

SUMMARY
GPCR functional selectivity open new opportunities for the design of safer drugs. Ligands orchestrate GPCR signaling cascades by modulating the receptor conformational landscape. Our study provides insights into the dynamic mechanism enabling opioid ligands to preferentially activate the G protein over the β-arrestin pathways through the μ-opioid receptor (μOR). We combine functional assays in living cells, solution NMR spectroscopy and enhanced-sampling molecular dynamic simulations to identify the specific μOR conformations induced by G protein-biased agonists. In particular, we describe the dynamic and allosteric communications between the ligand-binding pocket and the receptor intracellular domains, through conserved motifs in class A GPCRs. Most strikingly, the biased agonists trigger μOR conformational changes in the intracellular loop 1 and helix 8 domains, which may impair β-arrestin binding or signaling. The findings may apply to other GPCR families and provide key molecular information that could facilitate the design of biased ligands.
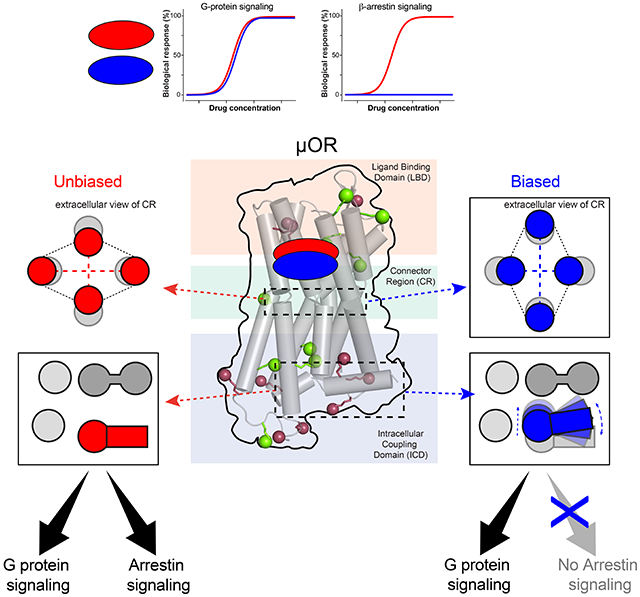
Graphical Abstract
eTOC blurb
Biased ligands of GPCRs offer new drug design strategies to enhance beneficial drug actions while reducing side effects. Cong et al. combined molecular simulations, NMR spectroscopy and functional assays to uncover the molecular mechanism of ligand bias in the μ-opioid receptor, which provides structural basis for designing better opioid analgesics.
INTRODUCTION
Cell signaling relies on second messenger systems that are modulated by G protein-coupled receptors (GPCRs) in a ligand-specific manner. GPCRs are known for the complexity of their signaling pathways and conformational landscape. Ligands may preferentially activate or inhibit distinct signaling pathways, by changing the conformations of the GPCR (Weis and Kobilka, 2018). This is known as functional selectivity (or ligand bias), which provides fine regulations of GPCR functions and new drug design opportunities. Functional selectivity of the μ-opioid receptor (μOR) is among the most studied, in a global effort to develop safer analgesics. Opioid analgesics are efficacious and inexpensive, however, their severe side effects caused the ongoing opioid epidemic starting from the 1990s. A number of studies from 2005 to 2010 associated major opioid side effects with the β-arrestin signaling pathways (reviewed in (Raehal et al., 2011)), which has driven over a decade of research for G protein-biased μOR agonists. This led to the discovery of oliceridine (TRV130) (DeWire et al., 2013) and PZM21 (Manglik et al., 2016), two G protein-biased agonists showing less side effects than morphine in early studies. Oliceridine was approved by the FDA in 2020 for pain management, however, it still has typical opioid side effects such as nausea, vomiting, dizziness, headache, and constipation. There are high expectations on PZM21, which outperformed oliceridine and morphine in mice (Manglik et al., 2016). Nevertheless, recent findings argue that G-protein selectivity may improve analgesia and tolerance but not necessarily the side effects (Kliewer et al., 2019). Opioid-induced respiratory depression and constipation could be independent of β-arrestin signaling (Kliewer et al., 2020). It was suggested that the favorable therapeutic profiles of oliceridine and PZM21 are due to low efficacy rather than functional selectivity (Gillis et al., 2020; Yudin and Rohacs, 2019). Yet, G protein selectivity correlates with broader therapeutic windows, enabling better separations of the beneficial and adverse opioid effects (Schmid et al., 2017). A potential explanation for these discrepancies is the weak selectivity of these ligands compared to the complexity of the GPCR signaling network (Conibear and Kelly, 2019). The diversity of the systems used may also be a source of inconsistency. There is a pressing need of strongly biased and highly specific ligands, to probe the μOR signaling network for more insights. Finding such probes demands understanding the molecular mechanism of functional selectivity, which is intrinsic to the GPCR. However, GPCRs are not static on-off switches but complex molecular machines which operate through strictly regulated motions (Weis and Kobilka, 2018). Functional selectivity relies on the dynamic equilibrium of GPCR conformations, which is so far poorly understood.
X-ray crystallography and cryoEM have successfully captured inactive and active μOR states (Huang et al., 2015; Koehl et al., 2018; Manglik et al., 2012). Yet, they represent essentially a few snapshots of the vast landscape of GPCR conformations. The active states of ternary complexes exhibit a large outward displacement of the transmembrane helix 6 (TM6), which requires stabilization by G proteins or G protein-mimetics (reviewed in (Weis and Kobilka, 2018)). Conformational changes induced by ligand-binding, however, are very subtle and dynamic, as revealed by recent NMR studies on the β2-adrenergic receptor (β2AR) (Manglik et al., 2015; Nygaard et al., 2013) and on μOR (Okude et al., 2015; Sounier et al., 2015). Agonist binding initiate slight conformational changes in the ligand-binding domain (LBD), sufficient to trigger long-range conformational changes via the connector region (CR) until the intracellular coupling domains (ICD), in an allosteric and dynamic manner. Signaling partners (e.g. G proteins or β-arrestins) couple to the activated ICD and induce further large-scale opening of ICD to a fully-active state. This activation process is common in various GPCRs such as the leukotriene B4 receptor BLT2 (Casiraghi et al., 2016), the β1-adrenergic receptor (β1AR) (Isogai et al., 2016), the adenosine A2A receptor (A2AR) (Clark et al., 2017; Eddy et al., 2018; Ye et al., 2016) and the muscarinic M2 receptor (M2R) (Xu et al., 2019). However, detailed activation dynamics, especially the mechanism of functional selectivity, are difficult to capture by X-ray crystallography or cryoEM. NMR spectroscopy has proven to be particularly suitable for monitoring subtle dynamic conformational transitions during GPCR activation (Kofuku et al., 2014; Liang et al., 2018; Shimada et al., 2019). MD on the other side has provided atomic-level detailed insights (Latorraca et al., 2017), such as long-timescale MD of β2AR deactivation and activation (Dror et al., 2011; Kohlhoff et al., 2014), as well as enhanced-sampling MD of the activation of M2R (Miao et al., 2013) and A2AR (Lovera et al., 2019). Here, we combined a thorough functional investigation of agonists, NMR spectroscopy and molecular dynamics (MD) simulations to obtain atomistic-level descriptions on μOR agonism. To this end, we have established a dual isotope labeling NMR scheme for μOR based on our previous study (Sounier et al., 2015). For the MD, we used the REST2 enhanced sampling scheme (REST2-MD, see METHOD DETAILS), which has proven efficient in monitoring GPCR activation (Cong et al., 2019; Cong et al., 2018; Cong and Golebiowski, 2018; Sena et al., 2017).
We studied five chemically distinct μOR agonists (Figure 1A): DAMGO, a well-characterized μOR-specific peptide (Emmerson et al., 1994; Handa et al., 1981); buprenorphine, a semi-synthetic thebaine analogue and partial agonist (Cowan et al., 1977a; Cowan et al., 1977b); BU72, a potent buprenorphine derivative (Neilan et al., 2004); as well as the aforementioned biased agonists, oliceridine and PZM21. By comparing the effects of these agonists on the μOR conformational dynamics, we provide here insights into the molecular mechanism of μOR functional selectivity.
Figure 1. Functional characterizations of the μOR agonists.

(A) Chemical structures of the five agonists. (B—F) Dose-dependent response curves of the agonists in (B) competitive binding against fluorescent naltrexone in living cells, (C) activating Gαi1, (D) inducing GRK2 recruitment, (E) inducing β-arrestin1 recruitment, and (F) inducing μOR internalization. Color code in (B—F) is the same than (A). Data shown are the means ± S.D. of a representative experiment performed in triplicates normalized to the maximal response induced by DAMGO and fitted using an operational model of agonism.
RESULTS
Opioids signaling in living cells
We first characterized the functional profiles of the five opioids in cell-based assays for their abilities to bind the target, to activate G proteins (Gi/o) and GPCR kinase 2/5 (GRK2/5), to recruit β-arrestins 1 and 2, and to trigger μOR internalization. Using advanced fluorescence methods, we probed i) competitive ligand binding to the target cells and μOR against naltrexone, ii) dissociation of the G protein heterotrimer, iii) ligand-induced μOR interactions with GRK2/5 or iv) with β-arrestins-1/2, and v) diminution of cell-surface μORs (internalization) (Figures 1B-1F and S1; Table 1).
