

Abstract
Cardiovascular disease is the leading cause of morbidity and mortality world-wide. Recently, the role of inflammation in the progression of diseases has significantly attracted considerable attention. In addition, various comorbidities, including diabetes, obesity, etc. exacerbate inflammation in the cardiovascular system, which ultimately leads to heart failure. Furthermore, cytokines released from specialized immune cells are key mediators of cardiac inflammation. Here, in this review article, we focused on the role of selected immune cells and cytokines (both pro-inflammatory and anti-inflammatory) in the regulation of cardiac inflammation and ultimately in cardiovascular diseases. While IL-1β, IL-6, TNFα, and IFNγ are associated with cardiac inflammation; IL-10, TGFβ, etc. are associated with resolution of inflammation and cardiac repair. IL-10 reduces cardiovascular inflammation and protects the cardiovascular system via interaction with SMAD2, p53, HuR, miR-375 and miR-21 pathway. In addition, we also highlighted recent advancements in the management of cardiac inflammation, including clinical trials of anti-inflammatory molecules to alleviate cardiovascular diseases.
Keywords: Inflammation, cytokines, interferons, TNFα, immune cells, cardiac diseases
Graphical Abstract
Introduction
Despite significant therapeutic advances, the highest rate of morbidity and mortality is observed due to cardiovascular diseases both in developed and developing countries, therefore, it is necessary to evaluate additional treatment options to manage cardiovascular diseases that are often associated with inflammation. Interestingly, inflammation is a normal reparative response to tissue injury and is a critical factor in the body’s ability to heal itself or to fight off infection (1–4), which involves the activation of leukocytes and is mediated, in part, by a family of cytokines and chemokines. Although self-limiting acute cardiovascular inflammation is meant to resolve the adverse cardiac injury, unrestrained chronic inflammation can cause severe tissue damage if not resolved as soon as the tissue repair process is completed (5–8). Experimental evidence suggests that inflammation plays a critical role in the progression of cardiovascular diseases, including myocardial infarction (9, 10), hypertension (11), atherosclerosis (12, 13), and hypertrophic heart failure (14, 15). Furthermore, a robust association exists between metabolic preconditioning and cardiovascular diseases (16, 17). Epidemiologic associations relating inflammation with obesity and diabetes have been reported since the 1960s, when concentrations of circulating fibrinogen and other acute-phase reactants were shown to be elevated in these conditions (18, 19). Framingham study has drew our attention to the increased incidence of heart failure in diabetic patients independent of age, coronary disease, hypertension, and body mass index (20). Furthermore, an increase in hemoglobin A1C (HbA1c) level enhances the risk of heart failure significantly in non-diabetic patients, which suggests that chronic hyperglycemia in pre-diabetic population increases the probability of heart failure (21). In addition to diabetes, obesity also increases cardiovascular risk and is mostly associated with an increase in reactive oxygen species and inflammation (22, 23). Cardiovascular inflammation is often associated with an increase in the level of inflammatory cytokines, including IL-1β (24, 25), IL-6 (25, 26) and TNFα (27, 28) secreted from resident and infiltrating immune cells with a parallel decrease of anti-inflammatory cytokines, including IL-10 and TGFβ (29). Although immune cells are key drivers, inflammatory cytokines are also produced by cardiac cells, including cardiomyocytes (30–32), cardiac fibroblasts (33, 34), and endothelial cells (35, 36) following ischemic or hypertrophic stress. Despite inflammation at the cellular and systemic levels in cardiovascular diseases, the heart has inherent and endogenous mechanisms to reduce inflammation. Either reducing the level of pro-inflammatory mediators or augmenting the level of anti-inflammatory mediators would reduce cardiac inflammation and improve cardiac function following hypertrophic or ischemic stress (14, 37).
Though numerous studies have proven the association of inflammation in cardiovascular diseases (38, 39), a standalone anti-inflammatory medicine regimen is not available to manage inflammation and alleviate cardiovascular diseases which could be due to multiple etiologies involved in cardiovascular diseases. Therefore, introducing anti-inflammatory therapy early in cardiovascular diseases to balance the healing response would be beneficial. Interestingly, several recombinant antibodies and medicines inhibit cardiac inflammation and improve cardiac function in animal models and patients (24, 39–42). These immune-modulatory substances can be used with standard of care medicines to alleviate cardiovascular diseases. In the later sections, we discuss a few important pharmacological targets to manage cardiovascular inflammation.
1. Inflammation in cardiovascular and metabolic diseases
Multiple diseases are associated with inflammation, including diabetes, obesity, and even general infection. In addition, inflammation is also increased in both acute and chronic cardiac diseases, such as myocardial infarction and hypertrophic heart failure. In addition, inflammatory mediators, such as TNFα, IL-1β, IL6, PAI-1, CRP, adiponectin, etc. are involved in the chronic inflammatory response and promote lipid accumulation with consequent development of atherosclerosis and cardiovascular diseases (43). In the following sections, we will discuss the contribution of inflammation in the progress of various cardiovascular and associated diseases.
1.1. Inflammatory signaling in diabetics and its impact on cardiovascular diseases
Diabetes is a prime risk factor for cardiovascular diseases. In the USA, diabetic adults experience two to four times higher mortality rate due to heart diseases and stroke as compared to non-diabetic patients. In diabetes, mortality is mostly associated with left ventricular dysfunction, atherosclerosis, coronary heart disease, or hypertension (6). Diabetic cardiomyopathy is defined as a disorder of the heart muscle caused by diabetes. The exact cellular events involved during diabetic cardiomyopathy are complex and remain unclear. In mice diabetic cardiomyopathy models, an impairment of systolic, diastolic, and cardiac performance are reported (6, 44). Diabetes has long been considered as a state of chronic, low-level inflammation. Recent evidence suggests that immune cell activation precedes insulin resistance in diabetic condition and may play an important role in the progression of cardiovascular diseases (44). Streptozotocin-induced diabetes is associated with an increase in the level of inflammatory cytokines, including TNFα and IL-1β in the heart. In addition, streptozotocin enhances cardiac remodeling and fibrosis as shown by increased expression of MMP9 and collagens (6). These data suggest that diabetic cardiomyopathy is associated with inflammation and fibrosis that hampers cardiac function. The level of potent vasodilators (like NO) decreases while the level of endothelin-1, a vasoconstrictor, increases in the patients with metabolic syndrome (45). These physiological changes are associated with the release of pro-inflammatory cytokines (46, 47). Dysregulated pro-inflammatory cytokines production exacerbates tissue injury by a variety of mechanisms including leukocyte recruitment, ROS production, mitochondrial dysfunction, fibrosis, and cell death. Diabetic mice exhibit contractile dysfunction, reduced ejection fraction and an increase in left ventricular end-diastolic pressure (48). Furthermore, oxidative stress, nitrosative stress, fibrosis, and apoptosis are increased in diabetic myocardium (48). Ares-Carrasco et al. studied the effect of diabetes together with hypertension in the progress of cardiac inflammation, apoptosis, and fibrosis. Streptozotocin-mediated short-term (6 weeks) activation of diabetic signaling in spontaneously hypertensive (SH) rats induces inflammation and apoptosis in the heart (49). Furthermore, the long-term streptozotocin treatment (22 weeks) increases cardiac fibrosis in the heart of SH rat. These data confirm that cardiac inflammation and apoptosis precede cardiac hypertrophy and fibrosis; therefore, blocking inflammation at the early stage may ameliorate cardiac remodeling (49). In the study by Ares-Carrasco et al., diabetes and hypertension caused cardiomyopathy independently and induction of diabetes in hypertensive animals had the only additive, but no significant effect on the cause of cardiopathy (49). Overall, diabetes increases cardiac inflammation and induces cardiac dysfunction. Novel strategies to reduce hyperglycemia-mediated inflammatory signaling would be a potential approach to ameliorate cardiac dysfunction in diabetic patients.
1.2. Obesity-induced inflammatory signaling and its impact on cardiovascular diseases
The prevalence of obesity is increasing worldwide and lately it has become a major public health issue due to its association with many comorbidities, such as diabetes, hypertension, dyslipidemia, etc. In the past decades, a dramatic increase in obesity has occurred both in children and adults world-wide. Obese MI patients exhibit a higher degree of systemic inflammation characterized by an increase in inflammatory cytokines (TNFα, IL-1β, IL-6, IL-8, IL-12, MCP-1), but a decrease in the level of IL-10 compared to non-obese MI patients. This suggests that there is a correlation between obesity and inflammation, including cardiac inflammation (50). Recently, the association of obesity with inflammation is a prime area of investigation. Obesity, mostly with visceral fat deposition, is associated with both systemic and cardiovascular low-grade inflammation, which results in a significant increase in morbidity and mortality due to cardiovascular diseases (11, 22, 23, 50–52). Obesity-associated systemic inflammation may be responsible for an increase in blood pressure as well. There is a strong correlation between obesity, and the level of IL-6 and CRP (53). IL-6 activates CRP production in the liver, suggesting, how cytokines regulate inflammation (54). IL-6 is also associated with increases in both systolic and diastolic blood pressure (53). Regardless of the mechanisms involved, weight loss in obese individuals reduces inflammation and lowers blood pressure (53, 55). Furthermore, a low IL-10 level is observed in the serum of obese women with metabolic syndrome (56) suggesting systemic inflammation in these populations. In addition, the level of IL-10 is decreased, but the level of IL-6 is increased in the serum of HFD (12 weeks)-induced obese mice (22). High-fat diet also induces fibrosis in the left atrium of WT and IL-10 knockout mice with a higher degree of atrial fibrosis in IL-10 KO mice (22). The levels of monocytes and macrophages increase in the left atrium of obese and IL-10 KO mice with a parallel increase in the levels of IL-1β and TNFα (22). The degree of monocyte and macrophage infiltration and release of pro-inflammatory cytokines are more in the atrium of obese IL-10 KO mice compared to their WT counterpart. In addition, the level of atrial MCP-1 increases in the atrium of obese IL-10 KO mice, but not in WT mice. IL-10 treatment decreases TGFβ level, but increases adiponectin level in the epicardial and pericardial tissue in IL-10 KO mice with a parallel decrease in monocyte infiltration and pro-inflammatory cytokines (IL-1β, TNFα and MCP-1) expression in the atrium (22). These data suggest, how managing inflammation can reduce cardiac hypertrophy and remodeling in obese patients (22, 23). These research support the notion that obesity-induced cardiac inflammation is detrimental to cardiac function. Thus, managing inflammation along with visceral fat would be a good strategy to reduce cardiovascular complications in obese patients.
1.3. Inflammation in Myocardial Infarction and Repair
Myocardial infarction (MI) is a pathological condition, where cardiac muscle cells die due to obstruction in blood flow to the heart muscle, generally, due to blockage of coronary arteries. The levels of TNFα, GMCSF, GCSF, MCSF, IFNα, and IFNβ increase in plasma of the patients with acute myocardial infarction (57). Neutrophils, macrophages, and mast cells mediate acute cardiac inflammation by releasing pro-inflammatory cytokines during myocardial infarction (58). Infiltration of neutrophils increases within 24 hrs. at the infarct site following MI, which releases IL-1β via the alarmin (S100A8/9)-TLR4-inflammasome pathway. The IL-1β via its interaction with receptor 1 on hematopoietic stem and progenitor cell, stimulates granulopoiesis which impairs cardiac function (59). Apart from production of cytokines, mast cells also release histamine which activates local cardiac inflammation following MI (58). In addition, macrophages also play a critical role in the regulation of inflammation following MI by releasing inflammatory and anti-inflammatory cytokines (60–62). For example, the pro-inflammatory macrophages are predominant in the heart 1–3 days after MI and release pro-inflammatory mediators, including GMCSF, IFN-γ, TNF-α and IL-1β, while anti-inflammatory macrophages predominate the heart 5–10 days after MI and promote the tissue repair by releasing IL-10 (anti-inflammatory), VEGF (angiogenic), and TGF-β1 (fibrotic) (60). Macrophages are polarized to different states i.e. pro-inflammatory vs anti-inflammatory via different cytokines. While IFNγ and GMCSF are pro-inflammatory mediators; cytokines, such as IL-4, IL-10, IL-13, and TGFβ1, are anti-inflammatory mediators of macrophage polarization (60). Apart from resident macrophages in the heart, remote monocytes also regulate cardiac inflammation following MI. Ly6Chigh monocytes differentiate to the inflammatory macrophages in a CCR2 dependent manner, while Ly6Clow monocytes differentiate to the anti-inflammatory macrophages via a CX3CR1 dependent manner following acute cardiac stress (60). Study by Jung et al (61) and Yang et al (62) demonstrated how IL-10 alleviates cardiac inflammation and dysfunction via polarizing macrophages to anti-inflammatory phenotype. In short, the pro-inflammatory macrophages dominate infarcted area following MI, but anti-inflammatory macrophages prevail the infarcted area during healing phase with a parallel increase in the level of IL-10 (62). Similarly, the treatment with IL-10 following MI in mice improved cardiac repair and function due to polarization of macrophages to an anti-inflammatory phenotype (61). In addition to macrophages and neutrophils, the infiltration of T-cell, including cytotoxic CD8+ T-cell and CD4+ helper T-cell, increase in ischemic and hypertrophic failing hearts. Depletion of CD4+ T-cells via antibody treatment reduces the severity of heart failure following MI, whereas adoptive transfer of splenic CD4+ cells (along with CD3+ T cells) causes fibrosis and hypertrophy in healthy mice (63). Hypoxia and reperfusion are also known to promote the release of inflammatory cytokines from leukocytes, including macrophages and neutrophils, during myocardial infarction (10, 64, 65). Treatment with IL-1β antibody reduces incidence of MI and angina in atherosclerosis patients (66) recommending inflammation would alleviate cardiovascular disease. Overall infiltration of different immune cells which release cytokines following MI drives cardiac remodeling. Thus, strategies to reduce immune cells from infiltrating the heart or reducing their activation in the failing heart could be a potential therapeutic strategy to manage ischemic heart injury.
1.4. Inflammation in Cardiac Hypertrophy and Remodeling
Cardiac hypertrophy and remodeling are characterized by increased cardiomyocytes size and fibrosis, which ultimately lead to heart failure. Recent studies suggest important roles of immune cells and inflammatory signaling in the progress of non-ischemic heart failure in patients (67). Recruitment of leukocytes into the myocardium is responsible for hypertrophic cardiac remodeling (68). Minimizing recruitment of monocytes in the heart reduces chronic remodeling following chronic stress. MCP-1 gene deletion (69) or administration of MCP-1 neutralizing antibody (70) inhibits chronic hypertrophic cardiac remodeling. IFNγ+ T cells (Th1)-bind to cardiac fibroblast via integrin α4 to induce TGFβ suggesting a coordinated role of cytokines (IFNγ) and immune cells (T-cell) in cardiac fibrosis (67). As expected, genetic deletion of TCR-α ameliorates cardiac fibrosis and heart failure (67). In the myocardium, TAC-induced reactive oxygen species increase the isolevuglandin (IsoLGs)-modified cardiac proteins level, which help in the proliferation of T-cells via activation of T-cell receptors. Intriguingly, administration of IsoLGs scavenger minimizes T-cell proliferation and thus ameliorates cardiac dysfunction suggesting the contribution of ROS-mediated activation of T-cells in impaired cardiac function following TAC (71). Interestingly, microbiomes, via their metabolites, regulate cardiac remodeling in a T-cell dependent manner (72). The expression of pro-inflammatory cytokines (IL-1α, IL-2, IL-6, TNFα, etc.) increase in the patients with dilated cardiomyopathy (57). We have previously shown that pro-inflammatory cytokines (IL-1β and TNFα) expression were increased in the isoproterenol (ISO)-treated cardiomyocytes and IL-10 treatment significantly reduced the levels of these cytokines via STAT-3-mediated NF-κB inhibition (14) (Figure 1). In a nutshell, infiltration of inflammatory cells and pro-inflammatory cytokines play important role in the progress of hypertrophic cardiac remodeling and thus use of anti-inflammatory therapies, such as IL-10, may improve cardiac function.
