

Abstract
Understanding the principles of colonization resistance of the gut microbiome to the pathogen Clostridioides difficile will enable the design of defined bacterial therapeutics. We investigate the ecological principles of community resistance to C. difficile using a synthetic human gut microbiome. Using a dynamic computational model, we demonstrate that C. difficile receives the largest number and magnitude of incoming negative interactions. Our results show that C. difficile is in a unique class of species that display a strong negative dependence between growth and species richness. We identify molecular mechanisms of inhibition including acidification of the environment and competition over resources. We demonstrate that Clostridium hiranonis strongly inhibits C. difficile partially via resource competition. Increasing the initial density of C. difficile can increase its abundance in the assembled community, but community context determines the maximum achievable C. difficile abundance. Our work suggests that the C. difficile inhibitory potential of defined bacterial therapeutics can be optimized by designing communities featuring a combination of mechanisms including species richness, environment acidification, and resource competition.
Keywords: Clostridioides difficile, computational modeling, ecological interactions, pathogen invasion, systems biology
Subject Categories: Biotechnology & Synthetic Biology; Microbiology, Virology & Host Pathogen Interaction
A combination of bottom‐up community assembly and computational modeling reveals determinants of Clostridioides difficile growth in synthetic human gut communities.
Introduction
Interaction with native members of human gut microbiota inhibits the ability of gastrointestinal pathogenic strains of Clostridioides difficile, Salmonella enterica, and Escherichia coli to secure an ecological niche and cause infection (Buffie & Pamer, 2013). The importance of colonization resistance by gut microbiota has been particularly highlighted in C. difficile infections, where treatment with fecal microbiota transplants (FMT) from healthy donors has proven astonishingly effective in eliminating the symptoms of C. difficile (Dowle, 2016). Because FMT has notable risks including the transfer of antibiotic‐resistant organisms, potential associations with flares of inflammatory bowel disease, and in rare cases death (Wang et␣al, 2016; Chen et␣al, 2018; DeFilipp et␣al, 2019), defined bacterial therapeutics that have been well‐characterized and standardized are needed to improve the safety and reproducibility of living bacterial therapeutic treatments. However, a key challenge to the design of effective and safe bacterial therapeutics is the vast design space of presence and absence of hundreds to thousands of potential organisms. Improving our understanding of the ecological principles of community resistance to C. difficile invasion could guide the design of maximally effective and safe therapeutics.
Multiple synthetic communities that inhibit C. difficile either in␣vitro or in␣vivo using murine models have been identified (Tvede & Rask‐Madsen, 1989; Lawley et␣al, 2012; Petrof et␣al, 2013; Buffie et␣al, 2014; Ghimire et␣al, 2020; Pereira et␣al, 2020). The majority of the defined communities are found by screening reduced complexity communities composed of isolates from a stool sample. The isolates are combined either randomly or selected based on phylogenetic diversity (Lawley et␣al, 2012; Petrof et␣al, 2013; Ghimire et␣al, 2020). Other C. difficile inhibiting communities have been more rationally designed based on predicted mechanisms of resource competition (Pereira et␣al, 2020) or statistical analyses of human and murine gut microbiome data that identify taxa that correlate with infection resistance (Buffie et␣al, 2014). However, the design process for therapeutic synthetic microbial communities frequently does not exploit information about interspecies interactions or molecular mechanisms. A deeper understanding of the ecological and molecular principles of communities that inhibit C. difficile could inform the rational design of therapeutic consortia.
In macroecology, there is a long history investigating principles of invasion that has been more recently applied to microbial systems (Mallon, van Elsas, et␣al, 2015). Invasion theory has identified four fundamental processes that determine the outcome of an invasion: dispersal, selection, drift, and diversification (Kinnunen et␣al, 2016). Biotic selection has been shown to be a key determinant of the outcome of an invasion, wherein higher diversity communities can competitively exclude an invader by reducing the availability of ecological niches and efficiently utilizing resources (Dillon et␣al, 2005; van Elsas et␣al, 2012; Ketola et␣al, 2017). However, community biodiversity does not always correlate with invasion outcome, as other biotic interactions (e.g., production of antimicrobial molecules), abiotic selection factors (e.g., environmental pH, resource availability) and dispersal, drift, and diversification processes each contribute to the outcome of invasion. For instance, in the case of a plant pathogen, the structure of the resource competition network was a better predictor of invasion outcome than biodiversity (Wei et␣al, 2015). In multiple invasions of microbial communities, the dispersal factor of the initial invader abundance (i.e., propagule pressure) was found to be the key determinant of the outcome of invasion (Acosta et␣al, 2015; Ketola et␣al, 2017; Kinnunen et␣al, 2018).
Synthetic communities composed of known organisms can be used to investigate the driving factors of invasion outcome (Wei et␣al, 2015; Ketola et␣al, 2017). Synthetic communities enable control of initial inoculum (i.e., organism presence/absence and initial abundance), which can be manipulated to understand the ecological and molecular mechanisms influencing invader growth. Dynamic computational models informed by the experimental measurements such as the generalized Lotka–Volterra (gLV) model can be used to decipher microbial interactions and predict community assembly (Mounier et␣al, 2008; Marino et␣al, 2014; Gonze et␣al, 2018). Previous modeling efforts with synthetic communities have revealed that pairwise interactions are informative of community assembly, making the characterization of lower‐order assemblages a powerful way to predict the behaviors of multispecies communities (Venturelli et␣al, 2018).
In this work, we use a defined synthetic gut community that represents the phylogenetic diversity of natural gut microbiota to study the determinants of C. difficile invasion success. To decipher microbial interactions and make predictions of community assembly and invasion, we use our data to construct a gLV model of our system and demonstrate that our model can accurately predict community assembly. Based on the inferred gLV interaction network, we demonstrate that negative interactions dominate the growth of C. difficile, which is a unique feature compared with all other species in our system. We identify multiple mechanisms that contribute to the inhibition of C. difficile growth including resource competition and external pH modification, highlighting that the mechanisms of inhibition of C. difficile vary across community contexts. Guided by our model, we identify a key closely related species, Clostridium hiranonis, that inhibits C. difficile growth in different synthetic communities. To investigate the ecological factors influencing invasion, we study the effect of propagule pressure and species richness on C. difficile growth. Our results show that C. difficile abundance exhibits a strong inverse relationship with species richness across a wide range of community contexts, and that this relationship is not universal to all species in our community. While increasing the propagule pressure of C. difficile can increase its abundance in the community, the sensitivity of each community to propagule pressure and the maximum saturating abundance of C. difficile are dictated by the microbial interaction network. We show that microbial communities feature a wide range of resistances to C. difficile and multiple mechanisms of C. difficile inhibition. Therefore, information about ecological and molecular mechanisms could be exploited to design bacterial therapeutics to inhibit C. difficile.
Results
C. difficile coexists in coculture with a subset of gut microbes
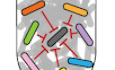
We sought to understand the ecological principles of C. difficile invasion using synthetic gut communities (Fig 1A). As a representative community, we chose a consortium of 13 prevalent gut microbes spanning the major human gut phyla Bacteroidetes, Firmicutes, Actinobacteria, and Proteobacteria (Forster et␣al, 2019). The community features Clostridium scindens, a species previously shown to inhibit growth of C. difficile in gnotobiotic mice (Buffie et␣al, 2014), and a well‐characterized set of 12 diverse species whose interactions on community assembly have been previously studied and computationally modeled (Venturelli et␣al, 2018) (Fig 1B).
Figure 1. Investigating the ecological principles of C. difficile invasion using a diverse synthetic human gut community.
- C. difficile (CD) invasibility is hypothesized to depend on initial invader density, species richness, environmental pH, and resource availability.
- Phylogenetic tree of 13‐member resident synthetic gut community and C. difficile based on concatenated alignment of 37 marker genes.
- Schematic of experimental and modeling workflow. Synthetic communities are cultured in microtiter plates in anaerobic conditions and incubated at 37°C. The absolute abundance of each species is determined by measuring cell density at 600 nm (OD600) and community composition using multiplexed 16S rRNA sequencing. Absolute abundance data are used to infer the parameters of a generalized Lotka–Volterra (gLV) model.
- Absolute abundance (OD600) of monospecies over time for three growth cycles. Datapoints indicate experimental biological replicates. Lines indicate simulations using the generalized Lotka–Volterra Full Model (trained on monospecies, pairs, and multispecies data, see Materials and Methods). Thin horizontal gray line indicates abundance threshold of 0.05 OD600.
- Absolute abundance (calculated OD600) of pairwise communities containing C. difficile over time for three growth cycles. Species were inoculated at an equal abundance ratio of C. difficile to resident species based on OD600 measurements. Datapoints indicate experimental data replicates. Lines indicate simulations using the generalized Lotka–Volterra Full Model (trained on monospecies, pairs, and multispecies data, see Materials and Methods). Thin horizontal gray line indicates abundance threshold of 0.05 OD600. Thick horizontal gray line indicates C. difficile monospecies maximal OD600 of 0.36. Calculated OD600 is the product of 16S relative abundance and community OD600.
Data information: In D and E, n = 1–3 biological replicates (See Appendix␣Table S4 for replicate information of each condition).
We used this synthetic gut community to investigate interspecies interactions influencing C. difficile growth. To decipher interspecies interactions driving C. difficile growth, we assembled combinations of species in microtiter plates in an anaerobic chamber and measured cell density by absorbance at 600 nm (OD600) and community composition by 16S rRNA gene sequencing at time points of interest (Materials and Methods). The absolute abundance of each species was calculated by multiplying its relative abundance from 16S rRNA gene sequencing by OD600 (“calculated OD600”). Time‐series measurements of species absolute abundance were used to infer the parameters of the gLV model (Fig 1C). The gLV model is a system of coupled ordinary differential equations that captures the growth rate and intra‐species interactions of single‐species and interspecies interactions that modify the growth dynamics of each species. The gLV model can be used to decipher interspecies interactions and predict the dynamics of all possible subcommunities within a larger system (Venturelli et␣al, 2018; Clark et␣al, 2021) and thus can be used to study the interspecies interactions between C. difficile and the resident gut community (i.e., all species excluding C. difficile).
We first characterized the temporal behavior of pairwise communities of C. difficile with each resident gut bacteria since we hypothesized that these direct interactions would have the largest impact on C. difficile growth compared with the interactions between resident gut bacteria. To this end, each resident species was grown alone and in coculture with C. difficile, specifically the R20291 reference strain of the epidemic ribotype 027 (Exp1, Fig 1D and E). A summary of all experiments throughout this work can be found in Appendix␣Table S1. Since variation in initial species proportions have been shown to influence community assembly (Wright & Vetsigian, 2016; Venturelli et␣al, 2018), we inoculated the pairs at 1:1 and 1:9 ratios of C. difficile to resident species based on OD600 values (Fig 1E, Appendix␣Fig S1). The communities were passaged using a 1:20 dilution at 26 and 52 h to observe community assembly over three batch culture growth cycles to understand the longer‐term behavior of the consortia.
Over this period of time, C. difficile and the resident species coexisted (both species present at greater than 0.05 OD600 after 78 h) in 19 of 33 (56%) conditions of 1:1 initial ratio, and 15 of 31 (48%) conditions of 1:9 initial ratio (Fig 1E, Appendix␣Fig S1). Although C. difficile and Bacteroides species coexisted in coculture over this period, the abundance of C. difficile was reduced compared with its abundance in monospecies. Bacteroides thetaiotaomicron and Bacteroides ovatus strongly inhibited C. difficile, reducing C. difficile’s carrying capacity in the final growth passage to 17 and 42% of its monospecies carrying capacity, while Bacteroides uniformis and Bacteroides vulgatus moderately inhibited C. difficile’s carrying capacity to 72 and 73% of its monospecies carrying capacity (Fig 1D and E). Bacteroides species have been shown to inhibit C. difficile growth (Mullish et␣al, 2019; Ghimire et␣al, 2020; Pereira et␣al, 2020) via suggested mechanisms of competition for mucosal carbohydrates or toxicity due to secondary bile acids (Mullish et␣al, 2019; Pereira et␣al, 2020). Because our media does not contain mucins or bile acids, the observed inhibition indicates a separate inhibition mechanism of C. difficile by Bacteroides species. We also identified closely related species that inhibit C. difficile including Clostridium hiranonis, the closest relative to C. difficile in the system (Fig 1B), which reduced C. difficile carrying capacity in the first growth passage to 59% of its monospecies carrying capacity (in the second and third passages, C. hiranonis became extinct and the inhibition was relieved). The next closest relative, Eubacterium rectale, reduced C. difficile’s carrying capacity in the third growth passage to 38% of its monospecies carrying capacity (Fig 1D and E). In sum, these data show that C. difficile can coexist over multiple batch culture cycles with a subset of species in our community.