All the five opioids behaved as agonists in the Gi1-2-3 and Goa-b activation assays (Figures 1C and S1A—S1D). PZM21 and oliceridine behaved as full agonists in our G protein assays, similar to the data by Ehrlich et al. using the same assays (Ehrlich et al., 2019). However, other studies described them as partial agonists (Gillis et al., 2020; Hill et al., 2018; Yudin and Rohacs, 2019). These discrepancies highlight how functional assay outcomes may vary due to overexpression, assay conditions, assay readout amplification, or the presence of high receptor reserve (Kelly 2013), whereby an agonist may achieve maximal response by occupying only a fraction of the existing receptor population. Compared to DAMGO (our reference ligand), buprenorphine was the only partial agonist in our assays, showing 63 ± 9% to 83 ± 10 % of efficacy even at saturating concentration (Figures 1B—1C and S1A—S1D; Table 1). This agrees with previous cell signaling assays (Ehrlich et al., 2019; McPherson et al., 2010; Traynor and Nahorski, 1995). Concerning the recruitments of GRK2/5 and β-arrestin-1/2, and μOR internalization, DAMGO and BU72 showed comparable efficacies with the latter being more potent, whereas oliceridine, PZM21 and buprenorphine showed nearly no response (Figures 1D-1F and S1E; Table 1). Therefore, oliceridine, PZM21 and buprenorphine were clearly G protein biased. Note that the bias factors could not be determined since no transduction coefficient (log(τ/KA) values could be calculated due to their lack of measurable response in the β-arrestin-1/2, GRK2/5 and internalization assays. Because our functional assays were to be compared with the NMR and REST2-MD data which provide rather qualitative information, we did not further quantify the ligands’ efficacy or bias. For the purpose of this study, we simply classified DAMGO and BU72 as unbiased full agonists, oliceridine and PZM21 as G protein-biased full agonists, and buprenorphine as a G protein-biased partial agonist.
Development of multidomain NMR sensors for allosteric GPCR motions.
We used a previously established mouse μOR construct for NMR spectroscopy, which contained an M721.36T mutation (superscript refers to the Ballesteros-Weinstein numbering) that increased the expression level (Sounier et al., 2015) and reduced the NMR peak overlaps. The N-terminus was truncated before residue G52. This μOR construct maintained intact μOR functions and was stable for the duration of the NMR experiments (Sounier et al., 2015) (Figures S1F and S2A). Our previous approach used lysine sensors to probe μOR motions in the solvent-accessible domains (Sounier et al., 2015). Here we developed a dual-isotope methyl labeling scheme to monitor the solvent-accessible domains and the 7-transmembrane helices (7TM) simultaneously. For this purpose, we introduced NMR-active 13C probes into methionines (biosynthetically) and lysines (through reductive methylation) of the μOR construct. The heteronuclear multiple-quantum coherence (HMQC) pulse sequences were used to obtain 2D 1H-13C chemical shift correlation maps (see METHOD DETAILS). The 13CH3-ε-methionine peak assignments were obtained by mutagenesis of individual methionine residues and the dimethylamine peak assignments from our previous work (Sounier et al., 2015). We unambiguously assigned twenty sensors in the unliganded (apo) μOR (Figures 2A-2C and S2-S3; see Methods). The G52 backbone amine was at the N-terminus. Three lysines (K209ECL2, K2335.39, K3036.58), and four methionines (M651.29, M1302.66, M2034.61, M2054.63) were located in the ligand binding domain (LBD). In the intracellular coupling domain (ICD), seven lysines (K98ICL1, K100ICL1, K174ICL2, K2605.66, K2696.24, K2716.26 and K3448.51) and four methionines (M1613.46, M2555.61, M264ICL3, M2816.36) were assigned. Only one methionine M2435.49 was assigned to the connector region (CR).
Figure 2. Development of multi-domain NMR sensors.

(A) Location of the NMR sensors in a cartoon representation of μOR in inactive form. The NMR sensors, ε-CH3 of methionine (green) and ε-NH2 of lysine (raspberry), are shown in balls, in the ligand binding domain (LBD, pale orange), the connector region (CR, pale green) and the intracellular coupling domain (ICD, blue). (B and C) Extracted 2D HMQC spectra of the methionine and lysine sensors, as well as the backbone amine of the N-terminal residue G52, in apo μOR. Asterisk indicates the peak positions of residual resonances of the N-terminal methionine in a small amount of untruncated μOR.
To monitor agonist-induced μOR activation, we collected the HMQC spectra of μOR bound to each agonist, with and without the G protein-mimetic nanobody, Nb33 (Figures 2A-2C and S2D-S2F). Nb33 was necessary to reach the fully active state of μOR (Sounier et al., 2015), whereas without Nb33 we obtained the effects of the agonist binding, which more likely reflect the intrinsic mechanism of ligand bias. Taking into account the receptor concentration and the ligand depletion (Hulme and Trevethick, 2010), we estimated that 99% of the μORs should be in complexes during the NMR measurements.
Conformational link between the ligand binding domain and the intracellular partner protein binding site
Upon binding of the agonists alone, the LBD sensors M651.29 and M2054.63 exhibited multiple peaks with different intensities, which varied among the five agonists (Figure S4D-S4E and S4M-S4N). This indicates intermediate-to-slow exchanges among multiple conformations of the receptor and/or the agonists on the NMR timescales. The N-terminal sensor G52 showed a remarkable peak appearance for all the five agonists (Figure S4G-S4H), which was further increased upon Nb33 binding (Figure S4I). This suggests that ligand binding stabilized the N-terminus, which was enhanced upon Nb33 binding. Indeed, the N-terminus formed a pocket lid in the crystal structure of BU72-μOR-Nb33 (PDB 5C1M). Nb33 binding at the ICD also caused spectra changes in the extracellular loop 2 (ECL2) in a ligand-dependent manner (Figure S4A-S4C). These results suggest that i) Nb33 binding at the ICD has a long-range allosteric modulation of the LBD conformations; ii) Nb33 triggers cooperative interactions among the N-terminus, ECL2 and the orthosteric pocket; and iii) the agonists alone do not stabilize the fully active state even in the LBD. Importantly, these data are in line with the allosteric link between the LBD and the ICD recently reported for other class A GPCRs (Eddy et al., 2018; Liu et al., 2017; Liu et al., 2019a).
At the G protein binding site (at TM5/ICL3/TM6 in the ICD), the five agonists induced only small conformational changes (Figures S5B, S5F and S5J). Nevertheless, the small changes were sufficient for the binding of Nb33, which in turn induced large conformational changes in TM5/TM6 as seen in the crystal structures of active μOR (Huang et al., 2015) (Figures S5C, S5G and S5K). The magnitudes of the changes in the ternary μOR-agonist-Nb33 complexes correlated with the G-protein efficacies of the agonists (Figure S5D and S5H). This suggests that the capacity of an agonist to cooperate with the G proteins allosterically determines its efficacy to activate the canonical G protein signaling pathway.
Biased, unbiased or partial agonists exhibit different binding poses
To investigate the molecular mechanism of the discrete agonist activities, we performed REST2-MD simulations (see METHOD DETAILS and Figure S6A) to examine how each agonist modulates the μOR conformational ensemble. All the simulations were initiated by docking the agonist to the crystal structure coordinates of μOR in an inactive form (PDB: 4DKL) (Manglik et al., 2012) without Nb33. The REST2-MD reproduced the binding poses of DAMGO and BU72 in the cryoEM/crystal structures of μOR in active forms (PDBs 6DDE (Koehl et al., 2018) and 5C1M (Huang et al., 2015), respectively, Figure S6B). This confirmed sufficient conformational sampling by the REST2-MD in our protocol. The biased agonists, oliceridine, PZM21 and buprenorphine, turned out to bind deeper into the CR than DAMGO and BU72. They inserted between W2936.48 and TM2, whereas DAMGO and BU72 bound on top of W2936.48 (Figure 3A). The REST2-MD also captured alternative, short-lived binding poses of oliceridine and PZM21, in which the ligands bound on top of W2936.48 (Figures S6C). These are likely transient poses in the binding process. Indeed, the HMQC spectra in the LBD indicated exchanges between different receptor/ligand conformations for all the five agonists (Figure S4) Whether such transient binding poses contribute to the receptor activation in the ICD is difficult to determine, since the LBD and the ICD are loosely coupled. Therefore, we focus on the ensemble changes in the receptor conformational equilibrium, without interpreting the roles of the transient binding poses.
Figure 3. Distinct binding patterns of biased, unbiased and partial agonists in comparison with NMR.

(A) Unbiased agonists (DAMGO and BU72) bind on top of W2936.48, whereas biased ones (oliceridine, PZM21 and buprenorphine) insert between W2936.48 and TM2. The conserved “PIF” motif (P2445.50, I1553.40 and F2896.44) and the NMR sensor M2435.49 in the CR are shown in sticks. (B) Ligand-dependent rotamers of W2936.48, as measured by the distribution of the dihedral angle χ2 during the REST2-MD simulations. (C) Extracted HMQC spectra of M2435.49 resonances in apo μOR (black) and at saturating concentration of DAMGO (red), BU72 (orange), oliceridine (blue), PZM21 (green) and buprenorphine (magenta). Dashed black lines indicate the position of the cross-sections shown above the spectra.
In the case of buprenorphine, the partial agonist, W2936.48 switched between two rotamers, whereas the full agonists maintained mostly one of the W2936.48 rotamers like in apo μOR (Figure 3B). W2936.48 is part of the conserved CW6.48xP motif in class A GPCRs, known as the “toggle switch” of receptor activation. It is located at the bottom of the orthosteric pocket, on top of the conserved “PIF” motif (P2445.50, I1553.40, and F2896.44) in the CR. Together, they form the so-called “connector region” which mediates the allosteric communications between the LBD and the ICD (Latorraca et al., 2017). W2936.48 plays important roles in μOR activation or inhibition (Huang et al., 2015; Yuan et al., 2015). Therefore, the different agonist binding poses with respect to W2936.48 may be the initial trigger of the different signaling outcomes.
The above REST2-MD observations were confirmed by the NMR sensor M2435.49 in the CR. We spotted a distinct pattern in the buprenorphine-μOR complex than in the other systems: the peak of M2435.49 shifted upfield in both the 1H and 13C dimensions, as expected from the rotation of W2936.48 toward M2435.49 upon buprenorphine binding (Figure 3C). Interestingly, during an experiment using the antagonist naloxone, the M2435.49 peak shifted slightly in the same directions as in the case of buprenorphine (Figure S2G). The shift is likely associated with some constitutively active conformations in apo μOR that were diminished upon naloxone binding. Given the well-documented “toggle switch” role of W2936.48 in μOR activation (Huang et al., 2015; Yuan et al., 2015), we conclude that the low efficacy of buprenorphine is associated with its low capability to stabilize the required W2936.48 rotameric state for activation.
