental abnormalities in mouse offspring. Nature 549, 528–532, doi: 10.1038/nature23910 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Lammert CR et al. Cutting Edge: Critical Roles for Microbiota-Mediated Regulation of the Immune System in a Prenatal Immune Activation Model of Autism. J Immunol 201, 845–850, doi: 10.4049/jimmunol.1701755 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Stellwagen D & Shatz CJ An instructive role for retinal waves in the development of retinogeniculate connectivity. Neuron 33, 357–367, doi: 10.1016/s0896-6273(02)00577-9 (2002). [DOI] [PubMed] [Google Scholar]
- 234.Spinelli L, Black FM, Berg JN, Eickholt BJ & Leslie NR Functionally distinct groups of inherited PTEN mutations in autism and tumour syndromes. J Med Genet 52, 128–134, doi: 10.1136/jmedgenet-2014-102803 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Tilot AK et al. Germline disruption of Pten localization causes enhanced sex-dependent social motivation and increased glial production. Human molecular genetics 23, 3212–3227, doi: 10.1093/hmg/ddu031 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Sarn N et al. Cytoplasmic-predominant Pten increases microglial activation and synaptic pruning in a murine model with autism-like phenotype. Molecular psychiatry, doi: 10.1038/s41380-020-0681-0 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Lord C, Elsabbagh M, Baird G & Veenstra-Vanderweele J Autism spectrum disorder. Lancet 392, 508–520, doi: 10.1016/S0140-6736(18)31129-2 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Logan MA & Freeman MR The scoop on the fly brain: glial engulfment functions in Drosophila. Neuron glia biology 3, 63–74, doi: 10.1017/S1740925X07000646 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Awasaki T et al. Essential role of the apoptotic cell engulfment genes draper and ced-6 in programmed axon pruning during Drosophila metamorphosis. Neuron 50, 855–867, doi: 10.1016/j.neuron.2006.04.027 (2006). [DOI] [PubMed] [Google Scholar]
- 240.Fuentes-Medel Y et al. Glia and muscle sculpt neuromuscular arbors by engulfing destabilized synaptic boutons and shed presynaptic debris. PLoS Biol 7, e1000184, doi: 10.1371/journal.pbio.1000184 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Hakim Y, Yaniv SP & Schuldiner O Astrocytes play a key role in Drosophila mushroom body axon pruning. PloS one 9, e86178, doi: 10.1371/journal.pone.0086178 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Cherra SJ 3rd & Jin Y A Two-Immunoglobulin-Domain Transmembrane Protein Mediates an Epidermal-Neuronal Interaction to Maintain Synapse Density. Neuron 89, 325–336, doi: 10.1016/j.neuron.2015.12.024 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Boulanger A et al. Axonal chemokine-like Orion induces astrocyte infiltration and engulfment during mushroom body neuronal remodeling. Nature communications 12, 1849, doi: 10.1038/s41467-021-22054-x (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Yu XM et al. Plum, an immunoglobulin superfamily protein, regulates axon pruning by facilitating TGF-beta signaling. Neuron 78, 456–468, doi: 10.1016/j.neuron.2013.03.004 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Ellis HM & Horvitz HR Genetic control of programmed cell death in the nematode C. elegans. Cell 44, 817–829, doi: 10.1016/0092-8674(86)90004-8 (1986). [DOI] [PubMed] [Google Scholar]
- 246.Nakajima YI & Kuranaga E Caspase-dependent non-apoptotic processes in development. Cell Death Differ 24, 1422–1430, doi: 10.1038/cdd.2017.36 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Meng L et al. The Cell Death Pathway Regulates Synapse Elimination through Cleavage of Gelsolin in Caenorhabditis elegans Neurons. Cell reports 11, 1737–1748, doi: 10.1016/j.celrep.2015.05.031 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Miller-Fleming TW et al. The DEG/ENaC cation channel protein UNC-8 drives activity-dependent synapse removal in remodeling GABAergic neurons. eLife 5, doi: 10.7554/eLife.14599 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Raiders S et al. Glia actively sculpt sensory neurons by controlled phagocytosis to tune animal behavior. eLife 10, doi: 10.7554/eLife.63532 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]