

Summary
Starch is the most abundant storage carbohydrate in maize kernels and provides calories for humans and other animals as well as raw materials for various industrial applications. Decoding the genetic basis of natural variation in kernel starch content is needed to manipulate starch quantity and quality via molecular breeding to meet future needs. Here, we identified 50 unique single quantitative trait loci (QTLs) for starch content with 18 novel QTLs via single linkage mapping, joint linkage mapping and a genome‐wide association study in a multi‐parent population containing six recombinant inbred line populations. Only five QTLs explained over 10% of phenotypic variation in single populations. In addition to a few large‐effect and many small‐effect additive QTLs, limited pairs of epistatic QTLs also contributed to the genetic basis of the variation in kernel starch content. A regional association study identified five non‐starch‐pathway genes that were the causal candidate genes underlying the identified QTLs for starch content. The pathway‐driven analysis identified ZmTPS9, which encodes a trehalose‐6‐phosphate synthase in the trehalose pathway, as the causal gene for the QTL qSTA4−2, which was detected by all three statistical analyses. Knockout of ZmTPS9 increased kernel starch content and, in turn, kernel weight in maize, suggesting potential applications for ZmTPS9 in maize starch and yield improvement. These findings extend our knowledge about the genetic basis of starch content in maize kernels and provide valuable information for maize genetic improvement of starch quantity and quality.
Keywords: starch content, linkage analysis, association mapping, pathway‐driven, trehalose‐6‐phosphate synthase
Introduction
Maize (Zea mays L.) is the most widely cultivated crop globally on account of its wide variety of uses involving human food, animal fodder, biofuel, starch, sweetener production and so on. Its starch, one of the three major chemical components in maize kernels, constitutes approximately 70% of the dry weight of maize seeds (Hannah and Boehlein, 2017; Hannah and James, 2008). As a result, increasing starch content has great potential for breeding high‐yielding maize cultivars. In addition to its critical role in providing calories for humans and other animals, starch is also a highly valued commodity and is processed by a multibillion‐dollar industry for applications in biofuels, paper manufacturing, high‐fructose corn syrup and pharmaceuticals (Egharevba, 2020; Ranum et al., 2014; Smith, 2008; Zeeman et al., 2010). Therefore, enhancing starch biosynthesis is of great importance for improving grain yields and starch quality in maize, which underpin efforts to feed a growing human population.
In maize kernels, starch is deposited in endosperm cells as insoluble granules in the amyloplast stroma and consists of linear amylose and branched amylopectin (Hannah and Boehlein, 2017; Seung and Smith, 2019). In contrast to the simplicity of the structure of starch, its synthesis is perhaps unexpectedly complex after the point at which sucrose enters the endosperm from photosynthetic tissues (Burrell, 2003; Stitt and Zeeman, 2012). Many carbohydrate metabolic processes—such as sucrose metabolism, sugar metabolism (glycolysis, the pentose phosphate pathway, pyruvate dehydrogenase complex, the tricarboxylic acid cycle) and the trehalose metabolic pathway—either directly or indirectly influence starch synthesis because of some common intermediate products or feedback regulation of some metabolite across these metabolic pathways (Beloff‐Chain and Pocchiari, 1960; Fichtner and Lunn, 2021; Li et al., 2019). For instance, trehalose‐6‐phosphate (T6P) in the trehalose pathway can activate adenosine diphosphate glucose pyrophosphorylase (AGPase) in the starch pathway and interfere with carbon allocation to the sink tissues by inducing starch synthesis in the source tissues (Gomez et al., 2006; Kolbe et al., 2005; Wingler et al., 2000). T6P is synthesized from uridine diphosphate glucose (UDPG) and glucose‐6‐phosphate (G6P) via a trehalose‐6‐phosphate synthase (TPS) and is degraded into trehalose via a trehalose‐6‐phosphate phosphatase (TPP) (Fichtner and Lunn, 2021; Paul et al., 2008). The substrates UDPG and G6P are the common raw materials shared among the starch pathway and other carbohydrate metabolism pathways, and consequently, the alternation of TPS or TPP would change starch content.
As a consequence of advances in mutant characterization and biochemical analysis over the past three decades, much is now known about the starch biosynthetic pathway, especially the clear role of starch synthases (e.g. GBSS, SSI, SSII, SSIII and SSIV), branching enzymes (e.g. BEI, BEIIa and BEIIb) and debranching enzymes (e.g. ISA and PUL) in starch synthesis (Jeon et al., 2010; Nelson and Pan, 1995; Smith and Zeeman, 2020). Mutations in the genes that encode these enzymes could increase sugar content, amylopectin or amylose and have been used in specialty maize breeding programmes. These genes cannot, however, be used for breeding high‐yield maize lines due to their pleiotropic effects associated with defective kernel phenotypes and negative yield performance. Furthermore, a few transcription factors, such as Opaque2 (O2), prolamine‐box binding factor (PBF), ZmNAC128 and ZmNAC130, have been reported to affect starch accumulation in maize endosperm (Zhang et al., 2019; Zhang et al., 2016). Thus, the clarification of the genetic architecture of starch content and subsequent identification of natural variants for starch content would allow breeders to more efficiently design breeding schemes to manipulate this trait in maize kernels.
To dissect the genetic architecture of starch content in maize kernels, many QTL mapping studies have been carried out to identify QTLs associated with starch content in maize kernels using different QTL mapping methods in various bi‐parent populations (Clark et al., 2006; Cook et al., 2012; Dong et al., 2015; Dudley et al., 2007; Guo et al., 2013; Karn et al., 2017; Li et al., 2009; Lin et al., 2019; Liu et al., 2008; Wang et al., 2010; Wassom et al., 2008; Yang et al., 2013; Zhang et al., 2008). These studies isolated a handful of single QTLs, as well as a limited number of epistatic QTLs, that contribute to the genetic basis of starch variation in the bi‐parent populations. Yet, none of them are responsible for the causative variation of starch content in maize kernels. Thanks to advances in next‐generation sequencing (NGS) technologies, the genome‐wide association study (GWAS) has become a powerful tool to effectively and efficiently identify genotype–phenotype associations (Xiao et al., 2017). Indeed, a GWAS identified 4 and 27 loci significantly associated with starch and amylose content, respectively (Li et al., 2018; Liu et al., 2016). However, GWAS often causes false positives due to population structure and may have reduced statistical power for detecting rare alleles because the power for detecting a QTL is determined by the frequency of its associated alleles (Bazakos et al., 2017; Myles et al., 2009; Pritchard et al., 2000; Zhao et al., 2007).
To overcome the disadvantages of GWAS, multi‐parent designs have recently been developed, including nested association mapping (NAM) and multi‐parent advanced generation intercrosses (MAGIC), and have emerged as an efficient way to identify QTLs for complex quantitative traits in plants (Buckler et al., 2009; Cavanagh et al., 2008; Dell Acqua et al., 2015; Huang et al., 2011; Yu et al., 2008). These genetic designs strengthen the mapping power and resolution based on high minor allele frequencies (MAFs) and the rapid decay of linkage disequilibrium, allowing for the discovery of QTLs with greater precision and accuracy (Glowinski and Flint‐Garcia, 2018). For instance, 21 QTLs that co‐localized with fewer than one‐half of previously reported QTLs were identified for starch content in the maize NAM population (Cook et al., 2012). However, this design is time‐consuming and detects fewer QTLs because of less extreme variation among the parental phenotypes (Yu et al., 2008). A more recent multi‐parent design called random‐open‐parent association mapping (ROAM) was developed, in which recombinant inbred line (RIL) populations are derived from several inbred lines crossed in combinations without an a priori requirement to interconnect across populations (Pan et al., 2016, 2017; Xiao et al., 2016). Compared with NAM and MAGIC, the obvious advantage of ROAM is that it saves time in developing mapping populations and takes advantage of existing genetic resources for dissecting the genetic architecture of complex traits of interest in plants. With these advantages, this approach has unravelled the genetic architecture of a wide range of complex traits, such as ear traits, kernel size and weight, and plant architecture (Liu et al., 2017; Pan et al., 2017; Xiao et al., 2016).
To deeply understand the genetic basis of starch synthesis and accumulation in maize kernels, we used six RIL populations that exhibit abundant diversity in starch content, a subset of the ROAM population (Xiao et al., 2016), to dissect the genetic architecture for starch content in maize kernels. Subsequently, we performed regional association mapping using a set of 508 diverse maize inbred lines (AMP508) and a co‐localization analysis between the identified QTLs and the known genes in the carbohydrate metabolism pathway to resolve the candidate genes underlying the detected QTLs. Out of 33 candidate genes, we cloned and characterized ZmTPS9 for qSTA4–2, which encodes a TPS involved in the trehalose metabolism. These findings provide insights into the genetic architecture of starch content in maize kernels and provide informative clues for improving starch quantity and quality via molecular breeding.
Results
Phenotypic variation in starch content
To dissect the genetic architecture of starch content in maize kernels, we used seven inbred lines to develop six RIL populations, DAN340/K22, K22/CI7, K22/BY815, DE3/BY815, BY815/KUI3 and KUI3/B77, with population sizes varying from 176 to 207 lines (Table S1). These seven parental lines exhibit substantial variation in starch content, with a range of 54.0−69.7% (Figure 1a, Table S1). Consequently, all six RIL populations displayed abundant phenotypic variation for starch content (with most populations following normal distributions), especially for the three populations developed from the common parent BY815 which showed up to a 10% range in starch content (Figure 1a, Table S1). An analysis of variance (ANOVA) showed that genotype variance was greater than environmental variance in nearly all populations (Table S1), indicating that phenotypic variations were mainly controlled by genetic factors. In the entire population, starch content exhibited an average broad‐sense heritability of 79.8%, ranging from 71.2% to 87.2%. Thus, the abundant phenotypic variation and high heritability of starch content among the six RIL populations provide a genetic basis for identifying new QTLs in maize.
Figure 1.