Conformational changes in the intracellular β-arrestin binding site
The biased agonists oliceridine and PZM21 produced distinct μOR conformations in the lower half of TM7, the intracellular loop 1 (ICL1) and the helix 8 (H8) during the REST2-MD. The simulations revealed an allosteric communication from W2936.48 to ICL1 and H8, through the conserved motifs G1.49N1.50 and N7.49P7.50xxY7.53. Namely, the biased agonists inserted between W2936.48 and A1172.53 in the CR and split TM6 and TM2, which let TM7 to approach TM3 (Figures 4A-4C). The N7.49P7.50xxY7.53 motif in TM7 thus moved toward TM3, away from the G1.49N1.50 motif in TM1 (Figures 4C-4E). This led to remarkable inward movements of TM7 and H8 toward TM3 in the ICD, closing the cleft between H8 and ICL1 (Figures 5A-5C). In the case of the unbiased agonists (DAMGO and BU72), W2936.48 interacted with A1172.53 while N7.49P7.50xxY7.53 remained in close contact with G1.49N1.50. Residues N3327.49 and Y3367.53 formed a hydrophilic cluster with N861.50 and D1142.50. D2.50 is the sodium binding site conserved in 90% of non-olfactory class A GPCRs (Katritch et al., 2014) and an important microswitch of receptor activation (Vanni et al., 2010). This configuration resembled those in the initial crystal structure (4DKL) and apo μOR (Figure 4), so did ICL1 and H8 (Figures 5A-5C). The partial biased agonist buprenorphine showed an in-between behavior. In the CR and the N7.49P7.50xxY7.53-G1.49N1.50 motifs, buprenorphine showed similar but weaker impacts than oliceridine and PZM21 (Figures 4B and 4E). The impacts barely reached the ICL1/H8 domains in the simulation timescale (Figures 5A-5C).
Figure 4. Biased agonists induced conformational changes in the CR and the lower half of TM7.

(A) In the CR, biased agonist binding (blue) splits the side chains of W2936.48 and A1172.53, which allows TM7 to approach TM3. The movements are measured by (B) the minimum side-chain distances between W2936.48 and A1172.53, against the Cα distances between I1553.40 and N3287.45. (C) Schematic presentation of the inter-helical movements. Arrows indicate the direction of the movements and dashed lines indicates the distances measured for (B and E). (D) In the lower half of TM7, the N7.49P7.50xxY7.53 motif moves away from the G1.49N1.50 motif in TM1 only for biased agonists. This is measured by (E) the side-chain distances between N861.50, N3327.49 and Y3367.53.
Figure 5. Biased agonists induced new clusters of μOR conformations in ICL1 and H8.

(A) Biased agonist binding (blue) triggered inward movements of TM7-H8 toward TM3 and ICL1, respectively, closing the ICL1-H8 cleft. (B) Schematic presentation of the movements in (A) from the extracellular view, as measured by (C) the Cα distances between the NMR sensors K98ICL1, K100ICL1 and K3448.51. Density maps of the measured distances illustrate a new cluster of conformations associated with the biased agonists oliceridine and PZM21. (D) Extracted HMQC spectra of K3448.51/K98ICL1 resonances of μOR in apo form (black) and at saturating concentrations of DAMGO (red), BU72 (orange), oliceridine (blue), PZM21 (green) and buprenorphine (magenta). Dashed black lines indicate the position of the cross-sections shown above the spectra. Black dots indicate the peak centers in apo μOR. Red arrows indicate the changes upon agonist binding.
The NMR spectra in the ICL1/H8 domain confirmed the above findings. DAMGO, BU72 and buprenorphine binding results in the small loss of signal intensity of these sensors in H8/ICL1 (Figure 5D). Upon binding the biased agonists oliceridine and PZM21, the lysine sensors K98ICL1 and K3448.50 showed multiple peaks with change in signal intensity, indicating a much more complex conformational equilibrium (Figure 5D). This suggests that the local conformations became more dynamic. Binding of Nb33 led to signal intensity decrease for all the ligands (Figure S5Q and S7A). We performed REST2-MD simulations on the five receptor-agonist-Nb33 complexes and found similar patterns at ICL1 and H8 (Figure S7B). The distinct conformations associated with oliceridine and PZM21 were still evident in the presence of Nb33 but less remarkable. Nb33 inserts slightly between ICL1 and H8. Thus, it increased the ICL1-H8 distances in all the five complexes, while in the case of oliceridine and PZM21, it also reduced the conformational dynamics in this domain.
Overall, the results suggest that the biased agonists act on the toggle switch W2936.48 to trigger conformational changes in TM7, ICL1 and H8 in the ICD. This occurs in an allosteric manner, via the conserved motifs N7.49P7.50xxY7.53 and G1.49N1.50. The conformational changes persist even when the receptor is coupled to Nb33. Although the binding sites of G proteins and arrestins largely overlap, only arrestins interact with ICL1/H8 in the structures of arrestin in complex with rhodopsin, neurotensin receptor 1, β1AR and M2R (Huang et al., 2020; Lee et al., 2020; Staus et al., 2020; Yin et al., 2019). Therefore, the distinct TM7-ICL1-H8 conformations generated by the biased agonists likely inhibit the binding of β-arrestins but not the G proteins (Figure 6).
Figure 6. Scheme of the proposed allosteric mechanism of μOR functional selectivity.

(A) Binding of the unbiased agonists cause W2936.48 in TM6 to approach TM2 in (B) the connector region. (C) Biased agonists bind deeper in the pocket and separate TM6 from TM2 in (D) the connector region, letting TM7 to approach TM3. This disrupts the interactions between the NPxxY motif, D1142.50 and the GN motif (C). The lower half of TM7 moves toward TM3, closing the cleft between H8 and ICL1, which may inhibit arrestin signaling (C). Blue arrows indicate the movements associated with allosteric functional selectivity. Conserved residues/motifs involved are highlighted in yellow. The transmembrane helices are represented by two adjoining cylinders colored in grey. The top cylinder of TM1 and TM7 are transparent for clarity.
DISCUSSION
The pharmacological outcome of a ligand varies with the test systems and conditions, which have led to contradictory findings and the ongoing debate on whether μOR functional selectivity can separate analgesia from opioid side effects. Even the characterization of functional selectivity itself is debated, due to the lack of deep comprehensions and field standards. In this study, we provide the basic molecular mechanism of biased and partial agonism in μOR, which is intrinsic to the receptor-ligand interactions and should be consistent under different test conditions. The approach and findings can serve as high-resolution monitors for the design and evaluation of μOR ligands with specific functions, e.g. partial or biased agonism/antagonism/inverse agonism for a specific signaling pathway. Such ligands may serve to pinpoint specific aspects of the μOR signaling network, helping resolve the ongoing debates from bottom up. GPCR ligand design is challenging despite the growing number of high-resolution structures. While structure-based design and screening have lifted the hit rate, there lacks tools to design or predict biased ligands. REST2-MD can identify biased μOR agonists and thus be used for ligand screening. Although it is more costly than docking or standard MD, it provides mechanistic and dynamics insights into the ligand actions, which is essential for the subsequent ligand optimization.
This study highlights the dynamic and allosteric details of μOR pre-activation upon agonist binding, prior to the coupling of intracellular signaling partners. The pre-activation stage is crucial for drug design because it is dictated by the ligands. Nevertheless, it is highly dynamic and inaccessible to X-ray crystallography or CryoEM. We took advantage of 2D HMQC NMR and enhanced-sampling MD to capture conformational dynamic patterns during the pre-activation, which differentiate the partial and the biased agonists from the full unbiased ones. The phenomenon that biased or partial ligands stabilize distinct receptor conformations has been reported for the angiotensin II receptor 1 (during pre-activation) (Wingler et al., 2019), the glucagon-like peptide-1 receptor (in fully active state) (Liang et al., 2018), the β1AR (Moukhametzianov et al., 2011; Solt et al., 2017) the A2AR (Huang et al., 2021; Susac et al., 2018; Ye et al., 2016) and the β2AR, either in pre-active state (biased agonism) (Liu et al., 2012) or fully active state (partial and biased agonism) (Masureel et al., 2018). Interestingly, Susac et al. also found different inward/outward movements of TM7 on the intracellular side upon agonist and antagonist binding, which is consistent with available A2AR crystal structures. However, it is unclear whether the TM7 movements of A2AR were associated with ligand bias. One drawback of REST2-MD is the loss of temporal information. Nevertheless, it has been shown that biased agonism in μOR is not controlled by binding or signaling kinetics, suggesting a mechanism dictated by receptor conformations (Pedersen et al., 2020)
Most of the findings here are independent of Nb33, except for the interplay observed between the agonists and Nb33 (between the LBD and the ICD), the mechanism of which remains obscure. The process of G protein binding likely determines the G protein subtype selectivity through transient GPCR-G protein interactions or intermediate conformations (Du et al., 2019; Liu et al., 2019b), but this is beyond the scope of the current study. Without Nb33 or G proteins, however, our NMR and REST2-MD results coherently and robustly illustrated the specific conformational dynamics underlying the partial and biased agonisms. The mechanism relies on highly-conserve amino-acid motifs, which may be common for other class A GPCRs.
