

Abstract
Objective:
Low molecular weight compounds that reduce the expression of MMP13 at the mRNA level might serve as disease-modifying osteoarthritis (OA) drugs (DMOADs). The objective of this study was to identify a candidate DMOAD that targets MMP13 expression.
Design:
A high-throughput screen was performed to identify compounds that suppress inflammatory cytokine-induced MMP13 expression. Ingenuity pathway analysis (IPA) using iTRAQ-based proteomic analysis was conducted to identify signaling pathways related to cytokines. MMP13 expression in chondrocytes was evaluated by RT-qPCR and western blot. Additionally, 10-week-old mice underwent destabilization of the medial meniscus (DMM) surgery to induce OA and were sacrificed 12 weeks post-surgery for pathological examination. OA was evaluated using the OARSI scoring system.
Results:
Colchicine was identified as a DMOAD candidate as it inhibited inflammatory cytokine-induced MMP13 expression in vitro, and the colchicine-administered DMM mice had significantly lower OARSI scores (adjusted P: 0.0242, mean difference: 1.6, 95% CI of difference: 0.1651–3.035) and significantly lower synovial membrane inflammation scores (adjusted P: 0.0243, mean difference: 0.6, 95% CI of difference: 0.06158–1.138) than DMM mice. IPA further revealed that components of the Rho signaling pathways are regulated by cytokines and colchicine. IL-1β and TNF-α activate RAC1 and SRC signals, respectively, leading to PLC-γ1 phosphorylation and synergistic induction of MMP13 expression. Most notably, colchicine abrogates inflammatory cytokine-induced phosphorylation of PLC-γ1, leading to induction of MMP13 expression.
Conclusions:
Colchicine is a promising candidate DMOAD that inhibits MMP13 expression and consequent cartilage degradation by disrupting the SRC/RAC1-phospho-PLCγ1-Ca2+ signaling pathway.
Keywords: osteoarthritis, colchicine, disease-modifying osteoarthritis drugs, matrix metalloproteinase13, drug screening, PLC-γ1
Introduction
Osteoarthritis (OA) is a degenerative joint disease characterized by cartilage destruction, wherein articular chondrocytes are stimulated upon exposure to abnormal environmental insults, such as inflammatory cytokines and high-magnitude mechanical stress1,2. Among the inflammatory cytokines, interleukin-1β (IL-1β) and tumor necrosis factor-α (TNF-α) are the principal mediators of cartilage destruction3–5, and they regulate the expression of catabolic proteins, such as matrix metalloproteinases (MMPs)6 as well as a disintegrin and metalloproteinase with thrombospondin motifs-4,5 (ADAMTS-4,5)7.
Although various proteinases are involved in cartilage destruction, particular attention has been paid to MMP13 owing to its overexpression in the joints and articular cartilage in patients with OA, as well as its negligible levels in normal adult tissues8. Moreover, Mmp13-deficient mice are resistant to osteoarthritic cartilage erosion9, whereas cartilage-specific overexpression of Mmp13 induces precocious arthritis with cartilage erosion in mice10. Therefore, selective inhibition of MMP13 expression might prevent cartilage destruction, and low molecular weight compounds that reduce the expression of MMP13 at the mRNA level could serve as disease-modifying osteoarthritis drugs (DMOADs). Accordingly, the objective of this study was to identify a candidate DMOAD that targets MMP13 expression.
Methods
Generation of SW1353-MMP13p-GFP cells.
Using bacterial artificial clone (BAC) recombination technique, GFP was recombined with the region downstream of the MMP13 promoter (MMP13p) in the BAC RP11–155B17 (The BACPAC Resources Center, http://bacpac.chori.org) that contains human MMP13 plus sequences 66 kb upstream and 88 kb downstream from the transcription start site. This recombinant BAC was transfected into SW1353 chondrosarcoma cells (ATCC: https://www.atcc.org/Products/All/HTB-94.aspx), and a SW1353-MMP13p-GFP cell line was established [Fig. S1 and S2(A–D)]. Primer sequences used for BAC recombination are presented in Table S1.
Small molecule library high-throughput screening.
The small molecule library (validated library: 3,399 compounds) was provided under the Drug Discovery Initiative, The University of Tokyo, Japan. SW1353-MMP13p-GFP cells were seeded at a density of 2.0 × 104 cells/well in black flat-bottom 96-well plates (Corning, NY, USA). After 24 h, the cells were treated with the test compounds (2 μM) along with IL-1β (30 ng/ml) and TNF-α (30 ng/ml). Each 96-well plate contained eight background wells with the cells, and eight control wells with the cells, IL-1β, and TNF-α. After 72 h, the medium was replaced with PBS(−), and fluorescence intensity was measured using a microplate reader (2104 EnVision; PerkinElmer) at an excitation wavelength of 485 nm and emission wavelength of 535 nm. The inhibition rate (InH(%)) for each compound was calculated as follows:
Cell culture.
The cells were seeded at a density of 5.0 × 105 cells per well in a 6-well plate containing DMEM/F-12 supplemented with 10% FBS and penicillin-streptomycin. In the cytokine stimulation experiment, either IL-1β (30 ng/ml) or TNF-α (30 ng/ml), or both were added, and the cells were harvested 24 h later for RT-qPCR or western blotting. Small molecules used for cell culture included colchicine, verapamil hydrochloride (Sigma-Aldrich), cytochalasin A, SU6656 (Santa Cruz Biotechnology, CA, USA), AZA (Merck Millipore, MA, USA), BAPTA, BAPTA-AM (ThermoFisher), 2ABP, and thapsigargin (Abcam). In plasmid expression experiments, SW1353 cells were transfected with plasmids expressing the constitutively active form of SRC (pLNCX chick src E378G, Addgene, USA), RAC1 (pcDNA3-EGFP-Rac1-Q61L, Addgene), or empty vector (pcDNA3) using Lipofectamine 3000 (ThermoFisher). In the siRNA knockdown experiment, SW1353 cells were transfected with Silencer Select siRNA (Ambion, USA) using Lipofectamine RNAiMAX (ThermoFisher). siRNAs were purchased from Ambion and targeted either SRC (#s13414), LCK (#s8108), FYN (#s5436), LYN (#s8356), FGR (#s5185), YES1 (#s14957), BLK (#s1994), HCK (#s6479), or PLCG1 (#s10633); negative control siRNA (#4390843) were also used.
Mice.
We manually assigned 10-week-old male C57BL/6J mice to the following three groups: a non-surgical control group (n = 10), a sham operation group (n = 10), and a destabilization of the medial meniscus (DMM) surgery group (n= 20). The mice in the DMM surgery group were further randomly assigned to a DMM group (n = 10) or DMM + colchicine group (n = 10). Experimental OA was induced by DMM surgery in the right knee, as previously reported11. Sham control surgery (capsulotomy only) was also performed in the right knee. Five days after surgery, the mice in the DMM + colchicine group were provided drinking water containing colchicine (0.02 mg/ml). The drinking water was changed three times per week. The knee joints of all mice were harvested 12 weeks after DMM surgery.
Isobaric tags for relative and absolute quantification (iTRAQ) sample labeling, mass spectrometry analysis, peptide identification, and pathway analysis.