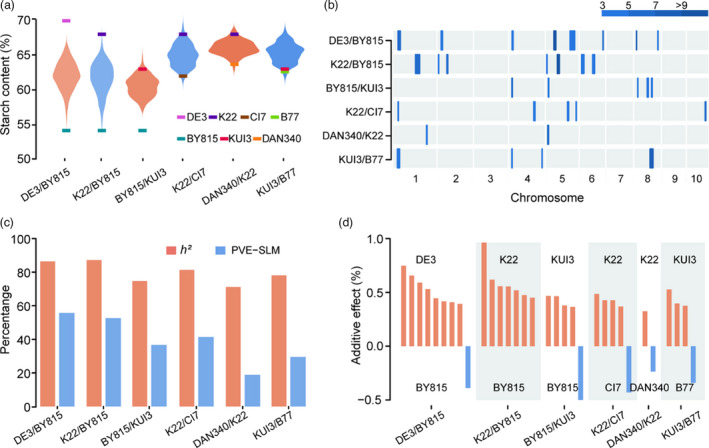
Phenotypic variation in starch content and summary of single QTLs for starch content identified by SLM analysis in six RIL populations. (a) Phenotypic variation in starch content among the six RIL populations. The short, horizontal bars of different colours indicate the starch content values for the seven parental lines. (b) Distribution of single QTLs on chromosomes. QTL regions across the maize genome are represented by confidence intervals, and LOD values are scaled by colour. (c) Broad‐sense heritability (h 2) and total PVE for single QTLs for starch content in each population. (d) Effect size and the origin of the increasing alleles of the identified single QTLs. Orange and blue bars indicate that increasing alleles come from the parents with high and low starch content, respectively, in a given population.
Genetic architecture of starch content dissected via three methods
To systematically identify single QTLs for starch content in maize kernels, we carried out three analyses for detecting QTLs, including single linkage mapping (SLM), joint linkage mapping (JLM) and GWAS. Using six genetic maps with the total length ranging from 1,670.4 to 1,958.6 cM (Pan et al., 2016; Xiao et al., 2016), we performed SLM analysis in each RIL population with the composite interval mapping method (Zeng, 1994). In total, 26 QTLs for starch content were detected, with the QTL number in a population ranging from two to nine (Figure 1b; Table 1; Data set S1). The 2.0‐LOD supporting QTL interval averaged 8.6 cM (8.8 Mb), with a range of 0.9−21.0 cM (0.7−31.6 Mb). The total phenotypic variation explained (PVE) by all identified QTLs in a population averaged 39.1% and ranged from 18.9% (DAN340/K22) to 55.7% (DE3/BY815), far less than broad‐sense heritability (Figure 1c; Table 1). These findings suggest that some minor QTLs for starch content cannot be detected in bi‐parent populations. Of these QTLs, most had moderate additive effects of 0.24−0.96% starch content (Figure 1d). The PVE for each QTL ranged from 3.8% (qSTA5−7 in DE3/BY815) to 18.8% (qSTA5−4 in DE3/BY815), and only 19.2% (5/26) of the QTLs had large effects with PVE ≥ 10% (Figure S1a). These results are consistent with the quantitative nature of starch content, which is known to be controlled by a large number of genes/QTLs with small effects (Glowinski and Flint‐Garcia, 2018). As expected, for 80.8% (21/26) of the QTLs, the alleles from the parent with high starch content in a population had additive effects for increasing starch content (Figure 1d; Data set S1). In addition, the QTL co‐localization analysis among these six populations showed only four QTLs (qSTA1−1, qSTA4−1, qSTA5−2, qSTA8−4) that were present among more than two populations. The high percentage (84.6%, 22/26) of QTLs uniquely detected in a given population underscored the genetic diversity of the founders of the RIL population (Data set S1).
Table 1.
Summary of QTLs for starch content identified via three methods using the BLUP values
| Method | Single QTLs | Epistasis analysis | ||
|---|---|---|---|---|
| QTL number* | Total PVE (%) † | Pairs of epistatic QTLs | Total PVE (%) † | |
| SLM | 26 (2–9) | 18.9–55.7 | None available | None available |
| JLM | 28 | 79.8 | 7 | 2.0 |
| GWAS | 22 | 81.0 | 2 | 1.7 |
SLM, Single Linkage Mapping; JLM, Joint Linkage Mapping.
The number of all QTLs identified via SLM in six RIL populations is shown before brackets, while the range of QTLs identified in a given RIL population are shown within the brackets.
Phenotypic variance explained (PVE) by all single QTLs or all epistatic QTLs.
Subsequently, we identified a total of 28 QTLs (likelihood ratio test, LRT ≥ 2.99) for starch content using the JLM analysis (Figure 2a; Table 1; Data set S1), which collectively explained 79.8% of the phenotypic variation. Compared with the SLM results, the QTL interval was expectedly small, with 82.1% (23/28) of the QTL intervals falling within 5 Mb. These detected QTLs had small estimated effects with each explaining 0.4−8.7% of the phenotypic variance (Data set S1). Three of five QTLs with large effects (PVE ≥ 5%) coincided with large‐effect QTLs detected by SLM. The distribution of allelic effects in the founders varied widely among the QTLs (Data set S1). The effect direction (positive or negative) of most alleles was associated with the starch content of the founders (Figure 2b). When the founder had a high starch content, it had more positive alleles and vice versa. When the founder had a moderate starch content, the number of positive and negative alleles was similar. Notably, BY815 carried the most negative alleles at half of the detected QTLs, which provides a genetic basis for BY815 having the lowest starch content among these founders.
Figure 2.
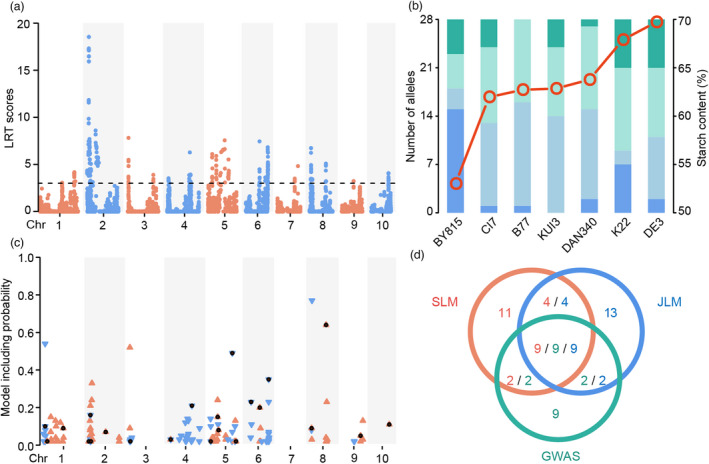
Overview of QTLs for starch content identified by JLM and GWAS methods. (a) Manhattan plot resulting from the JLM results for starch content in maize kernels. The horizontal dashed line shows the threshold of the likelihood ratio test (LRT = 2.99). (b) Distribution of allelic effects on starch content from seven founders at the QTLs identified by JLM. The columns show the number of alleles from the seven founders ordered according to the values of starch content. The red circles represent the starch content of each parent. The dark green and blue shadings show the number of alleles from a founder with the largest positive and negative effects at a given locus, respectively, whereas the light green and blue shadings show the number of alleles from a founder with moderate positive and negative effects, respectively. (c) Chromosome distribution of significant SNPs via GWAS. The orange upward‐facing triangles indicate that the major allele increased starch content relative to the minor allele, the blue downward‐facing triangles indicate the opposite effect, and the black dots indicate the candidate SNPs identified by the backward regression model. (d) Venn diagram of co‐localization between QTLs detected by the three models. Orange, blue and green numbers represent the number of QTLs detected by SLM, JLM and GWAS, respectively.
Finally, we identified 137 SNPs significantly associated with starch content via GWAS using model resampling techniques (Figure 2c; Data set S1). To solve the problem of the redundancy of the significant SNPs caused by linkage disequilibrium among physically close SNPs, a backward regression procedure was performed for the significant SNPs. Ultimately, 22 significant candidate SNPs were retained in the model, of which 59.1% (13/22) were located within a QTL support interval identified by SLM or JLM (Figure 2d). Each of the candidate SNPs explained a small fraction of the phenotypic variation (0.2−8.9%) (Data set S1), consistent with the findings from the JLM analysis. However, all candidate SNPs jointly explained a large portion of phenotypic variation (81.0%) (Table 1). Taken together, we identified 50 unique single QTLs (see the definition of single QTLs in the Methods) for starch content in the six RIL populations via three methods, with nine QTLs identified via three methods and eight QTLs identified via two methods. To test the QTL stability in different environments, we repeated all three statistical analyses in each environment (Data set S2). We found most of the identified QTLs are stable across the environments with 73.1% (19/26), 85.7% (24/28) and 77.2% (17/22) of the QTLs identified using the BLUP values overlapping with the QTLs identified in the individual environment using SLM, JLM and RIL‐GWAS, respectively (Data set S1).
In addition to single QTLs for starch content, we also tested pairwise epistatic effects on starch content using the identified QTLs and candidate SNPs via SLM, JLM and GWAS. We identified only seven pairs of epistatic QTLs between QTLs from the JLM results (PVE = 1.8−2.4%) and two pairs of epistatic QTLs between candidate SNPs from the GWAS results (PVE = 1.7% and 1.8%) (Data set S1). No epistatic effects were detected between the SLM‐identified QTLs. The extremely small PVE percentage explained by all epistatic QTLs (Table 1) indicates that epistasis does not substantially contribute to the genetic basis of starch content variation relative to the additive effects in these RIL populations. Collectively, starch content in maize kernels is controlled by a few large‐effect and many small‐effect additive QTLs as well as a few pairs of epistatic QTLs with small effects.
Regional association mapping identifies candidate genes underlying the detected QTLs
To identify the candidate genes for the detected QTLs, we extracted 86,290 SNPs across the QTL intervals with minor allele frequency (MAF) ≥ 0.05 from ~0.56 million SNPs in AMP508 (Fu et al., 2013; Yang et al., 2011; Yang et al., 2014). The marker‐trait association mapping identified 22 SNPs that were significantly associated with starch content at P ≤ 1.0 × 10−4, which were resolved to five candidate genes (Table 2; Figure 3a−d; Figure S2a,b). All these candidate genes fell within the 2‐LOD supporting interval of the QTLs identified via SLM, and four of them co‐located with significant loci identified via RIL‐GWAS (Figure 3e−g, Figure S2c,d). In addition, the polymorphisms including SNPs and Insertion–Deletions (InDels) among the seven parents of six RIL populations further supported their roles as candidate genes for the detected QTLs (Figure 3b−d, Figure S2a,b, Table S2). The lead SNP (chr1.S_55704731) at the ZmABCI6 (Zm00001d029039) locus, which fell within qSTA1–1 in the DE3/BY815 RIL population and co‐located with chr1.S_56040444 detected via RIL‐based GWAS, showed the most significant association with starch content (Figure 3b,e). The inbred lines carrying the C allele of the lead SNP chr1.S_55704731 had a 3.1% increase in starch content in comparison with those inbred lines carrying T alleles (Figure 3h). ZmABCI6 encodes a putative family member of ABC transporter I, of which the Arabidopsis ortholog (AtNAP7) is essential for Arabidopsis embryogenesis (Widiez et al., 2017; Xu and Moller, 2004). This suggested that ZmABCI6 may affect maize embryogenesis and, in turn, endosperm development and the synthesis and accumulation of kernel chemical composition. The remaining four genes, ZmPSKR1 (Zm00001d001877), ZmACP3 (Zm00001d012818), ZmCPR (Zm00001d010607) and ZmGTL3 (Zm00001d025917), are annotated as non‐starch‐pathway genes (Table 2), and the favourable alleles at the lead SNPs increased starch content by 1.5% (chr2.S_2222544), 1.6% (chr5.S_845872), 6.0% (chr8.S_121752963) and 1.7% (chr10.S_133752084), respectively, in comparison with unfavourable alleles (Figure 3i,j; Figure S2e,f). Notably, ZmACP3 fell within qSTA5−1 in the K22/BY815 RIL population (Figure 3f). It encodes a putative acyl carrier protein in the oil metabolism pathway, which shares a common carbon flux with the starch pathway (Chan and Vogel, 2010; Johnson and Alric, 2013). Zm CPR fell within qSTA8−4 identified in both BY815/KUI3 and KUI3/B77 RIL populations and co‐localized with chr8.S_121752642 detected by RIL‐GWAS (Figure 3g). ZmCRP encodes a putative cysteine‐rich protein containing the PLAC8 conserved motif, which might affect kernel development (Guo et al., 2010; Libault and Stacey, 2010) and, in turn, starch content in maize. These findings provide evidence that non‐starch‐pathway genes contain natural genetic variations and may contribute to phenotypic diversity with respect to maize starch content.
