

Summary:
Mammalian cells employ diverse pathways to prevent deleterious consequences during DNA replication, yet the mechanism by which cells survey individual replisomes to detect spontaneous replication impediments at the basal level, and their accumulation during replication stress, remains undefined. Here, we utilized Single-Molecule Localization Microscopy coupled with High-Order-Correlation image-mining algorithms, to quantify the composition of individual replisomes in single cells during unperturbed replication and under replicative stress. We identified a basal-level activity of ATR that monitors and regulates the amounts of RPA at forks during normal replication. Replication-stress amplifies the basal activity through the increased volume of ATR-RPA interaction and diffusion-driven enrichment of ATR at forks. This localized crowding of ATR enhances its collision probability, stimulating the activation of its replication-stress response. Finally, we provide a computational model describing how the basal activity of ATR is amplified to produce its canonical replication-stress response.
Keywords: DNA replication, ATR activity, Super-Resolution Imaging, High-Content Image Mining
eTOC:
Yin et al. use single-molecule imaging to visualize replisomes at the nanoscale, revealing that ATR continuously monitors the levels of RPA within replisomes during normal replication. This process supports ATR-CHK1 activation upon replicative stress, where accumulation of RPA at forks induces localized ATR crowding, and amplifies its encounter with activators.
Graphical Abstract
Introduction
Duplication of the eukaryotic genome is performed by multiple replication forks initiated from origins that are spatially distributed throughout the genome, and tightly regulated in an orderly fashion(Fragkos et al., 2015). While replication forks are highly coordinated to ensure an efficient and accurate duplication process, they are also stochastically challenged by various intrinsic replication impediments such as DNA repeats(Chandok et al., 2012) and secondary structures(Leon-Ortiz et al., 2014), replication-transcription conflicts(Hamperl et al., 2017), and DNA lesions(Chang et al., 2019). These intrinsic basal-level blockages spontaneously occur during normal replication, and results in fluctuations and obtrusions in replication fork progression, challenging the efficiency and accuracy of replication, and leading to genome instability(Saldivar et al., 2017; Zeman and Cimprich, 2014).
When cells encounter wide-ranging slowing and stalling of replication forks, they transit into a state of global replicative-stress. A primary response to replication-stress is the activation of ATR (Ataxia-Telangiectasia and Rad3-related, Mec1 in yeast), a replication checkpoint kinase that plays a central role in signaling to its various downstream effectors, including the cell cycle checkpoint kinase Chk1 (Rad53 in yeast), in the prevention of DNA replication catastrophe upon conditions of replication stress(Hustedt et al., 2013; Saldivar et al., 2017). Upon activation, ATR orchestrates two major processes to prevent new replication liabilities and protect ongoing replisomes: 1) The ATR-mediated global inhibition of origin firing(Chen et al., 2015; Ge and Blow, 2010; Randell et al., 2010; Santocanale and Diffley, 1998; Toledo et al., 2013). Although mild replication-stress conditions lead to the compensatory firing of dormant origins within already active replication factories(Chen et al., 2015; Ge and Blow, 2010), late origin firing is largely inhibited to ensure a sufficient RPA pool to protect single-stranded DNA (ssDNA) formed upon stress. Failure to suppress late origin firing results in exhaustion of the RPA pool and leads to replication catastrophe(Toledo et al., 2013). 2) ATR mediated stabilization and remodeling of stalled replication forks via numerous downstream regulators and effectors(Cobb et al., 2005; Couch et al., 2013; Han et al., 2015; Lossaint et al., 2013; Lucca et al., 2004). For example, ATR phosphorylates SMARCAL1 and thereby suppresses its activity in fork regression(Couch et al., 2013); ATR mediates the recruitment of FANCD2 to stalled forks to slow replication progression to minimize ssDNA exposure (Lossaint et al., 2013); and in yeast, Mec1 and Sgs1 cooperate to stabilize the polymerases at stalled replication forks (Cobb et al., 2005).
Despite the central importance of ATR in response to replication-stress, the mechanism of ATR activation is not fully understood. In vertebrates, ATR is activated via at least two distinct parallel pathways. During replication stress, RPA accumulates at stress-induced ssDNA at forks, recruiting the ATR-ATRIP (Mec1-Ddc2 in yeast) complex (Ball et al., 2005; Biswas et al., 2019; Deshpande et al., 2017; Zou and Elledge, 2003). ATR is then activated via interactions with either ETAA1 or TOPBP1 (Dpb11 in yeast) through their ATR-activation domains (AADs). While ETAA1 directly binds to RPA and stimulates ATR activation(Bass et al., 2016; Haahr et al., 2016), TOPBP1 is recruited at the 5’-ended-ssDNA-dsDNA fork junction through the RAD9-RAD1-HUS1 (9–1-1) clamp complex together with other components(Cotta-Ramusino et al., 2011; Duursma et al., 2013; Lee and Dunphy, 2013; Puddu et al., 2008) for ATR-Chk1 activation(Kumagai et al., 2006; Mordes et al., 2008). Although studies have identified the basic components in these separate ATR activation pathways, the preceding mechanism and activities of ATR prior to its engagement with TOPBP1 / ETAA1 remain unclear. In particular, it is unknown how ATR measures the amount of RPA at individual replication forks to recognize RPA accumulation at stressed forks and distinguish these from the unstressed forks that carry relatively less RPA. It is also unclear whether and how such stress-recognition, which is expected to be constantly implemented during both normal replication and replication stress, is linked to the activation of the canonical ATR-Chk1 stress responding activity.
Beyond its activities in replication-stress response, several studies have also been focusing on ATR activity during unperturbed replication. Quantitative phosphoproteomic analysis in budding yeast have identified an activity of Mec1, that correlates to normal replication and distinguishable from its canonical Mec1-Rad53 stress-response activity(Bastos de Oliveira et al., 2015). In human cells, the use of clinical ATR inhibitors (ATRi) in the absence of external replication-stress resulted in unscheduled origin firing(Moiseeva et al., 2017; Petermann et al., 2010; Syljuasen et al., 2005) that was accompanied by reduced fork progression(Couch et al., 2013; Moiseeva et al., 2017), suggesting that ATR and Chk1 also negatively regulate origin firing even during unperturbed replication. Despite these efforts, it remains unclear what are the specific basal-level activities of ATR in the absence of global replication-stress, and whether and how such activities are connected or converge to its global replication stress activity.
A major limitation towards a comprehensive understanding of the specific activities of ATR during normal replication and in response to global replication-stress is posed by the inadequacy in quantitative analysis with sufficient resolution. Previously, we have developed a multi-color Single-Molecule Localization Microscopy (SMLM)-based image mining technique that enables quantification of replication factors at the single-replisome level(Lee et al., 2021; Yin et al., 2019; Yin and Rothenberg, 2016). Here, we applied this approach to map the molecular configurations of individual replisomes within single cells as a function of ATR activity and its effectors during unperturbed replication, and in response to replication-stress. We identified a basal-level activity of ATR by which the specific levels of RPA at replisomes are surveyed via the diffusion-controlled ATR-RPA interaction during normal replication. We found that this basal ATR-RPA interaction regulates the level of RPA at forks by facilitating the removal of RPA via phosphorylation of RPA2 at Ser33 which alters its coordination on ssDNA. Importantly, this ATR activity is separable from the canonical role of ATR-Chk1-mediated origin firing. We propose that this mode of basal-level ATR-RPA interaction provides a unique diffusion-driven interface between ATR and RPA by which ATR surveys the RPA content of replisomes. When replication stress occurs, this ATR-RPA interface is amplified as RPA accumulates at stalled forks, forming localized concentration gradients of ATR that enable the subsequent activation of the canonical replication stress response. Based on our findings we have generated a computational model that describes how the localized basal activity of ATR develops into its canonical activation of ATR-Chk1 signaling in response to the global replication stress.
Results
Inhibition of ATR during unperturbed replication leads to RPA accumulation at replication forks
The inhibition of ATR activity during unperturbed replication was previously shown to induce new origin firing(Moiseeva et al., 2017; Petermann et al., 2010; Syljuasen et al., 2005), yet it is unclear whether and how ATR could also impact active replication forks at the basal-level. To determine this, we sought to quantify specific spatial features of individual replication forks along with their local density of RPA, as a function of ATR activity. To resolve replication forks in cells, we utilized multi-color Single-Molecule Localization Microscopy (SMLM)(Eric Betzig et al., 2006; Rust et al., 2006) of which the increased sensitivity and spatial resolution provides nanoscale features of the molecular complexes that cannot be resolved via diffraction-limited fluorescence microscopy (Figure S1A). The SMLM of U2OS cells immunostained against PCNA, RPA, and MCM reveals that PCNA, RPA and MCM spatially organized into fork-like configurations (Figure 1A and 1B). We also used SMLM to examine the organization of nascent DNA (pulse labeled with EdU, STAR Methods), PCNA, and MCM, which displays similar fork-like arrangements (Figure S1A). To ensure that the identification and analysis of individual forks within the densely populated SMLM images are unbiased, and robust, we performed multiplexed pattern recognition by using a Triple Correlation Function (TCF) based multi-color image mining algorithm(Lee et al., 2021; Yin et al., 2019), whose scheme is illustrated in Figure 1C-E and Methods S1. This approach computes the probability density for the occurrence of all possible 3-color molecular-patterns (or triplets), generating a probability density plot as a function of the lengths of the three edges that define a triangular configuration (Figure 1D (ii)). Figure 1E shows the most frequent PCNA–RPA–MCM configuration derived from the TCF of a single nucleus, which is comparable to a single replisome with respect to its morphology and scale(Goswami et al., 2018; Xiang et al., 2018). To display the pooled replication fork configurations from multiple S-phase cells, we overlaid the TCF-resolved fork patterns of each cell (Figure 1F, and S1B) providing a statistical representation that incorporates intrinsic variations factors (Methods S1). The average molecular density of RPA within each replisome-pattern, termed RPAfork, was derived from the probability density computed via TCF (STAR Methods, Methods S2)(Lee et al., 2021; Yin et al., 2019) and encoded into the size of the circles in Figure 1G. We note that in the early S-phase nucleus, MCM is overloaded and generally more abundant on chromatin at unfired and dormant origins (Figure S1C - S1E)(Ge et al., 2007). As these MCMs are not spatially associated with active replisomes, they do not affect the TCF quantification of RPAfork (see simulations in Figure S1F, S1G, and Methods S2). The number of replication foci (PCNA foci) and chromatin bound MCMs were estimated via Auto Correlation Function (ACF) analysis of the SMLM data (Sengupta et al., 2011; Veatch et al., 2012) (Figure S1C-S1E, and Methods S4), and was in good agreement with the estimates provided by previous studies (Table S1) (Baddeley et al., 2010; Chagin et al., 2016; Cseresnyes et al., 2009; Su et al., 2020).
Figure 1. ATR prevents RPA accumulation at replication forks during unperturbed replication.

