

Abstract
Ischemic stroke is amongst the leading causes of death and disabilities. The available treatments are suitable for only a fraction of patients and thus novel therapies are urgently needed. Blockage of one of the cerebral arteries leads to massive and persisting inflammatory reaction contributing to the nearby neuronal damage. Targeting the detrimental pathways of neuroinflammation has been suggested to be beneficial in conditions of ischemic stroke. Nuclear receptor 4A-family (NR4A) member Nurr1 has been shown to be a potent modulator of harmful inflammatory reactions, yet the role of Nurr1 in cerebral stroke remains unknown. Here we show for the first time that an agonist for the dimeric transcription factor Nurr1/retinoid X receptor (RXR), HX600, reduces microglia expressed proinflammatory mediators and prevents inflammation induced neuronal death in in vitro co-culture model of neurons and microglia. Importantly, HX600 was protective in a mouse model of permanent middle cerebral artery occlusion and alleviated the stroke induced motor deficits. Along with the anti-inflammatory capacity of HX600 in vitro, treatment of ischemic mice with HX600 reduced ischemia induced Iba-1, p38 and TREM2 immunoreactivities, protected endogenous microglia from ischemia induced death and prevented leukocyte infiltration. These anti-inflammatory functions were associated with reduced levels of brain lysophosphatidylcholines (lysoPCs) and acylcarnitines, metabolites related to proinflammatory events. These data demonstrate that HX600 driven Nurr1 activation is beneficial in ischemic stroke and propose that targeting Nurr1 is a novel candidate for conditions involving neuroinflammatory component.
Keywords: Stroke, Neuroinflammation, Microglia, Nuclear receptors, Metabolic profiling
1. Introduction
Ischemic stroke is a severe cerebrovascular disease caused by blockage of blood flow into the brain. It is one of the leading causes of death (GBD, 2015) influencing mainly the elderly, yet a growing portion of individuals affected are still at working age (Kissela et al., 2012; Béjot et al., 2014). If not causing imminent death, stroke is likely to lead to permanent disabilities markedly diminishing the quality of life of the affected individuals and their families. Thus, stroke is causing an enormous burden for the society and economy (Stroke Facts, 2017). While mechanical thrombectomy is highly beneficial in large vessel occlusions in the anterior circulation (Evans et al., 2017), recombinant tissue plasminogen activator (r-tPA) still remains the only clinically approved pharmacological treatment for cerebral stroke, yet it is suitable for only a fraction of patients due to its very narrow therapeutic time window and severe side effects (Quain et al., 2008). Thus, there is a pressing need for new stroke therapies.
Blockage of blood flow into the brain parenchyma initiates a complex cascade of events leading to subsequent inflammatory reaction. This is evident by the activation of resident immune cells, especially microglia, accumulation of various inflammatory mediators in the ischemic tissue and infiltration of peripheral immune cells into the affected area (Jin et al., 2010). Microglia play important roles in the normal brain physiology, enhance neurogenesis, produce neurotrophic factors (Raivich et al., 1999) and clear the dying debris after ischemic insult, yet their activation also promotes nearby neuronal death by secretion of pro-inflammatory cytokines such as tumor necrosis factor (TNF), interleukins and nitric oxide (Banati et al., 1993).
Nuclear receptors are ligand-activated transcription factors and key regulators of a wide range of homeostatic processes from development to different areas of metabolism (Chawla et al., 2001). They can be divided into two categories, where type 1 nuclear receptors include well-known steroid receptors such as glucocorticoid and estrogen receptors. Function of type 2 nuclear receptors is based on their ability to form heterodimers with the retinoid X receptor (RXR), and this RXR heterodimer activation is known to have neuroprotective effects in neurodegenerative diseases such as Alzheimer’s disease (AD) and Parkinson’s disease (PD) (Skerrett et al., 2014). The nuclear receptor 4A -family (NR4A) differs from the other type 2 nuclear receptors in a way that they lack the ligand binding pocket, thus functioning as ligand-independent transcription factors. A member of this receptor family (along with NR4A1 or Nurr77 and NR4A3 or Nor-1) is NR4A2, also referred to as nuclear receptor related 1 protein or Nurr1. These receptors are constitutively active but can also signal in complex with RXRs. Thus, the transcriptional activity of these receptors can be induced by agonists which recognize the RXR-NR4A heterodimer complex. 4-[5H-2,3-(2,5-Dimethyl-2,5-hexano)-5-methyldibenzo[b,e][1,4] diazepin-11-yl]benzoic acid, HX600, is a synthetic agonist for the RXR-NR4A heterodimer complex (Umemiya et al., 1997; Morita et al., 2005). These receptors are abundantly expressed in the brain in different neuronal cell types (Xiao et al., 1996; Yasuyoshi et al., 2003; Kadkhodaei et al., 2013), microglia and astrocytes (Saijo et al., 2009), and as well in peripheral immune cells (Sekiya et al., 2013) and other tissues in the periphery (Pearen and Muscat, 2010).