Table 2.
Summary of loci significantly associated with starch content identified by region‐association mapping
| Candidate gene* | Lead SNP † | Chr | Position ‡ | Allele § | MAF | P‐value | Description |
|---|---|---|---|---|---|---|---|
| Zm00001d029039 | chr1.S_55704731 | 1 | 55,704,731 | C /T | 0.16 | 2.7 × 10−6 | ZmABCI6, ABC transporter I family member 6 chloroplastic |
| Zm00001d001877 | chr2.S_2222544 | 2 | 2,222,544 | A /G | 0.34 | 9.2 × 10−6 | ZmPSKR1, Phytosulfokine receptor 1 |
| Zm00001d012818 | chr5.S_845872 | 5 | 845,872 | A/ G | 0.17 | 2.5 × 10−5 | ZmACP3, Acyl carrier protein 3 |
| Zm00001d010607 | chr8.S_121752963 | 8 | 121,752,963 | C /T | 0.06 | 4.2 × 10−5 | ZmCPR, PLAC8 family protein |
| Zm00001d025917 | chr10.S_133752084 | 10 | 133,752,084 | G/ T | 0.20 | 7.1 × 10−5 | ZmGTL3, Probable galacturonosyltransferase‐like 3 |
MAF, minor allele frequency.
A plausible biological candidate gene at the identified locus or the annotated gene nearest to the lead associated SNP.
The SNP with the most significant association at a given locus.
Physical position for SNPs according to the B73 reference genome Version 4.
The underlined nucleotide in a bold font is the favourable allele for each SNP.
Figure 3.
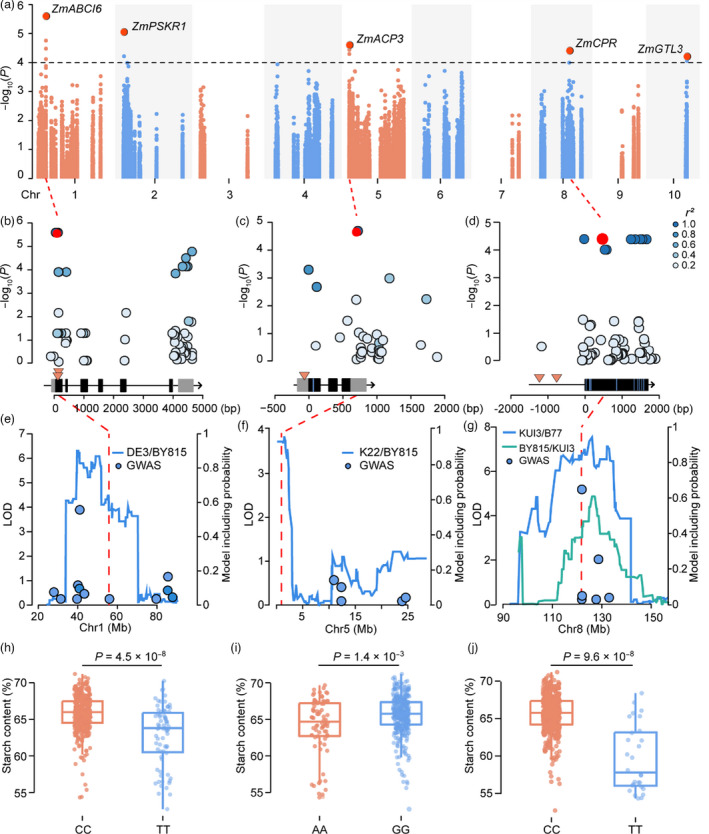
Associations between starch content and ZmABCI6, ZmACP3, and ZmCPR. (a) Manhattan plot of the results from the regional association mapping for starch content in AMP508. The black horizontal dashed line indicates the Bonferroni‐adjusted significance threshold (P = 1.0 × 10–4). The red dots indicate the lead SNPs at each significant locus; the plausible biological candidate gene at each of these loci is shown. (b–d) Associations between the SNPs at the ZmABCI6 (b), ZmACP3 (c) and ZmCPR (d) loci and starch content. The red dots show the lead SNP with the most significant association. Colour coding of the remaining SNPs reflects their extent of linkage disequilibrium (r 2) with the lead SNP. The black and grey boxes above the x axis represent exons and UTRs, respectively. The InDels (orange triangles) and nonsynonymous SNPs (blue vertical lines) in the promoter, UTRs and exons are shown. (e–g) Co‐localization of candidate genes and the QTL or SNPs identified via SLM and RIL‐GWAS. Blue and green lines show the LOD profile of the QTL identified via SLM for the indicated RILs, whereas the blue dots represent the RIL‐GWAS results. The red dashed vertical lines indicate the position of candidate genes. (h–j) Genetic effects of the lead SNPs at the ZmABCI6 (h), ZmACP3 (i) and ZmCPR (j) loci on starch content. The P‐values were calculated based on a two‐tailed Student’s t‐test.
Pathway‐driven analysis discovers candidate genes underlying the detected QTLs
Metabolic pathways related to carbohydrates other than starch, such as sucrose metabolism, sugar metabolism (glycolysis, the pentose phosphate pathway, pyruvate dehydrogenase complex, the tricarboxylic acid cycle) and the trehalose metabolic pathway, also affect starch synthesis indirectly, as these pathways share the same raw material, that is sucrose, and some intermediate metabolites with starch metabolism (Dos et al., 2018; Glawischnig et al., 2002; Spielbauer et al., 2006). Consequently, we identified 471 genes that encode enzymes in maize carbohydrate metabolism pathways from the MaizeCyc database (Data set S3). Of these genes, 28 were located in the QTL intervals identified by at least two of the methods described above, including six genes involved in starch metabolism, seven genes involved in sucrose metabolism, 18 genes involved in sugar metabolism, and two genes involved in trehalose metabolism with three genes shared in two metabolism pathways and one gene shared in three metabolism pathways (Figure S3; Data set S3), suggesting their possibility as candidate genes for these QTLs. For instance, ZmSBEⅠ (Zm00001d014844) on chromosome 5 is located within the QTL qSTA5−3 that was detected by both SLM and JLM (Figure S3) and encodes a 1,4‐α ‐glucan branching enzyme (Fisher et al., 1995) that has a preference for amylose as a substrate by catalysing the formation of α‐1,6‐branch points (Kuriki et al., 1997; Tetlow and Emes, 2014).
Hierarchical clustering analysis using expression data from 78 tissues (Chen et al., 2014) showed that these 28 pathway genes were clustered into four groups (Figure S4a). Nearly, all genes show constitutive expression except three genes in class 3, Zm00001d039066 (starch and sucrose metabolism), Zm00001d008816 (sugar metabolism‐glycolysis) and Zm00001d052060 (trehalose metabolism), which were specifically and highly expressed in maize developing kernels or endosperms (Figure S4b−d), suggesting that there is a high possibility that they represent the candidate genes underlying the corresponding identified QTLs.
ZmTPS9, involved in trehalose metabolism, affects kernel starch content
Zm00001d052060 mapped within the QTL identified via three methods, namely qSTA4–2 (SLM) with 7.4% of PVE in the K22/CI7 RIL population, JLM14 (JLM) with 1.2% of PVE and GWAS070 (GWAS) with 1.6% of PVE (Figure 4a). Its specifically high expression in the endosperm and increased expression during grain filling stages (Figure S4d) indicate that Zm00001d052060 might be a candidate gene for the identified QTL. Zm00001d052060 encodes a putative TPS, and hereafter names ZmTPS9. Amino acid sequence alignment showed that ZmTPS9 protein has a glycosyltransferase domain, a TPS‐like domain, which is required for T6P synthesis from UDPG and G6P, and a C‐terminal TPP‐like domain, which contains two conserved phosphatase boxes and might convert T6P to trehalose (Fichtner and Lunn, 2021; Van Dijck et al., 2002) (Figure S5), indicating that ZmTPS9 is a bifunctional synthase‐phosphatase enzyme. The previous finding that overexpressing otsA encoding for an Escherichia coli TPS in Arabidopsis leads to the enhancement of starch synthesis in leaves (Kolbe et al., 2005) suggests the possibility that ZmTPS9 might affect starch synthesis in maize.
Figure 4.