Limitations of the study
The current study mainly focused on the inherent mechanism of μOR ligand bias, prior to G protein or arrestin binding. One important question is how G proteins or arrestins respond to the distinct μOR conformations associated with the biased agonists. However, investigations on this aspect have been very challenging technically for both MD and NMR. The size of GPCR-agonist-G protein/arrestin ternary complexes are the major limit. Their conformational changes take place in timescales that challenge today’s all-atom MD simulations. Few high-resolution structures are available, especially for the complexes with arrestins. Such ternary complexes are also unstable under the NMR experiment conditions due to the destabilizing effect of the detergents. Therefore, studies in this aspect have been limited to G protein or arrestin surrogates (e.g. modified Gα subunits and nanobodies). The aspect of G protein/arrestin binding thus remains to be further explored.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Rémy Sounier (remy.sounier@igf.cnrs.fr)
Materials availability
DNA constructs generated by the authors and from ARPEGE can be obtained upon request from the Lead Contact, but we may require a payment and/or a completed Materials Transfer Agreement if there is potential for commercial application.
Data and code availability
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report any original codes.
Any additional information required to reproduce this work is available from the Lead Contact.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Bacterial cells
E. coli BL21(DE3) were grown in LB at 37°C.
Insect cells
Recombinant baculoviruses were generated using the BestBac or pFastBac baculovirus system according to manufacturer’s instructions (Expression Systems, ThermoFischer, respectively). High titer baculoviruses encoding genes were used to infect Sf9 cells at a cell density of 4 × 106 cells per ml in suspension in methionine deficient media (Expression System) in the presence of 3 μM naloxone with 13C methyl labelled methionine (Cambridge Isotope) added into the media at 250 mg·L−1 concentration. Cells were harvested by centrifugation 48 h post-infection and stored at −80 °C until purification.
Cell lines and transfection
HEK293 cells were grown in Dulbecco's Modified Eagle's Medium (DMEM, Life Technologies) supplemented with 10% fetal bovine serum (FBS, Life Technologies) without antibiotics at 37°C, 5% CO2.
METHOD DETAILS
Cell transfection
Transient transfection was performed using lipofectamine2000 (Invitrogen Life Technologies). Depending on the assay, HEK293 cells were stimulated 24h or 48h after transfection. For G protein activation and GRK2/5 recruitment, cells were seeded into a 6-well plate for 24h at a density of 750,000 cells per well. Cells were then detached, seeded and incubated for 24h in a 96-well white plate coated with poly-L-Ornithine at a density of 40,000 cells per well. For the signaling assays, we used three different constructs of full-length μOR (SNAP-μOR, μOR-2x-M72T, and μOR-YFP) depending on the experiment to be conducted (i.e. TR-FRET, BRET Gi and arrestin recruitment, respectively). For binding, β-arrestins recruitment and internalization assays, cells were directly transfected into a 96-well white plate coated with poly-L-Ornithine at a density of 40,000 cells per well following manufacturer's recommended protocol. For binding assay on membrane and solubilized receptors cells were transfected using electroporation. Electroporation was performed in a volume of 400 μL with a total 5 μg SNAP-μOR plasmid and 20,000,000 cells in electroporation buffer (50 mM K2HPO4, 20 mM CH3COOK, and 20 mM KOH, pH 7.4). After electroporation (260 V, 1 mF, Bio–Rad Gene Pulser electroporator), cells were resuspended in 15 mL DMEM supplemented with 10% FBS in T150 culture dishes (pretreated with Poly-L-Ornithine) for 24 h.
G protein activation assay
The cells were transfected with 4 plasmids encoding the mouse μOR receptor (Flag-μOR-2x), the β2 and Venus-γ2 G protein subunits and the Gα protein fused with a donor at a 1:1:1:1 ratio. Gαi1, Gαi2, Gαi3, GαOA and GαOB was fused to Renilla Luciferase2 (Rluc8 provided by the ARPEGE platform). 24 h after transfection, cells were washed twice with PBS complemented with 0.9 mM CaCl2 and 0.5 mM MgCl2. Basal conditions were achieved by the addition of PBS solutions, followed by Coelenterazine H at a final concentration of 5 μM. To evaluate the effects of the μOR agonists, the addition of Coelenterazine H was followed by stimulation with different agonists. Activation of the μOR promotes dissociation of the Gαβγ protein complex resulting in the Bioluminescence resonance energy transfer (BRET) signal decay. BRET between Rluc8 and Venus was measured after the addition of the Rluc8 substrate Coelenterazine H. BRET readings were collected using a Mithras LB940 plate reader (Berthold technologies, Rluc8 485 ± 20 nm; YFP 530 ± 25 nm) and the reading chamber was maintained at 37 °C throughout the entire reading time. The BRET signal was calculated by the ratio of emission of Venus (535 nm) to Rluc8 (480 nm):
GRK2 and GRK5 recruitment assay
The cells were transfected with 2 plasmids encoding the human μOR receptor fused to YFP at the C-terminal, and GRK2/5 was fused to Renilla Luciferase2 (provided by the ARPEGE platform). 24 h after transfection, cells were washed twice with PBS complemented with 0.9 mM CaCl2 and 0.5 mM MgCl2. The cells were incubated with Coelenterazine H at a final concentration of 5 μM, followed by stimulation with agonists before the BRET readings were captured. Recruitment of GRK2/5 is assessed by an increase in the BRET signal.
β-arrestins recruitment assay
The cells were transfected with 2 plasmids encoding the human μOR receptor fused to YFP at the C-terminal, and β-arrestin-1/β-arrestin-2 was fused to Renilla Luciferase1 at the N-terminal (provided by Dr. M. Scott to the ARPEGE platform). 24 h after transfection, cells were washed twice with PBS complemented with 0.9 mM CaCl2 and 0.5 mM MgCl2. The cells were incubated with Coelenterazine H at a final concentration of 5 μM, followed by stimulation with agonists before the BRET readings were captured. Recruitments of β-arrestin-1 and β-arrestin-2 are assessed by an increase in the BRET signal.
Internalization assay
The cells were transfected with one plasmid encoding the SNAP-μOR receptor. 24 hours after transfection, SNAP-μOR cells were washed with Tag-Lite buffer (PerkinElmer/CisBio Bioassays) and incubated at 37°C with benzylguanine-Lumi4-Tb (SNAP-Lumi4-Tb) at a concentration of 100 nM during 1 hour. The cells were washed 4 times with Tag-Lite buffer and then incubated with μOR agonists diluted in fluorescein buffer (24 μM). Reading was performed at 37°C on an Infinite F500® plate reader (TECAN, Lumi4-Terbium-criptate: 620 ± 10 nm; Fluorescein: 520 ± 10 nm) with an excitation at 337 nm and emission at 620 nm and 520 nm. Receptor internalization was monitored by time-resolved fluorescent resonance energy transfer (TR-FRET) at 37 °C during 70–80 min. The signal was calculated by the ratio of emission of terbium cryptate (620 nm) to fluorescein (520 nm): ΔR = (Ratio 620/520) X 10,000.
Fluorescent ligand-binding assay on living cells
HEK293 cells transfected with SNAP-μOR plasmid were seeded at a density of 40,000 cells per well in 96-well white plates coated with poly-L-Ornithine. 24h after transfection, SNAP-μOR was labeled 1 h at 37°C with 100nM SNAP-Lumi4-Tb diluted in Tag-lite labeling buffer. Fluorescent naltrexone and agonists were diluted in Tag-lite labeling buffer. A fixed concentration of fluorescent naltrexone-d2 was determined (0.5 nM = Kd) and used. Increasing concentration of agonists was added prior to the addition of fluorescent naltrexone-d2 in the plates containing labeled cells. Plates were incubated overnight at 4°C before homogenous time-resolved Fluorescent (HTRF) signal detection. HTRF detection was performed on a PHERAstar (BMG labtechnologies). The signal was collected both at 665 nm and 620 nm. HTRF ratio was obtained by dividing the acceptor signal at 665 nm by the donor signal at 620 nm and multiplying this value by 10,000.
Membrane preparation and SNAP labeling
Twenty-four hours after transfection cells were washed once with PBS solution, scraped and then collected by centrifugation 5 minutes at 300 g. The cell pellet was resuspended in 20 mL lysis buffer (10 mM HEPES, 1 mM EDTA) and homogenized using an electric homogenizer on ice. After centrifugation 5 min at 1,000g at 4 °C, the pellet was discarded and the supernatant was centrifuged at 30,000 g for 30 min at 4 °C. The resulting pellet was resuspended in 2 mL of Tag-Lite buffer containing 300 nM BG-Lumi4-Tb and incubated for 1 hour at 4 °C under circle rotator. Membranes were then resuspensed in 2 mL PBS to remove the excess of BG-Lumi4-Tb and centrifuged at 30,000 g for 30 min on a bentchtop centrifuge. This step was reproduced twice. Protein concentration was determined by BCA using BSA as standard. SNAP-μOR membranes were aliquoted and stored at −80 °C.
Fluorescent ligand-binding assay on solubilized receptors
SNAP-μOR membranes were resuspended in solubilization buffer (see below for composition details) and stirred 1 hour at 4 °C. After centrifugation at 36,000 g for 20 min at 4 °C, the solute material was complemented with 2 mM CaCl2 and loaded on M1 antibody Resin. Detergent exchange protocol was performed (see below for details) and solubilized SNAP-μOR was eluted from M1 antibody resin. Naltrexone-d2 binding affinity towards solubilized SNAP-μOR was determined at 3 nM against 0.5 nM on living cells and 1 nM HEK293 cell membranes. Freshly prepared solubilized SNAP-μOR were incubated with increasing concentrations of agonists and a fixed concentration of fluorescent naltrexone-d2 (12 nM = Kd) before HTRF signal detection.
(Met-ε)-[13CH3]-μOR-2x M72T expression
We generated a μ-OR mouse construct with features designed to enhance stability for NMR spectroscopy. A tobacco etch virus (TEV) protease recognition site was introduced after residue 51, and a human rhinovirus 3C protease site after residue 358. Receptor expression was largely improved by using a M721.36T single-point mutation as previously reported by naloxone binding (Sounier et al., 2015). A FLAG tag was added to the amino terminus and an 8 × His tag was appended to the carboxy terminus (μOR-2x). Recombinant baculoviruses were generated using the BestBac baculovirus system according to manufacturer’s instructions (Expression Systems). For the assignment of all other mutants, we used the pFastBac baculovirus system (ThermoFischer). High titer baculoviruses encoding μOR-2x genes were used to infect Sf9 cells at a cell density of 4 × 106 cells per ml in suspension in methionine deficient media (Expression System) in the presence of 3 μM naloxone with 13C methyl labelled methionine (Cambridge Isotope) added into the media at 250 mg·L−1 concentration. Cells were harvested by centrifugation 48 h post-infection and stored at −80 °C until purification.