Nurr1 activation has been shown to confer neuroprotection (Barneda-Zahonero et al., 2012; Hammond and Safe, 2015; Volakakis et al., 2010; Hammond et al., 2018) and is anti-inflammatory through its ability to suppress nuclear factor kappaB (NFκB) activation (Saijo et al., 2009; De Miranda et al., 2015), yet there are no reports on whether Nurr1 induction is neuroprotective in models of ischemic stroke. Ischemia induced Nurr1 mRNA upregulation has been reported in global ischemia in gerbils (Honkaniemi and Sharp, 1996) and in rats with permanent middle cerebral artery occlusion (Honkaniemi et al., 1997), whereas protein levels of Nurr1 were shown to be decreased in mice subjected to transient ischemia (Erdö et al., 2004).
Our hypothesis was that HX600 facilitates neuronal survival by inhibiting inflammation via activation of Nurr1. We tested this hypothesis in an in vitro – model of inflammation induced neuronal death and in mouse model of permanent middle cerebral artery occlusion using several analytical methods.
2. Materials and methods
2.1. Primary microglial culture
Primary microglial cultures were prepared from C57BL/6J mice (Jackson laboratories) of postnatal day 0–3 as described previously (Malm et al., 2015). Briefly, brains of decapitated mice were dissected and tissue was incubated in DMEM-F12 supplemented with 1% penicillin-streptomycin and Trypsin-EDTA after mechanical dissociation (all ThermoFisher Scientific, Waltham, MA, USA). The trypsin was inactivated with complete media (DMEM-F12, 10% fetal bovine serum, 1% penicillin-streptomycin, all ThermoFisher Scientific, Waltham, MA, USA), the tissue homogenized, plated on 15 cm dishes and grown at +37 °C 5% CO2 for three weeks. The astrocyte layer from the mixed glial culture was trypzinized and the remaining microglia removed, counted and plated on 48-well-plates at the density of 125 000 cells/well. Microglia were pre-treated with 1 μM HX600 for 24 h, after which the cells were exposed to 50 ng/ml lipopolysaccharide (LPS L2630, serotype O111:B4, Sigma Aldrich, St. Louis, MO, USA) for 3 h, while keeping the concentration of HX600 (synthetized by H. Kagechika) unaltered.
2.2. Primary cortical neuron culture
Primary cortical neuron cultures were prepared from mouse embryos of embryonic day 15 as described by Malm et al. (2015). Briefly, the cortices were dissected and tissue dissociated with 0.0125% trypsin (for 15 min at 37 °C, Sigma-Aldrich, St. Louis, MO, USA). After trypsin inactivation and washing the cells were counted and plated on 48-well-plates (coated with poly-D-lysine, Sigma-Aldrich, St. Louis, MO, USA) at a density of 125 000 cells/well in Neurobasal media supplemented with 2% B27 and 500 μM L-glutamine (all ThermoFisher Scientific, Waltham, MA, USA) and 10 μg/ml of gentamicin (Sigma Aldrich, St. Louis, MO, USA). On day 5 in vitro (DIV) 50% of media was changed, and on day 6 in vitro neurons were treated with 100 nM or 1 μM HX600 in the presence of 500 μM glutamate for 24 h. Alternatively, on DIV 6 the neurons were pre-exposed to HX600 for 24 h and followed by treatment with glutamate (500 μM) for another 24 h, without changing the HX600 concentration (100 nM or 1 μM). Neuronal viability was assessed using MTT assay.