Abundance of C. difficile in multispecies communities is inversely related to species richness
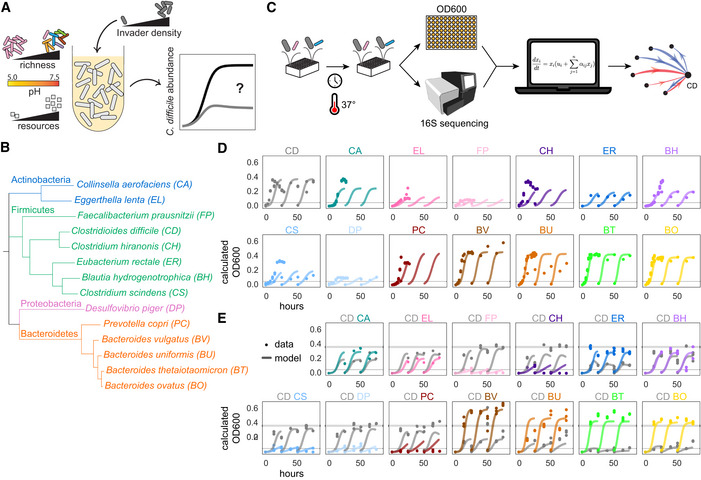
We next sought to understand whether the growth inhibition of C. difficile observed in a subset of pairwise communities persisted in multispecies communities and to identify the ecological principles governing C. difficile’s growth in multispecies communities. In order to design multispecies communities to experimentally characterize, we created a gLV model of our system trained on our monospecies data (Fig 1D), pairs data (Fig 1E), and previously published data of resident species pairs (Venturelli et␣al, 2018). We inferred an initial set of parameters of the gLV model (“Preliminary Model”, Appendix␣Fig S2A, Dataset EV1) based on these data (Table 1) and used the model to predict the abundance of C. difficile at 48 h in all possible 2–13 member resident communities (8,178 total communities, Appendix␣Fig S2B). Using the predictions from the Preliminary Model, we selected a set of 94 2–13 member communities whose C. difficile abundance spanned the full range of predicted C. difficile abundances and featured approximately equal representation of species at various initial species richness (number of species in the resident community).
Table 1.
Data used for gLV models.
| Model | Data |
|---|---|
| Preliminary Model |
Exp1 Exp2 Pairwise communities from Venturelli et al (2018) |
| Full Model |
Exp1 Exp2 Exp3 Exp5 Exp6 (except for MS002, MS010, MS011 data) Exp8 |
We experimentally assembled these communities with an equal initial abundance of all species (including C. difficile) and measured the composition of communities after 48 h (Exp2). We added C. difficile to communities at 0 h to investigate interspecies interactions in a perturbed, low‐density environment that could mimic a disturbance such as antibiotic treatment. We measured the community composition at 48 h as the Preliminary Model predicted that the majority of communities had reached stationary phase by this time. It is possible that the measured community compositions after this single batch culture cycle (short‐term dynamics) could differ from the composition of the communities after multiple dilution cycles (long‐term dynamics). However, measuring the community composition at the end of a single batch culture cycle allowed us to investigate the ecological and molecular factors influencing C. difficile growth in a wide range of community contexts that varied in the presence/absence of species and species richness levels.
We first looked at the relationship between initial species richness and C. difficile abundance in this dataset. The biodiversity–invasibility hypothesis holds that species‐rich communities have a higher fraction of ecological niches occupied, which reduces the availability of niches for invader species and thus enhances resistance to invasion relative to low‐richness communities (Elton, 1958). In agreement with the ecological theory, the mean C. difficile abundance across different communities decreased with species richness (Fig 2A). The negative relationship between species richness and C. difficile abundance remained the same whether richness was evaluated at the initial or final time point (Fig 2A, Appendix␣Fig S3A). Notably, C. difficile did not establish in any communities with richness greater than eight. The full community (13 resident members) excluded C. difficile from the community by 48 h. This resistance of the full community was observed not only with the ribotype 027 strain, but also for three individual clinical isolates of C. difficile that originated from patients within 72 h of their Clostridioides difficile Infection (CDI) diagnosis (Watson et␣al, 2019) (Exp6, Appendix␣Fig S3B, Materials and Methods).
Figure 2. Growth of C. difficile decreases with community richness.
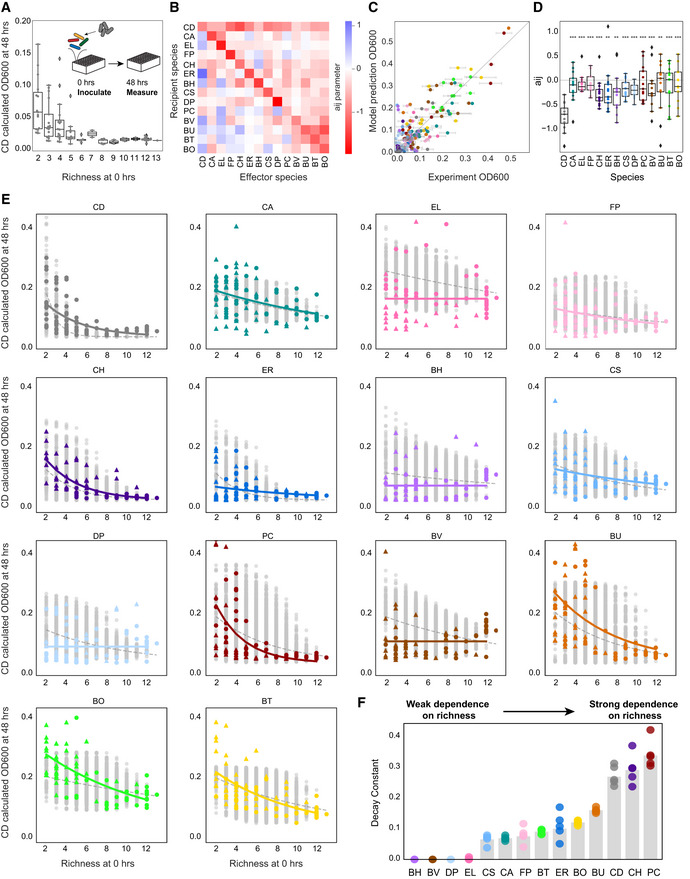
- Swarmplot of C. difficile (CD) absolute abundance (calculated OD600) at 48 h in 94 subcommunities as a function of initial species richness. Datapoints indicate mean of biological replicates. Line represents median, box edges represent first and third quartiles, and whiskers indicate the minimum and maximum. Outliers are denoted by diamonds. Calculated OD600 is the product of 16S relative abundance and community OD600.
- Heatmap of interspecies interaction coefficients of the generalized Lotka–Volterra model (gLV) Full Model.
- Scatterplot of absolute abundance (calculated OD600) versus predicted absolute abundance by the gLV Full Model in 24 held‐out communities (Pearson r = 0.84, P = 6*10−52). Error bars represent one SD from the mean of biological replicates. Datapoint color indicates species identity. Gray line indicates y = x, or 100% prediction accuracy. Calculated OD600 is the product of 16S relative abundance and community OD600.
- Box␣plot of incoming interspecies interactions for each species in gLV Full Model. Stars represent statistical significance between C. difficile and each resident species: *P < 0.05, **P < 0.01, ***P < 0.001 according to an unpaired t‐test. Line represents median, box edges represent first and third quartiles, and whiskers indicate the minimum and maximum. Outliers are denoted by diamonds.
- Subplot of the absolute abundance of each species at 48 h as a function of initial species richness in all 16,370 possible subcommunities of 2–13 species simulated by the gLV Full Model (gray data points) and in 204 experimentally measured subcommunities (mean value of biological replicates, colored data points). Circles represent subcommunities included in Full Model training dataset. Triangles represent subcommunities not included in Full Model training dataset. Lines display exponential decay model (y = ae −bx) fit to simulated data (gray dashed line) and experimental data (solid colored line). Calculated OD600 is the product of 16S relative abundance and community OD600.
- Barplot of decay constants b from exponential decay fit to experimental data in E. Colored datapoints are best fit parameters from five models, where each model was trained on a randomly sampled subset of the data consisting of 4/5 of the experimental datapoints. Gray bar indicates best fit parameter value from model trained on all data.
Data information: In A, C, and E, n = 1–3 biological replicates.
We wanted to understand whether the strong inverse relationship between species richness and C. difficile abundance could be explained by its interactions with the community. To investigate this question, we inferred a new set of gLV model parameters (“Full Model”, Fig 2B, Dataset EV2) using measurements of monospecies, pairwise and multispecies consortia (Table 1) and found that the Full Model had a high goodness of fit to the training data (Fig EV1A, Pearson r = 0.89, P = 0.0). To validate the predictive capability of the Full Model, we held out 24 randomly sampled communities from the training data set that spanned a broad range of species richness and C. difficile abundance (Fig EV1B) and found that the model predicted the community composition of the held‐out dataset with high accuracy (Fig 2C, Pearson r = 0.84, P = 6*10−52). In contrast, the Preliminary Model trained on monospecies and pairs was substantially less predictive of these 24 multispecies communities, indicating that the model required information from the multispecies experiments (Fig EV1C, Pearson r = 0.52, P = 1*10−14). We performed parameter uncertainty analysis to determine whether the parameters were sufficiently constrained by the data using Metropolis–Hastings Markov chain Monte Carlo (MCMC) (Materials and Methods). The coefficient of variation (CV) of 82% of the parameters was < 0.05 (CV ranged from 0.006 to 0.06), indicating that the parameters were sufficiently constrained by the data (Fig EV1D).
Figure EV1. Analysis of parameter uncertainty and predictive capability of generalized Lotka–Volterra models.
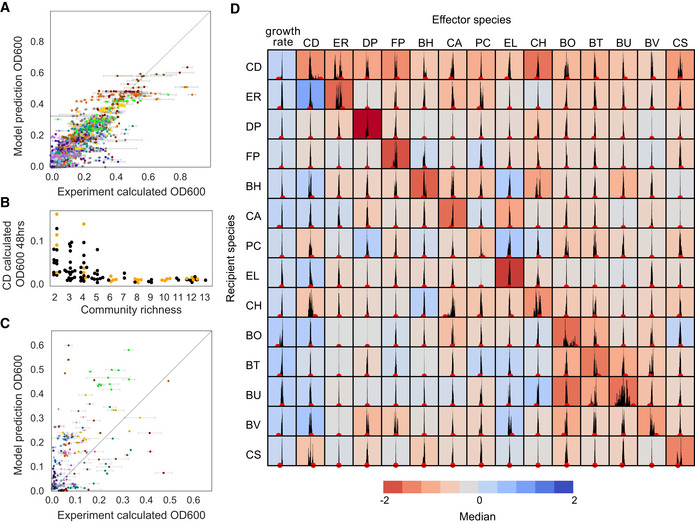
- Scatterplot of goodness of fit of experimental absolute abundance (calculated OD600) versus simulated species absolute abundance using the Full Model for the communities in the training data set (Pearson r = 0.89, P = 0.0). Error bars represent one SD from the mean of biological replicates. Gray line indicates y = x, or 100% prediction accuracy. Calculated OD600 is the product of 16S relative abundance and community OD600.
- Swarmplot highlighting 24 communities chosen as the held‐out set (data also shown in Fig 2A). Orange datapoints represent held‐out communities from training set. Black datapoints indicate communities from Fig 2A in training data set. Calculated OD600 is the product of 16S relative abundance and community OD600.
- Scatterplot of experimental absolute abundance (calculated OD600) versus predicted species absolute abundance (OD600) using the Preliminary Model for the 24 held‐out communities (Pearson r = 0.52, P = 1*10−14). Error bars represent one SD from the mean of biological replicates. Gray line indicates y = x, or 100% prediction accuracy. Calculated OD600 is the product of 16S relative abundance and community OD600.
- Histograms of parameter values determined using Metropolis–Hastings Monte Carlo (MCMC) analysis. Red dots indicate the parameter value in the Full Model (Materials and Methods). The black histograms indicate the MCMC distribution. The x‐axis is scaled to median of MCMC distribution +/− 0.25. The color of each subplot denotes the median value of the MCMC distribution.
Data information: In A‐C, n = 1–3 biological replicates.
Strikingly, in the interspecies interaction network (Fig 2B), all species inhibited C. difficile. C. difficile positively impacted most species in the community, which combines with the negative incoming interactions to generate multiple negative feedback loops on the growth of C. difficile. Increasing species richness increases the number of negative feedback loops on C. difficile’s growth, providing insight into the negative relationship between C. difficile abundance and species richness. C. difficile is unique in its large number and magnitude of incoming negative interactions in the system (Fig 2D). Because of this, we hypothesized that other species may not display the same strong inverse relationship between abundance in communities and richness. For example, a species with many positive incoming interactions may have a growth benefit in high richness communities containing growth‐promoting species.
We analyzed the relationship between abundance and richness for each species in multispecies communities with and without C. difficile (Exp2 and Exp3, Fig 2E). Additionally, we used the Full Model to simulate the abundance of each species in all possible communities (16,383 total communities) to supplement our experimental data (Fig 2E, gray points). To quantify the relationship between abundance and richness for each species, we fit an exponential decay model (y = ae −bx) to the data (solid colored lines) and simulations (dashed gray lines). The decay constant of the experimental data fits reveals that C. difficile, C. hiranonis, and Prevotella copri have strong negative relationships between abundance and species richness (b > 0.2, Fig 2F). However, Blautia hydrogenotrophica, B. vulgatus, Desulfovibrio piger, and Eggerthella lenta displayed no relationship between abundance and richness (b < 0.05). The decay constants fit to the simulated data (Fig EV2A) showed agreement with those fit to the experimental data (Fig 2F). For example, in the model, C. difficile and C. hiranonis were strongly dependent on richness (b > 0.2) and E. lenta and B. hydrogenotrophica displayed no relationship (b < 0.05). The decay constants did not correlate with species growth rate, so the relationship between abundance and richness cannot be explained by whether a species was fast‐ or slow‐growing (Fig EV2B). C. hiranonis also features a large number of negative incoming interactions, suggesting that the mechanism of C. hiranonis’s dependence on richness could be similar to that of C. difficile (Fig 2D). Overall, our data and model analysis shows that the abundance of C. difficile has a strong inverse relationship with species richness and this relationship is not universal to all species.
Figure EV2. Analysis of simulated and measured decay constants quantifying the relationship between species richness and abundance.
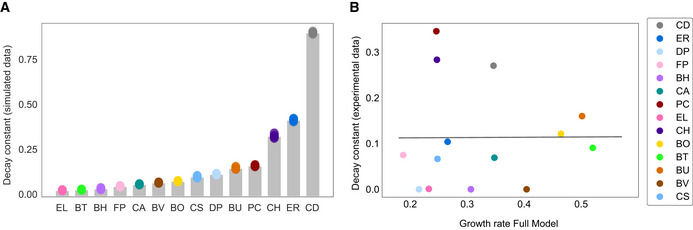
- Barplot of decay constants b from exponential decay fit to simulated data in 2e. Colored datapoints are best fit parameters from five models, where each model was trained on a subsample of the data consisting of 4/5 of the experimental datapoints. Gray bar indicates best fit parameter from model trained on all data.
- Scatterplot of relationship between species growth rate in the Full Model and decay constant fit to experimental data shown in Fig 2F. Gray line indicates linear regression (y = 0.006x+0.111, Pearson r = 0.01, P = 0.98).
Increasing propagule pressure increases C. difficile abundance in synthetic communities
The propagule–pressure hypothesis dictates that increasing propagule pressure, or the amount of invader (a product of its dispersal frequency and abundance), increases the chance of a successful invasion (Lockwood et␣al, 2005). To characterize the effect of propagule pressure in our system, we next looked at the relationship between the propagule pressure of C. difficile and its abundance at 48 h. In our system, we add C. difficile to the system a single timepoint, so the propagule pressure of C. difficile is only affected by initial abundance of C. difficile. In our experiments, we define propagule pressure as the initial fraction of C. difficile. We analyzed the relationship between initial fraction of C. difficile and final abundance of C. difficile in the 2–13 member multispecies communities (Exp2, gray data points in Fig 3A) in addition to measurements of 15 3–4 member resident communities (Exp4, Appendix␣Table S2). We focused on 3–4 member communities because communities in this narrow richness range featured a wide range of C. difficile abundances at 48 h (Fig 2A). We chose 15 communities with a wide range of predicted C. difficile abundances. We inoculated these communities at multiple species ratios and measured the composition over time (colored data points in Figs 3A and EV3A). In agreement with the theory, the final abundance of C. difficile correlated with the initial fraction of C. difficile in the community (Fig 3A, Pearson r = 0.75, P = 1*10−23). In all 15 3–4 member communities, the abundance of C. difficile at 48 h was higher in communities inoculated with a high initial fraction of C. difficile (approximately 65% of total community biomass) compared with a low initial fraction of C. difficile (approximately 10% of total community biomass) (Fig 3A, inset). This indicates that increasing propagule pressure of C. difficile can increase its abundance in the community at 48 h.
Figure 3. Impact of initial density on the growth of C. difficile .
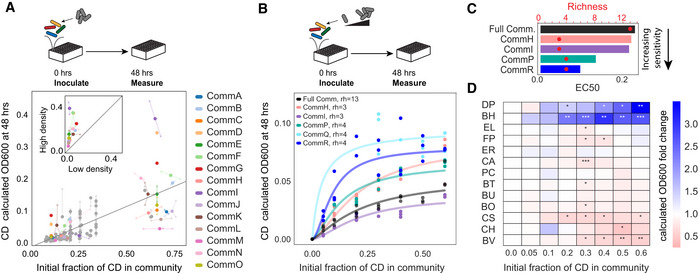
- Scatterplot of C. difficile (CD) absolute abundance (calculated OD600) at 48 h in communities as a function of the initial fraction of C. difficile. C. difficile was introduced into the communities at 0 h. Gray data points are 2–13 member resident communities measured in Fig 2A. Colored data points are 3–4 member communities measured at two initial conditions: low density (approximately 10% of total community OD600) or high density (approximately 65% total community OD600). Gray line indicates a linear regression (y = 0.25x‐0.01, Pearson r = 0.75, P = 1*10−23). Transparent data points indicate biological replicates and are connected to the corresponding mean values by transparent lines. Inset: Abundance of C. difficile at 48 h in communities invaded with low density or high density. Gray y = x line indicates no change in abundance. Calculated OD600 is the product of 16S relative abundance and community OD600.
- Absolute abundance (ODO600) of C. difficile at 48 h as a function of the initial fraction of C. difficile in different synthetic communities. C. difficile was added to communities at 0 h. Datapoints indicate biological replicates. Lines indicate Hill function fits (Materials and Methods). Resident species richness (rh) at 0 h is indicated in legend. Calculated OD600 is the product of 16S relative abundance and community OD600.
- Initial fraction of C. difficile corresponding to the half‐maximum abundance (EC50) inferred based on the fitted Hill functions in B for a subset of communities with sufficient measurements to constrain the function parameters. Red circles indicate the resident species richness at 0 h.
- Heatmap of the fold change of species absolute abundance (mean value of biological replicates, n = 3) in the full community with 5–60% initial C. difficile compared to 0% initial C. difficile condition. Calculated OD600 is the product of 16S relative abundance and community OD600. Stars represent statistical significance: *P < 0.05, **P < 0.01, ***P < 0.001 according to an unpaired t‐test.
Data information: In A, n = 1–3 biological replicates. In B, n = 1–3 biological replicates (see Appendix␣Table S4 for replication information of each condition). In D, n = 3 biological replicates.
Figure EV3. Dependence of C. difficile abundance on propagule pressure over time.
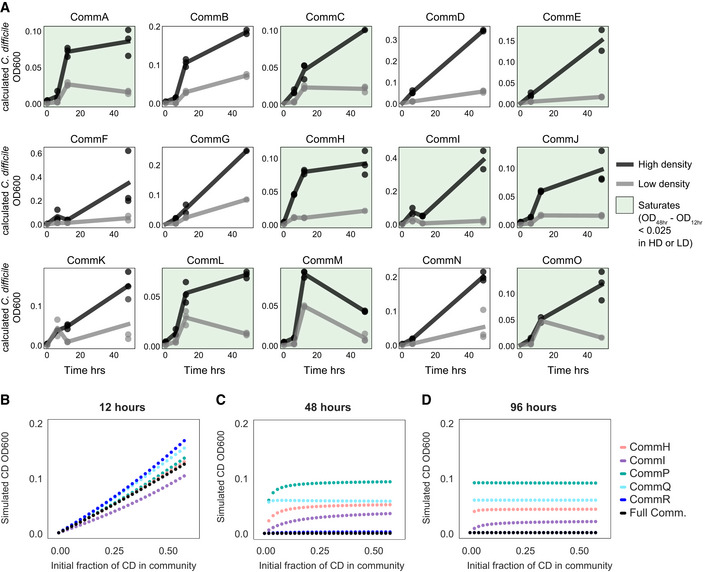
-
ALineplots of C. difficile (CD) abundance over time in CommA‐CommO communities. Final timepoint is same as shown in Fig 3A. In low‐density conditions, C. difficile inoculated at 10% of total community OD600 at 0 h. In high‐density conditions, C. difficile inoculated at 65% of total community OD600 at 0 h. Data points indicate biological replicates and lines indicate mean value of biological replicates. Communities where C. difficile abundance saturates by 48 h in either high‐density or low‐density conditions are highlighted in green (saturation defined as difference in C. difficile OD600 between 48 and 12 h is less than 0.025).
-
B–DLineplots of predicted absolute abundance (OD600) of C. difficile at 12, 24, and 96 h as a function of the initial fraction of C. difficile as simulated by the Full Model.
Data information: In A, n = 1–3 biological replicates (See Appendix␣Table S4 for replicate information of each condition).
For a set of 3–4 member communities, we performed a similar experiment but increased the number of tested initial fractions to better resolve the relationship between propagule pressure and abundance at 48 h (Exp5, Fig 3B). Increasing the propagule pressure of C. difficile yielded higher C. difficile abundance in the assembled community within a given range. However, the abundance of C. difficile approached a similar maximum abundance for high initial fractions beyond a threshold. The abundance of C. difficile at saturation in the community varied in different subcommunities, suggesting that the microbial interaction network determined the maximum abundance of C. difficile. We defined the sensitivity to propagule pressure as the initial invader fraction that resulted in the half‐maximal abundance of the invader at 48 h, analogous to the EC50 of a dose–response curve (Fig 3C). The communities displayed different sensitivities to the initial fraction of C. difficile, with the EC50 ranging between 0.1–0.2. Community R (Appendix␣Table S2) was the most sensitive to invasion by C. difficile. Therefore, while increasing propagule pressure of C. difficile can increase its abundance to a maximum threshold, the sensitivity to propagule pressure and maximum saturating abundance of C. difficile are dictated by the microbial interaction network.
In the experiments and simulations, the total initial OD600 was held constant, resulting in lower initial OD600 of each species with increasing richness (Materials and Methods). Therefore, we considered the possibility that C. difficile’s low abundance in high richness communities (Fig 2A) could be a result of lower initial abundance. To test this possibility, we introduced C. difficile into the full community (richness of 13) at a range of initial fractions. We observed that C. difficile grew to a higher abundance in the full community when propagule pressure was increased, although the maximum abundance was lower than in the majority of 3–4 member communities (Fig 3B). Therefore, while increasing propagule pressure can partially overcome the inhibiting effect of species richness on C. difficile growth in the linear regime of the dose response, high richness still reduces the maximum saturating C. difficile abundance.
The variation in C. difficile abundance at 48 h with propagule pressures in our experiments could be a transient effect as a consequence of being further away from the long‐term composition of the assembled community or history‐dependent behavior that persists long term. Our time‐series data of 3–4 member communities indicated that the abundance of C. difficile approached saturation by 48 h in most of the communities (Figs 3A and EV3A). This suggests that inoculating communities with high or low density of C. difficile yielded distinct long‐term abundances in these communities. In contrast, stability analysis of our model (Materials and Methods) found that all subcommunities are monostable and therefore have no long‐term history dependence. For example, our model simulations for the six communities in Fig 3B predict that the strong dependence of C. difficile abundance on propagule pressure is transient, as the dependence at early times (12 h) is reduced at longer timescales and converges to a final abundance independent of propagule pressure since these communities are monostable (Fig EV3B–D). Therefore, the gLV model may be missing information about the long‐term history‐dependent behaviors of these communities.
When increasing the propagule pressure of C. difficile in the six resident communities in Fig 3B, the composition of the resident communities at 48 h varied with initial C. difficile abundance. To quantify this variation, we computed the normalized Euclidean distance between the resident community composition in the presence and absence of C. difficile (Materials and Methods). The Euclidean distance correlated with the abundance of C. difficile in the community in our experimental data (Appendix␣Fig S4A, Pearson’s r = 0.61, P = 6*10−13) as well as in simulations of all 1–13 member resident communities invaded with C. difficile 6 h after inoculation (Appendix␣Fig S4B, Pearson’s r = 0.58, P = 0.0). These data indicate that in general, higher abundance of C. difficile results in a larger impact on the composition of the resident community. However, the unexplained variation in Euclidean distance for a fixed C. difficile initial abundance suggests that interspecies interactions also impact the extent to which the resident community is altered by the presence of C. difficile.
In the full community, we observed that the abundance of D. piger and B. hydrogenotrophica significantly increased in communities with higher C. difficile, while the abundance of B. vulgatus significantly decreased (Fig 3D). These trends were observed in the full community with the ribotype 027 strain of C. difficile as well as the full community with individual clinical C. difficile isolates (Fig EV4A). The interaction network from our model (Fig 2B) features a positive interaction between C. difficile and B. hydrogenotrophica, suggesting that increasing initial C. difficile abundance directly promotes the growth of B. hydrogenotrophica. However, the interspecies interaction coefficients impacting D. piger and B. vulgatus were not consistent with the observed trends with these two species. These data suggest that the gLV model may not capture the effects of high initial C. difficile density on the growth of all resident gut species. While at high initial densities C. difficile significantly increased the abundance of␣B. hydrogenotrophica in the full community (Fig 3D), B. hydrogenotrophica abundance was not affected in the 3‐member communities F, G, and N (Fig EV4B), highlighting that C. difficile’s impact on a given species depends on the community context in addition to its initial abundance. We note that B. hydrogenotrophica and D. piger share a similar metabolic niche as hydrogen consumers (Bernalier et␣al, 1996; Loubinoux et␣al, 2002), suggesting C. difficile could enhance their growth through a shared mechanism.
Figure EV4. Impact of C. difficile on resident species abundances.
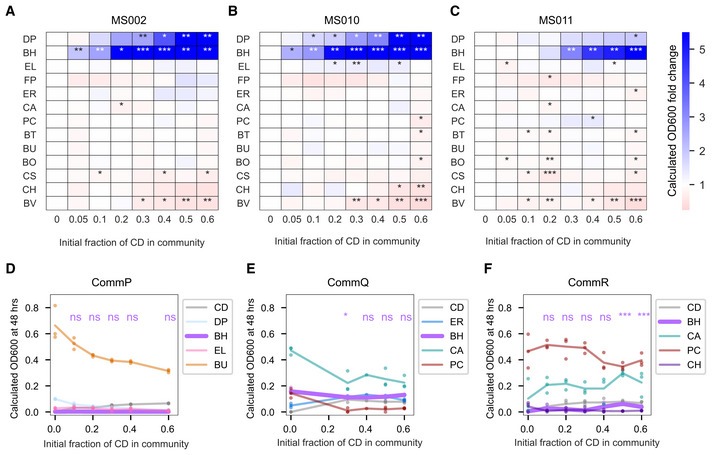
-
A–CHeatmap of the fold change of species absolute abundance (mean value of biological replicates) in full community with 5–60% initial C. difficile (CD) compared to the 0% initial C. difficile condition. Stars represent statistical significance: *P < 0.05, **P < 0.01, ***P < 0.001 according to an unpaired t‐test. A: C. difficile strain MS002, B: C. difficile strain MS010, C: C. difficile strain MS011.
-
D–FLineplots of species absolute abundance (calculated OD600) at 48 h as a function of initial C. difficile fraction. Datapoints indicate biological replicates and lines indicate the mean. Calculated OD600 is the product of 16S relative abundance and community OD600. Stars indicate a statistically significant difference in the absolute abundance of B. hydrogenotrophica compared to the absolute abundance of B. hydrogenotrophica in 0% initial C. difficile condition: *P < 0.05, **P < 0.01, ***P < 0.001, ns = no significant difference according to an unpaired t‐test. D: CommP, E: CommQ, F: CommR.
Data information: In A‐C, D, F n = 3 biological replicates. In E, n = 1 or n = 3 biological replicates (see Appendix␣Table S4 for replicate information of each condition).
A subset of synthetic communities inhibits C. difficile via acidification of the environment
While the community experiments revealed the importance of species richness and propagule pressure on the establishment of C. difficile in multispecies communities, there remains unexplained variation in the data. For example, communities with the same richness invaded with equal abundances of C. difficile showed a wide range of C. difficile abundances at 48 h (Fig 2A). Since environmental pH has been shown to influence C. difficile’s growth in previous studies (Wetzel & McBride, 2020; Yuille et␣al, 2020), we turned next to investigate how biotic modification of the environment alters the growth of C. difficile. To this end, we grew the set of 15 3–4 member communities for 6 h before invading with low or high initial densities of C. difficile to give the communities time to modify the environment. At the time of invasion, we measured the composition of the resident community and the pH of the media (Exp6, Fig 4A). To understand the role of invasion timing on the growth of C. difficile, we compared the C. difficile abundance in these communities invaded at 6 h with communities invaded at 0 h (Exp5) (Fig 4B). C.␣difficile’s ability to establish in multiple communities significantly depended on the timing of introduction (Fig 4B), indicating that biotic modification of the environment during those 6 h altered C. difficile’s ability to grow.
Figure 4. Impact of environmental factors on C. difficile invasion.
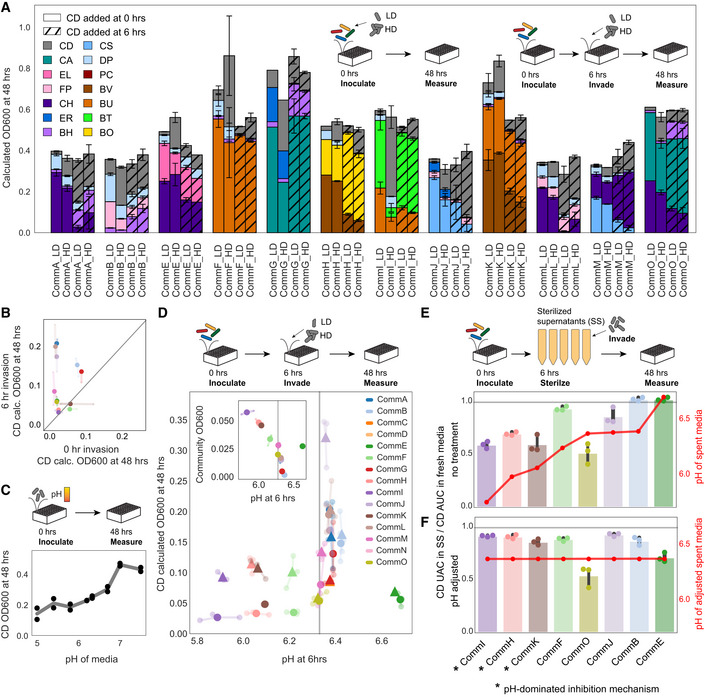
-
ABarplot of composition of communities invaded with C. difficile (CD) at low density (“LD”) or high density (“HD”). Color indicates species identity. Hash indicates invasion time. Error bars represent one SD from the mean of biological replicates. Calculated OD600 is the product of 16S relative abundance and community OD600.
-
BScatterplot of the absolute abundance (calculated OD600) of C. difficile at 48 h in communities when introduced at 0 h versus 6 h at low density (approximately 10% community OD600). Transparent data points indicate biological replicates and are connected to the corresponding mean values by transparent lines. Line denotes the x = y line corresponding to no change in growth. Color indicates community, see legend in D. Calculated OD600 is the product of 16S relative abundance and community OD600.
-
CLineplot of C. difficile OD600 at 48 h as a function of the initial environmental pH. Datapoints indicate biological replicates and line indicates mean value.
-
DScatterplot of the absolute abundance (calculated OD600) of C. difficile at 48 h in invaded communities as a function of the environmental pH at time of invasion. Fifteen 3–4 member communities were invaded with (▲) high‐density C. difficile (approximately 33% community OD600) or (●) low‐density C. difficile (approximately 10% community OD600) at 6 h. Color indicates community. Vertical gray line indicates pH of fresh media. Inset: Scatterplot of environmental pH and total community OD600 at 6 h. Transparent data points indicate biological replicates and are connected to the corresponding mean values by transparent lines. Vertical gray line indicates environmental pH of fresh media. Calculated OD600 is the product of 16S relative abundance and community OD600.
-
E,FBar plot of fold change of C. difficile growth in sterilized supernatants (E) or supernatants where the pH was adjusted to the pH of fresh media (F) compared to the growth of C. difficile in fresh media. Growth was quantified as area under the curve (AUC) of OD600 from 0 to 20 h. Datapoints indicate biological replicates, bars indicate mean value, and error bars represent one SD from the mean of biological replicates. Red line shows pH of community supernatants collected at 6 h (top) and pH adjusted supernatants (bottom). Horizontal gray line indicates no change in growth compared to fresh media.
Data information: In A, B, and D, n = 1–3 biological replicates (see Appendix␣Table S4 for replication information of each condition). In C, n = 2 biological replicates. In E and F, n = 3 biological replicates.
Communities that lowered the pH of the media during the first 6 h featured lower C. difficile abundance (Fig 4D). However, communities with lower pH at the time of invasion also had higher total biomass (Fig 4D, inset). Since these variables are related due to growth‐coupled production of acidic fermentation end products, either pH or resource competition could be responsible for inhibition of C. difficile. Because C. difficile abundance increased with environmental pH (Fig 4C), we hypothesized that growth inhibition was due to changes in media pH. To test our hypothesis, we grew a set of the communities and harvested and sterilized the community supernatants after 6 h. We grew C. difficile in either the sterile supernatant (Fig 4E) or a modified sterile supernatant wherein the pH was adjusted to the pH of the fresh media to eliminate the impact of pH on growth (Fig 4F). We quantified the growth of C. difficile as the area under the curve (AUC) of C. difficile OD600 over 20 h, which is influenced by both growth rate and carrying capacity. In Communities H, I, and K, which strongly inhibit C. difficile in both coculture and supernatant, increasing the supernatant pH to the pH value of fresh media eliminated the inhibition of C. difficile (Fig 4F), indicating that pH was the driving factor of C. difficile inhibition in these community supernatants. Each of these communities contained an abundant Bacteroides species (Appendix␣Table S2) whose fermentation end products acidify the media, suggesting Bacteroides species are responsible for the inhibition via pH modification. We observed the pH‐dependent inhibition of C. difficile by CommI in several other medias with varying buffering capacity and available substrates, indicating this result is not specific to our media (Appendix␣Fig S5).
In contrast to this pH‐dependent inhibition, the sterile supernatant of Community O (CommO) composed of C. hiranonis, Collinsella aerofaciens, and B. hydrogenotrophica, whose pH did not significantly differ from the pH of fresh media, inhibited C. difficile regardless of pH adjustment (Fig 4E and F). This implies that this community inhibits C. difficile via a pH‐independent mechanism. C. difficile was not inhibited by the sterile supernatant of Community E (CommE) composed of C. hiranonis, D. piger, and E. lenta, which uniquely had a higher pH than fresh media (Fig 4E). However, C. difficile’s growth was inhibited when the pH of the CommE sterile supernatant was reduced to the pH of fresh media (Fig 4F). This suggests that the sterile supernatant promotes C. difficile’s growth by enhancing environmental pH and the community inhibits C. difficile’s growth by a separate pH‐independent mechanism. The growth inhibition by CommE was only revealed when the pH increase of the media was eliminated, demonstrating an interplay of different mechanisms influencing C. difficile growth within the same community.
We explored if the sensitivity of a species to environmental pH correlated with its dependence on species richness as determined in Fig 2E. We measured the AUC of each species as a function of initial environmental pH in monospecies and determined the slope of the line fit to these data (Fig EV5A), representing the sensitivity of species growth to external pH. Our results demonstrated that there is no significant correlation between sensitivity to species richness and sensitivity to pH (Fig EV5C). Therefore, while acidification of the media is one mechanism by which communities can inhibit C. difficile in our system, our results suggest that there are also pH‐independent mechanisms that underlie C. difficile's strong inverse relationship between abundance and species richness.
Figure EV5. Resident gut species pH sensitivity in monoculture.
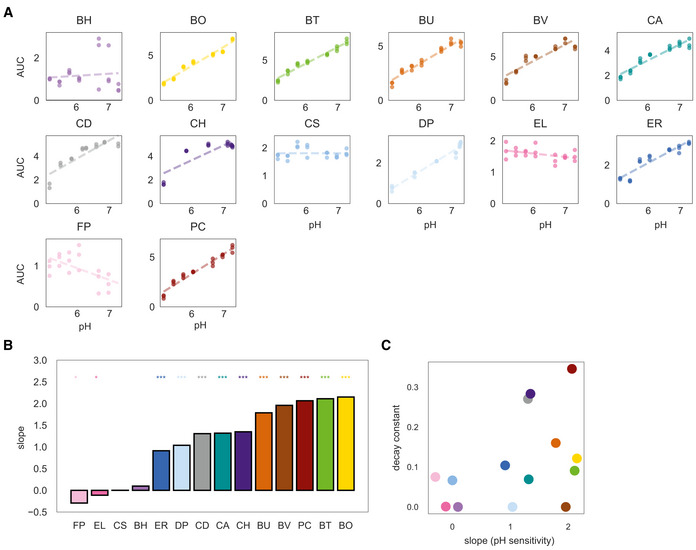
- Lineplots of monospecies growth as a function of the initial environmental pH of adjusted fresh media. Growth is quantified as area under the curve (AUC) from 0 to 20 h. Datapoints indicate biological replicates and dashed lines indicate linear regression fits.
- Barplot of slopes of linear regression fit to data in A. Stars denote statistical significance: *P < 0.05, **P < 0.01, ***P < 0.001 according to an unpaired t‐test.
- Scatterplot of decay constants from Fig 2F as a function of pH sensitivity slopes in B. Linear regression y = 0.06x+0.05, Pearson r = 0.44, P = 0.11).
Data information: In A, n = 2–3 biological replicates (See Appendix␣Table S4 for replicate information of each condition).
C. hiranonis inhibits C. difficile through a pH‐independent mechanism
We noted that the two communities that displayed pH‐independent growth inhibition (CommE and CommO) contained C. hiranonis, which has a strong bidirectional negative interaction with C. difficile in our Full Model (Fig 2B). Our model predicts that the abundance of C. difficile at 48 h decreases with increasing initial abundance of C. hiranonis in CommE, CommO, and the C. difficile‐C. hiranonis pair (Fig 5A). We confirmed this prediction experimentally (Exp7, Fig 5B). C. difficile was inhibited even by low initial amounts of C. hiranonis, featuring a significant decrease in growth between 0 and 10% initial C. hiranonis in CommE (> 4‐fold decrease) and CommO (> 1.5‐fold decrease) (Fig 5B). The strength of inhibition of C. difficile as a function of the C. hiranonis abundance was substantially higher in CommE and CommO than in the C. hiranonis‐C. difficile pair (Fig 5B). This result indicates that the other species in the communities enhanced the inhibitory effect of C. hiranonis on C. difficile growth.
Figure 5. C. hiranonis inhibits the growth of C. difficile .
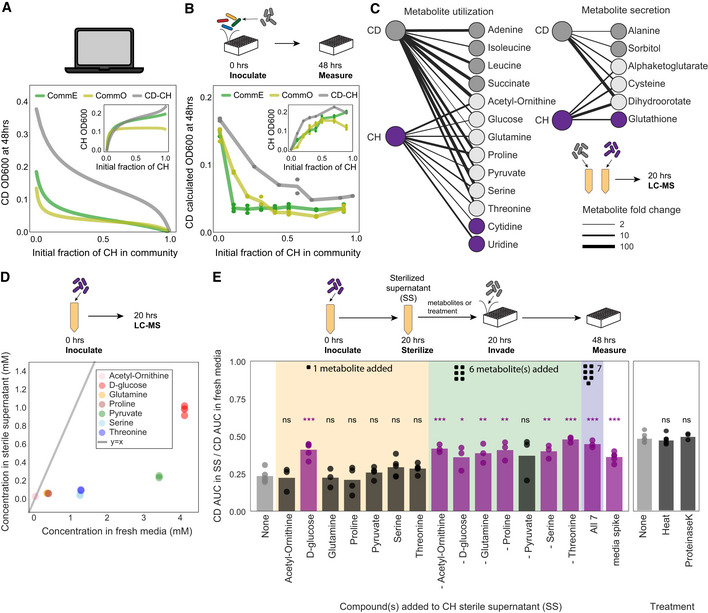
- Lineplot of simulated C. difficile (CD) absolute abundance (OD600) at 48 h using the generalized Lotka–Volterra (gLV) Full Model as a function of the initial fraction of C. hiranonis (CH) in different communities. Inset: Lineplot of simulated C. hiranonis absolute abundance (OD600) at 48 h in the gLV Full Model as a function of initial fraction of C. hiranonis in the community.
- Lineplot of C. difficile absolute abundance (calculated OD600) at 48 h as a function of the initial fraction of C. hiranonis in the community. Inset: Lineplot of C. hiranonis absolute abundance (calculated OD600) at 48 h in community as a function of initial fraction of C. hiranonis in the community. Datapoints indicate biological replicates, and lines indicate mean values. Calculated OD600 is the product of 16S relative abundance and community OD600.
- Bipartite network of metabolite utilization and secretion by C. difficile and C. hiranonis monospecies after 20 h of growth. Metabolites that changed by at least twofold compared to media control are shown. Edge width is proportional to the metabolite fold change.
- Scatterplot of concentration of seven metabolites in fresh media compared to C. hiranonis sterile supernatant.
- Bar plot of fold change of C. difficile growth in sterilized supernatants compared to the growth of C. difficile in fresh media. Growth was quantified as the area under the curve (AUC) of OD600 from 0 to 20 h. Datapoints indicate biological replicates, and bars indicate mean value. Media spike condition is the addition of 1X fresh media concentrated in a small volume. Stars represent statistical significance between condition and unmodified C. hiranonis supernatant: *P < 0.05, **P < 0.01, ***P < 0.001, and ns= not significant, according to an unpaired t‐test. Bars of all significant conditions are shaded purple.
Data information: In C and D, n = 3 biological replicates. In B, n = 1–3 and in E, n = 3–5 biological replicates (see Appendix␣Table S4 for replication information of each condition).
We next considered the mechanism of C. difficile inhibition by C. hiranonis. C. hiranonis is known to convert primary bile acids into secondary bile acids which are inhibitory to C. difficile (Kitahara et␣al, 2001). However, with no primary bile acids in our media we turned to other possible inhibition mechanisms. C. difficile was inhibited by the sterile supernatants of CommE and CommO (Fig 4E, Appendix␣Fig S6), indicating the inhibition effect does not require direct cell contact. These mechanisms could include production of antibiotics or toxic metabolic byproducts, modification of pH, or competition over resources. C. hiranonis has been shown to consume a broad range of metabolites in our media conditions (more than any of the other species in our system) (Venturelli et␣al, 2018) and is known to contain genes for Stickland amino acid fermentations, a major process for energy generation in C. difficile (Ridlon et␣al, 2020). Therefore, C. hiranonis could inhibit C. difficile by utilizing multiple resources that C. difficile consumes. We performed exo‐metabolomics using liquid chromatography–mass spectrometry (LC‐MS) of C. hiranonis and C. difficile supernatants harvested at 20 h and found that the two species utilize an overlapping set of metabolites in our media (Fig 5C). We identified seven metabolites that significantly decreased (> 2‐fold) in both supernatants: acetyl‐ornithine, glucose, glutamine, proline, pyruvate, serine, and threonine.
We designed an experiment to provide insight into potential resource competition between C. difficile and C. hiranonis over these metabolites in coculture. We first determined the concentration of these metabolites in fresh media and in the supernatant of C. hiranonis at 20 h using LC‐MS with a standard curve of known concentrations of each of the seven metabolites (Fig 5D). We then adjusted the concentration of each of the seven metabolites in C. hiranonis sterile supernatant to match the concentration in fresh media. We grew C. difficile in these modified supernatants, as well as supernatants modified to adjust all combinations of six or seven of the metabolites. We quantified the growth of C. difficile as the AUC of C. difficile OD600 over 48 h (Fig 5E). The fold change between C. difficile growth in C. hiranonis supernatant compared to fresh media was 23%, displaying strong inhibition consistent with the inhibition observed in coculture (Fig 5B) and our interaction network (Fig 2B). C. difficile grew significantly better in glucose‐adjusted supernatant (41% of fresh media growth) compared to unmodified supernatant (23% of fresh media growth), but there was no significant difference for any of the other six individual metabolite adjustments. Replenishing all seven metabolites in the supernatant significantly increased C. difficile’s growth to a similar level as the glucose addition (45% of fresh media growth). Interestingly, there was no significant difference between C. difficile growth in the seven‐compound supernatant and the six‐compound supernatant excluding glucose. Therefore, the metabolites excluding glucose did not significantly impact C. difficile growth when added individually but they did as a set. The fact that C. difficile grew equally in all of the six‐compound supernatants suggests metabolic redundancy among this set of compounds. In sum, these data show that replenishing the seven metabolites can partially relieve the observed C. hiranonis‐C. difficile inhibition. This suggests that C. difficile is inhibited in C. hiranonis supernatant due to low concentration of some subset of these metabolites which were consumed by C. hiranonis. This is supported by our data that showed these metabolites are also consumed by C. difficile in monospecies. However, we cannot rule out that the addition of these metabolites could compensate for the effects of different compound(s) that inhibits C. difficile.
Because restoration of these compounds only partially relieved the inhibition by C. hiranonis, we were curious about the mechanism of the remaining inhibition. We considered the possibility that C. hiranonis and C. difficile compete over other resources in addition to the seven identified above. To test whether the restoration of other compounds could further relieve the inhibition, we supplemented concentrated media to the C. hiranonis supernatant (10X concentrated ABB spiked in a 1:10 dilution into C. hiranonis supernatant). After concentrated media supplementation, compounds that were completely consumed by C. hiranonis were at 1× of their original media concentration, and compounds not utilized by C. hiranonis were at 2× of their original media concentration. Therefore, if resource competition alone was responsible for the inhibition, the growth of C. difficile would be fully rescued in the supplemented condition to the level of growth in fresh media. However, in the supplemented supernatant, C. difficile exhibited 36% of its growth in fresh media (Fig 5E). Therefore, these data suggest that the inhibitory effect of C. hiranonis conditioned media is due to a combination of resource competition and a separate unknown inhibitory mechanism. Further, the pH of C. hiranonis supernatant was not significantly different from fresh media, eliminating pH as a potential mechanism. To test whether a protein was responsible for C. difficile inhibition, we inactivated proteins in the C. hiranonis supernatant by either denaturing with heat (95°C for 1 h) or digesting with the broad‐spectrum protease Proteinase K. Our results showed no significant difference in C. difficile growth in these two treatments compared to no‐treatment control, suggesting the inhibition by C. hiranonis is not due to proteinaceous antibiotics or toxins. In sum, our results suggest that C. hiranonis partially inhibited C. difficile via resource competition, but also inhibits C. difficile via a protein‐independent mechanism.
Discussion
We combined bottom‐up construction of microbial communities with dynamic computational modeling to investigate microbial interactions impacting the growth of C. difficile. Our work demonstrates that microbial communities feature a wide range of resistances to C. difficile invasion. This variability in invasion outcome as a function of community context indicates that the choice of organisms is a major design factor that can be optimized to treat C. difficile infections and motivates exploiting information about ecological and molecular interactions in the design process. Previous efforts to design defined consortia for C. difficile inhibition used top‐down selections by reducing the complexity of cultured fecal samples alone or combined with screening of antibiotic resistance phenotypes (Lawley et␣al, 2012; Petrof et␣al, 2013). These methods rely on a trial‐and‐error approach to discover inhibitory consortia instead of testing consortia rationally designed to be inhibitory. Some consortia have been rationally designed by combining selected species in a bottom‐up approach, but we note that these selections use a single design criterion (Buffie et␣al, 2014; Pereira et␣al, 2020). We identified principles of C. difficile invasion that could be used as multiple criteria for designing inhibitory consortia in future studies. Previous work has demonstrated mechanisms of C. difficile inhibition by bile acid transformations (Buffie et␣al, 2014) and mucosal sugar competition (Pereira et␣al, 2020). Our results demonstrate that communities with high richness, communities that acidify the environment, and communities that compete over limiting resources utilized by C. difficile are promising candidates for inhibiting C. difficile. In sum, these results suggest multiple target phenotypes that could be combined to design an optimal defined bacterial therapeutic to inhibit C. difficile.
To collect data on many multispecies communities to train our model, we cultured communities for a single batch culture cycle. This experimental design is informative for deciphering microbial interactions since the system is farthest away from steady state in the first batch culture cycle and thus has rich dynamic behaviors. Although we did not characterize the long‐term dynamics of multispecies communities using multiple dilution cycles, we are able to draw many insights into C. difficile’s interactions with synthetic gut communities on the shorter timescale. Notably, C. difficile was the only species that was inhibited by all other community members. Infection by C. difficile disrupts the environment of gut bacteria by causing diarrhea (i.e., reduces residence time for gut bacteria), inducing intestinal inflammation, and altering the resource landscape (Fletcher et␣al, 2021), suggesting the possibility that gut bacteria have evolved to negatively impact the growth of C. difficile in order to promote their fitness in the gut.
Studies have shown that gut microbiomes of patients with CDI have significantly lower richness than healthy controls (Chang et␣al, 2008; Antharam et␣al, 2013), but this association does not distinguish whether CDI reduces the richness of gut microbiomes or low‐richness microbiomes are more susceptible to CDI. The striking trend between richness and C. difficile abundance in our data (Fig 2A) suggests that low‐richness microbiomes are more susceptible to CDI. Supporting this hypothesis, the susceptibility of low‐richness communities to invasion has been demonstrated in other microbial systems (van Elsas et␣al, 2012; Mallon, Poly, et␣al, 2015). This suggests that the reduction in gut microbiota richness by antibiotic treatment (Dethlefsen & Relman, 2011) could underlie the increased CDI risk that occurs after antibiotic use (Eze et␣al, 2017). Additionally, the efficacy of FMTs may be due in part to the high richness of stool samples which are estimated to have greater than one hundred species (Qin et␣al, 2010).
Based on our work, high richness communities would be the most effective bacterial therapeutics to inhibit C. difficile colonization. The scalable manufacturing of high richness bacterial therapeutics is challenging, indicating the need for new manufacturing techniques to reliably culture communities that maintain all species as opposed to standard culturing of single species. Nevertheless, if manufacturing of high richness communities remains a challenge, our work suggests it is possible to design low‐richness inhibitory communities. While all high richness communities (eight species or more) excluded C. difficile in our system, we found low‐richness communities that excluded C. difficile. For example, the 3–member Community I excluded C. difficile as effectively as the full community (Fig 3B). Corroborating these results, low‐richness communities as small as 5–7 members have been shown to inhibit C. difficile in␣vitro and in murine models (Lawley et␣al, 2012; Buffie et␣al, 2014; Pereira et␣al, 2020).
We demonstrated significant variation in the relationship between species abundance and richness across species in our community (Fig 2E and F). These trends are not explained by species growth rate (Fig EV2B) or sensitivity to pH (Fig EV5C). In addition, this variation in sensitivity to richness is not fully explained by patterns in the inferred interspecies interaction network. In our system, the abundance of B. hydrogenotrophica and E. lenta did not vary with richness. Notably, these species are unique in their ability as an acetogen to utilize hydrogen and carbon dioxide (Bernalier et␣al, 1996) and utilize arginine (Sperry & Wilkins, 1976), respectively. Therefore, orthogonal ecological niches may represent one mechanism of low sensitivity in abundance to richness. Further, the plasticity of a species’ ecological niche in response to competition may influence its relationship between abundance and richness. Future work will investigate these potential mechanisms determining the relationship between abundance and richness in larger communities and in␣vivo.
Bacteroides have been found to both inhibit and promote C. difficile growth in different environments (Ferreyra et␣al, 2014; Ghimire et␣al, 2020; Pereira et␣al, 2020; Hassall et␣al, 2021), but in our system all Bacteroides species inhibited C. difficile. We did not observe a strong inhibition of C. difficile by C. scindens which has been previously shown to occur via production of secondary bile acids that inhibit C. difficile germination (Buffie et␣al, 2014) presumably due to the absence of bile acids in our media. Instead, in our system the closest relative of C. difficile, C. hiranonis, was the strongest inhibitor of C. difficile abundance. Addition of one of the metabolites utilized by both C. difficile and C. hiranonis, glucose, partially rescued C. difficile growth in C. hiranonis spent media (Fig 5E), indicating glucose competition could be one mechanism of C. difficile inhibition. Inhibitory consortia could be designed to maximize resource competition over carbohydrates such as glucose between resident members and C. difficile. Because an estimated 20% of carbohydrates such as glucose escape absorption by the host and are present in the colon (Stephen et␣al, 1983; Anderwald et␣al, 2011), carbohydrate competition is likely relevant in the colon environment.
We note that three of the co‐utilized metabolites (proline, serine, and threonine, Fig 5C) are amino acids used in Stickland fermentation, a major energy generation process found in C. difficile and many other Clostridia (Mead, 1971). Recent work shows that the Stickland fermenting Clostridium bifermentans suppressed C. difficile abundance and prevented mortality in gnotobiotic mice while the non‐Stickland fermenter Clostridium sardiniense did not suppress abundance or prevent mortality (preprint: Girinathan et␣al, 2020). The study indicates that introducing competition over Stickland metabolites is a relevant mechanism for C. difficile inhibition in␣vivo. Our data suggest introduction of the gut microbe C. hiranonis into a community could potentially intensify competition over the utilized amino acids. Our data also indicate that C. hiranonis has an additional unknown mechanism of C. difficile inhibition beyond resource competition. The additional mechanism is not due to pH change and does not involve an extracellular protein (Fig 5E). This leads us to speculate that the inhibition could be due to a non‐protein antibiotic produced by C. hiranonis that is similar to the tryptophan‐derived antibiotics produced by C. scindens and Clostridium sordellii (Kang et␣al, 2019).
We showed that communities that reduce the external pH below 6.2 inhibit C. difficile in a pH‐dependent manner, consistent with studies showing that C. difficile has lower viability and rates of sporulation in acidic environments (Wetzel & McBride, 2020; Yuille et␣al, 2020). We note that our in␣vitro system differs from the human gut, lacking the pH‐buffering secretion of bicarbonate by host intestinal epithelial cells. However, the amount of bicarbonate buffer in our media (4.8 mM) is within the estimated range in the gastrointestinal tract (2–20 mM) (Litou et␣al, 2020). Additionally, the changes in pH that we observe in our in␣vitro measurements are within the variation seen in the colon, which has been shown to fluctuate between pH 5 and pH 8 (Koziolek et␣al, 2015). This suggests the observed pH changes in our experiments could be physiologically relevant. We also note an intriguing human cohort study that found a strong association between alkaline fecal pH and CDI (Gupta et␣al, 2016). Although it is not known whether alkaline pH is a cause of consequence of CDI, this study together with our data suggests that strategies for pH‐based inhibition of C. difficile in the colon are worth further investigation. If reduced pH can inhibit C. difficile in the colon, manipulation of the pH of the gut environment is a potential microbiome intervention strategy to inhibit C. difficile. The pH could be manipulated by bacterial therapeutics containing strong fermenters or dietary substrates that increase fermentation (Chung et␣al, 2007).
While propagule pressure has been shown to determine invasion success in microbial invasions (Acosta et␣al, 2015; Ketola et␣al, 2017; Kinnunen et␣al, 2018), we demonstrate that this applies to C. difficile in synthetic gut communities. Propagule pressure is known to be important in murine C. difficile infections, where mice cohoused with supershedders containing 108 CFU/g C. difficile in their feces became colonized with C. difficile, whereas mice cohoused with low shedders containing 102 CFU/g C. difficile did not become colonized (Lawley et␣al, 2009). However, the relationship between C. difficile dosage and incidence of CDI in humans is unknown. Our results suggest that the density of C. difficile could be an important variable in the outcome of C. difficile invasions in a clinical setting. While our data suggest the variation in C. difficile abundance with propagule pressure can result in long‐term history dependence (Fig EV3A), the effects of propagule pressure are transient in the model due to the absence of multistability (Fig EV3B). Our model is not informed by long‐term multispecies community measurements and therefore may not accurately capture the long‐term dynamics of the system. Future work will determine the long‐term effects of propagule pressure on C. difficile abundance using passaging or continuous culture and develop computational models that can accurately predict these history‐dependent behaviors.
Our absolute abundance method combines OD600 measurements and 16S rRNA gene sequencing to determine the absolute abundance of each species in multispecies communities. Biases in genome extraction efficiency, 16S rRNA gene copy number, and PCR amplification can impact measurements based on 16S rRNA gene sequencing (Crosby & Criddle, 2003; Laursen et␣al, 2017; Lim et␣al, 2018). We tested for potential bias in our workflow by measuring the relative abundance of mixed cultures containing 10% C. difficile based on OD600 measurements (Appendix␣Fig S7). These results indicate there was no significant bias in our method for these communities. Previous work using this absolute abundance method found that 75% of interactions were in qualitative agreement with sterile supernatant experiments (Venturelli et␣al, 2018). Further, 85% of our inferred interspecies interactions in the Full Model were in qualitative agreement with this previous study that studied a 12‐member subset of this community (Appendix␣Fig S8). Taken together, these results indicate that our absolute abundance method is reproducible across multiple studies and can decipher biologically meaningful interspecies interactions despite any potential biases.
In sum, we identified ecological and molecular mechanisms of resistance to invasion by C. difficile using a synthetic gut microbiome. While our system lacks the full diversity of the human gut microbiome and a host–interaction component, many of our results support principles of invasion theory based on a broad range of systems, suggesting that some of these principles could be generalized to the mammalian gut environment. Future work could create panels of gut microbial communities that feature different weightings of the inhibitory mechanisms identified in this work. These panels could be tested in␣vitro for inhibition of C. difficile growth and promising candidates could be introduced into germ‐free mouse models to evaluate their C. difficile inhibitory potential as bacterial therapeutics.
Materials and Methods
Strain and media information
The strains used in this work were obtained from the sources listed in Appendix␣Table S3. The three clinical C. difficile isolates (MS002, MS010, and MS011) were C. difficile NAAT (GeneXpert) positive via admission stool sample and toxin A (tcdA) and toxin B (tcdB) positive via in‐house research PCR. Each patient was diagnosed with and treated for CDI. Single‐use glycerol stocks were prepared as described previously (Clark et␣al, 2021). The media used in this work are anaerobic basal broth (ABB, Oxoid), clostridial reinforced medium (CRM, Difco), YP broth (Geva‐Zatorsky et␣al, 2015), and YBHI. YBHI broth recipe: BHI broth (Accumedia), 5 g/l yeast extract (BD Bacto), 1 g/l d‐cellobiose (Chem‐Impex), 1 g/l d‐maltose monohydrate (Sigma‐Aldrich), and 0.5 g/l l‐cysteine (Sigma‐Aldrich).
Starter cultures
Cells were cultured in an anaerobic chamber (Coy Lab products) with an atmosphere of 2.5 ± 0.5% H2, 15 ± 1% CO2, and balance N2. Single‐species starter cultures were inoculated by adding 100 µl of a single‐use 25% glycerol stock to 5 ml of anaerobic basal broth media (ABB). E. rectale starter cultures were supplemented with 33 mM sodium acetate (Sigma‐Aldrich) and D. piger starter cultures were supplemented with 28 mM sodium lactate (Sigma‐Aldrich) and 2.7 mM magnesium sulfate (Sigma‐Aldrich). To begin experiments with organisms in similar growth phases, starter cultures were inoculated either 16 or 41 h prior to experimental setup, depending on the growth rate of the organism (Appendix␣Table S3).
Inoculation of monospecies and pairs experiments
Starter cultures were diluted to 0.0022 OD600 in ABB (Tecan Infinite Pro F200). For monospecies in Exp1, diluted cultures were added directly to 96 deep well plates for final OD600 of 0.0022. For pairs in Exp2, diluted cultures were combined into pairs in 96 deep well plates at 1:1 or 1:10 volume ratios for final OD600 of 0.0011 or 0.00022 and 0.00198. Cultures were combined using a liquid handling robot (Tecan Evo 100). Plates were covered with gas‐permeable seal (BreatheEasy) and incubated at 37°C with no shaking.
Inoculation of multispecies community experiments
Starter cultures were diluted to 0.0066 OD600. Diluted cultures were combined into communities in 96 deep well plates using a liquid handling robot (Tecan Evo). The communities in Exp3 and Exp4 were created by combining equal volumes of each diluted starter culture, so the initial OD600 of each species in the community was 0.0066 divided by the number of species. The communities in Exp5 and Exp7 were combined so that all non‐C. difficile species had an initial OD600 of 0.00165, and C. difficile had an initial OD600 of 0.00055 (10% of community) in the low‐density condition of Exp5 and 0.009 (65% of community) in the high‐density condition of Exp5. In Exp7, the community OD600 was measured after 6 h of incubation and C. difficile was added so that its OD600 was 10% (low‐density condition) or 33% (high‐density condition) of the community. The 3–4 member communities in Exp6 were combined such that all non‐C. difficile species had an initial OD600 of 0.00165, and C. difficile had an initial OD600 of 0, 0.00026, 0.00055, 0.0012, 0.0021, 0.0033, 0.00495, and 0.0074 in the 3 member communities and 0, 0.00035, 0.00073, 0.00165, 0.0028, 0.0044, 0.0066, and 0.0099 in the 4 member communities for initial fractions 0, 0.1, 0.2, 0.3, 0.4, 0.5, and 0.6, respectively. The full community in Exp6 was combined so that all non‐C. difficile species had an initial OD600 of 0.00047, and C. difficile had an initial OD600 of 0, 0.00032, 0.0015, 0.0026, 0.0041, 0.0061, 0.0092 for initial fractions 0, 0.1, 0.2, 0.3, 0.4, 0.5, and 0.6, respectively. The C. hiranonis titration communities in Exp8 were combined so that all non‐C. hiranonis species had an initial OD600 of 0.00165, and C. hiranonis had an initial OD600 of 0, 0.00055, 0.00012, 0.0021, 0.0033, 0.0050, 0.012, and 0.045 for initial fractions 0, 0.1, 0.2, 0.3, 0.4, 0.5, 0.7, and 0.9, respectively. Plates were covered with gas‐permeable seals (BreatheEasy) and incubated at 37°C with no shaking.
Culture sample collection
At each timepoint, samples were mixed and aliquots were removed for sequencing and for measuring OD600. We measured OD600 of two dilutions of each sample and selected the value that was within the linear range of the instrument (Tecan Infinite Pro F200). Sequencing aliquots were spun down aerobically at 1,739 g for 15 min and stored at −80°C. For timepoints with dilutions, samples were mixed and aliquots were collected for sequencing and OD600 measurements before the samples were diluted 1:20 into fresh media. Abundance of the diluted sample was calculated by dividing the undiluted measurements by the dilution factor of 20.
pH measurements and adjustments
The pH of each community in Fig 4D was measured using a phenol red assay as described previously (Clark et␣al, 2021). The pH of each supernatant in Fig 4E was measured using a pH probe (Mettler Toledo). The pH of each supernatant was adjusted to the pH of fresh media by adding small volumes of sterile 5 M NaOH and 5 M HCl.
Supernatant experiments
Starter cultures were diluted to 0.0066 OD600. Diluted cultures were combined into communities in 96 deep well plates using a liquid handling robot (Tecan Evo). Communities were created by combining equal volumes of each species, so the final OD600 of each species in the community was 0.0066 divided by the number of species. Plates were covered with gas‐permeable seal (BreatheEasy) and incubated at 37°C with no shaking. After incubation time of 6 h (Fig 4E and F) or 20 h (Fig 5C), cultures were spun down aerobically at 3,500 rpm for 15 min and sterile filtered using Steriflip 0.2‐µM filters (Millipore‐Sigma) before returning to anaerobic chamber. Media controls were spun down and filtered aerobically in parallel with samples. C. difficile was inoculated in the sterilized supernatants to a final OD600 of 0.0022 in 96‐well microplates that were covered with gas‐permeable seals (BreatheEasy), incubated at 37°C with shaking, and OD600 was measured every 2 h (Tecan Infinite Pro F200).
Quantification of metabolites in C. difficile and C. hiranonis supernatants
Clostridioides difficile and C. hiranonis starter cultures were diluted to 0.0022 OD600 in ABB in triplicate. After 20 h of incubation, an aliquot of each culture and an aliquot of a media control were spun down aerobically at 3,500 rpm for 15 min and sterile filtered using Steriflip 0.2‐µM filters. The samples were stored at 4C overnight. The supernatants were then diluted 1:50 into water and analyzed by LC‐MS. For Fig 5D and E, a five‐point standard curve of mixed metabolite standard containing acetyl‐ornithine, glucose, glutamine, proline serine, sodium pyruvate, and threonine was prepared in water day‐of and analyzed alongside samples.
LC‐MS analysis was conducted as described previously (27, 80) using a Vanquish UPLC system (Thermo Scientific) coupled to a Q Exactive Orbitrap high‐resolution mass spectrometer (Thermo Scientific) using an electrospray ionization source operating in negative mode. Separation was achieved using a 2.1‐ by 100‐mm Acquity UHPLC BEH C18 column with 1.7‐µm particle size (Waters) at 25°C. Solvent A was 97:3 H2O‐methanol with 10 mM tributylamine adjusted to pH 8.2 by the addition of acetic acid to ˜10 mM final concentration. Solvent B was 100% methanol. The following gradient was used for separation: 0 to 2.5 min, 5% B; 2.5–17 min, linear gradient from 5% B to 95% B; 17–19.5 min, 95% B; 19.5–20 min, linear gradient from 95% B to 5% B; 20–25 min, 5% B. Mass spectrometry parameters were full MS‐SIM (single‐ion monitoring) scanning between 70 and 1,000 m/z, automatic control gain (ACG) target of 1e6, maximum injection time (IT) of 40 ms, and resolution of 70,000 full width at half‐maximum (FWHM). MAVEN software suite was used to analyze the data (81, 82). Compounds were identified by retention time matching to pure standards and monoisotopic mass.
For Fig 5C, secreted metabolites were defined as metabolites whose average signal in the three supernatant replicates was at least twofold greater than the signal of the media control. Utilized metabolites were defined as metabolites whose average signal in the three supernatant replicates was at least twofold less than the signal of the media control.
For Fig 5D, metabolite concentrations were calculated using the linear region of the standard curve. The difference in concentration between the supernatant and media control was used to calculate the amount of metabolite needed to add to the supernatant to adjust to media level. The supernatants in Fig 5E were adjusted by adding 20 µl of additive solution to 180 µl supernatant. The additive solutions contained between 1 and 7 metabolites diluted to the necessary concentrations in water. For the no‐treatment control, 20 µl of water was added to 180 µl supernatant. For the "1× media” condition, 20 µl of 10× media was added to 180 µl supernatant.
Genome extractions
Genomic DNA was extracted using a method adapted from previous work (Clark et␣al, 2021). Briefly, cell pellets were resuspended in 180‐µl enzymatic lysis buffer containing 20 mg/ml lysozyme (Sigma‐Aldrich), 20 mM Tris–HCl pH 8 (Invitrogen), 2 mM EDTA (Sigma‐Aldrich), and 1.2% Triton X‐100 (Sigma‐Aldrich). Samples were incubated at 37°C at 600 RPM for 30 min. Samples were treated with 25 µl 20 mg/ml Proteinase K (VWR) and 200 µl buffer AL (Qiagen), mixed by pipette and incubated at 56°C at 600 RPM for 30 min. Samples were treated with 200 µl 200 proof ethanol (Koptec), mixed by pipette, and transferred to 96‐well nucleic acid binding plates (Pall). After washing with 500 µl buffer AW1 and AW2 (Qiagen), a vacuum was applied for 10 min to dry excess ethanol. Genomic DNA was eluted with 110 µl buffer AE (Qiagen) preheated to 56°C and then stored at −20°C.
Genomic DNA was quantified using Sybr Green fluorescence assay with a 6‐point DNA standard curve (0, 0.5, 1, 2, 4, 6 ng/µl biotium). 1 µl of samples and 5 µl of standards were diluted into 95 µl of 1× SYBR green (Invitrogen) in TE buffer and mixed by pipette. Fluorescence was measured with an excitation/emission of 485/535 nm (Tecan Spark). Genomic DNA was normalized to 1 ng/µl in molecular grade water using a liquid handling robot (Tecan Evo 100). Samples < 1 ng/µl were not diluted. Diluted genomic DNA was stored at −20°C.
Primer design, library preparation, and sequencing
Dual‐indexed primers for multiplexed amplicon sequencing of the 16S v3‐v4 region were designed as described previously (Venturelli et␣al, 2018; Clark et␣al, 2021). Briefly, oligonucleotides (Integrated DNA Technology) were arrayed into 96‐well plates using an acoustic liquid handling robot (Echo LabCyte) and stored at −20°C. Genomic DNA was PCR‐amplified using Phusion High‐Fidelity DNA Polymerase (Thermo Fisher) for 25 cycles with 0.05 µM of each primer. Samples were pooled by plate, purified (Zymo Research), quantified by NanoDrop, and combined in equal proportions into a library. The library was quantified using Qubit 1× HS Assay (Invitrogen), diluted to 4.2 nM, and loaded at 21 pM onto Illumina MiSeq platform for 300‐bp paired end sequencing.
Data analysis
Sequencing data were analyzed using a method adapted from previous work (Venturelli et␣al, 2018). Basespace Sequencing Hub’s FastQ Generation demultiplexed the indices and generated FastQ files. FastQ files were analyzed using custom python scripts. Paired reads were merged using PEAR (Paired‐End reAd mergeR) v0.9.0 (Zhang et␣al, 2014). A reference database containing 16S v3‐v4 region of each species in the study was created by assembling consensus sequence based on sequencing results of each monospecies. Reads were mapped to the reference database using the mothur v1.40.5 command classify.seqs using the Wang method with bootstrap cutoff value of 60% (Wang et␣al, 2007; Schloss et␣al, 2009). Relative abundance was calculated by dividing the read counts mapped to each organism by the total reads in the sample. Absolute abundance was calculated by multiplying the relative abundance of an organism by the OD600 of the sample. Samples were excluded from further analysis if > 1% of the reads were assigned to a species not expected to be in the community (indicating contamination).
Generalized Lotka–Volterra Model
The gLV model is a set of N coupled first‐order ordinary differential equations:
where N is the number of species, the parameter Xi is the abundance of species i, the parameter ri is the basal growth rate of species i, the parameter αij , called the interaction parameter, is the growth modification of species i by species j and the parameter Xj is the abundance of species j. The parameter αij is constrained to be negative when i = j, representing intra‐species competition.
Parameter estimation
The gLV model parameters were estimated from time‐series measurements of single‐species and multispecies cultures using the nonlinear programming solver FMINCON in MATLAB, which finds the optimal set of parameters that minimizes a given cost function. The estimation was implemented using previously developed custom MATLAB scripts (Clark et␣al, 2021). The cost (C) of the optimization algorithm was computed by (i) simulating each species m in each community k with an ODE solver and summing the mean‐squared error between the abundance of each species in the simulation Xmodel and data Xexp at each timepoint n (ii) adding the sum each parameter θ squared multiplied by a regularization coefficient λ:
The second step is a L2 regularization, which penalizes the magnitude of the parameter vector to prevent overfitting the data. The optimization was repeated with a range of regularization coefficients. The regularization coefficient that resulted in a parameter set with a mean‐squared error of 110% of the non‐regularized parameter set was selected, which was λ = 0.5 for the Preliminary model and λ = 0.1 for the Full Model. The data used for parameter estimation for the Preliminary model and Full Model are given in Table 1. To validate the predictive ability of the model, 24 2–13 member resident communities (Appendix␣Fig S4A) were left out from the training data set and a set of parameters was inferred from this reduced data set using λ = 0.1 for the regularization coefficient. The community compositions of the 24 held‐out communities were simulated with this parameter set to evaluate the predictive capability of the model on held‐out data (Fig 2C).
Analysis of gLV model
To quantify the uncertainties in gLV parameters, an adaptive Markov chain Monte Carlo (MCMC) method was used to sample from the posterior gLV parameter (θ) distribution P(θ|y) given a sequence of m abundance measurements y = (y 1,…,y m). In particular, for the k‐th measurement, y k is a vector that concatenates all abundance measurements collected from all sub‐community experiments. Uncertainty for the k‐th measurement was modeled by an additive and independent noise, which is distributed according to N(0, ), where is the diagonal covariance matrix for experimental data collected in the k‐th measurement. Given a fixed parameter θ, the gLV model was simulated to obtain the model predicted abundance at every instant k. The likelihood to observe a sequence of abundance measurements y was then computed as:
where is the probability density function for the normal distribution . The posterior distribution was then described according to Bayes rule as , where P(θ) is the prior parameter distribution. Normal priors were used for the parameters. The means of the normal distributions were set to the parameters estimated by the FMINCON method and the coefficients of variation were set to 5%.
An adaptive, symmetric, random‐walk Metropolis MCMC algorithm (Haario et␣al, 2001) was then used to draw samples from this posterior distribution. Specifically, given the current sample θ( n ) at step n of the Markov chain, the proposed sample for step (n + 1) is θ( n +1) = θ( n )+δ( n ), where δ( n ) is drawn from a normal distribution. The algorithm is adaptive in the sense that the covariance of this normal distribution is given by , where is the covariance of θ(1),…, θ( n ) and α is a positive parameter. The proposed sample is accepted with probability 1 if P(θ( n +1)|y)/P(θ( n )|y) > 1, and it is accepted with probability β if P(θ( n +1)|y)/P(θ( n )|y) = β ≤ 1.
The algorithm described above was implemented using MATLAB R2020a, where the gLV models were solved using variable step solver ode23s. 120,000 MCMC samples were collected after a burn‐in period of 10,000 samples. The Gelman–Rubin potential scale reduction factor (PSRF) was used to evaluate convergence of the posterior distribution estimates, where a PSRF closer to 1 indicates better convergence. The average PSRF is 1.31 and 80% of the parameters have a PSRF < 1.5. The medians of the marginal distributions of all parameters correlated strongly with parameters estimated by the FMINCON method (Pearson r = 0.99).
To evaluate multistability in our gLV model, we enumerated all non‐negative equilibria of the subcommunities and then evaluated their local stability by checking the eigenvalues of the Jacobian at the respective equilibria.
Hill equation␣and exponential decay model fits
The community sensitivity to C. difficile initial abundance was quantified by fitting the data to the Hill equation:
where E is 48‐h abundance of C. difficile, Emax is the maximum 48‐h abundance of C. difficile across all initial fractions, A is the initial fraction of C. difficile, EC50 is the initial fraction that produces 50% of Emax value, and n is a measure of ultrasensitivity. The data were fit using custom python scripts implementing the curve_fit function of the scipy package optimization module.
The relationship between OD600 and species richness was quantified by fitting the experimental data or simulated data to an exponential decay function:
where y is calculated OD600, a is initial calculated OD600, b is the exponential decay constant, and x is species richness. The OD600 data were normalized to be between 0 and 1 for all species. The data were fit using custom python scripts implementing the curve_fit function of the scipy package optimization module.
Normalized Euclidean distances
The normalized Euclidean distance (D) between uninvaded resident community R and C. difficile‐invaded community V is calculated using.
here R is the 48‐h timepoint of the uninvaded resident community and V is the 48‐h timepoint of the resident community invaded with C. difficile. Ri is the relative abundance of species i in the uninvaded resident community, equal to reads of species i divided by the total community reads. Vi is the normalized relative abundance of species i in the invaded community, equal to reads of species i divided by the resident community reads (total community reads minus C. difficile reads).
Author contributions
OSV and SH conceived the study. SH carried out the experiments. SH, OSV, TBJ, and DA‐N designed and SH and TBJ implemented the metabolomic measurements. SEH and YQ performed computational modeling and analysis. RLC and YQ wrote customized scripts to perform modeling analyses. SH and OSV analyzed and interpreted the data. OSV secured funding. NS and LW isolated clinical C.difficile strains and provided information on these strains. SEH and OSV wrote the paper and all authors provided feedback on the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
Acknowledgements
We would like to acknowledge David M. Stevenson for sharing software to convert raw output from the mass spectrometer for downstream analysis. Research was sponsored by the National Institutes of Health and was accomplished under grant number R35GM124774, National Institute of Biomedical Imaging and Bioengineering under grant number R01EB030340 and the Army Research Office under grant number W911NF1910269. S.E.H. was supported in part by the National Institute of General Medical Sciences of the National Institutes of Health under Award Number T32GM008349. R.L.C. was supported in part by a National Human Genome Research Institute training grant to the Genomic Sciences Training Program under Award Number T32HG002760.
Mol Syst Biol. (2021) 17: e10355
Data availability
The datasets and computer code produced in this study are available in the following databases:
Community composition data: Dataset EV4 in this manuscript.
Metabolomics data: Dataset EV3 in this manuscript.
Modeling computer scripts: https://github.com/SusanHromada/NegativeInteractionsDetermineCdifficileGrowth.
