

Abstract
Purpose:
Preclinical and clinical data suggest that downstream inhibition with a MEK inhibitor, such as binimetinib, might be efficacious for NRAS-mutated cancers.
Patients and Methods:
Patients enrolled in the NCI-MATCH trial master protocol underwent tumor biopsy and molecular profiling by targeted next-generation sequencing. Patients with NRAS-mutated tumors, except melanoma, were enrolled in subprotocol Z1A, a single-arm study evaluating binimetinib 45 mg twice daily. The primary endpoint was objective response rate (ORR). Secondary endpoints included progression-free survival (PFS) and overall survival (OS). A post-hoc analysis examined the association of NRAS mutation type with outcome.
Results:
In total, 47 eligible patients with a refractory solid tumor harboring a codon 12, 13, or 61 NRAS mutation were treated. Observed toxicity was moderate, and 30% of patients discontinued treatment because of binimetinib-associated toxicity. The ORR was 2.1% (1 of 47 patients). A malignant ameloblastoma patient harboring a codon 61 NRAS mutation achieved a durable partial response (PR). A NRAS codon 61 mutated colorectal cancer patient had an unconfirmed PR, and two other NRAS codon 61 mutated colorectal patients had stable disease for at least 12 months. In an exploratory analysis, colorectal cancer patients bearing a NRAS codon 61 mutation (n = 8) had a significantly longer OS (p = 0.03) and PFS (p = 0.007) than those with codon 12 or 13 mutations (n = 16).
Conclusions:
Single-agent binimetinib did not show promising efficacy in NRAS-mutated cancers. The observation of increased OS and PFS in codon 61 NRAS-mutated colorectal cancer patients merits further investigation.
Keywords: MEK inhibitor, NRAS, targeted therapy, ameloblastoma, colorectal cancer, cholangiocarcinoma
Introduction
RAS mutations result in upregulation of the mitogen-activated protein kinase (MAPK) pathway and are thought to be key driver mutations in many malignancies (1). The importance of RAS mutations is underscored by their high prevalence in human malignancies. RAS, which has three highly homologous isoforms (KRAS, NRAS, and HRAS), is mutated in approximately 19% of human malignancies (2). NRAS-mutated tumors are less common than KRAS-mutated malignancies and are found in approximately 8% of human cancers (3,4). Although the NRAS mutation is most frequently found in melanoma, these mutations are seen in many other solid malignancies including colorectal, thyroid, biliary tract, endometrial, and ovarian cancers (5–11).
While recently there has been some progress in developing G12C KRAS inhibitors, by and large, efforts to directly target RAS have been unsuccessful (4,12,13). Clinical trials testing numerous strategies have not demonstrated biological activity (4). Interestingly, preclinical studies have shown NRAS-mutated cell lines to be more sensitive to MEK inhibition than KRAS-mutated cell lines (14–16). For example, one study found that 5 of 6 tested NRAS-mutated lung cancer cell lines were sensitive to MEK inhibitors (14).
In agreement with in vitro observations, the most successful effort in targeting RAS mutations has been in NRAS-mutated melanoma. The randomized phase 3 NEMO trial (17) compared the efficacy of binimetinib (MEK162, ARRY-162), a potent oral inhibitor of MEK1 and MEK2 (18,19), with that of dacarbazine in chemotherapy-naïve advanced melanoma patients with a codon 61 NRAS-mutated tumor. The NEMO trial demonstrated that binimetinib-treated patients, compared with those treated with dacarbazine, had an improved median PFS (2.8 months verse 1.5 months) and objective response rate (ORR) (15% verse 9%) (17). However, there was no difference in overall survival (OS) between the two arms (17).
On the basis of these preclinical and clinical data, we hypothesized that the binimetinib MEK inhibitor might be efficacious in other NRAS-mutated malignancies. Here, we report the results of the subprotocol of the NCI-MATCH basket trial that evaluated the anti-tumor efficacy of single agent binimetinib in patients with a refractory NRAS-mutated malignancy.
METHODS
NCI-MATCH Trial Study Design
The NCI-MATCH trial (NCT02465060) is a multicenter open-label phase 2 trial evaluating targeted therapy directed by molecular profiles. Eligible patients have histologically documented solid tumor, lymphoma, or multiple myeloma who require therapy after progression on at least one line of standard systemic therapy. Patients were required to have an Eastern Cooperative Oncology Group (ECOG) performance status ≤1 and measurable disease. Adequate renal, hematologic, and liver function were required.
Patients enrolled in the NCI-MATCH trial master protocol underwent a tumor biopsy and molecular profiling with the following investigational assays: an adapted Oncomine AmpliSeq™ (OCP) panel (Thermo Fisher Scientific, Waltham, MA) and immunohistochemistry assays for PTEN, MLH2, MSH2, and Rb expression in protocol-designated CLIA-accredited laboratories (20,21). Patients whose tumor had a molecular alteration targeted by one of the treatments included in the trial were offered enrollment onto a subprotocol according to the NCI-MATCH treatment assignment algorithm (MATCHbox). The treatment-assignment algorithm was designed to enroll patients in the treatment subprotocol that had the highest level of evidence for their therapeutic agent and alteration.
This study was conducted according to the principles of the Declaration of Helsinki and the International Conference on Harmonization Good Clinical Practice guidelines. The study was approved by the NCI Central Institutional Review Board (CIRB). All patients were provided with and signed CIRB-approved consent forms before enrollment.
Study Population for the NRAS Subprotocol Arm
Patients eligible for the NRAS subprotocol had a tumor harboring a codon 12, 13, or 61 NRAS-mutation. Melanoma patients were excluded from this subprotocol because binimetinib has already been extensively investigated in this population. Exclusion criteria included prior treatment with a MEK inhibitor, a history of retinal pathology and left ventricular ejection fraction < 50%. Patients needed to have completed chemotherapy, radiation, or surgery ≥ 4 weeks before starting this subprotocol.
Treatment and Evaluations for NRAS Subprotocol
Patients assigned to the NRAS subprotocol were treated with open label, orally administered binimetinib 45 mg twice daily continuously until disease progression or the development of unacceptable toxicity. A cycle was defined as 28 days. Decrease in the binimetinib dose below 30 mg twice daily was not allowed. Safety assessments performed included MUGA/echocardiography at the end of the second cycle and every 4 cycles thereafter. Retinal examinations were performed after the first cycle and then every 2 cycles.
Radiological tumor assessments, evaluated according to RECIST, version 1.1, were performed every 8 weeks (2 cycles) for the first 4 cycles and every 3 cycles thereafter (22). Adverse events assessment was performed according to the Common Toxicity Criteria for Adverse events (CTCAE), version 4.0.