2.3. Neuron-BV2 co-culture and viability assay
For neuron-BV2 co-culture experiment primary cortical neuron culture was first prepared as described above. On DIV 5 negative control siRNA (ThermoFisher Scientific, Waltham, MA, USA) or Nurr1 siRNA (Silencer® select siRNA for Nr4a2, ThermoFisher Scientific, Waltham, MA, USA) transfected BV2 cells were seeded on top of neurons at density of 1:5 (25 000 BV2 cells per 125 000 neurons) and left for 2 h to allow attachment. The cells were pretreated with 1 μM HX600 for 6 h, after which they were exposed to 100 ng/ml LPS (L2630, serotype O111:B4, Sigma Aldrich, St. Louis, MO, USA) and 30 ng/ml interferon gamma (IFNγ, Sigma-Aldrich, St. Louis, MO, USA) for 48 h. Concentration of HX600 was kept constant (1 μM) during the LPS/IFNγ exposure. After the exposure the cells were rinsed with PBS and fixed with 4% PFA for 20 min. The cells were washed again in PBS (pH 7.4) and neuronal viability in co-culture was assessed as described previously (Gresa-Arribas et al., 2012). Briefly, the co-cultures were stained with MAP-2 peroxidase labelling and the ABTS peroxidase substrate kit (Vector) was used to develop the color according to manufacturer’s instruction. Green colored product was formed after incubation for 30 min with ABTS kit in the dark. The plate was gently shaked and 150 μL of the substrate solution was then transferred to the 96 well plate and the absorbance was measured with microtiter plate reader Victor 2.0 (Perkin Elmer, MA, USA) at 405 nm. Results were calculated as relative absorbances compared to control wells. Alternatively, the co-cultures were stained with MAP-2 antibody (Sigma-Aldrich, St. Louis, MO, USA) followed by incubation with Alexa-488 conjugated secondary antibody. The extent of MAP-2 immunoreactivity as indicative of neuronal viability was imaged under fluorescent microscope and quantified using ImagePro software (Media Cybernetics Inc., Rockville, MD, USA).
2.4. Animals
For the permanent ischemia studies, adult (3–7 months old) male Balb/cOlaHsd mice (Harlan Laboratories B.V., An Venrey, Netherlands) were used. Total of 108 mice were used in the study. Mice were individually housed in controlled humidity, temperature and light conditions, and they had ad libitum access to food and water. Randomization to treatment groups was carried out by using GraphPad QuickCalcs (GraphPad Software, San Diego, CA, USA), and mice of different ages were distributed randomly to all groups. All data analyzes were performed blinded to the experimental groups. All the animal experiments followed the Council of Europe Legislation and Regulation for Animal Protection and were approved by the National Animal Experiment Board of Finland. Every effort was made to minimize the harm and suffering of the animals.
2.5. Ischemia surgeries and treatment of mice with HX600
For permanent occlusion of the middle cerebral artery (pMCAo), the anesthesia was induced using 5% isoflurane in 30% O2 and 70% N2O and maintained at 2% isoflurane during the surgery. The left MCA was permanently occluded as previously described (Dhungana et al., 2013). Briefly, a hole (1 mm in diameter) was drilled on the temporal bone to expose the MCA. The dura was removed, after which the artery was lifted and occluded using a thermocoagulator (Aaron Medical Industries Inc., Clearwater, FL, USA). MCA occlusion was confirmed by cutting the artery after which the temporal muscle was replaced and the wound was sutured. The body temperature of the animals was maintained at 37 ± 0.5 °C during the whole procedure by using a heating pad. The respiratory rate and temperature were monitored throughout the surgery. The sham operated mice underwent the same procedures except the occlusion of the MCA. Mice with hemorrhages visible in magnetic resonance imaging (MRI), bleeding during the surgery and unsuccessful induction of ischemia were excluded from further analysis. Altogether 8 mice were excluded from the study.
The mice were treated with HX600 dissolved in standard suspension vehicle (SSV) solution (containing 0.9% (w/v) NaCl, 0.5% (w/v) sodium carboxymethylcellulose, 0.5% (v/v) benzyl alcohol and 0.4% (v/v) Tween®80, all Sigma-Aldrich, St. Louis, MO, USA), or SSV alone by oral gavage. The dose of 60 mg/kg was administered immediately after pMCAo surgery and every 24 h thereafter. Mice were sacrificed at 1 or 3 days post injury (dpi) and the tissues were collected as described below.
2.6. Post-surgery evaluation of locomotor activity
Latency-to-move is a simple test which has been shown to be able to reveal motor deficits induced by pMCAo (Lubjuhn et al., 2009). We utilized this test to assess motor function of the mice at 1 day after the ischemic insult. The mice (7–12 in each treatment group) were placed on a flat surface and the time to move one body length (7 cm) was recorded. Two trials were allowed for each mouse. The person performing the test was blinded to the treatment groups.
