

Central Illustration
Key Words: amyloidosis, heart failure, treatment
Abbreviations and Acronyms: ATTR-wt, wild-type transthyretin subtype; ATTR-v, variant (hereditary) transthyretin subtype; AL, light chain subtype; ASCT, autologous stem cell transplantation; MCS, mechanical circulatory support
Highlights
-
•
Cardiac transplantation for amyloidosis was once considered contraindicated owing to unacceptably high morbidity/mortality rates.
-
•
Increased therapeutic options for AL and ATTR amyloidosis and improved pre-transplantation screening practices have led to markedly improved transplant outcomes over the past 10-15 years.
-
•
Mechanical circulatory support options remain limited but can be considered in selected patients, particularly for those with larger ventricular cavities.
-
•
Transplant prioritization rules may need to be reconsidered for amyloidosis patients to adequately prioritize AL amyloidosis patients, who are at increased risk of pre-transplantation mortality.
Although cardiac transplantation has been performed for patients with end-stage cardiac amyloidosis for decades, only in recent years has it garnered widespread acceptance. This primer will review the evolution of outcomes in patients with amyloidosis, outline special considerations in the amyloidosis population, and explore the role of mechanical circulatory support (MCS).
Evolution of Transplant Outcomes
In the 1980s and 1990s, cardiac transplant outcomes in amyloidosis were poor, with survival consistently worse than in the nonamyloidosis transplant population (Table) (1, 2, 3). Several factors contributed to these poor outcomes:
-
•
Most transplantations were for light chain (AL) amyloidosis, which has a higher risk of recurrent amyloid deposition in the transplanted heart.
-
•
Patients were often transplanted with significant extracardiac organ involvement, and would often die from complications of multiorgan disease.
-
•
Chemotherapy options were very limited, typically to alkylator/steroid combinations. Most patients were unable to achieve good long-term pathologic light chain control, and progressive amyloidosis in the transplanted heart (or in other vital organs) was common.
Table 1.
Cardiac Transplantation Outcomes in Amyloidosis
| Institution or Country (Ref. #) | Years | n (Population) | 1-Year Survival, % | 5-Year Survival, % | Median Survival, y | Comments |
|---|---|---|---|---|---|---|
| UK Natl Amyloidosis Centre (21) | 1984-2009 | 14 (all AL) | 86 | 45 | 7.5 | |
| Mayo (7) | 1994-2005 | 11 (all AL) | 82 | 65 | 6.3 | All underwent SCT; 2 died from complications of the SCT, 3 died from progressive amyloidosis |
| Mayo (13) | 1992-2011 | 23 (all AL) | 77 | 43 | 3.5 | 5-y survival less than for nonamyloid patients (43% vs 85%); 12/20 deaths from progressive amyloidosis |
| Mayo (12) | 2007-2015 | 7 (all ATTR-wt) | 100 | NR | NR | Reported 1 nonamyloid-related death at 3.8 y |
| Columbia (15) | 1997-2004 | 12 (10 AL, 2 ATTR-v) |
75 | NR | NR | Short-term survival not different from nonamyloid patients transplanted using extended donor criteria |
| Columbia (5) | 2001-2018 | 39 (18 AL, 16 ATTR-v, 5 ATTR-wt) |
Era 1: 75 AL, 100 ATTR; Era 2: 100 AL and ATTR |
Era 1: 33 AL, 67 ATTR; Era 2: 100 AL and ATTR |
NR | Survival worse than nonamyloid patients in Era 1, similar in Era 2 |
| Stanford (9) | 2004-2017 | 31 (13 AL, 18 ATTR) |
92 | 92 | NR | No differences in survival between amyloid and nonamyloid patients |
| Cedars-Sinai (10) | 2010-2018 | 46 (12 AL, 34 ATTR) |
91 (83 AL, 94 ATTR) |
NR | NR | After 3.7 y mean follow-up, 76% survival; 7 patients transplanted after MCS device bridging |