Statistical Analyses
This subprotocol was designed to accrue 35 patients; to ensure the enrollment of 31 eligible patients. Unexpectedly, 18 of the first 25 subjects enrolled in this subprotocol were colorectal cancer patients. Because of the predominance of colorectal cancer patients on this subprotocol, enrollment of patients with this diagnosis was halted after 24 had been registered to increase the number of non-colorectal cancer patients. The subprotocol was amended so that after the first 35 patients were enrolled, non-colorectal cancer patient accrual was allowed to continue for up to 6 more months to enroll a maximum of 35 additional patients or until response data were available on at least 31 patients, whichever came first. A maximum of 10 patients per tumor type was enforced during accrual beyond the first 35 patients.
This subprotocol’s primary objective was assessment of the ORR (complete response + PR) according to RECIST, version 1.1. If an objective response was detected in ≥ 5/31 patients (16%), the agent was considered promising and worthy of further testing. The subprotocol had 91.8% power to conclude that the agent is promising if the true ORR was 25%. The type I error rate (1-sided) was 1.8% under a null response rate of 5%.
Secondary objectives included assessment of PFS, 6-month PFS, and OS. PFS was defined as the time between the start of binimetinib treatment and disease progression or death from any cause, censored at the date of last disease assessment for patients who had not progressed. OS was defined as the time between initiation of binimetinib and death or the patient was censored at the date of last contact. Kaplan-Meier methodology was used to estimate survival distributions (23).
Exploratory analyses to assess differences in ORR and PFS by mutation codon were conducted using Fisher’s exact test (ORR) or log-rank (PFS) test. Testing was two-sided, level 0.05 without adjustment for multiple comparisons due to the exploratory nature of these analyses.
Analysis of Databases Containing Genomically Annotated Cancer Patient Survival Data and Cancer Cell Line Drug Sensitivity
Survival data on NRAS-mutated colorectal cancer patients was obtained from the Cancer Genome Atlas (TCGA) database on cBioPortal (www.cbioportal.org) (24,25). MEK inhibitor sensitivity data from the Genomics of Drug Sensitivity in Cancer Database and the Cancer Cell line Encyclopedia database was obtained from the Cancer Dependency Map portal (https://depmap.org/portal/) (26–28). Data from codon 61 and codon 12/13 NRAS-mutated cell lines were analyzed for sensitivity to MEK inhibitors by analyzing the area under the fitted dose response curve (AUC). Statistical significance was determined using two-tailed Welch’s t-test.
RESULTS
Between June 6, 2016 and July 24, 2017, 4,889 patients were screened for the MATCH trial (Figure 1). Of those patients, 114 had tumors harboring an NRAS mutation, and 53 of the 114 were subsequently enrolled in the NRAS subprotocol. Of the 53 patients enrolled, 50 patients started binimetinib treatment. Ultimately, 3 patients were excluded because they were found ineligible for the study, leaving 47 eligible patients who were treated on this subprotocol (Figure 1). The median age of the 47 eligible patients was 60 years (Table 1). In total, 31 patients (66%) had an ECOG performance status of 1 (Table 1). Most patients had been heavily pretreated, and 53% had received four or more prior lines of treatment (Table 1).
Figure 1:

CONSORT diagram
Table 1:
Baseline Patient Characteristics for Eligible Patients Who Started Protocol Treatment
| Characteristic | Total (n=47) |
|---|---|
| Median Age, yr (range) | 60 (30–84) |
| Sex | |
| Female | 29 (62%) |
| Male | 18 (38%) |
| ECOG Performance Status | |
| 0 | 16 (34%) |
| 1 | 31 (66%) |
| Prior Lines of Therapy | |
| 0–1 | 11 (23%) |
| 2 | 4 (9%) |
| 3 | 7 (15%) |
| >3 | 25 (53%) |
| Race | |
| White | 40 (85%) |
| Black | 3 (6%) |
| Asian | 2 (4%) |
| Not reported | 2 (4%) |
| Tumor Type | |
| Gastrointestinal tract malignancies | |
| Colorectal adenocarcinoma | 24 (51%) |
| Right colon (n=3) | |
| Left colon and rectum (n=21) | |
| Cholangiocarcinoma | 7 (15%) |
| Intrahepatic (n=6) | |
| Not specified (n=1) | |
| Gynecologic malignancies | |
| Low-grade papillary serous carcinoma of ovary | 3 (6%) |
| Endometrioid endometrial adenocarcinoma | 3 (6%) |
| Granulosa cell tumor of ovary, juvenile type | 1 (2%) |
| Head/neck and respiratory tract malignancies | |
| Thyroid carcinoma | 2 (4%) |
| Papillary thyroid cancer (n=1) | |
| Follicular thyroid cancer (n=1) | |
| Adenoid cystic carcinoma of maxillary sinus | 1 (2%) |
| Malignant ameloblastoma of mandible | 1 (2%) |
| Respiratory tract tumor | |
| Epithelioid mesothelioma of pleura | 1 (2%) |
| Adenoid cystic carcinoma of trachea | 1 (2%) |
| Urinary tract malignancies | |
| Mucinous adenocarcinoma of urinary bladder | 1 (2%) |
| Unknown primary site | 2 (4%) |
More than half the patients had metastatic colorectal adenocarcinoma (24/47, 51.1%). In agreement with other studies, our data demonstrated that NRAS-mutated colorectal cancers are most frequently mutated in codon 12 (13/24, 54.1%) (Table 2), and they occur mainly in left-sided colon and rectal tumors (21/24, 87.5%) (Table 1) (29). There were no significant differences in age, gender, race, performance status or prior lines of therapy between colorectal cancer patients harboring codon 12/13 and codon 61 NRAS mutations (Supplemental Table 1).
