

Abstract
Background
Cardiac amyloidosis (CA) has been associated with poor outcomes. Screening studies suggest that CA is overlooked—especially in the elderly. Recent advances in treatment have brought attention to the disease, but data on temporal changes in CA epidemiology are sparse.
Objectives
The aim of this work was to describe all patients with CA in Denmark, examining changes in patient characteristics from 1998 to 2017.
Methods
All patients with any form of amyloidosis diagnosed from 1998 to 2017, as well as their comorbidities and pharmacotherapy, were identified in Danish nationwide registries. CA was defined as any diagnosis code for amyloidosis combined with a diagnosis code for heart failure, cardiomyopathy, or atrial fibrillation or a procedural code for pacemaker implantation, regardless of the order. The index date was defined as the date of meeting those criteria. Patients were divided into 5-year periods by index date. For comparison, we also included control subjects (1:4 ratio) from the general population.
Results
CA criteria were met by 619 patients. Comparing 1998-2002 vs 2013-2017, the median age at baseline increased from 67.4 years (interquartile range [IQR]: 53.9-75.2 years) to 72.3 years (IQR: 66.0-79.3 years). The frequency of male patients increased from 62.1% to 66.2%. The incidence of CA rose from 0.88 to 3.56 per 100,000 person-years in the Danish population aged ≥65 years, and the 2-year mortality decreased from 82.6% (IQR: 69.9%-90.5%) to 50.2% (IQR: 43.1%-56.9%). Compared with control subjects, the mortality among CA patients was significantly higher (log-rank test: P < 0.0001).
Conclusions
CA, as defined in this study, was increasingly diagnosed on a national scale. The increasing frequency of male patients and median age suggest that wild-type transthyretin amyloidosis is driving this increase. Greater recognition of earlier, less advanced cases might explain decreasing mortality.
Key Words: cardiac amyloidosis, epidemiology, heart failure, outcomes, temporal changes
Abbreviations and Acronyms: AL, light chain amyloid; ATTR, transthyretin amyloid; CA, cardiac amyloidosis; CTS, carpal tunnel syndrome; wtATTR, wild-type amyloid transthyretin
Central Illustration

Cardiac amyloidosis (CA) has been associated with a high 5-year mortality of 44%-65% after diagnosis (1). Screening studies show higher prevalence of occult CA than previously expected, indicating that the burden of CA is underestimated (2, 3, 4, 5, 6). Recent advances in diagnostic modalities, such as bone tracer scintigraphy, cardiac magnetic resonance imaging, and mass spectrometry (7, 8, 9), as well as state-of-the-art pharmacotherapy have brought increased attention to identifying patients with transthyretin amyloidosis (10). Indeed, the possibility of noninvasive diagnosis and emergence of novel specific therapies are beginning to shift the perception of the disease from rare and incurable to less rare and treatable (11,12). Advances in immunomodulating drugs, proteasome inhibitors, steroid therapy, and autologous stem cell transplantation have significantly improved survival in light chain (AL) amyloidosis, especially if initiated early (13,14). Indeed, in 2021, daratumumab (in combination with bortezomib, cyclophosphamide, and dexamethasone) was approved by the U.S. Food and Drug Administration as first-line therapy for newly diagnosed AL amyloidosis after studies showed rapid hematologic and organ responses (15).
CA patients who are diagnosed often experience a considerable diagnostic delay (16). Receiving the correct diagnosis is important not just to receive disease-modifying therapy, but because patients with CA poorly tolerate many of the drugs commonly used in the treatment of heart failure and atrial fibrillation, resulting in possible worsening of symptoms or serious adverse reactions (17). In 2019, Gilstrap et al (18) published the first contemporary estimate of the incidence and prevalence of CA among Medicare beneficiaries in the United States, concluding that in hospitalized patients >65 years of age, the incidence rate of CA was 17 per 100,000 person-years and the prevalence rate was 55 per 100,000 person-years. However, no unselected national data exist. Our objective was therefore to describe the temporal trends in a Danish population of CA patients and to examine the changes in patient characteristics over the past 2 decades.
Methods
Regulatory approval
The study complies with the Declaration of Helsinki. Registry studies do not require regulatory approval in Denmark. The study received the mandated approval by the Danish Data Protection Agency (approval no.: P-2019-348).
Data sources
Data were extracted from Danish national registries, where all inpatient and outpatient diagnoses, admissions, procedures, and filled prescriptions, as well as income and municipality of residence, are registered. The registries have been validated and previously described in detail (19, 20, 21, 22).
Study patients
All patients diagnosed with amyloidosis from 1998 to 2017 were assessed for inclusion in the study. CA was defined as any International Classification of Diseases-10th Revision (ICD-10) diagnosis code for amyloidosis, combined with 1 possible cardiac manifestation of amyloidosis (diagnosed heart failure, cardiomyopathy, or atrial fibrillation or procedural code for pacemaker implantation), whichever was registered first. The ICD-10 codes used are listed in the Supplemental Appendix. Index date was defined as the date when the patient met both of these criteria (amyloidosis and a relevant cardiovascular diagnosis). This definition of CA has been used previously by Gilstrap et al (18), with the addition of atrial fibrillation and pacemaker implantation, as suggested in the subsequent editorial by Griffin et al (23).
For comparison, we also included control subjects from the general population in a 1:4 ratio. The control subjects were assigned the same index dates as cases (with the requirement of being alive and living in Denmark at the time) and were thereafter matched by sex, age (±1 year), and index year, with the use of risk-set matching. To assess differences over time, the 20-year study period was divided into four 5-year periods. Furthermore, the population of amyloidosis patients who did not meet criteria for CA were analyzed as well. The index date for patients with noncardiac amyloidosis was defined as the date of diagnosed amyloidosis.