(A and B) Representative multi-color SMLM images of replication factors PCNA, RPA, and MCM in S phase cells that were either non-treated (A, NT) or treated with ATR inhibitor (B, ATRi). Magnified panels for each image show representative triplet patterns of individual replisomes.
(C and D) Schematic illustration of the data mining workflow using TCF based approach for recognition of triplet patterns. Any molecule i from the Red, j from the Green, and k from the Blue channel can form a triplet , defined by their distances . The occurrence of all these triplets (e.g. , …) is then counted as a function of their triple-wise distances (i). When many molecules of the three species form a similar triplet pattern (i.e. many triplets with similar ), the probability density distribution of such triplet rises at from the level of uncorrelated random patterns (ii) (Methods S1).
(E) The most populated triplet pattern as derived from the single-cell TCF distribution (D, ii).
(F and G) Workflow for pooled representation of the TCF-resolved single-replisome configurations; The single-replisome configuration is resolved for each cell (F) and the configurations for multiple cells are overlaid (G).
(H) Quantification of the levels of RPAfork for the TCF-resolved single replisome configurations shown in (G) reveals an ATRi-induced RPA accumulation at forks. Mean values are shown as the black bar, N = 193 and 177 for NT and ATRi, respectively.
(I and J) Kinetics of ATRi-mediated RPA accumulation at forks. Mean values and the 1st and 3rd quartile are marked as black and colored bars, respectively. N = 193, 53, 70, and 177 for NT, 60, 120, and 180 minutes treatment in (I), respectively; N = 193, 177, 72, 68, and 79 for NT, 0, 60, 90, 120 minutes release in (J), respectively.
To determine whether ATR activity contributes to the levels of RPA at forks during unperturbed (basal-level) replication, we quantified RPAfork in cells that were treated with ATR inhibitor (ATRi, VE-821, 2 μM) for three hours. This analysis revealed that the inhibition of ATR activity induces an increase in RPAfork (Figure 1G - 1I) during basal-level replication, where the accumulation of RPA at forks is reversible and gradually decreases following removal of ATRi (Figure 1J). We note that the removal of ATRi by releasing the cells into fresh culture medium without ATR inhibitors might not immediately lead to the full recovery of ATR activity, resulting in slower recovery kinetics in both RPAfork and ATR-mediated origin firing as indicated by the overall RPA density within a nucleus (RPAglobal, Figure S2). To exclude the possibility that the observed accumulation of RPA at forks is due to an ATRi-induced fork damage in the form of collapsed or recessed replication forks(Whelan et al., 2020; Whelan et al., 2018), we examined the levels of both γH2AX and pRPA-Ser4/Ser8, which were unchanged following ATRi treatment (Figure S3). These measurements indicate that the observed increase in the levels of RPA at forks resulted from inhibition of ATR activity, and is not due to fork damage.
ATR prevents RPA accumulation at replication forks separable from its activity in limiting origin firing
In mammalian cells, inhibition of ATR can cause excess origin firing (Moiseeva et al., 2017) and thereby consumes more dNTPs, and in yeast inhibition of Mec1 can downregulate the dNTP pools (Zhao; et al., 2001). We therefore sought to examine whether the ATRi-induced increase in RPAfork is due to the ATRi-induced overconsumption of the dNTPs. To this end, we blocked origin firing by inhibiting CDC7 (CDC7i, PHA767491, 20 μM)(Dungrawala et al., 2015; Moiseeva et al., 2019; Toledo et al., 2013), and measured RPAfork with or without inhibition of ATR (Figure 2A – 2C). We found that inhibition of CDC7 did not affect the levels of RPAfork (Figure 2D, CDC7i+ATRi- vs. CDC7i-ATRi-), nor prevent RPA from accumulating at forks when ATR was also inhibited (Figure 2D, CDC7i+ATRi+ vs. CDC7i+ATRi-), suggesting that the ATRi-induced increase in RPAfork is not due to the ATRi-induced excessive consumption of dNTPs, and that the ATR activity of limiting RPA accumulation at forks is separable from its activity in limiting origin firing. We note that while we did not observe significant reduction in origin firing when acute CDC7i was applied, such CDC7i treatment did drastically block the origin firing induced by ATRi (Figure 2E). These results agree with previous studies that CDC7i does not significantly affect basal-level replication but strongly limit ATRi / Chk1i-induced origin firing(Ercilla et al., 2020; Moiseeva et al., 2017; Rainey et al., 2017; Toledo et al., 2013).
Figure 2. ATR prevents RPA accumulation at replication forks independent of ATR activity in limiting origin firing.

(A and B) Representative SMLM images of replication fork factors PCNA, RPA, and MCM in S phase cells that were treated with CDC7i (A) and CDC7i+ATRi (B). Magnified panels for each image show representative triplet patterns of individual replisomes.
(C) Overlaid PCNA-RPA-MCM TCF-resolved single-replisome configurations from SMLM images of single cells under different treatments. Circle size represents the average density of RPA at each fork within a nucleus.
(D) Quantification of the levels of RPAfork for the TCF-resolved single replisome configurations shown in (C) reveals that ATRi induces RPA accumulation even when excessive origin firing is suppressed via CDC7i. Mean values and the 1st and 3rd quartile were marked as black and colored bars, respectively, N = 193, 177, 67, and 97 for CDC7i-ATRi-, CDC7i-ATRi+, CDC7i+ATRi-, and CDC7i+ATRi+, respectively. Right panel: quantification of the abundance ratio between data with ATRi treatment and w/o ATRi treatment (ATRi / no ATRi). Error bar in log (Abundance Ratio) is the propagated SEM.
(E) Quantifications of the levels of RPAglobal within each single cell at different ATRi and CDC7i treatment conditions, indicating that inhibition of CDC7i suppresses the ATRi-induced origin firing. Mean values and the 1st and 3rd quartile were marked as black and colored bars, respectively, N = 1863, 1267, 277, and 483 for CDC7i-ATRi-, CDC7i-ATRi+, CDC7i+ATRi-, and CDC7i+ATRi+, respectively. Right panel: quantification of the abundance ratio between data with ATRi treatment and w/o ATRi treatment (ATRi / no ATRi). Error bars in log (Abundance Ratio) is the propagated SEM.
Chk1 is the main propagator of the canonical ATR-Chk1 activity in limiting origin firing during unperturbed replication(Moiseeva et al., 2017) and in response to replication stress(Toledo et al., 2013). We therefore examined whether Chk1 contributes to the basal-level ATR activity in regulating the accumulation of RPA at forks. As shown in Figure S4, inhibition of Chk1 did not result in an increase in RPAfork, but contributed to a rise in new origins firing as previously reported(Moiseeva et al., 2017; Toledo et al., 2013). These results further indicate that the basal activity of ATR that regulates the levels of RPA at forks during unperturbed replication is separable from its canonical ATR-Chk1 activity in limiting origin firing.
The physical interaction between ATR and RPA is mediated by the ATR co-factor ATRIP (Ddc2 in yeast), and is necessary for stimulating ATR-Chk1 signaling during replication stress(Cortez et al., 2001; Deshpande et al., 2017; Zou and Elledge, 2003). We therefore sought to explore whether ATRIP is also required for the basal-level activity of ATR for regulating RPA accumulation at forks. To determine this, we measured the levels of RPA at forks in cells where ATRIP expression was reduced (~50%) via siRNA. As shown in Figure 3A and 3B, the reduction of ATRIP resulted in a detectable increase in the levels of RPAfork, suggesting that the basal-level activity of ATR that regulate RPAfork requires a physical interface between ATR-ATRIP complex and RPA-ssDNA.
Figure 3. ATRIP is necessary for the ATR basal-activity in regulating the levels of RPA at forks.

(A) Overlaid PCNA-RPA-MCM TCF-resolved single-replisome configurations from SMLM images of single cells under different treatments as indicated. Circle size represents the average density of RPA at each fork within a nucleus.
(B) Quantification of the levels of RPAfork for the TCF-resolved single replisome configurations shown in (A) reveals that ATRIP is necessary for the ATR basal activity in regulating the RPA level at forks. Mean values and the 1st and 3rd quartile were marked as black and colored bars, respectively, N = 38, 46, 29, and 47 for siCTRL-NT, siCTRL-ATRi, siATRIP-NT, and siATRIP-ATRi, respectively. Right panel: quantification of the abundance ratio between data as indicated. Error bars in log (Abundance Ratio) is the propagated SEM.
(C) Schematic illustration of ATR basal activity mediated by ATRIP for regulating RPA levels at replication forks.
Phosphorylation of RPA2 Ser-33 prevents RPA from accumulating at replication forks
Our observation that the basal activity of ATR is independent of ATR-Chk1 signaling (Figure 2D), but relies on a physical interaction between ATR-ATRIP and RPA (Figure 3B), led us to the hypothesis that a direct interaction between ATR and RPA may contribute to the removal of RPA from ssDNA at forks. Ser33 on the N-terminal domain of RPA2 is amongst the primary phosphorylation sites for ATR kinase(Liu et al., 2012), and the phosphorylation of the N-terminal of RPA2 was suggested to affect RPA-ssDNA affinity(Fanning et al., 2006). To test this, we largely depleted endogenous RPA2 via siRNA and supplemented the cells with exogenous expression of wild-type (WT) RPA2, or RPA2 mutants Ser33-to-Ala (S33A, phospho-dead) and Ser33-to-Asp (S33D, phospho-mimic) (Figure 4A). Cells were characterized using flow cytometry showing that cell cycle progression was severely impaired upon depletion of RPA2, but recovered in cells that were supplemented via expression of the exogenous wildtype or mutant RPA2 (Figure S5A). Next, we preformed SMLM imaging and TCF analyses of RPAfork in these cells, revealing an appreciable increase in the levels of RPAfork in cells expressing RPA2-S33A as compared to cells expressing RPA2-S33D (Figure 4B and 4C). This trend indicates that the coordination of the RPA trimer on ssDNA may improve when the polar Ser33 or the negatively charged Asp33 is replaced by a hydrophobic alanine. Of note, we did not observe an obvious reduction in RPAfork for cells expressing RPA2-S33D as compared to RPA2-WT, suggesting that while the phosphorylation of Ser33 is necessary for the removal of RPA from ssDNA, it is not sufficient in-itself to facilitate RPA’s removal.
Figure 4. ATR regulates the accumulation of RPA at forks via phosphorylation of RPA2 Ser-33.

(A) Western-blot analysis of the expression of RPA2 and RPA2 mutants for RPA2 siRNA and RPA2 siRNA + transfection of myc-tagged wild-type (WT) RPA and S33A, S33D RPA2 mutants.
(B) Overlaid PCNA-RPA-MCM TCF-resolved single-replisome configurations from SMLM images of single cells harboring RPA2 WT and RPA2 phosphor-mutants and under CDC7i treatment conditions. Circle size represents the average density of RPA at each fork within a nucleus.