Table 2:
Distribution of NRAS-Mutations in Eligible Patients Who Started Protocol Treatment
| Colorectal (n=24) | Cholangiocarcinoma (n=7) | Other (n=16) | Total (n=47) | |
|---|---|---|---|---|
| Codon 12 | 13 (54.2%) | 0 (0%) | 4 (25%) | 17 (36.2%) |
| Gly12Asp | 8 (33.3%) | 0 (0%) | 3 (18.8%) | 11 (23.4%) |
| Gly12Cys | 1 (4.2%) | 0 (0%) | 0 (0%) | 1 (2.1%) |
| Gly12Ser | 1 (4.2%) | 0 (0%) | 0 (0%) | 1 (2.1%) |
| Gly12Val | 3 (12.5%) | 0 (0%) | 1 (6.2%) | 4 (8.5%) |
| Codon 13 | 3 (12.5%) | 2 (28.6%) | 3 (18.8%) | 8 (17%) |
| Gly13Arg | 1 (4.2%) | 2 (28.6%) | 1 (6.2%) | 4 (8.5%) |
| Gly13Asp | 0 (0%) | 0 (0%) | 1 (6.2%) | 1 (2.1%) |
| Gly13Cys | 1 (4.2%) | 0 (0%) | 1 (6.2%) | 2 (4.3%) |
| Gly13Val | 1 (4.2%) | 0 (0%) | 0 (0%) | 1 (2.1%) |
| Codon 61 | 8 (33.3%) | 5 (71.4%) | 9 (56.2%) | 22 (46.8%) |
| Gln61Arg | 1 (4.2%) | 3 (42.9%) | 6 (37.5%) | 10 (21.3%) |
| Gln61His | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) |
| Gln61Leu | 2 (8.3%) | 0 (0%) | 0 (0%) | 2 (4.3%) |
| Gln61Lys | 5 (20.8%) | 2 (28.6%) | 3 (18.8%) | 10 (21.3%) |
The most common non-colorectal cancers were cholangiocarcinoma (15%), low-grade papillary serous carcinoma of the ovary (6.4%), and endometrioid endometrial adenocarcinoma (6.4%) (Table 1). In contrast to colorectal adenocarcinoma, other cancers harbored predominantly codon 61 mutations (14/23, 60.9%) compared with codon 13 (5/23, 21.7%) and codon 12 (4/23, 17.4%) mutations (p = 0.03) (Table 2). Analysis of genomic alterations co-occurring with an NRAS mutation revealed that the TP53 mutation was most common (23/47, 48.9%) (Supplemental Figure 1). APC genomic alterations were also frequent (19/47, 40.4%), but these were observed only in colorectal cancers (19/24, 79.2%).
As of the data cut-off of May 3, 2019, all 47 patients had discontinued study treatment. The most common reason for withdrawal was progressive disease (62%). The median follow-up for patients was 24 months (range 4–30 months).
Efficacy
The study failed to demonstrate a promising level of activity and did not meet the primary endpoint. The observed ORR was 2.1% (90% CI 0.1–9.7%), and the null hypothesis of 5% ORR (deemed a non-promising level of activity) could not be rejected (Figure 2A). For the efficacy population (n = 47), the 6-month PFS was 29.2% (90% CI 19.4–44.0%), the median PFS was 3.5 months (95% CI 1.8–5.8 months), and the median OS was 10.5 months (95% CI 5.3–13.2 months) (Supplemental Figure 2A and 2B).
Figure 2:
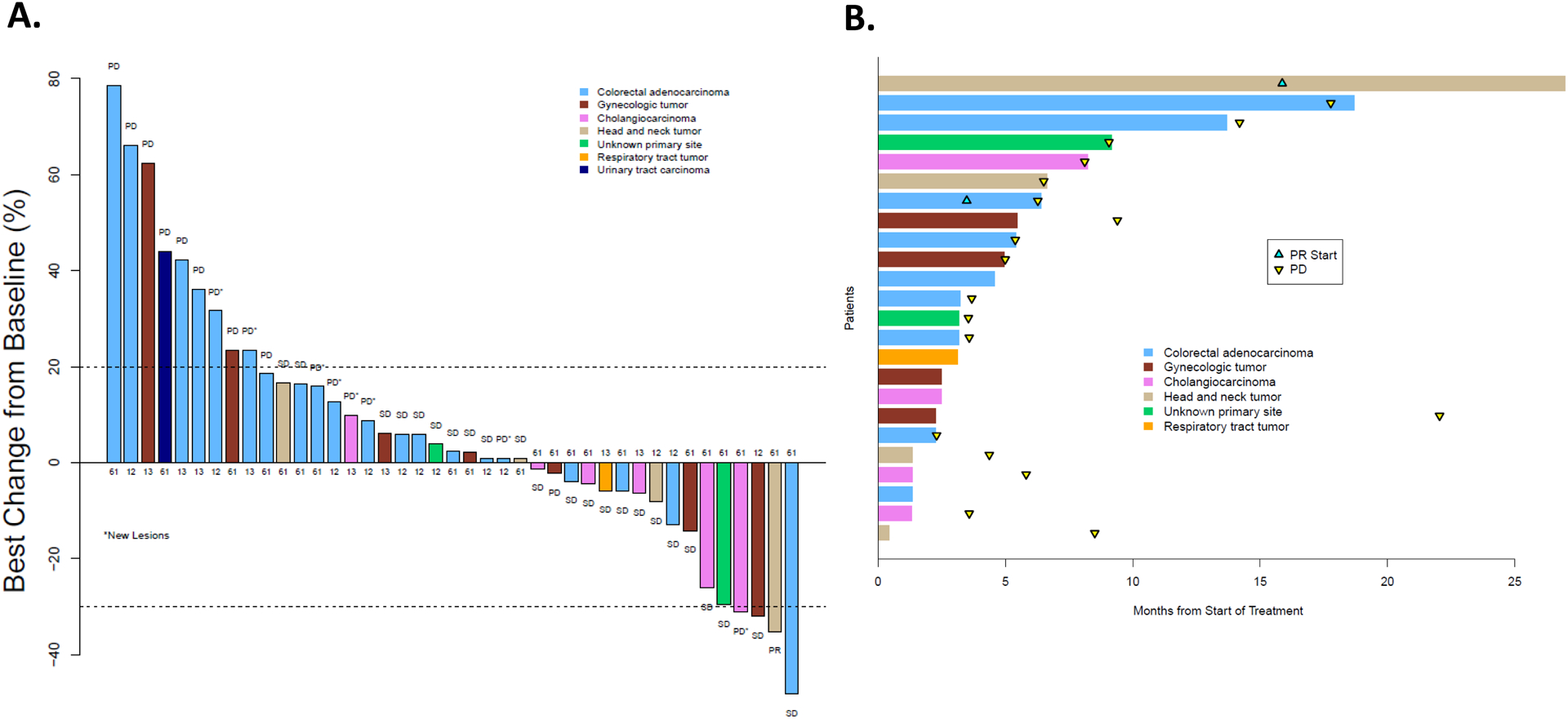
(A) Best overall response according to RECIST criteria in the 41 evaluable patients who remained on this subprotocol. The number associated with each tumor designates the NRAS codon that was mutated. (B) treatment duration of the 24 patients whose best response was stable disease or a partial response. Abbreviations: Progressive disease (PD), stable disease (SD), and PR (partial response).