Study covariates
Preexisting comorbidities were assessed via hospital admissions and outpatient diagnoses from 10 years before the index date. Concomitant pharmacotherapy was assessed through filled prescriptions 6 months before the index date. The ICD-10 and Anatomical Therapeutic Chemical Classification System codes used are listed in the Supplemental Appendix.
Statistics
Descriptive statistics were presented as percentages for categoric variables and as median (interquartile range [IQR]) for continuous variables. Differences between CA patients and control subjects were tested by means of chi-square test and Student's t-test where appropriate. Incidences of CA were calculated with the use of official annual population data from Statistics Denmark, whereby the entire Danish population is accounted for. The incidences of CA were calculated for each 5-year period of the study and tested for trend using the Cochrane-Armitage test for trend, adjusted for population growth per each time period. Mortality was described with the use of Kaplan-Meier estimates, comparing cumulative incidences of mortality over time periods and between patient groups with the use of log-rank test.
Statistical analyses were performed with the use of the SAS statistical software (version 9.4) as well as R statistical software (R Core Team). Level of statistical significance was defined as a P < 0.05.
Results
A total of 1,572 patients were diagnosed with amyloidosis during the study period, of whom 619 met the criteria for CA. Figure 1 shows a flow chart for patient selection. Matching in a 1:4 ratio to patients from the general population yielded 2,476 control subjects. The majority (63%) of CA patients were coded as unspecified amyloidosis, while hereditary forms accounted for about 10% (Supplemental Table 1 details further distribution of amyloidosis ICD codes among the study population). Approximately 62% of the CA patients were first diagnosed with heart failure, cardiomyopathy, or atrial fibrillation or underwent pacemaker implantation before being diagnosed with amyloidosis and 7% were diagnosed with a cardiac manifestation and amyloidosis the same day.
Figure 1.

Flow Chart of Patient Selection
All patients with diagnosed amyloidosis from 1998 to 2017 were identified in Danish nationwide registries. Cardiac amyloidosis (CA) was defined using a combination of International Classification of Diseases-10th Revision diagnosis and procedural codes. A total of 1,572 amyloidosis patients were identified, ∼40% of which met criteria for CA. CA, as defined in this study, is increasingly being diagnosed in Denmark.
Temporal changes in age, sex, and diagnosis
During each 5-year period of the study, the number of patients who were diagnosed with CA according to our study definition continuously increased. This finding was consistent when adjusted for population growth (Cochran-Armitage trend test: P < 0.0001). The incidence of CA among the entire Danish population ≥65 years of age rose from 0.88 to 3.56 per 100,000 person-years and from 1.21 to 5.19 per 100,000 person-years in men ≥65 years of age (Figure 2). The median age at baseline increased from 67.4 years (IQR: 53.9-75.2 years) in the first evaluated time period (1998-2002) to 72.3 years (IQR: 66.0-79.3 years) in the last time period (2013-2017). The median age at time of diagnosis of amyloidosis increased from 67.4 years (IQR: 53.9-75.1 years) in the first time period to 71.6 years (IQR: 64.7-79.0 years) in the last time period. For the patients who presented with a cardiac manifestation before being diagnosed with amyloidosis, the median time between first cardiac manifestation and a subsequently diagnosis of amyloidosis varied between 0.63 years (IQR: 0.20-2.17 years) and 1.95 years (IQR: 0.24-5.04 years). Over time, the frequency of male patients rose from 62.1% to 66.2%. Among patients aged ≥75 years, the frequency of male patients increased from 50% to 72.7%. The frequency of outpatient diagnosis of amyloidosis increased from 25.9% to 51.7%.
Figure 2.
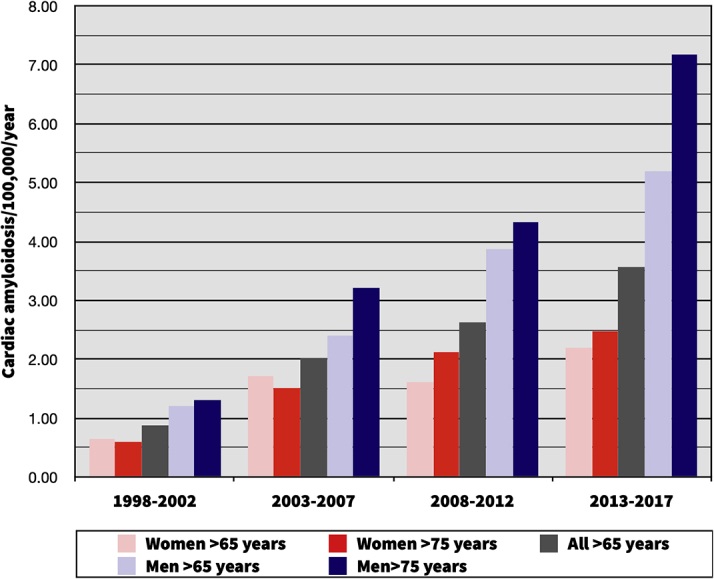
Temporal Changes in Cardiac Amyloidosis
Investigating the changes in incidence of cardiac amyloidosis (CA). The incidence of CA was calculated for each 5-year period of the study, adjusted for population growth per each time period. Between the first and last 5-year period (1998-2002 vs 2013-2017) the incidence of CA increased from 0.88 to 3.56 per 100,000 person-years in the entire Danish population, from 1.21 to 5.19 in men aged ≥65 years and from 1.30 to 7.18 in men aged ≥75 years. The increasing incidence of CA was most pronounced among elderly men, indicating that wild-type transthyretin amyloidosis is driving this rise.
Temporal changes in comorbidities and pharmacotherapy
Comorbidities and concomitant pharmacotherapy generally increased over time, as presented in Table 1 and Supplemental Tables 2 and 3, respectively. The prevalence of atrial fibrillation increased from 25.9% to 32.5% among CA patients, while concomitant anticoagulation therapy increased from 12.1% to 37.2%. Treatment with beta-blockers doubled (25.9% to 46.2%), while treatment with nondihydropyridine calcium channel blockers and digoxin decreased from 13.8% to 1.3% and from 29.3% to 5.1%, respectively.
