

Abstract
Throughout the central nervous system, astrocytes adopt precisely ordered spatial arrangements of their somata and arbors, which facilitate their many important functions. Astrocyte pattern formation is particularly important in the retina, where astrocytes serve as a template that dictates the pattern of developing retinal vasculature. Thus, if astrocyte patterning is disturbed, there are severe consequences for retinal angiogenesis and ultimately for vision – as seen in diseases such as retinopathy of prematurity. Here we discuss key steps in development of the retinal astrocyte population. We describe how fundamental developmental forces – their birth, migration, proliferation, and death – sculpt astrocytes into a template that guides angiogenesis. We further address the radical changes in the cellular and molecular composition of the astrocyte network that occur upon completion of angiogenesis, paving the way for their adult functions in support of retinal ganglion cell axons. Understanding development of retinal astrocytes may elucidate pattern formation mechanisms that are deployed broadly by other axon-associated astrocyte populations.
Introduction
The field of glial neurobiology is entering a golden age, thanks to new technologies and concepts that have driven widespread excitement about cells that were once dismissed as merely the “glue” of the brain. Progress in astrocyte neurobiology has been especially rapid in recent years, particularly when it comes to synapse-associated astrocytes such as those found in the gray matter of the cerebral cortex (Allen and Eroglu, 2017). By contrast, far less is known about astrocytes that reside in axon-rich regions such as the brain white matter (Köhler et al., 2021). Compared to their synapse-associated cousins, these axon-associated astrocytes remain relatively overlooked and poorly understood.
In recent years, the axon-associated astrocytes of the mammalian retina have emerged as a useful point of entry to identify new facets of astrocyte function. This enigmatic cell type resides within the retinal nerve fiber layer (RNFL) lining the inner surface of the retina. Compared to Müller glia, the major retinal astrocytic cell type, RNFL astrocytes receive much less attention. Perhaps this is for good reason: Müller glia are far more numerous and far more ubiquitous, interacting extensively with all retinal neuron types in all retinal layers. By contrast, retinal astrocytes are found exclusively in the RNFL, which contains only vasculature and the axons of retinal ganglion cells (RGCs) coursing towards the optic nerve head (Fig. 1A). Moreover, Müller glia are present in all vertebrate species, whereas retinal astrocytes are present only in mammals (Schnitzer, 1988). However, despite their small population size and restricted localization, RNFL astrocytes turn out to be extremely important. As we will see, they have critical roles in retinal development. They also likely have important axon-supporting functions (Büssow, 1980), although we do not yet understand these very well. By studying the RNFL astrocytes, we expect to uncover new types of astrocyte functions that are important not only in the retina, but may offer new insights into the broader function of axon-associated astrocytes throughout the central nervous system (CNS).
Figure 1: Development of the retinal nerve fiber layer.
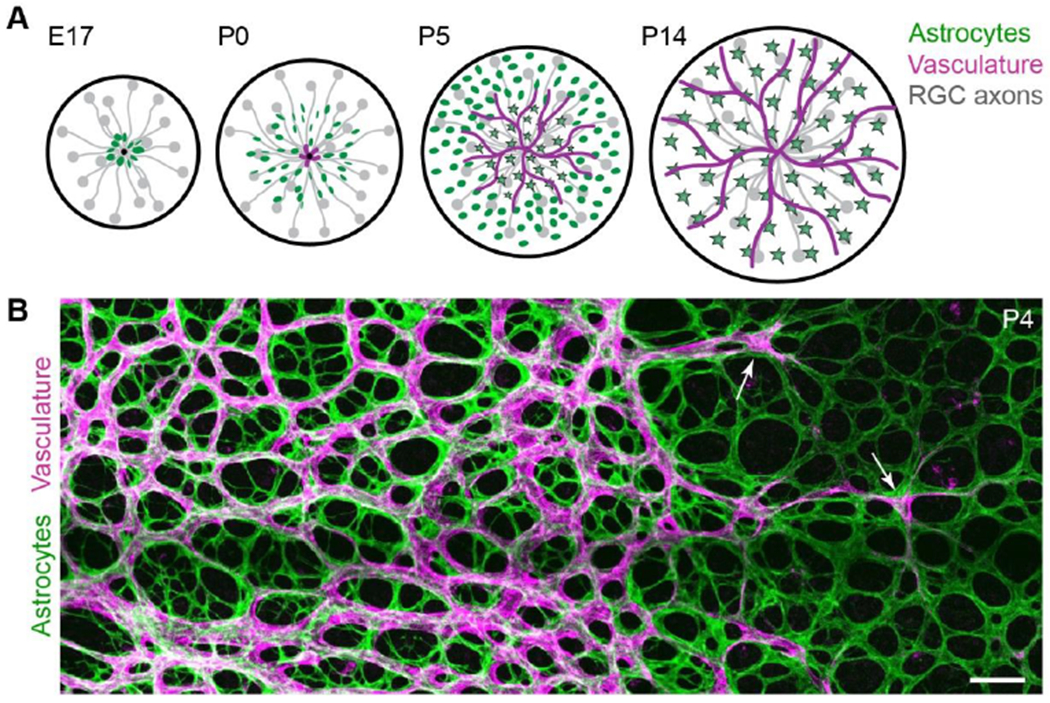
A) Schematic detailing sequential development of cell types comprising the RNFL. Before birth (E17 in mice), astrocytes begin to migrate into the RNFL using RGC axons as guides. Vessels enter postnatally (P0). Astrocytes mature as vessels pass over (stars, mature astrocytes).
B) En face confocal photomicrograph of flat-mounted retina depicting RNFL angiogenesis. Incoming vasculature (magenta; labeled by Griffonia simplicifolia Isolectin B4) progresses towards peripheral retina (at right), directly following the preformed astrocyte network (green, anti-PDGFRα). Arrows indicate tip cells at the angiogenic vanguard. Scale bar, 100 μm. (A) modified from Puñal et al. (2019); (B) modified from Perelli et al. (2021).
How does astrocyte development enable astrocyte function?
One of the most notable features of retinal astrocytes is their remarkably tight evolutionary and physical association with retinal blood vessels. Most vertebrate species do not have any intrinsic retinal vasculature, relying instead on extraretinal vessels to supply the neural tissue with oxygen. Intrinsic vasculature arose only within the mammalian lineage – and as we have noted above, RNFL astrocytes are also exclusive to mammals. In fact, even within the mammalian lineage there are some species, such as the horse and rabbit, in which the retina is only partially vascularized – and in these species RNFL astrocytes are present exclusively within the retinal subregions that contain vessels (Schnitzer, 1988, 1985; Stone and Dreher, 1987). The affinity between these two cell types can also be discerned in the primate fovea, the retinal region specialized for high-acuity vision. The fovea is the only region of the primate retina that lacks vasculature – and also lacks astrocytes (Schnitzer, 1988).
These observations from comparative neuroanatomy were the first hint that astrocytes might be important for guiding the development of retinal vasculature. We now know that this is indeed the case: Astrocytes serve as a template that patterns incoming vessels during retinal angiogenesis. During the perinatal period in mice, or the third trimester of development in humans, RNFL astrocytes arrange their cell bodies and arbors into a ring-like pattern that prefigures the eventual shape of the RNFL capillaries (Chu et al., 2001; Fruttiger et al., 1996; Fig. 1B). Endothelial cells then enter the retina at the optic nerve head and initiate angiogenesis, which spreads as a central-to-peripheral wave through the RNFL (Fig. 1A). During this process, angiogenic tip cells at the vascular wavefront extend filopodia along astrocyte arbors, using the astrocytes as a template to guide their growth (Dorrell et al., 2002; Gerhardt et al., 2003). As a result of this cellular behavior, retinal capillaries lie precisely on top of the pre-existing astrocyte arbor network (Fig. 1B).
In addition to physically patterning the vasculature by guiding tip cells, astrocytes are also the key source of molecular cues, such as vascular endothelial growth factor (VEGF)-A, which are critical for driving angiogenesis. Both the physical and molecular components of the astrocyte template exert powerful control over vascular development, as shown by genetic experiments in mice. Ablation of astrocytes before the onset of angiogenesis entirely prevents vasculature from entering the retina (O’Sullivan et al., 2017; Tao and Zhang, 2016). This is likely due to the absence of VEGF-A, since astrocyte-specific deletion of the Vegfa gene causes the same phenotype (Rattner et al., 2019). If astrocytes are ablated during angiogenesis, endothelial cells continue to grow towards the periphery but they do not form capillary-like loops, demonstrating a key role for the astrocyte physical template in vascular patterning (O’Sullivan et al., 2017). Physical and molecular guidance cues also become severely disrupted when astrocytes are overproduced. In mice with too many astrocytes, high VEGF-A levels and an overly dense physical template combine to delay angiogenesis and disrupt its patterning, leading to retinopathies and hemorrhage (Fruttiger et al., 1996; Gerhardt et al., 2003).
All of these phenotypes underscore the importance of astrocyte pattern formation in the subsequent development of vessels – a key step in forming a functional retina (for a comprehensive review of retinal angiogenesis see Selvam et al., 2018). And yet, basic steps in astrocyte development are only now beginning to be understood. Explaining these developmental phenomena is not just important for our basic knowledge of retinal development but also is directly relevant to disease, as disruptions of retinal angiogenesis underlie the childhood blinding disease known as retinopathy of prematurity (Hellström et al., 2013).
Here we review the developmental forces that shape the retinal astrocyte population: How do astrocytes become arranged into a template for vasculature, and how do they remodel into their adult configuration when angiogenesis is complete? To address these questions we will describe recent findings concerning how the RNFL astrocytes are specified, how they are produced in the correct numbers, and how they undertake their dramatic migration from the site of their birth at the optic nerve head to colonize the entire tissue (Stone and Dreher, 1987; Watanabe and Raff, 1988) (Fig. 2). We will highlight key cell-cell interactions that control these developmental events and the consequences for retinal development when they are perturbed. Finally, we note key open questions that must be addressed in order for us to fully understand development of this unusual astrocyte population.
Figure 2: Migration of retinal astrocytes precedes angiogenesis.
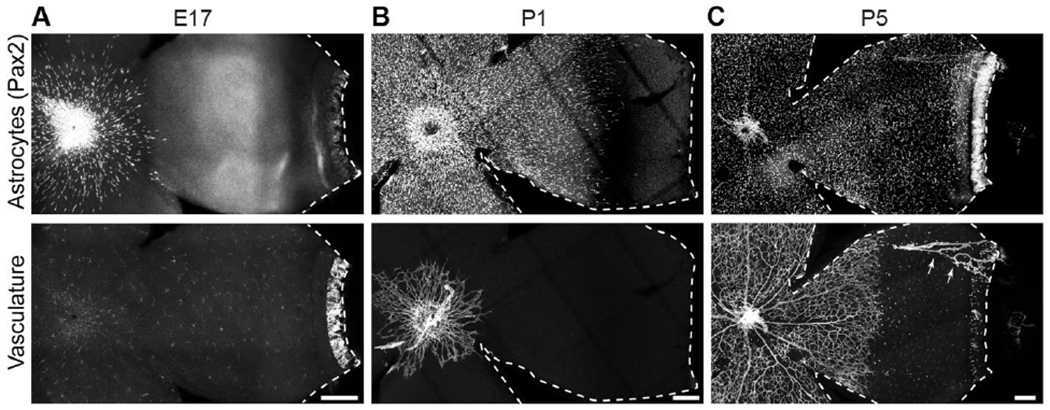
Flat-mounted mouse retinas labeled with markers of astrocyte nuclei (Pax2) and vasculature (A,C: Isolectin B4; B: anti-CD31).