(Met-ε)-[13CH3]-μOR-2x M72T purification
Cell pellets were resuspended in 10 mM Tris-HCl pH 7.5, 1 mM EDTA buffer containing 2 mg·mL−1 iodoacetamide and protease inhibitors without salt to lyse the cells by hypotonic lysis. Lysed cells were centrifuged (38,420 g) and the membranes were solubilized durinf 1h @ 4 °C using buffer containing 20 mM HEPES (pH 7.5), 200 mM NaCl, 0.5% (w/v) n-dodecyl-β-D-maltoside (DDM, Anatrace), 0.3% (w/v) CHAPS, 0.03% (w/v) choslesteryl-hemi-succinate (CHS, Sigma), 2 mg·mL−1 iodoacetamide and protease inhibitors. The solubilized receptor was loaded onto anti-Flag M1 column (Sigma) and washed thoroughly with DDM buffer containing 20 mM HEPES (pH 7.5), 100 mM NaCl, 0.1% (w/v) DDM, 0.03% (w/v) CHAPS, 0.015% (w/v) CHS and 2 mM CaCl2. While on the M1 antibody resin, the receptor was exchanged into lauryl-maltose-neopentyl-glycol (MNG-14, Anatrace) detergent-containing buffer composed of 20 mM HEPES (pH 7.5), 100 mM NaCl, 0.5% (w/v) MNG-14 and 0.01 % CHS. The detergent exchange was performed by washing the column with a series of seven buffers (3 CV each) made up of the following ratios (v/v) of MNG-14 buffer and DDM buffer: 0:1, 1:1, 4:1, 9:1, 19:1, 99:1 and MNG-14 exchange buffer alone. The column was then washed with 20x critical micelle concentration (cmc) MNG-14 buffer containing 20 mM HEPES (pH 7.4), 100 mM NaCl, 0.02% (w/v) MNG and 0.0004 % CHS and the bound receptor was eluted in the same buffer supplemented with 0.2 mg·mL−1 Flag peptide. To remove flexible amino and carboxy termini, TEV and 3C protease were added at a 1:5 and 1:10 protease:μOR-2x ratio by weight. The sample was incubated at 4 °C overnight in the presence of 100 μM of TCEP. We then used a negative Ni-NTA chromatography step to remove TEV and 3C proteases.
(Met-ε)-[13CH3]-μOR-2x M72T assignment procedure
To obtain sequence-specific assignments, we introduced single point mutations of 13 methionines in μOR by site-directed mutagenesis (Genecust). To identify suitable methionine substitutions, we performed a sequence alignment of μOR homologs and selected the most common amino acid for each position: M65T, M72T, M90I, M99L, M130L, M151I, M161I, M203I, M205L M243V, M255I, M264L and M281L. The mutants were expressed and purified as described above, except for M151I which was unstable in the detergent micelles. Ten methionines were unambiguously assigned by comparing the spectra of μOR and the μOR mutants in both apo and fully active states (Figure S3). M90 and M99 could not be assigned (Figure S3).
(Met-ε)-[13CH3]-μOR-2x M72T reductive methylation
Receptor preparation from the Ni-NTA flow through were incubated at 4 °C overnight with 10 mM 13C-formaldehyde and 10 mM NaBH3CN. Excess of reagent was eliminated by dialysis and (Met-ε)-[13CH3],(Lys-Nε,Nε)[13CH3, 13CH3]-μOR ((13C-Mε,Kme2)-μOR) was further purified by SEC chromatography in a buffer containing 0.01% MNG, 0.001% CHS, 20 mM HEPES pH 7.4 and 40 mM NaCl. The monodisperse peak was then concentrated to 30 to 60 μM final, and dialysed in 98.85% D2O buffer with 0.01% MNG, 0.0004% CHS, 20 mM HEPES-d18 pH 7.4 (uncorrected) and 40 mM NaCl.
Nanobody Nb33 expression and purification
The Nb33 was expressed and purified as described in our previous work (Sounier et al., 2017). Briefly, The DNA sequence of Nb33 was subcloned into a pMalp2x vector containing an N-terminal, 3C protease-cleavable maltose binding protein (MBP) tag and a C-terminal 8 × His tag. Plasmids were transformed into BL21(DE3) cells and protein expression induced in liquid broth (LB) by addition of IPTG to 0.5 mM at an OD600 of 0.6. Cells were harvested after overnight growth at 20°C by centrifugation at 6,000 g for 30 min. Cells were resuspended in 20 mM HEPES buffer (pH 7.5), 500 mM NaCl, 0.1 mg·mL−1 lysozyme and PMSF was added as a protease inhibitor before lysis by sonication. The cell lysate was centrifuged at 38,420 g for 30 min at 4 °C. The soluble fraction was isolated and was supplemented with imidazole to a final concentration of 20 mM. MBP–nanobody fusions were purified by Ni-NTA chromatography and MBP was removed using 3C protease. Cleaved MBP was separated from the nanobody by additional amylose purification and size exclusion chromatography in a buffer containing 20 mM HEPES pH 7.4 and 0.1 M NaCl.
NMR Spectroscopy
Final samples (~270 μl at 30–60 μM) were loaded into Shigemi microtubes susceptibility matched to D2O. All data for ligands and mutant studies were acquired on 700 MHz Bruker Avance III spectrometers (Bruker, Rheinstetten, Germany), equipped with 5 mm cryogenic H/C/N/D probes with z axis gradient. 1H-13C correlation spectra were recorded using heteronuclear multiple-quantum coherence (HMQC) experiments in echo anti-echo mode. 13C and 1H chemical shifts and peak line widths in the HMQC spectra reveal the chemical and magnetic environments of the 13C-methyl probes as well as their dynamic properties. Spectral widths in ω1 and ω2 were 8,417.5 Hz and 3,518.6 Hz at 700 MHz centred at 40 p.p.m. or 20 p.p.m. in the 13C dimension. 13C decoupling was performed with a GARP4 sequence. Typically, 134 complex points with 32–48 scans per FID were recorded, to ensure a 27-Hz resolution per point at 700 MHz before zero filling. The relaxation delay was set to 1.5 s. Thirty-two steady-state scans preceded data acquisition. Total collection time varied between 3 and 4 h, depending on the sample concentration. The spectra were visualized using CCPNMR (Vranken et al., 2005). All ligands were dissolved in perdeuterated dimethyl d6-sulfoxide (d6-DMSO, Cambridge Isotope) to 100 mM and directly added to the sample in the Shigemi tube at a final concentration five-fold of receptor. Nb33 were concentrated to 0.6 mM and dialysed in 100% D2O buffer with 0.01% MNG, 0.001% CHS, 20 mM HEPES-d18 pH 7.4 (uncorrected) and 40 mM NaCl. The nanobodies were added directly in the Shigemi tubes at a final concentration of two-fold of the receptor before data acquisition.
Molecular dynamics simulations
The initial coordinates of μOR were from the crystal structure of an inactive form (PDB: 4DKL). The ligands were docked to the initial μOR structure using Autodock Vina (Trott and Olson, 2010). Residues in the putative ligand-binding pocket were set flexible during docking. The protonation state of titrable residues were predicted at pH 7.4 using the H++ server (Gordon et al., 2005). The receptor-odorant complexes were embedded in a bilayer of POPC using PACKMOL-Memgen (Schott-Verdugo and Gohlke, 2019). Each system was solvated in a periodic 75 × 75 × 105 Å3 box of explicit water and neutralized with 0.15 M of Na+ and Cl− ions. Effective point charges of the ligands were obtained by RESP fitting (Wang et al., 2000) of the electrostatic potentials calculated with the HF/6-31G* basis set using Gaussian 09 (Frisch et al., 2009). The Amber 99SB-ildn (Lindorff-Larsen et al., 2010), lipid 14 (Dickson et al., 2014) and GAFF (Wang et al., 2004) force fields were used for the proteins, the lipids and the ligands, respectively. The TIP3P (Jorgensen et al., 1983) and the Joung-Cheatham (Joung and Cheatham, 2008) models were used for the water and the ions, respectively.
After energy minimization, all-atom MD simulations were carried out using Gromacs 5.1 (Van Der Spoel et al., 2005) patched with the PLUMED 2.3 plugin (Tribello et al., 2014). Each system was gradually heated to 310 K and pre-equilibrated during 10 ns of brute-force MD in the NPT-ensemble. The replica exchange with solute scaling (REST2) (Wang et al., 2011) technique was employed to enhance the sampling with 48 replicas in the NVT ensemble. REST2 is a type of Hamiltonian replica exchange simulation scheme, which performs many replicas of the same MD simulation system simultaneously. The replicas have modified free energy surfaces, in which the barriers are easier to cross than in the original system (Figure S6A). By frequently swapping the replicas during the MD, the simulations “travel” on different free energy surfaces and easily visit different conformational zones. Finally, only the samples on the original free energy surface are collected. The replicas are artificial and are only used to overcome the energy barriers. REST2, in particular, modifies the free energy surfaces by scaling (reducing) the force constants of the “solute” molecules in the simulation system. The protein and the ligands were considered as “solute”–the force constants of their van der Waals, electrostatic and dihedral terms were subject to scaling–in order to facilitate their conformational changes. The effective temperatures used here for generating the REST2 scaling factors ranged from 310 K to 700 K, following a distribution calculated with the Patriksson-van der Spoel approach (Patriksson and van der Spoel, 2008). Exchange between replicas was attempted every 1000 simulation steps. This setup resulted in an average exchange probability of ~40%. We performed 50 ns × 48 replicas of MD in the NVT ensemble for each system. The first 20 ns were discarded for equilibration. From our past experiences on REST2-MD of GPCR conformational changes (Cong et al., 2019; Cong et al., 2018; Cong and Golebiowski, 2018; Sena et al., 2017), we estimated that 50 ns should achieve millisecond timescale sampling. The original unscaled replica (at 310 K effective temperature) was collected and analyzed. Cluster analysis of the ligand binding pose was carried out on the non-restrained trajectory using the Gromacs Cluster tool. The middle structure of the most populated cluster was selected as the final binding pose.