| UK (1) | 1982-2002 | 24 (17 AL, 3 ATTR-v, 2 ATTR-wt, 2 ApoA1) |
63 | 38 | 2.4 | Nonamyloid 5-y survival 67%; survival greater for non-AL vs AL |
| France (22) | 2001-2006 | 8 (AL) | 86 | NR | NR | 75% alive after median 2.2 y follow-up |
| Germany (23) | 2001-2007 | 12 | 83 | NR | NR | 1- and 3-y survival rates similar to nonamyloid transplant patients |
| Germany (4) | 2002-2017 | 48 (32 AL, 16 ATTR) |
Era 1: 69 AL, 75 ATTR; Era 2: 85 AL, 75 ATTR |
Era 1: 31 AL, 50 ATTR; Era 2: 77 AL, 75 ATTR |
NR | Median survival 61% after 3.5 y; Survival worse than nonamyloid patients in Era 1, but equivalent in Era 2 |
| Spain (3) | 1984-2008 | 25 (13 AL, 10 ATTR-v, 2 AA) |
62 | 36 | NR | 5-y survival significantly worse than nonamyloid patients (36% vs 64%) |
| USA (2) | 1987-2002 | 69 | 75 | 54 | NR | Nonamyloid transplant patients survived longer (P = 0.03) |
| USA (24) | 1987-2010 | 142 | 79 | 47 | NR | Decreased survival vs nonamyloid transplants and nonamyloid restrictive cardiomyopathy transplants |
| USA (6) | 1987-2013 | 188 | NR | NR | NR | Mortality hazard ratios 2.08 in Era 1 vs other RCM and 1.84 vs all other diagnoses; no significant difference vs RCM or all other diagnoses in Era 2 |
| USA (11) | 1987-2018 | 313 | NR | NR | 10.2 | Median survival shorter for amyloid (10.2 y) vs nonamyloid (12.5 y); did not include multiorgan transplant recipients |
AL = light chain subtype; ApoA1 = apolipoprotein A1 subtype; ATTR-wt = wild-type transthyretin subtype; ATTR-v = variant (hereditary) transthyretin subtype; Era 1 = before 2008; Era 2 = since 2008; NR = not reported; RCM = restrictive cardiomyopathy; SCT = stem cell transplantation.
Although transplant rates slowly increased in the early 2000s, outcomes remained generally poor, and chemotherapy options for AL amyloidosis had not significantly changed. The landscape evolved considerably in the late 2000s, with 2007-2008 often regarded as a cutoff between an older “era 1” and a newer “era 2” (4, 5, 6) based on 2 factors:
-
•
Availability of new chemotherapy options for light chain control in AL amyloidosis, including bortezomib and lenalidomide.
-
•
Publication of a protocol emphasizing both extensive pre-transplantation screening for extracardiac organ involvement and aggressive post-transplantation plasma cell–directed therapy, typically including autologous stem cell transplantation (ASCT) (7). Other centers subsequently adopted similar approaches (5,8,9).
By the 2010s, 2 factors led to further improvements in outcomes:
-
•
Increased diagnosis of transthyretin (ATTR) amyloidosis, driven by the ability to make a noninvasive diagnosis with the use of bone scintigraphy and by newly approved therapeutic options (tafamidis, patisiran, inotersen).
-
•
Rapidly expanding options for plasma cell–directed therapies in AL amyloidosis (eg, daratumumab).
With these advances, transplant outcomes have markedly improved, now approaching or equaling nonamyloidosis transplants in multiple studies (Table 1) (4, 5, 6,8, 9, 10, 11, 12). At the same time, the frequency of transplants for amyloidosis has increased both in absolute numbers and as a percentage of total transplantations performed in the United States (0.3% in era 1 versus 1.2% in era 2) (6).
Special Considerations in Amyloidosis
Several considerations are critical for successful cardiac transplantation in amyloidosis, largely depending on the amyloidosis subtype.
For wild-type ATTR amyloidosis (ATTR-wt), there are no additional considerations for most patients, given the lack of other vital organ involvement typical of the disease, although cardiac transplantation is not a realistic option for most patients due to advanced patient age.
In variant/hereditary ATTR amyloidosis (ATTR-v), the situation is more complex and varies markedly by genotype. For genotypes that cause a predominantly “mixed” phenotype of cardiomyopathy and neuropathy (eg, V30M, T60A), cardiac transplantation alone will not prevent a progressive disabling polyneuropathy. For those patients, concomitant liver transplantation can be considered, with the goal being the removal of the source of the variant transthyretin protein. However, in the present era of effective pharmacologic therapies (eg, patisiran, inotersen), it is unclear if liver transplantation is still needed—versus an alternative approach of cardiac transplantation plus pharmacologic therapy. Before cardiac transplantation is performed in patients with mutations associated with mixed-phenotype disease, it is important to perform a thorough neurologic evaluation to exclude clinically significant neuropathy. Further evaluations may be needed for patients with mutations characterized by other organ involvement, such as gastrointestinal evaluation in patients with the T60A mutation.
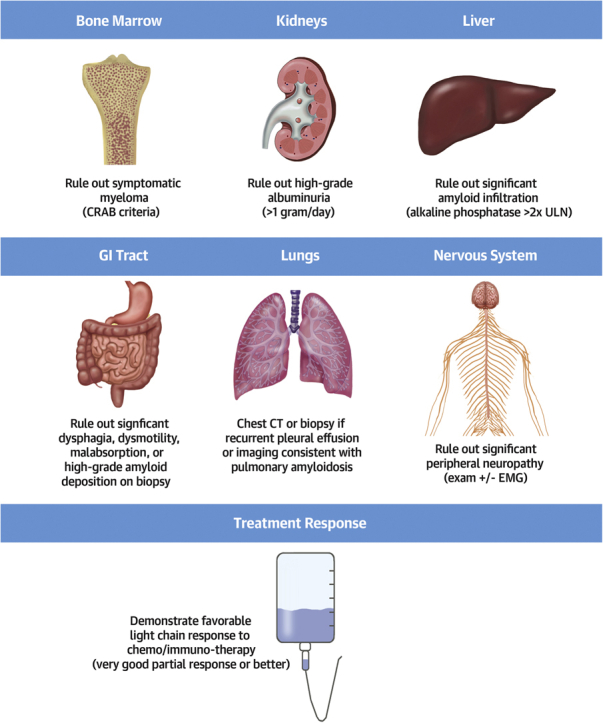
Because multiorgan amyloid infiltration is a common characteristic of AL amyloidosis, it is crucial to screen for the presence of significant extracardiac organ involvement. While thresholds of “too much” involvement vary among institutions, the principle of extensive screening is widely accepted, with several protocols published by large amyloid centers (Figure 1) (7, 8, 9, 10). Because progressive amyloid deposition is one of the most common causes of death after transplantation, it is important to show an adequate hematologic response to light chain suppressive therapy before transplantation whenever possible.
Figure 1.
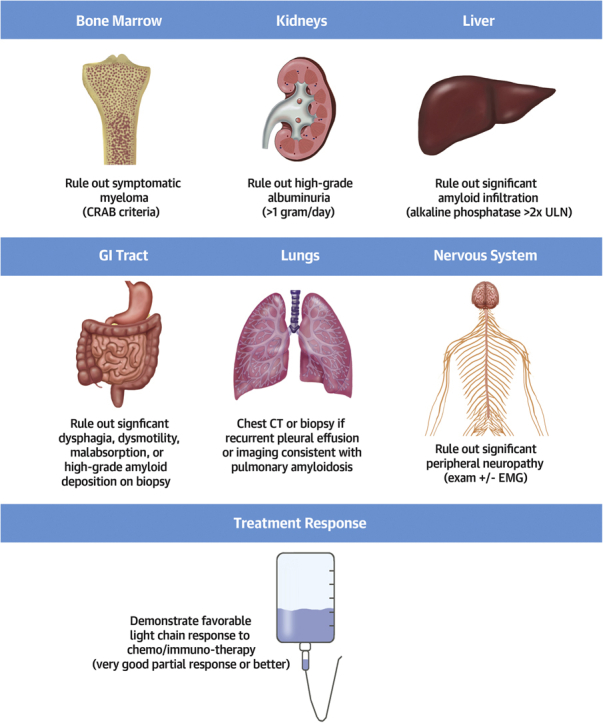
Assessment of Extracardiac Organs During Transplant Evaluation for AL Amyloid Cardiomyopathy
Very good partial response is defined as the difference between involved and uninvolved free light chain <40 mg/L. CRAB = hypercalcemia, renal dysfunction, anemia, destructive bone lesions; CT = computed tomography; EMG = electromyography; ULN = upper limit of normal.
Post-transplantation Chemotherapy/Immunotherapy
An extra consideration for patients transplanted for AL amyloidosis is the interaction of plasma cell–directed therapy with post-transplantation immunosuppression and rejection risks. Although there is no clear signal in published case series (Table 1), concomitant therapy with chemotherapy would be expected to raise the risk of infection. At centers that routinely use ASCT after cardiac transplantation, complications (including infection) have contributed to mortality in some series (7,13).
Treatment with light chain–directed therapies may serve to decrease the risk of rejection owing to their effects on immunoglobulin production. Indeed, the proteasome inhibitor bortezomib has been used in both the treatment of antibody-mediated rejection and for antibody desensitization before transplantation in patients without amyloidosis. On the other hand, treatment with the anti–plasma cell immunomodulatory agent lenalidomide (and others in the “imid” class) has been temporally associated with rejection episodes in multiple case reports and should be avoided after transplantation if possible (14).
Although the importance of effective light chain control after cardiac transplantation in AL amyloidosis is widely acknowledged, the best means of achieving control is unclear. During era 1, when chemotherapy/immunotherapy options were limited, ASCT was typically the standard (7,13,15). With increased options available for light chain control (eg, daratumumab) and the nontrivial ASCT-related mortality rates reported, other centers have moved away from ASCT as a standard, reserving it for patients who have inadequate light chain control with other approaches (9).
Mortality on the Transplant Waiting List
One important consideration for cardiac amyloidosis patients—particularly AL amyloidosis patients—is an extremely high mortality rate while on the transplant waiting list. Columbia University reported a 40% mortality rate while on the waiting list, and Massachusetts General Hospital reported a risk of death of 24% per month, 4.7 times the rate for nonamyloidosis patients (15,16). This high mortality rate was recognized in the 2018 changes to the heart allocation system by granting amyloidosis patients listed for transplantation a higher status (status 4) than most other patients; notably, there is no differentiation made between AL and ATTR amyloidosis for this criteria, and amyloidosis is grouped with hypertrophic and other restrictive cardiomyopathies (17). Early results from these changes reveal higher transplantation rates and lower waiting list mortality for the infiltrative cardiomyopathy population (5).
Mechanical Circulatory Support
Patients with cardiac-amyloidosis have multiple challenges for successful durable MCS. Factors include:
-
•
Small ventricular cavities leading to difficult inflow cannula placement and high risks for suction events.
-
•
Biventricular dysfunction leading to a high risk for right ventricular failure if left ventricular support devices are used alone.
-
•
Higher risk for infection for AL amyloidosis patients on active chemotherapy/immunotherapy.
Despite these limitations, MCS can be an option for selected patients. One study reported outcomes of 28 patients with restrictive cardiomyopathies who received left ventricular assist devices, including 10 patients with cardiac amyloidosis (1 AL, 9 ATTR) (18). Mean survival was reported to be 536 days, with the notable difference that patients with larger left ventricular cavities (left ventricular end-diastolic diameter >46 mm) had markedly longer survival, suggesting that they may be a subset of cardiac amyloidosis patients who may benefit from MCS. Another single-institution study evaluated 11 amyloidosis patients who received durable MCS as a bridge to transplant. In that cohort, all patients received biventricular support (total artificial heart or biventricular assist devices), and 4 received extracorporeal membrane oxygenation as a bridge to MCS; by 1 year, 9 patients had been transplanted and 2 had died (19). Recent INTERMACS data suggest that MCS outcomes in amyloid cardiomyopathy are worse than for dilated and other restrictive cardiomyopathies—particularly with left ventricle–only support—with higher rates of complications including gastrointestinal bleeding, renal dysfunction, and neurologic dysfunction (20). Overall, despite limited success, the optimal use of MCS in this population remains to be better defined.
Conclusions
Though cardiac transplantation poses unique challenges in systemic amyloidosis, tremendous improvements have been made over the past decade. With careful patient selection, and with a focus on effective plasma cell–directed therapies before and after transplantation, outcomes in multiple institutional case series and national transplant databases have improved, approaching parity with outcomes for patients transplanted for other indications. With the growing numbers of patients diagnosed with ATTR amyloidosis, and with continued improvement in pharmacologic therapy options for both AL and ATTR amyloidosis, cardiac transplantation for amyloidosis is likely to become increasingly common in the coming years.
Funding Support and Author Disclosures
Dr Witteles has served on advisory boards for Pfizer, Alnylam, Eidos, Ionis/Akcea, and Regeneron.
Footnotes
Anju Nohria, MD, served as Guest Editor for this paper.
The author attests they are in compliance with human studies committees and animal welfare regulations of the author’s institution and Food and Drug Administration guidelines, including patient consent where appropriate. For more information, visit the Author Center.
References
- 1.Dubrey S.W., Burke M.M., Hawkins P.N., Banner N.R. Cardiac transplantation for amyloid heart disease: the United Kingdom experience. J Heart Lung Transplant. 2004;23:1142–1153. doi: 10.1016/j.healun.2003.08.027. [DOI] [PubMed] [Google Scholar]
- 2.Kpodonu J., Massad M.G., Caines A., Geha A.S. Outcome of heart transplantation in patients with amyloid cardiomyopathy. J Heart Lung Transplant. 2005;24:1763–1765. doi: 10.1016/j.healun.2004.08.025. [DOI] [PubMed] [Google Scholar]
- 3.Roig E., Almenar L., Gonzalez-Vilchez F. Outcomes of heart transplantation for cardiac amyloidosis: subanalysis of the Spanish registry for heart transplantation. Am J Transplant. 2009;9:1414–1419. doi: 10.1111/j.1600-6143.2009.02643.x. [DOI] [PubMed] [Google Scholar]
- 4.Kristen A.V., Kreusser M.M., Blum P. Improved outcomes after heart transplantation for cardiac amyloidosis in the modern era. J Heart Lung Transplant. 2018;37:611–618. doi: 10.1016/j.healun.2017.11.015. [DOI] [PubMed] [Google Scholar]
- 5.Griffin J.M., Chiu L., Axsom K.M. United Network for Organ Sharing outcomes after heart transplantation for AL compared to ATTR cardiac amyloidosis. Clin Transplant. 2020;34:e14028. doi: 10.1111/ctr.14028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Davis M.K., Lee P.H., Witteles R.M. Changing outcomes after heart transplantation in patients with amyloid cardiomyopathy. J Heart Lung Transplant. 2015;34:658–666. doi: 10.1016/j.healun.2014.09.006. [DOI] [PubMed] [Google Scholar]
- 7.Lacy M.Q., Dispenzieri A., Hayman S.R. Autologous stem cell transplant after heart transplant for light chain (AL) amyloid cardiomyopathy. J Heart Lung Transplant. 2008;27:823–829. doi: 10.1016/j.healun.2008.05.016. [DOI] [PubMed] [Google Scholar]
- 8.Davis M.K., Kale P., Liedtke M. Outcomes after heart transplantation for amyloid cardiomyopathy in the modern era. Am J Transplant. 2015;15:650–658. doi: 10.1111/ajt.13025. [DOI] [PubMed] [Google Scholar]
- 9.Barrett C.D., Alexander K.M., Zhao H. Outcomes in patients with cardiac amyloidosis undergoing heart transplantation. J Am Coll Cardiol HF. 2020;8:461–468. doi: 10.1016/j.jchf.2019.12.013. [DOI] [PubMed] [Google Scholar]
- 10.Chen Q., Moriguchi J., Levine R. Outcomes of heart transplantation in cardiac amyloidosis patients: a single center experience. Transplant Proc. 2021;53:329–334. doi: 10.1016/j.transproceed.2020.08.020. [DOI] [PubMed] [Google Scholar]