(C) Quantification of the levels of RPAfork for the TCF-resolved single replisome configurations shown in (B) reveals that the expression of the phosphor-dead RPA2 S33A results in an accumulation of RPA at forks that is independent of origin firing. Mean values and the 1st and 3rd quartile were marked as black and colored bars, respectively, N = 43, 48, 52, 41, 39, and 56 for WT-CDC7i-, S33D-CDC7i-, S33A-CDC7i-, WT-CDC7i+, S33D-CDC7i+, and S33A-CDC7i+, respectively.
(D) Quantification of the abundance ratio between data as indicated. Error bars in log (Abundance Ratio) is the propagated SEM.
(E) Schematic illustration of the regulation of RPAfork levels by ATR via phosphorylation of RPA2 at Ser33.
To gain further mechanistic insights into the contribution of Ser33 phosphorylation on RPA2 to the removal of RPA, we sought to understand how the coordination of RPA binding to ssDNA is affected by phosphorylation of Ser33. To measure this, we designed an in-vitro single-molecule FRET (smFRET) assay that utilizes a 32-nt-long ssDNA 3’ tail as the ssDNA substrate, ensuring that binding of RPA is limited to a single RPA trimer. To monitor the specific coordination of RPA2 on the ssDNA, a FRET pair (Cy3-Donor and Cy5-Acceptor) were positioned 12-nt apart at the 3’-end of the ssDNA tail as depicted in Figure 5A. Since ssDNA alone favors a coil-like collapse conformation in physiological conditions, this FRET pair normally resides close to each other and yields high FRET efficiency (Figure 5B, DNA Only). Upon addition of RPA and its binding to ssDNA, the RPA-bound ssDNA is extended, which increases the distance between the FRET pair, and results in lower FRET values (Figure 5B, DNA + RPA). Using this approach, we measured the FRET values of ssDNA bound by recombinant wild-type RPA trimers or by mutant RPA with S33A or S33D mutant. The coordination of the ssDNA segment bound by WT RPA2 or the S33A mutant showed persistent and tight binding as exemplified by the real-time smFRET trajectories showing stable low FRET (RPA2-bound) state (Figure 5C) and the resulting single peak distribution in the FRET histograms (Figure 5D). The FRET histograms for the S33D mutant displayed two district distributions, consisting of a low FRET (RPA2-bound) state (~85%), and a high FRET (DNA-Only) state (~15%), indicating that the binding coordination of S33D RPA2 onto the ssDNA segment is less stable than that of WT or S33A RPA2. Indeed, we observed corresponding trends for a portion of individual smFRET trajectories in which the ssDNA segment displayed binding-unbinding fluctuations when interacting with the S33D mutant, in contrast to the persistent low FRET state observed in the smFRET trajectories of WT and S33A RPA2 (Figure 5D and 5E, respectively).
Figure 5. RPA2 S33 phosphorylation affects the coordination of ssDNA bound RPA.

(A) Schematic illustration of the single-molecule FRET assay for probing RPA2 coordination for ssDNA bound RPA trimer. The Cy3-Cy5 FRET reporter was labeled at the 3’-end of the 32nt-long ssDNA, with 12nt distancing in between, so that the changes in FRET efficiency represents altered coordination of RPA2 on ssDNA.
(B) Representative smFRET trajectories for DNA-Only (left panel) and with RPA with WT RPA2 bound (right panel).
(C) Representative smFRET trajectories of ssDNA bound by WT RPA (top panel) or by RPA with RPA2 S33A (middle panel) and RPA2 S33D (bottom panel). The smFRET trajectory for RPA2 S33D displays fluctuation in FRET signal indicating that phosphorylation of RPA2 at S33 reduces its coordination on ssDNA.
(D) FRET histograms for ssDNA bound RPA showing the specific FRET states (in red) for WT (top), S33A (middle) and S33D (bottom) as compared to ssDNA only (blue), revealing that S33D also populates unbound state corresponding to binding fluctuations of the phosphorylated RPA2 onto ssDNA.
(E) The Transition Density Plot (TDP) generated from HMM analysis of smFRET trajectories for the ssDNA bound RPA with WT (top), S33A (middle) and S33D (bottom) RPA2. This shows specific transitions for S33D corresponding to RPA2-ssDNA binding fluctuations. N provides the number of trajectories analyzed by HMM to generate the TPD analysis for each condition.
(F) Schematic illustration of the observed binding fluctuations of RPA2 domain onto ssDNA via Ser-33 phosphorylation.
To further probe the effect of RPA2 Ser33 on the coordination between the RPA trimer and ssDNA, we designed a similar ssDNA oligo substrate with the FRET pair positioned relatively distal to the 3’ end, thereby monitoring the coordination of RPA1 on ssDNA (Figure S6A). The binding of RPA to this substrate resulted in extended coordination of ssDNA by RPA1 displaying a single peak distribution at the low FRET (RPA1-bound) state for the WT and S33A RPA as well as for the S33D mutants (Figure S6B-S6D). The absence of the FRET population at the high FRET (DNA Only) state, or any binding-unbinding fluctuations in smFRET trajectories, indicates that the coordination of RPA1 with ssDNA is consistent and unaffected by the phosphorylation state of RPA2 Ser33, providing further validation that the two FRET substrates used here can distinctly probe the ssDNA coordination of RPA2 and RPA1. These findings suggest that phosphorylation of Ser33 does not directly cause RPA to dissociate from ssDNA, but rather ease the coordination of RPA and its unloading, likely by other fork-associated factors (Figure 5F).
ATR basal-level activity supports the formation of an ATR concentration gradients at replication forks during replication stress
The basal-level activity of ATR that we have identified limits the accumulation of RPA at replications forks during unperturbed replication, and is separable from its role in regulating origin activation. We therefore sought to understand whether and how the basal-level ATR activity is related to its canonical ATR-Chk1 activity in responding to replication stress. To determine this, we induced varied levels of replication stress by exposing cells to low and medium concentrations of hydroxyurea (HU) for a short duration (2hrs), and measured the levels of either RPA or ATR at forks (ATRfork) using TCF analysis, as a function of replication stress and ATR activity. These experiments revealed an increase in the local concentration of RPA and ATR at forks as replication stress increases (Figure 6A and 6B), suggesting that the rising levels of replication-stress lead to an increase in the amount of RPA localized at forks, and such stress-driven accumulation of RPA expands the interaction interface between ATR and RPA, thereby amplifies the basal ATR-RPA interaction. We postulate that this amplified basal interaction at forks generates diffusion-controlled concentration gradients that act as a precursor, providing a crowding environment that serves as platform for global ATR signaling and the activation of its canonical replication stress response.
Figure 6. Replication-stress amplifies the basal ATR-RPA interface and enriches ATR at forks.

(A and B) Quantification of the effect of replication stress and ATR inhibition on the levels of RPAfork (A) and ATRfork (B) for the TCF-resolved single-replisome configurations for the different treatment conditions as indicated. Replication stress induces a modest increase in the level of RPA and ATR at forks, and the levels of RPA are further amplified upon ATRi treatment. Mean values and the 1st and 3rd quartile were marked as black and colored bars, respectively. For RPAfork, N = 193, 35, 44, 177, 58, and 62 for HU0_ATRi-, HU50_ATRi-, HU500_ATRi-, HU0_ATRi+, HU50_ATRi+, and HU500_ATRi+, respectively; For ATRfork, N = 85, 46, 47, 46, 45, and 52 for HU0_ATRi-, HU50_ATRi-, HU500_ATRi-, HU0_ATRi+, HU50_ATRi+, and HU500_ATRi+, respectively. We note that the inhibition of ATR and replication stress have both led to an increase in the levels of RPAfork and ATRfork though their specific trends differ. Note that while the ATRi-accumulated RPA at forks could harbor more ATR (as compared to NT ATRfork), the overall transient nature of ATR-RPA interaction along with the direct inhibition of ATR can also reduce the association of ATR.
(C) Schematic illustration of replication stress induced enrichment of ATR at forks driven by the basal level ATR-RPA interface.
(D) Computational model for the replication-stress induced amplification of ATR basal activity with increase of RPAfork levels thereby leading to the canonical ATR-chk1 activation. Simulation workflow: ATR (green particle) were randomly seeded in a reaction cube and exhibit Brownian diffusion. RPA (red particle) were initialized at the fork fixed at the center of the cube, and increased at each iteration as replication stress is applied (i); the increase in RPA at the fork amplifies the frequency of the diffusion-driven ATR-RPA contact, resulting in enrichment of ATR at the fork (ii); To incorporate the basal activity of ATR that limits RPA accumulation at forks, RPA were simulated to decrease exponentially with the rate proportional to the number of ATR interacting with RPA (ATR-in-contact, iii).
(E - G) Trajectories of ATR-fork and ATR-ATR reaction kinetics derived from the Monte Carlo simulation in (D) for increasing levels of replication stress: (E) The enrichment of ATR at the fork as a result of the diffusion controlled ATR-RPA contacts, (F) Frequency of diffusion-mediated encounters of ATR with replication fork due to the replication-stress induced formation of a localized concertation gradient, which enhances the probability of ATR engaging with its activators at the fork, (G) Frequency of diffusion-mediated encounters of ATR with other ATR molecules at the vicinity of the replication fork increases due to the ATR concertation gradient. Mean ± SD, N = 10 simulations for each scenario/condition.
To further verify that the amplification of the basal ATR-RPA interaction leads to the concentration gradient of ATR around replication forks, we devised a computational approach that simulates the diffusion-controlled interaction between ATR and RPA at different stress levels and measures the concentration of ATR around the stressed replication fork. As illustrated in Figure 6D, we simulated a certain number of ATR molecules (green particles) freely diffusing within a reaction cube at the initial time point (t = 0), where RPA molecules residing at a replication fork were positioned at the center. The model incorporates the diffusion dynamics of ATR and monitors the temporal profile of the reaction at different levels of replication stress by considering and deriving the following independent parameters as the simulation progresses through each time point (t = 1, 2 …): i) the number of RPA at the fork linearly increased with a slope depending on the stress level applied; ii) ATR that diffuses into the collision radius of RPA at the fork establishes ATR-RPA contacts; and iii) the number of RPA at the fork decreases as ATR facilitates RPA removal, following a single-exponential decay with the rate proportional to the number of ATR-in-contact. Figure 6E displays the resulting number of ATR-in-contact with the fork-bound RPA at different stress levels of replication-stress for 100 iterations, providing a robust reproduction of the diffusion-controlled amplification of the basal ATR-RPA contact and formation of concentration gradients with increased replication stress. We further used this simulation to model how the localization of ATR at the fork affects its collision frequency with other ATR molecules and with its activators, and thereby stimulates the trans-activation of the ATR-Chk1 signaling. For this analysis, within each iteration the ATR molecules that establish contacts with RPA at the fork (Figure 6D(ii)) were set to diffuse locally for a certain duration, and the frequency of ATR diffusing into the collision radius of other ATR molecules or its activators (set at the fork) were recorded. As shown in Figure 6F and 6G, the increased concentration of ATR at the vicinity of the fork (Figure 6E) resulted in enhanced collisions amongst ATR molecules and between ATR and its activators, which together guarantees sufficient probabilities for the activator- and auto-phosphorylation-mediated ATR activation, respectively(Bass et al., 2016; Haahr et al., 2016; Kumagai et al., 2006; Liu et al., 2011; Mordes et al., 2008).