QUANTIFICATION AND STATISTICAL ANALYSIS
For the assays described in the section “G protein activation assay”, “GRK2 and GRK5 recruitment assay”, “β-arrestins recruitment assay”, “Internalization assay”, “Fluorescent ligand-binding assay on living cells” and “Fluorescent ligand-binding assay on solubilized receptors”, all experiments were conducted independently, at least three times. Data obtained were then plotted and analyzed using an operational model of agonism with GraphPad Prism Ver. 9.1.2 (GraphPad Software, Inc., San Diego, CA).
All the NMR data were processed in the same manner using NMRPipe/NMRDraw (Delaglio et al., 1995). Prior to Fourier transformation, the data matrices were zero-filled to 1024 (t1) x 4096 (t2) complex points and multiplied by a sine-bell window function in each dimension. Peak fitting analysis was performed with the program nlinLS (part of the NMRDraw package) using the same approach as previously described (Sounier et al., 2015). Briefly, Gaussian models were used for the fitting in each dimension, starting from values obtained from the peakpeaking routine in nmrDraw. The quality of the fits was examined visually by estimating the residual difference between the experimental data and the results of the model calculations. Peak volumes in apo-state, ligands alone and ternary complexes spectra were extracted from the peak fitting. Errors in the peak volume were calculated based on the effect of random noise for the peak height estimated by nlinLS. The peak intensities were normalized to the volume difference between the apo state and the ternary complex condition (DAMGO-Nb33) as the 100%.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| E. coli cells BL21(DE3) | NEB | C2527 |
| E. coli cells DH5α | NEB | C2987 |
| E. coli cells DH10Bac | invitrogen | 10361012 |
| Chemicals, peptides, and recombinant proteins | ||
| pFastBac vector | Thermofisher | Cat#10712024 |
| pCDNA3.1 vector | Thermofisher | Cat#V79020 |
| pVL1392 vector | Expression Systems | 91-030 |
| Tag-Lite buffer 5x | PerkinElmer/Cisbio | Cat#LABMED |
| SNAP-Lumi4-Tb | PerkinElmer/Cisbio | Cat#SSNPTBX |
| SNAP-red | PerkinElmer/Cisbio | Cat#SSNPREDF |
| Coelenterazine H | invitrogen | Cat#C6780 |
| Naltrexone-d2 | PerkinElmer/Cisbio | Cat#L0005RED |
| Iodoacetamide | Sigma | Cat#I6125 |
| Leupetin | Euromedex | Cat#SP-04-2217 |
| Benzamidine | Sigma | Cat#B6506 |
| Phenylmethanesulfonyl fluoride (PMSF) | Sigma | Cat#P7626 |
| n-dodecyl-beta-D-maltopyranoside (DDM) | Anatrace | Cat#D310 |
| Lauryl Maltose Neopentyl (MNG) | Anatrace | Cat#NG310 |
| Cholesterol hemisucinate (CHS) | Sigma | Cat#C6512 |
| Sodium Cholate hydrate | Sigma | Cat#C1254 |
| ANTI-FLAG® M1 Agarose Affinity Gel | Sigma-Aldrich | Cat#A4596 |
| ANTI-FLAG® M2 Agarose Affinity Gel | Sigma-Aldrich | Cat#A2220 |
| FLAG peptide (DYKDDDDK) | Covalab | https://www.covalab.com/peptide-synthesis |
| Monoclonal ANTI-FLAG® M2 antibody | Sigma-Aldrich | Cat#F3165 |
| ESF921 culture medium, Methionine Deficient | Expression Systems | Cat#96-200 |
| EX-CELL420 Serum-Free Medium | Sigma-Aldrich | Cat#14420 |
| L-Methionine (methyl-13C, 99%) | Eurisotop | Cat#DLM-206-0 |
| Deuterium oxide (D2O) | Eurisotop | Cat#D214 |
| 2,2-dimethyl-2-silapentane-5-sulfonic acid (DSS) | Eurisotop | Cat#DLM-32 |
| HEPES (D18, 98%) | Eurisotop | Cat#DLM-3786 |
| Dimethyl sulfoxide-d6 | Sigma-Aldrich | Cat#151874 |
| Formaldehyde (13C, 99%) 20 wt.% | Sigma-Aldrich | Cat#489417 |
| Sodium cyanoborohdride (NaBH3CN) | Sigma-Aldrich | Cat#156159 |
| Buprenorphine hydrochloride | Sigma-Aldrich | Cat#B9275 |
| TRV130 hydrochloride | Cliniscience | Cat#HY-16655A |
| [D-Ala2, N-Me-Phe4, Gly5-ol]-Enkephalin acetate salt (DAMGO) | Sigma-Aldrich | Cat#E7384 |
| BU72 | Prof. Peter Gmeiner | N/A |
| PZM21 | Prof. Peter Gmeiner | N/A |
| Experimental models: cell lines | ||
| insect cell line Sf9 | Life Technologies | Cat#11496015 |
| HEK293 human cells | ATCC | CRL-1573 |
| Recombinant DNA | ||
| pVL1392-μOR-2x-M72T | Sounier et al., 2015 | N/A |
| pFastBac-μOR-2x-M72T | Sounier et al., 2015 | N/A |
| pFastBac-μOR-2x-M72T-mutant | This study | N/A |
| pcDNA-μOR-YFP | This study | N/A |
| pcDNA-SNAP-μOR | This study | N/A |
| pcDNA-μOR-2x-M72T | This study | N/A |
| pcDNA-GRK2-RLuc | ARPEGE | https://www.arpege.cnrs.fr/ |
| pcDNA-FLAG-Gβ2 | ARPEGE | https://www.arpege.cnrs.fr/ |
| pcDNA-Gγ1-Venus | ARPEGE | https://www.arpege.cnrs.fr/ |
| pcDNA-Venus-Gγ2 | ARPEGE | https://www.arpege.cnrs.fr/ |
| pcDNA-Gαi1-RLuc8 | ARPEGE | https://www.arpege.cnrs.fr/ |
| pcDNA-Gαi2-RLuc8 | ARPEGE | https://www.arpege.cnrs.fr/ |
| pcDNA-Gαi3-RLuc8 | ARPEGE | https://www.arpege.cnrs.fr/ |
| pcDNA-GαOA-RLuc8 | ARPEGE | https://www.arpege.cnrs.fr/ |
| pcDNA-GαOB-RLuc8 | ARPEGE | https://www.arpege.cnrs.fr/ |
| pcDNA-arrestin1-RLuc | ARPEGE | https://www.arpege.cnrs.fr/ |
| pcDNA-arrestin2-RLuc | ARPEGE | https://www.arpege.cnrs.fr/ |
| pMal-p2x-Nb33 | Sounier et al., 2017 | https://doi.org/10.13018/BMR26936 |
| Software and algorithms | ||
| PyMOL | Schrödinger | https://pymol.org/2/ |
| Prism v.6.0 | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| NMRpipe | Delaglio et al., 1995 | https://www.ibbr.umd.edu/nmrpipe/install.html |
| CCPNMR Analysis v3.0 | Vranken et al., 2005 | https://www.ccpn.ac.uk/v2-software/software/analysis |
| Autodock Vina | Trott and Olson, 2010 | http://vina.scripps.edu/ |
| H++ server | Gordon et al., 2005 | http://biophysics.cs.vt.edu/ |
| GROMACS 5.1 | Van Der Spoel et al., 2005 | https://www.gromacs.org |
| PLUMED 2.3 | Tribello et al., 2014 | https://www.plumed.org/ |
| PACKMOL-Memgen | Schott-Verdugo and Gohlke, 2019 | https://github.com/alanwilter/acpype/blob/master/amber19-0_linux/bin/packmol-memgen |
Highlights.