The median PFS for patients with NRAS-mutated colorectal cancer was 1.8 months (90% CI 1.7–3.7 months) compared with 4.4 months (90% CI 3.6–8.5 months) for patients who had other cancer types (p = 0.07) (Supplemental Figure 3). The median PFS for patients with cholangiocarcinoma (n = 7) was 3.6 months (90% CI 1.8–not reached months) (Supplemental Figure 4).
The sole confirmed PR was observed in a patient with a Q61R mutation NRAS-mutated malignant ameloblastoma. This patient had developed the ameloblastoma 19 years prior to enrollment and had metastatic lung lesions measuring up to 6 cm. The patient received binimetinib for 26 months before discontinuing the subprotocol because of grade 2 myalgias. There was also one unconfirmed PR in a colorectal cancer patient whose tumor harbored an NRAS codon Q61R mutation along with a TP53 and APC mutation. This patient had a 48.2% tumor reduction according to RECIST criteria after cycle 4 (Supplemental Figure 5). Restaging scans at cycle 7 showed new metastatic lesions, and the patient was removed from this subprotocol. Two additional colorectal cancer patients harboring NRAS Q61K mutations remained on this subprotocol for 12 and 17 months with stable disease before ultimately developing progressive disease.
In a post-hoc analysis, patients with colorectal cancer harboring NRAS codon 61 mutations who were treated with binimetinib (n = 8) had a significantly longer OS (hazard ratio [HR] 0.34, 95% CI 0.12–0.95; p = 0.03) and PFS (HR 0.23, 95% CI 0.07–0.74; p = 0.007) than those with NRAS codon 12/13 mutations (n = 16) (Figure 3A and 3B). Similarly, when all tumor types were examined, binimetinib-treated patients with codon 61-mutated tumors also had significantly longer OS and PFS than those with codon 12/13-mutated tumors. Binimetinib-treated patients with codon 61 NRAS-mutated tumors had a median OS of 13.1 (90% CI 9.1–not reached) months compared to a median OS of 5.5 (90% CI 4.7–11.6) months for tumors harboring a codon 12 or 13 NRAS mutation (p = 0.04) (Supplemental Figure 6A). The median PFS for binimetinib-treated patients with codon 12/13 NRAS-mutated tumors was 1.8 months (90% CI 1.8–3.7 months) compared with 5.8 months (90% CI 2.5–9.1 months) for binimetinib-treated patients whose tumor harbored an NRAS codon 61 mutation (p = 0.006) (Supplemental Figure 6B). In contrast to colorectal cancer patients, binimetinib-treated patients with other tumor types with codon 61 NRAS mutations (n = 14) did not have a significantly longer PFS (HR 0.67, 95% CI 0.25–1.76; p = 0.4) and OS (HR 0.84, 95% CI 0.31–2.27; p = 0.70) than those with codon 12/13 NRAS mutations (n = 9). (Supplemental Figure 7A and 7B).
Figure 3:

(A) Kaplan-Meier estimates of OS; (B) PFS comparing binimetinib-treated colorectal cancer patients with tumors harboring codon 61 NRAS mutations to colorectal cancer patients with tumors harboring codon 12 or 13 NRAS mutations.
One explanation for the improved clinical outcomes of binimetinib-treated codon 61 NRAS-mutated colorectal cancer patients is that codon 61 NRAS-mutated colorectal cancers have a more indolent natural history than codon 12/13 NRAS-mutated colorectal cancers. We examined survival data of NRAS-mutated colorectal cancer patients (Supplemental Table 2) in the TCGA database (24,25) and found that codon 61 NRAS-mutated colorectal cancer patients (n=14) did not have an improvement in OS compared to codon 12/13 NRAS-mutated colorectal cancer patients (n=16) (HR 1.06, 95% CI 0.23–4.94; p=0.94) (Supplemental Figure 8).
We next examined the hypothesis that codon 61 NRAS-mutated tumors are more sensitive to MEK inhibition by examining MEK inhibitor drug sensitivity testing performed in large cancer cell line collections. Using NRAS-mutated cell lines from the Genomics of Drug Sensitivity in Cancer database (28), we found that codon 61 NRAS-mutated cancer cell lines were significantly more sensitive than codon 12/13 NRAS-mutated cancer cell lines to 5 different MEK inhibitors (refametinib, selumetinib, trametinib, PD-0325901, and PD-184352) (Figure 4), although more limited data from the Cancer Cell Line Encyclopedia did not demonstrate a statistically significant difference (Supplemental Figure 9) (27).
Figure 4:
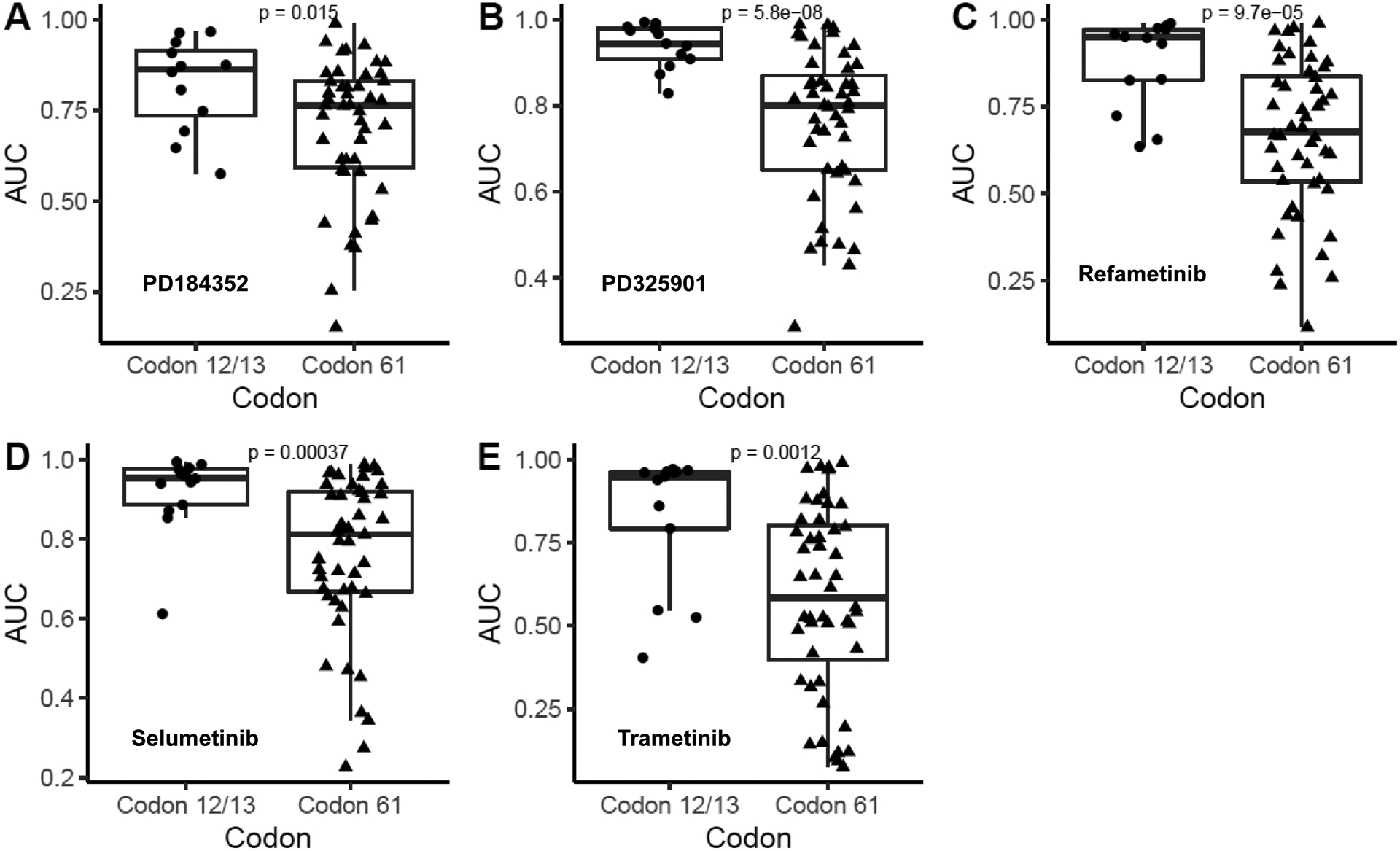