Fig. 1. IL-10 regulation of cardiac cell biology:
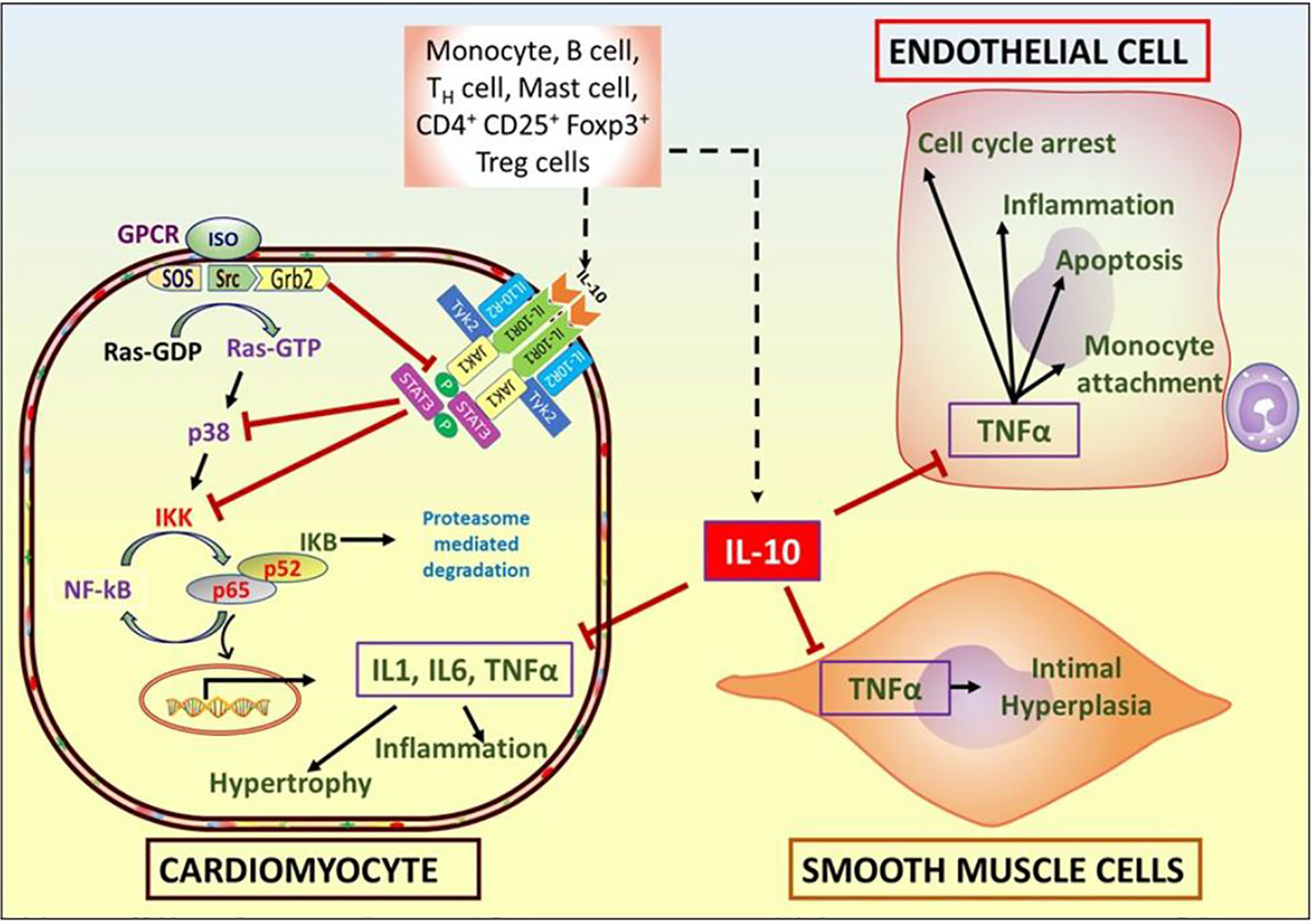
IL-10, an anti-inflammatory cytokine, is secreted from different immune and lymph cells, which inhibits isoproterenol (ISO)-induced hypertrophy and TNFα-mediated signaling, including inflammation. ISO-mediated activation of G-protein coupled receptor (GPCR) results in generation of Ras-GTP from Ras-GDP. Ras-GTPase activates inhibitor of nuclear factor-κB (IκB) kinase (IKK) via p38 which results in upregulation of hypertrophic and inflammatory genes expression. Activation of IL-10 receptor 1 (IL-10R1) and 2 (IL-10R2) by IL-10, results in phosphorylation of STAT3 which inhibits p38 and IKK, thus blunting ISO-induced hypertrophic signal in the cardiac muscle cells. Furthermore, IL-10 also protects endothelial cells from inflammatory injury following damage to the carotid artery and inhibits intimal hyperplasia. In vitro, IL-10 inhibits TNFα induced-apoptosis and cell cycle arrest of endothelial cells, and adhesion of monocytes to endothelial cells.
2. Inflammation in the aging heart
Inflammaging is characterized by chronic low-grade sterile inflammation in the aged people. It is associated with mitochondrial dysfunction, low-grade endotoxinemia, metabolic inflammation, gut dysbiosis, immune cell senescence, genomic instability etc. and ultimately leads to age-related chronic diseases such as heart disease (73–75). During aging, many factors/by-products including metabolites, altered proteins, mitochondrial DNA, micro RNAs, immune cells and cytokines are released, which trigger inflammation (73, 76–78). In the heart, inflammaging is associated with many cardiovascular diseases, such as atherosclerosis, high blood pressure and hypertrophic cardiac remodeling (79). Different immune cells are involved in the progress of inflammation in the aging, including neutrophils, macrophages, dendritic cells, B-cells, and T-cells (73–75, 80–83). However, role of only monocytes/macrophages and T-cells are largely studied in the inflammaging of heart.
With an increase in age, the level of resident anti-inflammatory macrophages (F4/80+CD206+) decreases in heart, while the level of monocyte-derived pro-inflammatory macrophages (F4/80+CD206−) increases (84, 85). The level of MCP-1, which regulates macrophage recruitment, increases in the left ventricle of the aged mice along with MMP-9, which is expressed by macrophages and is responsible for cardiac hypertrophy and fibrosis (83). MMP9 and MCP-1 are plasma biomarker of cardiac aging in mice and their role in cardiac aging in human should be studied (83). The level of inflammatory markers released by macrophages, including TNFα, that are associated with cardiac inflammation, increase in the plasma of aged mice (83). The plasma levels of MCP-1, MCP-2, and MIP-2, which correlate with left ventricular end-diastolic dimensions, also increase in the aged mice (83). Genetic deletion of MMP9 increases the level of anti-inflammatory macrophages in aged mice heart (85) and ameliorates myocardial fibrosis and diastolic dysfunction in aged mice (86). Interestingly, treatment with atorvastatin, a lipid lowering medicine, decreases MMP9 level and minimizes increase in the left ventricle thickness, diameter of cardiomyocytes, collagen deposition, and type I/III collagen ratio in aged rats (87). In the similar line, overexpression of MMP9 in macrophage augments ventricular hypertrophy, vessel rarefaction, inflammation and cardiac fibrosis in the aged mice (88). These data suggest the role of macrophage-derived MMP9 in the pathogenesis of cardiac aging. Genetic deletion of MMP28, which polarizes macrophages towards anti-inflammatory phenotype, increases the level of MMP9 in the aged left ventricle and augments inflammation (89). The expression of genes responsible of inflammatory response decreases while the level of genes responsible for fibrosis increases in the cardiac macrophages of aged mice, suggesting how macrophages play crucial role in inflammaging of heart (90).
T-cells are most studied immune cells in the context of cardiac inflammaging. When CD4+ T cells from older persons are activated in vitro, these cells demonstrate inflammatory Th17 phenotype with mitochondrial dysfunction and defects in autophagy. Metformin treatment normalizes the Th17 phenotype to a healthy CD4+ T cell phenotype found in young people with improving mitochondrial function and ameliorating autophagy defects (91). Inhibition of ROS production minimizes Th17 profile, suggesting an interplay of mitochondrial stress, oxidative stress and Th17 in inflammaging. It would be interesting to see if metformin reduces cardiac inflammation in humans and its effect on other immune cells, including monocytes/macrophages. A shorter telomere length is observed in the aged population and a shorter leukocyte telomere length is a risk factor for coronary heart disease (92) suggesting a link between old age, abnormal leukocyte production and heart disease. Dendritic cells from lymph nodes and spleen of aged mice are found to be activated and promote Th1 (releasing IFNγ) and Th17 (releasing IL-17) phenotype of CD4+ T cells. Activated dendritic cells transplantation from old mice to young mice, 7 days before heart transplantation, reduces the chances of allograft survival. Interestingly, senolytics (dasatinib+quercetin) inhibits dendritic cells activation and thus reduces Th1/Th17 immunogenicity, resulting in prolonged cardiac allograft survival (75). The level of effector memory and terminally differentiated CD4+ T-cells are increased in the blood of aged humans and mice. In contrast, the level of naive CD4+ T-cells is reduced in the aged people (80). Transplantation of senescent like human CD4+ T-cells into immunodeficient NSG-DR1 mice increases cardiac inflammation and fibrosis with a parallel increase in inflammatory signaling pathways, including cytokine-cytokine receptor interaction, IL-17 signaling pathway, chemokine signaling pathway, NOD-like receptor signaling pathway, TNF signaling pathway, complement and coagulation cascade, etc. (80). Transfer of human T-cells into the mice also increases the infiltration of monocytes, macrophages and dendritic cells in the mice heart suggesting how T-cells orchestrate immune cell infiltration and inflammation (80). These lines of research suggest the extensive role of immune cells, including T-cells and dendritic cells, in cardiac inflammaging and the immune cell-based approach may be a good option to manage inflammaging in the heart.
3. Regulators of cardiac inflammation and its resolution:
Cardiac inflammation is mediated and resolved by various enzymes, hormones, immune cells, cytokines, receptors, etc. Even if inflammation is governed by multiple factors, generally, the degree of inflammation is characterized by the level of immune cells in the inflamed area and quantifying the level of pro-inflammatory and anti-inflammatory cytokines in the tissue or in systemic circulation. Here, we discuss various immune cells and cytokines released by these cells that affect cardiac inflammation and its resolution.
3.1. Immune cells
The immune cells involved in the myocardial inflammation are neutrophils, monocytes/macrophages, eosinophils, mast cells, natural killer cells, T cells, and B cells (93, 94). Immune cells infiltrate in the heart in disease or pathological condition and release cytokines to initiate repair process (95). White blood cells can be classified as innate immune cells and adaptive immune cells based on the mechanisms of their action (96).
3.1.1. Innate immune cells
Innate immune cells are involved in rapid response, while adaptive immune cells are involved in slow response (97). Monocytes/ macrophage, mast cells, dendritic cells, natural killer cells and granulocytes (neutrophils, eosinophil and basophils) are considered as innate immune cells without antigen specificity (97). Innate immune cells infiltrate heart after MI to increase or resolute inflammation via releasing cytokines. For example, infiltration of CD45+ leukocytes (WBC) increase in the myocardium (95) following myocardial infarction which involves inflammation as discussed earlier. The number of Ly-6Chigh positive monocytes, which are pro-inflammatory, phagocytic and proteolytic, increases in the heart gradually and peaks on 3rd day and deceases gradually and normalized by 7th day after MI. However, the number of Ly-6Clow positive monocytes, which are anti-inflammatory, pro-angiogenic and reparative, increases in the heart gradually after 2 days of MI and peaks on 7th day after MI (98). Following MI, the levels of various leukocytes, including macrophage, dendritic cell, NK cells, increase in the heart till 7th day and decrease by 14th day though the levels of leukocytes are still higher than baseline level (95). The inflammatory genes expression (IL-1β, IL-6, NOS2 etc.) increases in the macrophages one day post-MI, in contrast, the level of IL-10 increases in the macrophages following 7 days of MI and remains increased till 14-day post-MI (95). This suggests that pro-inflammatory macrophages, expressing inflammatory cytokine, predominate at the injury site immediately after MI, while anti-inflammatory macrophages, expressing anti-inflammatory cytokines, predominate during the reparative phage of the cardiac inflammation (95). The expression of TLR4, which regulates inflammation, and co-stimulation molecule (CD80) increase in the macrophages isolated from heart, 1 day after MI (95). In addition, the expression of inflammasome gene (Pycard) and chemokine receptors (CCR2 and CXCR7), increase in the resident macrophages, 1–3 days post-MI. These studies demonstrate how cardiac macrophages mediate inflammation via TLR4, inflammasome, cytokines, and chemokines following MI (95). The traditional classification of macrophage is based on the cytokines released by macrophages in vitro and in vivo. Though the role of macrophages in initiation and resolution of cardiac inflammation is established, the classification of macrophages needs to be extended beyond traditional pro-inflammatory and anti-inflammatory phenotypes (99). The ratio of macrophage to leukocyte increases in the heart 3 days following MI and remains increased till 14th, suggesting important role of macrophages in initiation and resolution of inflammation in the heart (95). Neutrophils can convert Pro-IL-1β to IL-1β via releasing proteases. Thus, neutrophils can induce cardiac inflammation. In addition, neutrophils can destroy cardiac tissue via releasing myeloperoxidase. The ratio of neutrophil to leukocyte is higher in heart 1 day after MI, but decreases till 14th days after MI. This suggest that neutrophils first arrive at the injury site, before macrophages, following MI which initiates inflammation, but the level of neutrophils decreases over time along with resolution of cardiac inflammation (95).
3.1.2. Adaptive immune cells
Lymphocytes [B-cells and T-cells (CD4+ T-cell and CD8+ T-cells)] are considered as adaptive/acquired immune cells with characteristic antigen specificity and immunogenic memory (97). In contrast, Natural Killer (NK) cells lack antigen specificity, therefore, are considered as both innate and adaptive immune cells (80). Growing body of evidence advocates the contribution of adaptive immune response in the progress of cardiovascular diseases. After activation, B-cells generate antibody, which induces myocardial cell apoptosis, and increases expression of proinflammatory cytokines, including IL-1, IL-6 and TNFα (100). In addition, B-cells activates T-cells which ultimately can cause cardiac injury as described in preceding sections (100). Following MI, the level of B-cells in the heart increases gradually till 7th day and then decreases by 14th days though the level of cardiac B cells on 14th day post-MI is still higher than baseline. B-cells can also promote resolution of inflammation via releasing IL-10 in the pericardial adipose tissue following acute myocardial infarction (101). Therefore, management of B-cell mediated regulation of inflammation could be a therapeutic strategy to regulate heart failure. T-cells, on the other hand, are well studied in cardiac inflammation as discussed in previous section. The CD4 T helper cells such as Th1 cell (releases IFNγ which activates monocytes) and Th17 (releasing IL-17) cells induce cardiac inflammation. Further, the level of CD4+T helper cells, CD8+T cytotoxic cells, and γδT-cells is gradually increased in the heart till 7th day post-MI (95). The levels of Th1 and Treg cells also increase in the myocardium of heart 3 days following infarction and remain higher till 14th days. Similarly, the levels of NK and NKT cells increase in the heart 3 days following MI and gradually increase till 7th day. After 7th day, the level of these cells decrease by 14th day, but the level remains higher than baseline. These observations suggest how T cells and NK cells are associated with cardiac inflammation (95).