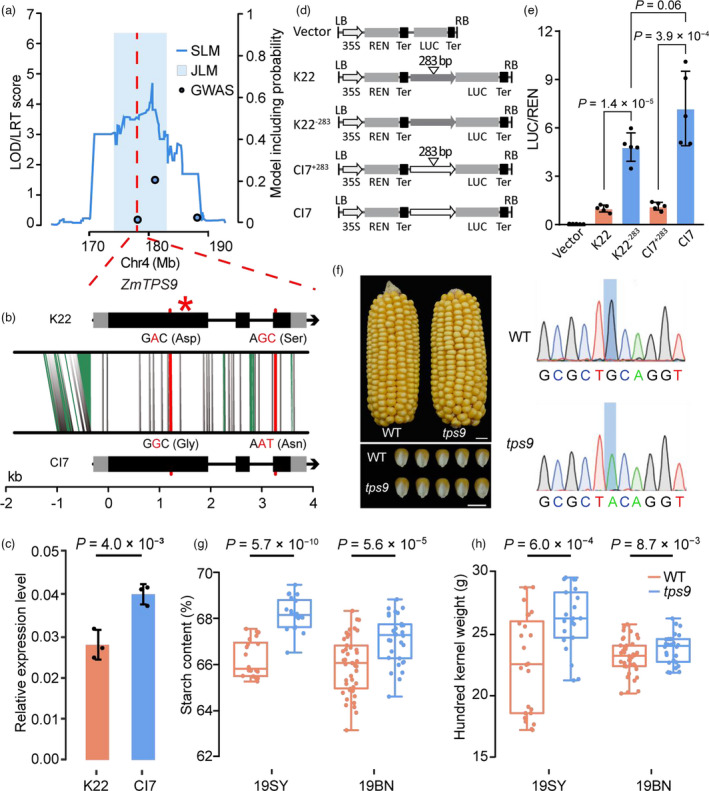
ZmTPS9 affected starch content and kernel weight. (a) LOD profile of qSTA4−2 in the K22/CI7 RIL population, JLM and RIL‐GWAS results at this locus. The red dashed lines indicate the position of ZmTPS9. The blue shading depicts the support interval from JLM with its height indicating the LRT score. (b) Gene structure of ZmTPS9 and sequence comparisons between K22 and CI7. Black and grey boxes represent exons and UTRs, respectively. The grey vertical lines show SNPs in non‐coding regions and synonymous SNPs in exons; the green segments show InDels; the red lines show nonsynonymous SNPs in exons. The red star indicates the mutation position of tps9. The red nucleotides indicate nonsynonymous SNPs between K22 and CI7. (c) Expression pattern of ZmTPS9 in developing kernels at 20 DAP. (d) Constructs used to test the effect of InDel283 on ZmTPS9 expression in transient expression assays in maize leaf protoplasts. K22, K22−283, CI7 and CI7+283 constructs harbour the promoter and 5’UTR of different ZmTPS9 alleles, including 1290 bp from K22, 1007 bp from K22 without a 283‐bp insertion, 1019 bp from CI7 and 1302 bp from CI7 with a 283‐bp insertion. (e) The alleles without a 283‐bp insertion are associated with increased LUC activity in comparison with the alleles with a 283‐bp insertion. The data were normalized with respect to the average values of the K22 construct. (f) Ear and kernel morphologies and genotype of wild type (WT) and tps9. Scale bars: 1 cm. The blue shading indicates the mutated nucleotide. (g–h) Quantification of starch content and hundred kernel weight between WT and tps9. 19SY: Sanya in 2019; 19BN: Bayan Nur in 2019. In c, e, g and h, the P‐values were based on a two‐tailed Student’s t‐test.
ZmTPS9 consists of 3,171 nucleotides with a 2,568‐bp open reading frame, a 293‐bp 5’ untranslated region (UTR) and a 310‐bp 3’UTR with two introns in the B73 reference genome (Figure 4b). Genomic sequencing identified 22 SNPs and seven InDels in the promoter and 5’UTR, three nonsynonymous SNPs (SNP chr4.S_178021716: A‐to‐G change resulting in Asp‐to‐Gly change; SNP chr4.S_178019585 and 178019586: GC‐to‐AT change resulting in Ser‐to‐Asn change) and 15 synonymous SNPs in the coding regions, as well as one SNP and two InDels in 3’UTR between K22 and CI7 (Figure 4b, Figure S6). These variations suggest that ZmTPS9 might affect starch content via coding region changes or transcriptional regulation, whereas no significant association signals were detected at the ZmTPS9 locus when we performed regional‐association mapping in AM508 (Figure 3a). Real‐time quantitative PCR (qPCR) analysis in kernels at 20 days after pollination found that ZmTPS9 expression was high in CI7 (Figure 4c), which allele at qSTA4‐2 increased starch content (Data set S1). This result suggests that the TPS domain in ZmTPS9 might be a positive regulator for starch content. Subsequently, we re‐sequenced the promoter of ZmTPS9 in AM508 and identified 63 polymorphic sites including all SNPs and InDels identified between K22 and CI7. No significant associations were again detected in the promoter and 5’UTR regions (Table S3). These findings suggest that some rare variants such as InDel283 around the transcription initiation site (TSS) of ZmTPS9, for which only one of the 416 tested inbred lines harboured the 283‐bp insertion (Table S3), might be the causal variants for starch content. To test this hypothesis, we performed transient expression assays in maize leaf protoplasts, in which two pairs of fragments with or without the 283‐bp insertion (~1 kb) upstream of the start codon of ZmTPS9 were fused upstream of the luciferase (LUC) gene (Figure 4d). All fragments without 283‐bp insertions exhibited higher LUC activity than did the corresponding fragments with 283‐bp insertions (Figure 4e), suggesting that InDel283 accounts for ZmTPS9 expression differences between K22 and CI7. Considering the K22 allele at qSTA4‐2 of decreasing starch content (Data set S1), we inferred that the 283‐bp insertion around the TSS of ZmTPS9 might decrease starch content via weakening the TPS enzyme activity of ZmTPS9.
To further determine the function of ZmTPS9, we identified an ethyl methanesulphonate (EMS) mutant (ems0390, designated as tps9) carrying a C‐to‐T substitution in the first exon that results in CAG becoming the stop codon TAG, thus introducing a premature stop codon and leading to loss of TPP enzyme activity (Figure 4f; Figure S6). Phenotypic analysis showed that starch content in homozygous tps9 plants was significantly greater than that in their homozygous wild‐type siblings (67.1% versus 66.0%, P = 5.6 × 10−5; 68.2% versus 66.3%, P = 5.7 × 10−10), with an increase in starch content of about 1.1% and 1.9% in Bayan Nur and Sanya, respectively, in 2019 (Figure 4g). Consequently, the hundred kernel weight (HKW) of tps9 increased by 0.9 g (P = 8.7 × 10−3) and 3.5 g (P = 6.0 × 10−4), respectively (Figure 4h). These results indicate that ZmTPS9 function affects starch content and, in turn, kernel weight.
Discussion
Starch content in maize kernels is a complex quantitative trait. Identification of the QTLs or genes controlling the variation in starch content aids in understanding the genetic basis of starch quantity and quality and facilitates genetic improvement of starch content. In this study, we identified 50 unique single QTLs and nine pairs of epistatic QTLs for starch content via SLM, JLM and GWAS using the BLUP values in the six RIL populations. Consistent with previous studies (Dong et al., 2015; Wang et al., 2010; Yang et al., 2013), the small effects of epistatic QTLs and most single additive QTLs indicate that the summation of many QTLs with small effects is responsible for the main contribution to the genetic basis of the variations in kernel starch content. When we consider the QTLs described here and previously identified QTLs for starch content (Alves et al., 2019; Cook et al., 2012; Dong et al., 2015; Guo et al., 2013; Karn et al., 2017; Li et al., 2018; Li et al., 2009; Lin et al., 2019; Liu et al., 2008; Liu et al., 2016; Wang et al., 2010; Wassom et al., 2008; Yang et al., 2013; Zhang et al., 2008), we note that 18 of 50 QTLs (36.0%) were detected only in this study. This finding could be the result of differences in genetic backgrounds, population size, captured recombinant events, environmental effects and QTL analysis methods. In general, QTL mapping populations developed from multiple parents capture greater levels of genetic diversity and recombination events and consequently have higher mapping power and resolution relative to bi‐parent populations to disclose the genetic architecture of complex quantitative traits (Glowinski and Flint‐Garcia, 2018). The fact that more QTLs with relatively narrow genetic intervals were identified by JLM and GWAS in multi‐parent populations than in bi‐parent populations in both our current study and previous studies (Cook et al., 2012; Liu et al., 2017; Pan et al., 2016; Xiao et al., 2016) strongly supports this conclusion. In addition, we identified only five candidate genes underlying the identified QTLs for starch content using regional association mapping with AMP508. This is not surprising, as association mapping using a natural population lacks the power to detect rare alleles caused by less extreme level of the starch content distribution in the population (Auer and Lettre, 2015). Joint linkage association mapping using multi‐parent populations avoids the potentially confounding influence of population structure, raises the allele frequency of a subset of alleles that are rare in the association population and thus is powerful for identifying rare‐allele and small‐effect loci (Auer and Lettre, 2015; Liang et al., 2021). We did identify ZmTPS9 as the causal gene for qSTA4–2, which was further validated by mutant analysis. In addition, the genotype–phenotype association analysis showed no common variants that were significantly associated with starch content, further supporting the possibility that the causative variants of ZmTPS9 might be rare alleles.
Among the five candidate genes that affect starch content identified via regional association mapping, none directly affected starch synthesis but instead affected metabolites related to starch synthesis, for example ZmACP3 and ZmGTL3, or the development of the starch storage organ, for example ZmABCI6 and ZmCRP. In addition to these two kinds of effectors, previous studies have already identified several transcription factors, such as those encoded by the aforementioned O2, PBF, ZmNAC128 and ZmNAC130, that regulate starch synthesis (Zhang et al., 2019; Zhang et al., 2016). O2 and PBF can regulate the expression of critical components in the starch biosynthetic enzyme complex, such as pyruvate orthophosphate dikinases and starch synthase III, and their down‐regulation or loss of function consequently decreases starch content via the down‐regulation of genes in the starch pathway and pentose phosphate pathway (Hennen‐Bierwagen et al., 2009; Zhang et al., 2016). Similarly, ZmNAC128 and ZmNAC130 can regulate the transcription of Bt2, which encodes a small subunit of AGPase, an enzyme responsible for initiating starch synthesis, and down‐regulation of ZmNAC128 and ZmNAC130 results in a significant reduction in starch content by affecting metabolic pathways related to non‐starch carbohydrates (Zhang et al., 2019). These findings suggest that non‐starch‐pathway genes play a critical role in affecting starch synthesis and accumulation in maize kernels in addition to genes encoding the key enzymes in the starch metabolic pathway.
Due to the complex network of many interconnected reactions and metabolites in carbohydrate metabolism, their alteration will affect starch metabolism. A GWAS for amylose content found that enzymes upstream of the starch pathway, such as the sucrose transporter, invertase, phosphoglucomutase, Nudix hydrolase, glycosyltransferases and glycosidases, are responsible for the precursors for starch biosynthesis (Li et al., 2018). Similarly, OsPK2, which encodes a pyruvate kinase involved in glycolysis, affects starch content, starch physicochemical properties and grain weight in rice (Cai et al., 2018). There is also more direct evidence that extensive recycling of glucose occurs before its incorporation into starch via the enzymes of the glycolytic and pentose phosphate pathway, among others, which is based on isotopologue abundance measurements from labelling experiments of developing maize kernels (Glawischnig et al., 2002). The mutations in the starch pathway could also lead to alterations in metabolite levels and enzyme activities in the primary carbohydrate metabolic pathways, that is glycolysis, the pentose phosphate pathway and the tricarboxylic acid cycle (Doehlert and Kuo, 1990; Tobias et al., 1992). These studies also indicate that central carbohydrate metabolism can affect starch content in maize kernels. Consequently, the 28 genes involved in maize carbohydrate metabolism that co‐localized with the identified QTLs (Data set S3) are likely to be candidate genes for these QTLs and might indirectly participate in the starch pathway. We should, however, note that our prediction of candidate genes underlying the detected QTLs was based on the co‐localization analysis; whether these are the causal genes for the QTL requires additional experiments, such as fine mapping, functional validation via overexpression and knocking out the target gene.