A) Astrocytes begin to enter the retina before birth (E17), prior to endothelial cell entry. No vasculature is present although lectin-labeled microglia are visible.
B) By P1, astrocytes cover over half of the RNFL. Vasculature is just beginning to exit the optic nerve head.
C) At P5, astrocytes have reached the periphery and vasculature covers approximately half of the retina. Arrows, hyaloid vessels that were not completely removed during retinal dissection. Ahead of the angiogenic wavefront astrocyte somata are arranged in a circular pattern, mirroring astrocyte arbors (Fig. 1B).
Dotted lines delineate edge of retinal tissue. Scale bars, 100 μm.
Genesis of retinal astrocytes
Compared to other retinal cell types, astrocytes have a unique lineage. The retina and other eye structures are derived from neuroepithelial cells of the embryonic eye primordium, which has two major progenitor zones: 1) the optic cup, which will produce the neural retina; and 2) the optic stalk, which is fated to generate astroglia restricted to the optic nerve. Optic cup progenitors are multipotent, giving rise to all of the numerous types of retinal neurons as well as Müller glia (Holt et al., 1988; Turner and Cepko, 1987). The only retinal cell type that these progenitors cannot make is the RNFL astrocyte (Watanabe and Raff, 1988). Instead, these astrocytes are generated by the optic disc progenitor zone – a small, specialized ring of neuroepithelial cells located at the boundary between the optic cup and stalk (Fig. 3). These cells line the head of the optic nerve at its junction with the neural retina, encircling the nascent nerve like a cuff (Dakubo et al., 2003; Morcillo et al., 2006).
Figure 3: Retinal astrocytes are produced by specialized neuroepithelial progenitors.
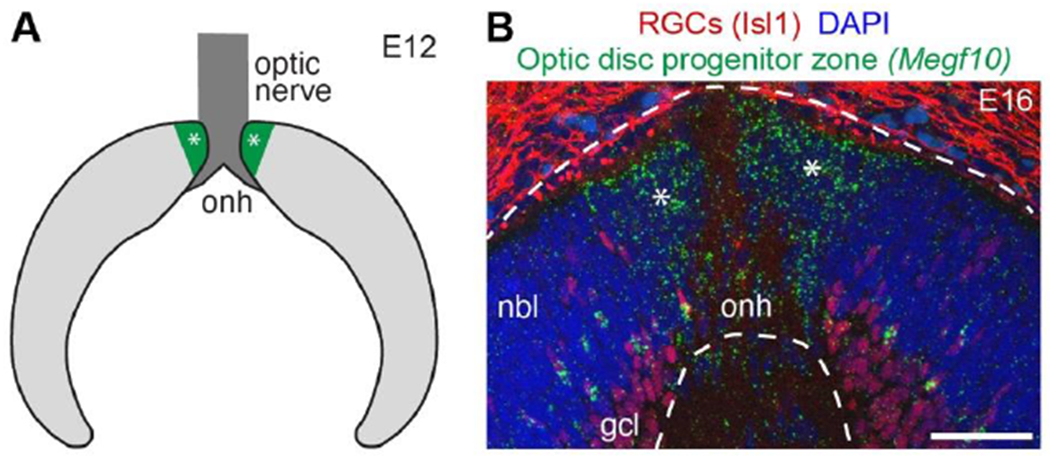
A) Schematic depicting cross-section of embryonic mouse retina. The optic disc progenitor zone (green, asterisks), which gives rise to retinal astrocytes, surrounds the optic nerve head (dark gray). Light gray, retinal progenitors derived from the optic cup; these give rise to retinal neurons and Müller glia.
B) Photomicrograph of E16 mouse retinal cross section. In situ hybridization for Megf10 gene, an astrocyte marker, reveals location of optic disc progenitor zone (asterisks). Dashed lines demarcate neural retina. Anti-Isl1 staining shows location of RGC cell bodies (also note non-specific staining outside neural retina).
Abbreviations: onh, optic nerve head; nbl, neuroblast layer containing retinal progenitors; gcl, ganglion cell layer. Scale bar, 50 μm.
The astrocyte progenitor zone forms in this location due to early patterning events that establish the proximal-distal axis of the eye primordium. These patterning events have been reviewed in detail elsewhere (Tao and Zhang, 2014), but we will summarize the key points here. Prior to neurogenesis, patterning cues from the ventral midline define the regions of the eye primordium that will become the optic cup and stalk. Sonic hedgehog (Shh) is a particularly important midline-derived cue. Regions close to the midline receive a high Shh signal and become optic stalk cells, defined by expression of the transcription factor Pax2. Meanwhile, distal regions receive a low signal and adopt the optic cup fate, defined by expression of Pax6. As eye morphogenesis proceeds, the border between optic stalk and cup progenitor zones is refined and sharpened through several different mechanisms (Tao and Zhang, 2014). One critical mechanism is cross-repression between Pax2 and Pax6. These transcription factors directly inhibit each other’s expression, leading to mutually exclusive domains of Pax2 and Pax6 expression in the stalk and cup, respectively (Schwarz et al., 2000). Each transcription factor then serves a key role in the function of its respective progenitor cells: Pax6 defines optic cup cells as multipotent neural progenitors (Marquardt et al., 2001), whereas Pax2 restricts the fate potential of optic stalk progenitors to generation of optic nerve astrocytes (Bosze et al., 2021; Soukkarieh et al., 2007).
The optic disc progenitor zone, which gives rise to retinal astrocytes, lies precisely at the junction between the optic stalk and cup (Fig. 3). Consistent with their gliogenic potential, the progenitors in this zone express optic stalk markers such as Pax2; however, they also express certain markers of the ventral neural retina that are absent from the optic stalk, such as Vax2 and Raldh2 (Morcillo et al., 2006). Thus, the astrocyte progenitor zone has its own gene expression profile, clearly distinguishing it from both of its neighboring regions. Formation of this small boundary region depends on the same patterning forces that establish the optic cup and stalk territories. In mouse mutants lacking key patterning genes such as Shh, Pax2, and Pax6, formation of the astrocyte progenitor zone is disrupted (Morcillo et al., 2006; Schwarz et al., 2000). It is also disrupted when Shh is deleted from RGC axons, suggesting that multiple Shh sources are required to establish this progenitor zone (Dakubo et al., 2003). In each of these mutants, the boundary between optic cup and stalk fails to form properly – there is a blurring of gene expression and anatomical features where the boundary should have formed. Thus, failure to establish a sharp cup/stalk boundary eliminates the conditions necessary to establish the astrocyte progenitor zone.
What mechanisms control the cell fate choices of the optic disc astrocyte progenitors? One molecular player is Pax2: Similar to its role in the optic stalk, Pax2 also appears to be a key determinant of astrocyte fate selection within optic disc progenitors. Loss of Pax2 function leads to ectopic expression of neural progenitor genes, suggesting that Pax2 normally represses neural genes as part of an astroglial fate selection program (Bosze et al., 2021). However, apart from the broad neuron-vs.-glia choice, there are many questions still unanswered. For example: Why are the optic disc progenitors restricted to generating RNFL astrocytes rather than optic nerve astrocytes or Müller glia? What factors endow these Pax2+ cells with the ability to generate migratory astrocytes that will enter the retina, instead of astrocytes that will remain local to the optic nerve? Future work will be needed to answer these questions.
Migration of astrocytes
Following their birth at the optic disc progenitor zone, astrocytes face a daunting developmental challenge: In order to arrange themselves into a template for vasculature, they must first migrate radially outward from the optic nerve head to colonize the entire retinal surface (Fig. 1A; Fig. 2). And they must do this in a controlled manner that generates an appropriate local astrocyte density at each retinal location. As we will see later in this review, migration is not the only developmental mechanism that regulates local astrocyte density and patterning. However, it is certainly the most dramatic: No other retinal cell type migrates over such a long distance. Here we review what is known about the cues that guide astrocyte migration, and we highlight the many open questions that will need to be answered before we can fully understand the mechanisms that control this key patterning event.
Migration of astrocytes into the retina from the optic nerve head was first described in the 1980s (Ling et al., 1989; Ling and Stone, 1988; Stone and Dreher, 1987; Watanabe and Raff, 1988). Initially it was thought that astrocytes enter the retina with the vasculature: Staining with astrocyte markers available at the time – primarily GFAP and vimentin – suggested that astrocyte and vascular immunoreactivity advance together across the developing rodent and cat retina (Ling et al., 1989; Ling and Stone, 1988). Subsequent work showed, however, that astrocyte migration is independent of vasculature (Chan-Ling and Stone, 1991a; Fruttiger, 2002). During their migration, astrocytes are still at an immature precursor stage: They are committed to an astrocyte fate, expressing cell-type-specific markers such as Pax2 and platelet-derived growth factor receptor α (PDGFRα), but they do not yet express GFAP or other markers of mature astrocytes (Chan-Ling et al., 2009; Chu et al., 2001; Fruttiger, 2002). Maturation of astrocyte precursors – including GFAP upregulation – is triggered by arrival of patent vessels (West et al., 2005; see section below on astrocyte differentiation). Thus, while a wave of GFAP expression spreads across the retina together with the vasculature, migration of astrocyte precursors happens substantially earlier. For example, in the mouse, Pax2+ PDGFRα+ astrocytes begin migrating in late embryogenesis and complete their migration by postnatal day (P)4 (Fig. 2). By contrast, RNFL angiogenesis does not begin until birth and is not completed until P8 (Fruttiger, 2002; O’Sullivan et al., 2017; West et al., 2005).
Since astrocytes do not hitch a ride with the vasculature, they must use other cues within the RNFL environment to guide their migration toward the retinal periphery. These cues are of two main varieties: 1) those that promote motility; and 2) those that provide directional information, orienting astrocytes onto a centrifugal migratory path. While many of the molecules underlying these guidance functions are still unknown, recent studies have begun to identify cellular and molecular cues that support astrocyte migration.