SW1353 cells (1.0 × 106) were seeded in a 15 cm dish. After achieving subconfluency was achieved, the cells were treated with cytokines and colchicine (1 μM). After 24 h, the samples were collected using a Nuclear/Cytosolic Fractionation Kit (Cell Biolabs Inc., CA, USA). Analysis of proteins using isobaric tags for relative and absolute quantification (iTRAQ)—a chemical label detected by mass spectrometry—was performed as previously described12,13. The protein profiles obtained through the iTRAQ-based proteomic analysis were subjected to ingenuity pathway analysis (IPA) (https://digitalinsights.qiagen.com/products-overview/discovery-insights-portfolio/analysis-and-visualization/qiagen-ipa/, QIAGEN, Redwood City, CA, USA).
Additional Methodology.
Reagents and protocols related to RT-qPCR, western bloting, histology and immunohistochemistry, coimmunoprecipitation, isolation of mouse primary epiphyseal chondrocytes and human articular chondrocytes, TUNEL assay, and immunofluorescence analysis of the articular cartilage and chondrocytes, are presented in the Supplemental Methods.
Statistics.
All data sets were compared using GraphPad Prism (GraphPad Software, La Jolla, CA) version 9.0. Data are shown as mean with 95% confidence interval (CI). For in vitro studies, the one-way analysis of variance (ANOVA) followed by Student’s t-test was used. For in vivo studies, analysis for multiple samples was carried out using the one-way ANOVA with Tukey’s post-hoc test. Values of p < 0.05 were considered to indicate significant differences.
Study approval.
All patient samples were obtained from subjects who provided informed consent for articular cartilage collection, in accordance with the Declaration of Helsinki and with approval from the ethical review board of Gifu University (IRB approval no. 2018–009), Graduate School of Medicine. All animal experiments were performed according to the guidelines of the Animal Care and Experimentation Committee of Gifu University (no. 27–87).
Results
IL-1β and TNF-α synergistically induce MMP13 expression.
SW1353 cells were stimulated with IL-1β, TNF-α, or both compounds. IL-1β and TNF-α induced MMP13 expression, and synergistically induced MMP13 expression at both the mRNA and protein levels [Fig. 1(A, B)], suggesting a crosstalk between IL-1β and TNF-α signaling for enabling MMP13 production.
Figure 1.

Colchicine is a candidate DMOAD that inhibits MMP13 expression. (A, B) Expression level of MMP13 mRNA (A) and protein (B) in SW1353 cells were determined by RT-qPCR and western blotting, respectively (n = 3; **p < 0.01; Student’s t test). The image is representative of n = 3 western blots. (C) The first round of screening of 3,399 compounds based on the inhibition rate of GFP fluorescence intensity in SW1353-MMPp13-GFP cells. (D) Expression level of MMP13 in SW1353 pretreated with colchicine before costimulation with IL-1β and TNF-α, determined by RT-qPCR (n = 3; **p < 0.01; Student’s t test). (E) Expression level of ADAMTS-4 and −5 and MMP13 in SW1353 cells pretreated with colchicine (0.1, 1, and 10 μM), followed by costimulation with IL-1β and TNF-α, determined by RT-qPCR (n = 3; *p < 0.05, **p < 0.01; Student’s t test). (F) Expression level of MMP13 protein in SW1353 cells determined by western blotting (n = 3; **p < 0.01; Student’s t test). Image is representative of n = 3 western blots. (G) Cell viability calculated by TUNEL staining (n = 3; **p < 0.01; Student’s t test). The data are expressed as mean with 95%CI (A, B, D–G).
High-throughput screening shows that colchicine is a DMOAD candidate.
We performed high-throughput screening using SW1353-MMP13p-GFP cells. The first screen of 3,399 compounds yielded 125 hits (3.7%) that inhibited GFP fluorescence intensity induced upon costimulation with IL-1β and TNF-α more than 60% [Fig. 1(C)]. We selected 12 out of 125 compounds and performed a second round of screening to examine whether these 12 compounds similarly inhibit the expression of endogenous MMP13 [Table S2]. The second screening yielded six hits that significantly inhibited MMP13 expression in SW1353 cells [Fig. S3(A)]. Furthermore, four of these six compounds significantly inhibited cytokine-induced Mmp13 expression in mouse primary epiphyseal chondrocytes [Fig. S3(B)]. Among the four compounds, colchicine (compound #5), which is approved for human administration by the Food and Drug Administration (FDA), was subjected to further experiments. We found that colchicine significantly dampened MMP13 induction upon costimulation with IL-1β and TNF-α in a dose-dependent manner [Fig. 1(D)]. Additionally, colchicine decreased the expression of ADAMTS-4 and ADAMTS-5; however, it did not affect the expression of MMP3 [Fig. 1(E)]. Colchicine reduced the intracellular accumulation of MMP13 protein [Fig. 1(F)]. Meanwhile, cell viability testing revealed that 10 μM colchicine induced cell death as a toxic effect [Fig. 1(G)].
In addition, MMP13 expression was significantly higher in chondrocytes isolated from similarly treated human articular cartilage explants than in that controls; however, the increase in MMP13 expression was significantly abrogated upon pretreatment with colchicine in a dose-dependent manner [Fig. S3(C)].
Self-oral administration of colchicine inhibits Mmp13 expression and delays cartilage destruction in a surgically induced murine model of OA.
In spite of colchicine administration, the mice in each group did not exhibit any abnormality in appearance. Mice weights (mean ± SEM) at 10 weeks of age were 27.0 ± 1.2 g, 26.2 ± 1.8 g, 25.5 ± 0.5 g and 27.5 ± 1.0 g in the control, sham, DMM, and DMM + colchicine groups, respectively, whereas those at 22 weeks were 30.5 ± 0.8 g, 31.4 ± 1.5 g, 30.8 ± 0.8 g, and 32.0 ± 1.9 g, respectively. Colchicine administration decreased cartilage destruction and Mmp13 expression 12 weeks after DMM surgery [Fig. 2(A, B)]. Moreover, immunohistochemistry demonstrated that colchicine administration both suppressed the decrease in the expression of type II collagen, which is the target of MMP13 [Fig. 2(B)], and blocked the increase in type X collagen expression and the number of TUNEL-positive cells, indicating hypertrophy and apoptosis, respectively [Fig. S4].
Figure 2.