3.1.3. Platelets
Though the role of platelets in thrombosis (102), heart failure (103) and cardiac inflammation (104) has been known for a long time, the immunogenic property of the platelets and its interaction with other immune cells are realized lately (105–107). Platelets interact with cardiac cells and other immune cells via platelet adhesion molecules (P-selectin, PSGL-1, CD154/CD40L, ICAM-2, etc.) and surface receptors (TLRs and chemokine receptors) (105). Platelets express a variety of CC chemokine (CCR1–10) and CXC chemokine receptors (CXCR1–5 and others), and release various chemokine (CXCL4) and cytokines, including IL-1β and IL-8 (have neutrophil chemotactic activity) (105). ICAM-2 is involved in the recruitment of leukocytes, including neutrophils, by platelets and sialylation of the ICAM-2 impairs adhesion of leukocytes (108, 109). Platelets interact with monocytes (110–112), T-cells (113) and eosinophils (114) via RANTES. Another cytokine, IL-8, released by platelets activates neutrophils (115) to mediate innate immune reaction. Platelets are considered as a part of the innate immune system due to their ability to generate TNFα via a TLR4 dependent mechanism (116) and IL-1β which help in adhesion of neutrophils on endothelial cells (117). In addition, platelets also influence the adaptive immune system via CD154 which help in dendritic cell maturation, B cell isotype switching and augment CD8+ T cell responses (118). Platelet activation is increased in the patients with inflammatory cardiomyopathy (104) and these patients are vulnerable for thrombus formation (119). A high platelet to lymphocyte ratio is a poor prognostic factor for survival in heart failure patients (103).
Immune cells, including monocytes/macrophages, B-cells, T-cells, platelets, and neutrophils, are the major mediators of cardiovascular inflammations via releasing inflammatory cytokines. Recent advances in understanding the role of immune cells in cardiac inflammation helped to develop medicines based on cytokines which are discussed later in this article.
3.2. Cytokines:
Cytokines released by immune cells are well known to promote or resolute cardiac inflammation. Among immune cells, pro-inflammatory macrophage is associated with cardiac inflammation while anti-inflammatory macrophage is associated with resolution of inflammation and tissue repair (120, 121). DeLeon-Pennell et al demonstrated that infection-derived LPS decreases the number of reparative anti-inflammatory macrophages in the infarcted myocardium while increasing the level of pro-inflammatory macrophages (121). Monocytes, macrophages, B-cells, T-cells, platelets, and neutrophils are major sources of various cytokines in heart and other organs. We encourage our readers to read the reviews about the role of these white blood cells in inflammation, including cardiac inflammation (58, 64, 122). Briefly, monocytes are originated in the bone marrow and circulate to the target organs and differentiate into macrophages. Mouse Ly-6Chigh monocytes (equivalent to human CD16negative) are pro-inflammatory and release pro-inflammatory cytokines, including TNFα and IL-1β, while mouse Ly-6Clow monocytes (equivalent to human CD16positive) resolve inflammation via releasing IL-10 in addition to releasing TGFβ and VEGF. Neutrophils are accumulated at the site of injury within an hour of myocardial infarction and attach to endothelium using selectins present on endothelial cells membrane. The interaction of neutrophil’s integrin with endothelium is essential for strong adhesion and cellular migration. Following neutrophil infiltration, monocytes and macrophages arrive at the site of inflammation and present there till inflammation get resolved. In addition to releasing cytokines, the neutrophils release proteolytic enzymes and reactive oxygen species that can harm cardiac cells, including cardiomyocytes. Interleukins, interferons and TNFα are primarily studied pro-inflammatory cytokines in the context of cardiac inflammation. A cytokine can regulate the expression of other cytokines and modulate a wide array of cellular functions via multiple signaling pathways, mitochondrial dysfunction, and endoplasmic reticulum stress in cardiac cells (123). In the following subsections, we discuss the role of key cytokines associated with cardiovascular disorder.
3.2.1. IL-1β:
IL-1β released from immune cells, injured endothelial cells and cardiac cells is associated with further cardiac inflammation. Acute myocardial infarction induces IL-1β-mediated cardiac inflammation which further damages the cardiac cells due to caspase-3-mediated apoptosis (24). Inhibition of IL-1 signaling using IL-1 receptor antagonist (IL-1ra) gene transfer suppresses the M-variant of encephalomyocarditis virus (EMCV)-induced myocarditis in mice with a parallel decrease of TNFα and iNOS (25). Platelet microparticles-conjugated with IL-1β antibody, gevokizumab, reduce cardiac inflammation and apoptosis, increase cardiac cells viability, and improve cardiac function following acute myocardial infarction. Gevokizumab treatment reduces pro-inflammatory cytokine (IL-1β, IL-5 and IL-6) expression and apoptosis in the hearts subjected to MI (24) suggesting that reducing inflammation can alleviate cardiac cell death. Furthermore, blocking IL-1β receptors via anakinra reduces pericardial inflammation in patient-associated with Erdheim–Chester disease. Erdheim–Chester disease patients are characterized by inflammation due to excessive production and accumulation of histiocytes (large phagocytic macrophages) (124). Further, anakinra also reduces inflammation in the patients with pulmonary arterial hypertension and right ventricular failure characterized by higher level of high-sensitive CRP and IL-6 (125) (Fig. 2). Even though blocking IL-1β can reduce cardiovascular inflammation significantly, it may not completely stop inflammation. For example, though treatment with canakinumab, an anti-IL-1β monoclonal antibody, reduces cardiovascular events in patients with atherothrombosis, still some of the patients are vulnerable for future adverse cardiovascular events (126). Although canakinumab is known to reduce IL6 level, MI patients treated with canakinumab still show higher IL-6 level. Further, canakinumab does not regulate level of other inflammatory cytokines, such as IL-18, in patients with MI. Thus, targeting multiple cytokines or inflammasomes that regulate the activation of IL-1 and IL-18 could be a good strategy to reduce cardiovascular events in MI patients (111).
Fig. 2: IL-1β augments cardiac inflammation.
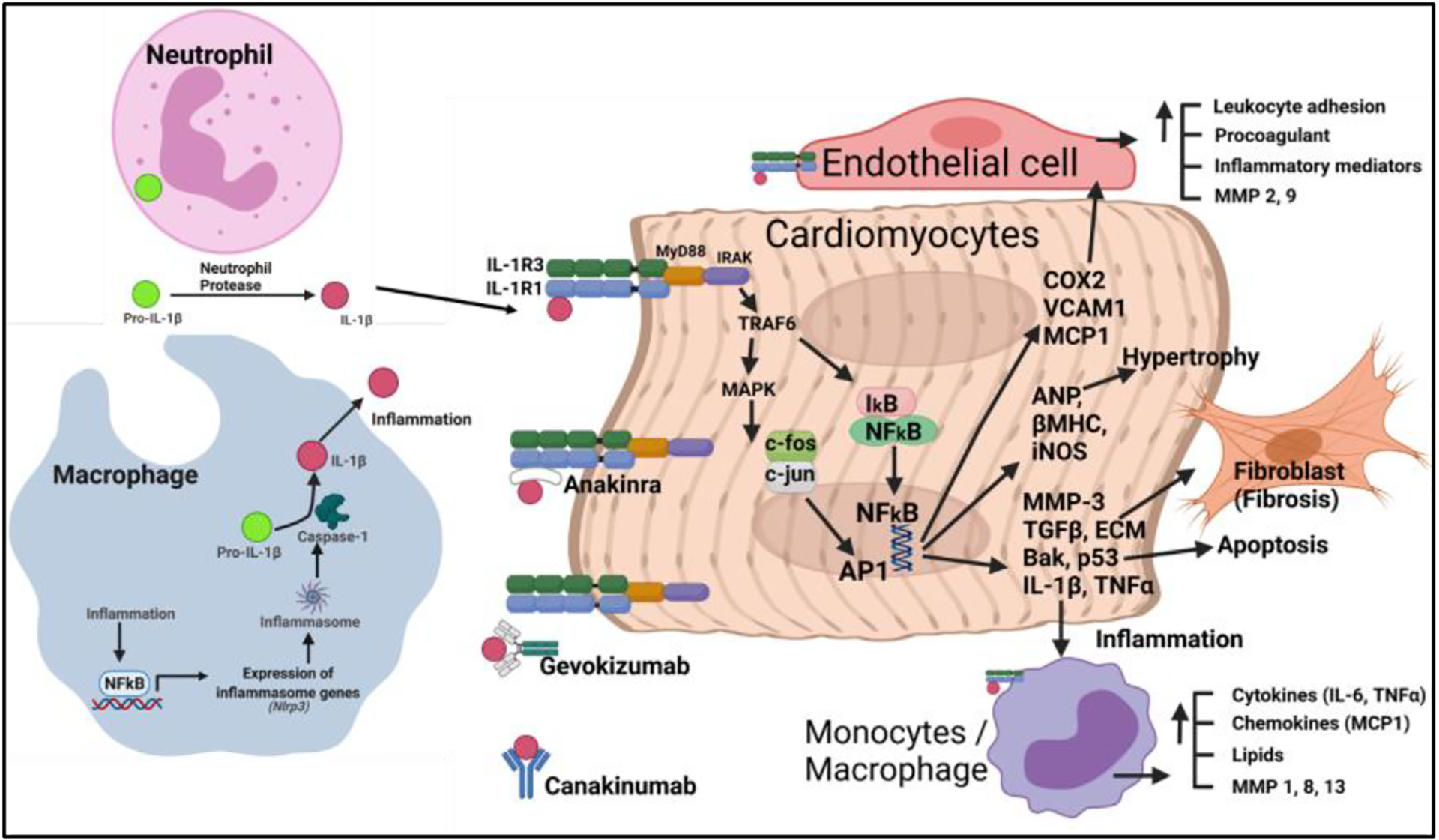
Inflammation augments NFkB mediated inflammatory genes expression, including Nlrp3 (inflammasome). Further, inflammasome-mediated caspase-1 activation produces IL-1β from pro-IL-1β (127). Neutrophils help in the production of IL-1β via releasing protease (128). In general, IL-1β via acting through its receptor (IL-1R) activates NFkB and AP1 signaling, which leads to the generation of inflammatory mediators (iNOS, IL-6, TNFα, MCP1, VCAM1, COX2, etc.), apoptosis (p53, Bak, etc.), hypertrophy (ANP, βMHC, etc.) and fibrosis (MMPs, TGFβ, ECM, etc.) in heart (129–131). VCAM1, MCP1 and COX2 derived lipids help in the leucocyte adhesion and favors blood clot formation via their interaction with endothelial cell (131). Anakinra (IL-1R blocker) and antibodies against IL-1β (gevokizumab and canakinumab) inhibits inflammatory activity of IL-1β.
3.2.2. IL-6
IL-6 is one of the most studied interleukins in the context of cardiovascular inflammation. It is a pro-inflammatory cytokine and is significantly involved in cardiac tissue damage following acute ischemic stress. It’s secretion is a major determinant of the production of acute-phase proteins that ultimately cause tissue damage and organ failure (132). Increased level of CRP and IL-6 are associated with an acute ischemic condition and are predictors of heart failure recurrence events (133, 134). Obese patients with cardiovascular risk have increased serum IL-6 level (135). Similarly, an increased serum IL-6 level is associated with endothelial dysfunction (26). Furthermore, chronic inflammation leads to increased expression of CRP, IL-6 and other cytokines in older patients and is one of the leading causes of mortality (136). The increased IL-6 level in patients following cardiac surgery is a strong predictor of mortality (137). Interestingly, sirukumab, an anti-IL-6 antibody, alleviates vascular inflammation and may provide vascular protection in patients (138). A clinical trial with anti-IL-6 receptor antibody, tocilizumab, is under investigation to study whether reducing systemic inflammation would reduce cardiac events in the patients. This trial will evaluate the effect of tocilizumab on the expression of pro-inflammatory and anti-inflammatory cytokines in the patients with cardiac arrest (139). In summary, as the expression of IL-6 is increased in multiple cardiovascular diseases, IL-6 can be a biomarker for heart failure. Furthermore, use of recombinant IL-6 antibody is shown to reduce cardiovascular inflammation, thus it could be a potential therapeutic molecule to manage heart failure.
3.2.3. IL-10
IL-10 is a pleiotropic anti-inflammatory cytokine, secreted by many immune cells including, Th2 cells, Treg cells, B-cells, mast cells and monocytes/macrophages. The anti-inflammatory property of IL-10 in cardiovascular biology is well established. A polymorphism at position - 1082 of the IL-10 promoter sequence reduces the production of IL-10 and increases the cardiac inflammation in diabetic patients (140). Similarly, during cardiac inflammation, the level of IL-10 decreases in the heart (29). In contrast, recombinant IL-10 treatment reduces cardiac inflammation via decreasing the level of IL-1, IL-6, IFNγ and iNOS (25, 141). In addition, the cardioprotective effect of IL-10 may also be mediated via an increase in macrophages polarization to an anti-inflammatory phenotype (61, 62) and/or upregulation of heme clearance pathway in the heart (141). Here, we discuss how IL-10 reduces cardiac inflammation and improves cardiac function in different heart failure models.
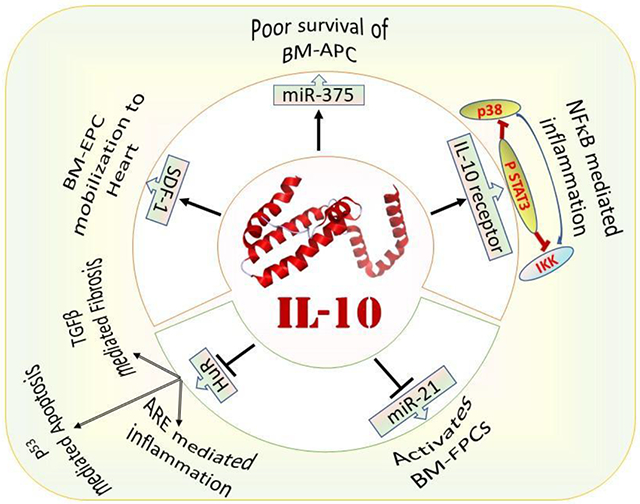
IL-10 protects the heart following MI via recruiting reparative stem/progenitor cells from the bone marrow to the heart. For example, IL-10 enhances the mobilization and homing of BM-derived endothelial progenitor cells to the heart following MI. SDF1 (CXCL12) which promotes movement of CXCR4 (CD184) positive stem cells from bone marrow to heart is decreased in IL-10 KO mouse following MI and intraperitoneal administration of SDF-1 increases the mobilization of stem cells to the heart of IL-10 KO mice suggesting IL-10 regulates stem cell mobilization via SDF-1 mediated mechanism (142). Further, reduced CXCR4 expression in IL-10 KO bone marrow endothelial progenitor cells (BM-EPCs) increases the susceptibility of BM-EPCs to death. Administration of IL-10 improves the retention of the transplanted BM-EPCs in the infarcted heart and thus improves cardiac function. Interestingly, WT bone marrow transplantation in IL-10 KO mice improves the cardiac function in IL-10 KO mice (142). Apart from protecting the heart by mobilizing endothelial progenitor cells, IL-10 also protects the heart by minimizing mobilization of fibroblast progenitor cells from bone marrow in the TAC model of cardiac hypertrophy via inhibiting miR-21 (143). Transplanting bone marrows from the WT mice in IL-10 KO mice alleviates TAC-induced cardiac fibrosis and improves heart function (143). Myocardial injection of BM-MNC expressing IL-10 into infarcted heart improves left ventricular function (stroke volume and +dp/dt) via decreasing T-lymphocyte accumulation, reactive hypertrophy and collagen deposition compared to BM-MNC lacking IL-10 (144).