One important source of molecular cues promoting astrocyte motility is the inner limiting membrane (ILM), a specialized band of extracellular matrix that is localized to the basal endfeet of the retinal neuroepithelium (Halfter et al., 2000; Libby et al., 2000). From this position, the ILM serves as the basement membrane for the RNFL. During development, the ILM has several critical roles in formation of the RNFL. First, the ILM serves as a substrate for RGC axon growth cones as they emerge from the cell body and navigate toward the optic disc (Randlett et al., 2011). Later, it serves as the migratory substrate for astrocytes traveling in the opposite direction, outwards from the optic disc. In mutant mice with impairments in ILM assembly or maintenance, astrocyte migration is severely disturbed. For example, deletion of the ILM matrix proteins laminin α1, β2, and γ3 causes ILM fragmentation, allowing neuronal bodies from the ganglion cell layer to protrude into the RNFL. A similar phenotype results from retina-specific disruption of cellular receptors for ILM matrix proteins, loss of which prevents ILM assembly (Clements et al., 2017; Tao and Zhang, 2016). In each of these mutants, astrocyte colonization of peripheral retina is profoundly disturbed; however, astrocytes are still able to migrate a short distance away from the optic nerve head. A close examination of the colonized region reveals that astrocytes are able to migrate into RNFL regions where the ILM remains intact, although they stop once they reach a hole in the fragmented matrix (Gnanaguru et al., 2013; Tao and Zhang, 2016). These findings indicate that the ILM functions as a permissive substrate for astrocyte migration.
While the ILM matrix is necessary for astrocyte motility, it does not appear to provide guidance cues that orient astrocytes away from the optic disc and towards the periphery. What cues are responsible for centrifugal guidance? During the time when astrocytes are migrating, RGC axons are the most prominent cellular feature within the RNFL. Because the axons converge at the optic nerve head, they are ideally positioned to guide centrifugal dispersion of astrocytes away from their optic disc progenitor zone (Fig. 1A). Indeed, immature astrocytes become closely associated with RGC axons, adopting an anatomy consistent with migration along the axon tracts (Fig. 4A). This interaction is maintained even in axon guidance mutants, in which RGC axons travel orthogonal to their usual trajectory (O’Sullivan et al., 2017). In such mutants, astrocytes polarize inappropriately along the misrouted axons and form migratory chains along them (Fig. 5), strongly suggesting that axons dictate the trajectory of astrocyte migration. To further test this model, O’Sullivan et al. (2017) examined astrocyte migration in Atoh7 (Math5) mutant mice, which lack RGCs. This analysis revealed that directional cues from RGC axons are necessary for long-distance astrocyte migration. When RGCs are absent, migrating astrocytes fail to polarize their cell bodies along the centrifugal axis. Instead they polarize in random directions, suggesting that astrocytes in mutant retina are unable to discern which way they should go. Consequently, astrocytes fail to colonize peripheral retina, leading to an increased density of astrocytes surrounding their origin at the optic nerve head (O’Sullivan et al., 2017). Even within the colonized region, the anatomy of the astrocyte network is irregular and dysmorphic (Edwards et al., 2012; O’Sullivan et al., 2017), likely due to the accumulation of excess astrocytes that were not able to migrate away (Fig. 5). Altogether, the data indicate that interactions with axons are critical for orienting migratory astrocytes and guiding them along the correct route so they can successfully colonize peripheral retina.
Figure 4: Cell-cell interactions of retinal astrocytes.
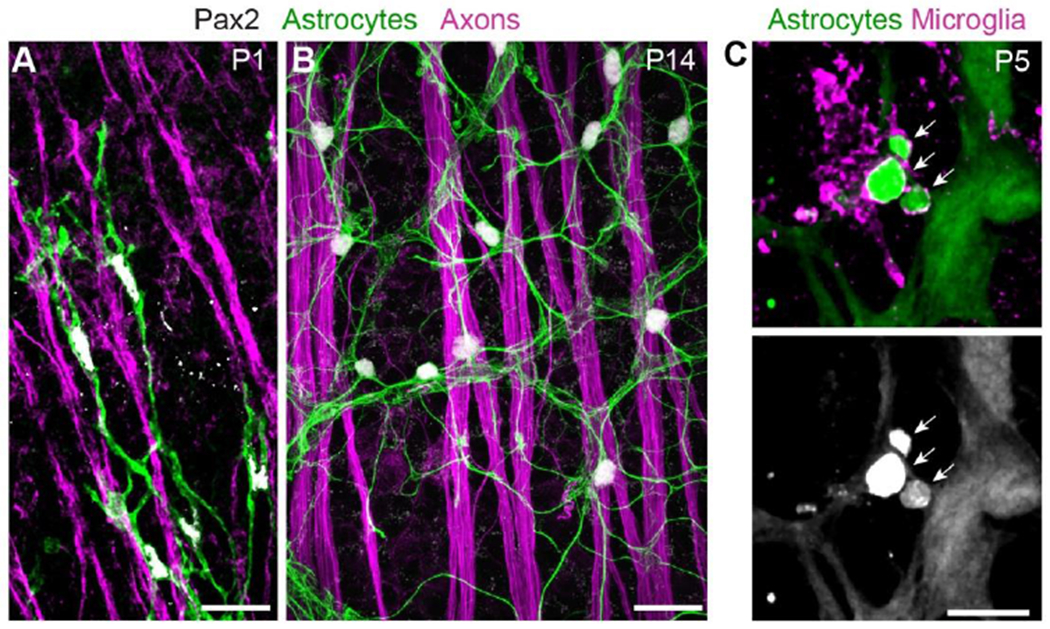
A,B) Interactions with axons (labeled by anti-Neurofilament medium chain).
At P1 (A), astrocyte precursor cells climb along RGC axons as they migrate towards peripheral retina. Note polarization of astrocyte arbors (revealed by Pax2-Cre-driven membrane-GFP reporter) along the axon bundles.
At P14 (B), mature astrocytes are no longer closely associated with RGC axons, instead showing uniform “mosaic” spacing. Astrocyte arbors labeled by anti-GFAP. See O’Sullivan et al. (2017) for further details on astrocyte-axon interactions during migration.
C) Interactions with microglia. At P5, astrocyte debris (arrows) can be found within microglia. Astrocytes were labeled with GFAP-Cre-driven TdTomato reporter; microglia were labeled with a combination of antibodies to Iba1 and P2Y12. At this age, GFAP-Cre is selective for astrocytes and not yet expressed by Müller glia.
Scale bars: 25μm (A,B), 10μm (C). (C) modified from Puñal et al. (2019).
Figure 5: Retinal ganglion cell axons guide astrocyte migration.
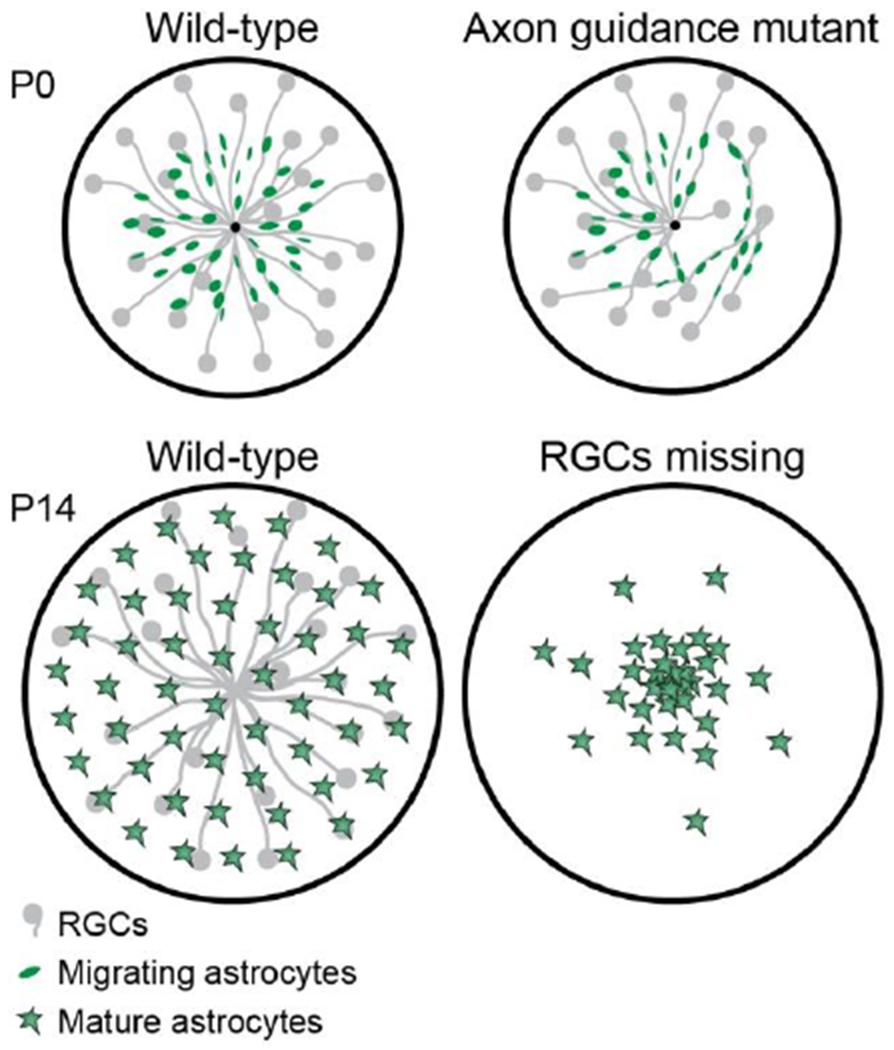
Schematic illustrating effects of RGC axon manipulations on astrocyte migration. Top: In retinas lacking both Robo1 and Robo2 genes, axon navigation is disrupted. Instead of projecting directly to the optic nerve head, as in wild-type retina (left), many mutant axons take meandering routes. Migrating astrocytes follow mistargeted axons regardless of their trajectories. Bottom: Atoh7 mutant mice lack RGCs. In these mutants, astrocytes can migrate a short distance from their origin but fail to reach peripheral retina. As a result their density in central retina is abnormally high. For further details see O’Sullivan et al. (2017).
We do not yet know the molecular nature of the astrocyte guidance cues provided by RGC axons. During migration, astrocytes may associate with cell-surface receptors on the axons themselves, or they may migrate along axon-derived cues that are deposited locally into the ILM matrix. Additionally, axons likely express attractive cues that act over short distances to recruit migratory astrocytes. One interesting candidate in this regard is platelet-derived growth factor (PDGF)-A, which has long been known to mediate paracrine signals between RGCs and astrocytes. Its best known function is as a mitogen: PDGF-A, secreted by RGCs, binds to astrocyte PDGFRα receptors to promote proliferation (Fruttiger et al., 1996; Mudhar et al., 1993). But recently, this interaction was shown to have another function as well. In an elegant live imaging study using retinal explants, Tao and Zhang (2016) showed that astrocytes are strongly attracted to a point source of PDGF-A. Therefore, PDGF-A is well positioned to serve as an attractive cue that recruits astrocytes to RGC axons. Once they have followed PDGF-A to the nearest axon, astrocytes would then be able to take advantage of the centrifugal directional information encoded by the axon’s trajectory. It will be interesting to see whether further experiments confirm this model.