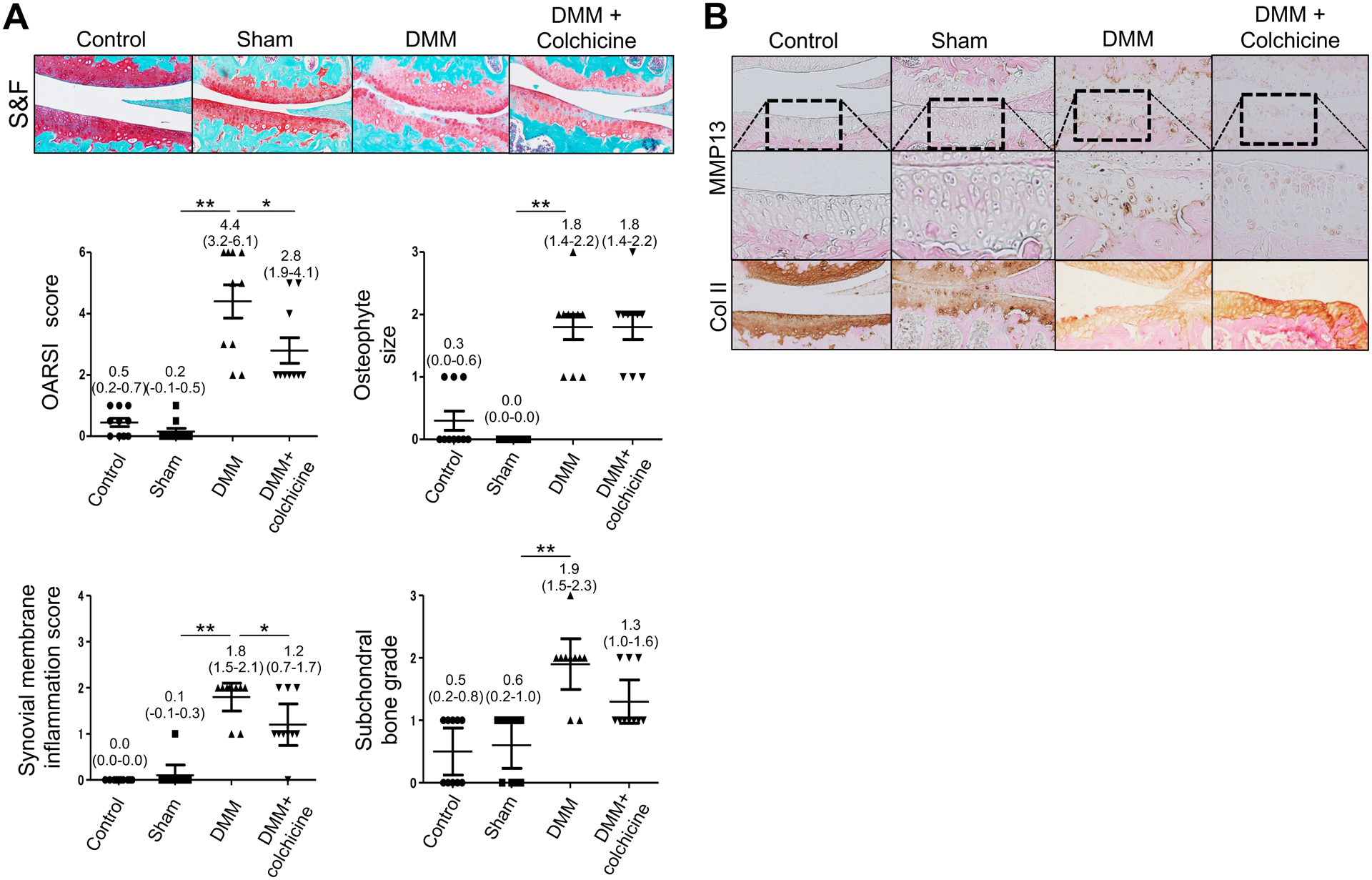
Colchicine delays cartilage destruction after DMM in mice. (A, B) The right knees of the control, sham, DMM, and DMM + colchicine mice were collected 12 weeks post-surgery. Colchicine administration was started 5 days post surgery. Sections were subjected to Safranin O-fast green staining (A) and immunohistochemistry (B). Images are representative of control, n = 5; sham, n = 10; DMM, n = 10; DMM + colchicine, n = 10. OARSI score, osteophyte size, synovium membrane inflammation score, and subchondral bone graded are evaluated. Data represent mean with 95%CI. Data were analyzed using the one-way ANOVA with Tukey’s post-hoc test. *adjusted p < 0.05, **adjusted p < 0.01.
The colchicine-administered mice with DMM exhibited significantly lower OARSI scores (adjusted P: 0.0242, mean difference: 1.6, 95% CI of difference: 0.1651–3.035) and synovial membrane inflammation scores (adjusted P: 0.0243, mean difference: 0.6, 95% CI of difference: 0.06158–1.138) than the mice with DMM [Fig. 2(A)]. However, no significant differences were observed in osteophyte size or subchondral bone grade between the groups [Fig. 2(A)].
Ingenuity Pathway Analysis (IPA) using iTRAQ-based proteomic analysis reveals that the Rho signaling pathways are regulated by both inflammatory cytokines and colchicine.
The iTRAQ analyses were performed to assess the protein profiles associated with the inflammatory cytokines and colchicine (control, cytokine, and cytokine+colchicine); additionally, statistical comparisons were performed for the protein profiles resulting from each treatment [Tables S3–5]. IPA of the protein profiles obtained through the iTRAQ-based proteomic analysis identified 39 pathways that were regulated by cytokine treatment (versus no treatment control), 67 pathways regulated by pretreatment with colchicine before cytokine treatment (versus cytokine treatment alone), and 232 pathways regulated by treatment with both colchicine and cytokine (versus no treatment). Notably, eight ingenuity signaling pathways, including Rho-related signaling pathways, that were regulated by treatment with inflammatory cytokines, and counteracted by colchicine, were identified [Fig. 3(A), Table 1]. Consistent with this result, treatment with either an actin polymerization inhibitor (cytochalasin A) or a RAC1 inhibitor (AZA1) completely blocked the induction of MMP13 expression by inflammatory cytokines [Fig. 3(B)]. However, transfection of constitutively active RAC1 into SW1353 cells failed to induce MMP13 expression; whereas colchicine inhibited MMP13 expression even in the presence of a constitutively active form of RAC1 [Fig. 3(C)].
Figure 3.

Colchicine rescues the pathological signaling pathway induced by inflammatory cytokines via the activation of RAC1 and/or SRC. (A) IPA showing signaling pathways that are significantly altered in response to all combinations of control, cytokine, and cytokine+colchicine. (B) MMP13 expression in SW1353 cells pretreated with either cytochalasin A (25 μM), AZA (25 μM), or SU6656 (500 nM), determined by RT-qPCR (n = 3; **p < 0.01; Student’s t test). (C) RT-qPCR showed MMP13 expression in SW1353 cells transfected with a constitutively active form of SRC (CA-Src), RAC (CA-Rac), or empty vector as the control, followed by colchicine (1 μM) treatment (n = 3; *p < 0.05, **p < 0.01; Student’s t test). (D) RT-qPCR showed MMP13 expression in SW1353 cells treated with colchicine (1 μM) after transfection with siRNA targeting SRC family members (n = 3; *p < 0.05, **p < 0.01; Student’s t test). (E) RT-qPCR showed MMP13 expression in SW1353 cells pretreated with either AZA (25 μM), SU6656 (500 nM), or colchicine (1 μM), before stimulation with either IL-1β or TNF-α (n = 3; **p < 0.01; Student’s t test). The data are expressed as mean with 95%CI (B–E).
Table 1.
Signaling pathways altered by cytokines that are counteracted by colchicine
| Ingenuity Canonical Pathways |
|---|
|
Colchicine inhibits cytokine-induced MMP13 expression by affecting the crosstalk point between IL-1β and TNF-α signals.