Comparison of MEK inhibitor sensitivity between codon 61 NRAS-mutated cell lines and codon 12/13 NRAS-mutated cell lines in the Genomics of Drug Sensitivity in Cancer Database. MEK inhibitors evaluated include PD184352 (A), PD325901 (B), Refametinib (C), Selumetinib (D), Trametinib (E). Drug sensitivity was measured by analyzing the area under the fitted dose response curve (AUC) and significance was determined using two-tailed Welch’s t-test.
Safety
Toxicities that are known to be caused by MEK inhibitors, including rash, diarrhea, retinal abnormalities, decreased ejection fraction, elevated creatinine phosphokinase levels, and hypertension, occurred in 43 (86%) of the 50 patients who started treatment. Grade 3 or 4 toxicities that were considered to be possibly, probably, or definitely binimetinib-related are shown in Table 3. One death, which was from multiorgan failure, was assessed as possibly related to binimetinib.
Table 3:
Grade 3 to 5 Adverse Events Possibly, Probably, or Definitely Associated with Study Treatment
| Toxicity Type | Subprotocol | ||
|---|---|---|---|
| EAY131-Z1A (n=50) | |||
| Grade | |||
| 3 | 4 | 5 | |
| (n) | (n) | (n) | |
| Heart failure | 1 | - | - |
| Myocardial infarction | 1 | - | - |
| Eye disorders | 1 | - | - |
| Mucositis oral | 1 | - | - |
| Nausea | 1 | - | - |
| Small intestinal obstruction | 1 | - | - |
| Fatigue | 1 | - | - |
| Multi-organ failure | - | - | 1 |
| Edema limbs | 1 | - | - |
| Urinary tract infection | 1 | - | - |
| Alanine aminotransferase increased | 1 | - | - |
| Alkaline phosphatase increased | 1 | - | - |
| Aspartate aminotransferase increased | 1 | - | - |
| CPK increased | 2 | - | - |
| Lymphocyte count decreased | 2 | - | - |
| White blood cell decreased | 1 | - | - |
| Ejection fraction decreased | 1 | - | - |
| Anorexia | 1 | - | - |
| Dehydration | 1 | - | - |
| Hypoalbuminemia | 1 | - | - |
| Hyponatremia | 1 | - | - |
| Hypophosphatemia | 1 | - | - |
| Muscle weakness lower limb | 1 | - | - |
| Muscle weakness upper limb | 1 | - | - |
| Syncope | 1 | - | - |
| Rash acneiform | 3 | - | - |
| Skin and subcutaneous tissue disorders | 1 | - | - |
| Hypertension | 6 | - | - |
Among the 50 patients treated on this subprotocol, 30% discontinued treatment because of adverse events. Binimetinib dose reduction was required in 44% of patients. The first dose reductions occurred at a median of 2.6 weeks into the trial. There was no difference in the rate of early discontinuation of binimetinib, for reasons other than progressive disease or death, between colorectal cancer patients harboring codon 12/13 and codon 61 NRAS mutations. Among the 24 binimetinib-treated colorectal patients, the vast majority of patients (21) discontinued therapy due to disease progression or death. Two colorectal cancer patients discontinued therapy because of adverse events (both tumors harbored a codon 12/13 NRAS mutation) and one colorectal cancer patient withdrew consent (this patient’s tumor harbored a codon 61 NRAS mutation).
Discussion
Targeted therapy for RAS-mutated malignancies represents an enormous yet critical challenge in clinical oncology. This subprotocol evaluated whether a single-agent MEK inhibitor, binimetinib, might be an effective therapy in patients with non-melanoma NRAS-mutated malignancies. Similarly to the lack of efficacy of MEK inhibitor monotherapy in KRAS-mutated cancers (19,30,31), single-agent binimetinib did not show a promising ORR in patients with NRAS-mutated solid tumors. The modest efficacy of binimetinib monotherapy was disappointing. In the NEMO trial, while binimetinib did not improve OS compared to dacarbazine in codon 61 NRAS-mutated melanoma, it did result in a modest increase in PFS over dacarbazine, and it had an ORR of 15% (17). Possible reasons for the lower ORR in this subprotocol compared to the NEMO trial include lineage or allelic differences between the two study populations. Melanoma patients who participated in the NEMO trial were treatment-naïve, with the exception of immunotherapy, while more than half the patients registered on this subprotocol had received four or more prior lines of therapy. However, taken together, data from the NEMO trial and this subprotocol clearly indicate that MEK inhibitor monotherapy is inadequate to provide clinical benefit to most patients with NRAS-mutated tumors.