While clearly important, axon-derived cues are probably not sufficient to explain astrocyte migratory choices. Once they have reached an axon, astrocytes could, in principle, migrate either towards central retina or the periphery. How do they know which way to go? One straightforward possibility is that there are long-range guidance cues expressed in gradients along the center-to-peripheral axis, which could serve to orient astrocytes in the correct direction. Because PDGF-A can attract astrocytes, several recent papers have proposed PDGF-A as such a cue (Selvam et al., 2018; Tao and Zhang, 2016). We think this is unlikely, however, because RGCs – the major source of PDGF-A – are most abundant in central retina, not the periphery. This is true not only for RGC somata (Rodriguez et al., 2014; Wang et al., 2017), but also for RGC axons due to their convergence at the optic nerve head. Thus, if astrocytes were to migrate towards the retinal region with the highest PDGF-A levels, they would migrate backwards, towards central retina. It therefore seems more likely that PDGF-A serves as a short-range cue promoting axon-astrocyte associations, as proposed above, rather than serving as a long-range cue.
Although PDGF-A attraction is unlikely to be responsible for orienting astrocytes towards the periphery, there are other molecules expressed in centrifugal gradients that could do so. One example is netrin-1, which is expressed by optic nerve head progenitors and is known to guide RGC axons (Dakubo et al., 2003; Deiner et al., 1997). Netrin-mediated repulsion could in principle drive astrocytes outward from the optic nerve head, but this idea remains to be tested. Homotypic repulsion among astrocytes would also be a viable strategy for oriented migration: Perhaps astrocytes seek to migrate away from crowded central retina towards uncolonized regions that lack astrocyte-derived repulsive cues. Such an idea, while plausible, is entirely hypothetical at this point.
Clearly, there is much work still to be done before we understand the molecular basis for astrocyte guidance and migration. In addition to identifying the cues that orient migrating astrocytes, another critical open question is the identity of cues that tell astrocytes to stop migrating. Why do some cells cease migrating in central retina while others continue towards the periphery? How individual cells decide between stopping and continuing is entirely unknown. Some hints may be provided by mice lacking the transcription factor Tlx (encoded by the Nr2e1 gene), which is expressed by many proliferative retinal cells including immature astrocytes. As we will see in the following sections, Tlx is important for just about all facets of astrocyte development, serving to promote multiple aspects of the immature astrocyte phenotype while suppressing astrocyte maturation. During migration, Tlx appears to be involved in start-stop decisions because, in Tlx mutant mice, astrocytes stop migrating prematurely – they fail to migrate beyond central retina (Miyawaki et al., 2004; Uemura et al., 2006). Because Tlx mutant astrocytes are accelerated in their maturation, this phenotype suggests that the decision to stop migrating and the decision to commence differentiation may be linked. Even if this model turns out to be true, how do individual astrocytes make these decisions? Further work illuminating the signals that induce and prevent astrocyte migration will be needed to answer this important question.
Proliferation of astrocyte precursors
Once astrocytes finish migrating they arrange their somata and arbors into ring-like shapes and await the arrival of vasculature (Fig. 2C). The patterning of these astrocytic rings becomes profoundly disturbed when local astrocyte density is altered (Fruttiger et al., 1996; O’Sullivan et al., 2017; Perelli et al., 2021). Therefore, tight regulation of proliferation and total astrocyte number is important for establishing the vascular template (Fig. 1B).
Astrocyte precursors remain capable of proliferating during their migration and prior to the arrival of vasculature (Chan-Ling et al., 2009; Morita et al., 2017; West et al., 2005). Once vessels arrive, immature astrocytes undergo differentiation and maturation, characterized by changes in gene expression and morphology (see the next section for further details). This key developmental transition brings the period of astrocyte proliferation to an end. While they are competent to divide, several extrinsic and intrinsic molecular factors are known to regulate their rate of proliferation, thereby controlling astrocyte cell numbers. First, PDGF-A released by RGCs not only acts as an attractant for astrocytes, as discussed in the migration section above, but it also functions as a mitogen. Overexpression of PDGF-A causes a massive overproduction of astrocytes, which in turn disrupts angiogenesis (Fruttiger et al., 1996; West et al., 2005). Second, astrocytes require the transcription factor Tlx for normal proliferation. Tlx promotes proliferation by regulating cell cycle proteins, such as cyclin D1. In Tlx mutant mice the number of proliferating astrocytes is reduced, which leads to an overall impairment in astrocyte network formation in combination with the migratory defects discussed above (Miyawaki et al., 2004).
A third molecular regulator of astrocyte proliferation is the hypoxia-inducible factor (HIF) signaling pathway. This evolutionarily conserved pathway, which is activated by local hypoxia, is the main molecular mechanism by which cells respond to changes in tissue oxygenation (Wilson et al., 2020). In the developing retina, tissue ahead of the angiogenic wavefront is hypoxic due to the lack of an intrinsic blood supply (Stone et al., 1995). While in principle any retinal cell could activate HIF signaling in response to this hypoxia, there is strong evidence that astrocytes are uniquely situated to be hypoxia sensors. Astrocytes are the only cells in the avascular hypoxic zone that express high levels of VEGF-A, which is a direct transcriptional target of the HIF pathway (Gerhardt et al., 2003; Morita et al., 2017; Stone et al., 1995; West et al., 2005). This suggests that astrocytes not only activate the HIF pathway, but they do so more effectively than neighboring cells. One reason for this distinctive response may be that astrocytes express a distinct HIF effector: the HIF2α transcription factor is selectively expressed by retinal astrocytes, whereas retinal neurons and progenitors cells express HIF1α (Nakamura-Ishizu et al., 2012; Perelli et al., 2021). The ability of astrocytes to serve as cell-autonomous hypoxia sensors is tied closely to their role in promoting angiogenesis. As we have seen, astrocyte-derived VEGF-A is critical for RNFL angiogenesis (Rattner et al., 2019). When astrocytic HIF signaling is suppressed, either by rearing animals in a high oxygen environment or by deleting the gene encoding HIF2α, astrocytes fail to express VEGF-A and angiogenesis is blocked (Duan et al., 2014; Morita et al., 2016; Perelli et al., 2021; Stone et al., 1995; West et al., 2005). Thus, astrocytic HIF signaling has a major non-autonomous impact on vasculature. But until recently it was unclear whether HIF signals also have a cell-autonomous effect on astrocyte development.
Several recent studies have clarified the role of hypoxia and HIF signaling within developing astrocytes. Astrocyte-specific deletion of HIF2α causes a cell-autonomous proliferation defect, which leads to a striking reduction in astrocyte numbers (Duan et al., 2014; Perelli et al., 2021). Conversely, mutations that elevate HIF signaling increase astrocyte numbers (Duan and Fong, 2019). These findings suggest that astrocyte proliferation is under the control of the HIF pathway. Supporting this idea, manipulations of environmental oxygen levels also have a major impact on astrocyte proliferation. Rearing neonatal mice in a high oxygen environment suppresses astrocyte proliferation, whereas exposure to a hypoxic environment increases astrocyte mitotic activity in a HIF2α-dependent manner (Perelli et al., 2021). Together these findings indicate that astrocytes use HIF2α to monitor local oxygen levels and adjust their proliferation rate accordingly. Hypoxia can also affect astrocyte proliferation indirectly, signaling through HIF1α in the neural retina to induce expression of the mitogen PDGF-A. When HIF1α is deleted from retinal progenitors, PDGF-A is downregulated and astrocyte cell numbers are diminished (Nakamura-Ishizu et al., 2012).
Together, these studies highlight the importance of oxygen sensing in the control of astrocyte numbers. Proliferation is an energetically costly activity, so it is somewhat surprising that astrocytes would proliferate more when oxygen is scarce. Why might astrocytes do this? We propose that this response is a key part of the feedback loop controlling retinal angiogenesis. When the retina is hypoxic, the HIF-VEGF pathway serves the critical role of recruiting formation of vasculature that will relieve tissue hypoxia. The HIF-dependent proliferation mechanism fits nicely into this model: it ensures that hypoxia will not only upregulate VEGF-A expression but will also enhance production of the cells that are best able to supply VEGF-A.
How might HIF2α regulate proliferation? While Tlx has not yet been proven to be a HIF target, it has many hallmarks of a HIF-regulated transcript. For example, Tlx is upregulated selectively in the astrocytes of avascular retina, where tissue hypoxia is greatest. Furthermore, Tlx expression is downregulated upon exposure to high oxygen (Uemura et al., 2006). Therefore, it seems highly likely that Tlx expression is regulated by HIF2α, or at the very least that it is a downstream effector of the HIF pro-proliferation signal. In the future, it will be interesting to see if there are any other proliferation effectors downstream of HIF, and how astrocytes integrate the various proliferation signals to ensure appropriate cell numbers. Failure to control cell numbers could have severe pathobiological consequences, as we will see below in the section on developmental retinopathies, so the precise integration of proliferation signals is likely to be of high importance for ensuring normal development.
Astrocyte differentiation: The role of vasculature
After astrocytes have completed their mission as angiogenesis guides, they undergo rapid molecular and anatomical changes. Anatomically, the astrocyte network experiences a dramatic reorganization once vessels have passed through. This reorganization involves disassembly of the circular astrocyte soma arrangement that is used during the vascular template phase. Following this rearrangement, a subset of astrocytes stays closely associated with major vessels (Stone and Dreher, 1987), while the rest of the astrocyte population becomes uniformly spaced across the adult retina (Fig. 2B). This spacing is achieved through an as-yet undiscovered repulsive “contact-spacing” mechanism (Chan-Ling and Stone, 1991b). Remodeling of the astrocyte network is accompanied by molecular changes, including: 1) Cessation of proliferative activity (as noted in the previous section above). This includes loss of sensitivity to mitogens such as PDGF-A (West et al., 2005). 2) Upregulation of GFAP. This is the canonical marker for maturation of retinal astrocytes (Duan et al., 2017; West et al., 2005). 3) Downregulation of immature astrocyte markers such as Tlx and VEGF-A (Gerhardt et al., 2003; Uemura et al., 2006; West et al., 2005). No doubt there are other significant molecular changes needed to implement this reorganization, as well as additional molecules that are induced once the astrocyte network has taken on its new configuration; however, these remain poorly characterized.
How does the arrival of vessels promote astrocyte maturation? One way is through increased delivery of oxygen. Enhanced oxygen delivery downregulates HIF signaling in astrocytes, which directly suppresses VEGF-A expression (Stone et al., 1995; West et al., 2005). Oxygen also suppresses expression of the Tlx transcription factor, a recurring character in this review because of its many crucial functions within immature astrocytes. As we have seen in previous sections, Tlx is required for astrocyte migratory activity and for astrocyte proliferation – two fundamental cellular behaviors that distinguish immature astrocytes from mature ones. In addition, Tlx also maintains astrocytes in a GFAPlow state via direct transcriptional repression of the Gfap locus (Shi et al., 2004; Uemura et al., 2006). Because Tlx regulates so many aspects of astrocyte immaturity, oxygen-dependent Tlx downregulation is likely one of the key mechanisms by which arrival of vessels drives astrocyte maturation.