T6P, produced from UDPG and G6P via TPS, and degraded into trehalose via TPP, acts as a sugar signal that regulates plant growth and development and potentially regulates the utilization of sucrose for growth and accumulation of storage reserves (Fichtner and Lunn, 2021; Martins et al., 2013; Schluepmann et al., 2003). As expected, it can potentially act as an effective indicator of the pool size of UDPG and G6P (Paul et al., 2008), which are the common raw materials shared among the starch pathway and other carbohydrate metabolism pathways. Consequently, the enhancement of TPS enzyme activity or the loss of TPP enzyme activity in ZmTPS9 might increase the T6P concentration, which might result in more sucrose being transported to the endosperm and, in turn, an increase in starch content and kernel weight. Here, we did find that knockout of the TPP domain in ZmTPS9 resulted in higher starch content and grain yield. This finding suggests potential applications for ZmTPS9 with respect to maize improvement via gene‐editing, even though the molecular mechanism by which ZmTPS9 regulates starch content is currently unknown.
Starch is a key constituent of the mature maize kernel and accounts for ~70% of the grain endosperm. Therefore, it is reasonable that the genes that regulate starch content will also influence kernel weight. These genes include Mn1 (Chourey et al., 2012; Li et al., 2013); ZmDA1 and ZmDAR1 (Xie et al., 2018); AGPase (Li et al., 2011); and GBSS, BEI, and BEIIb (Jiang et al., 2013), as well as ZmTPS9 identified in our study. Overexpression of Mn1, which encodes an endosperm‐expressed cell wall invertase, leads to improved grain yield and starch content by increasing photosynthetic efficiency, accelerating carbohydrate flow from source to sink tissues and speeding up grain filling in transgenic plants (Li et al., 2013). Similarly, ZmDA1 and ZmDAR1 encode ubiquitin receptors that function as negative regulators of cell proliferation during development, and overexpression of mutated ZmDA1 or ZmDAR1 increases sugar imports into the sink organ and increases kernel yield via the up‐regulation of many genes related to starch synthesis, including Sh2, Bt2, and GBSSI (Xie et al., 2018). In addition, this increased expression of starch synthesis‐related genes such as Sh2 and Bt2 enhances seed weight and starch content in transgenic maize (Li et al., 2011). A multigene engineering approach that targeted Sh1, Sh2, Bt2, GBSSIIa, BEI and BEIIb resulted in increased total starch content as well as kernel weight (Jiang et al., 2013). In contrast, when we compared our SLM‐QTL results with a previous QTL study for HKW with four of the same populations (DAN340/K22, K22/CI7, K22/BY815, DE3/BY815), only 21.7% (5/23) of QTLs for starch content were co‐localized with QTLs for HKW. One possibility to explain this finding is that the HKW effects of the QTLs for starch content were too small to discover, for example qSTA4–2 and its causative gene ZmTPS9. This is reasonable, as a weak correlation was observed for starch content and HKW in the same bi‐parent populations (r = −0.02–0.15). These findings indicate that the identification of QTLs for traits involved in HKW, such as starch content, will allow causal variants of HKW to be identified.
Methods
Plant materials and genotyping
Six RIL populations (DAN340/K22, K22/CI7, K22/BY815, DE3/BY815, BY815/KUI3, KUI3/B77) with nearly 200 lines per population were previously developed from seven maize inbred lines (Pan et al., 2016; Xiao et al., 2016). All 1,141 lines together with their parents (Table S1) used in this study have been genotyped previously using the Illumina MaizeSNP50 BeadChip (Ganal et al., 2011), with each population having 11,360 to 14,024 polymorphic SNPs, which were then used to construct high‐density linkage maps that captured 2,100 to 2,683 bins (a genomic region in which no recombination exists) per population, with the total length of the genetic maps ranging from 1,670.4 to 1,958.6 cM (Pan et al., 2016; Xiao et al., 2016). In addition, 1.03 million SNPs within the seven founder lines had been obtained by RNA‐sequencing (RNA‐seq) (Fu et al., 2013) and were used to facilitate the RIL‐GWAS. The SNP positions in the B73 reference genome Version 2 were converted to those in Version 4 using CrossMap Version 2.0.5 (Zhao et al., 2014). The EMS mutant of ZmTPS9 (stock number: ems0390) was ordered from the Maize EMS induced Mutant Database (Lu et al., 2018) and was backcrossed with B73 three times. The mutation site was genotyped by sequencing with the primers listed in Table S4.
Field trials and starch measurement
All RIL lines together with their parents were planted in three environments: Sanya, Hainan (18.2°N, 109.1°E) in 2013 and 2015, and Bayan Nur, Neimeng (40.8°N, 107.4°E) in 2014. The lines were divided into six groups based on different crosses and planted in one‐row plots (2.5‐m rows, 0.67 m between rows) in a completely randomized block design within each group. All plants in each row were self‐pollinated and harvested at maturity. Fifty kernels were bulked for each row with equal numbers collected from the middle part of five well‐grown ears and were ground to a fine powder for starch measurement using a fermentable carbohydrate assay (Xiao et al., 2016; Zhou and Bao, 2012). In brief, 120–150 mg powder was digested with heat‐stable α‐amylase and glucoamylase. The starch was then fermented into ethanol and carbon dioxide (CO2) by yeast, following by heating. Finally, the starch content was calculated as the weight loss owing to fermentation and heat. All of the samples were measured with two sub‐samples analysed in parallel, and the average was used for subsequent analyses.
Phenotypic data analysis
All statistical analyses were performed using R Version 3.1.1 (www.R‐project.org). The R function ‘AOV’ was used to estimate the variances of the starch content. The model for the variance analysis was y = μ + αg + βe + ε, where αg is the effect of the gth line, βe is the effect of the eth environment, and ε is the error. All of the effects were considered to be random. These variance components were used to calculate the broad‐sense heritability as (Knapp et al., 1985), where is the genetic variance, is the residual error, and e is the number of environments. To eliminate the influence of environmental effects, the best linear unbiased predictor (BLUP) value for each line was calculated using a linear mixed model that considered both genotype and environment as random effects in the R function ‘LME4’. The model was yij = μ + ei + fj + εij , where yij is the phenotypic value of individual j in environment i, μ is the grand mean, ei is the effect of different environments, fj is the genetic effect, and εij is the random error. The BLUP values were used for phenotypic description statistics and QTL analysis.
Single linkage mapping
Using high‐density genetic linkage maps of six RIL populations, SLM was performed with composite interval mapping (Zeng, 1994) implemented in Windows QTL Cartographer 2.5 (Wang et al., 2012) for each RIL population. Model 6 of the Composite Interval Mapping module was used to detect QTLs throughout the genome by scanning with a 2.0‐cM interval between markers with a 10‐cM window size. Forward‐backward stepwise regression with five controlling markers was used to control the background from flanking markers. To determine the threshold logarithm of odds (LOD) value for putative QTLs, 1000 permutations were performed, and the resulting LOD score threshold ranged from 2.7 to 3.2 (α = 0.05). For simplicity, we used a LOD score of 3.0 as the global threshold. The confidence interval for the QTL position was estimated with the 2.0‐LOD support interval method according to the study of Liu et al., (2017). The R function ‘LM’ was used to determine total PVE by significant individual QTLs (R Core Team, 2020).
Joint linkage mapping
We combined six RIL populations to perform JLM, and a linear mixed model was used to detect significant recombination blocks (Xiao et al., 2016). The model is as follows: y = Xβ + Zγ + ξ + ε, where Xβ represents fixed effects, Z is an N × P matrix for the parental allelic genotype (N is the total number of lines in the six RIL populations and P is the number of lines used to construct RIL populations), γ is a vector that represents the genetic effects associated with the markers, ξ is a vector that represents the polygenic effects, and ε is a vector that represents the residual errors. The restricted maximum likelihood was used to estimate the parameters. A permutation test of 500 permutated samples was used to determine the threshold of likelihood ratio test scores, and the threshold of likelihood ratio test scores was 2.99 (α = 0.05).
SNP projection and genome‐wide association analysis
To perform a GWAS for starch content, we projected the 1.03 million SNP genotypes of the seven parental lines obtained by RNA‐seq (Fu et al., 2013) onto 1,141 offspring RILs using a two‐step imputation strategy as described (Xiao et al., 2016). Overall, 99,404 genetic blocks were available for the GWAS, which captured the current recombination occurring during the development of the RIL populations and the historical recombination in the seven founders. We carried out the GWAS using a modified stepwise regression method. To control for the effect of a polygenic background, the GWAS was performed one chromosome at a time. For each chromosome, we forced the population effects and the effects of QTLs detected by SLM and JLM on other chromosomes to be included in a general linear model. The residual of this model was then used as the dependent variable to test all SNPs on the current chromosome. We used both forward and backward regressions to select variables, and the cut‐off P‐value for SNPs entering or leaving the model was determined by 500 permutations. The SNPs in the final model were regarded as significant SNPs, and the P‐value was calculated from the marginal F‐values of the SNPs. To reduce SNP redundancy, we performed a final backward regression for the significant SNPs. For SNPs falling within the QTL regions, a backward regression was conducted one QTL at a time, in which the population effects and all other QTLs were fitted to the model. For the remaining SNPs falling outside the QTL regions, a backward regression was conducted by forcing population effects and all QTLs in the model. The median cut‐off P‐value of 10 chromosomes was used as the threshold of the marker resulting in the final backward model.
To take together all QTLs identified via SLM, JLM and GWAS, we defined a single QTL as being present if more than two loci identified via GWAS fell within a QTL interval identified via JLM or SLM, or if more than two QTLs identified via JLM fell within a QTL identified via SLM.
Epistasis analysis
Based on all significant QTLs or loci obtained as described above by SLM, JLM and GWAS, we extracted loci with the peak value LOD score for each QTL. The JLM used parental allelic genotype data, whereas GWAS used biallelic genotype data. For simplicity, all the heterozygous genotypes (<4%) were assigned as missing values to ensure that only homozygous allelic interactions were estimated and tested. Then, we performed epistatic analysis for every pair of peak loci using a two‐way ANOVA in the R environment and took P < 0.05/N (N = all pairwise significant epistatic interactions) as the threshold. Combined with the genotypic information concerning all significant single loci and two‐locus interactions, we used the R function ‘LM’ to estimate their contributions to the phenotypic variation.