Discussion
Model connecting ATR fork surveillance activity to its canonical replication-tress response
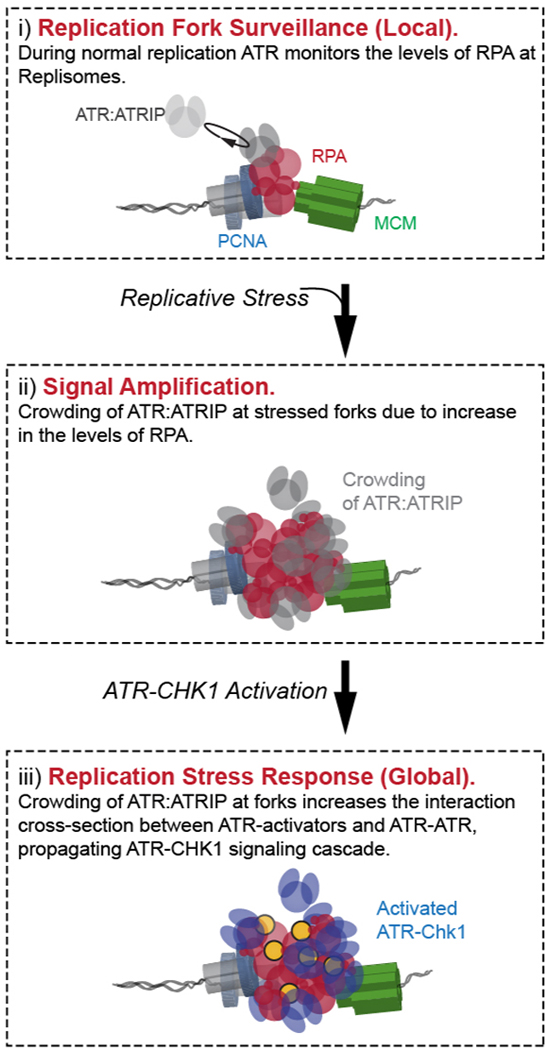
The ability to resolve and quantify the specific molecular and spatial features of individual replication-forks within single cells opens new avenues of research by providing previously undetectable information. Using this new approach, we have quantified the single-fork characteristics of ATR activities to identify a basal activity of ATR during unperturbed replication, and establish the mechanistic convergence from this basal activity to its response to replication stress. Based on our results, we propose a new integrated model for ATR roles during replication, as illustrated in Figure 7: During normal basal-level (unperturbed) replication, ATR stochastically and frequently contacts RPA at replication forks in a diffusion-controlled manner (Figure 7A). In active replication forks whose progression is unchallenged, the exchange of RPA is rapid such that the ATR-RPA interaction is too transient to allow for a productive reaction between ATR and RPA. At forks that encounter transient impediments that lead to accumulation of RPA, the prolonged physical contact between ATR and RPA will increase the probability of ATR to phosphorylate Ser33 on RPA2 and thereby facilitate RPA’s removal (Figure 7B). When cells encounter global replication stress leading to wide-ranging slowing or stalling of replication forks, the diffusion-controlled ATR-RPA contacts are amplified with the rapid accumulation of RPA at stressed forks, promoting local molecular crowding of ATR. The concentration gradients of ATR at the vicinity of forks substantially increases the probability for productive collisions between ATR and its activators TOPBP1(Kumagai et al., 2006; Mordes et al., 2008) / ETAA1(Bass et al., 2016; Haahr et al., 2016) for ATR activation, and between ATR molecules for ATR auto-phosphorylation(Liu et al., 2011) and overall ATR signal amplification. The amassment of these localized amplification events leads to the emergence of the canonical global replication-stress response and activation of checkpoint kinases, limiting origin firing and arresting active replicative helicases (Figure 7C). Combined, this model presents the mechanistic correlation between ATR surveying replication forks via ATR-RPA contact and ATR responding to replication stress via enrichment and crowding of ATR at forks (Figure 7D).
Figure 7. Model depicting the basal-level activity of ATR in replication-fork surveillance and as a platform for replication-stress induced activation of the canonical ATR-Chk1 signaling.

(A-B) Basal-level activity of ATR during normal replication: ATR monitor the level of RPA at replication forks via diffusion-controlled ATR-RPA contact, preventing RPA from accumulating at impeded forks via phosphorylation of Ser33 at RPA2.
(C) Gradual increase in global replication stress lead to the accumulation of RPA at forks, and the basal ATR-RPA contacts is amplified accordingly. This results in local crowding of ATR at the vicinity of forks. The formation of ATR concentration gradients around forks guarantees a high probability for the diffusion-mediated encounter of ATR with its activators at the fork, which drives the activation of the canonical ATR-Chk1 signaling.
(D) Schematic semi-quantitative illustration of the reaction coordinates for ATR-RPA interface at replication forks and its progression and expansion with increase replicative-stress, whereby the activation of the canonical ATR-Chk1 replication stress response emerges from the basal activity of ATR.
RPA as a versatile signaling platform
RPA is the primary respondent to newly exposed ssDNA, immediately binding and protecting it from potential degradation. The ssDNA-bound RPA must then be removed in a timely manner to allow the transient ssDNA to be later accessed by replicative polymerases during normal replication, or other proteins during Homologous Recombination (HR) mediated repair. It is therefore important to be able to efficiently alter the RPA-ssDNA affinity. Our data identified that during normal replication the basal activity of ATR is necessary for facilitating the removal of RPA via phosphorylation of RPA2 at Ser33. Indeed, ATR inhibition had been found to disrupt the recruitment of PALB2 and Rad51 at IR-induced RPA-positive DNA double strand break (DSB) foci(Buisson et al., 2017). We speculate that these could also partially be attributed to the ATRi-induced failure in removing RPA. In addition, it was found that RPA with hyper-phosphorylated N-terminal of RPA2 could not be recruited to replication sites(Vassin et al., 2009). This can be explained by our data that phosphorylation of RPA2 enhances the removal of RPA from replication forks, and this will also likely contribute to the exclusion of hyper-phosphorylated RPA. Furthermore, ATR-facilitated RPA removal could also serve as an auto-regulatory approach to limit ATR activation fluctuating at the basal levels during normal replication, since RPA removal would prevent further recruitment of ATR and amplification of ATR signaling at undisturbed forks. We note that while our data reveals that ATR activity supports the removal of RPA from replication forks by phosphorylating Ser33 on RPA2, it does not exclude the possibility that other factors also participate in this process and further alter RPA-ssDNA affinity for the full removal of RPA(Fanning et al., 2006; Yates et al., 2018).
The ATR-RPA interface at forks provides a unique mechanism of feedback
The basal activity of ATR facilitates the removal of RPA at forks, and is separable from its canonical ATR-Chk1 signaling in replication stress, yet it defines an interface that provides a unique feedback mechanism between ATR and RPA that enables recognition of stress for the canonical activation of ATR. Our data indicates that this unique feedback mode between ATR and RPA at forks is mediated via a diffusion-controlled ATR-RPA contact, whereby the accumulation of RPA at forks induces localized crowding of ATR, increasing the reaction cross-section of ATR with its activators. This feedback mechanism addresses the enduring question of how ATR ‘calculates’ the amount of RPA to determine if it reached the activation threshold(Saldivar et al., 2017), as it establishes an interface that directly connects ATR activity in surveying replication forks with the canonical activation of ATR in response to replication stress.
Another major question regarding ATR activation is why cells adopt two parallel activation pathways that are independently mediated by TOPBP1 and ETAA1(Saldivar et al., 2017). While both TOPBP1 and ETAA1 activate ATR in response to replication stress, it was reported that the ATR-mediated phosphorylation of Chk1 and other downstream effectors is highly dependent on TOPBP1 but hardly affected by ETAA1, whereas the ATR-mediated RPA phosphorylation is insensitive to TOPBP1 but strongly relies on ETAA1(Bass et al., 2016). Furthermore, it was reported that such ATR-mediated RPA phosphorylation is significantly influenced by ETAA1 even during unperturbed replication(Bass et al., 2016; Haahr et al., 2016). As our data identified a distinct activity of ATR phosphorylating RPA during normal replication, we speculate that the specific roles of TOPBP1 and ETAA1 correspond to the activation of different ATR functions, with TOPBP1 participates in the activation of ATR-Chk1 signaling in response to replication stress, while ETAA1 is responsible for ATR-mediated RPA phosphorylation during both unperturbed and stressed replication. Nevertheless, whether such a mechanistic correlation exists would benefit from future studies.
Replication related DNA damage due to inhibition of ATR
It is largely accepted that the major source of stress-induced DNA damage in cells with defective or inactive ATR stems from the excess origin firing in the absence of functional ATR-Chk1 signaling(Toledo et al., 2013) which then exhausts the pool of RPA and leads to replication catastrophe. However, overexpression of RPA or suppression of origin firing in ATR-inhibited cells only delayed but did not prevent the emergence of replication catastrophe and DNA damage(Dungrawala et al., 2015). Specifically, the Cortez group have used iPOND-MS to analyze ATR-inhibited cells upon HU treatment, which showed that the Non-Homologous End Joining (NHEJ) repair proteins are only enriched at ATRi-induced newly fired replication forks, whereas most DSB sensor and HR repair proteins are recruited to forks even when ATRi-induced excess origin firing was blocked via CDC7i(Dungrawala et al., 2015). This suggests that in addition to its role in limiting origin firing, ATR also protects the pre-existing replication forks(Dungrawala et al., 2015). Our data shows that inhibition of ATR results in extreme RPA accumulation at ongoing forks during replication stress, and explains how ATR protects the pre-existing forks since the ATRi-induced accumulated RPA could block the access of related repair and stress response proteins to the ssDNA(Myler et al., 2016). Therefore, the proper exchange of RPA at forks as mediated by ATR could prevent excessive replication stress events that can lead to undesired remodeling at ongoing forks.
In summary, our study defines the basal activity of ATR in replication fork surveillance that is independent of the canonical ATR-Chk1 signaling in replication stress. This distinct activity is mediated by ATR-RPA interface at replication fork, establishing a unique feedback mechanism that enables the full activation of the ATR-Chk1 signaling when replication stress occurs. The multi-color SMLM and TCF analysis applied here allows us to detect and quantify the fine features of individual replication forks in cells and provide unique mechanistic insights. We anticipate that the multi-color SMLM-TCF platform described here can be broadly utilized as a powerful approach for the identification and quantitative analyses of specific molecular patterns at crowded cellular locations. Finally, the newly defined activity of ATR along with the experimental approach can be used for establishing an accurate delineation of ATR-Chk1 signaling to offer precise therapeutic approach for ATRi related therapies.