NMR and MD simulations help defining the molecular mechanisms of μOR biased signaling
Biased, unbiased and partial agonists stabilize different μOR conformations
Biased agonists stabilize specific conformations in the TM7, ICL1 and H8 domains
The bias in conformation persists after binding to a G protein mimetic nanobody
ACKNOWLEDGEMENTS
We acknowledge support from INSERM (France, S.G.), CNRS (France, H.D.), the National Institutes of Health (USA, grant NIDA-DA036246 to S.G.), the German Research Foundation (Deutsche Forschungsgemeinschaft, grant CO 1715/1-1 to X.C.) and the UCA-Jedi program (France, grant ANR-15-IDEX-01 to X.C. and J.G.). The CBS is a member of the French Infrastructure for Integrated Structural Biology (FRISBI), supported by the National Research Agency (ANR-10-INBS-05) and is a GIS-IBIsA platform. We also thank Prof. Peter Gmeiner for providing Bu72 and PZM21, and GENCI-CINES (France) for providing access to the supercomputer OCCIGEN (grant A0080711387 to J.G). All cell-based assays were carried out at the ARPEGE facility (BioCampus, Montpellier).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Casiraghi M, Damian M, Lescop E, Point E, Moncoq K, Morellet N, Levy D, Marie J, Guittet E, Baneres JL, et al. (2016). Functional Modulation of a G Protein-Coupled Receptor Conformational Landscape in a Lipid Bilayer. J Am Chem Soc 138, 11170–11175. [DOI] [PubMed] [Google Scholar]
- Clark LD, Dikiy I, Chapman K, Rodstrom KE, Aramini J, LeVine MV, Khelashvili G, Rasmussen SG, Gardner KH, and Rosenbaum DM (2017). Ligand modulation of sidechain dynamics in a wild-type human GPCR. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong X, Cheron JB, Golebiowski J, Antonczak S, and Fiorucci S (2019). Allosteric Modulation Mechanism of the mGluR5 Transmembrane Domain. Journal of chemical information and modeling. [DOI] [PubMed] [Google Scholar]
- Cong X, Fiorucci S, and Golebiowski J (2018). Activation Dynamics of the Neurotensin G Protein-Coupled Receptor 1. Journal of chemical theory and computation 14, 4467–4473. [DOI] [PubMed] [Google Scholar]
- Cong X, and Golebiowski J (2018). Allosteric Na(+)-binding site modulates CXCR4 activation. Physical chemistry chemical physics : PCCP 20, 24915–24920. [DOI] [PubMed] [Google Scholar]
- Conibear AE, and Kelly E (2019). A Biased View of mu-Opioid Receptors? Mol Pharmacol 96, 542–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowan A, Doxey JC, and Harry EJ (1977a). The animal pharmacology of buprenorphine, an oripavine analgesic agent. Br J Pharmacol 60, 547–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowan A, Lewis JW, and Macfarlane IR (1977b). Agonist and antagonist properties of buprenorphine, a new antinociceptive agent. Br J Pharmacol 60, 537–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, and Bax A (1995). NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR 6, 277–293. [DOI] [PubMed] [Google Scholar]
- DeWire SM, Yamashita DS, Rominger DH, Liu G, Cowan CL, Graczyk TM, Chen XT, Pitis PM, Gotchev D, Yuan C, et al. (2013). A G protein-biased ligand at the mu-opioid receptor is potently analgesic with reduced gastrointestinal and respiratory dysfunction compared with morphine. J Pharmacol Exp Ther 344, 708–717. [DOI] [PubMed] [Google Scholar]
- Dickson CJ, Madej BD, Skjevik AA, Betz RM, Teigen K, Gould IR, and Walker RC (2014). Lipid14: The Amber Lipid Force Field. J Chem Theory Comput 10, 865–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dror RO, Arlow DH, Maragakis P, Mildorf TJ, Pan AC, Xu HF, Borhani DW, and Shaw DE (2011). Activation mechanism of the beta(2)-adrenergic receptor. P Natl Acad Sci USA 108, 18684–18689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Y, Duc NM, Rasmussen SGF, Hilger D, Kubiak X, Wang L, Bohon J, Kim HR, Wegrecki M, Asuru A, et al. (2019). Assembly of a GPCR-G Protein Complex. Cell 177, 1232–1242 e1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddy MT, Lee MY, Gao ZG, White KL, Didenko T, Horst R, Audet M, Stanczak P, McClary KM, Han GW, et al. (2018). Allosteric Coupling of Drug Binding and Intracellular Signaling in the A2A Adenosine Receptor. Cell 172, 68–80 e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich AT, Semache M, Gross F, Da Fonte DF, Runtz L, Colley C, Mezni A, Le Gouill C, Lukasheva V, Hogue M, et al. (2019). Biased Signaling of the Mu Opioid Receptor Revealed in Native Neurons. iScience 14, 47–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emmerson PJ, Liu MR, Woods JH, and Medzihradsky F (1994). Binding affinity and selectivity of opioids at mu, delta and kappa receptors in monkey brain membranes. J Pharmacol Exp Ther 271, 1630–1637. [PubMed] [Google Scholar]
- Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, et al. (2009). Gaussian 09, Revision A.02. (Wallingford CT: ). [Google Scholar]
- Gillis A, Gondin AB, Kliewer A, Sanchez J, Lim HD, Alamein C, Manandhar P, Santiago M, Fritzwanker S, Schmiedel F, et al. (2020). Low intrinsic efficacy for G protein activation can explain the improved side effect profiles of new opioid agonists. Sci Signal 13. [DOI] [PubMed] [Google Scholar]
- Gordon JC, Myers JB, Folta T, Shoja V, Heath LS, and Onufriev A (2005). H++: a server for estimating pKas and adding missing hydrogens to macromolecules. Nucleic Acids Res 33, W368–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handa BK, Land AC, Lord JA, Morgan BA, Rance MJ, and Smith CF (1981). Analogues of beta-LPH61-64 possessing selective agonist activity at mu-opiate receptors. Eur J Pharmacol 70, 531–540. [DOI] [PubMed] [Google Scholar]
- Hill R, Disney A, Conibear A, Sutcliffe K, Dewey W, Husbands S, Bailey C, Kelly E, and Henderson G (2018). The novel mu-opioid receptor agonist PZM21 depresses respiration and induces tolerance to antinociception. Br J Pharmacol 175, 2653–2661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang SK, Pandey A, Tran DP, Villanueva NL, Kitao A, Sunahara RK, Sljoka A, and Prosser RS (2021). Delineating the conformational landscape of the adenosine A2A receptor during G protein coupling. Cell 184, 1884–1894 e1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Manglik A, Venkatakrishnan AJ, Laeremans T, Feinberg EN, Sanborn AL, Kato HE, Livingston KE, Thorsen TS, Kling RC, et al. (2015). Structural insights into micro-opioid receptor activation. Nature 524, 315–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Masureel M, Qu Q, Janetzko J, Inoue A, Kato HE, Robertson MJ, Nguyen KC, Glenn JS, Skiniotis G, et al. (2020). Structure of the neurotensin receptor 1 in complex with beta-arrestin 1. Nature 579, 303–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulme EC, and Trevethick MA (2010). Ligand binding assays at equilibrium: validation and interpretation. Br J Pharmacol 161, 1219–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isogai S, Deupi X, Opitz C, Heydenreich FM, Tsai CJ, Brueckner F, Schertler GF, Veprintsev DB, and Grzesiek S (2016). Backbone NMR reveals allosteric signal transduction networks in the beta1-adrenergic receptor. Nature 530, 237–241. [DOI] [PubMed] [Google Scholar]
- Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, and Klein ML (1983). Comparison of Simple Potential Functions for Simulating Liquid Water. J Chem Phys 79, 926–935. [Google Scholar]
- Joung IS, and Cheatham TE (2008). Determination of alkali and halide monovalent ion parameters for use in explicitly solvated biomolecular simulations. J Phys Chem B 112, 9020–9041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katritch V, Fenalti G, Abola EE, Roth BL, Cherezov V, and Stevens RC (2014). Allosteric sodium in class A GPCR signaling. Trends Biochem Sci 39, 233–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kliewer A, Gillis A, Hill R, Schmiedel F, Bailey C, Kelly E, Henderson G, Christie MJ, and Schulz S (2020). Morphine-induced respiratory depression is independent of beta-arrestin2 signalling. Br J Pharmacol 177, 2923–2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kliewer A, Schmiedel F, Sianati S, Bailey A, Bateman JT, Levitt ES, Williams JT, Christie MJ, and Schulz S (2019). Phosphorylation-deficient G-protein-biased mu-opioid receptors improve analgesia and diminish tolerance but worsen opioid side effects. Nat Commun 10, 367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koehl A, Hu H, Maeda S, Zhang Y, Qu Q, Paggi JM, Latorraca NR, Hilger D, Dawson R, Matile H, et al. (2018). Structure of the micro-opioid receptor-Gi protein complex. Nature 558, 547–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kofuku Y, Ueda T, Okude J, Shiraishi Y, Kondo K, Mizumura T, Suzuki S, and Shimada I (2014). Functional dynamics of deuterated beta2 -adrenergic receptor in lipid bilayers revealed by NMR spectroscopy. Angew Chem Int Ed Engl 53, 13376–13379. [DOI] [PubMed] [Google Scholar]
- Kohlhoff KJ, Shukla D, Lawrenz M, Bowman GR, Konerding DE, Belov D, Altman RB, and Pande VS (2014). Cloud-based simulations on Google Exacycle reveal ligand modulation of GPCR activation pathways. Nat Chem 6, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latorraca NR, Venkatakrishnan AJ, and Dror RO (2017). GPCR Dynamics: Structures in Motion. Chem Rev 117, 139–155. [DOI] [PubMed] [Google Scholar]
- Lee Y, Warne T, Nehme R, Pandey S, Dwivedi-Agnihotri H, Chaturvedi M, Edwards PC, Garcia-Nafria J, Leslie AGW, Shukla AK, et al. (2020). Molecular basis of beta-arrestin coupling to formoterol-bound beta1-adrenoceptor. Nature 583, 862–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang YL, Khoshouei M, Glukhova A, Furness SGB, Zhao P, Clydesdale L, Koole C, Truong TT, Thal DM, Lei S, et al. (2018). Phase-plate cryo-EM structure of a biased agonist-bound human GLP-1 receptor-Gs complex. Nature 555, 121–125. [DOI] [PubMed] [Google Scholar]