Oxygen is not the only signal from blood vessels that drives astrocyte differentiation: They also bring along their own set of signaling molecules that can influence astrocyte development. Endothelial cell-derived leukemia inhibitory factor (LIF) serves as a maturation factor, which promotes downregulation of VEGF-A and suppresses proliferation. LIF knockout mice exhibit prolonged VEGF-A expression and proliferative capacity even after the vessels have colonized a given area (Kubota et al., 2008). A second signaling molecule produced by endothelial cells is apelin; this peptide acts in an autocrine fashion to induce the secretion of LIF, thereby enhancing astrocyte maturation (Sakimoto et al., 2012). Apelin also plays a role in promoting angiogenesis – it drives proliferation of endothelial tip cells, the specialized filopodial cells located at the angiogenic vanguard. These dual roles create a complex sequence of events: Apelin secretion causes a short-term increase in angiogenesis, but this is countered over the long-term by increased LIF signaling and enhanced astrocyte maturation, which diminish astrocyte-derived angiogenic cues (Sakimoto et al., 2012).
Despite the progress in identifying signals for astrocyte maturation, it remains unclear whether the cues identified so far are sufficient to explain why vasculature can induce such a rapid and dramatic phenotypic change in astrocytes. More research is needed to determine whether other signaling molecules are involved in promoting the transition to astrocyte maturity. It is also unclear how the different maturation signals from vasculature are integrated to drive distinct aspects of the mature astrocyte phenotype. Finally, almost nothing is known about the physical rearrangement of astrocyte somata – either how it is accomplished or why it is necessary. These will be important topics for future work.
Developmental death of retinal astrocytes
Naturally occurring cell death is a major contributor to developmental tissue patterning, especially in the nervous system (Cecconi et al., 1998; Raven et al., 2003; Yamaguchi and Miura, 2015). It would not be surprising if death were involved in sculpting the retinal astrocyte population into a template for angiogenesis, or in dismantling the template when angiogenesis is complete. However, until recently little was known about developmental astrocyte death – not only in the retina but also throughout the nervous system. Surprisingly few studies have explored whether astrocyte numbers decline over development. While astrocytes have been reported to die via apoptosis, both in the retina and in the brain (Bucher et al., 2013; Chan-Ling et al., 2009; Distler et al., 2000; Foo et al., 2011; Soriano et al., 1993), the impact of these apoptotic deaths on the size of the astrocyte population has not typically been evaluated – leaving open the question of whether developmental cell death is relevant to astrocyte patterning.
Puñal et al. (2019) showed that retinal astrocytes do indeed undergo naturally occurring cell death. The extent of cell loss is particularly dramatic, as astrocytes are overproduced by a factor of three before being culled to their final numbers during the first two weeks of mouse postnatal development. Consistent with past reports, apoptotic astrocytes were observed; however, modeling showed that the frequency of apoptosis could not account for the magnitude of astrocyte loss. Further, when apoptosis mechanisms were blocked using an astrocyte-specific Bax knockout mouse, there was no effect on astrocyte removal, suggesting that apoptosis is not the main mechanism through which astrocytes die during development. This was quite unexpected, since the vast majority of developmental cell death occurs by apoptosis.
Instead, astrocytes appear to be killed and engulfed by microglia, the resident immune cells and major phagocytic cells of the CNS. Astrocyte material is found within phagocytic cups and lysosomes of microglia, but only during the developmental period of astrocyte death (Fig. 4C). Furthermore, ablation of microglia leads to accumulation of excess astrocytes that survive, differentiate, and incorporate into the mature astrocyte network. This finding suggests microglia are active participants in astrocyte death. Microglia also participate in conventional apoptotic death, but in a much more limited and passive manner: When microglia are experimentally eliminated, the corpses of apoptotic neurons accumulate within the developing retina, but there is no increase in the number of viable neurons (Puñal et al., 2019). Thus, for apoptosis, microglia play the nonessential role of simply cleaning up the corpses. By contrast, microglia ablation increases astrocyte survival, demonstrating that microglia are key effectors of astrocyte death.
Two key findings about the developmental death of astrocytes illustrate how important this process is for the overall development of the retina. First, the underlying mechanisms are extremely redundant – it is very difficult to completely break this system. Puñal et al. (2019) used multiple mutant mouse strains to block the major microglial phagocytosis pathways, but no single pathway emerged as the main one responsible for astrocyte loss. This finding suggests a high degree of redundancy between these pathways, although it is always possible that some unique undescribed pathway could mediate astrocyte clearance. Furthermore, even fully removing microglia from the retina does not permanently block death. While death is initially delayed, astrocytes begin dying again approximately two days later, suggesting that a compensatory non-microglial mechanism has been activated to ensure astrocyte death. This compensatory mechanism appears to involve astrocytes taking over the microglial killing role, as astrocytes were observed to engulf each other when microglia were absent (Puñal et al., 2019).
A second indication of the importance of astrocyte death comes from observing the consequences for vasculature when death does not proceed normally. In Csflr−/− mice lacking microglia, the denser astrocyte network is accompanied by bleeding from RNFL vasculature (Puñal et al., 2019). The mechanism underlying this phenotype is still to be determined, but it is consistent with the phenotype of other mice in which astrocyte numbers are experimentally increased (Fruttiger et al., 1996; Perelli et al., 2021). Therefore, we favor the interpretation that RNFL bleeding in Csflr mutants is due to increased astrocyte survival, which leads either to an astrocyte patterning defect or an overabundance of an astrocyte-derived factor such as VEGF-A. This model remains to be tested.
Now that we know developing astrocytes die in large numbers, and we understand something of the mechanisms by which they die, it may soon become possible to manipulate death and study its role in astrocyte patterning. However, we are not quite there yet. Eliminating microglia entirely is a fairly blunt manipulation that likely has nonspecific effects. Identification of the molecular mechanisms by which microglia kill astrocytes would enable a more targeted approach, blocking microglia from killing astrocytes without otherwise disturbing their physiology. This approach would entail identification of the relevant “eat-me” signals on astrocytes, and/or their receptors on microglia, but for now these molecules remain unknown. In addition to enhanced specificity, there may be other advantages to blocking these pathways: If it turns out that astrocytes use the same receptor-ligand system to perform compensatory eating, blocking this molecular interaction might prove to have larger effects on astrocyte survival than simply eliminating microglia. Achieving a more complete blockade of astrocyte death will provide a clearer picture of the consequences for retinal development when death does not occur normally.
Aside from the molecular mechanism for astrocyte death, several other key open questions remain. What causes astrocyte removal to begin at a certain moment in development, and what causes it to stop? Does clearance continue until astrocytes reach a certain critical final cell number? Or are there specific signals that terminate the death period – e.g. developmental changes in astrocyte or microglial gene expression? There is evidence that astrocytes themselves may express cues that drive microglia to remove them. Across development, as astrocyte numbers first increase and then decline, there is a strikingly close correlation between astrocyte number and the phagocytic capacity of microglia (as determined by expression of the lysosomal marker CD68). This correlation supports a model in which either total astrocyte numbers or local astrocyte density is responsible for driving microglia to remove them (Puñal et al., 2019). However, the signaling molecules involved in this process remain a mystery. Further, the mechanism used to determine which astrocytes are removed and which remain alive is also unknown. Is an “eat-me” signal exposed by some astrocytes but not others? If so, what causes this dichotomy in astrocyte behavior? Answering these questions will yield a better understanding of the unusual mode of astrocyte death in the retina, with potential implications for understanding astrocyte death throughout the nervous system.
Role of astrocytes in developmental disorders of the retina
Retinopathy of prematurity (ROP) is a disorder of retinal vascular development that occurs exclusively in preterm children. With advances in neonatology that have improved survival rates of severely premature infants, ROP has become a worldwide leading cause of childhood blindness (Hartnett and Penn, 2012). The major risk factor for ROP is exposure to supplemental oxygen. While oxygen treatment is often a lifesaving necessity for these children, it can have unintended negative consequences for retinal blood vessel formation. Under high-oxygen conditions the hypoxic drive for angiogenesis is diminished, which halts the advance of vasculature across the retina. This is known as Phase I of ROP. In Phase II, return to normoxic conditions reactivates angiogenesis, but vessels do not resume growing in an orderly fashion along the astrocyte template. Instead, they engage in unpatterned neovascular growth that can lead to hemorrhage, inflammation, and ultimately retinal detachment (Hellström et al., 2013).
Given their importance in patterning the vasculature during angiogenesis, astrocytes are certainly positioned to play a role in the pathogenesis of ROP. If oxygen exposure were to disrupt development of the astrocyte template, this might contribute to disease processes such as the halting of angiogenesis in Phase I or the failure of orderly RNFL angiogenesis in Phase II. Few studies have probed this model in depth. However, histopathological studies suggest that ROP could involve defects in astrocyte development: Ahead of the angiogenic wavefront, the astrocyte layer becomes thicker and clumped in ROP tissue, suggesting an excess of astrocytes and/or altered astrocyte patterning (Foos, 1987; Sun et al., 2010). It is plausible that such changes could cause vascular defects: We know from mouse studies that overproduction of astrocytes can delay angiogenesis and alter vascular patterning (Duan and Fong, 2019; Fruttiger et al., 1996).
Nevertheless, prevailing models of ROP pathogenesis have emphasized direct effects of oxygen stress on the vasculature. One possible reason for this is that the dominant mouse model of ROP, known as the oxygen-induced retinopathy (OIR) model, is not designed to probe early stages of the disease process that may involve astrocytes. In OIR, mice are exposed to high oxygen at P7 and returned to room air at P12. This model has proven extremely valuable in understanding how relative hypoxia – i.e. the P12 return to room air – induces formation of neovascular tufts, a key pathological feature of ROP. However, the model is less suitable for investigating how oxygen stress affects development of the astrocyte template or the initial angiogenic events in the RNFL, which are largely complete by P7 (Fig. 2). Therefore, the insights emerging from this model have necessarily emphasized the pathobiology of Phase II ROP rather than the early stages that may involve astrocytes.
To test the impact of oxygen stress on formation of the astrocyte template, Perelli et al. (2021) shifted the timing of oxygen exposure so that it coincides with RNFL development. In the neonatal oxygen-induced retinopathy (NOIR) protocol, mice are exposed to high oxygen (75%) on the day of birth (P0) and returned to room air (21% oxygen) at P4. The decrement in oxygen levels at P4 triggers HIF-dependent astrocyte proliferation, leading to a massive overproduction of astrocytes and disruption of astrocyte patterning. These errors within the astrocyte template are present ahead of the vascular wavefront, positioning them to impede angiogenesis. Consequently, a variety of vascular developmental defects are seen in NOIR-treated mice including delayed RNFL angiogenesis, patterning errors within the RNFL vascular network, and hemorrhage. The severity of these vascular phenotypes in any given animal is proportional to the amount of excess astrocytes, strongly suggesting that exuberant astrocyte proliferation contributes to vessel defects (Perelli et al., 2021).