Our findings suggest that colchicine blocks RAC1 signaling, which is necessary, but insufficient, to induce MMP13 expression. A previous study has indicated that mechanical stress promotes matrix synthesis through the integrin β1-SRC-PLC-γ1/RAC1-ERK1/2 pathway14 and that PLC-γ1 and RAC1 coregulate EGF-induced cytoskeleton remodeling by direct functional interactions15. This motivated us to examine whether SRC and PLC-γ1 are also involved in the induction of MMP13 expression by IL-1β and TNF-α. Our results showed that MMP13 expression was attenuated by ~50% in the presence of the selective SRC family kinase inhibitor, SU6656, [Fig. 3(B)]. The SRC kinase family is a family of non-receptor tyrosine kinases including eight members in vertebrates, namely, SRC, LCK, FYN, LYN, FGR, YES1, BLK, and HCK16. Indeed, we found that the expression of siRNA directed against SRC attenuated cytokine-induced MMP13 expression by ~55% [Fig. 3(D)]. In contrast, the knockdown of the other SRC family members either failed to alter MMP13 expression by inflammatory cytokines (LCK, FYN, LYN, and FGR) or enhanced its expression in response to these signals (YES1, BLK, and HCK). Consistent with the necessity of SRC signaling for MMP13 expression, we found that overexpression of a constitutively active form of SRC promoted MMP13 expression, whereas SRC-induced MMP13 expression was completely abrogated by colchicine pretreatment [Fig. 3(C)]. Moreover, co-transfection of the constitutively active forms of SRC and RAC synergistically induced the expression of MMP13, which was abrogated by colchicine pretreatment [Fig. 3(C)]. In each inflammatory cytokine stimulation experiment, the SRC inhibitor, SU6656, modestly inhibited IL-1β-mediated induction of MMP13 expression, and moderately inhibited TNF-α-induced MMP13 expression [Fig. 3(E)], whereas the RAC1 inhibitor, AZA1, abolished the induction of MMP13 by either IL-1β or TNF-α [Fig. 3(E)]. In comparison, colchicine moderately inhibited the induction of MMP13 by either IL-1β or TNF-α [Fig. 3(E)].
Colchicine inhibits the PLC-γ1 phosphorylation on microtubules.
Consistent with the role of SRC in MMP13 expression, we found that cytokines increased phospho-SRC levels in a time-dependent manner, however, colchicine did not attenuate SRC phosphorylation [Fig. 4(A)]. As both SRC and RAC1 interact with PLC-γ114,15, we examined whether PLC-γ1 activity is involved in the signaling pathway associated with cytokine-induced MMP13 expression. Indeed, cytokine administration promoted the phosphorylation of PLC-γ1 (Tyr783) in a time-dependent manner. Notably, PLC-γ1 phosphorylation (Tyr783) was markedly suppressed in the presence of colchicine, whereas the steady-state level of PLC-γ1 was not affected [Fig. 4(A)]. Considering that the phosphorylation of PLC-γ1 on Tyr783 results in its activation17, these results imply that colchicine inhibits cytokine-induced MMP13 expression by acting both downstream of SRC and upstream of PLC-γ1 phosphorylation. Consistent with this notion, siRNA-mediated knockdown of PLC-γ1 attenuated cytokine-induced MMP13 expression [Fig. S5]. Cumulatively, these results strongly indicate that the phosphorylation and activation of PLC-γ1 are regulated by colchicine.
Figure 4.

Colchicine attenuates the phosphorylation of PLC-γ1 bound to microtubules. (A) Western blotting shows that the pretreatment with colchicine (1 μM) suppresses the phosphorylation of PLC-γ1, but not that of SRC, promoted by costimulation with IL-1β and TNF-α. The image is representative of n = 3 western blots. (B) Immunofluorescence showed cellular localization of phosphorylated PLC-γ1 in SW1353 cells in the presence of IL-1β and TNF-α and/or colchicine (1 μM). The image is representative of n = 3 immunofluorescence. (C) Coimmunoprecipitation of endogenous tubulin and PLC-γ1 in SW1353 cells treated with IL-1β, TNF-α, and/or colchicine (1 μM). Anti-tubulin, anti-p-PLC-γ1 antibodies, and non-immune IgG were used for immunoprecipitation (IP) and immunoblotting (IB). The image is representative of n = 3 IB. (D) RT-qPCR shows MMP13 expression in SW1353 cells pretreated with either BAPTA (10 μM), BAPTA-AM (10 μM), verapamil (100 μM), 2ABP (100 μM), or thapsigargin (1 μM), before stimulation with IL-1β and TNF-α (n = 3; **p < 0.01; Student’s t test). The data are expressed as mean with 95%CI (D).
As colchicine is known to inhibit microtubule polymerization18, we next sought to determine whether PLC-γ1 might also bind to microtubules. In fact, immunofluorescence showed that phospho-PLC-γ1 (Tyr783) colocalized with tubulin and the accumulation of tubulin and PLC-γ1 was increased by cytokine administration [Fig. 4(B)]. Notably, colchicine abrogated the accumulation of tubulin and phosphorylation of PLC-γ1 (Tyr783), although they remained colocalized [Fig. 4(B)]. Furthermore, immunoprecipitation experiments using SW1353 cell lysate and antibodies against phospho-PLC-γ1 (Tyr783) and β-tubulin showed that endogenous phospho-PLC-γ1 (Tyr783) coimmunoprecipitated with β-tubulin, with or without cytokines, and the reciprocal experiment showed the same results [Fig. 4(C)]. Colchicine significantly decreased the levels of coimmunoprecipitation between the proteins. PLC-γ1 generates inositol 1,4,5-trisphosphate (IP3), which releases Ca2+ from the endoplasmic reticulum (ER) via IP3 receptors19, and enhances calcium signaling to regulate MMP13 expression20,21. Finally, when SW1353 cells were treated with either inhibitors of extracellular calcium influx (BAPTA and verapamil) or with inhibitors of calcium release from the ER (BAPTA-AM, intracellular Ca2+ chelator; 2ABP, IP3 receptor inhibitor; thapsigargin, endoplasmic reticular Ca2+ -ATPase inhibitor), only the inhibitors of ER calcium release effectively inhibited MMP13 expression [Fig. 4(D)].
Colchicine abrogates the phosphorylation of PLC-γ1 in degenerated articular cartilage in vivo and ex vivo.
SRC and PLC-γ1 phosphorylation (Tyr783) was enhanced and the tubulin content was increased following DMM surgery, which correlated with increased expression of MMP13, compared with those in mice that received a sham surgery [Fig. 2(A) and 5]. The self-oral administration of colchicine markedly decreased the phosphorylated PLC-γ1 (Tyr783) and tubulin content; however, it did not affect the phosphorylated SRC content following DMM surgery [Fig. 5]. Similar results were obtained in human articular cartilage explants [Fig. S6].
Figure 5.