Table 1.
Baseline Characteristics
| Total | Time Period |
||||
|---|---|---|---|---|---|
| 1998-2002 | 2003-2007 | 2008-2012 | 2013-2017 | ||
| Demographics | |||||
| n | 619 | 58 | 144 | 183 | 234 |
| Male | 63.0 | 62.1 | 59.0 | 62.3 | 66.2 |
| Age at first cardiac manifestation, y | 68.2 (59.9-75.8) | 66.3 (53.8-74.6) | 65.7 (57.2-74.4) | 67.4 (59.7-75.9) | 70.4 (63.2-76.6) |
| Age at amyloidosis diagnosis, y | 69.9 (61.5-77.1) | 67.4 (53.9-75.1) | 66.0 (58.0-76.8) | 68.9 (61.2-76.3) | 71.6 (64.7-79.0) |
| Age at baseline, y | 70.0 (63.0-77.8) | 67.4 (53.9-75.2) | 66.9 (59.4-77.1) | 70.5 (61.4-76.7) | 72.3 (66.0-79.3) |
| Cardiac manifestation or amyloidosis first diagnosed? | |||||
| Cardiac manifestationa | 62.0 | 65.5 | 56.3 | 60.7 | 65.8 |
| Time between first cardiac manifestation and subsequently diagnosed amyloidosis, yb | 1.26 (0.20-4.70) | 0.63 (0.20-2.17) | 1.95 (0.24-5.04) | 0.92 (0.12-4.78) | 1.39 (0.24-5.03) |
| Comorbidities | |||||
| Ischemic heart disease | 24.9 | 22.4 | 25.0 | 27.9 | 23.1 |
| Acute myocardial infarction | 7.6 | 5.2 | 7.6 | 11.5 | 5.1 |
| Heart failure | 29.9 | 29.3 | 25.7 | 28.4 | 33.8 |
| Atrial fibrillation | 29.7 | 25.9 | 25.7 | 30.6 | 32.5 |
| Hypertension | 28.1 | 17.2 | 25 | 29 | 32.1 |
| Diabetes | 14.1 | 10.3 | 13.2 | 13.1 | 16.2 |
| Stroke | 7.3 | 6.9 | 3.5 | 9.3 | 8.1 |
| Chronic obstructive pulmonary disease | 9.4 | 6.9 | 6.9 | 9.3 | 11.5 |
| Chronic renal failure | 27.0 | 17.2 | 27.1 | 31.2 | 26.1 |
| MGUSc | 5.8 | 3.5 | 6.3 | 3.8 | 7.7 |
| Multiple myeloma | 17.3 | 15.5 | 16.0 | 17.5 | 18.4 |
| Treatment with melphalan ± concomitant corticosteroids, bortezomib, thalidomide, or daratumumab or received stem cell conditioning | 15.4 | 0 | 4.9 | 17.5 | 23.9 |
| Suspected amyloid light chain amyloidosisd | 23.4 | 15.5 | 18.8 | 26.8 | 25.6 |
| Education | |||||
| Masters/doctoral degree or equivalent tertiary education level (ISCED 7-8) | 3.1 | 1.7 | 2.8 | 3.8 | 3 |
| Income | |||||
| Highest quartile incomee | – | 24.1 | 27.1 | 25.7 | 23.1 |
| Inpatient vs outpatient diagnosis of amyloidosis | |||||
| Inpatient | 55.1 | 67.2 | 69.4 | 49.2 | 47.9 |
| Outpatient | 43.1 | 25.9 | 27.8 | 49.7 | 51.7 |
Values are n, median (interquartile range), or %, unless otherwise noted.
ISCED = International Standard Classification of Education.
Cardiac manifestation = heart failure, cardiomyopathy, atrial fibrillation, or pacemaker implantation.
Calculated only for those in whom cardiac manifestation was first.
Monoclonal gammopathy of unknown significance. Present at baseline, even if >10 years previous.
Defined as multiple myeloma and/or treatment with melphalan ± concomitant corticosteroids, bortezomib, thalidomide, or daratumumab or received stem cell conditioning.
Calculated using mean household income during the 5-year period before baseline.
Temporal changes in hematologic cancer and associated treatment
The frequency of concurrent malignancy increased over time. Although the frequency of multiple myeloma (MM), as well as monoclonal gammopathy of unknown significance (MGUS), was quite stable, treatment with melphalan ± concomitant corticosteroids, bortezomib, thalidomide, daratumumab. or stem cell transplantation rose from 0% to 23.9% during the study period, as presented in Table 1. The frequencies of MGUS and MM among control subjects were very low (Supplemental Table 4).
Extracardiac manifestations of amyloidosis
Previous surgery for carpal tunnel syndrome (CTS) was reported in 10.6% of patients with CA, compared with 1.8% of control subjects (Supplemental Table 5). Multiple CTS surgical procedures were more common among CA patients with CTS than in control subjects with CTS (59.2% vs 33.3%; P = 0.003). The median time from CTS surgery to the index date was 3.5 years (IQR: 0.65-8.13 years) in CA patients, and the median time from CTS surgery to the first cardiac manifestation or amyloidosis, whichever came first, was 2.11 years (IQR: −0.45 to 6.27 years) (Supplemental Table 6). Biceps tendon rupture was rare in both groups, and spinal stenosis was more prevalent among CA patients (7.3% vs 2.3%; P < 0.001).