References
- Acosta F, Zamor RM, Najar FZ, Roe BA, Hambright KD (2015) Dynamics of an experimental microbial invasion. Proc Natl Acad Sci USA 112: 11594–11599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderwald C, Gastaldelli A, Tura A, Krebs M, Promintzer‐Schifferl M, Kautzky‐Willer A, Stadler M, DeFronzo RA, Pacini G, Bischof MG (2011) Mechanism and effects of glucose absorption during an oral glucose tolerance test among females and males. J Clin Endocrinol Metab 96: 515–524 [DOI] [PubMed] [Google Scholar]
- Antharam VC, Li EC, Ishmael A, Sharma A, Mai V, Rand KH, Wang GP (2013) Intestinal dysbiosis and depletion of butyrogenic bacteria in clostridium difficile infection and nosocomial diarrhea. J Clin Microbiol 51: 2884–2892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernalier A, Willems A, Leclerc M, Rochet V, Collins MD (1996) Ruminococcus hydrogenotrophicus sp. nov., a new H2/CO2‐utilizing acetogenic bacterium isolated from human feces. Arch Microbiol 166: 176–183 [DOI] [PubMed] [Google Scholar]
- Buffie CG, Bucci V, Stein RR, McKenney PT, Ling L, Gobourne A, No D, Liu H, Kinnebrew M, Viale A et␣al (2014) Precision microbiome reconstitution restores bile acid mediated resistance to Clostridium difficile . Nature 517: 205–208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buffie CG, Pamer EG (2013) Microbiota‐mediated colonization resistance against intestinal pathogens. Nat Rev Immunol 13: 790–801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang JY, Antonopoulos DA, Kalra A, Tonelli A, Khalife WT, Schmidt TM, Young VB (2008) Decreased diversity of the fecal Microbiome in recurrent Clostridium difficile‐associated diarrhea. J Infect Dis 197: 435–438 [DOI] [PubMed] [Google Scholar]
- Chen T, Zhou Q, Zhang D, Jiang F, Wu J, Zhou J‐Y, Zheng X, Chen Y‐G (2018) Effect of faecal microbiota transplantation for treatment of Clostridium difficile infection in patients with inflammatory bowel disease: a systematic review and meta‐analysis of cohort studies. J Crohns Colitis 12: 710–717 [DOI] [PubMed] [Google Scholar]
- Chung Y‐C, Hsu C‐K, Ko C‐Y, Chan Y‐C (2007) Dietary intake of xylooligosaccharides improves the intestinal microbiota, fecal moisture, and pH value in the elderly. Nutr Res 27: 756–761 [Google Scholar]
- Clark RL, Connors BM, Stevenson DM, Hromada SE, Hamilton JJ, Amador‐Noguez D, Venturelli OS (2021) Design of synthetic human gut microbiome assembly and butyrate production. Nat Commun 12: 3254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crosby LD, Criddle CS (2003) Understanding bias in microbial community analysis techniques due to rrn operon copy number heterogeneity. Biotechniques 34: 790–802 [DOI] [PubMed] [Google Scholar]
- DeFilipp Z, Bloom PP, Torres Soto M, Mansour MK, Sater MRA, Huntley MH, Turbett S, Chung RT, Chen Y‐B, Hohmann EL (2019) Drug‐resistant E. coli bacteremia transmitted by fecal microbiota transplant. N Engl J Med 381: 2043–2050 [DOI] [PubMed] [Google Scholar]
- Dethlefsen L, Relman DA (2011) Incomplete recovery and individualized responses of the human distal gut microbiota to repeated antibiotic perturbation. Proc Natl Acad Sci USA 108: 4554–4561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillon RJ, Vennard CT, Buckling A, Charnley AK (2005) Diversity of locust gut bacteria protects against pathogen invasion. Ecol Lett 8: 1291–1298 [Google Scholar]
- Dowle C (2016) Faecal microbiota transplantation: a review of FMT as an alternative treatment for Clostridium difficile infection. Biosci Horizons Int J Student Res 9: hzw007 [Google Scholar]
- Elton CS (1958) The ecology of invasions by animals and plants. London: Methuen; [Google Scholar]
- Eze P, Balsells E, Kyaw MH, Nair H (2017) Risk factors for Clostridium difficile infections ‐ an overview of the evidence base and challenges in data synthesis. J Glob Health 7: 10417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreyra JA, Wu KJ, Hryckowian AJ, Bouley DM, Weimer BC, Sonnenburg JL (2014) Gut microbiota‐produced succinate promotes C. difficile infection after antibiotic treatment or motility disturbance. Cell Host Microbe 16: 770–777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher JR, Pike CM, Parsons RJ, Rivera AJ, Foley MH, McLaren MR, Montgomery SA, Theriot CM (2021) Clostridioides difficile exploits toxin‐mediated inflammation to alter the host nutritional landscape and exclude competitors from the gut microbiota. Nat Commun 12: 462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forster SC, Kumar N, Anonye BO, Almeida A, Viciani E, Stares MD, Dunn M, Mkandawire TT, Zhu A, Shao Y et␣al (2019) A human gut bacterial genome and culture collection for improved metagenomic analyses. Nat Biotechnol 37: 186–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geva‐Zatorsky N, Alvarez D, Hudak JE, Reading NC, Erturk‐Hasdemir D, Dasgupta S, von Andrian UH, Kasper DL (2015) In vivo imaging and tracking of host‐microbiota interactions via metabolic labeling of gut anaerobic bacteria. Nat Med 21: 1091–1100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghimire S, Roy C, Wongkuna S, Antony L, Maji A, Keena MC, Foley A, Scaria J (2020) Identification of Clostridioides difficile‐inhibiting gut commensals using culturomics, phenotyping, and combinatorial community assembly. mSystems 5: e00620–e719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girinathan BP, DiBenedetto N, Worley J, Peltier J, Lavin R, Delaney ML, Cummins C, Onderdonk AB, Gerber GK, Dupuy B et␣al (2020) In vivo commensal control of Clostridioides difficile virulence. bioRxiv 10.1101/2020.01.04.894915 [PREPRINT] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonze D, Coyte KZ, Lahti L, Faust K (2018) Microbial communities as dynamical systems. Curr Opin Microbiol 44: 41–49 [DOI] [PubMed] [Google Scholar]
- Gupta P, Yakubov S, Tin K, Zea D, Garankina O, Ghitan M, Chapnick EK, Homel P, Lin YS, Koegel MM (2016) Does alkaline colonic pH predispose to Clostridium difficile infection? South Med J 109: 91–96 [DOI] [PubMed] [Google Scholar]
- Haario H, Saksman E, Tamminen J (2001) An adaptive metropolis algorithm. Bernoulli 7: 223–242 [Google Scholar]
- Hassall J, Cheng JKJ, Unnikrishnan M (2021) Dissecting individual interactions between pathogenic and commensal bacteria within a multispecies gut microbial community. mSphere 6: e00013‐21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang JD, Myers CJ, Harris SC, Kakiyama G, Lee I‐K, Yun B‐S, Matsuzaki K, Furukawa M, Min H‐K, Bajaj JS et␣al (2019) Bile acid 7α‐dehydroxylating gut bacteria secrete antibiotics that inhibit Clostridium difficile: role of secondary bile acids. Cell Chem Biol 26: 27–34.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ketola T, Saarinen K, Lindström L (2017) Propagule pressure increase and phylogenetic diversity decrease community’s susceptibility to invasion. BMC Ecol 17: 15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinnunen M, Dechesne A, Proctor C, Hammes F, Johnson D, Quintela‐Baluja M, Graham D, Daffonchio D, Fodelianakis S, Hahn N et␣al (2016) A conceptual framework for invasion in microbial communities. ISME J 10: 2773–2775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinnunen M, Dechesne A, Albrechtsen H‐J, Smets BF (2018) Stochastic processes govern invasion success in microbial communities when the invader is phylogenetically close to resident bacteria. ISME J 12: 2748–2756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitahara M, Takamine F, Imamura T, Benno Y (2001) Clostridium hiranonis sp. nov., a human intestinal bacterium with bile acid 7alpha‐dehydroxylating activity. Int J Syst Evol Microbiol 51: 39–44 [DOI] [PubMed] [Google Scholar]
- Koziolek M, Grimm M, Becker D, Iordanov V, Zou H, Shimizu J, Wanke C, Garbacz G, Weitschies W (2015) Investigation of pH and temperature profiles in the GI tract of fasted human subjects using the Intellicap® system. J Pharm Sci 104: 2855–2863 [DOI] [PubMed] [Google Scholar]
- Laursen MF, Dalgaard MD, Bahl MI (2017) Genomic GC‐content affects the accuracy of 16S rRNA gene sequencing based microbial profiling due to PCR bias. Front Microbiol 8: 1934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawley TD, Clare S, Walker AW, Goulding D, Stabler RA, Croucher N, Mastroeni P, Scott P, Raisen C, Mottram L et␣al (2009) Antibiotic treatment of Clostridium difficile carrier mice triggers a supershedder state, spore‐mediated transmission, and severe disease in immunocompromised hosts. Infect Immun 77: 3661–3669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawley TD, Clare S, Walker AW, Stares MD, Connor TR, Raisen C, Goulding D, Rad R, Schreiber F, Brandt C et␣al (2012) Targeted restoration of the intestinal microbiota with a simple, defined bacteriotherapy resolves relapsing Clostridium difficile disease in mice. PLOS Pathog 8: e1002995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim MY, Song E‐J, Kim SH, Lee J, Nam Y‐D (2018) Comparison of DNA extraction methods for human gut microbial community profiling. Syst Appl Microbiol 41: 151–157 [DOI] [PubMed] [Google Scholar]
- Litou C, Psachoulias D, Vertzoni M, Dressman J, Reppas C (2020) Measuring pH and buffer capacity in fluids aspirated from the fasted upper gastrointestinal tract of healthy adults. Pharm Res 37: 42 [DOI] [PubMed] [Google Scholar]
- Lockwood JL, Cassey P, Blackburn T (2005) The role of propagule pressure in explaining species invasions. Trends Ecol Evol 20: 223–228 [DOI] [PubMed] [Google Scholar]
- Loubinoux J, Valente FMA, Pereira IAC, Costa A, Grimont PAD, Le Faou AE (2002) Reclassification of the only species of the genus Desulfomonas, Desulfomonas pigra, as Desulfovibrio piger comb. nov. Int J Syst Evol Microbiol 52: 1305–1308 [DOI] [PubMed] [Google Scholar]
- Mallon CA, van Elsas JD, Salles JF (2015) Microbial invasions: the process, patterns, and mechanisms. Trends Microbiol 23: 719–729 [DOI] [PubMed] [Google Scholar]
- Mallon CA, Poly F, Le Roux X, Marring I, van Elsas JD, Salles JF (2015) Resource pulses can alleviate the biodiversity–invasion relationship in soil microbial communities. Ecology 96: 915–926 [DOI] [PubMed] [Google Scholar]
- Marino S, Baxter NT, Huffnagle GB, Petrosino JF, Schloss PD (2014) Mathematical modeling of primary succession of murine intestinal microbiota. Proc Natl Acad Sci USA 111: 439–444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mead GC (1971) The amino acid‐fermenting clostridia. Microbiology 67: 47–56 [DOI] [PubMed] [Google Scholar]
- Mounier J, Monnet C, Vallaeys T, Arditi R, Sarthou A‐S, Hélias A, Irlinger F (2008) Microbial interactions within a cheese microbial community. Appl Environ Microbiol 74: 172–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullish BH, McDonald JAK, Pechlivanis A, Allegretti JR, Kao D, Barker GF, Kapila D, Petrof EO, Joyce SA, Gahan CGM et␣al (2019) Microbial bile salt hydrolases mediate the efficacy of faecal microbiota transplant in the treatment of recurrent Clostridioides difficile infection. Gut 68: 1791–1800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira FC, Wasmund K, Cobankovic I, Jehmlich N, Herbold CW, Lee KS, Sziranyi B, Vesely C, Decker T, Stocker R et␣al (2020) Rational design of a microbial consortium of mucosal sugar utilizers reduces Clostridiodes difficile colonization. Nat Commun 11: 5104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrof EO, Gloor GB, Vanner SJ, Weese SJ, Carter D, Daigneault MC, Brown EM, Schroeter K, Allen‐Vercoe E (2013) Stool substitute transplant therapy for the eradication of Clostridium difficile infection: ‘RePOOPulating’ the gut. Microbiome 1: 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T et␣al (2010) A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464: 59–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridlon JM, Devendran S, Alves JM, Doden H, Wolf PG, Pereira GV, Ly L, Volland A, Takei H, Nittono H et␣al (2020) The ‘in␣vivo lifestyle’ of bile acid 7α‐dehydroxylating bacteria: comparative genomics, metatranscriptomic, and bile acid metabolomics analysis of a defined microbial community in gnotobiotic mice. Gut Microbes 11: 381–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ et␣al (2009) Introducing mothur: open‐source, platform‐independent, community‐supported software for describing and comparing microbial communities. Appl Environ Microbiol 75: 7537–7541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperry JF, Wilkins TD (1976) Arginine, a growth‐limiting factor for Eubacterium lentum . J Bacteriol 127: 780–784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephen AM, Haddad AC, Phillips SF (1983) Passage of carbohydrate into the colon. Direct measurements in humans. Gastroenterology 85: 589–595 [PubMed] [Google Scholar]
- Tvede M, Rask‐Madsen J (1989) Bacteriotherapy for chronic relapsing Clostridium difficile diarrhoea in six patients. Lancet 333: 1156–1160 [DOI] [PubMed] [Google Scholar]
- van Elsas JD, Chiurazzi M, Mallon CA, Elhottovā D, Krištůfek V, Salles JF (2012) Microbial diversity determines the invasion of soil by a bacterial pathogen. Proc Natl Acad Sci USA 109: 1159–1164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venturelli OS, Carr AV, Fisher G, Hsu RH, Lau R, Bowen BP, Hromada S, Northen T, Arkin AP (2018) Deciphering microbial interactions in synthetic human gut microbiome communities. Mol Syst Biol 14: e8157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Garrity GM, Tiedje JM, Cole JR (2007) Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73: 5261–5267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Xu M, Wang W, Cao X, Piao M, Khan S, Yan F, Cao H, Wang B (2016) Systematic review: adverse events of fecal microbiota transplantation. PLoS One 11: e0161174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson L, Zimbric ML, Shaughnessy C, Kale S, Debad J, Navaratnam M, Safdar N (2019) 2359. Prospective feasibility study for novel ultrasensitive multiplexed immunoassay for Clostridiodes difficile toxins A and B. Open Forum Infect Dis 6: S812–S813 [Google Scholar]
- Wei Z, Yang T, Friman V‐P, Xu Y, Shen Q, Jousset A (2015) Trophic network architecture of root‐associated bacterial communities determines pathogen invasion and plant health. Nat Commun 6: 8413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wetzel D, McBride SM (2020) The impact of pH on Clostridioides difficile sporulation and physiology. Appl Environ Microbiol 86: e02706–e2719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright ES, Vetsigian KH (2016) Inhibitory interactions promote frequent␣bistability among competing bacteria. Nat Commun 7: 11274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuille S, Mackay WG, Morrison DJ, Tedford MC (2020) Drivers of Clostridioides difficile hypervirulent ribotype 027 spore germination, vegetative cell growth and toxin production in␣vitro. Clin Microbiol Infect 26: 941.e1–941.e7 [DOI] [PubMed] [Google Scholar]
- Zhang J, Kobert K, Flouri T, Stamatakis A (2014) PEAR: a fast and accurate illumina paired‐End reAd mergeR. Bioinformatics 30: 614–620 [DOI] [PMC free article] [PubMed] [Google Scholar]