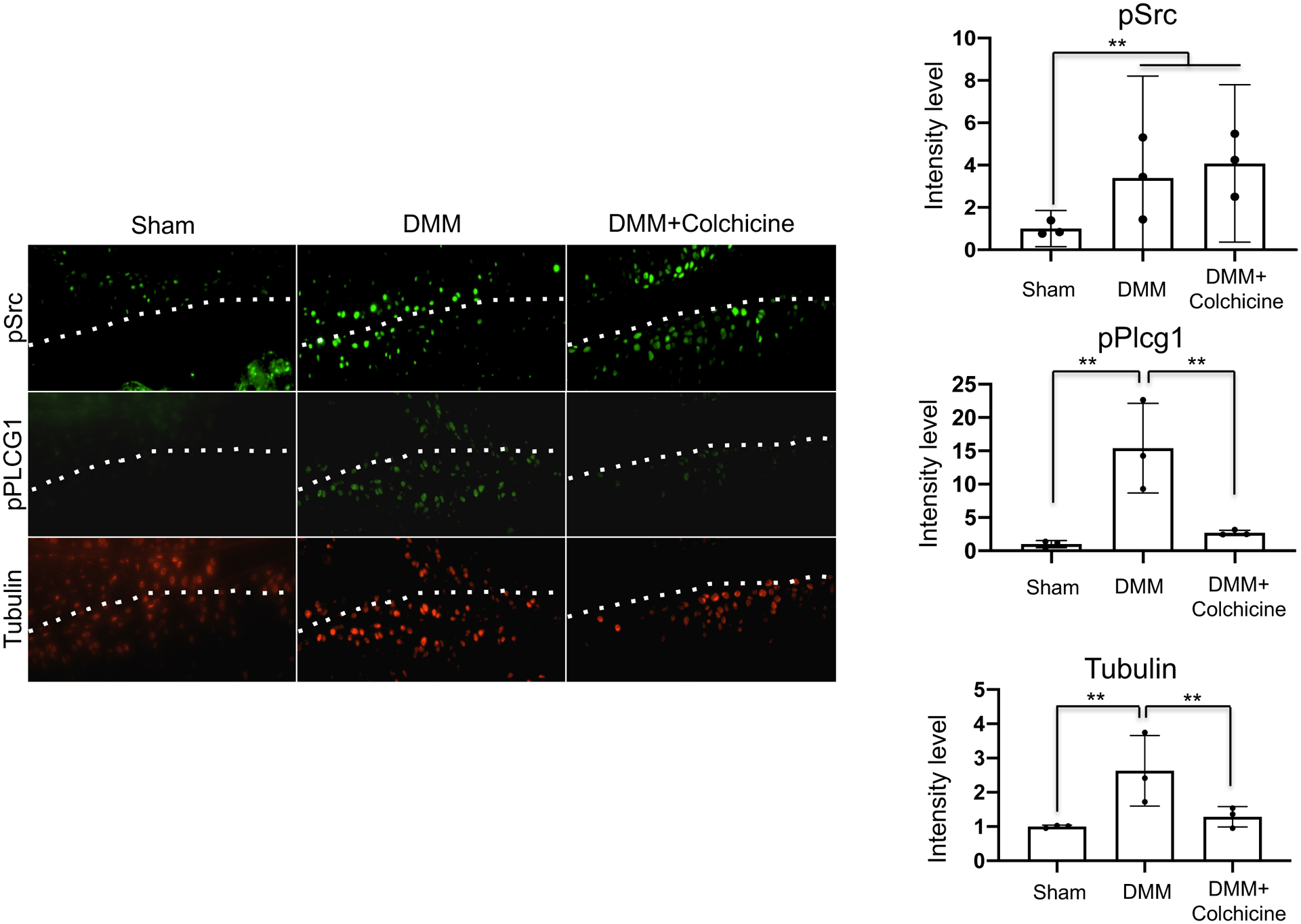
Colchicine decreases the phosphorylation of PLC-γ1 induced by the destabilization of medial meniscus (DMM) or inflammatory cytokines. The right knees of sham, DMM, and DMM + colchicine mice were collected 12 weeks post-surgery. Colchicine administration was initiated 5 days post surgery. Sections were subjected to immunofluorescence staining. Images show the cellular localization of either phospho-SRC, phospho-PLC-γ1, or tubulin in the articular cartilage. Images are representative of n = 3 mice per group. Immunofluorescence intensity was quantified using ImageJ software (n = 3; **p < 0.01). Data represent mean with 95%CI. Data were analyzed using the one-way ANOVA with Tukey’s post-hoc test.
Discussion
We performed a high-throughput screen for small molecules and identified colchicine as a candidate DMOAD. Furthermore, colchicine inhibited cytokine-induced MMP13 expression via the SRC/RAC1-phospho-PLCγ1-Ca2+ signaling pathway in vitro, whereas oral colchicine administration protected surgical OA model mice against cartilage degeneration in vivo.
Although colchicine is primarily indicated for gout and familial Mediterranean fever, recently, it has also been reported to be effective for the treatment of various inflammatory conditions, including pericarditis, atherosclerosis, and osteoarthritis, owing to its anti-inflammatory and immune-modulating effects22,23. Its anti-inflammatory effect is exerted by binding to tubulin to block caspase-1 activation, which is the enzymatic component of the nucleotide-binding oligomerization domain receptor family pyrin 3 (NLRP3) inflammasome24–26. In fact, in the current study, colchicine administration mitigated synovitis [Fig. 2(A)]; however, the suppression of NLRP3 inflammasome formation is not likely the mechanism by which colchicine suppresses MMP13 expression considering that the knockdown of the inflammasome elements, NLRP3 and Caspase1, did not significantly suppress MMP13 expression [Fig. S7], and Nlrp3 knockout mice did not exhibit protective effects against cartilage destruction in the OA mouse model27.
After IPA using iTRAQ-based proteomic analysis to identify the signaling pathways regulated by inflammatory cytokines and colchicine, we focused on the Rho signaling pathways, which have been implicated in the regulation of MMP13 expression in chondrocytes and cartilage degeneration28–30. In the current study, the RAC1-specific small molecule inhibitor, AZA, abolished the cytokine-induced expression of MMP13. In contrast, the constitutively active form of RAC1 alone, failed to induce MMP13 expression. These results suggest that RAC1 signaling is necessary but not sufficient, to induce MMP13 expression in chondrocytes. The activation of RAC1 and SRC is responsible for signal transduction in chondrocytes under conditions of mechanical stimulation14, and SRC is related to the activation of RAC1 in many other systems31,32. This suggests that SRC signaling may also be involved in cytokine-induced MMP13 expression. Indeed, the transfection of SW1353 cells with an expression vehicle encoding a constitutively active form of SRC, induced MMP13 expression. Conversely, an SRC-specific inhibitor inhibited cytokine-mediated induction of MMP13 expression. Importantly, co-transfection of plasmids expressing the active forms of SRC and RAC1 synergistically induced MMP13 expression whereas colchicine completely abrogated the activation of RAC1- and SRC-induced MMP13 expression. In addition, the administration of RAC1- and SRC-specific inhibitors with colchicine indicated that IL-1β and TNF-α activate RAC1 and SRC signaling, respectively, to promote MMP13 expression; whereas colchicine abrogates both signals. These results suggest that either microtubules, which are the target of colchicine, or microtubule-associated components, lie at a junction between the RAC1 and SRC signaling pathways.
In light of prior work, indicating that RAC1 and SRC often act in combination with PLC-γ1 to regulate cellular signaling events15,33–37, we explored whether PLC-γ1, encoded by the Plcg1 gene, was similarly involved in inflammatory cytokine-induced expression of MMP13. The transfection of SW1353 cells with siRNA targeting Plcg1 (encoding PLC-γ1) abolished cytokine-induced MMP13 expression. Moreover, colchicine administration abrogated the inflammatory cytokine-induced phosphorylation of PLC-γ1 (Tyr783), indicating that colchicine acts on PLC-γ1 downstream of SRC. Considering that the phosphorylation of PLC-γ1 at three tyrosine residues (Tyr771, Tyr 783, and Tyr 1248) induces increased enzymatic activity, whereas the phosphorylation of Tyr783 is essential for PLC-γ1 activation38, it was postulated that colchicine inactivates PLC-γ1. In addition, the results of immunofluorescence and coimmunoprecipitation of tubulin and phospho-PLC-γ1 (Tyr783) demonstrated that colchicine interaction with tubulin abrogates cytokine-induced phosphorylation of PLC-γ1 (Tyr783) bound to microtubules. Phosphorylated PLC-γ1 enhances calcium signaling through inositol 1,4,5-trisphosphate (IP3) that binds to IP3 receptors on the ER39, and calcium signaling is involved in MMP13 expression and cartilage degeneration40,41. Interestingly, inhibitors of intracellular calcium released from the ER, but not inhibitors of extracellular calcium influx, suppressed cytokine-dependent MMP13 expression. The administration of the Ca2+ ionophore, A2318 (compound #2) [Fig. S3(A) and S3(B), Table S2], also disrupts cytokine-dependent MMP13 expression; consistent with the notion that only the release of intracellular Ca2+ stores promotes MMP13 expression. Taken together, our results suggest that colchicine inhibits MMP13 expression by disrupting the SRC/RAC1-phospho-PLCγ1-Ca2+ signaling pathway. However, colchicine did not suppress osteophyte formation in the mouse OA model, suggesting that colchicine does not affect the TGF-β or BMP-2 signaling pathways responsible for osteophyte formation42,43.
Previously, results of clinical trials on colchicine in the context of osteoarthritis have reported44–46. Two of the trial demonstrated the efficacy and safety of colchicine for OA,45,46 whereas another found that colchicine reduces inflammation and high bone turnover biomarkers known to be associated with OA severity, but not OA symptoms, over a 16-week study period44. Our results further demonstrate that colchicine was effective for controlling synovitis [Fig. 1(A)]. Thus, colchicine is expected to have an anti-inflammatory effect, presumably caused by the inhibition of inflammasome formation26. In the current study, we demonstrate that colchicine can ameliorate cartilage degeneration in vivo. Although the long-term effects of colchicine on cartilage degeneration have not been evaluated, it has been proven efficacious and safe for the long-term treatment of familial Mediterranean fever47,48, which may also translate to OA; however, this requires further verification.
One of the primary limitations of this study is that the results do not negate the possibility that colchicine potency in OA is also associated with an MMP13-independent mechanism in addition to the described MMP13 dependent pathway. In fact, based on our pathway analysis results, colchicine clearly regulates several signaling pathways that may exert inhibitory effects on cartilage degradation. For example, colchicine was found to inhibit the expression of the aggrecan-degrading enzymes ADAMTS-4 and ADAMTS −5 [Fig. 1(E)] while consistently rescuing the diminished proteoglycan content in human articular cartilage explants [Fig. 2(C)]. Moreover, although we demonstrated that colchicine abrogates PLC-γ1 phosphorylation upon costimulation with IL-1β and TNF-α, while inhibiting MMP13 expression, further investigation is required to clarify the specific effects of colchicine on the IL-1β and TNF-α signaling pathways. Additionally, we did not report the serum levels of colchicine in the DMM mouse model following provision of 0.02 mg/ml in the drinking water; however, colchicine consumption was estimated to be approximately 0.08 mg/mouse/day based on the amount of colchicine-containing water consumption. Thus, it remains necessary to determine the optimal dose of colchicine for OA treatment. Finally, regarding pain-related behavior in the mice with DMM, we focused on cartilage degeneration without examining pain-related behavior or knee function. A long-term study is, therefore, warranted to investigate the slow-acting effect of colchicine on cartilage degeneration, and consequently, pain-related behavior and knee function.
In conclusion, via a high-throughput screening approach, we found that colchicine is a DMOAD candidate and provided in vitro and in vivo evidence suggesting that colchicine inhibits MMP13 expression by disrupting the SRC/RAC1-phospho-PLCγ1-Ca2+ signaling pathway [Fig. 6]. We expect that our findings provide a possible mechanism of cartilage destruction, while also raising the possibility that PLC-γ1 is a promising target to block inflammatory cytokine-induced MMP13 expression and prevent OA-related cartilage destruction.
Figure 6.

Colchicine blocks MMP13 expression in the articular cartilage via PLC-γ1 phosphorylation. Our results suggest that IL-1β and TNF-α promote the activation of RAC1 and phosphorylation of SRC, respectively. These two activated kinases cooperate to induce PLC-γ1 phosphorylation, which in turn activates intracellular calcium signaling, leading to MMP13 expression in articular chondrocytes.
Supplementary Material
Acknowledgements
We thank H. Yoshioka, T. Satake, S. Komura, T. Miyagawa, K. Kawashima, H. Asano, and Y. Nakamura for their thoughtful discussions, and T. Ishihara for his advice on statistical analysis.
Role of the funding source
This work was supported by JSPS KAKENHI Grant Numbers 26893104 and 16H06263, and JOSKAS Research Grant 2017 to HO. Generation of the SW1353-MMP13p-GFP cell line was supported by grants from NIAMS/NIH (AR055148, AR055552, and AR060735) to AL. This work was supported by the Platform Project for Supporting in Drug Discovery and Life Science Research from Japan Agency for Medical Research and Development (AMED).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
The authors have declared that no conflict of interest exists.
References
- 1.Heinegård D, Saxne T. The role of the cartilage matrix in osteoarthritis. Nat Rev Rheumatol 2011;7(1):50–6. doi: 10.1038/nrrheum.2010.198 [published Online First: 2010/11/30] [DOI] [PubMed] [Google Scholar]
- 2.Tetlow LC, Adlam DJ, Woolley DE. Matrix metalloproteinase and proinflammatory cytokine production by chondrocytes of human osteoarthritic cartilage: associations with degenerative changes. Arthritis Rheum 2001;44(3):585–94. doi: [DOI] [PubMed] [Google Scholar]
- 3.Goldring MB, Otero M. Inflammation in osteoarthritis. Curr Opin Rheumatol 2011;23(5):471–8. doi: 10.1097/BOR.0b013e328349c2b1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Martel-Pelletier J, Alaaeddine N, Pelletier JP. Cytokines and their role in the pathophysiology of osteoarthritis. Front Biosci 1999;4:D694–703. doi: 10.2741/martel [published Online First: 1999/10/15] [DOI] [PubMed] [Google Scholar]
- 5.Kobayashi M, Squires GR, Mousa A, Tanzer M, Zukor DJ, Antoniou J, et al. Role of interleukin-1 and tumor necrosis factor alpha in matrix degradation of human osteoarthritic cartilage. Arthritis Rheum 2005;52(1):128–35. doi: 10.1002/art.20776 [DOI] [PubMed] [Google Scholar]
- 6.Pelletier JP, Martel-Pelletier J, Abramson SB. Osteoarthritis, an inflammatory disease: potential implication for the selection of new therapeutic targets. Arthritis Rheum 2001;44(6):1237–47. doi: [DOI] [PubMed] [Google Scholar]
- 7.Verma P, Dalal K. ADAMTS-4 and ADAMTS-5: key enzymes in osteoarthritis. J Cell Biochem 2011;112(12):3507–14. doi: 10.1002/jcb.23298 [DOI] [PubMed] [Google Scholar]
- 8.Li H, Wang D, Yuan Y, Min J. New insights on the MMP-13 regulatory network in the pathogenesis of early osteoarthritis. Arthritis Res Ther 2017;19(1):248. doi: 10.1186/s13075-017-1454-2 [published Online First: 2017/11/10] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang M, Sampson ER, Jin H, Li J, Ke QH, Im HJ, et al. MMP13 is a critical target gene during the progression of osteoarthritis. Arthritis Res Ther 2013;15(1):R5. doi: ar4133 [pii] 10.1186/ar4133 [published Online First: 2013/01/10] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Neuhold LA, Killar L, Zhao W, Sung ML, Warner L, Kulik J, et al. Postnatal expression in hyaline cartilage of constitutively active human collagenase-3 (MMP-13) induces osteoarthritis in mice. J Clin Invest 2001;107(1):35–44. doi: 10.1172/JCI10564 [published Online First: 2001/01/03] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Glasson SS, Blanchet TJ, Morris EA. The surgical destabilization of the medial meniscus (DMM) model of osteoarthritis in the 129/SvEv mouse. Osteoarthritis Cartilage 2007;15(9):1061–9. doi: 10.1016/j.joca.2007.03.006 [published Online First: 2007/04/30] [DOI] [PubMed] [Google Scholar]
- 12.Ishii M, Suehara Y, Sano K, Kohsaka S, Hayashi T, Kazuno S, et al. Proteomic signatures corresponding to the SS18/SSX fusion gene in synovial sarcoma. Oncotarget 2018;9(101):37509–19. doi: 10.18632/oncotarget.26493 [published Online First: 2018/12/25] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ross PL, Huang YN, Marchese JN, Williamson B, Parker K, Hattan S, et al. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol Cell Proteomics 2004;3(12):1154–69. doi: 10.1074/mcp.M400129-MCP200 [published Online First: 2004/09/22] [DOI] [PubMed] [Google Scholar]
- 14.Ren K, Liu F, Huang Y, Liang W, Cui W, Wang Q, et al. Periodic mechanical stress activates integrinβ1-dependent Src-dependent PLCγ1-independent Rac1 mitogenic signal in rat chondrocytes through ERK1/2. Cell Physiol Biochem 2012;30(4):827–42. doi: 10.1159/000341461 [published Online First: 2012/08/10] [DOI] [PubMed] [Google Scholar]
- 15.Li S, Wang Q, Wang Y, Chen X, Wang Z. PLC-gamma1 and Rac1 coregulate EGF-induced cytoskeleton remodeling and cell migration. Mol Endocrinol 2009;23(6):901–13. doi: 10.1210/me.2008-0368 [published Online First: 2009/03/05] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thomas SM, Brugge JS. Cellular functions regulated by Src family kinases. Annu Rev Cell Dev Biol 1997;13:513–609. doi: 10.1146/annurev.cellbio.13.1.513 [DOI] [PubMed] [Google Scholar]
- 17.Poulin B, Sekiya F, Rhee SG. Intramolecular interaction between phosphorylated tyrosine-783 and the C-terminal Src homology 2 domain activates phospholipase C-gamma1. Proc Natl Acad Sci U S A 2005;102(12):4276–81. doi: 10.1073/pnas.0409590102 [published Online First: 2005/03/11] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Skoufias DA, Wilson L. Mechanism of inhibition of microtubule polymerization by colchicine: inhibitory potencies of unliganded colchicine and tubulin-colchicine complexes. Biochemistry 1992;31(3):738–46. doi: 10.1021/bi00118a015 [DOI] [PubMed] [Google Scholar]
- 19.Rameh LE, Rhee SG, Spokes K, Kazlauskas A, Cantley LC, Cantley LG. Phosphoinositide 3-kinase regulates phospholipase Cgamma-mediated calcium signaling. J Biol Chem 1998;273(37):23750–7. doi: 10.1074/jbc.273.37.23750 [DOI] [PubMed] [Google Scholar]
- 20.Zhu L, Jones C, Zhang G. The role of phospholipase C signaling in macrophage-mediated inflammatory response. J Immunol Res 2018;2018:5201759. doi: 10.1155/2018/5201759 [published Online First: 2018/02/08] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.He Z, Leong DJ, Zhuo Z, Majeska RJ, Cardoso L, Spray DC, et al. Strain-induced mechanotransduction through primary cilia, extracellular ATP, purinergic calcium signaling, and ERK1/2 transactivates CITED2 and downregulates MMP-1 and MMP-13 gene expression in chondrocytes. Osteoarthr Cartil 2016;24(5):892–901. doi: 10.1016/j.joca.2015.11.015 [published Online First: 2015/12/12] [DOI] [PubMed] [Google Scholar]
- 22.Siak J, Flint N, Shmueli HG, Siegel RJ, Rader F. The Use of Colchicine in Cardiovascular Diseases: A Systematic Review. Am J Med 2021. doi: 10.1016/j.amjmed.2021.01.019 [published Online First: 2021/02/21] [DOI] [PubMed] [Google Scholar]
- 23.Leung YY, Yao Hui LL, Kraus VB. Colchicine--Update on mechanisms of action and therapeutic uses. Semin Arthritis Rheum 2015;45(3):341–50. doi: 10.1016/j.semarthrit.2015.06.013 [published Online First: 2015/08/01] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dalbeth N, Lauterio TJ, Wolfe HR. Mechanism of action of colchicine in the treatment of gout. Clin Ther 2014;36(10):1465–79. doi: 10.1016/j.clinthera.2014.07.017 [published Online First: 2014/08/21] [DOI] [PubMed] [Google Scholar]
- 25.Taylor EW. The mechanism of colchicine inhibition of mitosis. I. Kinetics of inhibition and the binding of H3-colchicine. J Cell Biol 1965;25:SUPPL:145–60. doi: 10.1083/jcb.25.1.145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Misawa T, Takahama M, Kozaki T, Lee H, Zou J, Saitoh T, et al. Microtubule-driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome. Nat Immunol 2013;14(5):454–60. doi: 10.1038/ni.2550 [published Online First: 2013/03/17] [DOI] [PubMed] [Google Scholar]