Temporal changes in mortality
Cumulative mortality was assessed at 1, 2, and 5 years, revealing a steady decline over time by each time-period (log-rank test: P < 0.0001), as shown in Figure 3. From the first to the last time period (1998-2002 to 2013-2017), the cumulative 1-year mortality decreased from 74.1% (IQR: 60.5%-83.7%) to 39.2% (IQR: 32.8%-45.5%) and the cumulative 2-year mortality decreased from 82.6% (IQR: 69.9%-90.5%) to 50.2% (IQR: 43.1%-56.9%). The cumulative 5-year mortality decreased from 91.4% (IQR: 79.6%-96.5%) to 68.4% (IQR: 59.0%-76.2%); however, censoring of data at the end of the study period reduced the maximum follow-up time for the patients with index dates in the last 5 years of the study, permitting a maximum 4.8-year follow up, limiting comparisons. Still, between 1998-2002 and 2008-2012, the 5-year mortality decreased from 91.4% (IQR: 79.6%-96.5%) to 74.9% (IQR: 67.9%-80.6%). Compared with control subjects, the mortality among CA patients was significantly higher (log-rank test: P < 0.0001), as shown in Figure 4.
Figure 3.

5-Year Mortality in Patients With Cardiac Amyloidosis by Time Period
The mortality among patients with cardiac amyloidosis (CA)was described for each 5-year period of the study with the use of cumulative incidence functions. The mortality decreased significantly between the first and last 5-year periods. The improved survival might be explained by increased earlier detection of less severe cases of CA, a greater incidence of new cases of transthyretin amyloidosis vs amyloid light chain amyloidosis, and progress in the treatment of amyloid light chain amyloidosis.
Figure 4.

5-Year Mortality in Patients With Cardiac Amyloidosis vs Age- and Sex-Matched Control Subjects From the General Population
Comparison of mortality between patients with cardiac amyloidosis (CA) and age- and sex-matched control subjects. The mortality among patients with CA vs age- and sex-matched control subjects from the general population was described with the use of cumulative incidence functions. The mortality among patients with CA was significantly higher than among control subjects. CA is associated with a high mortality after diagnosis.
Noncardiac amyloidosis
A total of 936 amyloidosis patients did not meet criteria for CA. Among patients with noncardiac amyloidosis, cardiovascular comorbidities and mortality were generally lower than in cases of CA. The median age at amyloidosis diagnosis increased from 59.6 years (IQR: 46.3-69.3 years) to 65.0 years (IQR: 53.3-73.4 years) during the study period. Over time, the incidence of noncardiac amyloidosis rose from 1.61 to 2.36 per 100,000 person-years in the Danish population ≥65 years of age, from 1.52 to 2.93 in men ≥65 years of age and from 0.91 to 1.89 in women ≥65 years of age. Previous surgery for CTS was present in 3.0% of patients with noncardiac amyloidosis, and spinal stenosis was present in 2.2%. Supplemental Table 7 presents characteristics of the patients with noncardiac amyloidosis.
Discussion
This study comprehensively examined the landscape of amyloidosis in Denmark during 1998-2017. Three main trends in the temporal changes were noted: 1) CA is being increasingly diagnosed in Denmark; 2) in patients with CA, the median diagnostic delay from overt heart disease to subsequent amyloidosis diagnosis was approximately 1 year; and 3) mortality decreased significantly over the study period. It should be noted that these observed trends are reported for time periods before the national approval of disease-modifying drugs for amyloid transthyretin (ATTR) CA. Attention to this disease is likely to increase as effective pharmacologic treatment options are being increasingly made available.
CA is increasingly diagnosed
Amyloidosis and CA, as defined in this study, are being increasingly diagnosed in Denmark (Central Illustration). Over the course of the past 20 years, the number of diagnosed CA patients per year quadrupled. At the same time, the median age at the time of amyloidosis diagnosis and the frequency of male patients has been rising, indicating that the diagnosis of wild-type (wt) ATTR is likely driving this increase, because wtATTR usually manifests in the elderly and is a predominantly male condition (25-50:1 male:female ratio) (24, 25, 26, 27), and hereditary ATTR is rare in Denmark. Although CA is increasingly diagnosed, this study shows that it remains rare in Denmark. The incidence of CA in our study was 3.6/100,000/year in the Danish population ≥65 years of age, contrasting with the 55/100,000 in the Medicare-derived U.S. population >65 years of age, as described by Gilstrap et al (18). The observed difference may be due to our data including both inpatient and outpatient diagnoses from the entire Danish population, in contrast to data from hospitalized Medicare patients only. Furthermore, differences in the median age between patients in our data and in the data used by Gilstrap et al (18) might also contribute to observed differences. Finally, the Danish population is almost entirely White, and in other populations the prevalence of hereditary ATTR is much higher; for example, Black Americans have a 4% prevalence of V122I (28).
Central Illustration.

Danish Nationwide Registry Analysis of Cardiac Amyloidosis: Study Design and Main Results
Diagnostic delay
The median diagnostic delay from overt heart failure, cardiomyopathy, or atrial fibrillation or pacemaker implantation to subsequent diagnosed amyloidosis, was approximately 15 months, with 75% of the patients being diagnosed within 56 months. These findings resonate well with a recently published retrospective study by Ladefoged et al (29), where clinical and echocardiographic patient characteristics from 50 consecutive patients with wtATTR were analyzed, concluding that the median diagnostic delay was 13 months (IQR: 2-47 months).
Extracardiac manifestations
Previous CTS surgery was prevalent in 10% (n = 62) of the CA patients at baseline, and in 12.3% (n = 76) of the CA patients during the entire study period. Among CA patients with CTS, the median time from CTS surgery to diagnosed amyloidosis was 2.9 years (IQR: 0.28-7.83 years), the median time from CTS surgery to diagnosed heart failure was 4.26 years (IQR: 1.21-8.94 years). These results are consistent with data from a large registry study on the association of CTS and heart failure by Fosbøl et al (30), where the median time elapsed from CTS surgery to heart failure diagnosis was 3.7 years (IQR: 1.5-6.6 years). Milandri et al (4) recently published on the prevalence of CTS in 538 subjects with AL or ATTR amyloidosis, establishing a 20.3% prevalence of CTS in ATTR patients, while the prevalence in AL patients was similar to that of the comparator control population (4.1%). Our findings of the prevalence of CTS are significantly lower, presumably owing to the mixed etiology of CA in our study.
AL amyloidosis
Patients in our cohort could not be coded specifically as AL amyloidosis, but one can theorize about the prevalence in our cohort based on the presence of conditions and therapies associated with AL amyloidosis. A total of 21% of the CA patients had a concomitant lymphoid/hematologic cancer (DC81-DC96) at baseline, of which the vast majority was MM. Coexisting AL amyloidosis is present in 10%-15% of MM patients (31), however the presence of coexistent cardiac manifestations in our study population most likely increases the probability of concurrent AL amyloidosis. Furthermore, 7.4% of the patients had procedural codes for treatment with melphalan ± concomitant corticosteroids, bortezomib, thalidomide, or daratumumab or had received stem cell conditioning, without having MM—indicating treatment for AL amyloidosis. Combining all of the CA patients with either MM or the above-mentioned treatment for AL amyloidosis yields a subpopulation of 23.4% (n = 145) patients that might be interpreted as suspected of having AL amyloidosis. This estimate is imprecise and undoubtedly overestimates the frequency of true AL amyloidosis, but regarded as such, it still provides insight in the changing characteristics in patients with amyloidosis. The rise in pharmacotherapy indicating treatment against AL amyloidosis, was similar in patients with CA and noncardiac amyloidosis, making the frequencies of suspected AL amyloidosis similar between the 2 groups. Previous studies describe cardiac involvement being present in 50%-70% of AL amyloidosis (18,32); in our study material, cardiac involvement was present in approximately 45% of suspected AL amyloidosis.
Noncardiac amyloidosis
Approximately 60% of the patients with amyloidosis did not meet the criteria for CA and were considered as having noncardiac amyloidosis. Patients with noncardiac amyloidosis were younger at time of amyloidosis diagnosis, and the frequency of male patients was lower. Given the definition of CA, the cardiovascular comorbidities were naturally lower among patients with noncardiac amyloidosis. MM and MGUS were lower among patients with noncardiac amyloidosis, in line with the younger age of the patients (33,34). Mortality in patients with noncardiac amyloidosis appears significantly lower than in patients with CA; however, because of separate definitions of index date (time at amyloidosis diagnosis vs time at meeting criteria for CA), comparison between the two groups is complicated (Supplemental Figures 1 and 2). In AL amyloidosis, cardiac involvement is the major determinant of survival (32), possibly explaining the discrepancy in mortality between patients with noncardiac amyloidosis and CA, even though the frequencies of suspected AL amyloidosis were similar.
Pharmacologic treatment
While the development of comorbidities and socioeconomic factors were similar between patients with CA and control subjects, the changes in pharmacotherapy over time differed significantly. In patients with CA, there was a dramatic decline in treatment with digoxin and nondihydropyridine calcium channel blockers. Current guidelines state that digoxin use in CA generally should be avoided (or used carefully), owing to increased sensitivity for digoxin toxicity and subsequent risk of arrhythmia, likely explained by binding of digoxin by amyloid fibrils. However, recent data suggest that with rigorous patient selection and monitoring, digoxin may be a therapeutic option for some patients with CA (35, 36, 37). Nondihydropyridine calcium channel blockers are known to cause worsening heart failure in patients with CA and are contraindicated (38,39). In contrast, a large increase in treatment with beta-blockers over the study period was apparent. High-dose beta-blockers often worsen heart failure and aggravate hypotension in CA and are generally avoided (40), but they may be cautiously attempted in low doses in the setting of atrial fibrillation to help rate control. The increase in beta-blocker treatment observed in the present study might be due to an increase in prevalence of atrial fibrillation and decrease in digoxin treatment. Amiodarone treatment was infrequent but stable during the study period and is usually well tolerated in CA (41). Oral anticoagulation therapy (OAC) increased during the 2 decades of the study, reflecting treatment guideline changes and the introduction of direct oral anticoagulants (42,43). Oral anticoagulants are indicated in CA complicated by AF, regardless of CHA2DS2-VASc score, and may be considered even in sinus rhythm depending on echocardiographic findings (44,45). As such, the temporal changes in pharmacotherapy observed in this study are seemingly in line with the current understanding of proper pharmacotherapy in CA.
Comorbidities
Because of our definition of CA, cardiovascular comorbidities (such as ischemic heart disease) and associated conditions and pharmacotherapy are overrepresented in the CA cohort compared with only age- and sex-matched control subjects. The increased burden of coexisting diseases among cases is reflected, to some extent, in the striking and contrasting mortality among case and control subjects (Figure 4).
Mortality
During the study period, mortality decreased significantly. This could be due to a shift in the type of CA, from AL-CA early in the time period, to more ATTR-CA later, particularly wtATTR. Other possible explanations might be a disparity in frequency of “true CA” over time due to changes in coding practice over the years, or perhaps earlier detection of CA in recent years. However, earlier diagnosis of CA is partially contradicted by an increasing age at diagnosis and stable time elapsed between first cardiac manifestation and subsequent diagnosed amyloidosis. Although contemporary studies report improved survival in heart failure over the last decades (46, 47, 48, 49), the observed magnitude in the present study is beyond comparison, thereby presumably reflecting changes in patient characteristics rather than treatment advances. Although great breakthroughs in disease modifying drugs for the treatment of ATTR-CA have been made recently (11,12), their availability in Denmark is limited to clinical trials and their effects on mortality are likely not observable in this study population. Recent progresses in treatment for AL-CA may account for some of the improved survival in the most recent time period (14). Increased detection of less severe cases of CA is another possible explanation, well fitting both the observed improved short-term survival and the recent increased attention to the disease. Indeed, as CA is being increasingly recognized and treated with disease-modifying drugs, further studies on epidemiology and outcomes in CA are compelling.
Study Strengths and limitations
The national registries provide an essentially complete follow-up in all in- and outpatient diagnoses. The procedure codes and cardiovascular diagnosis codes in the registries are validated and have high positive predictive values (21,22,50). Although ICD-10 allows for increased specificity in coding of the type of amyloidosis, the use of these codes is not yet well implemented in clinical practice, as was apparent in this study, where the majority of patients were coded as unspecified amyloidosis (DE859). To circumvent this, we created a surrogate measure for CA (code for amyloidosis + code for one of heart failure/cardiomyopathy/atrial fibrillation or procedural code for pacemaker implantation), based on previous work by Gilstrap et al (18) and the review by Griffin et al (23). This definition is neither 100% sensitive or specific, so it will both include patients without CA and miss patients with CA. Thus, a portion of the data will not be representative of “true” CA.
Because of the definition of CA used, cardiovascular comorbidities and associated pharmacotherapy are logically more prevalent in cases than in age- and sex-matched control subjects. However, the temporal changes of these variables among case and control subjects are interesting to compare because they reflect introduction of novel therapeutic options and revisions of guidelines. Comparison between patients with CA and patients with noncardiac amyloidosis is limited by inherently different definitions of index date (date at meeting criteria for CA vs date of diagnosed amyloidosis).
Due to the descriptive nature of the study, causal relationships cannot be assessed. Important information such as body mass index, smoking, family history, left ventricular ejection fraction, electrocardiography readings, and blood samples were not available for analysis. Data on race are not available in the registries that we used.
Conclusions
CA, as defined in this study, is increasingly diagnosed in Denmark. The increasing frequency of male patients and median age at the time of diagnosis along with the lower associated mortality might suggest that wtATTR is driving this increase. Mortality is decreasing, possibly due to increased earlier recognition of less advanced cases. The observed trends are seen before national approval of disease-modifying drugs for ATTR-CA and are expected to continue as effective pharmacologic treatment options are made more widely available.
Perspectives.
COMPETENCY IN MEDICAL KNOWLEDGE: CA is increasingly diagnosed in Denmark, and the increasing frequency of male patients and median age at diagnosis suggests that wtATTR is driving the increase. Greater early recognition and resultant less advanced cases might explain decreasing mortality, as the trends uncovered in this retrospective study are observed before national approval of disease-modifying drugs for ATTR-CA.
TRANSLATIONAL OUTLOOK: Future studies on clinical outcomes in CA are needed as novel disease-modifying drugs are approved for use in treatment of both AL-CA and ATTR-CA, revolutionizing the therapeutic possibilities and further drawing important attention to the disease.
Funding Support and Author Disclosures
Dr Westin has received a grant from the Erik og Susanna Olesens Almenvelgørende Fond during the conduct of the study. Dr Westin has received grants from Pfizer, Arvid Nilssons Fond, Højmosegård-legatet, Frimodt-Heineke Fonden, and Hjertecentrets Forskningsudvalg, Rigshospitalet, outside the submitted work. Dr Gustafsson has received personal fees from Abbott, AstraZeneca, Pfizer, Boehringer Ingelheim, Novartis, and Orion Pharma, and other compensation from Corvia, outside the submitted work. Dr Salomo has received consulting income from Takeda, Bristol Myers Squibb, Janssen, and Amgen, outside the submitted work. Dr Køber has received speakers honoraria from Novartis, AstraZeneca, Novo, and Boehringer, outside the submitted work. Dr Maurer has received grant support from National Institutes of Health (R01HL139671, R21AG058348, and K24AG036778); has received consulting income from Pfizer, Eidos, Prothena, Akcea, and Alnylam; and his institution received clinical trial funding from Pfizer, Prothena, Eidos, and Alnylam. Dr Fosbøl has received an independent research grant from Novo Nordisk Foundation, outside the submitted work. All other authors have reported that they have no relationships relevant to the contents of this paper to disclose.
Acknowledgment
The data underlying this article were provided by Statistics Denmark, under permission from the Danish Data Protection Agency. Data will be shared on request to the corresponding author with permission from the Danish Data Protection Agency.
Footnotes
The authors attest they are in compliance with human studies committees and animal welfare regulations of the authors’ institutions and Food and Drug Administration guidelines, including patient consent where appropriate. For more information, visit the Author Center.
Appendix
For supplemental tables, figures, and the ICD codes, please see the online version of this paper.
Appendix
References
- 1.Escher F., Senoner M., Doerler J. When and how do patients with cardiac amyloidosis die? Clin Res Cardiol. 2020;109:78–88. doi: 10.1007/s00392-019-01490-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Treibel T.A., Fontana M., Gilbertson J.A. Occult transthyretin cardiac amyloid in severe calcific aortic stenosis. Circ Cardiovasc Imaging. 2016;9 doi: 10.1161/CIRCIMAGING.116.005066. [DOI] [PubMed] [Google Scholar]
- 3.Castaño A., Narotsky D.L., Hamid N. Unveiling transthyretin cardiac amyloidosis and its predictors among elderly patients with severe aortic stenosis undergoing transcatheter aortic valve replacement. Eur Heart J. 2017;38:2879–2887. doi: 10.1093/eurheartj/ehx350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Scully P.R., Treibel T.A., Fontana M. 1 A multi-centre study of cardiac amyloidosis in TAVI patients. Heart. 2018;104:A15. [Google Scholar]
- 5.González-López E., Gallego-Delgado M., Guzzo-Merello G. Wild-type transthyretin amyloidosis as a cause of heart failure with preserved ejection fraction. Eur Heart J. 2015;36:2585–2594. doi: 10.1093/eurheartj/ehv338. [DOI] [PubMed] [Google Scholar]
- 6.Tanskanen M., Peuralinna T., Polvikoski T. Senile systemic amyloidosis affects 25% of the very aged and associates with genetic variation in alpha2-macroglobulin and tau: a population-based autopsy study. Ann Med. 2008;40:232–239. doi: 10.1080/07853890701842988. [DOI] [PubMed] [Google Scholar]
- 7.Agha A.M., Parwani P., Guha A. Role of cardiovascular imaging for the diagnosis and prognosis of cardiac amyloidosis. Open Heart. 2018;5 doi: 10.1136/openhrt-2018-000881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bokhari S., Shahzad R., Castaño A. Nuclear imaging modalities for cardiac amyloidosis. J Nucl Cardiol Off Publ Am Soc Nucl Cardiol. 2014;21:175–184. doi: 10.1007/s12350-013-9803-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sharpley F.A., Manwani R., Mahmood S. A novel mass spectrometry method to identify the serum monoclonal light chain component in systemic light chain amyloidosis. Blood Cancer J. 2019;9:1–4. doi: 10.1038/s41408-019-0180-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Witteles R.M., Bokhari S., Damy T. Screening for transthyretin amyloid cardiomyopathy in everyday practice. J Am Coll Cardiol HF. 2019;7:709–716. doi: 10.1016/j.jchf.2019.04.010. [DOI] [PubMed] [Google Scholar]
- 11.Maurer M.S., Schwartz J.H., Gundapaneni B. Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N Engl J Med. 2018;379:1007–1016. doi: 10.1056/NEJMoa1805689. [DOI] [PubMed] [Google Scholar]
- 12.Ruberg F.L., Grogan M., Hanna M. Transthyretin amyloid cardiomyopathy: JACC state-of-the-art review. J Am Coll Cardiol. 2019;73:2872–2891. doi: 10.1016/j.jacc.2019.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Izumiya Y., Takashio S., Oda S. Recent advances in diagnosis and treatment of cardiac amyloidosis. J Cardiol. 2018;71:135–143. doi: 10.1016/j.jjcc.2017.10.003. [DOI] [PubMed] [Google Scholar]
- 14.Barrett C.D., Dobos K., Liedtke M. A changing landscape of mortality for systemic light chain amyloidosis. J Am Coll Cardiol HF. 2019;7:958–966. doi: 10.1016/j.jchf.2019.07.007. [DOI] [PubMed] [Google Scholar]
- 15.Palladini G., Kastritis E., Maurer M.S. Daratumumab plus CyBorD for patients with newly diagnosed AL amyloidosis: safety run-in results of ANDROMEDA. Blood. 2020;136:71–80. doi: 10.1182/blood.2019004460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bishop E., Brown E.E., Fajardo J. Seven factors predict a delayed diagnosis of cardiac amyloidosis. Amyloid. 2018;25:174–179. doi: 10.1080/13506129.2018.1498782. [DOI] [PubMed] [Google Scholar]
- 17.Hassan W., Al-Sergani H., Mourad W. Amyloid heart disease. Tex Heart Inst J. 2005;32:178–184. [PMC free article] [PubMed] [Google Scholar]
- 18.Gilstrap L.G., Dominici F., Wang Y. Epidemiology of cardiac amyloidosis-associated heart failure hospitalizations among fee-for-service Medicare beneficiaries in the United States. Circ Heart Fail. 2019;12 doi: 10.1161/CIRCHEARTFAILURE.118.005407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gaist D., Sørensen H.T., Hallas J. The Danish prescription registries. Dan Med Bull. 1997;44:445–448. [PubMed] [Google Scholar]
- 20.Schmidt M., Schmidt S.A.J., Sandegaard J.L. The Danish National Patient Registry: a review of content, data quality, and research potential. Clin Epidemiol. 2015;7:449–490. doi: 10.2147/CLEP.S91125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Adelborg K., Sundbøll J., Munch T. Positive predictive value of cardiac examination, procedure and surgery codes in the Danish National Patient Registry: a population-based validation study. BMJ Open. 2016;6 doi: 10.1136/bmjopen-2016-012817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sundbøll J., Adelborg K., Munch T. Positive predictive value of cardiovascular diagnoses in the Danish National Patient Registry: a validation study. BMJ Open. 2016;6 doi: 10.1136/bmjopen-2016-012832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Griffin J.M., Maurer M.S. Cardiac amyloidosis a rare disease in older adults hospitalized for heart failure? Circ Heart Fail. 2019;12 doi: 10.1161/CIRCHEARTFAILURE.119.006169. [DOI] [PubMed] [Google Scholar]
- 24.Gertz M.A., Benson M.D., Dyck P.J. Diagnosis, prognosis, and therapy of transthyretin amyloidosis. J Am Coll Cardiol. 2015;66:2451–2466. doi: 10.1016/j.jacc.2015.09.075. [DOI] [PubMed] [Google Scholar]
- 25.González-López E., Gagliardi C., Dominguez F. Clinical characteristics of wild-type transthyretin cardiac amyloidosis: disproving myths. Eur Heart J. 2017;38:1895–1904. doi: 10.1093/eurheartj/ehx043. [DOI] [PubMed] [Google Scholar]