- Lindorff-Larsen K, Piana S, Palmo K, Maragakis P, Klepeis JL, Dror RO, and Shaw DE (2010). Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins 78, 1950–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JJ, Horst R, Katritch V, Stevens RC, and Wuthrich K (2012). Biased signaling pathways in beta2-adrenergic receptor characterized by 19F-NMR. Science 335, 1106–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Ahn S, Kahsai AW, Meng KC, Latorraca NR, Pani B, Venkatakrishnan AJ, Masoudi A, Weis WI, Dror RO, et al. (2017). Mechanism of intracellular allosteric beta2AR antagonist revealed by X-ray crystal structure. Nature 548, 480–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Masoudi A, Kahsai AW, Huang LY, Pani B, Staus DP, Shim PJ, Hirata K, Simhal RK, Schwalb AM, et al. (2019a). Mechanism of beta2AR regulation by an intracellular positive allosteric modulator. Science 364, 1283–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Xu X, Hilger D, Aschauer P, Tiemann JKS, Du Y, Liu H, Hirata K, Sun X, Guixa-Gonzalez R, et al. (2019b). Structural Insights into the Process of GPCR-G Protein Complex Formation. Cell 177, 1243–1251 e1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovera S, Cuzzolin A, Kelm S, De Fabritiis G, and Sands ZA (2019). Reconstruction of apo A2A receptor activation pathways reveal ligand-competent intermediates and state-dependent cholesterol hotspots. Sci Rep 9, 14199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manglik A, Kim TH, Masureel M, Altenbach C, Yang Z, Hilger D, Lerch MT, Kobilka TS, Thian FS, Hubbell WL, et al. (2015). Structural Insights into the Dynamic Process of beta2-Adrenergic Receptor Signaling. Cell 161, 1101–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manglik A, Kruse AC, Kobilka TS, Thian FS, Mathiesen JM, Sunahara RK, Pardo L, Weis WI, Kobilka BK, and Granier S (2012). Crystal structure of the μ-opioid receptor bound to a morphinan antagonist. Nature 485, 321–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manglik A, Lin H, Aryal DK, McCorvy JD, Dengler D, Corder G, Levit A, Kling RC, Bernat V, Hubner H, et al. (2016). Structure-based discovery of opioid analgesics with reduced side effects. Nature 537, 185–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masureel M, Zou Y, Picard LP, van der Westhuizen E, Mahoney JP, Rodrigues J, Mildorf TJ, Dror RO, Shaw DE, Bouvier M, et al. (2018). Structural insights into binding specificity, efficacy and bias of a beta2AR partial agonist. Nat Chem Biol 14, 1059–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPherson J, Rivero G, Baptist M, Llorente J, Al-Sabah S, Krasel C, Dewey WL, Bailey CP, Rosethorne EM, Charlton SJ, et al. (2010). mu-opioid receptors: correlation of agonist efficacy for signalling with ability to activate internalization. Mol Pharmacol 78, 756–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao YL, Nichols SE, Gasper PM, Metzger VT, and McCammon JA (2013). Activation and dynamic network of the M2 muscarinic receptor. P Natl Acad Sci USA 110, 10982–10987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moukhametzianov R, Warne T, Edwards PC, Serrano-Vega MJ, Leslie AG, Tate CG, and Schertler GF (2011). Two distinct conformations of helix 6 observed in antagonist-bound structures of a beta1-adrenergic receptor. Proc Natl Acad Sci U S A 108, 8228–8232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neilan CL, Husbands SM, Breeden S, Ko MC, Aceto MD, Lewis JW, Woods JH, and Traynor JR (2004). Characterization of the complex morphinan derivative BU72 as a high efficacy, long-lasting mu-opioid receptor agonist. Eur J Pharmacol 499, 107–116. [DOI] [PubMed] [Google Scholar]
- Nygaard R, Zou Y, Dror RO, Mildorf TJ, Arlow DH, Manglik A, Pan AC, Liu CW, Fung JJ, Bokoch MP, et al. (2013). The dynamic process of beta(2)-adrenergic receptor activation. Cell 152, 532–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okude J, Ueda T, Kofuku Y, Sato M, Nobuyama N, Kondo K, Shiraishi Y, Mizumura T, Onishi K, Natsume M, et al. (2015). Identification of a Conformational Equilibrium That Determines the Efficacy and Functional Selectivity of the mu-Opioid Receptor. Angewandte Chemie 54, 15771–15776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patriksson A, and van der Spoel D (2008). A temperature predictor for parallel tempering simulations. Phys. Chem. Chem. Phys 10, 2073–2077. [DOI] [PubMed] [Google Scholar]
- Pedersen MF, Wrobel TM, Marcher-Rorsted E, Pedersen DS, Moller TC, Gabriele F, Pedersen H, Matosiuk D, Foster SR, Bouvier M, et al. (2020). Biased agonism of clinically approved mu-opioid receptor agonists and TRV130 is not controlled by binding and signaling kinetics. Neuropharmacology 166, 107718. [DOI] [PubMed] [Google Scholar]
- Raehal KM, Schmid CL, Groer CE, and Bohn LM (2011). Functional selectivity at the mu-opioid receptor: implications for understanding opioid analgesia and tolerance. Pharmacological reviews 63, 1001–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid CL, Kennedy NM, Ross NC, Lovell KM, Yue Z, Morgenweck J, Cameron MD, Bannister TD, and Bohn LM (2017). Bias Factor and Therapeutic Window Correlate to Predict Safer Opioid Analgesics. Cell 171, 1165–1175 e1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schott-Verdugo S, and Gohlke H (2019). PACKMOL-Memgen: A Simple-To-Use, Generalized Workflow for Membrane-Protein-Lipid-Bilayer System Building. J Chem Inf Model 59, 2522–2528. [DOI] [PubMed] [Google Scholar]
- Sena DM Jr., Cong X, Giorgetti A, Kless A, and Carloni P (2017). Structural heterogeneity of the mu-opioid receptor's conformational ensemble in the apo state. Scientific reports 8, 45761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada I, Ueda T, Kofuku Y, Eddy MT, and Wuthrich K (2019). GPCR drug discovery: integrating solution NMR data with crystal and cryo-EM structures. Nat Rev Drug Discov 18, 59–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solt AS, Bostock MJ, Shrestha B, Kumar P, Warne T, Tate CG, and Nietlispach D (2017). Insight into partial agonism by observing multiple equilibria for ligand-bound and Gs-mimetic nanobody-bound beta1-adrenergic receptor. Nat Commun 8, 1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sounier R, Mas C, Steyaert J, Laeremans T, Manglik A, Huang W, Kobilka BK, Demene H, and Granier S (2015). Propagation of conformational changes during mu-opioid receptor activation. Nature 524, 375–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sounier R, Yang Y, Hagelberger J, Granier S, and Demene H (2017). (1)H, (13)C and (15)N backbone chemical shift assignments of camelid single-domain antibodies against active state micro-opioid receptor. Biomol NMR Assign 11, 117–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staus DP, Hu H, Robertson MJ, Kleinhenz ALW, Wingler LM, Capel WD, Latorraca NR, Lefkowitz RJ, and Skiniotis G (2020). Structure of the M2 muscarinic receptor-beta-arrestin complex in a lipid nanodisc. Nature 579, 297–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susac L, Eddy MT, Didenko T, Stevens RC, and Wuthrich K (2018). A2A adenosine receptor functional states characterized by (19)F-NMR. Proc Natl Acad Sci U S A 115, 12733–12738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traynor JR, and Nahorski SR (1995). Modulation by mu-opioid agonists of guanosine-5'-O-(3-[35S]thio)triphosphate binding to membranes from human neuroblastoma SH-SY5Y cells. Mol Pharmacol 47, 848–854. [DOI] [PubMed] [Google Scholar]
- Tribello GA, Bonomi M, Branduardi D, Camilloni C, and Bussi G (2014). PLUMED 2: New feathers for an old bird. Computer Physics Communications 185, 604–613. [Google Scholar]
- Trott O, and Olson AJ (2010). AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 31, 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, and Berendsen HJ (2005). GROMACS: fast, flexible, and free. J Comput Chem 26, 1701–1718. [DOI] [PubMed] [Google Scholar]
- Vanni S, Neri M, Tavernelli I, and Rothlisberger U (2010). A conserved protonation-induced switch can trigger "ionic-lock" formation in adrenergic receptors. J Mol Biol 397, 1339–1349. [DOI] [PubMed] [Google Scholar]
- Vranken WF, Boucher W, Stevens TJ, Fogh RH, Pajon A, Llinas M, Ulrich EL, Markley JL, Ionides J, and Laue ED (2005). The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins 59, 687–696. [DOI] [PubMed] [Google Scholar]
- Wang JM, Cieplak P, and Kollman PA (2000). How well does a restrained electrostatic potential (RESP) model perform in calculating conformational energies of organic and biological molecules? J Comput Chem 21, 1049–1074. [Google Scholar]
- Wang JM, Wolf RM, Caldwell JW, Kollman PA, and Case DA (2004). Development and testing of a general amber force field. J Comput Chem 25, 1157–1174. [DOI] [PubMed] [Google Scholar]
- Wang L, Friesner RA, and Berne BJ (2011). Replica exchange with solute scaling: a more efficient version of replica exchange with solute tempering (REST2). J Phys Chem B 115, 9431–9438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weis WI, and Kobilka BK (2018). The Molecular Basis of G Protein-Coupled Receptor Activation. Annu Rev Biochem 87, 897–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wingler LM, Elgeti M, Hilger D, Latorraca NR, Lerch MT, Staus DP, Dror RO, Kobilka BK, Hubbell WL, and Lefkowitz RJ (2019). Angiotensin Analogs with Divergent Bias Stabilize Distinct Receptor Conformations. Cell 176, 468–478 e411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Hu Y, Kaindl J, Risel P, Hubner H, Maeda S, Niu X, Li H, Gmeiner P, Jin C, et al. (2019). Conformational Complexity and Dynamics in a Muscarinic Receptor Revealed by NMR Spectroscopy. Molecular cell. [DOI] [PubMed] [Google Scholar]
- Ye L, Van Eps N, Zimmer M, Ernst OP, and Prosser RS (2016). Activation of the A2A adenosine G-protein-coupled receptor by conformational selection. Nature 533, 265–268. [DOI] [PubMed] [Google Scholar]
- Yin W, Li Z, Jin M, Yin YL, de Waal PW, Pal K, Yin Y, Gao X, He Y, Gao J, et al. (2019). A complex structure of arrestin-2 bound to a G protein-coupled receptor. Cell Res 29, 971–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan S, Palczewski K, Peng Q, Kolinski M, Vogel H, and Filipek S (2015). The mechanism of ligand-induced activation or inhibition of mu- and kappa-opioid receptors. Angew Chem Int Ed Engl 54, 7560–7563. [DOI] [PubMed] [Google Scholar]
- Yudin Y, and Rohacs T (2019). The G-protein-biased agents PZM21 and TRV130 are partial agonists of mu-opioid receptor-mediated signalling to ion channels. Br J Pharmacol 176, 3110–3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report any original codes.
Any additional information required to reproduce this work is available from the Lead Contact.