2.7. Measurement of lesion volume
The lesion volumes were determined by magnetic resonance imaging (MRI) at 48 h after pMCAO. A vertical 9.4 T Oxford NMR 400 magnet (Oxford Instrument PLC, Abington, UK) was used for visualizing the lesion as described previously (Dhungana et al., 2013). The mice were anaesthetized with 5% isoflurane in 30% O2 and 70% N2O, and multislice T2-weighted images (repetition time 3000 ms, echo time 40 ms, matrix size 128 × 256, field of view 19.2 × 19.2 mm2, slice thickness 0.8 mm and number of slices 12) were obtained. The images were analyzed by utilizing in-house made software Aedes in the Matlab environment (Math-works, Natick, MA, USA). The infarction volume was calculated by using the following formula: Infarct volume = (volume of left hemisphere − (volume of right hemisphere − measured infarct volume))/volume of left hemisphere (Shuaib et al., 2002).
2.8. Immunohistochemistry
Mice were euthanized at 1 or 3 dpi (5–8 mice per group) for tissue collection. Mice were anesthetized with 250 mg/kg Avertin® (Sigma-Aldrich, St. Louis, MO, USA) and perfused transcardially with saline containing 2500 IU/L heparin (Heparin LEO 5000 IU/ml, Leo Pharma A/S, Ballerup, Denmark). The brains were removed and post-fixed in 4% paraformaldehyde (PFA, Sigma-Aldrich, St. Louis, MO, USA) for 20 h, followed by cryoprotection in 30% sucrose for 48 h. The brains were frozen on liquid nitrogen and cut into 20 μm thick sections with a cryostat (Leica Microsystems, Wetzlar, Germany). All immunohistochemical stainings were carried out on six consecutive sections at 400 μm intervals.
The brain sections were incubated overnight at room temperature with primary antibodies (Leucocyte common antigen [CD45] 1:100 dilution, Bio-Rad, Hercules, CA, USA; Cluster of differentiation 68 [CD68] 1:2000 dilution, Bio-Rad, Hercules, CA, USA; Glial fibrillary acidic protein [GFAP] 1:500 dilution, ABR Affinity BioReagents, Golden, CO, USA; Ionized calcium-binding adapter molecule 1 [Iba-1] 1:250 dilution, Wako Chemicals, Tokyo, Japan; Lymphocyte antigen 6 complex locus G6D [Ly6-G neutrophil] 1:100 dilution, BioLegend, San Diego, CA, USA; Phosphorylated P38 mitogen-activated protein kinase [phospho-p38] 1:100 dilution, Cell Signaling Technology Inc., Danvers, MA, USA; Triggering receptor expressed on myeloid cells 2 [TREM2] 1:100 dilution, Lifespan Biosciences, Seattle, WA, USA). All stainings except the Ly6-G neutrophil stain required antigen retrieval in 10 mM aqueous solution of sodium citrate dihydrate (pH 6, preheated to +92 °C, Sigma-Aldrich, St. Louis, MO, USA) before application of primary antibodies. Secondary antibodies were applied on sections after three washes in 0.05% Tween®20 (Sigma-Aldrich, St. Louis, MO, USA) in PBS. Fluorescent Alexa 488 or 568-conjugated secondary antibodies (1:200 dilution, Abcam, Cambridge, UK) were used for visualization of the immunoreactivities. For phospho-p38 and TREM2 stainings biotinylated secondary antibody (1:200 dilution, Vector Laboratories, Burlingame, CA, USA) was used, followed by reaction with avidin-biotin complex reagent (1:200 dilution, Vector Laboratories, Burlingame, CA, USA) according to the instructions provided by the manufacturer. The bound phospho-p38 immunoreactivity was then visualized by development with nickel-enhanced 3,3′-diaminobenzidine, and TREM2 was visualized following the instructions of Tyramide Signal Amplification -kit (Perkin Elmer, MA, USA).
For quantification of the immunoreactivities in the peri-ischemic area, a 718 × 532 μm cortical area adjacent to the infarct border was imaged using 10× magnification on an AX70 microscope (Olympus corporation, Tokyo, Japan) with an adjacent digital camera (Color View 12 or F-View, Soft imaging system, Muenster, Germany) running AnalySis software (Soft Imaging System). For CD45 and neutrophil stainings, images of equal size were taken from the lesion area, where the majority of the immunoreactivity was observed. Quantification of the immunoreactivities was performed using ImagePro Plus software (Media Cybernetics Inc., Rockville, MD, USA) at a predefined range and presented as relative immunoreactive area. All analyses were performed blinded to the study groups.