Limitation of the Study
Beyond the general challenges in multi-color SMLM imaging, there are also major limitations in utilizing image analyses approaches for extraction of information from the crowded and seemingly chaotic multi-color SMLM images. The TCF analysis is one of the few endeavors in multiplexed SMLM image mining, but it also has clear limitations. For example, while the TCF plots the probability density distribution of triplet patterns (the individual replisomes in this study), it lacks a physical model to analytically describe the TCF profile for in-depth quantifications of the underlying heterogeneity amongst the obtained patterns. Although in this study the dispersion of the TCF distribution profile is guaranteed to be comparable across different experiments by analyzing only the early-S phase cells and maintaining identical treatment conditions (cellular fixation, IF staining, etc.), it is still limited in formulating or refining the mathematical structure of such TCF distribution, and instead the most frequent patterns (the maximum of the TCF profile) is utilized as the only metric in the current format of this approach for further interpretation. Ongoing and future efforts in our and other labs are aimed at further development of rigorous and comprehensive mathematical models that can be applied for refining the interpretation of the TCF distribution in a more precise manner.
STAR Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Eli Rothenberg (Eli.Rothenberg@nyulangone.org).
Materials Availability
Plasmids generated in this study are available upon request from the lead contact.
Data and Code Availability
Reconstructed SMLM images, TCF configurations, RPAfork and RPAglobal, etc. are available on Mendeley Data at DOI: 10.17632/s4b8ztvnxd.1
Matlab code for TCF analysis is available on Github at Codes for the Triple- and Pair-Correlation algorithms, as well as a testing demo (with simulation codes) are available at DOI: 10.17632/s4b8ztvnxd.1
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Culture
For SMLM imaging in Figs. 1, 2, 6, S1, S2, and S4, U2OS cells (ATCC HTB-96) were grown on glass coverslips (Fisher Scientific, 12–548-B) for 24 hours in DMEM media (ThermoFisher 11965) with 10% FBS (Gemini Bio. 100–106) and 100 U/mL Penicillin-Streptomycin (ThermoFisher 15140). The cells were synchronized to G0/G1 interface via 48 – 72 hours Serum withdrawal, followed by 17 hours incubation with full medium for entry into S-phase. Different inhibitors were introduced at 14 hours after release (early S-phase) for 3 hours prior if otherwise not stated, while HU (0 – 500 μM) was added during the last 2 hours of kinase inhibitors treatment.
Small Interfering RNA (siRNA)
For SMLM imaging in Figure 3, 4, and S5, ATRIP and RPA2 were depleted via siRNA reverse transfection. siRNA reverse transfections were performed using Lipofectamine RNAiMax (ThermoFisher) following the manufacturer’s instructions. STORM experiments were done 72 hours after transfection. The siRNAs used in this study are: ATRIP siRNA (Qiagen, siATRIP, SI03071523): CAGGGTCATCCCTAAGCCTTT; RPA2 siRNA (Dharmacon, siRPA2, J-017058-09-0010): AACAUGAAGUU-CUGCGGUA.
Transient Transfection
For SMLM imaging in Figure 4 and S5, cells were also transfected with different myc-RPA plasmids using Lipofectamine 3000 (ThermoFisher) for 48 hours according to manufacturer’s protocol (see Figure 4A for Western Blot validation of efficiency of RPA2 replacement). Such transfection was performed 24 hours after siRPA2 transfection. The S33A-RPA2 and S33D-RPA2 mutants were generated based on pEF6-RPA2-Myc(Vassin et al., 2009). DNA fragments with S33A or S33D site-directed mutagenesis were obtained by overlap polymerase chain reaction (PCR). RPA2-Myc fragment in pEF6-RPA2-Myc was then replaced with S33A-RPA2-Myc or S33D-RPA2-Myc using XbaI-PmeI site.
METHOD DETAILS
Computation of the TCF
Typically, the TCF, as well as other correlation functions, could be computed in the frequency domain in their Fourier forms . The Fourier form of the TCF, termed as the bispectrum , could be calculated as Equation (1):
| (1) |
where * denotes conjugate. However, such computation involves 4D Fourier Transform and therefore infeasible for even high-performance cluster computers. However, since SMLM data consists of localization coordinates (rather than intensity values at each pixel across the entire image canvas), we directly calculated the TCF from the obtained coordinates by visiting each coordinate in the first channel, and calculating and in the second and third channels at r1, and r2 displaced from the location of the visited coordinate, respectively(Yin et al., 2019). Moreover, since the triplets are randomly oriented in the ROI, the TCF at , was averaged along , and was thus transformed to where .
Post-Processing of the TCF
The TC profiles as the function of the triple-wise distances were Gaussian smoothed (σ = 15 nm), followed by recording of all the local maxima that were greater than mean + 2.5SD, where the mean and the SD are the expectation and standard deviation of the smoothed TC profile (Methods S1). The configuration was then determined as . The resolved triangular configurations with the perimeter of ≥ 180 nm were rejected. The PCNA-MCM axis of the resolved triangle configurations were aligned onto the same horizontal line with their mid-points fixed at the origin, and overlaid together so that the statistical relative position of RPA1 to PCNA-MCM is displayed. For EdU-PCNA-MCM pattern, the EdU-MCM axis were aligned onto the same horizontal with the position of MCM fixed at the origin. The local density of RPA1 within each fork pattern is calculated by dividing the Cross-Correlation-Function (CCF) of PCNA-MCM at from the recognized local maximum of the TCF, followed by multiplying the average global density of RPA1 (Methods S2). Nuclei with unrecognizable local maximum of the TCF or nonpositive CCF of PCNA-MCM were not considered in our statistics.
Microscope setup for SMLM imaging
For SMLM-SR imaging we used a custom-built optical imaging platform based on a Leica DMI 300 inverse microscopy(Yin et al., 2019). Different fluorophores were excited by different laser lines. Briefly, a 639 nm laser (CNI), 561 nm laser (Cobolt), and 488 nm Laser (OBIS) were aligned and reflected into an HCX PL APO 63X NA = 1.47 OIL CORR TIRF Objective (Zeiss) by a penta-edged dichroic beam splitter (FF408/504/581/667/762-Di01-22×29). The 488, 561, and 639 nm laser lines were adjusted to ~ 0.8, 1.0, and 1.5 kW/cm2, and excited cell samples at Highly Inclined and Laminated Optical sheet (HILO) illumination mode. A 405 nm laser line (Applied Scientific Pro., SL-405 nm-150 mW) was also introduced to drive Alexa Fluor 647 fluorophores back to their ground state.
Photons from different fluorophores were sequentially collected for multi-color imaging. The emitted photons were first expanded by a 2X lens tube (Diagnostic Instrument) and filtered by a single-band pass filter (Alexa Fluor 647: FF01–676/37, Semrock; Alexa Fluor 568: FF01–607/36, Semrock; Alexa Fluor 488: FF01–531/40, Semrock) that switched in a filter wheel (ThorLabs, FW102C) in accordance with the sequentially switched illumination. The fluorophores were then modulated with a chromatic aberration correction lens (Thorlabs, AC354–300-A, see below for channel alignment and chromatic aberration correction) before collected on a sCMOS camera (Photometrics, Prime 95B). All raw image stacks were acquired at 33 Hz for a minimum of 2000 frames.
SMLM localization and Reconstruction
Given the nature of the patterned noise of the sCMOS Camera, each pixel of the images was weighted by the inverse of its variance, followed by box-filtering with a box size of 4 times of the FWHM of a 2D Gaussian PSF. The filtered image was then extracted from the raw image, followed by roughly localizing the local maximums. The local maximums from all the frames of the image stack were then submitted for 2D-Gaussian multi-PSF fitting (DAOSTORM)(Holden et al., 2011). The 2D-Gaussian multi-PSF fitting was achieved through the Maximum Likelihood Estimation (MLE) with up to 4 PSFs. In brief, the likelihood function at each pixel was a convolution of a Poisson distribution of the shot noise governed by the photons emitted from fluorophores nearby, and a gaussian distribution of the readout noise that characterized by the expectation, variance, and the analog-to-digital conversion factor. These factors describe the pattern noise of the camera and were pre-calibrated(Huang et al., 2013). The fitting accuracy was estimated by Cramér-Rao lower bound (CRLB), and the distribution of the accuracy of all successive localizations were fitted into a skew-Gaussian distribution. In addition, the localizations appeared in consecutive frames within 2.5 times of the localization precision were considered as one blinking event. Such localizations were weighted by the inverse of its own CRLB determined variance and averaged into one localization(Huang et al., 2013) for the purpose of Auto-PC computation. We note that the raw coordinate lists from this DAOSTORM fitting were then directly submitted to Auto- and Triple-Correlation analyses. For display purpose, the representative images were generated by rendering the raw coordinates into 10 nm pixel canvas and convolved with a 2D-Gaussian (σ = 10 nm) kernel.
Microscope setup for smFRET
For smFRET we used a custom-built optical imaging platform based on an Olympus IX70 inverse microscopy. A 532 nm laser (UltraLaser, MGL-FN-532-1000) was collimated and reflected into a 100× NA = 1.49 TIRF Objective (Olympus) by a dichroic beam splitter (Semrock, Di03-405/488/532/635-t1) at a center-off position to achieve a TIRF illumination. A 639 nm laser (ultraLaser, MRL-FN-639-800) was also aligned to examine number of molecules immobilized onto the surface. Photons from Cy3 or Cy5 were first split by a 635 nm edged beam splitter (Semrock, Di03-635-t1) and filtered by single-band pass filters 582 ± 32 nm (Semrock, FF01-582/64) and 680 ± 21 nm (Semrock, FF01-680/42), respectively. The fluorescence splitter and filters were integrated in an achromatic beam-splitting device (Photometrics, OptoSplit II). The photons form Cy3 and Cy5 were then collected on to an EMCCD camera side-by-side (Andor, iXon 897). All raw image stacks were acquired at 33 Hz for 4000 frames, with the EM gain was set to 300. The apparent pixel size with this configuration is ~ 160×160 nm2.
smFRET analysis
The single molecule PSFs in both donor and acceptor channels were localized and measured independently. Similarly, each PSF, 7×7 pixel2 square cropped from each local maximum, was localized by fitting into a 2D Gaussian profile via MLE, followed by aligning the localizations from the two channels together (see below for channel alignment). A FRET pair was identified if the localizations of a donner and an acceptor are not further than 2 pixels (~300 nm), and their MLE-fitted intensities were readout throughout the entire time stacks. Note that if a submitted PSF failed for MLE fitting (i.e. a PSF bleached from a time point), the intensity was calculated by subtracting the background of the PSF from its maximum pixel. The raw FRET trajectories, excluding the bleaching (of either the donor or the acceptor) part were then submitted to the vbFRET algorithm for Hidden-Markov-Model (HMM) analysis(Bronson et al., 2009). Note that 1 to 4 states were attempted for each trajectory and the best fitted number-of-states was determined via Maximum Evidence(Bronson et al., 2009). The FRET histograms (Fig. 4E) and the Transition Density Plot (TDP, Figure. 5E and S6D) were then calculated from the HMM-idealized FRET trajectories and normalized by the number of molecules.