Furthermore, in another elegant study, we have shown that IL-10 protects heart by inhibiting HuR, which is known to stabilize mRNAs of IL-1β and TNFα after binding to AU-rich elements (ARE) of these mRNAs, and thus reduce inflammation, apoptosis and fibrosis following MI (Fig. 3) (145, 146). Deletion of HuR in mouse macrophage minimizes the levels of TFGβ, TNFα and P53, after inflammatory insult (LPS) compared to control. In a mouse model of Trypanosoma cruzi infection, T-cells are reported to be the major source of IL-10 which reduces cardiac inflammation (147). Deletion of IL-10 upregulates iNOS expression in the cardiac tissue and causes myocarditis in mice infected with CVB3. Considering the role of macrophages in generating reactive nitric oxide from iNOS, IL-10 might suppress myocarditis via polarization of cardiac macrophages in CVB3-infected mice (29). In addition, IL-10 reduces inflammation via STAT-3 dependent inhibition of NF-κB (14), IL-10 affects cardiovascular signaling in multiple pathways, both directly and indirectly. For example, IL-10 increases the angiogenic and survival of bone marrow angiogenic progenitor cell (BMAPC) via inhibition of microRNA-375 (miR-375) and increases the regeneration of cardiac cells after myocardial infarction (148) (Fig. 3).
Fig. 3. IL-10 helps in cardiac regeneration and protects the heart via alleviating inflammation and apoptosis.
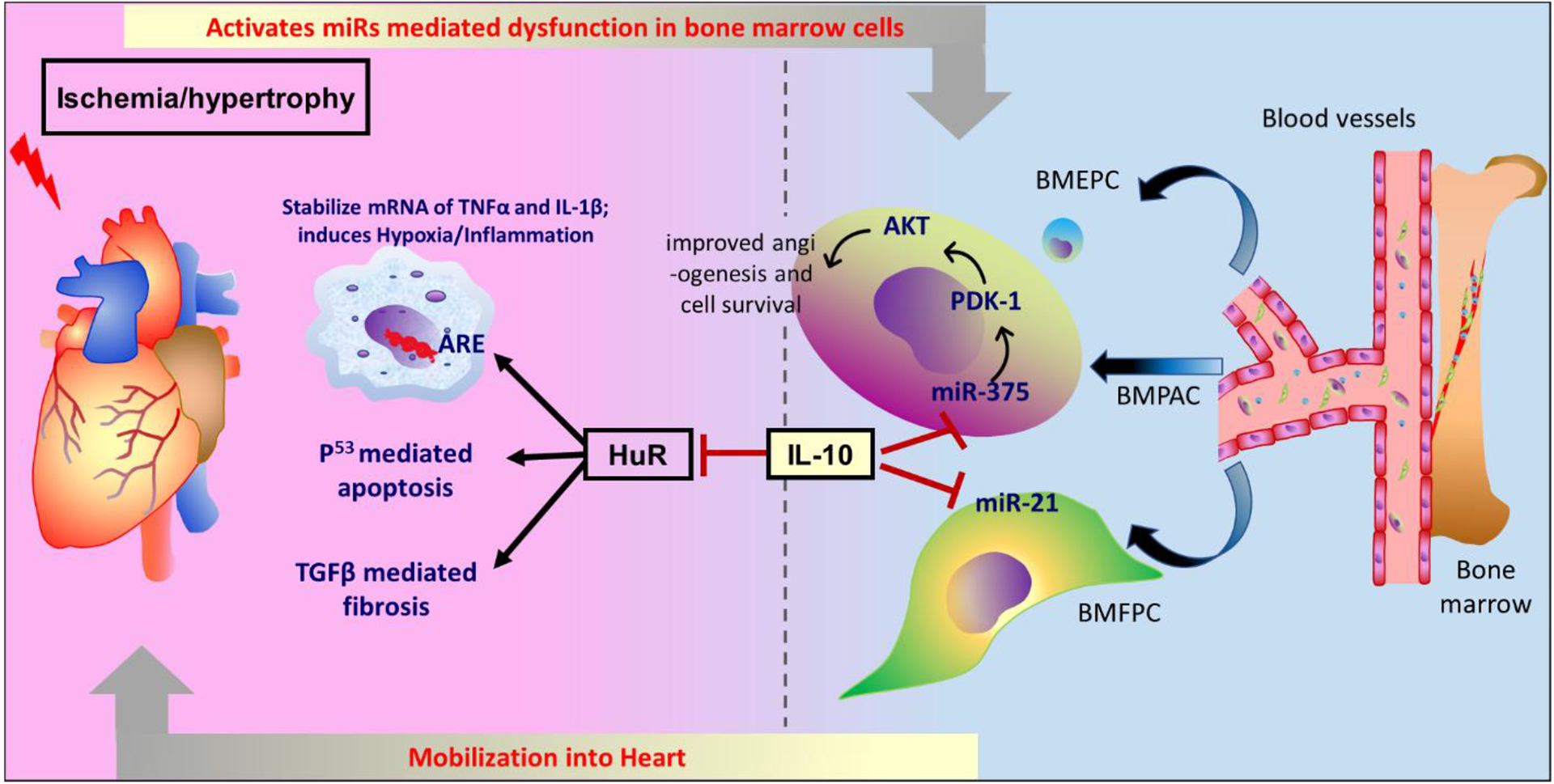
IL-10 inhibits miR-375 which upregulates PDK1/AKT pathway and increases angiogenesis and cell survival of bone marrow angiogenic progenitor cells (BMAPC). The miR-375 inhibition in BMAPC increases its survival after transplantation in the heart following MI. This suggests how IL-10 can protect the heart following MI via downregulating miR-375. Following MI, the level of SDF-1 decreases which reduces bone marrow progenitor cell mobilization and homing to the heart that impairs cardiac regeneration. Exogenous administration of IL-10 increases the expression of SDF-1 which helps in the movement of bone marrow progenitor cells and improves cardiac function in an infarcted heart. The deletion of HuR protein in macrophage reduces the level of TNFα, p53 and TGF-β following inflammatory insult. Upon binding of HuR to AU-rich elements (AREs) in the 3 untranslated regions of mRNA of inflammatory cytokine, including IL-1β and TNFα, stabilizes the cytokines which augments cardiac inflammation. HuR also promotes apoptosis via p53 signaling and augments fibrosis via activation of TGF-β. IL-10 inhibits HuR and, therefore, reduces cardiac inflammation, apoptosis and fibrosis. In IL-10 KO mice, the deletion of HuR alleviates cardiac dysfunction. IL-10 also inhibits the movement of bone marrow fibroblast progenitor cells following pressure overload-induced hypertrophy and minimizes cardiac fibrosis.
The increase in the angiogenic potential of BMAPC by inhibition of miR-375 is due to upregulation of 3-phosphoinositide-dependent protein kinase 1 and subsequent activation of protein kinase B (also known as RAC-alpha serine/threonine-protein kinase). Interestingly, the level of miR-375 increases in the left ventricle within 5 days after myocardial infarction and treatment with IL-10 reduces the level of miR-375 in the mice subjected to MI (148). Similarly, the level of miR-375 is elevated in the patients of the heart suggesting clinical utility of their previous finding in the mice model (149). Further, myocardial deletion of miR-375 using AAV6/9 system augments the neovascularization, decreases cardiac fibrosis, minimizes inflammation and improves cardiac function after myocardial infarction with a parallel increase of anti-inflammatory macrophage polarization. Notably, inhibition of miR-375 minimizes the LPS-induced increase in the pro-inflammatory cytokines expression, including TNFα and IL-1β in macrophages (149). IL-10 mediated inhibition of miR-375 also affects proliferation of endothelial progenitor cells and endothelial cell growth (150), which are important for cardiac regeneration following MI. For example, exosomes isolated from IL-10 KO mice have lesser potential to repair and proliferate endothelial progenitor cells compared to exosomes isolated from WT mice (150). Interestingly, deletion of miR-375 augments the reparative potential of exosomes isolated from the IL-10 KO mice (150) suggesting that IL-10 promotes endothelial cell growth via inhibition of miR-375. Further, IL-10 minimizes inflammation of endothelial cells following damage to the carotid artery and reduces the development of neointimal growth (36) probably by interfering with activity of TNFα because IL-10 reduces TNFα-promoted adhesion of monocytes to endothelial cells, apoptosis and cell cycle arrest of endothelial cells, in vitro (36). Therefore, IL-10 may reduce stenosis-induced arterial inflammation and the development of thrombus in patients.
Recent studies have demonstrated that the cardioprotective role of IL-10 is, at least partly, mediated by regulating immune cell functions (61, 62). The level of pro-inflammatory macrophages (characterized by release of iNOS) significantly increase in myocardial tissues by day 7 following MI and then gradually decreases, while the level of anti-inflammatory macrophages (characterized by arginase expression) increases gradually from 7th day to day 28 with a parallel increase of IL-10 in the blood. Thus, IL-10 alleviates cardiac inflammation via polarizing anti-inflammatory macrophages (62). IL-10 treatment to mice following MI promotes polarization of macrophage to an anti-inflammatory phenotype and is associated with fibroblast activation resulting in better cardiac repair, improved left ventricular function (61). Interestingly, IL-10 treatment does not affect the level of pro-inflammatory macrophage (releasing/expressing CCL3, CCL5, IL1β, IL-6 and TNFα), but increases the level of anti-inflammatory macrophages (releasing/expressing arginase, mannose receptor 1/CD206, TGFβ1) in left ventricular infarct region of mice, in vivo. Similarly, when the macrophages isolated from the left ventricular infarction region are stimulated with IL-10, it displays an anti-inflammatory phenotype. IL-10 treatment improves proliferation and migration of post-MI cardiac fibroblast and reduces extracellular matrix accumulation 7 days after MI, which is required for prevention of tissue rupture and excessive fibrosis (61). Interestingly, expression of IL-10 on bone marrow derived stem cells negatively regulates cardiac inflammation and myocardial infarction (151). However, the expression of IL-10 is reduced in the bone marrow derived stem cells of diabetic mice (151). Delivery of bone marrow derived mesenchymal stem cell from diabetic mice overexpressing IL-10 via CRISPR activation system in the heart after MI improves survival of stem cells and reduces inflammatory cells infiltration and inflammatory cytokine production with a parallel increase of angiogenesis which favors reduction of MI and improvement of cardiac function (151). In this context, IL-10 also may minimize graft versus host disease via type-1 regulatory T-cell (152) and B-cell (153) mediated mechanisms.
The anti-inflammatory and cardioprotective effect of IL-10 could also be mediated independent of immune cell biology. For example, IL-10 treatment minimizes infarct area and improves cardiac function due to improved capillary density, and reduced apoptosis via upregulation of heme clearance pathway (141) in the heart of diabetic mice subjected to MI. Thus, IL-10 can afford cardioprotection in diabetic mice by modulating iron biology. In this context, it would be interesting to study the effect of IL-10 on iron regulating hormones (hepcidin) and iron transporter (ferroportin) in diabetic heart failure.
In summary, IL-10 is an anti-inflammatory cytokine that has been studied extensively in the context of cardiac inflammation and alleviates cardiac inflammation via activating SDF1 and STAT3 while inhibiting HuR and microRNAs (miR-21 and miR375). It also reduces the polarization of pro-inflammatory macrophages but increases polarization of anti-inflammatory macrophages. IL-10 alleviates MI and cardiac hypertrophy associated with fibrosis.
3.2.4. TNFα
TNFα is released from mast cells and macrophages, and a potent inflammatory cytokine implicated in cardiovascular inflammation by inducing multiple inflammatory cytokines via NFkB mediated mechanism (28, 154, 155). The TNFα (RNA and protein) level and its receptors (I and II) increase in the myocarditis patient’s heart. The degree of cardiac dysfunction positively correlates with the level of cardiac TNFα in the patients with viral myocarditis (156). Similarly, the level of circulating TNFα and its receptors increase in chronic heart failure patient (154, 155) resulting in cardiac remodeling via TNFα dependent activation of MMP-3 signaling pathway (155). As expected, myocardial rupture rate is less in TNFα knockout mice following MI compared to WT mice (157). Furthermore, genetic deletion of TNFα reduces inflammatory cell infiltration, minimizes LV infarct size, and improves mice survival following MI (157). TNFα knockout mice also shows reduced cardiac fibroblast activation, fibrosis and improved cardiac function following TAC (158). These data suggest the important role of TNFα in the progress of both hypertrophic and ischemic heart failure. At the cellular level, TNFα regulates inflammation and phagocytosis of endothelial cells. For example, TNFα dependent activation of NF-κB signaling induces the expression of IL-8 (a neutrophil chemotactic factor) in human microvasculature endothelial cell (28). In addition, TNFα also induces intercellular adhesion molecule (ICAM, also known as CD45) on endothelial cells (159) that helps in the adhesion of leukocytes which drives further inflammation and phagocytosis of endothelial cells as well as nearby tissues. Treatment with anti-TNFα antibody decreases the level of cardiac inflammation and necrosis, and mortality in infected mice (27). Treatment with etanercept, a TNFα receptor blocker, alleviates cardiac damage and improves cardiovascular function with a parallel decrease in the level of TNFα and IL-17 in psoriasis patients (160). Thus, the use of TNFα inhibitor seems to be a promising medicine to alleviate cardiovascular dysfunction associated with inflammation in psoriasis patients. In summary, TNFα is another widely studied inflammatory cytokine released from multiple immune cells, including macrophages. It is associated with immune cell infiltration, inflammation, fibrosis and left ventricular dysfunction in models of heart failure. Blocking the action of TNFα reduces cardiac inflammation and dysfunction in animal models and patients.
3.2.5. Interferon
Interferon is also a group of cytokines associated with inflammation via their interaction with leukocytes, including macrophages and monocytes (161, 162) and IFN also drives inflammatory cell death (163). Originally, the interferon was reported to protect the host cells from viral infection by ‘interfering’ with viral replication. Among the 3 types of interferons, type II (interferon γ) is studied commonly in cardiovascular inflammation (164, 165).
IFNγ drives cardiovascular inflammation via stimulating macrophages (161). IFNγ induces multiple genes in monocytes, including C–X–C motif chemokine 10 [(CXCL10 or interferon γ-induced protein 10 kDa (IP10)], ISG54 and ICAM-1 via STAT1 mediated signal transduction (162). Interestingly, ISG54 induces apoptosis in a BAX and BAK dependent manner (166) and it is regulated by IFNγ (167). Thus, INFγ may facilitate cardiac apoptosis via activation of ISG54. Interferons regulates expression of other cytokines, and vice-versa. On the arterial wall, IL-12/IL-18-activated T-cells secrete IFNγ (Th1) (165), which suppress proliferation of anti-inflammatory Th2 cells that can be inhibited by IL-10 (162). Based on one school of thought, IFNγ-induced activation of macrophages releases reactive nitrate intermediates, including NO and peroxynitrite, which has an antiviral effect. Based on this hypothesis, researchers reported a low and delayed release of IFNγ in the heart of mice susceptible to CVB3 infection (29). The level of IFN-γ in the patients with coronary artery disease is higher and it decreases with exercise due to a parallel increase in the level of anti-inflammatory IL-10 and a decrease of cardiac damage marker, CRP (168). Therefore, exercise may alleviate IFNγ associated coronary artery disease via alleviating inflammation. These reports suggest that genetic deletion of IFN-γ may be helpful in alleviating cardiac inflammation. However, global genetic deletion of IFNγ augments autoimmune myocarditis (169). It is evident that though IFN-γ is associated with cardiac inflammation, complete deletion of IFNγ would be deleterious to the heart. In summary, IFNγ is an inflammatory cytokine which activates monocytes/macrophages to induce cardiac inflammation. Though genetic knockout of IFNγ may not help in cardiac inflammation, reducing the level of IFNγ or interfering with the activity of IFNγ by various means, including exercise or IL-10 treatment, could be helpful in cardiac inflammation.
As discussed above, antagonizing inflammatory cytokines would reduce cardiovascular inflammation and improve cardiovascular function. Some of the cytokines, including IL-1β and IL-18 are activated by inflammasome. Therefore, targeting the inflammasome is also a good option to reduce cardiovascular inflammation. The immunosuppressive drugs, such as cyclosporine, reduce the formation of inflammatory cytokines and are helpful in reducing inflammation (IL-6 and TNFα) and cardiac damage, and improve cardiovascular function (140), though care should be taken for unwanted adverse effects.
4. Molecules that alleviate cardiac inflammation
We have discussed how cytokines, including ILs, TNFα and IFNγ released from immune cells, promote/resolute cardiac inflammation. Among the discussed: IL-1, IL-6, TNFα and IFNγ promote cardiac inflammation. Fortunately, antibodies against IL-6 receptor (tocilizumab) (40, 41), IL-1β (canakinumab) (126), IL-1 receptor (anakinra) (39) and TNFα (etanercept) (160) reduce cardiac inflammation and adverse cardiac events. However, caution is required during the administration of these antibodies because of the increased incidence of infection. Inflammatory cytokines are helpful for killing invading pathological microorganisms, thus, neutralizing them can result in an increase in bacterial infection. In this context, using anti-inflammatory IL-10 seems a good option. IL-10 resolutes cardiac inflammation by various pathways. IL-10 alleviates cardiac inflammation and remodeling via inhibiting SMAD (143), miR-375 (148), miR21 (143), HuR (145, 170) and P53 (146), but stimulating SDF-1 (146). IL-10 also reduces cardiac inflammation via negatively regulating IL-1 (145), IL-6 (145) and TNFα (14, 171).
Some of the clinically used medicines to manage cardiac disease and infection also decrease inflammation. For example, angiotensin-converting enzyme (ACE) inhibitor-enalapril decreases cardiac inflammation in the heart of mice infected with T. cruzi with a collateral decrease of inflammatory cells in the heart compared with untreated mice (172). Similarly, lipid-lowering medicine, simvastatin, which inhibits 3-hydroxy-3-methylglutaryl (HMG) coenzyme A reductase, minimizes cardiac inflammation, improves left ventricular ejection fraction, reduces left ventricular mass, and alleviates fibrosis in T. cruzi infected dogs (173) by elevating the level of IL-10 in the plasma (173). Pretreatment of azithromycin, an antibiotic, reduces cardiac inflammation and cardiac remodeling after MI in mice (174). Pimobendan, a phosphodiesterase type III inhibitor with calcium sensitizing properties is used to manage cardiac failure in animals, also alleviate cardiac inflammation. Pimobendan reduces myocardiotrophic variant of encephalomyocarditis (EMC) virus-induced myocarditis that is associated with inflammatory cell infiltration in the heart. Pimobendan also reduces the level of TNFα and IL1β in the heart and decreases the level of iNOS and its enzymatic product nitric oxide (NO), which increases nitrosative stress (175). A reduction in the level of IL-1β, IL-6, and TNFα by the PDE inhibitor is not due to the canonical cAMP/ cGMP pathway as either cAMP or cGMP does not reduce the production of IL-1β, IL-6 and TNFα in peripheral blood mononuclear cells, in vitro (176). Therefore, these medicines may alleviate cardiac inflammation in patients or animals taking these medicines.
Apart from cytokine-based treatment, few clinically used medicines, including those used for managing hypertension (enalapril), high cholesterol (simvastatin), heart failure (pimobendan) and bacterial infection (azithromycin) decrease cardiac inflammation in animal models. Clinical studies to evaluate the role of these medicines to reduce cardiac inflammation are warranted so that these medicines may be used in patients/subjects with cardiovascular diseases or infection associated with cardiovascular inflammation.
5. Clinical trials of anti-inflammatory compounds for reducing cardiac inflammation
The level of inflammatory cytokines (IL-1β, IL-6 and IL-18) and sensitivity C-reactive protein, an inflammatory biomarker, increase in cardiac patients vulnerable to develop heart failure (177, 178). As we have discussed earlier, decreasing inflammation in the cardiovascular system would decrease cardiac remodeling and improve cardiac function. Therefore, many molecules are being evaluated in the clinical trials to alleviate cardiac inflammation and increase cardiac function.
Canakinumab, an IL-1β antibody, decreases recurrent cardiovascular events (MI and angina) in atherosclerosis patients harboring MI with a decrease in CRP. The cardioprotective effect is independent of lipid-level lowering property suggesting anti-inflammatory effect of the antibody medicine is helpful in reducing cardiovascular events (66). Thrombocytopenia, neutropenia, and incidence of sepsis are some of the side effects of canakinumab, which should be kept in mind while treating the patients with this medicine (66). Colchicine, an anti-inflammatory medicine to manage gout and pericarditis, when given with lipid lowering medicine (statin) reduces cardiovascular events in patients with stable coronary disease (179). When used independently, colchicine also reduces risk of ischemic cardiac events (42) in patients with MI which could be due to a decrease in local cardiac production of inflammatory cytokines, including IL-1β, IL-6, and IL-18 (180). The common adverse events observed with colchicine treatment are gastrointestinal events, diarrhea, nausea, and pneumonia (42). Anakinra, an anti-inflammatory recombinant IL-1R antagonist, reduces the level of C-reactive protein in patients with non-ST elevation acute coronary syndrome after 2 weeks of treatment suggesting cardioprotective effect. However, an increase in the incidence of major adverse cardiovascular events, including death, stroke, and new MI, are reported following the withdrawal of the treatment for up to 1 year. A detailed mechanistic study is required to elucidate the negative effect of anakinra on the cardiovascular system after its withdrawal. Inclacumab, an anti-inflammatory antibody against P-selectin, exerts cardioprotective effect via reducing troponin I and myocardial creatine kinase after non-ST elevation acute coronary intervention (181). However, inclacumab does not reduce saphenous vein graft disease following coronary artery bypass graft surgery (182). Even though we described clinical success of many anti-inflammatory molecules in heart disease, not all anti-inflammatory molecules reduce all cardiac events associated with inflammation. For example, inclacumab (182), darapladib (a lipoprotein-associated phospholipase A2 inhibitor) (183, 184), varespladib (an inhibitor of secretory phospholipase A2) (185), methotrexate (anticancer compound) (186), and succinobucol (antioxidant compound) (187) failed to show cardioprotective effect.
Pre-clinical data from our and others lab suggest that IL-10 could be helpful in reducing cardiac inflammation and remodeling. Though there are no clinical trials registered for evaluating the efficacy of IL-10 in cardiovascular diseases, its clinical trial is being continued in other inflammation associated diseases. A Phase 2 clinical trial is under progress to evaluate efficacy, safety and tolerability of a novel IL-10 conjugate (F8IL-10) in alleviating ulcerative colitis as an add-on therapy to infliximab (ClinicalTrials.gov Identifier: NCT03269695). Efficacy and safety of gastrointestinal selective IL-10 fusion protein (AMT-101) is being evaluated in ulcerative colitis (ClinicalTrials.gov Identifier: NCT04583358) and pouchitis (ClinicalTrials.gov Identifier: NCT04741087) as well. If these IL-10 conjugates are approved for clinical use, the effect of these agents can be evaluated on cardiac inflammation. Clinically approved anti-inflammatory compounds, which reduce cardiac inflammation by managing cytokines level and recruitment of leukocytes can be used to manage cardiovascular diseases.
6. Conclusion:
In conclusion, limiting cardiac inflammation would protect the heart in various pathological conditions by minimizing adverse cardiac remodeling. Minimizing activation of inflammatory immune cells, including T cells, neutrophils, and mast cells would reduce cardiac inflammation. Though few commercially available antibodies against inflammatory cytokines reduce cardiac inflammation, caution should be employed for administering these agents due to increased chances of infection. IL-10 remains a good target to manage cardiac inflammation. Other clinically used medicines are found to reduce cardiac inflammation. Though these medicines cannot be used only for reducing cardiac inflammation, these medicines may be preferred over other medicines with similar mechanisms of action in patients with systemic and cardiac inflammation.
Acknowledgements
The work presented here was supported by grant number HL135060 from National Institutes of Health to S.K.V. and Postdoctoral grant number 826859 from American Heart Association to P.R.
Abbreviations:
- ACE
angiotensin converting enzyme
- Akt
RAC-alpha serine/threonine-protein kinase
- BAK
Bcl-2 antagonist/killer
- BAX
Bcl-2 like protein 4
- Bcl-2
B-cell lymphoma 2
- BM
Bone marrow
- BM-MNC
Bone Marrow-Mononuclear cell
- BMPAC
Bone marrow progenitor angiogenic cell
- cAMP
cyclic adenosine monophosphate
- cGMP
cyclic guanosine monophosphate
- CCL
chemokine (C-C motif) ligand
- CCR2
C-C motif Chemokine receptor 2
- CD
Cluster of differentiation
- CRP
C reactive protein
- CVB
Coxsackie B Virus
- Cxcr
C-x-c motif chemokine receptor
- EMCV
Encephalomyocarditis virus
- ERA
estrogen replacement and atherosclerosis
- GCSF
granulocyte colony stimulating factor
- GMCSF
granulocyte macrophage colony stimulating factor
- HFD
high fat diet
- HuR
Hu family of RNA-binding protein
- ICAM
intercellular adhesion molecule
- IFN
Interferon
- IL
interleukin
- iNOS
inducible nitric oxide synthase
- ISG54
IFN-stimulated gene 54
- JAM-A
Junction adhesion molecule A
- KO
knockout
- LPS
lipopolysaccharide
- MIP
Macrophage inflammatory protein
- MCP
monocyte chemotactic protein
- MCSF
macrophage colony stimulating factor
- MI
myocardial infarction
- miR
microRNA
- MMP
matrix metalloproteinase
- PAI-1
Plasminogen activator inhibitor-1
- PDE
Phosphodiesterase
- PDK1
3-phisphoinositide-dependent protein kinase 1
- PSGL-1
P-selectin glycoprotein ligand 1
- ROS
reactive oxygen species
- SDF-1
stromal cell–derived factor 1
- SMAD
small mothers against decapentaplegic, a transcription factor protein
- STAT
signal transducers and activators of transcription
- STZ
streptozotocin
- s-VCAM
soluble vascular cell adhesion molecule
- TAC
Transverse aortic constriction
- TCR-α
T cell receptor α
- TLR
Toll like receptor
- TNF
tissue necrosis factor
- TGF
transforming growth factor
- VEGF
vascular endothelial growth factor
- WT
wild type
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest
The authors declare that there is no potential conflict of interest.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Reference:
- 1.Adams DH, Lloyd AR. Chemokines: leucocyte recruitment and activation cytokines. Lancet. 1997;349(9050):490–5. [DOI] [PubMed] [Google Scholar]
- 2.Turner MD, Nedjai B, Hurst T, Pennington DJ. Cytokines and chemokines: At the crossroads of cell signalling and inflammatory disease. Biochim Biophys Acta. 2014;1843(11):2563–82. [DOI] [PubMed] [Google Scholar]
- 3.Ono SJ, Nakamura T, Miyazaki D, Ohbayashi M, Dawson M, Toda M. Chemokines: roles in leukocyte development, trafficking, and effector function. J Allergy Clin Immunol. 2003;111(6):1185–99; quiz 200. [DOI] [PubMed] [Google Scholar]
- 4.Olson TS, Ley K. Chemokines and chemokine receptors in leukocyte trafficking. Am J Physiol Regul Integr Comp Physiol. 2002;283(1):R7–28. [DOI] [PubMed] [Google Scholar]
- 5.Maskrey BH, Megson IL, Whitfield PD, Rossi AG. Mechanisms of resolution of inflammation: a focus on cardiovascular disease. Arterioscler Thromb Vasc Biol. 2011;31(5):1001–6. [DOI] [PubMed] [Google Scholar]
- 6.Westermann D, Rutschow S, Jager S, Linderer A, Anker S, Riad A, et al. Contributions of inflammation and cardiac matrix metalloproteinase activity to cardiac failure in diabetic cardiomyopathy: the role of angiotensin type 1 receptor antagonism. Diabetes. 2007;56(3):641–6. [DOI] [PubMed] [Google Scholar]
- 7.Wang Z, Nakayama T. Inflammation, a link between obesity and cardiovascular disease. Mediators Inflamm. 2010;2010:535918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Martinez E, D’Albuquerque PM, Llibre JM, Gutierrez F, Podzamczer D, Antela A, et al. Changes in cardiovascular biomarkers in HIV-infected patients switching from ritonavir-boosted protease inhibitors to raltegravir. AIDS. 2012;26(18):2315–26. [DOI] [PubMed] [Google Scholar]
- 9.Frangogiannis NG. The inflammatory response in myocardial injury, repair, and remodelling. Nat Rev Cardiol. 2014;11(5):255–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Neumann FJ, Ott I, Gawaz M, Richardt G, Holzapfel H, Jochum M, et al. Cardiac release of cytokines and inflammatory responses in acute myocardial infarction. Circulation. 1995;92(4):748–55. [DOI] [PubMed] [Google Scholar]
- 11.Glowinska B, Urban M. [Selected cytokines (Il-6, Il-8, Il-10, MCP-1, TNF-alpha) in children and adolescents with atherosclerosis risk factors: obesity, hypertension, diabetes]. Wiad Lek. 2003;56(3–4):109–16. [PubMed] [Google Scholar]
- 12.Libby P, Ridker PM, Maseri A. Inflammation and atherosclerosis. Circulation. 2002;105(9):1135–43. [DOI] [PubMed] [Google Scholar]
- 13.Libby P Inflammation in atherosclerosis. Arterioscler Thromb Vasc Biol. 2012;32(9):2045–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Verma SK, Krishnamurthy P, Barefield D, Singh N, Gupta R, Lambers E, et al. Interleukin-10 treatment attenuates pressure overload-induced hypertrophic remodeling and improves heart function via signal transducers and activators of transcription 3-dependent inhibition of nuclear factor-kappaB. Circulation. 2012;126(4):418–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kishore R, Krishnamurthy P, Garikipati VN, Benedict C, Nickoloff E, Khan M, et al. Interleukin-10 inhibits chronic angiotensin II-induced pathological autophagy. J Mol Cell Cardiol. 2015;89(Pt B):203–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Williams LJ, Nye BG, Wende AR. Diabetes-Related Cardiac Dysfunction. Endocrinol Metab (Seoul). 2017;32(2):171–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Type Ziegler D. 2 diabetes as an inflammatory cardiovascular disorder. Curr Mol Med. 2005;5(3):309–22. [DOI] [PubMed] [Google Scholar]
- 18.Fearnley GR, Vincent CT, Chakrabarti R. Reduction of blood fibrinolytic activity in diabetes mellitus by insulin. Lancet. 1959;2(7111):1067. [DOI] [PubMed] [Google Scholar]
- 19.Ogston D, McAndrew GM. Fibrinolysis in Obesity. Lancet. 1964;2(7371):1205–7. [DOI] [PubMed] [Google Scholar]
- 20.Kannel WB, McGee DL. Diabetes and cardiovascular disease. The Framingham study. JAMA. 1979;241(19):2035–8. [DOI] [PubMed] [Google Scholar]
- 21.Matsushita K, Blecker S, Pazin-Filho A, Bertoni A, Chang PP, Coresh J, et al. The association of hemoglobin a1c with incident heart failure among people without diabetes: the atherosclerosis risk in communities study. Diabetes. 2010;59(8):2020–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kondo H, Abe I, Gotoh K, Fukui A, Takanari H, Ishii Y, et al. Interleukin 10 Treatment Ameliorates High-Fat Diet-Induced Inflammatory Atrial Remodeling and Fibrillation. Circ Arrhythm Electrophysiol. 2018;11(5):e006040. [DOI] [PubMed] [Google Scholar]
- 23.Kesherwani V, Chavali V, Hackfort BT, Tyagi SC, Mishra PK. Exercise ameliorates high fat diet induced cardiac dysfunction by increasing interleukin 10. Front Physiol. 2015;6:124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li Z, Hu S, Huang K, Su T, Cores J, Cheng K. Targeted anti-IL-1beta platelet microparticles for cardiac detoxing and repair. Sci Adv. 2020;6(6):eaay0589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nakano A, Matsumori A, Kawamoto S, Tahara H, Yamato E, Sasayama S, et al. Cytokine gene therapy for myocarditis by in vivo electroporation. Hum Gene Ther. 2001;12(10):1289–97. [DOI] [PubMed] [Google Scholar]
- 26.Esteve E, Castro A, Lopez-Bermejo A, Vendrell J, Ricart W, Fernandez-Real JM. Serum interleukin-6 correlates with endothelial dysfunction in healthy men independently of insulin sensitivity. Diabetes Care. 2007;30(4):939–45. [DOI] [PubMed] [Google Scholar]
- 27.Yamada T, Matsumori A, Sasayama S. Therapeutic effect of anti-tumor necrosis factor-alpha antibody on the murine model of viral myocarditis induced by encephalomyocarditis virus. Circulation. 1994;89(2):846–51. [DOI] [PubMed] [Google Scholar]
- 28.Lakshminarayanan V, Drab-Weiss EA, Roebuck KA. H2O2 and tumor necrosis factor-alpha induce differential binding of the redox-responsive transcription factors AP-1 and NF-kappaB to the interleukin-8 promoter in endothelial and epithelial cells. J Biol Chem. 1998;273(49):32670–8. [DOI] [PubMed] [Google Scholar]
- 29.Szalay G, Sauter M, Hald J, Weinzierl A, Kandolf R, Klingel K. Sustained nitric oxide synthesis contributes to immunopathology in ongoing myocarditis attributable to interleukin-10 disorders. Am J Pathol. 2006;169(6):2085–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aoyagi T, Matsui T. The Cardiomyocyte as a Source of Cytokines in Cardiac Injury. J Cell Sci Ther. 2011;2012(S5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jacoby JJ, Kalinowski A, Liu MG, Zhang SS, Gao Q, Chai GX, et al. Cardiomyocyte-restricted knockout of STAT3 results in higher sensitivity to inflammation, cardiac fibrosis, and heart failure with advanced age. Proc Natl Acad Sci U S A. 2003;100(22):12929–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhu X, Bagchi A, Zhao H, Kirschning CJ, Hajjar RJ, Chao W, et al. Toll-like receptor 2 activation by bacterial peptidoglycan-associated lipoprotein activates cardiomyocyte inflammation and contractile dysfunction. Crit Care Med. 2007;35(3):886–92. [DOI] [PubMed] [Google Scholar]
- 33.Wang C, Zhang C, Liu L, A X, Chen B, Li Y, et al. Macrophage-Derived mir-155-Containing Exosomes Suppress Fibroblast Proliferation and Promote Fibroblast Inflammation during Cardiac Injury. Mol Ther. 2017;25(1):192–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Martin R, Gutierrez B, Cordova C, Roman AS, Alvarez Y, Hernandez M, et al. Secreted Phospholipase A2-IIA Modulates Transdifferentiation of Cardiac Fibroblast through EGFR Transactivation: An Inflammation-Fibrosis Link. Cells. 2020;9(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang L, Gill R, Pedersen TL, Higgins LJ, Newman JW, Rutledge JC. Triglyceride-rich lipoprotein lipolysis releases neutral and oxidized FFAs that induce endothelial cell inflammation. J Lipid Res. 2009;50(2):204–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Verma SK, Garikipati VN, Krishnamurthy P, Khan M, Thorne T, Qin G, et al. IL-10 Accelerates Re-Endothelialization and Inhibits Post-Injury Intimal Hyperplasia following Carotid Artery Denudation. PLoS One. 2016;11(1):e0147615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kaur K, Dhingra S, Slezak J, Sharma AK, Bajaj A, Singal PK. Biology of TNFalpha and IL-10, and their imbalance in heart failure. Heart Fail Rev. 2009;14(2):113–23. [DOI] [PubMed] [Google Scholar]
- 38.Zhou X, Li J, Guo J, Geng B, Ji W, Zhao Q, et al. Gut-dependent microbial translocation induces inflammation and cardiovascular events after ST-elevation myocardial infarction. Microbiome. 2018;6(1):66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ikonomidis I, Lekakis JP, Nikolaou M, Paraskevaidis I, Andreadou I, Kaplanoglou T, et al. Inhibition of interleukin-1 by anakinra improves vascular and left ventricular function in patients with rheumatoid arthritis. Circulation. 2008;117(20):2662–9. [DOI] [PubMed] [Google Scholar]
- 40.Orrem HL, Nilsson PH, Pischke SE, Kleveland O, Yndestad A, Ekholt K, et al. IL-6 Receptor Inhibition by Tocilizumab Attenuated Expression of C5a Receptor 1 and 2 in Non-ST-Elevation Myocardial Infarction. Front Immunol. 2018;9:2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Interleukin-6 Receptor Mendelian Randomisation Analysis C, Swerdlow DI, Holmes MV, Kuchenbaecker KB, Engmann JE, Shah T, et al. The interleukin-6 receptor as a target for prevention of coronary heart disease: a mendelian randomisation analysis. Lancet. 2012;379(9822):1214–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tardif JC, Kouz S, Waters DD, Bertrand OF, Diaz R, Maggioni AP, et al. Efficacy and Safety of Low-Dose Colchicine after Myocardial Infarction. N Engl J Med. 2019;381(26):2497–505. [DOI] [PubMed] [Google Scholar]
- 43.Vicenova B, Vopalensky V, Burysek L, Pospisek M. Emerging role of interleukin-1 in cardiovascular diseases. Physiol Res. 2009;58(4):481–98. [DOI] [PubMed] [Google Scholar]
- 44.Festa A, D’Agostino R Jr., Howard G, Mykkanen L, Tracy RP, Haffner SM. Chronic subclinical inflammation as part of the insulin resistance syndrome: the Insulin Resistance Atherosclerosis Study (IRAS). Circulation. 2000;102(1):42–7. [DOI] [PubMed] [Google Scholar]
- 45.Tesauro M, Schinzari F, Rovella V, Di Daniele N, Lauro D, Mores N, et al. Ghrelin restores the endothelin 1/nitric oxide balance in patients with obesity-related metabolic syndrome. Hypertension. 2009;54(5):995–1000. [DOI] [PubMed] [Google Scholar]
- 46.Woods M, Mitchell JA, Wood EG, Barker S, Walcot NR, Rees GM, et al. Endothelin-1 is induced by cytokines in human vascular smooth muscle cells: evidence for intracellular endothelin-converting enzyme. Mol Pharmacol. 1999;55(5):902–9. [PubMed] [Google Scholar]
- 47.Westermann D, Van Linthout S, Dhayat S, Dhayat N, Escher F, Bucker-Gartner C, et al. Cardioprotective and anti-inflammatory effects of interleukin converting enzyme inhibition in experimental diabetic cardiomyopathy. Diabetes. 2007;56(7):1834–41. [DOI] [PubMed] [Google Scholar]
- 48.Rajesh M, Mukhopadhyay P, Batkai S, Patel V, Saito K, Matsumoto S, et al. Cannabidiol attenuates cardiac dysfunction, oxidative stress, fibrosis, and inflammatory and cell death signaling pathways in diabetic cardiomyopathy. J Am Coll Cardiol. 2010;56(25):2115–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ares-Carrasco S, Picatoste B, Benito-Martin A, Zubiri I, Sanz AB, Sanchez-Nino MD, et al. Myocardial fibrosis and apoptosis, but not inflammation, are present in long-term experimental diabetes. Am J Physiol Heart Circ Physiol. 2009;297(6):H2109–19. [DOI] [PubMed] [Google Scholar]
- 50.Gruzdeva O, Uchasova E, Dyleva Y, Akbasheva O, Matveeva V, Karetnikova V, et al. Relationship key factor of inflammation and the development of complications in the late period of myocardial infarction in patients with visceral obesity. BMC Cardiovasc Disord. 2017;17(1):36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chang JS, Chang CC, Chien E, Lin SS, Cheng-Shiuan T, Bai CH, et al. Association between interleukin 1beta and interleukin 10 concentrations: a cross-sectional study in young adolescents in Taiwan. BMC Pediatr. 2013;13:123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Poirier P, Giles TD, Bray GA, Hong Y, Stern JS, Pi-Sunyer FX, et al. Obesity and cardiovascular disease: pathophysiology, evaluation, and effect of weight loss: an update of the 1997 American Heart Association Scientific Statement on Obesity and Heart Disease from the Obesity Committee of the Council on Nutrition, Physical Activity, and Metabolism. Circulation. 2006;113(6):898–918. [DOI] [PubMed] [Google Scholar]
- 53.Frohlich M, Imhof A, Berg G, Hutchinson WL, Pepys MB, Boeing H, et al. Association between C-reactive protein and features of the metabolic syndrome: a population-based study. Diabetes Care. 2000;23(12):1835–9. [DOI] [PubMed] [Google Scholar]
- 54.Anty R, Bekri S, Luciani N, Saint-Paul MC, Dahman M, Iannelli A, et al. The inflammatory C-reactive protein is increased in both liver and adipose tissue in severely obese patients independently from metabolic syndrome, Type 2 diabetes, and NASH. Am J Gastroenterol. 2006;101(8):1824–33. [DOI] [PubMed] [Google Scholar]
- 55.Novi RF, Porta M, Lamberto M, Molinatti GM. Reductions of body weight and blood pressure in obese hypertensive patients treated by diet. A retrospective study. Panminerva Med. 1989;31(1):13–5. [PubMed] [Google Scholar]
- 56.Esposito K, Pontillo A, Giugliano F, Giugliano G, Marfella R, Nicoletti G, et al. Association of low interleukin-10 levels with the metabolic syndrome in obese women. J Clin Endocrinol Metab. 2003;88(3):1055–8. [DOI] [PubMed] [Google Scholar]
- 57.Matsumori A, Yamada T, Suzuki H, Matoba Y, Sasayama S. Increased circulating cytokines in patients with myocarditis and cardiomyopathy. Br Heart J. 1994;72(6):561–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Frangogiannis NG, Smith CW, Entman ML. The inflammatory response in myocardial infarction. Cardiovasc Res. 2002;53(1):31–47. [DOI] [PubMed] [Google Scholar]
- 59.Sreejit G, Abdel-Latif A, Athmanathan B, Annabathula R, Dhyani A, Noothi SK, et al. Neutrophil-Derived S100A8/A9 Amplify Granulopoiesis After Myocardial Infarction. Circulation. 2020;141(13):1080–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ma Y, Mouton AJ, Lindsey ML. Cardiac macrophage biology in the steady-state heart, the aging heart, and following myocardial infarction. Transl Res. 2018;191:15–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jung M, Ma Y, Iyer RP, DeLeon-Pennell KY, Yabluchanskiy A, Garrett MR, et al. IL-10 improves cardiac remodeling after myocardial infarction by stimulating M2 macrophage polarization and fibroblast activation. Basic Res Cardiol. 2017;112(3):33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yang W, Li S, Zhao Y, Sun Y, Huang Y, Cao X, et al. Changes in the expression of interleukin-10 in myocardial infarction and its relationship with macrophage activation and cell apoptosis. Ann Transl Med. 2020;8(10):643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bansal SS, Ismahil MA, Goel M, Patel B, Hamid T, Rokosh G, et al. Activated T Lymphocytes are Essential Drivers of Pathological Remodeling in Ischemic Heart Failure. Circ Heart Fail. 2017;10(3):e003688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nahrendorf M, Pittet MJ, Swirski FK. Monocytes: protagonists of infarct inflammation and repair after myocardial infarction. Circulation. 2010;121(22):2437–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Arango Duque G, Descoteaux A. Macrophage cytokines: involvement in immunity and infectious diseases. Front Immunol. 2014;5:491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N Engl J Med. 2017;377(12):1119–31. [DOI] [PubMed] [Google Scholar]
- 67.Nevers T, Salvador AM, Velazquez F, Ngwenyama N, Carrillo-Salinas FJ, Aronovitz M, et al. Th1 effector T cells selectively orchestrate cardiac fibrosis in nonischemic heart failure. J Exp Med. 2017;214(11):3311–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Damilano F, Franco I, Perrino C, Schaefer K, Azzolino O, Carnevale D, et al. Distinct effects of leukocyte and cardiac phosphoinositide 3-kinase gamma activity in pressure overload-induced cardiac failure. Circulation. 2011;123(4):391–9. [DOI] [PubMed] [Google Scholar]
- 69.Dewald O, Zymek P, Winkelmann K, Koerting A, Ren G, Abou-Khamis T, et al. CCL2/Monocyte Chemoattractant Protein-1 regulates inflammatory responses critical to healing myocardial infarcts. Circ Res. 2005;96(8):881–9. [DOI] [PubMed] [Google Scholar]
- 70.Kuwahara F, Kai H, Tokuda K, Takeya M, Takeshita A, Egashira K, et al. Hypertensive myocardial fibrosis and diastolic dysfunction: another model of inflammation? Hypertension. 2004;43(4):739–45. [DOI] [PubMed] [Google Scholar]
- 71.Ngwenyama N, Kirabo A, Aronovitz M, Velazquez F, Carrillo-Salinas F, Salvador AM, et al. Isolevuglandin-Modified Cardiac Proteins Drive CD4+ T-Cell Activation in the Heart and Promote Cardiac Dysfunction. Circulation. 2021;143(12):1242–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Carrillo-Salinas FJ, Anastasiou M, Ngwenyama N, Kaur K, Tai A, Smolgovsky SA, et al. Gut dysbiosis induced by cardiac pressure overload enhances adverse cardiac remodeling in a T cell-dependent manner. Gut Microbes. 2020;12(1):1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Liberale L, Montecucco F, Tardif JC, Libby P, Camici GG. Inflamm-ageing: the role of inflammation in age-dependent cardiovascular disease. Eur Heart J. 2020;41(31):2974–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Keshavarz-Bahaghighat H, Darwesh AM, Sosnowski DK, Seubert JM. Mitochondrial Dysfunction and Inflammaging in Heart Failure: Novel Roles of CYP-Derived Epoxylipids. Cells. 2020;9(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Iske J, Seyda M, Heinbokel T, Maenosono R, Minami K, Nian Y, et al. Senolytics prevent mt-DNA-induced inflammation and promote the survival of aged organs following transplantation. Nat Commun. 2020;11(1):4289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Franceschi C, Garagnani P, Parini P, Giuliani C, Santoro A. Inflammaging: a new immune-metabolic viewpoint for age-related diseases. Nat Rev Endocrinol. 2018;14(10):576–90. [DOI] [PubMed] [Google Scholar]
- 77.Franceschi C, Campisi J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J Gerontol A Biol Sci Med Sci. 2014;69 Suppl 1:S4–9. [DOI] [PubMed] [Google Scholar]
- 78.Olivieri F, Lazzarini R, Recchioni R, Marcheselli F, Rippo MR, Di Nuzzo S, et al. MiR-146a as marker of senescence-associated pro-inflammatory status in cells involved in vascular remodelling. Age (Dordr). 2013;35(4):1157–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Liu D, Richardson G, Benli FM, Park C, de Souza JV, Bronowska AK, et al. Inflammageing in the cardiovascular system: mechanisms, emerging targets, and novel therapeutic strategies. Clin Sci (Lond). 2020;134(17):2243–62. [DOI] [PubMed] [Google Scholar]
- 80.Delgobo M, Heinrichs M, Hapke N, Ashour D, Appel M, Srivastava M, et al. Terminally Differentiated CD4(+) T Cells Promote Myocardial Inflammaging. Front Immunol. 2021;12:584538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Frasca D, Romero M, Garcia D, Diaz A, Blomberg BB. Hyper-metabolic B cells in the spleens of old mice make antibodies with autoimmune specificities. Immun Ageing. 2021;18(1):9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hagen M, Derudder E. Inflammation and the Alteration of B-Cell Physiology in Aging. Gerontology. 2020;66(2):105–13. [DOI] [PubMed] [Google Scholar]
- 83.Chiao YA, Dai Q, Zhang J, Lin J, Lopez EF, Ahuja SS, et al. Multi-analyte profiling reveals matrix metalloproteinase-9 and monocyte chemotactic protein-1 as plasma biomarkers of cardiac aging. Circ Cardiovasc Genet. 2011;4(4):455–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Molawi K, Wolf Y, Kandalla PK, Favret J, Hagemeyer N, Frenzel K, et al. Progressive replacement of embryo-derived cardiac macrophages with age. J Exp Med. 2014;211(11):2151–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ma Y, Chiao YA, Clark R, Flynn ER, Yabluchanskiy A, Ghasemi O, et al. Deriving a cardiac ageing signature to reveal MMP-9-dependent inflammatory signalling in senescence. Cardiovasc Res. 2015;106(3):421–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chiao YA, Ramirez TA, Zamilpa R, Okoronkwo SM, Dai Q, Zhang J, et al. Matrix metalloproteinase-9 deletion attenuates myocardial fibrosis and diastolic dysfunction in ageing mice. Cardiovasc Res. 2012;96(3):444–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Han L, Li M, Liu Y, Han C, Ye P. Atorvastatin may delay cardiac aging by upregulating peroxisome proliferator-activated receptors in rats. Pharmacology. 2012;89(1–2):74–82. [DOI] [PubMed] [Google Scholar]
- 88.Toba H, Cannon PL, Yabluchanskiy A, Iyer RP, D’Armiento J, Lindsey ML. Transgenic overexpression of macrophage matrix metalloproteinase-9 exacerbates age-related cardiac hypertrophy, vessel rarefaction, inflammation, and fibrosis. Am J Physiol Heart Circ Physiol. 2017;312(3):H375–H83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ma Y, Chiao YA, Zhang J, Manicone AM, Jin YF, Lindsey ML. Matrix metalloproteinase-28 deletion amplifies inflammatory and extracellular matrix responses to cardiac aging. Microsc Microanal. 2012;18(1):81–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Pinto AR, Godwin JW, Chandran A, Hersey L, Ilinykh A, Debuque R, et al. Age-related changes in tissue macrophages precede cardiac functional impairment. Aging (Albany NY). 2014;6(5):399–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bharath LP, Agrawal M, McCambridge G, Nicholas DA, Hasturk H, Liu J, et al. Metformin Enhances Autophagy and Normalizes Mitochondrial Function to Alleviate Aging-Associated Inflammation. Cell Metab. 2020;32(1):44–55 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Opstad TB, Kalstad AA, Holte KB, Berg TJ, Solheim S, Arnesen H, et al. Shorter Leukocyte Telomere Lengths in Healthy Relatives of Patients with Coronary Heart Disease. Rejuvenation Res. 2020;23(4):324–32. [DOI] [PubMed] [Google Scholar]
- 93.Strassheim D, Dempsey EC, Gerasimovskaya E, Stenmark K, Karoor V. Role of Inflammatory Cell Subtypes in Heart Failure. J Immunol Res. 2019;2019:2164017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wang J, Duan Y, Sluijter JP, Xiao J. Lymphocytic subsets play distinct roles in heart diseases. Theranostics. 2019;9(14):4030–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Yan X, Anzai A, Katsumata Y, Matsuhashi T, Ito K, Endo J, et al. Temporal dynamics of cardiac immune cell accumulation following acute myocardial infarction. J Mol Cell Cardiol. 2013;62:24–35. [DOI] [PubMed] [Google Scholar]
- 96.Epelman S, Liu PP, Mann DL. Role of innate and adaptive immune mechanisms in cardiac injury and repair. Nat Rev Immunol. 2015;15(2):117–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Dranoff G Cytokines in cancer pathogenesis and cancer therapy. Nat Rev Cancer. 2004;4(1):11–22. [DOI] [PubMed] [Google Scholar]
- 98.Hilgendorf I, Gerhardt LM, Tan TC, Winter C, Holderried TA, Chousterman BG, et al. Ly-6Chigh monocytes depend on Nr4a1 to balance both inflammatory and reparative phases in the infarcted myocardium. Circ Res. 2014;114(10):1611–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Nahrendorf M, Swirski FK. Abandoning M1/M2 for a Network Model of Macrophage Function. Circ Res. 2016;119(3):414–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Cordero-Reyes AM, Youker KA, Torre-Amione G. The role of B-cells in heart failure. Methodist Debakey Cardiovasc J. 2013;9(1):15–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wu L, Dalal R, Cao CD, Postoak JL, Yang G, Zhang Q, et al. IL-10-producing B cells are enriched in murine pericardial adipose tissues and ameliorate the outcome of acute myocardial infarction. Proc Natl Acad Sci U S A. 2019;116(43):21673–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zhang S, Liu Y, Wang X, Yang L, Li H, Wang Y, et al. SARS-CoV-2 binds platelet ACE2 to enhance thrombosis in COVID-19. J Hematol Oncol. 2020;13(1):120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ye GL, Chen Q, Chen X, Liu YY, Yin TT, Meng QH, et al. The prognostic role of platelet-to-lymphocyte ratio in patients with acute heart failure: A cohort study. Sci Rep. 2019;9(1):10639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Weikert U, Kuhl U, Schultheiss HP, Rauch U. Platelet activation is increased in patients with cardiomyopathy: myocardial inflammation and platelet reactivity. Platelets. 2002;13(8):487–91. [DOI] [PubMed] [Google Scholar]
- 105.von Hundelshausen P, Weber C. Platelets as immune cells: bridging inflammation and cardiovascular disease. Circ Res. 2007;100(1):27–40. [DOI] [PubMed] [Google Scholar]
- 106.Mezger M, Nording H, Sauter R, Graf T, Heim C, von Bubnoff N, et al. Platelets and Immune Responses During Thromboinflammation. Front Immunol. 2019;10:1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Morrell CN, Aggrey AA, Chapman LM, Modjeski KL. Emerging roles for platelets as immune and inflammatory cells. Blood. 2014;123(18):2759–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kuijper PH, Gallardo Tores HI, Lammers JW, Sixma JJ, Koenderman L, Zwaginga JJ. Platelet associated fibrinogen and ICAM-2 induce firm adhesion of neutrophils under flow conditions. Thromb Haemost. 1998;80(3):443–8. [PubMed] [Google Scholar]
- 109.Weber KS, Alon R, Klickstein LB. Sialylation of ICAM-2 on platelets impairs adhesion of leukocytes via LFA-1 and DC-SIGN. Inflammation. 2004;28(4):177–88. [DOI] [PubMed] [Google Scholar]
- 110.von Hundelshausen P, Weber KS, Huo Y, Proudfoot AE, Nelson PJ, Ley K, et al. RANTES deposition by platelets triggers monocyte arrest on inflamed and atherosclerotic endothelium. Circulation. 2001;103(13):1772–7. [DOI] [PubMed] [Google Scholar]
- 111.Schober A, Manka D, von Hundelshausen P, Huo Y, Hanrath P, Sarembock IJ, et al. Deposition of platelet RANTES triggering monocyte recruitment requires P-selectin and is involved in neointima formation after arterial injury. Circulation. 2002;106(12):1523–9. [DOI] [PubMed] [Google Scholar]
- 112.von Hundelshausen P, Koenen RR, Sack M, Mause SF, Adriaens W, Proudfoot AE, et al. Heterophilic interactions of platelet factor 4 and RANTES promote monocyte arrest on endothelium. Blood. 2005;105(3):924–30. [DOI] [PubMed] [Google Scholar]
- 113.Schall TJ, Bacon K, Toy KJ, Goeddel DV. Selective attraction of monocytes and T lymphocytes of the memory phenotype by cytokine RANTES. Nature. 1990;347(6294):669–71. [DOI] [PubMed] [Google Scholar]
- 114.Kameyoshi Y, Dorschner A, Mallet AI, Christophers E, Schroder JM. Cytokine RANTES released by thrombin-stimulated platelets is a potent attractant for human eosinophils. J Exp Med. 1992;176(2):587–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Baggiolini M, Walz A, Kunkel SL. Neutrophil-activating peptide-1/interleukin 8, a novel cytokine that activates neutrophils. J Clin Invest. 1989;84(4):1045–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Aslam R, Speck ER, Kim M, Crow AR, Bang KW, Nestel FP, et al. Platelet Toll-like receptor expression modulates lipopolysaccharide-induced thrombocytopenia and tumor necrosis factor-alpha production in vivo. Blood. 2006;107(2):637–41. [DOI] [PubMed] [Google Scholar]
- 117.Lindemann S, Tolley ND, Dixon DA, McIntyre TM, Prescott SM, Zimmerman GA, et al. Activated platelets mediate inflammatory signaling by regulated interleukin 1beta synthesis. J Cell Biol. 2001;154(3):485–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Elzey BD, Tian J, Jensen RJ, Swanson AK, Lees JR, Lentz SR, et al. Platelet-mediated modulation of adaptive immunity. A communication link between innate and adaptive immune compartments. Immunity. 2003;19(1):9–19. [DOI] [PubMed] [Google Scholar]
- 119.Bobbert P, Weikert U, Schmidt-Lucke C, Skurk C, Meyer A, Steffens D, et al. Platelet activation and thrombus formation relates to the presence of myocardial inflammation in patients with cardiomyopathy. J Cardiol. 2014;63(5):379–84. [DOI] [PubMed] [Google Scholar]
- 120.Lu W, Wang Q, Sun X, He H, Wang Q, Wu Y, et al. Qishen Granule Improved Cardiac Remodeling via Balancing M1 and M2 Macrophages. Front Pharmacol. 2019;10:1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.DeLeon-Pennell KY, Iyer RP, Ero OK, Cates CA, Flynn ER, Cannon PL, et al. Periodontal-induced chronic inflammation triggers macrophage secretion of Ccl12 to inhibit fibroblast-mediated cardiac wound healing. JCI Insight. 2017;2(18). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Tecchio C, Micheletti A, Cassatella MA. Neutrophil-derived cytokines: facts beyond expression. Front Immunol. 2014;5:508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Ye J, Huang Y, Que B, Chang C, Liu W, Hu H, et al. Interleukin-12p35 Knock Out Aggravates Doxorubicin-Induced Cardiac Injury and Dysfunction by Aggravating the Inflammatory Response, Oxidative Stress, Apoptosis and Autophagy in Mice. EBioMedicine. 2018;35:29–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Tomelleri A, Cavalli G, De Luca G, Campochiaro C, D’Aliberti T, Tresoldi M, et al. Treating Heart Inflammation With Interleukin-1 Blockade in a Case of Erdheim-Chester Disease. Front Immunol. 2018;9:1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Trankle CR, Canada JM, Kadariya D, Markley R, De Chazal HM, Pinson J, et al. IL-1 Blockade Reduces Inflammation in Pulmonary Arterial Hypertension and Right Ventricular Failure: A Single-Arm, Open-Label, Phase IB/II Pilot Study. Am J Respir Crit Care Med. 2019;199(3):381–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ridker PM, MacFadyen JG, Thuren T, Libby P. Residual inflammatory risk associated with interleukin-18 and interleukin-6 after successful interleukin-1beta inhibition with canakinumab: further rationale for the development of targeted anti-cytokine therapies for the treatment of atherothrombosis. Eur Heart J. 2020;41(23):2153–63. [DOI] [PubMed] [Google Scholar]
- 127.McDaniel MM, Kottyan LC, Singh H, Pasare C. Suppression of Inflammasome Activation by IRF8 and IRF4 in cDCs Is Critical for T Cell Priming. Cell Rep. 2020;31(5):107604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Clancy DM, Sullivan GP, Moran HBT, Henry CM, Reeves EP, McElvaney NG, et al. Extracellular Neutrophil Proteases Are Efficient Regulators of IL-1, IL-33, and IL-36 Cytokine Activity but Poor Effectors of Microbial Killing. Cell Rep. 2018;22(11):2937–50. [DOI] [PubMed] [Google Scholar]
- 129.Peiro C, Lorenzo O, Carraro R, Sanchez-Ferrer CF. IL-1beta Inhibition in Cardiovascular Complications Associated to Diabetes Mellitus. Front Pharmacol. 2017;8:363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Jain A, Kaczanowska S, Davila E. IL-1 Receptor-Associated Kinase Signaling and Its Role in Inflammation, Cancer Progression, and Therapy Resistance. Front Immunol. 2014;5:553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Libby P Interleukin-1 Beta as a Target for Atherosclerosis Therapy: Biological Basis of CANTOS and Beyond. J Am Coll Cardiol. 2017;70(18):2278–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Papanicolaou DA, Wilder RL, Manolagas SC, Chrousos GP. The pathophysiologic roles of interleukin-6 in human disease. Ann Intern Med. 1998;128(2):127–37. [DOI] [PubMed] [Google Scholar]
- 133.Ikonomidis I, Andreotti F, Economou E, Stefanadis C, Toutouzas P, Nihoyannopoulos P. Increased proinflammatory cytokines in patients with chronic stable angina and their reduction by aspirin. Circulation. 1999;100(8):793–8. [DOI] [PubMed] [Google Scholar]
- 134.Liuzzo G, Biasucci LM, Gallimore JR, Caligiuri G, Buffon A, Rebuzzi AG, et al. Enhanced inflammatory response in patients with preinfarction unstable angina. J Am Coll Cardiol. 1999;34(6):1696–703. [DOI] [PubMed] [Google Scholar]
- 135.Wainstein MV, Mossmann M, Araujo GN, Goncalves SC, Gravina GL, Sangalli M, et al. Elevated serum interleukin-6 is predictive of coronary artery disease in intermediate risk overweight patients referred for coronary angiography. Diabetol Metab Syndr. 2017;9:67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Aukrust P, Ueland T, Lien E, Bendtzen K, Muller F, Andreassen AK, et al. Cytokine network in congestive heart failure secondary to ischemic or idiopathic dilated cardiomyopathy. Am J Cardiol. 1999;83(3):376–82. [DOI] [PubMed] [Google Scholar]
- 137.Brocca A, Virzi GM, de Cal M, Giavarina D, Carta M, Ronco C. Elevated Levels of Procalcitonin and Interleukin-6 are Linked with Postoperative Complications in Cardiac Surgery. Scand J Surg. 2017;106(4):318–24. [DOI] [PubMed] [Google Scholar]
- 138.Feaver RCM, Hoang S, Berzin E, Armstrong A, Gardner D, Fisher P, Liu H, Mackey A, Manka D, Wamhoff B. The Anti-IL-6 Antibody Sirukumab Inhibits Vascular Inflammation in a Human Surrogate Model of Atherosclerosis. 2014: In Arthritis & Rheumatology 2014 Oct 1 (Vol. 66, pp. S187–S187). 111 RIVER ST, HOBOKEN 07030–5774, NJ USA: WILEY-BLACKWELL. [Google Scholar]
- 139.Meyer MAS, Wiberg S, Grand J, Kjaergaard J, Hassager C. Interleukin-6 Receptor Antibodies for Modulating the Systemic Inflammatory Response after Out-of-Hospital Cardiac Arrest (IMICA): study protocol for a double-blinded, placebo-controlled, single-center, randomized clinical trial. Trials. 2020;21(1):868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Girndt M, Kaul H, Sester U, Ulrich C, Sester M, Georg T, et al. Anti-inflammatory interleukin-10 genotype protects dialysis patients from cardiovascular events. Kidney Int. 2002;62(3):949–55. [DOI] [PubMed] [Google Scholar]
- 141.Gupta R, Liu L, Zhang X, Fan X, Krishnamurthy P, Verma S, et al. IL-10 provides cardioprotection in diabetic myocardial infarction via upregulation of Heme clearance pathways. JCI Insight. 2020;5(17). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Krishnamurthy P, Thal M, Verma S, Hoxha E, Lambers E, Ramirez V, et al. Interleukin-10 deficiency impairs bone marrow-derived endothelial progenitor cell survival and function in ischemic myocardium. Circ Res. 2011;109(11):1280–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Verma SK, Garikipati VNS, Krishnamurthy P, Schumacher SM, Grisanti LA, Cimini M, et al. Interleukin-10 Inhibits Bone Marrow Fibroblast Progenitor Cell-Mediated Cardiac Fibrosis in Pressure-Overloaded Myocardium. Circulation. 2017;136(10):940–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Burchfield JS, Iwasaki M, Koyanagi M, Urbich C, Rosenthal N, Zeiher AM, et al. Interleukin-10 from transplanted bone marrow mononuclear cells contributes to cardiac protection after myocardial infarction. Circ Res. 2008;103(2):203–11. [DOI] [PubMed] [Google Scholar]
- 145.Krishnamurthy P, Rajasingh J, Lambers E, Qin G, Losordo DW, Kishore R. IL-10 inhibits inflammation and attenuates left ventricular remodeling after myocardial infarction via activation of STAT3 and suppression of HuR. Circ Res. 2009;104(2):e9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Krishnamurthy P, Lambers E, Verma S, Thorne T, Qin G, Losordo DW, et al. Myocardial knockdown of mRNA-stabilizing protein HuR attenuates post-MI inflammatory response and left ventricular dysfunction in IL-10-null mice. FASEB J. 2010;24(7):2484–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Roffe E, Rothfuchs AG, Santiago HC, Marino AP, Ribeiro-Gomes FL, Eckhaus M, et al. IL-10 limits parasite burden and protects against fatal myocarditis in a mouse model of Trypanosoma cruzi infection. J Immunol. 2012;188(2):649–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Garikipati VN, Krishnamurthy P, Verma SK, Khan M, Abramova T, Mackie AR, et al. Negative Regulation of miR-375 by Interleukin-10 Enhances Bone Marrow-Derived Progenitor Cell-Mediated Myocardial Repair and Function After Myocardial Infarction. Stem Cells. 2015;33(12):3519–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Garikipati VNS, Verma SK, Jolardarashi D, Cheng Z, Ibetti J, Cimini M, et al. Therapeutic inhibition of miR-375 attenuates post-myocardial infarction inflammatory response and left ventricular dysfunction via PDK-1-AKT signalling axis. Cardiovasc Res. 2017;113(8):938–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Yue Y, Garikipati VNS, Verma SK, Goukassian DA, Kishore R. Interleukin-10 Deficiency Impairs Reparative Properties of Bone Marrow-Derived Endothelial Progenitor Cell Exosomes. Tissue Eng Part A. 2017;23(21–22):1241–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Meng X, Zheng M, Yu M, Bai W, Zuo L, Bu X, et al. Transplantation of CRISPRa system engineered IL10-overexpressing bone marrow-derived mesenchymal stem cells for the treatment of myocardial infarction in diabetic mice. J Biol Eng. 2019;13:49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Tawara I, Sun Y, Liu C, Toubai T, Nieves E, Evers R, et al. Donor- but not host-derived interleukin-10 contributes to the regulation of experimental graft-versus-host disease. J Leukoc Biol. 2012;91(4):667–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Rowe V, Banovic T, MacDonald KP, Kuns R, Don AL, Morris ES, et al. Host B cells produce IL-10 following TBI and attenuate acute GVHD after allogeneic bone marrow transplantation. Blood. 2006;108(7):2485–92. [DOI] [PubMed] [Google Scholar]
- 154.Levine B, Kalman J, Mayer L, Fillit HM, Packer M. Elevated circulating levels of tumor necrosis factor in severe chronic heart failure. N Engl J Med. 1990;323(4):236–41. [DOI] [PubMed] [Google Scholar]
- 155.Tziakas D, Chalikias G, Parissis JT, Hatzinikolaou H, Stakos D, Papadopoulou E, et al. Prolonged activation of tumor necrosis factor (TNF)-alpha and its soluble receptors in chronic heart failure patients both in the compensated and decompensated state. Interplay between their levels and metalloproteinase-3. Eur Cytokine Netw. 2004;15(3):231–9. [PubMed] [Google Scholar]
- 156.Calabrese F, Carturan E, Chimenti C, Pieroni M, Agostini C, Angelini A, et al. Overexpression of tumor necrosis factor (TNF)alpha and TNFalpha receptor I in human viral myocarditis: clinicopathologic correlations. Mod Pathol. 2004;17(9):1108–18. [DOI] [PubMed] [Google Scholar]
- 157.Sun M, Dawood F, Wen WH, Chen M, Dixon I, Kirshenbaum LA, et al. Excessive tumor necrosis factor activation after infarction contributes to susceptibility of myocardial rupture and left ventricular dysfunction. Circulation. 2004;110(20):3221–8. [DOI] [PubMed] [Google Scholar]
- 158.Awad AE, Kandalam V, Chakrabarti S, Wang X, Penninger JM, Davidge ST, et al. Tumor necrosis factor induces matrix metalloproteinases in cardiomyocytes and cardiofibroblasts differentially via superoxide production in a PI3Kgamma-dependent manner. Am J Physiol Cell Physiol. 2010;298(3):C679–92. [DOI] [PubMed] [Google Scholar]
- 159.Lakshminarayanan V, Beno DW, Costa RH, Roebuck KA. Differential regulation of interleukin-8 and intercellular adhesion molecule-1 by H2O2 and tumor necrosis factor-alpha in endothelial and epithelial cells. J Biol Chem. 1997;272(52):32910–8. [DOI] [PubMed] [Google Scholar]
- 160.Ikonomidis I, Papadavid E, Makavos G, Andreadou I, Varoudi M, Gravanis K, et al. Lowering Interleukin-12 Activity Improves Myocardial and Vascular Function Compared With Tumor Necrosis Factor-a Antagonism or Cyclosporine in Psoriasis. Circ Cardiovasc Imaging. 2017;10(9). [DOI] [PubMed] [Google Scholar]
- 161.Schroecksnadel K, Frick B, Winkler C, Fuchs D. Crucial role of interferon-gamma and stimulated macrophages in cardiovascular disease. Curr Vasc Pharmacol. 2006;4(3):205–13. [DOI] [PubMed] [Google Scholar]
- 162.Ito S, Ansari P, Sakatsume M, Dickensheets H, Vazquez N, Donnelly RP, et al. Interleukin-10 inhibits expression of both interferon alpha- and interferon gamma- induced genes by suppressing tyrosine phosphorylation of STAT1. Blood. 1999;93(5):1456–63. [PubMed] [Google Scholar]
- 163.Lee SH, Kwon JY, Kim SY, Jung K, Cho ML. Interferon-gamma regulates inflammatory cell death by targeting necroptosis in experimental autoimmune arthritis. Sci Rep. 2017;7(1):10133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.da Silva MV, de Almeida VL, de Oliveira WD, Matos Cascudo NC, de Oliveira PG, da Silva CA, et al. Upregulation of Cardiac IL-10 and Downregulation of IFN-gamma in Balb/c IL-4(−/−) in Acute Chagasic Myocarditis due to Colombian Strain of Trypanosoma cruzi. Mediators Inflamm. 2018;2018:3421897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Ranjbaran H, Sokol SI, Gallo A, Eid RE, Iakimov AO, D’Alessio A, et al. An inflammatory pathway of IFN-gamma production in coronary atherosclerosis. J Immunol. 2007;178(1):592–604. [DOI] [PubMed] [Google Scholar]
- 166.Stawowczyk M, Van Scoy S, Kumar KP, Reich NC. The interferon stimulated gene 54 promotes apoptosis. J Biol Chem. 2011;286(9):7257–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Guinn Z, Brown DM, Petro TM. Activation of IRF3 contributes to IFN-gamma and ISG54 expression during the immune responses to B16F10 tumor growth. Int Immunopharmacol. 2017;50:121–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Goldhammer E, Tanchilevitch A, Maor I, Beniamini Y, Rosenschein U, Sagiv M. Exercise training modulates cytokines activity in coronary heart disease patients. Int J Cardiol. 2005;100(1):93–9. [DOI] [PubMed] [Google Scholar]
- 169.Eriksson U, Kurrer MO, Sebald W, Brombacher F, Kopf M. Dual role of the IL-12/IFN-gamma axis in the development of autoimmune myocarditis: induction by IL-12 and protection by IFN-gamma. J Immunol. 2001;167(9):5464–9. [DOI] [PubMed] [Google Scholar]
- 170.Rajasingh J, Bord E, Luedemann C, Asai J, Hamada H, Thorne T, et al. IL-10-induced TNF-alpha mRNA destabilization is mediated via IL-10 suppression of p38 MAP kinase activation and inhibition of HuR expression. FASEB J. 2006;20(12):2112–4. [DOI] [PubMed] [Google Scholar]
- 171.Yamaoka M, Yamaguchi S, Okuyama M, Tomoike H. Anti-inflammatory cytokine profile in human heart failure: behavior of interleukin-10 in association with tumor necrosis factor-alpha. Jpn Circ J. 1999;63(12):951–6. [DOI] [PubMed] [Google Scholar]
- 172.Penitente AR, Leite AL, de Paula Costa G, Shrestha D, Horta AL, Natali AJ, et al. Enalapril in Combination with Benznidazole Reduces Cardiac Inflammation and Creatine Kinases in Mice Chronically Infected with Trypanosoma cruzi. Am J Trop Med Hyg. 2015;93(5):976–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Melo L, Caldas IS, Azevedo MA, Goncalves KR, da Silva do Nascimento AF, Figueiredo VP, et al. Low doses of simvastatin therapy ameliorate cardiac inflammatory remodeling in Trypanosoma cruzi-infected dogs. Am J Trop Med Hyg. 2011;84(2):325–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Al-Darraji A, Haydar D, Chelvarajan L, Tripathi H, Levitan B, Gao E, et al. Azithromycin therapy reduces cardiac inflammation and mitigates adverse cardiac remodeling after myocardial infarction: Potential therapeutic targets in ischemic heart disease. PLoS One. 2018;13(7):e0200474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Iwasaki A, Matsumori A, Yamada T, Shioi T, Wang W, Ono K, et al. Pimobendan inhibits the production of proinflammatory cytokines and gene expression of inducible nitric oxide synthase in a murine model of viral myocarditis. J Am Coll Cardiol. 1999;33(5):1400–7. [DOI] [PubMed] [Google Scholar]
- 176.Matsumori A, Ono K, Sato Y, Shioi T, Nose Y, Sasayama S. Differential modulation of cytokine production by drugs: implications for therapy in heart failure. J Mol Cell Cardiol. 1996;28(12):2491–9. [DOI] [PubMed] [Google Scholar]
- 177.Held C, White HD, Stewart RAH, Budaj A, Cannon CP, Hochman JS, et al. Inflammatory Biomarkers Interleukin-6 and C-Reactive Protein and Outcomes in Stable Coronary Heart Disease: Experiences From the STABILITY (Stabilization of Atherosclerotic Plaque by Initiation of Darapladib Therapy) Trial. J Am Heart Assoc. 2017;6(10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Bro-Jeppesen J, Kjaergaard J, Wanscher M, Nielsen N, Friberg H, Bjerre M, et al. Systemic Inflammatory Response and Potential Prognostic Implications After Out-of-Hospital Cardiac Arrest: A Substudy of the Target Temperature Management Trial. Crit Care Med. 2015;43(6):1223–32. [DOI] [PubMed] [Google Scholar]
- 179.Nidorf SM, Eikelboom JW, Budgeon CA, Thompson PL. Low-dose colchicine for secondary prevention of cardiovascular disease. J Am Coll Cardiol. 2013;61(4):404–10. [DOI] [PubMed] [Google Scholar]
- 180.Martinez GJ, Robertson S, Barraclough J, Xia Q, Mallat Z, Bursill C, et al. Colchicine Acutely Suppresses Local Cardiac Production of Inflammatory Cytokines in Patients With an Acute Coronary Syndrome. J Am Heart Assoc. 2015;4(8):e002128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Tardif JC, Tanguay JF, Wright SR, Duchatelle V, Petroni T, Gregoire JC, et al. Effects of the P-selectin antagonist inclacumab on myocardial damage after percutaneous coronary intervention for non-ST-segment elevation myocardial infarction: results of the SELECT-ACS trial. J Am Coll Cardiol. 2013;61(20):2048–55. [DOI] [PubMed] [Google Scholar]
- 182.Stahli BE, Gebhard C, Duchatelle V, Cournoyer D, Petroni T, Tanguay JF, et al. Effects of the P-Selectin Antagonist Inclacumab on Myocardial Damage After Percutaneous Coronary Intervention According to Timing of Infusion: Insights From the SELECT-ACS Trial. J Am Heart Assoc. 2016;5(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.O’Donoghue ML, Braunwald E, White HD, Lukas MA, Tarka E, Steg PG, et al. Effect of darapladib on major coronary events after an acute coronary syndrome: the SOLID-TIMI 52 randomized clinical trial. JAMA. 2014;312(10):1006–15. [DOI] [PubMed] [Google Scholar]
- 184.Investigators S, White HD, Held C, Stewart R, Tarka E, Brown R, et al. Darapladib for preventing ischemic events in stable coronary heart disease. N Engl J Med. 2014;370(18):1702–11. [DOI] [PubMed] [Google Scholar]
- 185.Nicholls SJ, Kastelein JJ, Schwartz GG, Bash D, Rosenson RS, Cavender MA, et al. Varespladib and cardiovascular events in patients with an acute coronary syndrome: the VISTA-16 randomized clinical trial. JAMA. 2014;311(3):252–62. [DOI] [PubMed] [Google Scholar]
- 186.Ridker PM, Everett BM, Pradhan A, MacFadyen JG, Solomon DH, Zaharris E, et al. Low-Dose Methotrexate for the Prevention of Atherosclerotic Events. N Engl J Med. 2019;380(8):752–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Tardif JC, McMurray JJ, Klug E, Small R, Schumi J, Choi J, et al. Effects of succinobucol (AGI-1067) after an acute coronary syndrome: a randomised, double-blind, placebo-controlled trial. Lancet. 2008;371(9626):1761–8. [DOI] [PubMed] [Google Scholar]