Preclinical data suggests that NRAS mutation allelic differences can generate functionally significant phenotypic distinctions (32–34). Mechanistically, codon 61 NRAS mutations activate NRAS differently than codon 12 and 13 NRAS-mutations. Codon 61 NRAS mutations block the hydrolysis of GTP to GDP (35), while codon 12 and 13 mutations interfere with the binding of GTPase-activating proteins, which ordinarily would accelerate the hydrolysis of GTP to GDP (36). We explored whether the outcomes of binimetinib-treated patients with codon 61 NRAS-mutated tumors were different than binimetinib-treated patients with codon 12/13 NRAS-mutated tumors. Binimetinib-treated colorectal cancer patients whose tumor harbored a codon 61 NRAS-mutated tumor had a significantly longer OS and PFS than binimetinib-treated colorectal cancer patients with a codon 12/13 NRAS-mutated tumor. Similarly, an analysis of all binimetinib-treated patients, regardless of tumor type, also demonstrated that patients with codon 61 NRAS-mutated tumors had a superior OS and PFS compared to patients with a codon 12/13 NRAS-mutated tumor. However, when colorectal cancer patients were excluded from the analysis, the improvement in OS and PFS was no longer statistically significant. Given the colorectal cancer patient predominance in this trial, it is unclear whether this improvement in OS and PFS was solely attributable to colorectal cancer or that it is more generalizable to other tumor types.
One possible explanation for the superior OS and PFS of binimetinib-treated colorectal cancer patients with codon 61 NRAS-mutated tumors is that these tumors have a more indolent natural history than codon 12/13 NRAS-mutated cancers. Using the TCGA database, where 80% of patients had pathologically staged I to III colorectal cancers, we found no difference in the OS of colorectal cancer patients with codon 12/13 NRAS mutations and codon 61 NRAS mutations. Cercek and colleagues performed a similar analysis and found that NRAS-mutated metastatic colorectal cancer patients with exon 3 (codon 60 and 61) NRAS mutations had a shorter OS than metastatic colorectal cancer patients with exon 2 (codon 12 and 13) NRAS mutations (37). Potential reasons for the differences observed between these two analyses include the different stage distribution of the two data sets and the relatively small sample size.
Since codon 61 NRAS-mutated colorectal cancer does not appear to have a more indolent natural history than codon 12/13 NRAS-mutated colorectal cancer, an alternative explanation is that codon 61 NRAS-mutated colorectal cancers are more susceptible to binimetinib. All the colorectal cancer patients that either achieved an unconfirmed PR or stable disease for longer than 12 months harbored a codon 61 NRAS mutation. Consistent with the findings observed in our trial, we found that codon 61 NRAS-mutated cell lines were more sensitive to MEK inhibitors compared to codon 12/13 NRAS-mutated cell lines in the Genomics of Drug Sensitivity in Cancer Database. While analysis of MEK inhibitor sensitivity in more limited data from the Cancer Cell Line Encyclopedia cell line collection did not replicate these findings, previous work has noted inconsistent results between these two large pharmacogenomic databases (38). To more rigorously assess the hypothesis that cancers with NRAS codon 61 mutations are more sensitive to MEK inhibition than cancers with NRAS codon 12/13 mutations, future preclinical studies could explore whether the differential sensitivity of MEK inhibitors is observed in experimental systems, such as isogenic cell lines or transgenic mice, that only differ in the NRAS codon that is mutated. Interestingly, a phosphoproteomic study demonstrated that while NRAS codon 61 mutated tumors exhibit hyperactivation of the MAPK pathway, tumors harboring NRAS codon 12 mutations were more reliant on the PI3K/AKT pathway (39). If this finding is confirmed, this differential downstream signaling could explain how NRAS codon 61 mutated cancer are more sensitive to MEK inhibition than NRAS codon 12/13 mutated cancers.
Although MEK inhibitor monotherapy is insufficient to treat NRAS-mutated cancers, the modest activity observed in this subprotocol and in NRAS codon 61-mutated melanoma suggests that MEK inhibitor combinations may potentially be an effective treatment strategy. In an NRAS-mutated murine model of melanoma, single-agent MEK inhibition was cytostatic, whereas combined MEK and CDK4/6 inhibition resulted in significant tumor regression (40). In this model, MEK inhibitor monotherapy induced apoptosis but did not block cell cycle arrest, while combined MEK and CDK4/6 inhibition was more potent because it stimulated both apoptosis and cell cycle arrest. Consequently, the strategy of combined MEK and CDK4/6 inhibition is undergoing clinical testing. In addition to MEK inhibitor-based strategies, encouraging anti-tumor activity has recently been reported in trials using MRTX849 and AMG510, which are novel covalent inhibitors of KRAS G12C (13,41). These data raise the possibility that similar compounds might have activity in NRAS G12C- or G13C-mutated tumors. Data from this subprotocol indicate that NRAS G12C mutations were present in 1 of 17 (5.8%) codon 12-mutated tumors and 2 of 8 (25%) codon 13-mutated tumors.
A patient with a codon 61 NRAS-mutated malignant ameloblastoma had a durable 26-month PR to binimetinib. Malignant ameloblastoma is a rare odontogenic tumor that appears to be driven by the MAPK pathway. More than 80% of malignant ameloblastomas harbor either an RAS or a BRAF mutation; most (46–62%) are V600E BRAF mutations, and the minority (6%) are NRAS mutations (42,43). V600E BRAF-mutated malignant ameloblastomas have been reported to have durable responses to BRAF inhibition (44,45). Although this patient had indolent tumor for 19 years prior to accrual to the trial, the patient’s 26-month response to binimetinib further supports the sensitivity of this NRAS-mutated tumor type to MAPK inhibition (44,45).
High percentages of patients in this subprotocol discontinued treatment (30%) and required dose reductions (44%) because of binimetinib-associated toxicity. This finding is similar to the experience reported for patients with melanoma treated with binimetinib in the NEMO trial, in which 25% of patients discontinued binimetinib because of toxicity (17).
In conclusion, MEK inhibitor monotherapy is an insufficient treatment for NRAS-mutated solid tumors. A post-hoc analyses suggested that binimetinib-treated colorectal cancer patients with an NRAS codon 61-mutated tumor had a significantly longer OS and PFS than binimetinib-treated colorectal cancer patients with an NRAS codon 12- or 13-mutated tumor. The design of future clinical trials targeting NRAS should carefully consider differentiating between codon 12/13 and codon 61 NRAS-mutated tumors as these two populations appear to have important biological differences and therapeutic sensitivities.
Supplementary Material
Statement of Translational Relevance.