- 11.Ohiomoba R.O., Youmans Q.R., Ezema A. Cardiac transplantation outcomes in patients with amyloid cardiomyopathy. Am Heart J. 2021;236:13–21. doi: 10.1016/j.ahj.2021.02.016. [DOI] [PubMed] [Google Scholar]
- 12.Rosenbaum A.N., AbouEzzeddine O.F., Grogan M. Outcomes after cardiac transplant for wild type transthyretin amyloidosis. Transplantation. 2018;102:1909–1913. doi: 10.1097/TP.0000000000002240. [DOI] [PubMed] [Google Scholar]
- 13.Grogan M., Gertz M., McCurdy A. Long term outcomes of cardiac transplant for immunoglobulin light chain amyloidosis: the Mayo Clinic experience. World J Transplant. 2016;6:380–388. doi: 10.5500/wjt.v6.i2.380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Qualls D.A., Lewis G.D., Sanchorawala V., Staron A. Orthotopic heart transplant rejection in association with immunomodulatory therapy for AL amyloidosis: a case series and review of the literature. Am J Transplant. 2019;19:3185–3190. doi: 10.1111/ajt.15499. [DOI] [PubMed] [Google Scholar]
- 15.Maurer M.S., Raina A., Hesdorffer C. Cardiac transplantation using extended-donor criteria organs for systemic amyloidosis complicated by heart failure. Transplantation. 2007;83:539–545. doi: 10.1097/01.tp.0000255567.80203.bd. [DOI] [PubMed] [Google Scholar]
- 16.Gray Gilstrap L., Niehaus E., Malhotra R. Predictors of survival to orthotopic heart transplant in patients with light chain amyloidosis. J Heart Lung Transplant. 2014;33:149–156. doi: 10.1016/j.healun.2013.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Organ Procurement and Transplantation Network US Department of Health and Human Services. Criteria requirements in adult heart allocation policy. https://optn.transplant.hrsa.gov/media/2413/adult_heart_criteria.pdf
- 18.Grupper A., Park S.J., Pereira N.L. Role of ventricular assist therapy for patients with heart failure and restrictive physiology: improving outcomes for a lethal disease. J Heart Lung Transplant. 2015;34:1042–1049. doi: 10.1016/j.healun.2015.03.012. [DOI] [PubMed] [Google Scholar]
- 19.Kittleson M.M., Cole R.M., Patel J. Mechanical circulatory support for cardiac amyloidosis. Clin Transplant. 2019;33:e13663. doi: 10.1111/ctr.13663. [DOI] [PubMed] [Google Scholar]
- 20.Michelis K.C., Zhong L., Tang W.H.W. Durable mechanical circulatory support in patients with amyloid cardiomyopathy: insights from INTERMACS. Circ Heart Fail. 2020;13:e007931. doi: 10.1161/CIRCHEARTFAILURE.120.007931. [DOI] [PubMed] [Google Scholar]
- 21.Sattianayagam P.T., Gibbs S.D., Pinney J.H. Solid organ transplantation in AL amyloidosis. Am J Transplant. 2010;10:2124–2131. doi: 10.1111/j.1600-6143.2010.03227.x. [DOI] [PubMed] [Google Scholar]
- 22.Mignot A., Varnous S., Redonnet M. Heart transplantation in systemic (AL) amyloidosis: a retrospective study of eight French patients. Arch Cardiovasc Dis. 2008;101:523–532. doi: 10.1016/j.acvd.2008.06.018. [DOI] [PubMed] [Google Scholar]
- 23.Kristen A.V., Sack F.U., Schonland S.O. Staged heart transplantation and chemotherapy as a treatment option in patients with severe cardiac light-chain amyloidosis. Eur J Heart Fail. 2009;11:1014–1020. doi: 10.1093/eurjhf/hfp121. [DOI] [PubMed] [Google Scholar]
- 24.DePasquale E.C., Nasir K., Jacoby D.L. Outcomes of adults with restrictive cardiomyopathy after heart transplantation. J Heart Lung Transplant. 2012;31:1269–1275. doi: 10.1016/j.healun.2012.09.018. [DOI] [PubMed] [Google Scholar]