How relevant is this NOIR model for understanding ROP pathogenesis? Perelli et al. (2021) found long-lasting retinal pathologies in NOIR-treated mice, many of which resemble pathological features of ROP (Foos, 1987). For example, NOIR mice exhibit persistence of the hyaloid vasculature, retinal detachment, and vitreous hemorrhage – all of which are key features of severe ROP that are absent from the standard OIR model (Hartnett and Penn, 2012). Similar pathologies are also generated upon treatment with high oxygen from P0-P7 (McMenamin et al., 2016). These findings strongly suggest that neonatal oxygen exposure provides a useful new model for certain features of ROP, particularly the early stages that are related to RNFL development. Of course, standard OIR remains an important model for other pathologies, such as neovascular tuft formation, that are not prominent features of the NOIR model (Perelli et al., 2021). As medical advances continue to improve survival of the most premature infants, models of the earliest pathological events may become increasingly relevant to clinical practice. Therefore, we envision that both mouse models will remain useful for dissecting distinct aspects of ROP pathobiological mechanisms.
The finding that oxygen stress can perturb development of the astrocyte template suggests intriguing new treatment possibilities for ROP. Today, the major ROP treatments are focused on minimizing neovascularization during Phase II by blocking VEGF-A function (Wood et al., 2021). This can be done by ablating the peripheral avascular retina, which is the major source of VEGF-A signals; or by administering anti-VEGF drugs. Both approaches have drawbacks: Surgery permanently destroys retinal tissue, while anti-VEGF therapy often prevents the completion of angiogenesis, leading to potential future complications within avascular retinal regions (Wood et al., 2021). Moreover, because of the pattern of angiogenic spread in human retina, the impact of anti-VEGF is not necessarily restricted to peripheral retina but may also delay angiogenesis within high-acuity macular regions (Chen et al., 2020). Off-target effects are an additional safety concern when anti-VEGF therapy is used in infants. Even with intraocular administration, these drugs are known to reach the circulatory system and to affect systemic VEGF signaling, raising the possibility that other VEGF-dependent developmental processes could be inadvertently impaired (Wood et al., 2021). Finally, neither surgery nor anti-VEGF approaches treat the underlying problems that cause arrest of vessel growth in the first place. If there were a way to prevent oxygen therapy from disrupting astrocyte development, this might allow angiogenesis to proceed more normally once the patient is returned to room air. Such an approach should reduce the formation of pathological neovascularizations that would require treatment via anti-VEGF therapy. The results of Perelli et al. (2021) suggest that preventing hypoxia-induced astrocyte hyperproliferation could be one way to normalize astrocyte development during oxygen stress. There has been some interest in drugs that block the HIF pathway itself (Hoppe et al., 2016; Lee et al., 2021; Sears et al., 2008); however, such an approach might produce too many side effects because, at least in mice, entirely blocking astrocyte HIF signaling also blocks VEGF-A production and prevents angiogenesis (Duan et al., 2014; Perelli et al., 2021). Further work will be needed to identify other ways of blunting astrocyte proliferation and to see whether these interventions might improve NOIR pathologies.
Retinal astrocytes and their commonalities with other CNS glia
Due to their selective interactions with axons, retinal astrocytes have much in common with the astroglia located in white matter tracts of the brain and spinal cord. Thus, in the mature CNS, these astrocyte populations likely all serve comparable functions related to the maintenance of axon health. While ROP is a retina-specific disease, there are other disorders affecting the brain that are also linked to defects in astrocyte development. For example, in the leukodystrophy known as vanishing white matter disease, astrocytes adopt an abnormal morphology and fail to mature fully – a phenotype that may be highly relevant to the astrocyte maturation signals we have discussed here. In this disease, astrocytic failure to mature also prevents the maturation of oligodendrocyte precursor cells, leading to a lack of myelin in affected white matter areas. Additionally, pathological astrocytes negatively affect the health of neurons in the area, with thinner axons and increased numbers of unmyelinated axons being present in vanishing white matter animal models (Bugiani et al., 2018; Dietrich et al., 2005; Dooves et al., 2016).
What exactly are astrocytes doing to keep axons happy, and which of these functions become impaired in disease? It has long been clear that astrocytes provide metabolic support to axons and make important contributions to blood-CNS barriers. Aside from these functions, however, the specific ways in which astrocytes support axons remain surprisingly murky. Recent studies in the retina have highlighted some intriguing new astrocyte functions that may turn out to be critical for maintaining axon health. One example is transcellular mitophagy: RGC axons appear to shed unneeded or dysfunctional mitochondria which are engulfed and degraded by astrocytes (Davis et al., 2014). This phenomenon was observed particularly frequently within the specialized population of mouse astrocytes that forms the glial lamina, the boundary between retina and optic nerve through which RGC axons pass as they exit the eye. However, it was also observed in other CNS regions such as layer 1 white matter in mouse cerebral cortex (Davis et al., 2014). Because astrocytes at the glial lamina are implicated in glaucoma pathogenesis, this mitochondrial engulfment phenomenon may turn out to be relevant to disease. However, this remains to be fully explored.
Another example of a supportive astrocyte function has also emerged from studies of mouse glaucoma models. Astrocytes are the major CNS site of glycogen storage, which can be deployed in times of high energy demand to support glycolytic ATP production. A recent study suggests that astrocytes can shuttle glycogen and other metabolites over long distances through connexin-43 gap junctions, thereby delivering metabolic resources to sites of axonal stress or injury (Cooper et al., 2020). Using a mouse glaucoma model that involves an acute increase in intraocular pressure, these authors found that glycogen shuttling occurs rapidly after the insult to RGC axons and serves to protect RGCs from dysfunction and death. However, this protective function comes at the cost of increased vulnerability to injury within the donating regions, presumably because local astrocytes have already given away their metabolic resources (Cooper et al., 2020). It is easy to envision how such a mechanism, if confirmed, could generalize to other CNS regions, promoting axon health in times of acute stress but contributing to disease pathogenesis when axonal insults are widely distributed or sustained over time.
As suggested by these two examples, illuminating the functions of axon-associated astrocytes will be important not only for understanding basic astrocyte biology, but also for insight into degenerative diseases. Retinal astrocytes have several key advantages as an experimental system to delve into this problem: Because they are easily accessible on the surface of the retina and exist in a two-dimensional network – unlike the complex, three-dimensional astrocyte network within brain white matter tracts – retinal astrocytes offer anatomical and experimental simplicity. Discovering more about the biology and functions of retinal astrocytes should illuminate shared functions of axon-associated astrocytes throughout the CNS.
One key function of retinal astrocytes that we have highlighted here – their role in angiogenesis – appears to work differently in the brain and spinal cord. Whereas retinal astrocytes dictate angiogenesis through patterning cues and angiogenic growth factors, these jobs are shared in white matter between astrocytes and another axon-associated glial population, the oligodendrocyte precursor cells (OPCs). Similar to retinal astrocytes, white matter OPCs physically interact with angiogenic tip cells and use HIF-VEGF (as well as Wnt) signaling to promote angiogenesis (Chavali et al., 2020; Yuen et al., 2014; Zhang et al., 2020). They also proliferate in a hypoxia and HIF-dependent manner, ensuring sufficient quantities of OPCs are present to drive angiogenesis (Chavali et al., 2020). Nevertheless, even though many of the key angiogenic functions are assumed by OPCs, astrocytes also contribute to angiogenesis via HIF signaling (Ma et al., 2012; Zhang et al., 2020). The involvement of multiple cell types and multiple molecular pathways (i.e. Wnt and HIF) may explain why mouse white-matter vascular phenotypes are typically more subtle than those obtained by manipulating retinal astrocytes. Because OPCs are absent from the retina, it seems that retinal astrocytes are forced to bear a greater responsibility for angiogenesis.
Conclusion
In this review, we have described the ways in which retinal astrocytes are essential for the development and function of the retina as a whole. Astrocytes provide both chemical (i.e. VEGF) and physical (i.e. template) cues to endothelial cells, which underscores the significant role of astrocytes in the recruitment and patterning of retinal vasculature. The formation of the astrocyte template and regulation of astrocyte number are tightly controlled to ensure the vasculature develops correctly. When these processes go awry, it can be devastating for the function of the retina, as in severe cases of ROP.
While we are continually learning more about astrocytes’ role in development, we know comparatively little about their function in the adult retina. Recent intriguing discoveries concerning transcellular mitophagy and long-range nutrient delivery highlight the surprisingly wide cell-biological behavioral repertoire of axon-associated astrocytes. Going forward, it will be important both to identify these new and unexpected astrocyte behaviors as well as to delineate the specific functions of the behaviors we already know about. Success in these endeavors will not only yield new insights into visual system physiology, as we begin to delineate the specific functions of retinal astrocytes, but it should also provide general insights into the function of astrocytes throughout the CNS, especially in the white matter tracts of the brain.
Highlights.