- 27.Bougault C, Gosset M, Houard X, Salvat C, Godmann L, Pap T, et al. Stress-induced cartilage degradation does not depend on the NLRP3 inflammasome in human osteoarthritis and mouse models. Arthritis Rheum 2012;64(12):3972–81. doi: 10.1002/art.34678 [DOI] [PubMed] [Google Scholar]
- 28.Long DL, Willey JS, Loeser RF. Rac1 is required for matrix metalloproteinase 13 production by chondrocytes in response to fibronectin fragments. Arthritis Rheum 2013;65(6):1561–8. doi: 10.1002/art.37922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hu X, Ji X, Yang M, Fan S, Wang J, Lu M, et al. Cdc42 is essential for both articular cartilage degeneration and subchondral bone deterioration in experimental osteoarthritis. J Bone Miner Res 2018;33(5):945–58. doi: 10.1002/jbmr.3380 [published Online First: 2018/02/15] [DOI] [PubMed] [Google Scholar]
- 30.Zhu S, Liu H, Wu Y, Heng BC, Chen P, Liu H, et al. Wnt and Rho GTPase signaling in osteoarthritis development and intervention: implications for diagnosis and therapy. Arthritis Res Ther 2013;15(4):217. doi: 10.1186/ar4240 [published Online First: 2013/07/11] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chaturvedi LS, Marsh HM, Shang X, Zheng Y, Basson MD. Repetitive deformation activates focal adhesion kinase and ERK mitogenic signals in human Caco-2 intestinal epithelial cells through Src and Rac1. J Biol Chem 2007;282(1):14–28. doi: 10.1074/jbc.M605817200 [published Online First: 2006/11/06] [DOI] [PubMed] [Google Scholar]
- 32.Gupta SK, Vlahakis NE. Integrin alpha9beta1 mediates enhanced cell migration through nitric oxide synthase activity regulated by Src tyrosine kinase. J Cell Sci 2009;122(Pt 12):2043–54. doi: 10.1242/jcs.041632 [published Online First: 2009/05/26] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rao JN, Liu SV, Zou T, Liu L, Xiao L, Zhang X, et al. Rac1 promotes intestinal epithelial restitution by increasing Ca2+ influx through interaction with phospholipase C-(gamma)1 after wounding. Am J Physiol Cell Physiol 2008;295(6):C1499–509. doi: 10.1152/ajpcell.00232.2008 [published Online First: 2008/10/15] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jones NP, Katan M. Role of phospholipase Cgamma1 in cell spreading requires association with a beta-Pix/GIT1-containing complex, leading to activation of Cdc42 and Rac1. Mol Cell Biol 2007;27(16):5790–805. doi: 10.1128/MCB.00778-07 [published Online First: 2007/06/11] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bourguignon LY, Singleton PA, Diedrich F. Hyaluronan-CD44 interaction with Rac1-dependent protein kinase N-gamma promotes phospholipase Cgamma1 activation, Ca(2+) signaling, and cortactin-cytoskeleton function leading to keratinocyte adhesion and differentiation. J Biol Chem 2004;279(28):29654–69. doi: 10.1074/jbc.M403608200 [published Online First: 2004/05/03] [DOI] [PubMed] [Google Scholar]
- 36.Zhou X, Izumi Y, Burg MB, Ferraris JD. Rac1/osmosensing scaffold for MEKK3 contributes via phospholipase C-gamma1 to activation of the osmoprotective transcription factor NFAT5. Proc Natl Acad Sci U S A 2011;108(29):12155–60. doi: 10.1073/pnas.1108107108 [published Online First: 2011/06/28] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lu Y, Xu Y, Yin Z, Yang X, Jiang Y, Gui J. Chondrocyte migration affects tissue-engineered cartilage integration by activating the signal transduction pathways involving Src, PLCγ1, and ERK1/2. Tissue Eng Part A 2013;19(21–22):2506–16. doi: 10.1089/ten.TEA.2012.0614 [published Online First: 2013/08/02] [DOI] [PubMed] [Google Scholar]
- 38.Kim HK, Kim JW, Zilberstein A, Margolis B, Kim JG, Schlessinger J, et al. PDGF stimulation of inositol phospholipid hydrolysis requires PLC-gamma 1 phosphorylation on tyrosine residues 783 and 1254. Cell 1991;65(3):435–41. doi: 10.1016/0092-8674(91)90461-7 [DOI] [PubMed] [Google Scholar]
- 39.Berridge MJ. Inositol trisphosphate and calcium signalling. Nature 1993;361(6410):315–25. doi: 10.1038/361315a0 [DOI] [PubMed] [Google Scholar]
- 40.Falconer AMD, Chan CM, Gray J, Nagashima I, Holland RA, Shimizu H, et al. Collagenolytic matrix metalloproteinases antagonize proteinase-activated receptor-2 activation, providing insights into extracellular matrix turnover. J Biol Chem 2019;294(26):10266–77. doi: 10.1074/jbc.RA119.006974 [published Online First: 2019/05/19] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Loeser RF, Forsyth CB, Samarel AM, Im HJ. Fibronectin fragment activation of proline-rich tyrosine kinase PYK2 mediates integrin signals regulating collagenase-3 expression by human chondrocytes through a protein kinase C-dependent pathway. J Biol Chem 2003;278(27):24577–85. doi: 10.1074/jbc.M304530200 [published Online First: 2003/04/30] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hashimoto S, Creighton-Achermann L, Takahashi K, Amiel D, Coutts RD, Lotz M. Development and regulation of osteophyte formation during experimental osteoarthritis. Osteoarthr Cartil 2002;10(3):180–7. doi: 10.1053/joca.2001.0505 [published Online First: 2002/03/01] [DOI] [PubMed] [Google Scholar]
- 43.Blaney Davidson EN, Vitters EL, van der Kraan PM, van den Berg WB. Expression of transforming growth factor-beta (TGFbeta) and the TGFbeta signalling molecule SMAD-2P in spontaneous and instability-induced osteoarthritis: role in cartilage degradation, chondrogenesis and osteophyte formation. Ann Rheum Dis 2006;65(11):1414–21. doi: 10.1136/ard.2005.045971 [published Online First: 2006/01/28] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Leung YY, Haaland B, Huebner JL, Wong SBS, Tjai M, Wang C, et al. Colchicine lack of effectiveness in symptom and inflammation modification in knee osteoarthritis (COLKOA): a randomized controlled trial. Osteoarthritis Cartilage 2018;26(5):631–40. doi: 10.1016/j.joca.2018.01.026 [published Online First: 2018/02/07] [DOI] [PubMed] [Google Scholar]
- 45.Aran S, Malekzadeh S, Seifirad S. A double-blind randomized controlled trial appraising the symptom-modifying effects of colchicine on osteoarthritis of the knee. Clin Exp Rheumatol 2011;29(3):513–8. [published Online First: 2011/06/29] [PubMed] [Google Scholar]
- 46.Das SK, Ramakrishnan S, Mishra K, Srivastava R, Agarwal GG, Singh R, et al. A randomized controlled trial to evaluate the slow-acting symptom-modifying effects of colchicine in osteoarthritis of the knee: a preliminary report. Arthritis Rheum 2002;47(3):280–4. doi: 10.1002/art.10455 [DOI] [PubMed] [Google Scholar]
- 47.Ben-Chetrit E, Levy M. Colchicine prophylaxis in familial Mediterranean fever: reappraisal after 15 years. Semin Arthritis Rheum 1991;20(4):241–6. [DOI] [PubMed] [Google Scholar]
- 48.Imazio M, Andreis A, Brucato A, Adler Y, De Ferrari GM. Colchicine for acute and chronic coronary syndromes. Heart 2020. doi: 10.1136/heartjnl-2020-317108 [published Online First: 2020/07/01] [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.