Alignment of different color channels
To ensure accurate alignment of the SMLM and smFRET data acquired in the different channels, we carried out an alignment/registration routine (see methods and Supporting Figure 6 in Ref. Yin et al., 2019). In brief, the red (639 nm excited) channel was set to the reference channel to which the other one / two channels (blue: 488 nm excited and green: 561 nm excited for SR or 532 nm excited for FRET) were aligned using polynomial mapping algorithm. To generate a map mapping the blue and green channel to the reference channel, we acquired diffraction-limited images of spatially separated fluorescent beads (ThermoFisher, T-7279) which emitted fluorescence spanning a broad range covering all the three channels. The mass center of the ith of N > 200 beads in the reference channel was recorded as () and that in the other two channels was recorded as (). The localizations were then submitted to a 2nd polynomial function
| (2) |
where , ; , denoting the maximum integer smaller than ; , denoting the modulo of ; ; and and .
and were optimized by resolving Equation (2) and then applied to warp the coordinates of experimental samples from the blue and green channel to the red channel using Equation (2).
The system mapping corrections in A, was derived to also incorporate axial calibration and other sources of mapping errors(Carlini et al., 2015; Li et al., 2019) described in Methods S3.
Monte Carlo Simulation
1000 ATR molecules were seeded in a cube measuring 1000 × 1000 × 1000 units (in 3 dimensions). Each ATR molecule exhibited Brownian diffusion with the step size randomly generated from a Gaussian distribution (σ = 5 units). ATR were reflected at the boundary of the cube to avoid loss of the molecules. A replisome was designed as a sphere with radius of 10 units and positioned at the center of the cube. The number of RPA molecules linearly grows with time to simulate the stress induced accumulation of RPA at forks, and such growth was assigned at the very beginning of each time point (iteration). Within each iteration, if ATR diffused onto the replisome and there were enough RPA to accommodate them, ATR would remain until RPA was removed. The number of RPA decreased exponentially at a speed proportional to the number of ATR in-contact with the replisome. 10 simulations were performed for each stress level and the number of RPA and ATR molecules were obtained.
Within each iteration as described above, the ATR-in-contact were also used for a nested simulation where the ATRs exhibited Brownian diffusion within the replisome with the step size randomly generated from a Gaussian distribution (σ = 2). 100 Brownian steps were performed for one simulation and the occurrence of ATR visit the center of the replisome (r = 2 units) and that of ATR collide with each other (collision radius = 2 units) were recorded. 10 simulations were performed for each stress level.
Cell pre-extraction, fixation, and fluorescent labeling
For specific analysis of only chromatin bound replisomes and RPA we carried out pre-extraction prior to fixation to achieve fine removal of nuclear proteins that are unbound to chromatin(Britton et al., 2013). Briefly, cells were permeabilized with 0.5% Triton X-100 in CSK buffer (10 mM Hepes, 300 mM Sucrose, 100 mM NaCl, 3 mM MgCl2, pH = 7.4) for 10 minutes and fixed with paraformaldehyde (4%) for 30 minutes. The cells were then washed twice with blocking buffer (2% glycine, 2% BSA, 0.2% geltin, and 50 mM NH4Cl in PBS). Note that due to ATR’s high diffusiveness, we fixed the cells before carrying out the permeabilization for imaging in Figure 6. Cells were treated with the click-reaction cocktail without fluorophore conjugated picolyl azide (Click-iT chemistry, ThermoFisher, C10640) for better staining of PCNA via the PC10 antibody(Ligasova et al., 2012). The cells were blocked with blocking buffer for 1 hour at room temperature (RT) or overnight at 4°C for further immunofluorescence staining (see below for antibody concentration and incubation durations). After immunofluorescence staining, the fixed cells were then mounted onto microscope glass for SMLM-SR imaging in freshly mixed imaging buffer (1 mg/mL glucose oxidase, 0.02 mg/mL catalase, 10% glucose, and 100 mM cysteamine (MEA)).
Epi-fluorescence
U2OS cells grown in presence of EdU (10 μM) for 3 hours were treated with HU (500 μM, 2 hours), ATRi (VE821 2μM, 3 hours), ATRi + HU (HU was introduced during the last 2 of the 3 hours ATRi treatment), Chk1i (UCN01, 300nM, 1 hour) and Chk1i (3 hours). Cells were washed twice with PBS, permeabilized with 0.5% Triton X-100 in CSK buffer (10 minutes) and fixed with paraformaldehyde (4%, 15 minutes). EdU incorporation was detected using the Click-iT Plus EdU Alexa Fluor 488 Imaging Kit (C10337; Molecular Probes) and then incubated in blocking buffer overnight. Cells were incubated with primary antibody to γH2AX (1:1000, Millipore, JBW301) and then Alexa Fluor 568 secondary antibodies (1:200, Molecular Probes) diluted in blocking buffer for 1 h each at room temperature. Nuclei were counterstained with Hoechst-33342, mounted, and images were captured using a ZEISS Axio Observer.D1/5 equipped with a Plan-apochromat 63×/1.4 N.A. oil immersion objective.
Flow Cytometry
At 48 h after the different myc-RPA transfection, cells were harvested and washed twice in ice-cold PBS, fixed in 70% iced ethanol, and treated with 50ug/ml propidium iodide (PI) along with 50ug/ml RNase A for 1 hr at room temperature. Cells were analyzed for DNA content by flow cytometry using a FACSCalibur™ cell analyzer (Becton Dickinson). The proportions for each cell population in G1, S, or G2/M phase of the cell cycle were then estimated by using Modfit cell cycle analysis software.
Western Blotting
Cells were harvested using Laemmli sample buffer (Bio-Rad) containing (final concentration) 2% SDS, 10% glycerol, 5% 2-mercaptoethanol, 0.002% bromphenol blue, and 60 mM Tris-HCl (pH ~ 6.8), and lysed by heating the samples at 95°C for 15 minutes. Protein extracts were calibrated and resolved by SDS-polyacrylamide gel electrophoresis on Nupage 4 – 12% Bis-Tris, 3 – 8% Tris-Acetate gels (Invitrogen), or 4 – 15% TGX gels (Bio-Rad) in 1 × Tris-Glycine-SDS buffer. Proteins were transferred onto polyvinylidene difluoride (PVDF) membrane (Millipore) and incubate in blocking buffer (5% milk in TBST) for 1 hour at room temperature or overnight at 4°C. The membrane was then incubated with primary antibodies for 1 hour at room temperature or overnight at 4°C, followed by incubation with secondary antibodies conjugated with horseradish peroxidase. Blots were detected using an Enhanced Chemiluminescence Detection Kit (GE Healthcare) and were developed with a LICOR Odyssey imager or Western Lightning Plus-ECL reagent.
DNA oligonucleotides, Inhibitors, and Antibodies
DNA oligonucleotides used in Figure 5 are:
5’-TGGCGACGGCAGCGAGGCTTTTTTTTTTTTTTTTTTTT-iCy3-TTTTTTTTTTTT-3’-Cy5 5’-GCCTCGCTGCCGTCGCCA-3’-biotin
DNA oligonucleotides used in Figure S6 are:
5’-TGGCGACGGCAGCGAGGCTTTTTTTTTTTTTTTT-iCy3-TTTTTTTTTTTTTT-3’ Cy5–5’-GCCTCGCTGCCGTCGCCA-3’-biotin
For the inhibitors used in this study, cells were treated with inhibitors 3 hours before fixation. ATRi: VE-821 (Selleckchem), 2 μM, 3 hours incubation; CDC7i: PHA767491 (Selleckchem), 20μM, 3 hours incubation; Chk1i: UCN01 (Sigma-Aldrich), 100 nM, 1 hour incubation; Chk2i: PV1019 (Sigma-Aldrich), 5 μM, 3 hours incubation; For Hydroxyurea treatments (HU, Sigma-Aldrich), cells were treated 2 hours before fixation.
Antibodies against the imaged proteins are (ms = mouse; rb = rabbit; AF = AlexaFluor): ms-anti-PCNA (SCBT sc56, 1:1,000), rb-anti-MCM6 (AF568 conjugated, Abcam ab211916, 1:1,000), rb-anti-RPA1 (AF647 conjugated, Abcam ab199240, 1:10,000), rb-anti-pS4/S8RPA2 (Abcam ab87277, 1:1000). Secondary antibodies are: goat-anti-ms (AF488 conjugated, ThermoFisher, 1:10,000), goat-anti-rb (AF647 conjugated, ThermoFisher, 1:10,000).
Antibodies against the blotted proteins are:
Ms-anti-RPA2 (Millipore, MABE285, 1:1000), rb-anti-c-myc (Bethyl, A190–105A, 1:1000), ms-anti-β-tubulin (Sigma-Aldrich T8328, 1:1000), Ms-anti-HRP (Abcam, ab181708, 1: 10000), Rb-anti-HRP (Abcam, ab34885, 1:10000).