Association mapping
An association mapping panel composed of 508 diverse inbred lines (Yang et al., 2011) was grouped into temperate and tropic/subtropic groups and planted in one‐row plots (2.5‐m rows, 0.67 m between rows) in a completely randomized block design within each group in Sanya, Hainan (18.2°N, 109.1°E) in 2009 and in Ya’an, Sichuan (30.0°N, 103.0°E) and again in Sanya, Hainan in 2010. More than six maize ears in each row were self‐pollinated for all lines, and five well‐grown ears in each plot were selected, from which 300 kernels were bulked for phenotyping. Fifty kernels were randomly selected from the bulked kernels to quantify starch content using the aforementioned method. The BLUP values for starch content for each line were used to perform association mapping. For regional association mapping, 86,290 SNPs with MAF of 0.05 and missing rate of 0.2 were extracted from 56,110 SNPs genotyped by the Illumina MaizeSNP50 BeadChip (Ganal et al., 2011; Li et al., 2012) and the 1.03 million SNPs previously identified by RNA‐seq (Fu et al., 2013). To investigate the genomic variants of the associated genes, the promoter, 5’UTR and coding regions were re‐sequenced in the seven parents of six RIL populations (Table S4). For ZmTPS9‐based association mapping, an ~1‐kb promoter fragments of ZmTPS9 were amplified by primers TPS9‐AMseq (Table S4), and the PCR products were sequenced. The sequences were aligned by MAFFT (Katoh et al., 2019) and edited by Bioedit (Hall, 1999). The SNPs and InDels were extracted by TASSEL 5 (Bradbury et al., 2007) and included with the existing SNPs in the coding regions for candidate‐gene association mapping. The marker‐trait association mapping was carried out by using a mixed linear model (MLM, Yu et al., 2006) accounting for population structure and relative kinship (Li et al., 2012; Yang et al., 2011) presented in TASSEL 5 (Bradbury et al., 2007).
RNA extraction and qPCR
Total RNA was isolated from maize kernels at 20 days after pollination using the Plant Total RNA Extraction kit (TianGen, Beijing, China). First‐strand cDNA was synthesized using M5 First Strand cDNA Synthesis Kit (Mei5bio, Beijing, China). qPCR was carried out with a Real‐time PCR Supermix kit (SYBRgreen, with anti‐Taq; Mei5bio) on a 7500 Real‐Time PCR System (Applied Biosystems). Expression levels of ZmTPS9 were normalized to that of maize Actin (Table S4). For each sample, three biological replicates and three technical replicates were used. The relative expression levels were determined using the 2−ΔΔCT method (Livak and Schmittgen, 2001). The primer information for qPCR is shown in Table S4.
Transient expression assays in maize protoplasts
The ~1‐kb promoter fragments (with or without a 283‐bp insertion) of ZmTPS9 were amplified from K22 and CI7 DNA, respectively, using specific primers (Table S4) and then inserted upstream of the LUC gene in vector pGreenII 0800‐LUC that had been cleaved with KpnI and PstI. The Renilla luciferase (REN) gene driven by the 35S promoter in these constructs was used as the internal control to evaluate protoplast transfection efficiency. The isolation of protoplasts from leaves of 14‐day‐old etiolated B73 seedlings, the transformation of constructs into protoplasts using polyethylene glycol‐mediated transformation, the culture of protoplasts and detection of the LUC signal were carried out as previously described (Huang et al., 2018). Relative LUC activity was calculated by normalizing LUC activity to REN activity. Five biological replicates were assayed per plasmid.
Accession numbers
Sequence data from this study can be found in the GenBank data libraries under accession number MW659943–MW660359 for promoter sequence in AMP508, and MW674929 and MW674930 for gene sequence of K22 and CI7 of ZmTPS9.
Conflict of interests
The authors declare that they have no competing interests.
Author contributions
X.Y. conceived the research and designed the experiments. S.H. performed most of the experiments. M.W. and S.H. analysed the data. X.Z., W.C. and Y.L. performed transient expression assays in maize protoplasts. X.S., X.F., H.F., J.X., Y.X. and G.B. performed field trials and phenotyping. S.H., M.W., X.Z. and X.Y. wrote the manuscript. J.L. edited the manuscript. All authors read and approve the manuscript.
Supporting information
Figure S1 Number of QTLs for starch content identified via the SLM method sorted according to PVE value
Figure S2 Associations between starch content and ZmPSKR1 and ZmGTL3
Figure S3 Co‐localization of the identified QTLs and 471 genes involved in carbohydrate metabolism
Figure S4 Expression patterns of 28 pathway genes co‐localized with the detected QTLs
Figure S5 The amino acid sequence alignment of TPS proteins from Arabidopsis thaliana, Oryza sativa, Sorghum bicolor, Triticum aestivum and Zea mays
Figure S6 Sequence comparisons of ZmTPS9 between CI7 and K22
Table S1 Statistical summary, broad‐sense heritability, and variances associated with starch content in six RIL populations.
Table S2 Summary of polymorphisms of five candidate genes in seven parents via resequencing.
Table S3 Associations between the polymorphic sites at the ZmTPS9 locus and starch content in maize kernels.
Table S4 Primers used in this study.
Data set S1 Summary of QTLs for starch content identified using the BLUP values.
Data set S2 Summary of QTLs for starch content identified in each environment.
Data set S3 List of 471 genes encoding enzymes involved in carbohydrate metabolism that are related to starch synthesis.
Acknowledgement
We greatly appreciate Dr. Jianbing Yan at Huazhong Agricultural University for sharing the high‐density genetic linkage maps for the six RIL populations. This study was supported by the National Key Research and Development Program of China (2016YFD0100503), the National Natural Science Foundation of China (31421005, 31722039) and the Beijing Outstanding Young Scientist Program (BJJWZYJH01201910019026).
Hu, S. , Wang, M. , Zhang, X. , Chen, W. , Song, X. , Fu, X. , Fang, H. , Xu, J. , Xiao, Y. , Li, Y. , Bai, G. , Li, J. and Yang, X. (2021) Genetic basis of kernel starch content decoded in a maize multi‐parent population. Plant Biotechnol. J. 10.1111/pbi.13645
References
- Alves, M.L. , Carbas, B. , Gaspar, D. , Paulo, M. , Brites, C. , Mendes‐Moreira, P. , Brites, C.M. et al. (2019) Genome‐wide association study for kernel composition and flour pasting behavior in wholemeal maize flour. BMC Plant Biol. 19, 123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auer, P.L. and Lettre, G. (2015) Rare variant association studies: considerations, challenges and opportunities. Genome Med. 7, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazakos, C. , Hanemian, M. , Trontin, C. , Jimenez‐Gomez, J.M. and Loudet, O. (2017) New strategies and tools in quantitative genetics: How to go from the phenotype to the genotype. Annu. Rev. Plant Biol. 68, 435–455. [DOI] [PubMed] [Google Scholar]
- Beloff‐Chain, A. and Pocchiari, F. (1960) Carbohydrate metabolism. Annu. Rev. Biochem. 29, 295–346. [DOI] [PubMed] [Google Scholar]
- Bradbury, P.J. , Zhang, Z. , Kroon, D.E. , Casstevens, T.M. , Ramdoss, Y. and Buckler, E.S. (2007) TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics, 23, 2633–2635. [DOI] [PubMed] [Google Scholar]
- Buckler, E.S. , Holland, J.B. , Bradbury, P.J. , Acharya, C.B. , Brown, P.J. , Browne, C. , Ersoz, E. et al. (2009) The genetic architecture of maize flowering time. Science, 325, 714–718. [DOI] [PubMed] [Google Scholar]
- Burrell, M.M. (2003) Starch: the need for improved quality or quantity—an overview. J. Exp. Bot. 54, 451–456. [DOI] [PubMed] [Google Scholar]
- Cai, Y. , Li, S. , Jiao, G. , Sheng, Z. , Wu, Y. , Shao, G. , Xie, L. et al. (2018) OsPK2 encodes a plastidic pyruvate kinase involved in rice endosperm starch synthesis, compound granule formation and grain filling. Plant Biotechnol. J. 16, 1878–1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavanagh, C. , Morell, M. , Mackay, I. and Powell, W. (2008) From mutations to MAGIC: Resources for gene discovery, validation and delivery in crop plants. Curr. Opin. Plant Biol. 11, 215–221. [DOI] [PubMed] [Google Scholar]
- Chan, D.I. and Vogel, H.J. (2010) Current understanding of fatty acid biosynthesis and the acyl carrier protein. Biochem. J. 430, 1–19. [DOI] [PubMed] [Google Scholar]
- Chen, J. , Zeng, B. , Zhang, M. , Xie, S. , Wang, G. , Hauck, A. and Lai, J. (2014) Dynamic transcriptome landscape of maize embryo and endosperm development. Plant Physiol. 166, 252–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chourey, P.S. , Li, Q. and Cevallos‐Cevallos, J. (2012) Pleiotropy and its dissection through a metabolic gene Miniature1 (Mn1) that encodes a cell wall invertase in developing seeds of maize. Plant Sci. 184, 45–53. [DOI] [PubMed] [Google Scholar]
- Clark, D. , Dudley, J.W. , Rocheford, T.R. and Ledeaux, J.R. (2006) Genetic analysis of corn kernel chemical composition in the random mated 10 generation of the cross of generations 70 of IHO × ILO. Crop Sci. 46, 807–819. [Google Scholar]
- Cook, J.P. , Mcmullen, M.D. , Holland, J.B. , Tian, F. , Bradbury, P. , Ross‐Ibarra, J. , Buckler, E.S. et al. (2012) Genetic architecture of maize kernel composition in the nested association mapping and inbred association panels. Plant Physiol. 158, 824–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dell Acqua, M. , Gatti, D.M. , Pea, G. , Cattonaro, F. , Coppens, F. , Magris, G. , Hlaing, A.L. et al. (2015) Genetic properties of the MAGIC maize population: a new platform for high definition QTL mapping in Zea mays . Genome Biol. 16, 167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doehlert, D.C. and Kuo, T.M. (1990) Sugar metabolism in developing kernels of starch‐deficient endosperm mutants of maize. Plant Physiol. 92, 990–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong, Y. , Zhang, Z. , Shi, Q. , Wang, Q. , Zhou, Q. and Li, Y. (2015) QTL identification and meta‐analysis for kernel composition traits across three generations in popcorn. Euphytica, 204, 649–660. [Google Scholar]