Therapeutic targeting of RAS-mutated malignancies is an elusive but highly sought- after goal in clinical oncology. Whereas MEK inhibitor monotherapy is ineffective in KRAS-mutated cancers, preclinical data and clinical studies in NRAS-mutated melanoma suggest that single-agent MEK inhibition may be efficacious in NRAS-mutated cancers. We treated 47 patients with NRAS-mutated non-melanoma cancers with the MEK inhibitor binimetinib. Although a malignant ameloblastoma patient had a durable response and two colorectal cancer patients remained on binimetinib for 12 months, the trial did not meet its primary endpoint. Subsequent analysis revealed a potentially important biological difference between codon 61 and codon 12/13 NRAS-mutated tumors. Colorectal cancer patients with codon 61 NRAS-mutated tumors had superior survival outcomes after binimetinib treatment than patients with tumors harboring codon 12/13 NRAS-mutations. Preclinical studies have also revealed significant biological differences between NRAS mutation alleles, and future studies should explore how to exploit these differences.
Funding:
This study was coordinated by the ECOG-ACRIN Cancer Research Group (Peter J. O’Dwyer, MD and Mitchell D. Schnall, MD, PhD, Group Co-Chairs) and was supported by the National Cancer Institute of the National Institutes of Health under the following award numbers: CA180820, CA180794, CA180858, CA180867, CA180870, and CA189997. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health, nor does mention of trade names, commercial products, or organizations imply endorsement by the U. S. government. Array BioPharma provided the study drug for this study. The work of JMC is supported by DFCI Hale Family Center for Pancreatic Cancer, Team Evan Schumacher, Haya Linde Memorial Fund, and the Pancreatic Cancer Collective New Therapies Challenge Grant, an initiative of the Lustgarten Foundation and Stand Up To Cancer, Grant Number SU2C-AACR-PCC-02-18. Stand Up To Cancer is a division of the Entertainment Industry Foundation. Research Grants are administered by the American Association for Cancer Research, the Scientific Partner of SU2C. Stanley R. Hamilton, M.D., is the recipient of the Frederick F. Becker Distinguished University Chair in Cancer Research at the University of Texas.
Authors’ Disclosures of Potential Conflicts of Interest:
James Cleary received research funding to his institution from Abbvie, Merus, Roche, and Bristol Myers Squib. He received research funding from Merck, Astrazeneca, Esperas Pharma, and Tesaro, received consulting fees from Bristol Myers Squibb, and received travel funding from Bristol Myers Squib. Rebecca Heist has received consulting fees from Boehringer Ingelheim, Novartis, Apollomics, EMD Serono/KgA and Daichii Sankyo and Tarveda. She received research funding to institution, not to self, from Novartis, Abbvie, Daichii Sankyo, Agios, Corvus, Genentech Roche, Mirati, Exelixis. Stanley R. Hamilton is a consultant to Bristol-Myers Squibb Company, Guardant, and LOXO-Oncology; an advisory board member for Fred Hutchinson Cancer Research Center and Halio DX; and a Scientific/Advisory Committee member for Thermo-Fisher Scientific and Merck & Co., Inc. Dr. Arteaga has received grant support from Pfizer, Lilly, and Takeda. He served in an advisory role to Novartis, Lilly, TAIHO Oncology, Daiichi Sankyo, Merck, AstraZeneca, OrigiMed, Immunomedics, PUMA Biotechnology, Arvinas and the Susan G. Komen Foundation. He holds minor stock options in Provista and Y-TRAP. Dr. Flaherty serves on the Board of Directors of Clovis Oncology, Strata Oncology, Vivid Biosciences, and Checkmate Pharmaceuticals: Corporate Advisory Board of X4 Pharmaceuticals; Scientific Advisory Boards of Sanofi, Amgen, Asana, Adaptimmune, Fount, Aeglea, Shattuck Labs, Tolero, Apricity, Oncoceutics, Fog Pharma, Neon, PIC Therapeutics, Tvardi, xCures, Monopteros, Vibliome; and, consultant to Novartis, Genentech, BMS, Merck, Takeda, Verastem, Boston Biomedical, Pierre Fabre, and Debiopharm.
References
- 1.Stephen AG, Esposito D, Bagni RK, McCormick F. Dragging Ras back in the ring. Cancer Cell 2014;25:272–81. [DOI] [PubMed] [Google Scholar]
- 2.Prior IA, Hood FE, Hartley JL. The frequency of Ras mutations in cancer. Cancer Res 2020;80:2969–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Prior IA, Lewis PD, Mattos C. A comprehensive survey of Ras mutations in cancer. Cancer Res 2012;72:2457–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Singh H, Longo DL, Chabner BA. Improving prospects for targeting RAS. J Clin Oncol 2015;33:3650–9. [DOI] [PubMed] [Google Scholar]
- 5.Berger MF, Hodis E, Heffernan TP, Deribe YL, Lawrence MS, Protopopov A, et al. Melanoma genome sequencing reveals frequent PREX2 mutations. Nature 2012;485:502–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.The Cancer Genome Atlas Research N. Comprehensive molecular characterization of gastric adenocarcinoma. Nature 2014;513:202–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.The Cancer Genome Atlas Research N. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014;511:543–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Agrawal N, Akbani R, Aksoy BA, Ally A, Arachchi H, Asa Sylvia L, et al. Integrated genomic characterization of papillary thyroid carcinoma. Cell 2014;159:676–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012;487:330–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kandoth C, Schultz N, Cherniack AD, Akbani R, Liu Y, et al. Integrated genomic characterization of endometrial carcinoma. Nature 2013;497:67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wardell CP, Fujita M, Yamada T, Simbolo M, Fassan M, Karlic R, et al. Genomic characterization of biliary tract cancers identifies driver genes and predisposing mutations. J Hepatol 2018;68:959–69. [DOI] [PubMed] [Google Scholar]
- 12.Cox AD, Fesik SW, Kimmelman AC, Luo J, Der CJ. Drugging the undruggable RAS: mission possible? Nat Rev Drug Discov 2014;13:828–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fakih M, O’Neil B, Price TJ, Falchook GS, Desai J, Kuo J, et al. Phase 1 study evaluating the safety, tolerability, pharmacokinetics (PK), and efficacy of AMG 510, a novel small molecule KRASG12C inhibitor, in advanced solid tumors. J Clin Oncol 2019;37:3003. [Google Scholar]