QUANTIFICATION AND STATISTICAL ANALYSIS
All statistical analyses are described in the figure legends. The student t-tests between two interested groups were performed in OriginLab. All experiments were completed at least three times unless otherwise indicated.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-PCNA (SR) | Santa Cruz | Cat# sc-56, RRID: AB_628110 |
| Rabbit monoclonal anti-MCM6 (AlexaFluor 568 conjugated, SR) | Abcam | Cat# ab211916, RRID: N/A |
| Rabbit monoclonal anti-RPA70 (AlexaFluor 647 conjugated, SR) | Abcam | Cat# ab199240, RRID: AB_2687566 |
| Rabbit polyclonal anti-RPA32(phosphor S4+S8) (SR) | Abcam | Cat# ab87277, RRID: AB_1952482 |
| Goat polyclonal anti-Mouse (AlexaFluor 488 conjugated, SR) | Thermal Fisher | Cat# A-11001, RRID: AB_2534069 |
| Goat polyclonal anti-Rabbit (AlexaFluor 647 conjugated, SR) | Thermal Fisher | Cat# A27040, RRID: AB_2536101 |
| Mouse monoclonal anti-γH2AX (IF) | Millipore | Cat# 16-193, RRID: AB_310795 |
| Goat polyclonal anti-Mouse (AlexaFluor 568 conjugated, IF) | Thermal Fisher | Cat# A-11004, RRID: AB_2534072 |
| Mouse monoclonal anti-RPA32 (WB) | Millipore | Cat# MABE285, RRID: AB_11205561 |
| Rabbit polyclonal anti-Myc (WB) | Bethyl | Cat# A190-105A, RRID: AB_67390 |
| Mouse monoclonal anti-β-Tublin (WB) | Sigma-Aldrich | Cat# T8328, RRID: AB_1844090 |
| Mouse polyclonal anti-HRP (WB) | Abcam | Cat# ab181708, RRID: N/A |
| Rabbit polyclonal anti-HRP (WB) | Abcam | Cat# ab34885, RRID: AB_732995 |
| Bacterial and Virus Strains | ||
| Biological Samples | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| ATR inhibitor, VE-821 | Selleckchem | Cat# S8007, CAS No. 1232410-49-9 |
| CDC7 inhibitor, PHA767491 | Selleckchem | Cat# S2742, CAS No. 942425-68-5 |
| Chk1 inhibitor, UCN-01 | Sigma-Aldrich | Cat# U6508, CAS No. 112953-11-4 |
| Chk2 inhibitor, PV1019 | Sigma-Aldrich | Cat# 220488, CAS No. 516480-79-8 |
| Hydroxyurea | Sigma-Aldrich | Cat# H8627, CAS No. 127-07-1 |
| Hoechst-33342 | Thermo Fisher | Cat# H3570, CAS No. 23491-52-3 |
| DMEM cell culture medium | Thermo Fisher | Cat# 11965 |
| FBS | Gemini Bio | Cat# 100-106 |
| Penicillin-Streptomycin | Thermo Fisher | Cat# 15140 |
| Critical Commercial Assays | ||
| Click reaction kit for EdU and Alexa Fluor 647 azide | Thermo Fisher | Cat# 10640 |
| Lipofectamine RNAiMax reagent | Thermo Fisher | Cat# 13778075 |
| Lipofectamine 3000 transfection kit | Thermo Fisher | Cat# L3000015 |
| Deposited Data | ||
| Representative images, uncropped gels, all the data | This study | DOI: 10.17632/s4b8ztvnxd.1 |
| Experimental Models: Cell Lines | ||
| U2OS | ATCC | Cat# HTB-96, RRID: CVCL_0042 |
| Experimental Models: Organisms/Strains | ||
| Oligonucleotides | ||
| ATRIP FlexiTube GeneSolution | QIAGEN | SI03072496 |
| ON-TARGET plus Human RPA2 siRNA | Dharmacon | SO-2916681G |
| 5′-TGGCGACGGCAGCGAGGCTTTTTTTTTTTTTTTTTTTT-iCy3-TTTTTTTTTTTT-3′-Cy5 (Figure 5) | IDT | N/A |
| 5′-GCCTCGCTGCCGTCGCCA-3′-biotin (Figure 5) | IDT | N/A |
| 5′-TGGCGACGGCAGCGAGGCTTTTTTTTTTTTTTTT-iCy3-TTTTTTTTTTTTTT-3′ (Figure S6) | IDT | N/A |
| Cy5-5′-GCCTCGCTGCCGTCGCCA-3′-biotin (Figure S6) | IDT | N/A |
| Recombinant DNA | ||
| Plasmid pEF6-RPA2-WT-Myc | This study | N/A |
| Plasmid pEF6-RPA2-S33D-Myc | This study | N/A |
| Plasmid pEF6-RPA2-S33A-Myc | This study | N/A |
| Software and Algorithms | ||
| Reconstruction of SMLM (CUDA + Matlab) | CUDA, MathWorks | MATLAB, RRID: SCR_001622 |
| TCF (Matlab) | Yin et al., 2019 | DOI: 10.17632/s4b8ztvnxd.1 |
| FACSCalibur | BD Biosciences | RRID: SCR_000401 |
| ImageJ | Schneider et al., 2012 | https://imagej.nih.gov/ij/ |
| Originlab | OriginLab | https://www.originlab.com/ |
| Other | ||
Highlights:
Inhibition of ATR (ATRi) results in RPA accumulation at forks during normal replication
ATRi-induced RPA accumulation is independent of Chk1 but requires ATR-RPA contacts
ATR-mediated phosphorylation of Ser33 at RPA2 facilitates RPA’s removal from forks
HU-induced RPA amplifies the diffusion-driven ATR-RPA contact, leading to crowding of ATR
Acknowledgement
This work was supported by NIH grants 1R35GM134947-01 (E.R.), 1R01AI153040-01 (E.R.), 1P01CA247773-01/549 (E.R.), R01ES031658 (T.T.H.); American Cancer Society grant RSG-16-241-01-DMC (E.R.); V Foundation BRCA Research grants (E.R. and T.T.H.). M.M. is supported by the French National Research Agency, the French National Cancer Institute and the French National League Against Cancer (équipe labellisée). We thank members of the Rothenberg laboratory for critical discussion. We are thankful for David Cortez and Marcus Smolka for their valuable inputs and helpful suggestions on this work.
Footnotes
Declaration of Interests
All authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference
- Baddeley D, Chagin VO, Schermelleh L, Martin S, Pombo A, Carlton PM, Gahl A, Domaing P, Birk U, Leonhardt H, et al. (2010). Measurement of replication structures at the nanometer scale using super-resolution light microscopy. Nucleic Acids Res 38, e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ball HL, Myers JS, and Cortez D. (2005). ATRIP binding to replication protein A-single-stranded DNA promotes ATR-ATRIP localization but is dispensable for Chk1 phosphorylation. Mol Biol Cell 16, 2372–2381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bass TE, Luzwick JW, Kavanaugh G, Carroll C, Dungrawala H, Glick GG, Feldkamp MD, Putney R, Chazin WJ, and Cortez D. (2016). ETAA1 acts at stalled replication forks to maintain genome integrity. Nat Cell Biol 18, 1185–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastos de Oliveira FM, Kim D, Cussiol JR, Das J, Jeong MC, Doerfler L, Schmidt KH, Yu H, and Smolka MB (2015). Phosphoproteomics reveals distinct modes of Mec1/ATR signaling during DNA replication. Mol Cell 57, 1124–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas H, Goto G, Wang W, Sung P, and Sugimoto K. (2019). Ddc2ATRIP promotes Mec1ATR activation at RPA-ssDNA tracts. PLoS Genet 15, e1008294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britton S, Coates J, and Jackson SP (2013). A new method for high-resolution imaging of Ku foci to decipher mechanisms of DNA double-strand break repair. J Cell Biol 202, 579–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronson JE, Fei J, Hofman JM, Gonzalez RL Jr., and Wiggins CH (2009). Learning rates and states from biophysical time series: a Bayesian approach to model selection and single-molecule FRET data. Biophys J 97, 3196–3205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buisson R, Niraj J, Rodrigue A, Ho CK, Kreuzer J, Foo TK, Hardy EJ, Dellaire G, Haas W, Xia B, et al. (2017). Coupling of Homologous Recombination and the Checkpoint by ATR. Mol Cell 65, 336–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlini L, Holden SJ, Douglass KM, and Manley S. (2015). Correction of a Depth-Dependent Lateral Distortion in 3D Super-Resolution Imaging. PLoS One 10, e0142949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chagin VO, Casas-Delucchi CS, Reinhart M, Schermelleh L, Markaki Y, Maiser A, Bolius JJ, Bensimon A, Fillies M, Domaing P, et al. (2016). 4D Visualization of replication foci in mammalian cells corresponding to individual replicons. Nat Commun 7, 11231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandok GS, Patel MP, Mirkin SM, and Krasilnikova MM (2012). Effects of Friedreich’s ataxia GAA repeats on DNA replication in mammalian cells. Nucleic Acids Res 40, 3964–3974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang S, Naiman K, Thrall ES, Kath JE, Jergic S, Dixon NE, Fuchs RP, and Loparo JJ (2019). A gatekeeping function of the replicative polymerase controls pathway choice in the resolution of lesion-stalled replisomes. Proc Natl Acad Sci U S A 116, 25591–25601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YH, Jones MJ, Yin Y, Crist SB, Colnaghi L, Sims RJ 3rd, Rothenberg E, Jallepalli PV, and Huang TT (2015). ATR-mediated phosphorylation of FANCI regulates dormant origin firing in response to replication stress. Mol Cell 58, 323–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobb JA, Schleker T, Rojas V, Bjergbaek L, Tercero JA, and Gasser SM (2005). Replisome instability, fork collapse, and gross chromosomal rearrangements arise synergistically from Mec1 kinase and RecQ helicase mutations. Genes Dev 19, 3055–3069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortez D, Guntuku S, Qin J, and Elledge SJ (2001). ATR and ATRIP: Partners in checkpoint signaling. Science 294, 1713–1716. [DOI] [PubMed] [Google Scholar]
- Cotta-Ramusino C, McDonald ER 3rd, Hurov K, Sowa ME, Harper JW, and Elledge SJ (2011). A DNA damage response screen identifies RHINO, a 9–1-1 and TopBP1 interacting protein required for ATR signaling. Science 332, 1313–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couch FB, Bansbach CE, Driscoll R, Luzwick JW, Glick GG, Betous R, Carroll CM, Jung SY, Qin J, Cimprich KA, and Cortez D. (2013). ATR phosphorylates SMARCAL1 to prevent replication fork collapse. Genes Dev 27, 1610–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cseresnyes Z, Schwarz U, and Green CM (2009). Analysis of replication factories in human cells by super-resolution light microscopy. BMC Cell Biol 10, 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshpande I, Seeber A, Shimada K, Keusch JJ, Gut H, and Gasser SM (2017). Structural Basis of Mec1-Ddc2-RPA Assembly and Activation on Single-Stranded DNA at Sites of Damage. Mol Cell 68, 431–445 e435. [DOI] [PubMed] [Google Scholar]
- Dungrawala H, Rose KL, Bhat KP, Mohni KN, Glick GG, Couch FB, and Cortez D. (2015). The Replication Checkpoint Prevents Two Types of Fork Collapse without Regulating Replisome Stability. Mol Cell 59, 998–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duursma AM, Driscoll R, Elias JE, and Cimprich KA (2013). A role for the MRN complex in ATR activation via TOPBP1 recruitment. Mol Cell 50, 116–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ercilla A, Benada J, Amitash S, Zonderland G, Baldi G, Somyajit K, Ochs F, Costanzo V, Lukas J, and Toledo L. (2020). Physiological Tolerance to ssDNA Enables Strand Uncoupling during DNA Replication. Cell Rep 30, 2416–2429 e2417. [DOI] [PubMed] [Google Scholar]
- Eric Betzig, Patterson George H., Rachid Sougrat, Lindwasser O. Wolf, Scott Olenych, Bonifacino Juan S., Davidson Michael W., Jennifer Lippincott-Schwartz, and Hess HF (2006). Imaging Intracellular Fluorescent Proteins at Nanometer Resolution. Science 313, 1642–1645. [DOI] [PubMed] [Google Scholar]
- Fanning E, Klimovich V, and Nager AR (2006). A dynamic model for replication protein A (RPA) function in DNA processing pathways. Nucleic Acids Res 34, 4126–4137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fragkos M, Ganier O, Coulombe P, and Mechali M. (2015). DNA replication origin activation in space and time. Nat Rev Mol Cell Biol 16, 360–374. [DOI] [PubMed] [Google Scholar]