New features of astrocyte biology are emerging from studies of retinal astrocytes
Astrocyte pattern formation enables retinal angiogenesis
Astrocytes undertake a long-distance migration to colonize the retinal tissue
Local oxygen levels control multiple steps in astrocyte development
Defects in astrocyte development may contribute to retinopathy of prematurity
Acknowledgements
This work was supported by the NIH (EY030611 to J.N.K.) and a Duke University Holland-Trice Graduate Student Award (to C.E.P.). We thank Vanessa Puñal for the images in Figure 2; Matthew O’Sullivan for the images in Figure 4A,B; and Robin Perelli for the image in Figure 1B. We thank members of the Kay lab for providing thoughtful comments on the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
References
- Allen NJ, Eroglu C, 2017. Cell Biology of Astrocyte-Synapse Interactions. Neuron 96, 697–708. 10.1016/j.neuron.2017.09.056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosze B, Suarez-Navarro J, Soofi A, Lauderdale JD, Dressler GR, Brown NL, 2021. Multiple roles for Pax2 in the embryonic mouse eye. Dev. Biol 472, 18–29. 10.1016/j.ydbio.2020.12.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bucher F, Stahl A, Agostini HT, Martin G, 2013. Hyperoxia causes reduced density of retinal astrocytes in the central avascular zone in the mouse model of oxygen-induced retinopathy. Mol. Cell. Neurosci 56, 225–33. 10.1016/j.mcn.2013.06.001 [DOI] [PubMed] [Google Scholar]
- Bugiani M, Vuong C, Breur M, van der Knaap MS, 2018. Vanishing white matter: a leukodystrophy due to astrocytic dysfunction. Brain Pathol 28, 408–421. 10.1111/bpa.12606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Büssow H, 1980. The astrocytes in the retina and optic nerve head of mammals: a special glia for the ganglion cell axons. Cell Tissue Res 206, 367–78. 10.1007/BF00237966 [DOI] [PubMed] [Google Scholar]
- Cecconi F, Alvarez-Bolado G, Meyer BI, Roth KA, Gruss P, 1998. Apaf1 (CED-4 homolog) regulates programmed cell death in mammalian development. Cell 94, 727–737. 10.1016/S0092-8674(00)81732-8 [DOI] [PubMed] [Google Scholar]
- Chan-Ling T, Chu Y, Baxter L, Weible Ii M, Hughes S, 2009. In vivo characterization of astrocyte precursor cells (APCs) and astrocytes in developing rat retinae: differentiation, proliferation, and apoptosis. Glia 57, 39–53. 10.1002/glia.20733 [DOI] [PubMed] [Google Scholar]
- Chan-Ling T, Stone J, 1991a. Factors determining the migration of astrocytes into the developing retina: migration does not depend on intact axons or patent vessels. J. Comp. Neurol 303, 375–86. 10.1002/cne.903030304 [DOI] [PubMed] [Google Scholar]
- Chan-Ling T, Stone J, 1991b. Factors determining the morphology and distribution of astrocytes in the cat retina: a “contact-spacing” model of astrocyte interaction. J. Comp. Neurol 303, 387–99. 10.1002/cne.903030305 [DOI] [PubMed] [Google Scholar]
- Chavali M, Ulloa-Navas MJ, Pérez-Borredá P, Garcia-Verdugo JM, McQuillen PS, Huang EJ, Rowitch DH, 2020. Wnt-Dependent Oligodendroglial-Endothelial Interactions Regulate White Matter Vascularization and Attenuate Injury. Neuron 108, 1130–1145.e5. 10.1016/j.neuron.2020.09.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Imperio R, Seely KR, Viehland C, Izatt JA, Prakalapakorn SG, Freedman SF, Toth CA, 2020. Slow progressive perifoveal vascular formation in an infant with aggressive posterior retinopathy of prematurity. J. AAPOS 24, 323–326. 10.1016/j.jaapos.2020.07.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu Y, Hughes S, Chan-Ling T, 2001. Differentiation and migration of astrocyte precursor cells and astrocytes in human fetal retina: relevance to optic nerve coloboma. FASEB J 15, 2013–5. 10.1096/fj.00-0868fje [DOI] [PubMed] [Google Scholar]
- Clements R, Turk R, Campbell KP, Wright KM, 2017. Dystroglycan Maintains Inner Limiting Membrane Integrity to Coordinate Retinal Development. J. Neurosci 37, 8559–8574. 10.1523/JNEUROSCI.0946-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper ML, Pasini S, Lambert WS, D’Alessandro KB, Yao V, Risner ML, Calkins DJ, 2020. Redistribution of metabolic resources through astrocyte networks mitigates neurodegenerative stress. Proc. Natl. Acad. Sci. U. S. A 117, 18810–18821. 10.1073/pnas.2009425117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dakubo GD, Wang YP, Mazerolle C, Campsall K, McMahon AP, Wallace VA, 2003. Retinal ganglion cell-derived sonic hedgehog signaling is required for optic disc and stalk neuroepithelial cell development. Development 130, 2967–80. [DOI] [PubMed] [Google Scholar]
- Davis CHO, Kim KY, Bushong EA, Mills EA, Boassa D, Shih T, Kinebuchi M, Phan S, Zhou Y, Bihlmeyer NA, Nguyen JV, Jin Y, Ellisman MH, Marsh-Armstrong N, 2014. Transcellular degradation of axonal mitochondria. Proc. Natl. Acad. Sci. U. S. A 111, 9633–9638. 10.1073/pnas.1404651111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deiner MS, Kennedy TE, Fazeli A, Serafini T, Tessier-Lavigne M, Sretavan DW, 1997. Netrin-1 and DCC mediate axon guidance locally at the optic disc: loss of function leads to optic nerve hypoplasia. Neuron 19, 575–89. 10.1016/s0896-6273(00)80373-6 [DOI] [PubMed] [Google Scholar]
- Dietrich J, Lacagnina M, Gass D, Richfield E, Mayer-Pröschel M, Noble M, Torres C, Pröschel C, 2005. EIF2B5 mutations compromise GFAP+ astrocyte generation in vanishing white matter leukodystrophy. Nat. Med 11, 277–283. 10.1038/nm1195 [DOI] [PubMed] [Google Scholar]
- Distler C, Kopatz K, Telkes I, 2000. Developmental changes in astrocyte density in the macaque perifoveal region. Eur. J. Neurosci 12, 1331–41. 10.1046/j.1460-9568.2000.00029.x [DOI] [PubMed] [Google Scholar]
- Dooves S, Bugiani M, Postma NL, Polder E, Land N, Horan ST, van Deijk A-LF, van de Kreeke A, Jacobs G, Vuong C, Klooster J, Kamermans M, Wortel J, Loos M, Wisse LE, Scheper GC, Abbink TEM, Heine VM, van der Knaap MS, 2016. Astrocytes are central in the pathomechanisms of vanishing white matter. J. Clin. Invest 126, 1512–24. 10.1172/JCI83908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorrell MI, Aguilar E, Friedlander M, 2002. Retinal vascular development is mediated by endothelial filopodia, a preexisting astrocytic template and specific R-cadherin adhesion. Invest. Ophthalmol. Vis. Sci 43, 3500–10. [PubMed] [Google Scholar]
- Duan L-J, Fong G-H, 2019. Developmental vascular pruning in neonatal mouse retinas is programmed by the astrocytic oxygen-sensing mechanism. Development 146, dev175117. 10.1242/dev.175117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan L-J, Takeda K, Fong G-H, 2014. Hypoxia inducible factor-2α regulates the development of retinal astrocytic network by maintaining adequate supply of astrocyte progenitors. PLoS One 9, e84736. 10.1371/journal.pone.0084736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan LJ, Pan SJ, Sato TN, Fong GH, 2017. Retinal Angiogenesis Regulates Astrocytic Differentiation in Neonatal Mouse Retinas by Oxygen Dependent Mechanisms. Sci. Rep 7, 1–16. 10.1038/s41598-017-17962-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards MM, McLeod DS, Li R, Grebe R, Bhutto I, Mu X, Lutty G a, 2012. The deletion of Math5 disrupts retinal blood vessel and glial development in mice. Exp. Eye Res 96, 147–56. 10.1016/j.exer.2011.12.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foo LC, Allen NJ, Bushong EA, Ventura PB, Chung W-S, Zhou L, Cahoy JD, Daneman R, Zong H, Ellisman MH, Barres BA, 2011. Development of a method for the purification and culture of rodent astrocytes. Neuron 71, 799–811. 10.1016/j.neuron.2011.07.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foos RY, 1987. Retinopathy of prematurity. Pathologic correlation of clinical stages. Retina 7, 260–76. [PubMed] [Google Scholar]
- Fruttiger M, 2002. Development of the mouse retinal vasculature: angiogenesis versus vasculogenesis. Invest. Ophthalmol. Vis. Sci 43, 522–7. [PubMed] [Google Scholar]
- Fruttiger M, Calver AR, Krüger WH, Mudhar HS, Michalovich D, Takakura N, Nishikawa S, Richardson WD, 1996. PDGF mediates a neuron-astrocyte interaction in the developing retina. Neuron 17, 1117–31. 10.1016/s0896-6273(00)80244-5 [DOI] [PubMed] [Google Scholar]
- Gerhardt H, Golding M, Fruttiger M, Ruhrberg C, Lundkvist A, Abramsson A, Jeltsch M, Mitchell C, Alitalo K, Shima D, Betsholtz C, 2003. VEGF guides angiogenic sprouting utilizing endothelial tip cell filopodia. J. Cell Biol 161, 1163–1177. 10.1083/jcb.200302047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gnanaguru G, Bachay G, Biswas S, Pinzón-Duarte G, Hunter DD, Brunken WJ, 2013. Laminins containing the β2 and γ3 chains regulate astrocyte migration and angiogenesis in the retina. Development 140, 2050–60. 10.1242/dev.087817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halfter W, Dong S, Schurer B, Osanger A, Schneider W, Ruegg M, Cole GJ, 2000. Composition, synthesis, and assembly of the embryonic chick retinal basal lamina. Dev. Biol 220, 111–28. 10.1006/dbio.2000.9649 [DOI] [PubMed] [Google Scholar]
- Hartnett ME, Penn JS, 2012. Mechanisms and management of retinopathy of prematurity. N. Engl. J. Med 367, 2515–26. 10.1056/NEJMra1208129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellström A, Smith LEH, Dammann O, 2013. Retinopathy of prematurity. Lancet (London, England) 382, 1445–57. 10.1016/S0140-6736(13)60178-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt CE, Bertsch TW, Ellis HM, Harris WA, 1988. Cellular determination in the Xenopus retina is independent of lineage and birth date. Neuron 1, 15–26. 10.1016/0896-6273(88)90205-x [DOI] [PubMed] [Google Scholar]
- Hoppe G, Yoon S, Gopalan B, Savage AR, Brown R, Case K, Vasanji A, Chan ER, Silver RB, Sears JE, 2016. Comparative systems pharmacology of HIF stabilization in the prevention of retinopathy of prematurity. Proc. Natl. Acad. Sci. U. S. A 113, E2516–25. 10.1073/pnas.1523005113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köhler S, Winkler U, Hirrlinger J, 2021. Heterogeneity of Astrocytes in Grey and White Matter. Neurochem. Res 46, 3–14. 10.1007/s11064-019-02926-x [DOI] [PubMed] [Google Scholar]
- Kubota Y, Hirashima M, Kishi K, Stewart CL, Suda T, 2008. Leukemia inhibitory factor regulates microvessel density by modulating oxygen-dependent VEGF expression in mice. J. Clin. Invest 118, 2393–403. 10.1172/JCI34882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee D, Miwa Y, Kunimi H, Ibuki M, Shoda C, Nakai A, Kurihara T, 2021. HIF Inhibition Therapy in Ocular Diseases. Keio J. Med 10.2302/kjm.2021-0004-IR [DOI] [PubMed] [Google Scholar]