- 14.Ohashi K, Sequist LV, Arcila ME, Lovly CM, Chen X, Rudin CM, et al. Characteristics of lung cancers harboring NRAS mutations. Clin Cancer Res 2013;19:2584–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, et al. The cancer cell line encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012;483:603–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vujic I, Posch C, Sanlorenzo M, Yen AJ, Tsumura A, Kwong A, et al. Mutant NRASQ61 shares signaling similarities across various cancer types–potential implications for future therapies. Oncotarget 2014;5:7936–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dummer R, Schadendorf D, Ascierto PA, Arance A, Dutriaux C, Di Giacomo AM, et al. Binimetinib versus dacarbazine in patients with advanced NRAS-mutant melanoma (NEMO): a multicentre, open-label, randomised, phase 3 trial. Lancet Oncol 2017;18:435–45. [DOI] [PubMed] [Google Scholar]
- 18.Lee PA, Wallace E, Marlow A, Yeh T, Marsh V, Anderson D, et al. Preclinical development of ARRY-162, a potent and selective MEK 1/2 inhibitor [abstract 2515]. Cancer Res 2010;70:2515. [Google Scholar]
- 19.Bendell JC, Javle M, Bekaii-Saab TS, Finn RS, Wainberg ZA, Laheru DA, et al. A phase 1 dose-escalation and expansion study of binimetinib (MEK162), a potent and selective oral MEK1/2 inhibitor. Br J Cancer 2017;116:575–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lih CJ, Takebe N. Considerations of developing an NGS assay for clinical applications in precision oncology: the NCI-MATCH NGS assay experience. Curr Probl Cancer 2017;41:201–11. [DOI] [PubMed] [Google Scholar]
- 21.Khoury JD, Wang WL, Prieto VG, Medeiros LJ, Kalhor N, Hameed M, et al. Validation of immunohistochemical assays for integral biomarkers in the NCI-MATCH EAY131 clinical trial. Clin Cancer Res 2018;24:521–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 2009;45:228–47. [DOI] [PubMed] [Google Scholar]
- 23.Kaplan EL, Meier P. Nonparametric estimation from incomplete observations. J Am Statist Assoc 1958;53:457–81. [Google Scholar]
- 24.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2012;2:401–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 2013;6:pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Seashore-Ludlow B, Rees MG, Cheah JH, Cokol M, Price EV, Coletti ME, et al. Harnessing connectivity in a large-scale small-molecule sensitivity dataset. Cancer Discov 2015;5:1210–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, et al. The cancer cell line encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012;483:603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang W, Soares J, Greninger P, Edelman EJ, Lightfoot H, Forbes S, et al. Genomics of Drug Sensitivity in Cancer (GDSC): a resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res 2013;41:D955–D61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cercek A, Braghiroli MI, Chou JF, Hechtman JF, Kemeny N, Saltz L, et al. Clinical features and outcomes of patients with colorectal cancers harboring NRAS mutations. Clin Cancer Res 2017;23:4753–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rosen LS, LoRusso P, Ma WW, Goldman JW, Weise A, Colevas AD, et al. A first-in-human phase I study to evaluate the MEK1/2 inhibitor, cobimetinib, administered daily in patients with advanced solid tumors. Invest New Drugs 2016;34:604–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Finn RS, Ahn DH, Javle MM, Tan BR Jr., Weekes CD, Bendell JC, et al. Phase 1b investigation of the MEK inhibitor binimetinib in patients with advanced or metastatic biliary tract cancer. Invest New Drugs 2018;36:1037–43. [DOI] [PubMed] [Google Scholar]
- 32.Burd CE, Liu W, Huynh MV, Waqas MA, Gillahan JE, Clark KS, et al. Mutation-specific RAS oncogenicity explains NRAS codon 61 selection in melanoma. Cancer Discov 2014;4:1418–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim J, Novak D, Sachpekidis C, Utikal J, Larribère L. STAT3 relays a differential response to melanoma-associated NRAS mutations. Cancers 2020;12:119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kong G, Chang YI, You X, Ranheim EA, Zhou Y, Burd CE, et al. The ability of endogenous Nras oncogenes to initiate leukemia is codon-dependent. Leukemia 2016;30:1935–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Frech M, Darden TA, Pedersen LG, Foley CK, Charifson PS, Anderson MW, et al. Role of glutamine-61 in the hydrolysis of GTP by p21H-ras: an experimental and theoretical study. Biochemistry 1994;33:3237–44. [DOI] [PubMed] [Google Scholar]
- 36.Adari H, Lowy D, Willumsen B, Der C, McCormick F. Guanosine triphosphatase activating protein (GAP) interacts with the p21 ras effector binding domain. Science 1988;240:518–21. [DOI] [PubMed] [Google Scholar]
- 37.Cercek A, Braghiroli MI, Chou JF, Hechtman JF, Kemeny N, Saltz L, et al. Clinical features and outcomes of patients with colorectal cancers harboring NRAS mutations. Clin Cancer Res 2017;23:4753–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Haibe-Kains B, El-Hachem N, Birkbak NJ, Jin AC, Beck AH, Aerts HJWL, et al. Inconsistency in large pharmacogenomic studies. Nature 2013;504:389–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Posch C, Sanlorenzo M, Vujic I, Oses-Prieto JA, Cholewa BD, Kim ST, et al. Phosphoproteomic analyses of NRAS(G12) and NRAS(Q61) mutant melanocytes reveal increased CK2α kinase levels in NRAS(Q61) mutant cells. J Invest Dermatol 2016;136:2041–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kwong LN, Costello JC, Liu H, Jiang S, Helms TL, Langsdorf AE, et al. Oncogenic NRAS signaling differentially regulates survival and proliferation in melanoma. Nat Med 2012;18:1503–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hallin J, Engstrom LD, Hargis L, Calinisan A, Aranda R, Briere DM, et al. The KRASG12C inhibitor MRTX849 provides insight toward therapeutic susceptibility of KRAS-mutant cancers in mouse models and patients. Cancer Discov 2020;10:54–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brown NA, Rolland D, McHugh JB, Weigelin HC, Zhao L, Lim MS, et al. Activating FGFR2–RAS–BRAF mutations in ameloblastoma. Clin Cancer Res 2014;20:5517–26. [DOI] [PubMed] [Google Scholar]
- 43.Sweeney RT, McClary AC, Myers BR, Biscocho J, Neahring L, Kwei KA, et al. Identification of recurrent SMO and BRAF mutations in ameloblastomas. Nat Genet 2014;46:722–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Broudic-Guibert M, Blay J-Y, Vazquez L, Evrard A, Karanian M, Taïeb S, et al. Persistent response to vemurafenib in metastatic ameloblastoma with BRAF mutation: a case report. J Med Case Rep 2019;13:245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fernandes GS, Girardi DM, Bernardes JPG, Fonseca FP, Fregnani ER. Clinical benefit and radiological response with BRAF inhibitor in a patient with recurrent ameloblastoma harboring V600E mutation. BMC Cancer 2018;18:887. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.