- Ge XQ, and Blow JJ (2010). Chk1 inhibits replication factory activation but allows dormant origin firing in existing factories. J Cell Biol 191, 1285–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge XQ, Jackson DA, and Blow JJ (2007). Dormant origins licensed by excess Mcm2–7 are required for human cells to survive replicative stress. Genes Dev 21, 3331–3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goswami P, Abid Ali F, Douglas ME, Locke J, Purkiss A, Janska A, Eickhoff P, Early A, Nans A, Cheung AMC, et al. (2018). Structure of DNA-CMG-Pol epsilon elucidates the roles of the non-catalytic polymerase modules in the eukaryotic replisome. Nat Commun 9, 5061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haahr P, Hoffmann S, Tollenaere MA, Ho T, Toledo LI, Mann M, Bekker-Jensen S, Raschle M, and Mailand N. (2016). Activation of the ATR kinase by the RPA-binding protein ETAA1. Nat Cell Biol 18, 1196–1207. [DOI] [PubMed] [Google Scholar]
- Hamperl S, Bocek MJ, Saldivar JC, Swigut T, and Cimprich KA (2017). Transcription-Replication Conflict Orientation Modulates R-Loop Levels and Activates Distinct DNA Damage Responses. Cell 170, 774–786 e719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han X, Mayca Pozo F, Wisotsky JN, Wang B, Jacobberger JW, and Zhang Y. (2015). Phosphorylation of Minichromosome Maintenance 3 (MCM3) by Checkpoint Kinase 1 (Chk1) Negatively Regulates DNA Replication and Checkpoint Activation. J Biol Chem 290, 12370–12378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holden SJ, Uphoff S, and Kapanidis AN (2011). DAOSTORM: an algorithm for high- density super-resolution microscopy. Nat Methods 8, 279–280. [DOI] [PubMed] [Google Scholar]
- Huang F, Hartwich TM, Rivera-Molina FE, Lin Y, Duim WC, Long JJ, Uchil PD, Myers JR, Baird MA, Mothes W, et al. (2013). Video-rate nanoscopy using sCMOS camera-specific single-molecule localization algorithms. Nat Methods 10, 653–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hustedt N, Gasser SM, and Shimada K. (2013). Replication checkpoint: tuning and coordination of replication forks in s phase. Genes (Basel) 4, 388–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumagai A, Lee J, Yoo HY, and Dunphy WG (2006). TopBP1 activates the ATR-ATRIP complex. Cell 124, 943–955. [DOI] [PubMed] [Google Scholar]
- Lee J, and Dunphy WG (2013). The Mre11-Rad50-Nbs1 (MRN) complex has a specific role in the activation of Chk1 in response to stalled replication forks. Mol Biol Cell 24, 1343–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee WTC, Yin Y, Morten MJ, Tonzi P, Gwo PP, Odermatt DC, Modesti M, Cantor SB, Gari K, Huang TT, and Rothenberg E. (2021). Single-molecule imaging reveals replication fork coupled formation of G-quadruplex structures hinders local replication stress signaling. Nat Commun 12, 2525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leon-Ortiz AM, Svendsen J, and Boulton SJ (2014). Metabolism of DNA secondary structures at the eukaryotic replication fork. DNA Repair (Amst) 19, 152–162. [DOI] [PubMed] [Google Scholar]
- Li Y, Wu YL, Hoess P, Mund M, and Ries J. (2019). Depth-dependent PSF calibration and aberration correction for 3D single-molecule localization. Biomed Opt Express 10, 2708–2718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ligasova A, Strunin D, Liboska R, Rosenberg I, and Koberna K. (2012). Atomic scissors: a new method of tracking the 5-bromo-2’-deoxyuridine-labeled DNA in situ. PLoS One 7, e52584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Opiyo SO, Manthey K, Glanzer JG, Ashley AK, Amerin C, Troksa K, Shrivastav M, Nickoloff JA, and Oakley GG (2012). Distinct roles for DNA-PK, ATM and ATR in RPA phosphorylation and checkpoint activation in response to replication stress. Nucleic Acids Res 40, 10780–10794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Shiotani B, Lahiri M, Maréchal A, Tse A, Leung, Charles Chung Y., Glover JNM, Yang, Xiaohong H, and Zou L. (2011). ATR Autophosphorylation as a Molecular Switch for Checkpoint Activation. Molecular Cell 43, 192–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lossaint G, Larroque M, Ribeyre C, Bec N, Larroque C, Decaillet C, Gari K, and Constantinou A. (2013). FANCD2 binds MCM proteins and controls replisome function upon activation of s phase checkpoint signaling. Mol Cell 51, 678–690. [DOI] [PubMed] [Google Scholar]
- Lucca C, Vanoli F, Cotta-Ramusino C, Pellicioli A, Liberi G, Haber J, and Foiani M. (2004). Checkpoint-mediated control of replisome-fork association and signalling in response to replication pausing. Oncogene 23, 1206–1213. [DOI] [PubMed] [Google Scholar]
- Moiseeva T, Hood B, Schamus S, O’Connor MJ, Conrads TP, and Bakkenist CJ (2017). ATR kinase inhibition induces unscheduled origin firing through a Cdc7-dependent association between GINS and And-1. Nature Communications 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moiseeva TN, Yin Y, Calderon MJ, Qian C, Schamus-Haynes S, Sugitani N, Osmanbeyoglu HU, Rothenberg E, Watkins SC, and Bakkenist CJ (2019). An ATR and CHK1 kinase signaling mechanism that limits origin firing during unperturbed DNA replication. Proc Natl Acad Sci U S A 116, 13374–13383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mordes DA, Glick GG, Zhao R, and Cortez D. (2008). TopBP1 activates ATR through ATRIP and a PIKK regulatory domain. Genes Dev 22, 1478–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myler LR, Gallardo IF, Zhou Y, Gong F, Yang SH, Wold MS, Miller KM, Paull TT, and Finkelstein IJ (2016). Single-molecule imaging reveals the mechanism of Exo1 regulation by single-stranded DNA binding proteins. Proc Natl Acad Sci U S A 113, E1170–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petermann E, Woodcock M, and Helleday T. (2010). Chk1 promotes replication fork progression by controlling replication initiation. Proc Natl Acad Sci U S A 107, 16090–16095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puddu F, Granata M, Di Nola L, Balestrini A, Piergiovanni G, Lazzaro F, Giannattasio M, Plevani P, and Muzi-Falconi M. (2008). Phosphorylation of the budding yeast 9–1-1 complex is required for Dpb11 function in the full activation of the UV-induced DNA damage checkpoint. Mol Cell Biol 28, 4782–4793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rainey MD, Quachthithu H, Gaboriau D, and Santocanale C. (2017). DNA Replication Dynamics and Cellular Responses to ATP Competitive CDC7 Kinase Inhibitors. ACS Chem Biol 12, 1893–1902. [DOI] [PubMed] [Google Scholar]
- Randell JC, Fan A, Chan C, Francis LI, Heller RC, Galani K, and Bell SP (2010). Mec1 is one of multiple kinases that prime the Mcm2–7 helicase for phosphorylation by Cdc7. Mol Cell 40, 353–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rust MJ, Bates M, and Zhuang X. (2006). Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat Methods 3, 793–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saldivar JC, Cortez D, and Cimprich KA (2017). The essential kinase ATR: ensuring faithful duplication of a challenging genome. Nat Rev Mol Cell Biol 18, 622–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santocanale C, and Diffley JFX (1998). A Mec1- and Rad53-dependent checkpoint controls late-firing origins of DNA replication. Nature 395, 615–618. [DOI] [PubMed] [Google Scholar]
- Sengupta P, Jovanovic-Talisman T, Skoko D, Renz M, Veatch SL, and Lippincott-Schwartz J. (2011). Probing protein heterogeneity in the plasma membrane using PALM and pair correlation analysis. Nat Methods 8, 969–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su QP, Zhao ZW, Meng L, Ding M, Zhang W, Li Y, Liu M, Li R, Gao YQ, Xie XS, and Sun Y. (2020). Superresolution imaging reveals spatiotemporal propagation of human replication foci mediated by CTCF-organized chromatin structures. Proc Natl Acad Sci U S A 117, 15036–15046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syljuasen RG, Sorensen CS, Hansen LT, Fugger K, Lundin C, Johansson F, Helleday T, Sehested M, Lukas J, and Bartek J. (2005). Inhibition of human Chk1 causes increased initiation of DNA replication, phosphorylation of ATR targets, and DNA breakage. Mol Cell Biol 25, 3553–3562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toledo LI, Altmeyer M, Rask MB, Lukas C, Larsen DH, Povlsen LK, Bekker-Jensen S, Mailand N, Bartek J, and Lukas J. (2013). ATR prohibits replication catastrophe by preventing global exhaustion of RPA. Cell 155, 1088–1103. [DOI] [PubMed] [Google Scholar]
- Vassin VM, Anantha RW, Sokolova E, Kanner S, and Borowiec JA (2009). Human RPA phosphorylation by ATR stimulates DNA synthesis and prevents ssDNA accumulation during DNA-replication stress. J Cell Sci 122, 4070–4080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veatch SL, Machta BB, Shelby SA, Chiang EN, Holowka DA, and Baird BA (2012). Correlation functions quantify super-resolution images and estimate apparent clustering due to over-counting. PLoS One 7, e31457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whelan DR, Lee WTC, Marks F, Kong YT, Yin Y, and Rothenberg E. (2020). Super-resolution visualization of distinct stalled and broken replication fork structures. PLoS Genet 16, e1009256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whelan DR, Lee WTC, Yin Y, Ofri DM, Bermudez-Hernandez K, Keegan S, Fenyo D, and Rothenberg E. (2018). Spatiotemporal dynamics of homologous recombination repair at single collapsed replication forks. Nat Commun 9, 3882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang W, Roberti MJ, Heriche JK, Huet S, Alexander S, and Ellenberg J. (2018). Correlative live and super-resolution imaging reveals the dynamic structure of replication domains. J Cell Biol 217, 1973–1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates LA, Aramayo RJ, Pokhrel N, Caldwell CC, Kaplan JA, Perera RL, Spies M, Antony E, and Zhang X. (2018). A structural and dynamic model for the assembly of Replication Protein A on single-stranded DNA. Nat Commun 9, 5447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Y, Lee WTC, and Rothenberg E. (2019). Ultrafast data mining of molecular assemblies in multiplexed high-density super-resolution images. Nat Commun 10, 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Y, and Rothenberg E. (2016). Probing the Spatial Organization of Molecular Complexes Using Triple-Pair-Correlation. Sci Rep 6, 30819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeman MK, and Cimprich KA (2014). Causes and consequences of replication stress. Nature Cell Biology 16, 2–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Chabes A, Domkin V, Thelander L, and Rothstein R. (2001). The ribonucleotide reductase inhibitor Sml1 is a new target of the Mec1/Rad53 kinase cascade during growth and in response to DNA damage. The EMBO Journal 20, 3544–3553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou L, and Elledge SJ (2003). Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 300, 1542–1548. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Reconstructed SMLM images, TCF configurations, RPAfork and RPAglobal, etc. are available on Mendeley Data at DOI: 10.17632/s4b8ztvnxd.1
Matlab code for TCF analysis is available on Github at Codes for the Triple- and Pair-Correlation algorithms, as well as a testing demo (with simulation codes) are available at DOI: 10.17632/s4b8ztvnxd.1
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.