- Libby RT, Champliaud MF, Claudepierre T, Xu Y, Gibbons EP, Koch M, Burgeson RE, Hunter DD, Brunken WJ, 2000. Laminin expression in adult and developing retinae: evidence of two novel CNS laminins. J. Neurosci 20, 6517–28. 10.1523/JNEUROSCI.20-17-06517.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling TL, Mitrofanis J, Stone J, 1989. Origin of retinal astrocytes in the rat: evidence of migration from the optic nerve. J. Comp. Neurol 286, 345–52. 10.1002/cne.902860305 [DOI] [PubMed] [Google Scholar]
- Ling TL, Stone J, 1988. The development of astrocytes in the cat retina: evidence of migration from the optic nerve. Brain Res. Dev. Brain Res 44, 73–85. 10.1016/0165-3806(88)90119-8 [DOI] [PubMed] [Google Scholar]
- Ma S, Kwon HJ, Huang Z, 2012. A functional requirement for astroglia in promoting blood vessel development in the early postnatal brain. PLoS One 7, e48001. 10.1371/journal.pone.0048001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquardt T, Ashery-Padan R, Andrejewski N, Scardigli R, Guillemot F, Gruss P, 2001. Pax6 is required for the multipotent state of retinal progenitor cells. Cell 105, 43–55. [DOI] [PubMed] [Google Scholar]
- McMenamin PG, Kenny R, Tahija S, Lim J, Naranjo Golborne C, Chen X, Bouch S, Sozo F, Bui B, 2016. Early Postnatal Hyperoxia in Mice Leads to Severe Persistent Vitreoretinopathy. Invest. Ophthalmol. Vis. Sci 57, 6513–6526. 10.1167/iovs.16-19928 [DOI] [PubMed] [Google Scholar]
- Miyawaki T, Uemura A, Dezawa M, Yu RT, Ide C, Nishikawa S, Honda Y, Tanabe Y, Tanabe T, 2004. Tlx, an orphan nuclear receptor, regulates cell numbers and astrocyte development in the developing retina. J. Neurosci 24, 8124–34. 10.1523/JNEUROSCI.2235-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morcillo J, Martínez-Morales JR, Trousse F, Fermin Y, Sowden JC, Bovolenta P, 2006. Proper patterning of the optic fissure requires the sequential activity of BMP7 and SHH. Development 133, 3179–90. 10.1242/dev.02493 [DOI] [PubMed] [Google Scholar]
- Morita A, Ushikubo H, Mori A, Arima S, Sakamoto K, Nagamitsu T, Ishii K, Nakahara T, 2017. A delay in vascularization induces abnormal astrocyte proliferation and migration in the mouse retina. Dev. Dyn 246, 186–200. 10.1002/dvdy.24484 [DOI] [PubMed] [Google Scholar]
- Morita A, Ushikubo H, Mori A, Sakamoto K, Nakahara T, 2016. Exposure to high-concentration oxygen in the neonatal period induces abnormal retinal vascular patterning in mice. Birth Defects Res. B. Dev. Reprod. Toxicol 107, 216–224. 10.1002/bdrb.21187 [DOI] [PubMed] [Google Scholar]
- Mudhar HS, Pollock RA, Wang C, Stiles CD, Richardson WD, 1993. PDGF and its receptors in the developing rodent retina and optic nerve. Development 118, 539–52. [DOI] [PubMed] [Google Scholar]
- Nakamura-Ishizu A, Kurihara T, Okuno Y, Ozawa Y, Kishi K, Goda N, Tsubota K, Okano H, Suda T, Kubota Y, 2012. The formation of an angiogenic astrocyte template is regulated by the neuroretina in a HIF-1-dependent manner. Dev. Biol 363, 106–14. 10.1016/j.ydbio.2011.12.027 [DOI] [PubMed] [Google Scholar]
- O’Sullivan ML, Puñal VM, Kerstein PC, Brzezinski JA, Glaser T, Wright KM, Kay JN, 2017. Astrocytes follow ganglion cell axons to establish an angiogenic template during retinal development. Glia 65, 1697–1716. 10.1002/glia.23189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perelli RM, O’Sullivan ML, Zarnick S, Kay JN, 2021. Environmental oxygen regulates astrocyte proliferation to guide angiogenesis during retinal development. Development 148. 10.1242/dev.199418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puñal VM, Paisley CE, Brecha FS, Lee MA, Perelli RM, Wang J, O’Koren EG, Ackley CR, Saban DR, Reese BE, Kay JN, 2019. Large-scale death of retinal astrocytes during normal development is non-apoptotic and implemented by microglia. PLoS Biol 17, e3000492. 10.1371/journal.pbio.3000492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randlett O, Poggi L, Zolessi FR, Harris WA, 2011. The oriented emergence of axons from retinal ganglion cells is directed by laminin contact in vivo. Neuron 70, 266–80. 10.1016/j.neuron.2011.03.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rattner A, Williams J, Nathans J, 2019. Roles of HIFs and VEGF in angiogenesis in the retina and brain. J. Clin. Invest 130, 3807–3820. 10.1172/JCI126655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raven M. a, Eglen SJ, Ohab JJ, Reese BE, 2003. Determinants of the exclusion zone in dopaminergic amacrine cell mosaics. J. Comp. Neurol 461, 123–36. 10.1002/cne.10693 [DOI] [PubMed] [Google Scholar]
- Rodriguez AR, de Sevilla Müller LP, Brecha NC, 2014. The RNA binding protein RBPMS is a selective marker of ganglion cells in the mammalian retina. J. Comp. Neurol 522, 1411–43. 10.1002/cne.23521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakimoto S, Kidoya H, Naito H, Kamei M, Sakaguchi H, Goda N, Fukamizu a, Nishida K, Takakura N, 2012. A role for endothelial cells in promoting the maturation of astrocytes through the apelin/APJ system in mice. Development 139, 1327–1335. 10.1242/dev.072330 [DOI] [PubMed] [Google Scholar]
- Schnitzer J, 1988. Astrocytes in the guinea pig, horse, and monkey retina: Their occurrence coincides with the presence of blood vessels. Glia 1, 74–89. 10.1002/glia.440010109 [DOI] [PubMed] [Google Scholar]
- Schnitzer J, 1985. Distribution and immunoreactivity of glia in the retina of the rabbit. J. Comp. Neurol 240, 128–142. 10.1002/cne.902400203 [DOI] [PubMed] [Google Scholar]
- Schwarz M, Cecconi F, Bernier G, Andrejewski N, Kammandel B, Wagner M, Gruss P, 2000. Spatial specification of mammalian eye territories by reciprocal transcriptional repression of Pax2 and Pax6. Development 127, 4325–34. [DOI] [PubMed] [Google Scholar]
- Sears JE, Hoppe G, Ebrahem Q, Anand-Apte B, 2008. Prolyl hydroxylase inhibition during hyperoxia prevents oxygen-induced retinopathy. Proc. Natl. Acad. Sci. U. S. A 105, 19898–903. 10.1073/pnas.0805817105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selvam S, Kumar T, Fruttiger M, 2018. Retinal vasculature development in health and disease. Prog. Retin. Eye Res 63, 1–19. 10.1016/j.preteyeres.2017.11.001 [DOI] [PubMed] [Google Scholar]
- Shi Y, Lie DC, Taupin P, Nakashima K, Ray J, Yu RT, Gage FH, Evans RM, 2004. Expression and function of orphan nuclear receptor TLX in adult neural stem cells. Nature 427, 78–83. 10.1038/nature02211 [DOI] [PubMed] [Google Scholar]
- Soriano E, Del Río JA, Auladell C, 1993. Characterization of the phenotype and birthdates of pyknotic dead cells in the nervous system by a combination of DNA staining and immunohistochemistry for 5′-bromodeoxyuridine and neural antigens. J. Histochem. Cytochem 41, 819–27. 10.1177/41.6.8315274 [DOI] [PubMed] [Google Scholar]
- Soukkarieh C, Agius E, Soula C, Cochard P, 2007. Pax2 regulates neuronal-glial cell fate choice in the embryonic optic nerve. Dev. Biol 303, 800–13. 10.1016/j.ydbio.2006.11.016 [DOI] [PubMed] [Google Scholar]
- Stone J, Dreher Z, 1987. Relationship between astrocytes, ganglion cells and vasculature of the retina. J. Comp. Neurol 255, 35–49. 10.1002/cne.902550104 [DOI] [PubMed] [Google Scholar]
- Stone J, Itin A, Alon T, Pe’er J, Gnessin H, Chan-Ling T, Keshet E, 1995. Development of retinal vasculature is mediated by hypoxia-induced vascular endothelial growth factor (VEGF) expression by neuroglia. J. Neurosci 15, 4738–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Dalal R, Gariano RF, 2010. Cellular composition of the ridge in retinopathy of prematurity. Arch. Ophthalmol. (Chicago, Ill. 1960) 128, 638–41. 10.1001/archophthalmol.2010.59 [DOI] [PubMed] [Google Scholar]
- Tao C, Zhang X, 2016. Retinal Proteoglycans Act as Cellular Receptors for Basement Membrane Assembly to Control Astrocyte Migration and Angiogenesis. Cell Rep 17, 1832–1844. 10.1016/j.celrep.2016.10.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao C, Zhang X, 2014. Development of astrocytes in the vertebrate eye. Dev. Dyn 243, 1501–10. 10.1002/dvdy.24190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner DL, Cepko CL, 1987. A common progenitor for neurons and glia persists in rat retina late in development. Nature 328, 131–6. 10.1038/328131a0 [DOI] [PubMed] [Google Scholar]
- Uemura A, Kusuhara S, Wiegand SJ, Yu RT, Nishikawa S, 2006. Tlx acts as a proangiogenic switch by regulating extracellular assembly of fibronectin matrices in retinal astrocytes. J. Clin. Invest 116, 369–77. 10.1172/JCI25964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, O’Sullivan ML, Mukherjee D, Puñal VM, Farsiu S, Kay JN, 2017. Anatomy and spatial organization of Müller glia in mouse retina. J. Comp. Neurol 525, 1759–1777. 10.1002/cne.24153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe T, Raff MC, 1988. Retinal astrocytes are immigrants from the optic nerve. Nature 332, 834–7. 10.1038/332834a0 [DOI] [PubMed] [Google Scholar]
- West H, Richardson WD, Fruttiger M, 2005. Stabilization of the retinal vascular network by reciprocal feedback between blood vessels and astrocytes. Development 132, 1855–62. 10.1242/dev.01732 [DOI] [PubMed] [Google Scholar]
- Wilson JW, Shakir D, Batie M, Frost M, Rocha S, 2020. Oxygen-sensing mechanisms in cells. FEBS J 287, 3888–3906. 10.1111/febs.15374 [DOI] [PubMed] [Google Scholar]
- Wood EH, Chang EY, Beck K, Hadfield BR, Quinn AR, Harper CA, 2021. 80 Years of vision: preventing blindness from retinopathy of prematurity. J. Perinatol 10.1038/s41372-021-01015-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi Y, Miura M, 2015. Programmed cell death in neurodevelopment. Dev. Cell 32, 478–490. 10.1016/j.devcel.2015.01.019 [DOI] [PubMed] [Google Scholar]
- Yuen TJ, Silbereis JC, Griveau A, Chang SM, Daneman R, Fancy SPJ, Zahed H, Maltepe E, Rowitch DH, 2014. Oligodendrocyte-encoded HIF function couples postnatal myelination and white matter angiogenesis. Cell 158, 383. 10.1016/J.CELL.2014.04.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Kim B, Zhu X, Gui X, Wang Y, Lan Z, Prabhu P, Fond K, Wang A, Guo F, 2020. Glial type specific regulation of CNS angiogenesis by HIFα-activated different signaling pathways. Nat. Commun 11, 2027. 10.1038/s41467-020-15656-4 [DOI] [PMC free article] [PubMed] [Google Scholar]