2.9. Brain cell isolation and flow cytometry
4–6 mice from each treatment group were perfused transcardially at 3 dpi with heparinized saline as above. Brains (cerebellum removed) were isolated and placed on ice in Hanks balanced salt solution (ThermoFisher Scientific, Waltham, MA, USA). The tissue was minced on ice in digest buffer with 0.5 mg/ml collagenase type IV (Worthington, Lakewood, NJ, USA), 25 u/ml DNAse I and RPMI-1640 (both from Sigma-Aldrich), and after that incubated for 20 min at +37 °C, 5% CO2. The tissue was put back on ice, triturated and passed through 70 μm and 40 μm cell strainers (Falcon, Corning, NY, USA). Homogenates were centrifuged at 450g for 5 min, resuspended in Miltenyi Myelin Debris Removal Beads II (Miltenyi, Cologne, Germany) and incubated at +4 °C for 15 min. After incubation the samples were washed with cold MACS buffer (PBS including 0.5% Bovine Serum Albumin, both from Sigma-Aldrich) and centrifuged at 400g for 10 min, after which the pellets were resuspended into cold MACS buffer. Samples were applied to magnetic LD columns (Miltenyi) using 50 μm CellTrics pre-filters (Sysmex, Norderstedt, Germany), and the flow-through was collected on ice, counted and spun at 400g for 10 min, and finally resuspended in RPMI-1640.
Cell surface staining was done using 200,000–300,000 cells per mouse in round-bottom polypropylene 96-well-plates (Corning). Cells were washed with PBS, stained with Zombie NIR fixable viability dye (BioLegend, San Diego, CA, USA) for 15 min at RT, then blocked with CD16/32 (clone 24G2, BD Biosciences, San Jose, CA, USA) and stained with the following antibodies: CD45 PerCP-Cy5.5 (clone 30 F11, eBioscience, San Diego, CA, USA), CD11b PECy7 (clone M1/70, eBioscience), Ly6G FITC (clone 1A8, BioLegend) F4/80 PE (clone A3–1, AbD Serotec, Oxford, UK), and Ly6C APC (clone AL-21, BD Biosciences) for 30 min at +4 °C. Cells were washed twice with PBS and fixed with 0.5% Ultra Pure formaldehyde (ThermoFisher Scientific, Waltham, MA, USA). Samples were acquired on BD FACSAria III equipped with 488- and 633-nm lasers with standard configuration. Data were analyzed with FCSExpress v5 (DeNovo Software Glendale, CA, USA).
2.10. Quantitative real-time PCR
The relative expression levels of inducible nitric oxide synthase iNOS (NOS2), macrophage receptor with collagenous structure (Marco), interleukin 1 beta IL-1β (Il1b), interleukin 6 IL-6 (Il6), matrix metallopeptidase 9 (Mmp9) and complement component 3 (C3) were measured from primary microglia exposed to LPS and HX600 using quantitative PCR (qPCR). Total RNAs were isolated using RNeasy mini kit (Qiagen, CA, USA) according to the manufacturer’s instructions. The concentration and purity of RNA samples were determined using a NanoDrop 2000 (Thermo Fisher Scientific). Complementary DNA (cDNA) was synthesized by using 500 ng of total RNA, Maxima reverse transcriptase, dNTP and random hexamer primers (Life technologies, CA, USA). The final concentration of cDNA was 2.5 ng/μl. qPCR was run using the StepOne Plus Real Time PCR system (Applied Bioscience, CA, USA). Gene expression analysis was carried out using comparative Ct method (ΔΔCT) where the threshold cycle of the target genes was normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH, ThermoFisher Scientific, Waltham, MA, USA) and ribosomal RNA (S18 endogenous calibrator, ThermoFisher Scientific, Waltham, MA, USA) internal housekeeping gene controls (ΔCT). The mRNA expression is presented as a fold change.
2.11. Microglia morphology measurement
The morphological analysis of Iba-1 expressing cells was done as previously described (Huuskonen et al., 2017) at one day after ischemia. Area and perimeter of the cells, along with other parameters, were analyzed using Matlab v2013b. Zeiss LSM 700 confocal microscope (Zeiss Inc., Maple Grove, USA) combined with digital camera (Color View 12 or F-View; Soft Imaging System, