

Abstract
Cowpea mosaic virus (CPMV), a non-enveloped plant virus, and empty CPMV (eCPMV), a virus-like particle (VLP) composed of CPMV capsid without nucleic acids, are potent in situ cancer vaccines when administered intratumorally (I.T.). However, it is unclear how immune cells recognize these nanoparticles and why they are immunogenic, which was investigated in this study. CPMV generated stronger selective induction of cytokines and chemokines in naïve mouse splenocytes and exhibited more potent anti-tumor efficacy than eCPMV. MyD88 is required for both CPMV- and eCPMV-elicited immune responses. Screening with human embryonic kidney (HEK)-293 cell toll-like receptor (TLR) reporter assays along with experiments in corresponding TLR−/− mice indicated CPMV and eCPMV capsids are recognized by MyD88-dependent TLR2 and TLR4. CPMV but not eCPMV is additionally recognized by TLR7. Secretion of type I interferons (IFNs), which requires the interaction between TLR7 and encapsulated single-stranded RNAs (ssRNAs), is critical to CPMV’s better efficacy. The same recognition mechanisms are also functional in human peripheral blood mononuclear cells (PBMCs). Overall, these findings link CPMV immunotherapy efficacy with molecular recognition, provide rationale for how to develop more potent viral particles, accentuate the value of multi-TLR agonists as in situ cancer vaccines, and highlight the functional importance of type I IFNs for in situ vaccination.
Keywords: cowpea mosaic virus, nanoparticle, virus-like nanoparticle, in situ vaccination, cancer immunotherapy, toll-like receptors
Graphic abstract
1. Introduction
Cancers are recognized by the patient’s immune system, but aggressive tumors have immunosuppressive tumor microenvironments (TMEs) that block anti-tumor immune responses. Cancer immunotherapies must overcome this immunosuppression to be effective [1, 2]. Disrupting tumor-mediated immunosuppression is important for improving responses to cancer immunotherapies, and there is intensive research in how to disrupt the immunosuppression and enable antitumor immunity to reduce and eliminate cancer. Immune checkpoint blockades (ICBs) targeting CTLA-4 and PD-1, or PDL-1 are the best-established strategies for disrupting the tumor-mediated immunosuppression and are now widely used [3, 4]. However, they often do not get a response or responses are transient in many patients, so further improved strategies are needed [5].
In situ vaccination (ISV) is a form of cancer immunotherapy specifically designed to reverse the immunosuppression in the TME and generate an immunostimulatory environment in directly treated tumors. This creates effective local innate anti-tumor immunity which in turn can generate strong systemic immunity (adaptive response) to eliminate or reduce untreated metastatic tumors. Unlike standard vaccines which carry antigens as well as adjuvant, ISV brings only the adjuvant to the tumor, and the tumor itself serves as the source of antigens. This avoids the complex challenge of identifying potential antigens and predicting which will be best presented on class I or II HLA of each specific patient by antigen presenting cells [6–8]. While there are many potential mechanisms of various ISV approaches, often it starts with the recognition by and stimulation of innate immune cells, like antigen presenting cells (APCs) in the tumor with associated cytokine secretion, leading to changes in the type, number and activation status of tumor-infiltrating lymphocytes (TILs), resulting in an effective anti-tumor immune response [9]. ISV often induces immunogenic cell death (ICD), which further stimulates innate immune cells, and releases tumor antigens to support an anti-tumor immune response [10].
Non-infectious, genetically and physically stable plant virus-based nanoparticles (PVNP) have potential as adjuvants in vaccines and immunotherapy due to their immunogenic properties [11]. The proteinaceous and multivalent capsid structures together with the nucelic acid cargos render PVNPs highly visible to the immune system; their nanoparticular size enables efficient trafficking throug the lymph and uptale by phagocytes of the innate immune system, including antigen presenting cells [11–13]. Cowpea mosaic virus (CPMV) is particularly immunostimulatory and effective for in situ vaccination to treat cancer. Previously, we have shown that in situ vaccination by intratumoral (I.T.) injection of either CPMV or eCPMV triggered potent anti-tumor immunity in both murine cancer models and dog patients with spontaneous cancer [14–18]. CPMV, which does not infect animals, is a plant picornavirus with two single-stranded + sense RNAs that are encapsidated seperately into identical icosahedral capsids measuring 28 nm in diameter; each capsid is made of 60 copies each of a large (42-kDa) and small (24-kDa) protein subunit. Empty CPMV (eCPMV) is a virus-like particle (VLP) with the identical capsid but without any RNA [19]. Published studies showed that CPMV and eCPMV can mediate potent antitumor efficacy when used as in situ vaccines in mouse cancer models and companion dogs with spontaneous tumors [15]. While the immune stimulation properties of (e)CPMV are clear, the mechanisms by which these viral particles are recognized by immune cells are poorly understood.
While it is established that viral RNAs as pathogen-associated molecular patterns (PAMPs) can be detected by various pattern recognition receptors (PRRs), such as TLRs [20–22], RIG-I-like receptors (RLRs) [23,24] and the cGAS-Sting pathway [25], the specific receptors recognizing most viruses, particularly the capsids, have not been adequately investigated. TLR2 has recently been suggested to recognize the repeating structure of some viral capsids [26, 27]. TLR4 is also able to recognize proteins in viruses, such as respiratory syncytial virus and mouse mammary tumor virus [28]. . In addition, viral nucleic acids are recognized by TLRs 3, 7, 8, and 9 to simulate IFN signaling [22, 29], which has marked immunostimulatory effects and been studied in anticancer immunotherapies. The examples include papaya mosaic virus and CpG Oligonucleotide-loaded PVNPs [30–33]. With the unique structure and composition of CPMV, we hypothesized that, although not infectious in mammals, CPMV is recognized through one or more known pathogen pattern recognition pathways expressed by mammals. To investigate this, we utilized TLR reporter cells, genetically modified mice and human peripheral blood mononuclear cells (PBMCs) to identify the recognition pathways and associated responses that mediate immune response to CPMV and eCPMV.
2. Materials and methods
2.1. Preparation of wild-type CPMV and eCPMV
CPMV and eCPMV nanoparticles were produced as previously described [16]. Levels of endotoxin were assayed by the Endozyme II Recombinant Factor C Endotoxin Detection Assay (Biovendor). A level of <50 endotoxin units (EU/mg protein) in a preparation was considered negligible.
2.2. Endotoxin removal and heat precipitation
If endotoxin levels in particle preparations exceeded 50 EU/mg, an endotoxin removal was performed by treating particles three times with 1% (v/v) Triton X-14, samples were incubated on a rolling platform at 4ºC for 20 min, then at 37ºC for 10 min before centrifuging at 20,000 x g for 10 min at room temperature. The clear layer of the phase separation was collected and transferred to a new tube. After the last step, the samples were centrifuged at 150,000 x g for 1 h, at 4ºC. The supernatants were discarded, and the pellets resuspended in 0.1 M potassium phosphate buffer (pH 7.2). We heated 10 μg of CPMV or eCPMV particles with or without LPS for 10 min at 90ºC followed by brief centrifugation, and 2 μg aliquots were fractionated by agarose gel electrophoresis in TBE containing GelRed nucleic acid stain. The gels were subsequently stained with Coomassie Brilliant Blue. To test for endotoxins, 10 μg of CPMV or eCPMV with and without LPS were diluted 1:10 in endotoxin-free water and assessed using the Endozyme II Recombinant Factor C Endotoxin Detection Assay (Biovendor) according to the manufacturer’s instructions.
2.3. Animals and in situ vaccination
Female C57BL/6J mice (6–8 weeks old) were purchased from The Jackson Laboratory. MyD88−/−, TRIF−/−, TLR2−/−, TLR4−/−, TLR5−/−, TLR7−/−, and TLR9−/− were originally purchased from The Jackson Laboratory and genotyped and bred at Dartmouth College. TLR2/4 double-gene knockout (DKO) mice were generated by backcross of TLR2 and TLR4 single KO mice and genotyped by PCR. Resiquimod (R848) was purchased from Invivogen (San Diego, CA, USA). All mouse studies were approved by the Institutional Animal Care and Use Committee of Dartmouth College. We implanted 1.25×105 live cells/30 μL PBS orthotopically into mice skin by intradermal injection and in situ vaccination was done by intratumorally injecting the noted mass of reagents in 30uL PBS. Tumor growth was monitored every other day by caliper. Weight was measured every other day. Mice were euthanized when the tumor size reached 200 mm2 or were observed till 30 days after treatment. Tumor size was scored by the simple formula of length x width to assess surface area, rather than the more common approach of extrapolating tumor volume from length and width measurements, since we have found that for dermal tumors the length x width measurement enables very reproducible measurement. A tumor of 200mm2 has an approximate volume of 1500 mm3.
2.4. Cell culture, cytokine quantification and Luminex assay
Supernatants from coculture of CPMV, eCPMV with mouse splenocytes or human PBMCs were tested by enzyme-linked immunosorbent assay (ELISA) to detect interleukin-6 (IL-6), interferon α (IFN-α), or interferon β (IFN-β) (BioLegend). Human PBMCs from 3 deidentified healthy donors were purchased from Dartmouth core facility. Mouse/human Luminex assays (Invitrogen, Life Technologies, Carlsbad, CA, USA) were assayed by Luminex MagPix® plate reader (Luminex Corp., Austin, TX) according to the manufacturer’s instructions. For ex vivo stimulation, 5×105 cells were stimulated in 200 μL with 12 μg/mL CPMV or eCPMV for 24 h in 96-well plates, and the supernatant was collected and stored frozen (−20oC) for cytokine quantification. These cells were cultured in RPMI1640 (Invitrogen, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum and antibiotics if not otherwise mentioned. All cell cultures were maintained in a 5% CO2 incubator at 37ºC.
2.5. TLR reporter cell culture and QuantiBlue assay
Human embryonic kidney-293 (HEK-293)-Blue Null-v and HEK-Blue TLR cells were purchased from InvivoGen (San Diego, CA) and cultured as instructed. In general, in a 96-well plate (200 μL total volume) containing the appropriate cells (50,000–75,000 cells/well), 20 μL of the test article or the positive control ligand or 12 ug/mL (e)CPMV (final concentration) is added to the wells. The media added to the wells is designed for the detection of NF-κB induced SEAP expression. After a 16–24hr incubation the optical density (OD) is read at 650nm on a Molecular Devices SpectraMax 340PC absorbance detector.
2.6. Statistics
Data are presented as the average means +/− standard error of the means (SEM). The results shown here are representative responses from multiple experiments which are repeated at least once with similar results. T-test p-values <0.05 were required to assign significance and are represented as an asterisk (*) in the figures and p <0.01 as **, and p <0.001 as ***. Flank tumor growth curves were analyzed using two-way ANOVA, with p>0.05 as ns, p <0.05 as *, p <0.01 as **, and p <0.001 as ***.
3. Results
3.1. MyD88, but not TRIF, is required for CPMV and eCPMV stimulation of cytokines and chemokines in vitro and anti-tumor immunity in vivo
To investigate this hypothesis, we first established an assay to measure the recognition of CPMV or eCPMV by primary immune cells. Splenocytes from C57BL/6J mice were cultured with CPMV and eCPMV for 24 hours and supernatants were analyzed for cytokines and chemokines by 32-panel Luminex and ELISAs (Fig 1A). 14 of the 32 luminex cytokines and chemokines were selectively increased 2-fold or more by both CPMV and eCPMV as compared to unstimulated splenocytes. (Fig. 1B). The majority of the upregulated cyto/chemokines are canonically pro-inflammatory. For example, tumor necrosis factor (TNF)-α, interleukin (IL)-6, MIP-1, and CXCL10, which stimulate the immune response and the recruitment of inflammatory cells [34–37], are upregulated by CPMV and eCPMV. IFN-γ, a key cytokine released by T cells to induce a Th1-type immune response, is also induced by both particles, although more by CPMV [38, 39]. These cyto/chemokines were generated in roughly equal amounts by both CPMV and eCPMV, with the exceptions of IFNγ, CXCL9 and MCP-1, which had at least 2-fold higher levels induced by CPMV than eCPMV. Uniquely, type I IFNs (IFNα and IFNβ were assayed interchangeably) are upregulated by CPMV but not eCPMV (Fig. 1B, S. Fig 1). This demonstrates that CPMV and eCPMV stimulate mouse splenocytes to release high levels of specific cyto/chemokines. Both CPMV and eCPMV stimulate similar but not identical patterns of cyto/chemokines, with CPMV stimulating higher levels of some cytokines and uniquely inducing type I IFNs.
Fig 1. MyD88 is required for CPMV and eCPMV-elicited upregulation of cytokines and chemokines in vitro and anti-tumor immunity in vivo.


A. Schematic of splenocyte assay design. Splenocytes were cultured for 24hr with (e)CPMV and cytokine levels were analyzed. B. Induction of cytokines and chemokines by CPMV and eCPMV did not occur in MyD88−/− mouse splenocytes and IFNβ was only induced by CPMV (n=3). C. Schematic of in situ vaccination strategy. 1.25×105 of B16F10 melanoma cells were injected intradermally on day −7. 100 ug of CPMV or eCPMV was injected intratumorally on D0 and D7. Tumor growth was monitored every other day till 30 days after first treatment and euthanized or when the tumor size reached 200mm2. D-F. MyD88, but not TRIF, is required for CPMV and eCPMV-elicited anti-tumor immunity in vivo (n=5). Data for bar graphs calculated using unpaired Student’s t-test. Tumor progression was monitored by measuring tumor surface area (length x width). Growth curves were analyzed using two-way ANOVA. survival curves were analyzed using log-rank (Mantel-Cox) test, with p>0.05 as ns, p <0.05 as *, p <0.01 as **, and p <0.001 as ***. All experiments are repeated at least once with similar results.
TLR signaling is mediated through two adaptor proteins, MyD88 and TRIF [22]. Thus, we assayed splenocytes from MyD88 knockout (KO) and TRIF KO mice to determine whether the recognition of CPMV and eCPMV require MyD88 and/or TRIF. Induced expression of cytokines and chemokines caused by CPMV and eCPMV did not occur in MyD88 KO splenocytes, demonstrating that all recognition of both reagents is mediated by MyD88 (Fig. 1B). This conclusion was supported by normal cyto/chemokine induction in TRIF KO splenocytes showing that TRIF is not mediating CPMV or eCPMV cytokine stimulation (S. Fig. 1).
To test the importance of MyD88 and/or TRIF for in situ vaccination response to CPMV and eCPMV, B16F10 dermal tumors were established and treated with 2 weekly injections of either of these reagents as previously established for treatment efficacy (Fig. 1C) [9]. CPMV and eCPMV both elicited strong anti-tumor response with a stronger anti-tumor efficacy by CPMV. (Fig. 1D, S. Fig 2). No significant weight loss or observable side effect was observed for either treatment (S. Fig. 3). However, the treatment had no efficacy in MyD88 KO mice but when tested in TRIF KO mice, efficacy was similar to the treatment response of wild type C57BL6 mice (Figs. 1D–F). Taken together, these results confirmed that CPMV and eCPMV require MyD88 to be recognized both in vitro and in vivo, and in situ vaccination efficacy requires MyD88 but not TRIF.
3.2. CPMV and eCPMV are both recognized by TLRs 2 and 4, but only CPMV is recognized by TLR 7.
To determine the specific TLRs recognizing CPMV and eCPMV, we used transgenic HEK-Blue™ TLR reporter cell lines, which stably express individual mouse/human TLR on the cell surface and relevant genes from the TLR signaling pathway. TLR stimulation generates an NF-κB-inducible secreted embryonic alkaline phosphatase (SEAP) reporter gene which is detectable by colorimetric assay. CPMV and eCPMV were incubated with these transgenic cell lines and, 12 hours later, SEAP activity was assayed. Data show that both CPMV and eCPMV are potent activators of TLR2 and TLR4, and CPMV, but not eCPMV, activates TLR7 in both mouse and human TLR reporter assays (Figs. 2A–B). TLRs other than 2, 4 and 7 do not recognize CPMV or eCPMV (S. Figs 2A–B). Negative results of TLR3 reporter assay are consistent with the findings that both particles require MyD88 but not TRIF, as TLR3 only signals through TRIF. This reporter gene assay suggested that both TLRs 2 and 4 can recognize CPMV and eCPMV; and only TLR7 can recognize CPMV.
Fig 2. In vitro, both CPMV and eCPMV are recognized by TLR2 and TLR4; however, CPMV is recognized only by TLR 7 to induce type I IFNs.

A, B. Screened by hTLR and mTLR HEK-Blue reporter cells (n=3), both CPMV and eCPMV showed induction of TLR2 and TLR4 transactivation; however, only CPMV shows induction of TLR7 transactivation. C, D. Knockout of either TLR2, TLR4 or TLR7 in mouse splenocytes (n=3) was reduced but did not completely eliminate induction of IL-6. E. Type I IFNs are only induced by CPMV but not eCPMV in a TLR7-dependent manner in mouse splenocyte assays (n=3). F, G. Knockout of both TLR2 and TLR4 in mouse splenocytes eliminated IL-6 induced by eCPMV and attenuated IL-6 induction but not IFNβ induction by CPMV (n=3). Data for bar graphs calculated using unpaired Student’s t-test with p>0.05 as ns, p <0.05 as *, p <0.01 as **, and p <0.001 as ***. All experiments are repeated at least once with similar results.
To investigate individual involvement of TLRs 2, 4, 7 in recognition of CPMV/eCPMV, splenocyte assays from TLR2, 4, and 7 KO mice were performed. Utilizing the assay introduced in Fig. 1, supernatant was analyzed by ELISA for IL-6, (as a representative inflammatory cytokine), and IFNβ since it is differentially expressed between CPMV and eCPMV. IL-6 generation by CPMV exposure was still induced but significantly attenuated in TLR2, 4, and 7 KO mouse splenocytes (Fig 2C). Similarly, induction of IL-6 by eCPMV was attenuated in TLR2 and TLR4 KO mouse splenocytes but not in TLR7 KO splenocytes, confirming that TLR7 is not stimulated by eCPMV (Fig. 2D). To verify that the HEK reporter assays accurately reflected insensitivity of other TLRs to CPMV, we tested mouse splenocytes from TLR5, and 9 KO mice. Loss of MyD88-dependent TLRs 5 or 9 did not significantly attenuate the induction of IL-6 by CPMV or eCPMV (S. Figs 3A–B). As hypothesized, type I IFNs are only induced by ssRNA-containing CPMV in a TLR7 dependent manner (Fig. 2E). Noticeably, knockout of either TLR2 or TLR4 did reduce IL-6 induction but did not completely abolish it. While TLR2 and TLR4 act in an additive manner to induce inflammatory cytokines, neither is individually required for inducing these cytokines.
We have shown that TLRs 2 and 4 both recognize the CPMV/eCPMV capsid. While deletion of either TLR2 or 4 reduced IL-6 induction, the splenocytes with these mutations still responded strongly to CPMV and eCPMV. To directly determine whether the capsid recognition is limited to TLRs 2 and 4, we generated and tested mice with homozygous deletion of both TLRs 2 and 4. Knockout of both these TLRs completely abolished the induction of IL-6 expression by eCPMV, but a small production of IL-6 and normal production of IFNβ occurred when cells were stimulated by CPMV (Figs. 2F–G). Taken together, these results confirmed that CPMV and eCPMV capsids are recognized by TLRs 2 and 4 and the ssRNAs in CPMV are recognized by TLR7 which is required to induce type I IFNs in vitro. TLR7 is fully responsible for type I IFN and contributes weakly to IL-6 production since TLR2/4 double KO (DKO) mice still have increased IL-6 compared to unstimulated cells when exposed to CPMV.
3.3. Neither TLR2 nor TLR4 are required for full in vivo antitumor immunity elicited by CPMV and eCPMV.
To determine the importance of each of TLR 2, 4, or 7 and the cyto/chemokines they induce for treating tumors by in situ vaccination, we conducted efficacy studies in different TLR KO mice using intradermal B16F10 melanoma I.T. injected with CPMV, eCPMV, or PBS. In situ vaccination with CPMV consistently slows tumor growth more than eCPMV demonstrating the importance of the viral RNA (Fig. 3A and 1D); this data is consistent with our previous work [16]. Efficacy of CPMV and eCPMV in TLR2 KO mice and TLR4 KO mice was not distinguishable (Figs. 3A–B) from the same reagent treating tumors in wild type mice (Fig. 1D). DKO of TLRs 2 and 4 completely abolished the efficacy of eCPMV, but CPMV retained weaker but recognizable ability to slow tumor growth (Fig. 3C). This confirmed our previous observation that neither TLR2 or TLR4 are required for cytokine generation in splenocytes and suggested that the likely reduced cytokine levels in TLR2 or TLR4 KO mice were still sufficient to support the full treatment effects.
Fig 3. TLR2 and TLR4 are complementary to each other for the antitumor efficacy by CPMV and eCPMV in vivo; ssRNAs in CPMV induce type I IFNs by TLR7 which improves efficacy.


ISV strategy to treat B16F10 intradermal tumors as shown in Fig. 1c. A, B. CPMV and eCPMV remained normally effective in TLR2 KO or TLR4 KO mice (n=5). C. Knockout of both TLR2 and TLR4 attenuated antitumor efficacy of CPMV and eliminated the antitumor efficacy of eCPMV. D, E. CPMV and eCPMV have equal antitumor efficacy in TLR7 KO and in IFNαR KO mice (n=5). Tumor progression was monitored by measuring tumor area (length x width). Tumor progression was monitored by measuring tumor surface area (length x width). Growth curves were analyzed using two-way ANOVA. survival curves were analyzed using log-rank (Mantel-Cox) test, p <0.05 as *, p <0.01 as **, and p <0.001 as ***. All experiments are repeated at least once with similar results.
CPMV demonstrated superior efficacy In treating B16F10 tumors compared to eCPMV (Fig 1. D); however, CPMV had lost this superior efficacy in TLR7 KO (Fig. 3D). This shows that elimination of only TLR7 reduces the efficacy of the response using CPMV, unlike individual elimination of either TLR2 or TRL4. As expected, in TLR5 KO mice or TLR9 KO mice, CPMV and eCPMV each had normal efficacy for treating B16F10 (S. Fig. 4).
3.4. ssRNAs in CPMV induce type I IFNs by TLR7 which improves efficacy
Despite clear efficacy of ISV using eCPMV, complete CPMV has greater ability to reduce or eliminate tumor growth (Fig. 1D). As noted, the difference of recognition between these reagents is viral ssRNA found in CPMV but not eCPMV. As shown above, TLR7 mediates recognition of the viral ssRNA and without TLR7, IL-6 induction is reduced, but type I IFN is not induced at all. When these findings are evaluated, it suggests that the weaker efficacy of eCPMV is due to lack of type I IFN production.
This central role of type I IFNs in ISV with CPMV is supported by finding that CPMV and eCPMV have equal treatment efficacy in TLR7 KO mice (Fig. 3D). Since we know that ssRNAs in CPMV is recognized by TLR7, this shows that TLR7 is uniquely contributing to treatment efficacy. The unique generation of type I IFNs by TLR7 stimulation suggested that the reduced response of CPMV in TLR7 mice was due to lack of type I IFNs. To test that hypothesis directly, efficacy of CPMV in situ vaccination was tested in IFNα receptor KO mice (IFNαR−/−) whose cells cannot respond to either IFNα or β. If the reduction seen by loss of TLR7 was solely due to lack of type I IFN generation, then anti-tumor efficacy of CPMV which stimulates TLR7 should be identical to the reduced efficacy of eCPMV. This identical response to CPMV and eCPMV is what is observed (Fig. 3E). This result highlights the value of type I IFN generation for ISV. This also demonstrates that in this situation the tumor cells, which have intact IFNα receptor are not mediating ISV effects through their own response to type I IFN.
Overall, these observations confirm that CPMV and eCPMV stimulate antitumor immunity through recognition by TLRs 2 and 4 and elimination of either TLR2 or 4 does not limit treatment efficacy, however elimination of both does. Additionally, ssRNAs in CPMV stimulate TLR7 and cause secretion of type I IFNs, which plays a unique and important role in generating antitumor efficacy by in situ vaccination using CPMV.
3.5. Human PBMCs recognize CPMV and eCPMV by human TLR2 and 4; CPMV by human TLR7.
Whether and with what receptors human leukocytes may respond to CPMV is still not fully understood. Previous work also showed that CPMV can activate purified human monocytes, which responded with elevated expression of HLA-DR, and CD86 [27]. Figure 2A shows that specific human TLRs recognize CPMV and eCPMV in HEK-blue reporter cells. Overall, this demonstrates that immune recognition of CPMV appears similar for mice, and humans, and likely for all mammalian species. To further determine how human cells recognize and respond to CPMV, what cytokines PBMCs generate in response, and whether in situ vaccination using CPMV could have similar efficacy treating human cancer, we investigated recognition and cytokine generation of CPMV and eCPMV by human PBMCs. CPMV or eCPMV were incubated with human PBMCs from different donors for 24 hours and cytokine/chemokine generation was assayed with human Luminex. We confirmed that CPMV and eCPMV were also immunogenic on human PBMCs and selectively increased cytokines and chemokines in a pattern similar to mouse splenocytes (Fig. 4A). CPMV induces type I IFNs by human PBMCs whereas eCPMV does not, which is identical to type I IFN induction in mouse splenocytes. Interestingly, in a pattern that is similar but not identical to mouse splenocytes, while CPMV and eCPMV all significantly increased expression of the same cytokines other than type I IFN, some were less strongly induced by eCPMV (Fig. 4A).
Fig 4. Human PBMC recognized and responded to CPMV and eCPMV in a similar manner as mouse splenocytes.

A. Selective upregulation of cytokines and chemokines by CPMV and eCPMV in human PBMCs compared to control, and IFNα was only induced by CPMV (n=3). B, C. Only blocking both TLR2 and TLR4 by antibodies could eliminate IL-6 induced by eCPMV/ attenuated IL-6 induced but not IFNβ by CPMV in human PBMC assay (data is the average of results from n=3 different PBMC donors). Data for bar graphs calculated using unpaired Student’s t-test with p>0.05 as ns, p <0.05 as *, p <0.01 as **, and p <0.001 as ***. All experiments are repeated at least once with similar results.
To determine whether human PBMCs recognize CPMV and eCPMV with the same TLRs as mouse splenocytes, we incubated human PBMCs with anti-hTLR2 blocking antibody and/or anti-hTLR4 blocking antibody 30 minutes prior to culturing with CPMV or eCPMV. Similar to findings using KO mice in Fig. 2, the induced expression of IL-6 by eCPMV were abolished only when both anti-hTLR2 and anti-hTLR4 blocking antibodies were applied simultaneously (Fig. 4B). However, intact CPMV is still capable of inducing cytokines and chemokines even if human PBMC is blocked by anti-TLR2 and anti-TLR4 antibodies (Fig. 4C). Although there is no hTLR7 specific blocking antibody available, these results still confirmed that human PBMCs recognize and respond to eCPMV similarly to mouse splenocytes; and infer that RNA carried by CPMV would be similarly recognized by TLR7 and generates type I IFNs. Together, this supports potential human clinical relevancy and highlights the potential to treat human tumors with in situ vaccination with CPMV as well as eCPMV.
3.6. Addition of R848 to eCPMV improved antitumor effect.
CPMV is an agonist to TLRs 2, 4, and 7 and has better efficacy in B16F10 than eCPMV, which suggests value in treating tumors with multiple TLR agonists concurrently as an in situ vaccination strategy, especially in order to stimulate type I IFNs. Previous study showed that i.t. injection of R848, a TLR7 agonist, had excellent efficacy in CT26 colon cancer mouse model [40]. We next investigated whether I.T. injection of R848 with eCPMV can generate a synergistic effect that matched CPMV efficacy since CPMV has a superior efficacy contributed by additional TLR7 recognition and stimulation. eCPMV and R848 were injected in B16F10 tumors concurrently and the combination did show improved efficacy compared to single agent treatment (Fig. 5A). This further confirms the importance of including TLR7 stimulation for tumor ISV, especially the type I interferons induced by TLR7, for ISV (Fig. 5B, C). The combination was more effective than mono-treatment with eCPMV using a relatively high dose of R848 that had no efficacy alone. However, the combination is still less effective than CPMV, further highlighting the potential of CPMV for single agent in situ vaccination.
Fig. 5. Improved efficacy is observed in the combination of eCPMV and R848.

A. Schematic of in situ vaccination strategy adapted from Fig 1c. 1.25×105 of B16F10 melanoma cells were injected intradermally on day −7. 100 ug of CPMV, eCPMV, or eCPMV + 40 ug of R848 was injected intratumorally on D0 and D7. Tumor growth was monitored every other day. B, C. ISV treatment with combination of eCPMV (100 ug) and R848 (40 ug) showed improved efficacy but is still inferior to CPMV (100 ug) in B16F10-tumor bearing mice (n=5). Tumor progression was monitored by measuring tumor surface area (length x width). Growth curves were analyzed using two-way ANOVA. survival curves were analyzed using log-rank (Mantel-Cox) test, p <0.05 as *, p <0.01 as **, and p <0.001 as ***. All experiments are repeated at least once with similar results.
4. Discussion
These findings demonstrate that CPMV and eCPMV are immunostimulatory because they are recognized by and stimulate TLRs 2, 4, and 7 (summarized in Fig. 6). We further show that type I IFN is particularly important for achieving optimal ISV results. CPMV and eCPMV are recognized in a similar manner and with similar response by both mouse splenocytes and human PBMCs. These studies support the application of CPMV as a promising in situ vaccine reagent to treat human cancers as well as provide rationale for concurrent application of multiple TLR agonists as in situ cancer vaccines. Our findings provide the theoretical backbone for future development of combination immunotherapies using multi-TLR agonist and non-enveloped plant viruses as promising adjuvants to enhance antitumor efficacy.
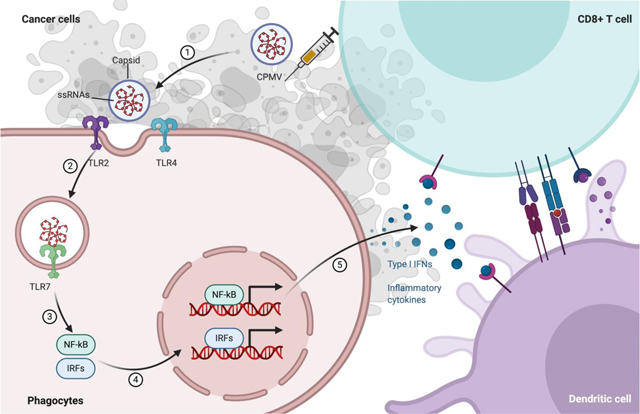
Fig 6: Schematic illustration of CPMV recognition by TLR2, TLR4 and TLR7.

Upper panel: CPMV is i.t. injected into a solid tumor, which is recognized by immune cells and stimulates proinflammatory cytokines and chemokines to recruit other immune cells and stimulate antitumor immunity. Lower panel: One APC (like dendritic cell) in the injected site is shown: the capsid of CPMV and eCPMV (not shown) is recognized by TLR2 and TLR4 located at plasma membrane. Upon entry by endocytosis, ssRNA of CPMV is recognized by TLR7. Downstream signaling cascade following TLR2/TLR4, and TLR7 activation is MyD88, which is the adaptor protein that initiates the different pathways activating IRF5, IRF7 (plasmacytoid DCs only), NF-κB, and AP-1 in order to induce the antiviral response. NF-κB and AP-1 combine to induce the transcription of inflammatory cytokines. IRF5/IRF7 in association with both NF-κB and AP-1 form the enhanceosome for the transcription of IFN-β/IFN-α, respectively. In addition, IRF7 also has IFN-β-stimulatory effect.
Here, we demonstrate that CPMV has superior antitumor efficacy in the B16F10 model compared to eCPMV. Similarly, previous studies in the ID8 ovarian mouse model showed that CPMV has better efficacy than eCPMV with more recruited and activated DCs and stronger inhibition of Treg cells when administered as ISV into the peritoneum which contained many dispersed small tumors.[16]. Results reported here extend this finding to direct injection into a solid tumor (melanoma). In mouse splenocytes, CPMV and eCPMV selectively increase proinflammatory cytokines and chemokines ex vivo.
CPMV is not the only plant virus that has been studied as an ISV immunostimulatory reagent although in our comparative experiments it has always had superior efficacy. Our group has performed experimental comparisons of CPMV with other plant viruses for immunogenicity and anti-tumor efficacy [18, 41–43]. CPMV had a more outstanding anti-tumor performance in efficacy studies compared with other plant viruses like tobacco mosaic virus (TMV), cowpea chlorotic mettle virus (CCMV), sesbania mosaic virus (SeMV), and physalis mosaic virus (PhMV) in B16F10 had reduced efficacy compared to CPMV in previous work [18, 42]. The basis for CPMV superior efficacy is not yet understood but it likely involves the multi-TLR activation it mediates. Our results agree with previous study which demonstrated that a combinatorial treatment of TLR agonists can have a profound effect on stimulation of innate responses [44]. It will be of interest to investigate whether other TLR-stimulating nanoparticles are more potent ISV reagents when multiple TLRs are concurrently stimulated by them. One example of other VLPs that are not infectious in mammals but stimulate the immune system is an M13 phage-based vaccine which is sensed by TLR9 and signals through MyD88 [45].
A relevant aspect of any TLR stimulation is that different leukocytes express different TLRs, express them at different levels, and produce different effector responses to TLR stimulation. This complexity of cell types, TLR expression patterns and response to stimulation of TLRs by different cell types means that a full understanding of the role of each of the TLRs involved in CPMV recognition has to investigate which cell types respond, through which TLRs and with what results of the stimulation. Of interest and not well understood are mechanisms of specific cell recruitment and activation following ISV with CPMV and eCPMV in vivo. TLR7 signals through MyD88 which leads to induction of NF-κB-dependent inflammatory cytokine production by conventional DCs (cDCs) and macrophages. When expressed by plasmacytoid DCs (pDCs), TLR7 can induce type I IFN secretion [46, 47]. Moreover, DCs, macrophages, and other APCs ingest CPMVs both in vitro and in vivo in murine models, causing activation of innate immunity [15, 48]. CPMV and eCPMV stimulate recruitment of innate immunostimulatory cells [14, 16]. Similarly, novel TLR9 agonists are also thought to induce a systemic effect by activating pDCs [49–51]. Whether CPMV can further synergize with TLR9 agonists and whether the pDCs trigger an immune response through direct antigen presentation versus the secretion of type 1 IFN alone and how this may impact T cell exhaustion in distant tumors are questions of significant interest for further development of ISV strategies.
These studies illustrate the importance of type I interferons for ISV. Our previous work has shown that the RNA genome of CPMV is not required to induce antitumor immunity [14], but this and previous data [16] demonstrates that the encapsulated RNA increases the efficacy of in situ vaccination with CPMV. The unique value of the RNA in this system is fully attributable to generation of type I IFN, although activation of TLR7 by the RNA does increase other cytokines and chemokines. The importance of TLR7 recognizing viral RNA for in situ vaccination is further supported by studies using papaya mosaic virus in which the efficacy of PapMV is TLR7-dependent [52–54]. TLR7-driven type I interferons were required for the superior antitumor potency of RNA-based vaccines [55]. Type I interferon also supports antigen-specific immunity against viruses by in part increasing major histocompatibility complex (MHC) expression, dendritic cell (DC) maturation, and functional inhibition of Treg cells [30, 56–57]. Induced by type I and II IFNs, CXCL9, which interacts with chemokine receptor 3 (CXCR3), is a chemoattractant for immune cells such as monocytes, DCs, natural killer (NK) cells and T-cells [58–65]. In addition, CXCL9 and 10 are critical to T-cell infiltration in solid tumors [66, 67]. The demonstration that the lack of IFNαR (the receptor for type I interferon) in normal host cells (Fig. 3E) was done against a background in which tumor cells still had the ability to express IFNαR. The complete elimination of the type I IFN effect in IFNαR KO mice, in which the only cells that can respond to type I IFNs, are the tumor cells shows that, in this model, the possible response of the tumor cells to type I IFN does not contribute a discernable amount to treatment efficacy. Type I IFNs are known to be secreted once the viral nucleic acids are recognized by TLRs [22]. This concept is utilized in clinical trials involving bacteriophage Qβ loaded with CpGs [68, NCT02680184, NCT03084640, and NCT03618641]. However, here we directly proved that the importance of type I by NP is critical for ISV treatment of tumors by viruses.
We also demonstrated that the lack of type I IFN generation by eCPMV could at least in part be remedied by application of R848, a TLR7 agonist, which improved the response. However, this synergy is still less effective than CPMV. This could be due to the protective capsid of CPMV may facilitate delivery of TLR7 agonist (ssRNA) to endosomes because of the recognized tendency of innate immune cells to phagocytose nanoparticles [69–72], or possibly the soluble TLR7 agonist has a shorter half-life in the tumor and draining lymph nodes than being loaded in NPs [32, 73, 74]. Our results also provide a rationale for using CPMV rather than combining individual purified TLR ligands. This advantage of CPMV could reflect the aggressive ingestion of nanoparticles by phagocytic cells which focuses the impact of CPMV in the TME on the innate immune cells [14, 75]. Moreover, a single CPMV particle is likely to interact with multiple molecules of TLR2 and 4 at the same time which could mediate a crosslinking of the receptors. While such crosslinking is often a very strong signal, it is not yet known whether that matters for TLRs or whether a single soluble ligand that interacts with a single receptor is just as strong at signaling. It is interesting to note that since TLRs are most commonly activated by high copy number molecules on microorganism pathogens [29], this cross-linking of TLRs may be occurring in the majority of TLR activation situations.
Our group has previously shown that ISV with CPMV has the ability to potentiate immune checkpoint blockade therapy (ICB) and this study provided the mechanism by which CPMV stimulates the immune system [76]. While ICB is approved for treating multiple solid tumor types, most treated patients still fail to respond [77]. Recent literature has also shown that suppression of type I IFN signaling in tumors mediates resistance to anti-PD-1 treatment [78]; and exposure to type I interferons can potentiate antitumor efficacy in combination with chemotherapy and ICB therapy [79–81]. The combination of anti-PD-1 and PapMV reduced tumor growth and increased survival and PapMV efficacy against tumors is dependent on type I interferons (IFNs) generated by RNA recognition by TLRs [81]. A number of publications clearly show synergy between ICB therapy and ISV in mouse models, as ISV can generate increased numbers of the tumor-recognizing effector T cells needed for ICB therapy [82, 83]. Since CPMV promotes the activation of multiple innate immune cells and the secretion of pro-inflammatory cytokines to induce T cell effector function [14, 18, 84], the potential of ISV with CPMV to remodel the TME into a proinflammatory environment supports potential combination with ICB in patients to improve antitumor responses.
Conclusions
As with most cancer therapies, combination of multiple approaches is rapidly becoming the standard for immunotherapy. Understanding the detailed mechanisms of immunotherapy reagents, like which receptors are activated will enable better rational therapy combinations. These implications extend further since these plant viruses can be readily modified to carry different cargoes inside the capsids. Examples include TMV-conjugated multiantigen subunit vaccine [85] and cucumber mosaic virus (CuMV) with adjuvant [86]. Similarly, the capsids of CPMV could be an excellent and readily modified drug delivery carrier for cancer vaccines that rely on intracellular reagent delivery and stimulation.In situ vaccination with CPMV treatment holds significant therapeutic potential for multiple reasons. The approach was well tolerated, with no discernable toxicity in the treated animals apart from rare vitiligo, although we did not conduct a systematic toxicology study of ISV using CPMV or eCPMV. Roughly 1 out of 50 treated mice with B16F10 tumors develop vitiligo and this often occurred in mice whose tumor was completely eliminated, just as vitiligo in melanoma patients is considered a positive sign that the immune system recognizes and is attacking the melanoma [87]. While the recognition mechanisms of CPMV and eCPMV are investigated in this study, the cellular mechanisms mediating response are not clear. One of our future goals includes detailed investigation of cellular mechanisms in hope to explain the superior efficacy of CPMV.
Supplementary Material
Acknowledgments:
This research was funded by the National Cancer Institute of the US National Institutes of Health, grant numbers U01CA218292, R01CA224605, R01CA253615 awarded to Drs. Steinmetz and Fiering. This study was supported by Dartmouth Mouse Modeling Shared Resource, Dartmouth Irradiation, Preclinical Imaging and Microscopy Shared Resource and Dartlab Immune Monitoring Shared Resource, which receive support from the Dartmouth Norris Cotton Cancer Center, through NCI funded grant 5P30CA023108.
Footnotes
Credit author statement:
Chenkai Mao, Conceptualization, Methodology, Investigation, Writing – original draft, review and editing. Veronique Beiss, Methodology, Investigation. Jennifer Fields, Investigation. Nicole F. Steinmetz: Supervision, Project administration, Funding acquisition, Writing – review and editing. Steve Fiering: Supervision, Project administration, Funding acquisition, Writing – review and editing.
Declaration of competing Interest:
Drs. Fiering and Steinmetz are co-founders of and have a financial relationship with Mosaic Immunoengineering Inc, which may be considered potential competing interest. The other authors declare no potential conflict of interest.
Data Availability:
All relevant data are available within the article and its supplementary information files, or available upon reasonable requests.
Reference
- [1].Rabinovich GA, Gabrilovich D, & Sotomayor EM (2007). Immunosuppressive strategies that are mediated by tumor cells. Annual review of immunology, 25, 267–296. 10.1146/annurev.immunol.25.022106.141609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Dougan M, & Dranoff G. (2009). Immune therapy for cancer. Annual review of immunology, 27, 83–117. 10.1146/annurev.immunol.021908.132544 [DOI] [PubMed] [Google Scholar]
- [3].Tang J, Yu JX, Hubbard-Lucey VM, Neftelinov ST, Hodge JP, & Lin Y. (2018). Trial watch: The clinical trial landscape for PD1/PDL1 immune checkpoint inhibitors. Nature reviews. Drug discovery, 17(12), 854–855. 10.1038/nrd.2018.210 [DOI] [PubMed] [Google Scholar]
- [4].Lu J, Lee-Gabel L, Nadeau MC, Ferencz TM, & Soefje SA (2015). Clinical evaluation of compounds targeting PD-1/PD-L1 pathway for cancer immunotherapy. Journal of oncology pharmacy practice : official publication of the International Society of Oncology Pharmacy Practitioners, 21(6), 451–467. 10.1177/1078155214538087 [DOI] [PubMed] [Google Scholar]
- [5].Yun S, Vincelette ND, Green MR, Wahner Hendrickson AE, & Abraham I. (2016). Targeting immune checkpoints in unresectable metastatic cutaneous melanoma: a systematic review and meta-analysis of anti-CTLA-4 and anti-PD-1 agents trials. Cancer medicine, 5(7), 1481–1491. 10.1002/cam4.732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Sagiv-Barfi I, Czerwinski DK, Levy S, Alam IS, Mayer AT, Gambhir SS, & Levy R. (2018). Eradication of spontaneous malignancy by local immunotherapy. Science translational medicine, 10(426), eaan4488. doi: 10.1126/scitranslmed.aan4488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Kreiter S, Vormehr M, van de Roemer N, Diken M, Löwer M, Diekmann J, … Sahin U. (2015). Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature, 520(7549), 692–696. doi: 10.1038/nature14426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Hammerich L, Marron TU, Upadhyay R. et al. Systemic clinical tumor regressions and potentiation of PD1 blockade with in situ vaccination. Nat Med 25, 814–824 (2019). 10.1038/s41591-019-0410-x [DOI] [PubMed] [Google Scholar]
- [9].Lee JM, Lee MH, Garon E, Goldman JW, Salehi-Rad R, Baratelli FE, Schaue D, Wang G, Rosen F, Yanagawa J, Walser TC, Lin Y, Park SJ, Adams S, Marincola FM, Tumeh PC, Abtin F, Suh R, Reckamp KL, Lee G, Wallace WD, Lee S, Zeng G, Elashoff DA, Sharma S, Dubinett SM (2017). Phase I Trial of Intratumoral Injection of CCL21 Gene-Modified Dendritic Cells in Lung Cancer Elicits Tumor-Specific Immune Responses and CD8+ T-cell Infiltration. Clinical cancer research : an official journal of the American Association for Cancer Research, 23(16), 4556–4568. 10.1158/1078-0432.CCR-16-2821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Galluzzi L, Vitale I. Oncogene-induced senescence and tumour control in complex biological systems. Cell Death Differ. (2018) 25:1005–6. doi: 10.1038/s41418-018-0102-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Lee KL, Twyman RM, Fiering S, & Steinmetz NF (2016). Virus-based nanoparticles as platform technologies for modern vaccines. Wiley interdisciplinary reviews. Nanomedicine and nanobiotechnology, 8(4), 554–578. 10.1002/wnan.1383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Shoeb E, & Hefferon K. (2019). Future of cancer immunotherapy using plant virus-based nanoparticles. Future science OA, 5(7), FSO401. 10.2144/fsoa-2019-0001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Gomes AC, Mohsen M, & Bachmann MF (2017). Harnessing Nanoparticles for Immunomodulation and Vaccines. Vaccines, 5(1), 6. 10.3390/vaccines5010006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Lizotte PH, Wen AM, Sheen MR, Fields J, Rojanasopondist P, Steinmetz NF, & Fiering S. (2016). In situ vaccination with cowpea mosaic virus nanoparticles suppresses metastatic cancer. Nature nanotechnology, 11(3), 295–303. 10.1038/nnano.2015.292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Hoopes PJ; Wagner RJ; Duval K; Kang K; Gladstone DJ; Moodie KL; Crary-Burney M; Ariaspulido H; Veliz FA; Steinmetz NF; Fiering SN Molecular Pharmaceutics 2018. 10.1021/acs.molpharmaceut.8b00126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Wang C, Beiss V, & Steinmetz NF (2019). Cowpea Mosaic Virus Nanoparticles and Empty Virus-Like Particles Show Distinct but Overlapping Immunostimulatory Properties. Journal of virology, 93(21), e00129–19. 10.1128/JVI.00129-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Cai H, Wang C, Shukla S, Steinmetz NF, Cowpea Mosaic Virus Immunotherapy Combined with Cyclophosphamide Reduces Breast Cancer Tumor Burden and Inhibits Lung Metastasis. Adv. Sci. 2019, 6, 1802281. 10.1002/advs.201802281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Murray AA, Wang C, Fiering S, & Steinmetz NF (2018). In Situ Vaccination with Cowpea vs Tobacco Mosaic Virus against Melanoma. Molecular pharmaceutics, 15(9), 3700–3716. 10.1021/acs.molpharmaceut.8b00316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Saunders K, Sainsbury F, & Lomonossoff GP (2009). Efficient generation of cowpea mosaic virus empty virus-like particles by the proteolytic processing of precursors in insect cells and plants. Virology, 393(2), 329–337. 10.1016/j.virol.2009.08.023 [DOI] [PubMed] [Google Scholar]
- [20].Lester SN, & Li K. (2014). Toll-like receptors in antiviral innate immunity. Journal of molecular biology, 426(6), 1246–1264. 10.1016/j.jmb.2013.11.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Boehme KW, & Compton T. (2004). Innate sensing of viruses by toll-like receptors. Journal of virology, 78(15), 7867–7873. 10.1128/JVI.78.15.7867-7873.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Kawasaki T, & Kawai T. (2014). Toll-like receptor signaling pathways. Frontiers in immunology, 5, 461. 10.3389/fimmu.2014.00461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M, Taira K, Akira S, & Fujita T. (2004). The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nature immunology, 5(7), 730–737. 10.1038/ni1087 [DOI] [PubMed] [Google Scholar]
- [24].Rehwinkel J, & Gack MU (2020). RIG-I-like receptors: their regulation and roles in RNA sensing. Nature reviews. Immunology, 20(9), 537–551. 10.1038/s41577-020-0288-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Ni G, Ma Z, & Damania B. (2018). cGAS and STING: At the intersection of DNA and RNA virus-sensing networks. PLoS pathogens, 14(8), e1007148. 10.1371/journal.ppat.1007148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Shepardson Kelly M., Schwarz Benjamin, Larson Kyle, Morton Rachelle V., Avera John, Kimberly McCoy Alayna Caffrey, Harmsen Ann, Douglas Trevor, Agnieszka Rynda-Apple. mBio Nov 2017, 8 (6) e01356–17; DOI: 10.1128/mBio.01356-17 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [27].Albakri MM, Veliz FA, Fiering SN, Steinmetz NF, & Sieg SF (2020). Endosomal toll-like receptors play a key role in activation of primary human monocytes by cowpea mosaic virus. Immunology, 159(2), 183–192. 10.1111/imm.13135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Uematsu S, & Akira S. (2008). Toll-Like receptors (TLRs) and their ligands. Handbook of experimental pharmacology, (183), 1–20. 10.1007/978-3-540-72167-3_1 [DOI] [PubMed] [Google Scholar]
- [29].Kawai T, & Akira S. (2011). Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity, 34(5), 637–650. 10.1016/j.immuni.2011.05.006 [DOI] [PubMed] [Google Scholar]
- [30].Zitvogel L, Galluzzi L, Kepp O, Smyth MJ, & Kroemer G. (2015). Type I interferons in anticancer immunity. Nature reviews. Immunology, 15(7), 405–414. 10.1038/nri3845 [DOI] [PubMed] [Google Scholar]
- [31].Lebel ME, Chartrand K, Tarrab E, Savard P, Leclerc D, Lamarre A. (2016) Potentiating Cancer Immunotherapy Using Papaya Mosaic Virus-Derived Nanoparticles. Nano letters 16, 1826–32 [DOI] [PubMed] [Google Scholar]
- [32].Cai H, Shukla S, Steinmetz NF, The Antitumor Efficacy of CpG Oligonucleotides is Improved by Encapsulation in Plant Virus-Like Particles. Adv. Funct. Mater. 2020, 30, 1908743. 10.1002/adfm.201908743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Rohovie MJ, Nagasawa M, & Swartz JR (2017). Virus-like particles: Next-generation nanoparticles for targeted therapeutic delivery. Bioengineering & translational medicine, 2(1), 43–57. 10.1002/btm2.10049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Grenier A; Dehoux M; Boutten A; Arce-Vicioso M; Durand G; Gougerot-Pocidalo M-A; Chollet-Martin S. Oncostatin M production and regulation by human polymorphonuclear neurtophis. Blood 1999, 93, (4), 1413. doi: 10.1182/blood.V93.4.1413 [DOI] [PubMed] [Google Scholar]
- [35].Queen MM, Ryan RE, Holzer RG, Keller-Peck CR, & Jorcyk CL (2005). Breast cancer cells stimulate neutrophils to produce oncostatin M: potential implications for tumor progression. Cancer research, 65(19), 8896–8904. 10.1158/0008-5472.CAN-05-1734 [DOI] [PubMed] [Google Scholar]
- [36].Nathan C. (2006). Neutrophils and immunity: challenges and opportunities. Nature reviews. Immunology, 6(3), 173–182. 10.1038/nri1785 [DOI] [PubMed] [Google Scholar]
- [37].Mauer J, Denson JL, & Brüning JC (2015). Versatile functions for IL-6 in metabolism and cancer. Trends in immunology, 36(2), 92–101. 10.1016/j.it.2014.12.008 [DOI] [PubMed] [Google Scholar]
- [38].Gajewski TF, Fuertes MB, & Woo SR (2012). Innate immune sensing of cancer: clues from an identified role for type I IFNs. Cancer immunology, immunotherapy : CII, 61(8), 1343–1347. 10.1007/s00262-012-1305-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Fuertes MB, Kacha AK, Kline J, Woo SR, Kranz DM, Murphy KM, & Gajewski TF (2011). Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8{alpha}+ dendritic cells. The Journal of experimental medicine, 208(10), 2005–2016. 10.1084/jem.20101159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Zofia Pilch & Tonecka ( Roszczenko ) & Agata Braniewska & a Sas & Marcin Skorzynski & Louis Boon& Jakub Golab & Linde Meyaard & Tomasz Rygiel. (2018). Antitumor Activity of TLR7 Is Potentiated by CD200R Antibody Leading to Changes in the Tumor Microenvironment. Cancer Immunology Research. 6. 10.1158/2326-6066.CIR-17-0454. [DOI] [PubMed] [Google Scholar]
- [41].Shukla S, Ablack AL, Wen AM, Lee KL, Lewis JD, & Steinmetz NF (2013). Increased tumor homing and tissue penetration of the filamentous plant viral nanoparticle Potato virus X. Molecular pharmaceutics, 10(1), 33–42. 10.1021/mp300240m [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Shukla S, Wang C, Beiss V, Cai H, Washington T 2nd, Murray AA, Gong X, Zhao Z, Masarapu H, Zlotnick A., Fiering S, & Steinmetz NF (2020). The unique potency of Cowpea mosaic virus (CPMV) in situ cancer vaccine. Biomaterials science, 8(19), 5489–5503. 10.1039/d0bm01219j [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Lee KL, Murray AA, Le D, Sheen MR, Shukla S, Commandeur U, Fiering S, & Steinmetz NF (2017). Combination of Plant Virus Nanoparticle-Based in Situ Vaccination with Chemotherapy Potentiates Antitumor Response. Nano letters, 17(7), 4019–4028. 10.1021/acs.nanolett.7b00107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Albin TJ, Tom JK, Manna S, Gilkes AP, Stetkevich SA, Katz BB, Supnet M, Felgner J, Jain A, Nakajima R, Jasinskas A, Zlotnik A, Pearlman E, Davies DH, Felgner PL, Burkhardt AM, & Esser-Kahn AP (2019). Linked Toll-Like Receptor Triagonists Stimulate Distinct, Combination-Dependent Innate Immune Responses. ACS central science, 5(7), 1137–1145. 10.1021/acscentsci.8b00823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Hashiguchi S, Yamaguchi Y, Takeuchi O, Akira S, & Sugimura K. (2010). Immunological basis of M13 phage vaccine: Regulation under MyD88 and TLR9 signaling. Biochemical and biophysical research communications, 402(1), 19–22. 10.1016/j.bbrc.2010.09.094 [DOI] [PubMed] [Google Scholar]
- [46].Gilliet M, Cao W, & Liu YJ (2008). Plasmacytoid dendritic cells: sensing nucleic acids in viral infection and autoimmune diseases. Nature reviews. Immunology, 8(8), 594–606. 10.1038/nri2358 [DOI] [PubMed] [Google Scholar]
- [47].Reizis B, Bunin A, Ghosh HS, Lewis KL, Sisirak V. Plasmacytoid dendritic cells: Recent Progress and open questions. Annual review of immunology. 2011;29:163–183. doi: 10.1146/annurev-immunol-031210-101345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Gonzalez MJ, Plummer EM, Rae CS, & Manchester M. (2009). Interaction of Cowpea mosaic virus (CPMV) nanoparticles with antigen presenting cells in vitro and in vivo. PloS one, 4(11), e7981. 10.1371/journal.pone.0007981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Krieg AM (2008). Toll-like receptor 9 (TLR9) agonists in the treatment of cancer. Oncogene, 27(2), 161–167. 10.1038/sj.onc.1210911 [DOI] [PubMed] [Google Scholar]
- [50].Parker BS, Rautela J, & Hertzog PJ (2016). Antitumour actions of interferons: implications for cancer therapy. Nature reviews. Cancer, 16(3), 131–144. 10.1038/nrc.2016.14 [DOI] [PubMed] [Google Scholar]
- [51].Ribas A, Medina T, Kummar S, Amin A, Kalbasi A, Drabick JJ, Barve M, Daniels GA, Wong DJ, Schmidt EV, Candia AF, Coffman RL, Leung A, & Janssen RS (2018). SD-101 in Combination with Pembrolizumab in Advanced Melanoma: Results of a Phase Ib, Multicenter Study. Cancer discovery, 8(10), 1250–1257. 10.1158/2159-8290.CD-18-0280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Lebel MÈ, Chartrand K, Leclerc D, & Lamarre A. (2015). Plant Viruses as Nanoparticle-Based Vaccines and Adjuvants. Vaccines, 3(3), 620–637. 10.3390/vaccines3030620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Lebel MÈ, Daudelin JF, Chartrand K, Tarrab E, Kalinke U, Savard P, Labrecque N, Leclerc D, & Lamarre A. (2014). Nanoparticle adjuvant sensing by TLR7 enhances CD8+ T cell-mediated protection from Listeria monocytogenes infection. Journal of immunology (Baltimore, Md. : 1950), 192(3), 1071–1078. 10.4049/jimmunol.1302030 [DOI] [PubMed] [Google Scholar]
- [54].Carignan D; Herblot S; Laliberté-Gagné M-È; Bolduc M; Duval M; Savard P; Leclerc D. Nanomedicine: Nanotechnology, Biology and Medicine 2018, 14, (7), 2317–2327. 10.1016/j.nano.2017.10.015 [DOI] [PubMed] [Google Scholar]
- [55].Kranz LM, Diken M, Haas H, Kreiter S, Loquai C, Reuter KC, Meng M, Fritz D, Vascotto F, Hefesha H, Grunwitz C, Vormehr M, Hüsemann Y, Selmi A, Kuhn AN, Buck J, Derhovanessian E, Rae R, Attig S, Diekmann J, … Sahin U. (2016). Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature, 534(7607), 396–401. 10.1038/nature18300 [DOI] [PubMed] [Google Scholar]
- [56].Diamond MS, Kinder M, Matsushita H, Mashayekhi M, Dunn GP, Archambault JM, Lee H, Arthur CD, White JM, Kalinke U, Murphy KM, & Schreiber RD (2011). Type I interferon is selectively required by dendritic cells for immune rejection of tumors. The Journal of experimental medicine, 208(10), 1989–2003. 10.1084/jem.20101158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Bacher N, Raker V, Hofmann C, et al. Interferon-α suppresses cAMP to disarm human regulatory T cells. Cancer Research. 2013. September;73(18):5647–5656. DOI: 10.1158/0008-5472.can-12-3788. [DOI] [PubMed] [Google Scholar]
- [58].Tensen CP, Flier J, Van Der Raaij-Helmer EM, Sampat-Sardjoepersad S, Van Der Schors RC, Leurs R, Scheper RJ, Boorsma DM, Willemze R (May 1999). “Human IP-9: A keratinocyte-derived high affinity CXC-chemokine ligand for the IP-10/Mig receptor (CXCR3)”. The Journal of Investigative Dermatology. 112 (5): 716–22. doi: 10.1046/j.1523-1747.1999.00581.x. PMID . [DOI] [PubMed] [Google Scholar]
- [59].Santodonato L, D’Agostino G, Nisini R, Mariotti S, Monque DM, Spada M, Lattanzi L, Perrone MP, Andreotti M, Belardelli F, & Ferrantini M. (2003). Monocyte-derived dendritic cells generated after a short-term culture with IFN-alpha and granulocyte-macrophage colony-stimulating factor stimulate a potent Epstein-Barr virus-specific CD8+ T cell response. Journal of immunology (Baltimore, Md. : 1950), 170(10), 5195–5202. 10.4049/jimmunol.170.10.5195 [DOI] [PubMed] [Google Scholar]
- [60].Siegal FP, Kadowaki N, Shodell M, Fitzgerald-Bocarsly PA, Shah K, Ho S, Antonenko S, & Liu YJ (1999). The nature of the principal type 1 interferon-producing cells in human blood. Science (New York, N.Y.), 284(5421), 1835–1837. 10.1126/science.284.5421.1835 [DOI] [PubMed] [Google Scholar]
- [61].Sun S, Zhang X, Tough DF, & Sprent J. (1998). Type I interferon-mediated stimulation of T cells by CpG DNA. The Journal of experimental medicine, 188(12), 2335–2342. 10.1084/jem.188.12.2335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Hilkens CM, Schlaak JF, & Kerr IM (2003). Differential responses to IFN-alpha subtypes in human T cells and dendritic cells. Journal of immunology (Baltimore, Md. : 1950), 171(10), 5255–5263. 10.4049/jimmunol.171.10.5255 [DOI] [PubMed] [Google Scholar]
- [63].Luster AD, & Ravetch JV (1987). Biochemical characterization of a gamma interferon-inducible cytokine (IP-10). The Journal of experimental medicine, 166(4), 1084–1097. 10.1084/jem.166.4.1084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Wendel M, Galani IE, Suri-Payer E, & Cerwenka A. (2008). Natural killer cell accumulation in tumors is dependent on IFN-gamma and CXCR3 ligands. Cancer research, 68(20), 8437–8445. 10.1158/0008-5472.CAN-08-1440 [DOI] [PubMed] [Google Scholar]
- [65].Costa C, Traves SL, Tudhope SJ, Fenwick PS, Belchamber KB, Russell RE, Barnes PJ, & Donnelly LE (2016). Enhanced monocyte migration to CXCR3 and CCR5 chemokines in COPD. The European respiratory journal, 47(4), 1093–1102. 10.1183/13993003.01642-2015 [DOI] [PubMed] [Google Scholar]
- [66].Mikucki M, Fisher D, Matsuzaki J. et al. Non-redundant requirement for CXCR3 signalling during tumoricidal T-cell trafficking across tumour vascular checkpoints. Nat Commun 6, 7458 (2015). 10.1038/ncomms8458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Musha H, Ohtani H, Mizoi T, Kinouchi M, Nakayama T, Shiiba K, Miyagawa K, Nagura H, Yoshie O, & Sasaki I. (2005). Selective infiltration of CCR5(+)CXCR3(+) T lymphocytes in human colorectal carcinoma. International journal of cancer, 116(6), 949–956. 10.1002/ijc.21135 [DOI] [PubMed] [Google Scholar]
- [68].Mohsen MO, Vogel M, Riether C, Muller J, Salatino S, Ternette N, Gomes AC, Cabral-Miranda G, El-Turabi A, Ruedl C, Kundig TM, Dermime S, Knuth A, Speiser DE, & Bachmann MF (2019). Targeting Mutated Plus Germline Epitopes Confers Pre-clinical Efficacy of an Instantly Formulated Cancer Nano-Vaccine. Frontiers in immunology, 10, 1015. 10.3389/fimmu.2019.01015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Yue H, Wei W, Yue Z, Lv P, Wang L, Ma G, & Su Z. (2010). Particle size affects the cellular response in macrophages. European journal of pharmaceutical sciences : official journal of the European Federation for Pharmaceutical Sciences, 41(5), 650–657. 10.1016/j.ejps.2010.09.006 [DOI] [PubMed] [Google Scholar]
- [70].Jia R, Guo JH, & Fan MW (2012). The effect of antigen size on the immunogenicity of antigen presenting cell targeted DNA vaccine. International immunopharmacology, 12(1), 21–25. 10.1016/j.intimp.2011.08.016 [DOI] [PubMed] [Google Scholar]
- [71].Rodell CB, Arlauckas SP, Cuccarese MF, Garris CS, Li R, Ahmed MS, Kohler RH, Pittet MJ, & Weissleder R. (2018). TLR7/8-agonist-loaded nanoparticles promote the polarization of tumour-associated macrophages to enhance cancer immunotherapy. Nature biomedical engineering, 2, 578–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Neek M, Kim TI, & Wang SW (2019). Protein-based nanoparticles in cancer vaccine development. Nanomedicine : nanotechnology, biology, and medicine, 15(1), 164–174. 10.1016/j.nano.2018.09.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Thauvin C, Widmer J, Mottas I, Hocevar S, Allémann E, Bourquin C, & Delie F. (2019). Development of resiquimod-loaded modified PLA-based nanoparticles for cancer immunotherapy: A kinetic study. European journal of pharmaceutics and biopharmaceutics : official journal of Arbeitsgemeinschaft fur Pharmazeutische Verfahrenstechnik e.V, 139, 253–261. 10.1016/j.ejpb.2019.04.007 [DOI] [PubMed] [Google Scholar]
- [74].Widmer J, Thauvin C, Mottas I, Nguyen VN, Delie F, Allémann E, & Bourquin C. (2018). Polymer-based nanoparticles loaded with a TLR7 ligand to target the lymph node for immunostimulation. International journal of pharmaceutics, 535(1–2), 444–451. 10.1016/j.ijpharm.2017.11.031 [DOI] [PubMed] [Google Scholar]
- [75].Gonzalez MJ, Plummer EM, Rae CS, & Manchester M. (2009). Interaction of Cowpea mosaic virus (CPMV) nanoparticles with antigen presenting cells in vitro and in vivo. PloS one, 4(11), e7981. 10.1371/journal.pone.0007981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Wang C, Steinmetz NF, A Combination of Cowpea Mosaic Virus and Immune Checkpoint Therapy Synergistically Improves Therapeutic Efficacy in Three Tumor Models. Adv. Funct. Mater. 2020, 30, 2002299. 10.1002/adfm.202002299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Wei SC, Duffy CR, & Allison JP (2018). Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer discovery, 8(9), 1069–1086. 10.1158/2159-8290.CD-18-0367 [DOI] [PubMed] [Google Scholar]
- [78].Ma C, & Fan R. (2014). Cancer immunotherapy and next-generation clinical immune assessment. Frontiers in oncology, 4, 265. 10.3389/fonc.2014.00265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Cauwels A. et al. Delivering Type I Interferon to Dendritic Cells Empowers Tumor Eradication and Immune Combination Treatments. Cancer Res January 15 2018. (78) (2) 463–474; DOI: 10.1158/0008-5472.CAN-17-1980 [DOI] [PubMed] [Google Scholar]
- [80].Minn AJ, & Wherry EJ (2016). Combination Cancer Therapies with Immune Checkpoint Blockade: Convergence on Interferon Signaling. Cell, 165(2), 272–275. 10.1016/j.cell.2016.03.031 [DOI] [PubMed] [Google Scholar]
- [81].Terawaki S, Chikuma S, Shibayama S. Hayashi T, Yoshita T, Okazaki T, Honjo T. IFN-α Directly Promotes Programmed Cell Death-1 Transcription and Limits the Duration of T Cell-Mediated Immunity, Seigo Terawaki, J Immunol. 2011, 186 (5) 2772–2779; DOI: 10.4049/jimmunol.1003208 [DOI] [PubMed] [Google Scholar]
- [82].Hammerich L, Binder A, Brody JD. In situ vaccination: cancer immunotherapy both personalized and off-the-shelf. Mol Oncol. (2015) 9:1966–81. doi: 10.1016/j.molonc.2015.10.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Hammerich L, Davis TA, Keler T, Salazar AM, & Brody J. (2016). Combining In Situ Vaccination with Immune Checkpoint Blockade Induces Long-Term Regression of Lymphoma Tumors. Blood, 128(22), 465-465. doi: 10.1182/blood.V128.22.465.46527471229 [DOI] [Google Scholar]
- [84].Wang C; Fiering SN; Steinmetz NF, Cowpea Mosaic Virus Promotes Anti-Tumor Activity and Immune Memory in a Mouse Ovarian Tumor Model. Advanced Therapeutics 2019, 0 (0), 1900003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Mansour AA, Banik S, Suresh RV, Kaur H, Malik M, McCormick AA, & Bakshi CS (2018). An Improved Tobacco Mosaic Virus (TMV)-Conjugated Multiantigen Subunit Vaccine Against Respiratory Tularemia. Frontiers in microbiology, 9, 1195. 10.3389/fmicb.2018.01195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Mohsen MO, Heath MD, Cabral-Miranda G, Lipp C, Zeltins A, Sande M, Stein JV, Riether C, Roesti E, Zha L, Engeroff P, El-Turabi A, Kundig TM, Vogel M, Skinner MA, Speiser DE, Knuth A, Kramer MF, & Bachmann MF (2019). Vaccination with nanoparticles combined with micro-adjuvants protects against cancer. Journal for immunotherapy of cancer, 7(1), 114. 10.1186/s40425-019-0587-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Byrne KT, & Turk MJ (2011). New perspectives on the role of vitiligo in immune responses to melanoma. Oncotarget, 2(9), 684–694. doi: 10.18632/oncotarget.323 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.