- Dos, A.L. , Pandey, P.K. , Moraes, T.A. , Feil, R. , Lunn, J.E. and Stitt, M. (2018) Feedback regulation by trehalose 6‐phosphate slows down starch mobilization below the rate that would exhaust starch reserves at dawn in Arabidopsis leaves. Plant Direct, 2, e78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudley, J.W. , Clark, D. , Rocheford, T.R. and Ledeaux, J.R. (2007) Genetic analysis of corn kernel chemical composition in the random mated 7 generation of the cross of generations 70 of IHP × ILP. Crop Sci. 47, 45–57. [Google Scholar]
- Egharevba, H.O. (2020) Chemical properties of starch and its application in the food industry. In Chemical Properties of Starch ( Emeje, M. , ed), pp. 63–88. London: IntecOpen. [Google Scholar]
- Fichtner, F. and Lunn, J.E. (2021) The role of trehalose 6‐Phosphate (Tre6P) in plant metabolism and development. Annu. Rev. Plant Biol. 72, 3.1–3.24. [DOI] [PubMed] [Google Scholar]
- Fisher, D.K. , Kim, K. , Gao, M. , Boyer, C.D. and Guiltinan, M.J. (1995) A cDNA encoding starch branching enzyme I from maize endosperm. Plant Physiol. 108, 1313–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu, J. , Cheng, Y. , Linghu, J. , Yang, X. , Kang, L. , Zhang, Z. , Zhang, J. et al. (2013) RNA sequencing reveals the complex regulatory network in the maize kernel. Nat. Commun. 4, 2832. [DOI] [PubMed] [Google Scholar]
- Ganal, M.W. , Durstewitz, G. , Polley, A. , Bérard, A. , Buckler, E.S. , Charcosset, A. , Clarke, J.D. et al. (2011) A large maize (Zea mays L.) SNP genotyping array: Development and germplasm genotyping, and genetic mapping to compare with the B73 reference genome. PLoS One, 6, e28334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glawischnig, E. , Gierl, A. , Tomas, A. , Bacher, A. and Eisenreich, W. (2002) Starch biosynthesis and intermediary metabolism in maize kernels. Quantitative analysis of metabolite flux by nuclear magnetic resonance. Plant Physiol. 130, 1717–1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glowinski, A. and Flint‐Garcia, S. (2018) Germplasm Resources for Mapping Quantitative Traits in Maize. Cham: Springer International Publishing. [Google Scholar]
- Gomez, L.D. , Baud, S. , Gilday, A. , Li, Y. and Graham, I.A. (2006) Delayed embryo development in the ARABIDOPSIS TREHALOSE‐6‐PHOSPHATE SYNTHASE 1 mutant is associated with altered cell wall structure, decreased cell division and starch accumulation. Plant J. 46, 69–84. [DOI] [PubMed] [Google Scholar]
- Guo, M. , Rupe, M.A. , Dieter, J.A. , Zou, J. , Spielbauer, D. , Duncan, K.E. , Howard, R.J. et al. (2010) Cell number regulator1 affects plant and organ size in maize: implications for crop yield enhancement and heterosis. Plant Cell, 22, 1057–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, Y. , Yang, X. , Chander, S. , Yan, J. , Zhang, J. , Song, T. and Li, J. (2013) Identification of unconditional and conditional QTL for oil, protein and starch content in maize. Crop J. 1, 34–42. [Google Scholar]
- Hall, T.A. (1999) BioEdit: A user‐friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Vol. 41, 95–98. [Google Scholar]
- Hannah, L.C. and Boehlein, S. (2017) Starch biosynthesis in maize endosperm. In Maize Kernel Development ( Larkins, B.A. ed), pp. 149–159. Wallingford: CABI PUBLISHING‐CAB INT. [Google Scholar]
- Hannah, L.C. and James, M. (2008) The complexities of starch biosynthesis in cereal endosperms. Curr. Opin. Biotechnol. 19, 160–165. [DOI] [PubMed] [Google Scholar]
- Hennen‐Bierwagen, T.A. , Lin, Q. , Grimaud, F. , Planchot, V. , Keeling, P.L. , James, M.G. and Myers, A.M. (2009) Proteins from multiple metabolic pathways associate with starch biosynthetic enzymes in high molecular weight complexes: a model for regulation of carbon allocation in maize amyloplasts. Plant Physiol. 149, 1541–1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, C. , Sun, H. , Xu, D. , Chen, Q. , Liang, Y. , Wang, X. , Xu, G. et al. (2018) ZmCCT9 enhances maize adaptation to higher latitudes. Proc. Natl Acad. Sci. USA, 115, E334–E341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, X. , Paulo, M.J. , Boer, M. , Effgen, S. , Keizer, P. , Koornneef, M. and van Eeuwijk, F.A. (2011) Analysis of natural allelic variation in Arabidopsis using a multiparent recombinant inbred line population. Proc. Natl Acad. Sci. USA, 108, 4488–4493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon, J. , Ryoo, N. , Hahn, T. , Walia, H. and Nakamura, Y. (2010) Starch biosynthesis in cereal endosperm. Plant Physiol. Biochem., 48, 383–392. [DOI] [PubMed] [Google Scholar]
- Jiang, L. , Yu, X. , Qi, X. , Yu, Q. , Deng, S. , Bai, B. , Li, N. et al. (2013) Multigene engineering of starch biosynthesis in maize endosperm increases the total starch content and the proportion of amylose. Transgenic Res. 22, 1133–1142. [DOI] [PubMed] [Google Scholar]
- Johnson, X. and Alric, J. (2013) Central carbon metabolism and electron transport in Chlamydomonas reinhardtii: metabolic constraints for carbon partitioning between oil and starch. Eukaryot Cell, 12, 776–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karn, A. , Gillman, J.D. and Flint‐Garcia, S.A. (2017) Genetic analysis of teosinte alleles for kernel composition traits in maize. Genes Genomes Genetics, 7, 1157–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh, K. , Rozewicki, J. and Yamada, K.D. (2019) MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 20, 1160–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knapp, S. , Stroup, W. and Ross, W. (1985) Exact confidence intervals for heritability on a progeny mean basis. Crop Sci. 25, 192–194. [Google Scholar]
- Kolbe, A. , Tiessen, A. , Schluepmann, H. , Paul, M. , Ulrich, S. and Geigenberger, P. (2005) Trehalose 6‐phosphate regulates starch synthesis via posttranslational redox activation of ADP‐glucose pyrophosphorylase. Proc. Natl Acad. Sci. USA, 102, 11118–11123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuriki, T. , Stewart, D.C. and Preiss, J. (1997) Construction of chimeric enzymes out of maize endosperm branching enzymes I and II: activity and properties. J. Biol. Chem. 272, 28999–29004. [DOI] [PubMed] [Google Scholar]
- Li, B. , Liu, H. , Zhang, Y. , Kang, T. , Zhang, L. , Tong, J. , Xiao, L. et al. (2013) Constitutive expression of cell wall invertase genes increases grain yield and starch content in maize. Plant Biotechnol. J. 11, 1080–1091. [DOI] [PubMed] [Google Scholar]
- Li, C. , Huang, Y. , Huang, R. , Wu, Y. and Wang, W. (2018) The genetic architecture of amylose biosynthesis in maize kernel. Plant Biotechnol. J. 16, 688–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, N. , Zhang, S. , Zhao, Y. , Li, B. and Zhang, J. (2011) Over‐expression of AGPase genes enhances seed weight and starch content in transgenic maize. Planta, 233, 241–250. [DOI] [PubMed] [Google Scholar]
- Li, Q. , Yang, X. , Xu, S. , Cai, Y. , Zhang, D. , Han, Y. , Li, L. et al. (2012) Genome‐wide association studies identified three independent polymorphisms associated with alpha‐tocopherol content in maize kernels. PLoS One, 7, e36807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Y. , Wang, W. , Feng, Y. , Tu, M. , Wittich, P.E. , Bate, N.J. and Messing, J. (2019) Transcriptome and metabolome reveal distinct carbon allocation patterns during internode sugar accumulation in different sorghum genotypes. Plant Biotechnol. J. 17, 472–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Y. , Wang, Y. , Wei, M. , Li, X. and Fu, J. (2009) QTL identification of grain protein concentration and its genetic correlation with starch concentration and grain weight using two populations in maize (Zea mays L.). J. Genet. 88, 61–67. [DOI] [PubMed] [Google Scholar]
- Liang, Y. , Liu, H. , Yan, J. and Tian, F. (2021) Natural variation in crops: realized understanding, continuing promise. Annu. Rev. Plant Biol. 72:7.1–7.29. [DOI] [PubMed] [Google Scholar]
- Libault, M. and Stacey, G. (2010) Evolution of FW2.2‐like (FWL) and PLAC8 genes in eukaryotes. Plant Signal. Behav. 5, 1226–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, F. , Zhou, L. , He, B. , Zhang, X. , Dai, H. , Qian, Y. , Ruan, L. et al. (2019) QTL mapping for maize starch content and candidate gene prediction combined with co‐expression network analysis. Theor. Appl. Genet. 132, 1931–1941. [DOI] [PubMed] [Google Scholar]
- Liu, J. , Huang, J. , Guo, H. , Lan, L. , Wang, H. , Xu, Y. , Yang, X. et al. (2017) The conserved and unique genetic architecture of kernel size and weight in maize and rice. Plant Physiol. 175, 774–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, N. , Xue, Y. , Guo, Z. , Li, W. and Tang, J. (2016) Genome‐wide association study identifies candidate genes for starch content regulation in maize kernels. Front. Plant Sci. 7, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Y.Y. , Dong, Y.B. , Niu, S.Z. , Cui, D.Q. , Wang, Y.Z. , Wei, M.G. , Li, X.H. et al. (2008) QTL identification of kernel composition traits with popcorn using both F2:3 and BC2F2 populations developed from the same cross. J. Cereal Sci. 48, 625–631. [Google Scholar]
- Livak, K.J. and Schmittgen, T.D. (2001) Analysis of relative gene expression data using real‐time quantitative PCR and the 2−ΔΔC T method. Methods, 25, 402–408. [DOI] [PubMed] [Google Scholar]
- Lu, X. , Liu, J. , Ren, W. , Yang, Q. , Chai, Z. , Chen, R. , Wang, L. et al. (2018) Gene‐indexed mutations in maize. Mol. Plant, 11, 496–504. [DOI] [PubMed] [Google Scholar]