- 26.Grogan M., Scott C.G., Kyle R.A. Natural history of wild-type transthyretin cardiac amyloidosis and risk stratification using a novel staging system. J Am Coll Cardiol. 2016;68:1014–1020. doi: 10.1016/j.jacc.2016.06.033. [DOI] [PubMed] [Google Scholar]
- 27.Martinez-Naharro A., Hawkins P.N., Fontana M. Cardiac amyloidosis. Clin Med. 2018;18:s30–s35. doi: 10.7861/clinmedicine.18-2s-s30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Connors L.H., Prokaeva T., Lim A. Cardiac amyloidosis in African Americans: comparison of clinical and laboratory features of transthyretin V122I amyloidosis and immunoglobulin light chain amyloidosis. Am Heart J. 2009;158:607–614. doi: 10.1016/j.ahj.2009.08.006. [DOI] [PubMed] [Google Scholar]
- 29.Ladefoged B., Dybro A., Povlsen J.A. Diagnostic delay in wild type transthyretin cardiac amyloidosis—a clinical challenge. Int J Cardiol. 2020;304:138–143. doi: 10.1016/j.ijcard.2019.12.063. [DOI] [PubMed] [Google Scholar]
- 30.Fosbøl E.L., Rørth R., Leicht B.P. Association of carpal tunnel syndrome with amyloidosis, heart failure, and adverse cardiovascular outcomes. J Am Coll Cardiol. 2019;74:15–23. doi: 10.1016/j.jacc.2019.04.054. [DOI] [PubMed] [Google Scholar]
- 31.Rajkumar S.V., Gertz M.A., Kyle R.A. Primary systemic amyloidosis with delayed progression to multiple myeloma. Cancer. 1998;82:1501–1505. [PubMed] [Google Scholar]
- 32.Grogan M., Dispenzieri A., Gertz M.A. Light-chain cardiac amyloidosis: strategies to promote early diagnosis and cardiac response. Heart. 2017;103:1065–1072. doi: 10.1136/heartjnl-2016-310704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kaseb H., Annamaraju P., Babiker H.M. Monoclonal gammopathy of undetermined significance. Treasure Island, FL: StatPearls. https://www.statpearls.com/ArticleLibrary/viewarticle/25248 Accessed April 10, 2021. [PubMed]
- 34.Weinhold N., Johnson D.C., Rawstron A.C. Inherited genetic susceptibility to monoclonal gammopathy of unknown significance. Blood. 2014;123:2513–2517. doi: 10.1182/blood-2013-10-532283. [DOI] [PubMed] [Google Scholar]
- 35.Donnelly J.P., Gabrovsek A., Sperry B.W. Digoxin use in cardiac amyloidosis. J Card Fail. 2019;25:S25–S26. [Google Scholar]
- 36.Rubinow A., Skinner M., Cohen A.S. Digoxin sensitivity in amyloid cardiomyopathy. Circulation. 1981;63:1285–1288. doi: 10.1161/01.cir.63.6.1285. [DOI] [PubMed] [Google Scholar]
- 37.Muchtar E., Gertz M.A., Kumar S.K. Digoxin use in systemic light-chain (AL) amyloidosis: contra-indicated or cautious use? Amyloid. 2018;25:86–92. doi: 10.1080/13506129.2018.1449744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gertz M.A., Falk R.H., Skinner M. Worsening of congestive heart failure in amyloid heart disease treated by calcium channel–blocking agents. Am J Cardiol. 1985;55:1645. doi: 10.1016/0002-9149(85)90995-6. [DOI] [PubMed] [Google Scholar]
- 39.Griffiths B.E., Hughes P., Dowdle R. Cardiac amyloidosis with asymmetrical septal hypertrophy and deterioration after nifedipine. Thorax. 1982;37:711–712. doi: 10.1136/thx.37.9.711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Quarta C.C., Kruger J.L., Falk R.H. Cardiac amyloidosis. Circulation. 2012;126:e178–e182. doi: 10.1161/CIRCULATIONAHA.111.069195. [DOI] [PubMed] [Google Scholar]
- 41.Siddiqi O.K., Ruberg F.L. Cardiac amyloidosis: an update on pathophysiology, diagnosis, and treatment. Trends Cardiovasc Med. 2018;28:10–21. doi: 10.1016/j.tcm.2017.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zoni-Berisso M., Lercari F., Carazza T. Epidemiology of atrial fibrillation: European perspective. Clin Epidemiol. 2014;6:213–220. doi: 10.2147/CLEP.S47385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kornej J., Börschel C.S., Benjamin E.J. Epidemiology of atrial fibrillation in the 21st century. Circ Res. 2020;127:4–20. doi: 10.1161/CIRCRESAHA.120.316340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dubrey S., Pollak A., Skinner M. Atrial thrombi occurring during sinus rhythm in cardiac amyloidosis: evidence for atrial electromechanical dissociation. Br Heart J. 1995;74:541–544. doi: 10.1136/hrt.74.5.541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Feng D., Edwards W.D., Oh J.K. Intracardiac thrombosis and embolism in patients with cardiac amyloidosis. Circulation. 2007;116:2420–2426. doi: 10.1161/CIRCULATIONAHA.107.697763. [DOI] [PubMed] [Google Scholar]
- 46.Buddeke J., Valstar G.B., van Dis I. Mortality after hospital admission for heart failure: improvement over time, equally strong in women as in men. BMC Public Health. 2020;20:36. doi: 10.1186/s12889-019-7934-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.MacIntyre K., Capewell S., Stewart S. Evidence of improving prognosis in heart failure. Circulation. 2000;102:1126–1131. doi: 10.1161/01.cir.102.10.1126. [DOI] [PubMed] [Google Scholar]
- 48.Joffe S.W., Webster K., McManus D.D. Improved survival after heart failure: a community-based perspective. J Am Heart Assoc. 2013;2:e000053. doi: 10.1161/JAHA.113.000053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Meta-analysis Global Group in Chronic Heart Failure (MAGGIC) The survival of patients with heart failure with preserved or reduced left ventricular ejection fraction: an individual patient data meta-analysis. Eur Heart J. 2012;33:1750–1757. doi: 10.1093/eurheartj/ehr254. [DOI] [PubMed] [Google Scholar]
- 50.Thygesen S.K., Christiansen C.F., Christensen S. The predictive value of ICD-10 diagnostic coding used to assess Charlson comorbidity index conditions in the population-based Danish National Registry of Patients. BMC Med Res Methodol. 2011;11:83. doi: 10.1186/1471-2288-11-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.