- Martins, M.C. , Hejazi, M. , Fettke, J. , Steup, M. , Feil, R. , Krause, U. , Arrivault, S. et al. (2013) Feedback inhibition of starch degradation in Arabidopsis leaves mediated by trehalose 6‐phosphate. Plant Physiol. 163, 1142–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myles, S. , Peiffer, J. , Brown, P.J. , Ersoz, E.S. , Zhang, Z. , Costich, D.E. and Buckler, E.S. (2009) Association mapping: critical considerations shift from genotyping to experimental design. Plant Cell, 21, 2194–2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson, O. and Pan, D. (1995) Starch synthesis in maize endosperms. Annu. Rev. Plant Physiol. Plant Mol. Biol. 46, 475–496. [Google Scholar]
- Pan, Q. , Li, L. , Yang, X. , Tong, H. , Xu, S. , Li, Z. , Li, W. et al. (2016) Genome‐wide recombination dynamics are associated with phenotypic variation in maize. New Phytol. 210, 1083–1094. [DOI] [PubMed] [Google Scholar]
- Pan, Q. , Xu, Y. , Li, K. , Peng, Y. , Zhan, W. , Li, W. , Li, L. et al. (2017) The genetic basis of plant architecture in 10 maize recombinant inbred line populations. Plant Physiol. 175, 858–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul, M.J. , Primavesi, L.F. , Jhurreea, D. and Zhang, Y. (2008) Trehalose metabolism and signaling. Annu. Rev. Plant Biol. 59, 417–441. [DOI] [PubMed] [Google Scholar]
- Pritchard, J.K. , Stephens, M. , Rosenberg, N.A. and Donnelly, P. (2000) Association mapping in structured populations. Am. J. Hum. Genet. 67, 170–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team . (2020) R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing. https://www.R‐project.org/ [Google Scholar]
- Ranum, P. , Rosas, J.P.P. and Garcia Casal, M.N. (2014) Global maize production, utilization, and consumption. Ann. N. Y. Acad. Sci. 1312, 105–112. [DOI] [PubMed] [Google Scholar]
- Schluepmann, H. , Pellny, T. , van Dijken, A. , Smeekens, S. and Paul, M. (2003) Trehalose 6‐phosphate is indispensable for carbohydrate utilization and growth in Arabidopsis thaliana . Proc. Natl Acad. Sci. USA, 100, 6849–6854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seung, D. and Smith, A.M. (2019) Starch granule initiation and morphogenesis–progress in Arabidopsis and cereals. J. Exp. Bot. 70, 771–784. [DOI] [PubMed] [Google Scholar]
- Smith, A.M. (2008) Prospects for increasing starch and sucrose yields for bioethanol production. Plant J. 54, 546–558. [DOI] [PubMed] [Google Scholar]
- Smith, A.M. and Zeeman, S.C. (2020) Starch: a flexible, adaptable carbon store coupled to plant growth. Annu. Rev. Plant Biol. 71, 217–245. [DOI] [PubMed] [Google Scholar]
- Spielbauer, G. , Margl, L. , Hannah, L.C. , Romisch, W. , Ettenhuber, C. , Bacher, A. , Gierl, A. et al. (2006) Robustness of central carbohydrate metabolism in developing maize kernels. Phytochemistry, 67, 1460–1475. [DOI] [PubMed] [Google Scholar]
- Stitt, M. and Zeeman, S.C. (2012) Starch turnover: pathways, regulation and role in growth. Curr. Opin. Plant Biol. 15, 282–292. [DOI] [PubMed] [Google Scholar]
- Tetlow, I.J. and Emes, M.J. (2014) A review of starch‐branching enzymes and their role in amylopectin biosynthesis. IUBMB Life, 66, 546–558. [DOI] [PubMed] [Google Scholar]
- Tobias, R.B. , Boyer, C.D. and Shannon, J.C. (1992) Alterations in carbohydrate intermediates in the endosperm of starch‐deficient Maize (Zea mays L.) Genotypes. Plant Physiol. 99, 146–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dijck, P. , Mascorro‐Gallardo, J.O. , De Bus, M. , Royackers, K. , Iturriaga, G. and Thevelein, J.M. (2002) Truncation of Arabidopsis thaliana and Selaginella lepidophylla trehalose‐6‐phosphate synthase unlocks high catalytic activity and supports high trehalose levels on expression in yeast. Biochem. J. 366, 63–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, S. , Basten, C.J. and Zeng, Z.B. (2012) Windows QTL Cartographer 2.5. Raleigh, NC: Department of Statistics, North Carolina State University. [Google Scholar]
- Wang, Y.Z. , Li, J.Z. , Li, Y.L. , Wei, M.G. , Li, X.H. and Fu, J.F. (2010) QTL detection for grain oil and starch content and their associations in two connected F2:3 populations in high‐oil maize. Euphytica, 174, 239–252. [Google Scholar]
- Wassom, J.J. , Wong, J.C. , Martinez, E. , King, J.J. , Debaene, J. , Hotchkiss, J.R. , Mikkilineni, V. et al. (2008) QTL associated with maize kernel oil, protein, and starch concentrations; kernel mass; and grain yield in Illinois high oil × B73 backcross‐derived lines. Crop Sci. 48, 243–252. [Google Scholar]
- Widiez, T. , Ingram, G.C. and Gutierrez‐Marcos, J.F. (2017) Embryo‐endosperm‐sporophyte interactions in maize seeds. In ( Larkins, B.A. , ed), pp. 95–107. Wallingford: CABI Publishing. [Google Scholar]
- Wingler, A. , Fritzius, T. , Wiemken, A. , Boller, T. and Aeschbacher, R.A. (2000) Trehalose induces the ADP‐glucose pyrophosphorylase gene, ApL3, and starch synthesis in Arabidopsis. Plant Physiol. 124, 105–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao, Y. , Liu, H. , Wu, L. , Warburton, M. and Yan, J. (2017) Genome‐wide association studies in maize: praise and stargaze. Mol. Plant, 10, 359–374. [DOI] [PubMed] [Google Scholar]
- Xiao, Y. , Tong, H. , Yang, X. , Xu, S. , Pan, Q. , Qiao, F. , Raihan, M.S. et al. (2016) Genome‐wide dissection of the maize ear genetic architecture using multiple populations. New Phytol. 210, 1095–1106. [DOI] [PubMed] [Google Scholar]
- Xie, G. , Li, Z. , Ran, Q. , Wang, H. and Zhang, J. (2018) Over‐expression of mutated ZmDA1 or ZmDAR1 gene improves maize kernel yield by enhancing starch synthesis. Plant Biotechnol. J. 16, 234–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, X.M. and Moller, S.G. (2004) AtNAP7 is a plastidic SufC‐like ATP‐binding cassette/ATPase essential for Arabidopsis embryogenesis. Proc. Natl Acad. Sci. USA, 101, 9143–9148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, G. , Dong, Y. , Li, Y. , Wang, Q. , Shi, Q. and Zhou, Q. (2013) Verification of QTL for grain starch content and its genetic correlation with oil content using two connected RIL populations in high‐oil maize. PLoS One, 8, e53770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, N. , Lu, Y. , Yang, X. , Huang, J. , Zhou, Y. , Ali, F. , Wen, W. et al. (2014) Genome wide association studies using a new nonparametric model reveal the genetic architecture of 17 agronomic traits in an enlarged maize association panel. PLoS Genet. 10, e1004573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, X. , Gao, S. , Xu, S. , Zhang, Z. , Prasanna, B.M. , Li, L. , Li, J. et al. (2011) Characterization of a global germplasm collection and its potential utilization for analysis of complex quantitative traits in maize. Mol. Breeding, 28, 511–526. [Google Scholar]
- Yu, J. , Holland, J.B. , Mcmullen, M.D. and Buckler, E.S. (2008) Genetic design and statistical power of nested association mapping in maize. Genetics, 178, 539–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, J.M. , Pressoir, G. , Briggs, W.H. , Bi, I.V. , Yamasaki, M. , Doebley, J.F. , Mcmullen, M.D. et al. (2006) A unified mixed‐model method for association mapping that accounts for multiple levels of relatedness. Nat. Genet. 38, 203–208. [DOI] [PubMed] [Google Scholar]
- Zeeman, S.C. , Kossmann, J. and Smith, A.M. (2010) Starch: its metabolism, evolution, and biotechnological modification in plants. Annu. Rev. Plant Biol. 61, 209–234. [DOI] [PubMed] [Google Scholar]
- Zeng, Z.B. (1994) Precision mapping of quantitative trait loci. Genetics, 136, 1457–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, J. , Lu, X.Q. , Song, X.F. , Yan, J.B. , Song, T.M. , Dai, J.R. , Rocheford, T. et al. (2008) Mapping quantitative trait loci for oil, starch, and protein concentrations in grain with high‐oil maize by SSR markers. Euphytica, 162, 335–344. [Google Scholar]
- Zhang, Z. , Dong, J. , Ji, C. , Wu, Y. and Messing, J. (2019) NAC‐type transcription factors regulate accumulation of starch and protein in maize seeds. Proc. Natl Acad. Sci. USA, 116, 11223–11228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Z. , Zheng, X. , Yang, J. , Messing, J. and Wu, Y. (2016) Maize endosperm‐specific transcription factors O2 and PBF network the regulation of protein and starch synthesis. Proc. Natl Acad. Sci. USA, 113, 10842–10847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, H. , Sun, Z. , Wang, J. , Huang, H. , Kocher, J. and Wang, L. (2014) CrossMap: A versatile tool for coordinate conversion between genome assemblies. Bioinformatics, 30, 1006–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, K. , Aranzana, M.J. , Kim, S. , Lister, C. , Shindo, C. , Tang, C. , Toomajian, C. et al. (2007) An Arabidopsis example of association mapping in structured samples. PLoS Genet. 3, e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, L. and Bao, X. (2012). High throughput method for measuring total fermentables in small amount of plant part. U.S. Patent 8, 329,426 P. 2012‐12‐11
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Number of QTLs for starch content identified via the SLM method sorted according to PVE value
Figure S2 Associations between starch content and ZmPSKR1 and ZmGTL3
Figure S3 Co‐localization of the identified QTLs and 471 genes involved in carbohydrate metabolism
Figure S4 Expression patterns of 28 pathway genes co‐localized with the detected QTLs
Figure S5 The amino acid sequence alignment of TPS proteins from Arabidopsis thaliana, Oryza sativa, Sorghum bicolor, Triticum aestivum and Zea mays
Figure S6 Sequence comparisons of ZmTPS9 between CI7 and K22
Table S1 Statistical summary, broad‐sense heritability, and variances associated with starch content in six RIL populations.
Table S2 Summary of polymorphisms of five candidate genes in seven parents via resequencing.
Table S3 Associations between the polymorphic sites at the ZmTPS9 locus and starch content in maize kernels.
Table S4 Primers used in this study.
Data set S1 Summary of QTLs for starch content identified using the BLUP values.
Data set S2 Summary of QTLs for starch content identified in each environment.
Data set S3 List of 471 genes encoding enzymes involved in carbohydrate metabolism that are related to starch synthesis.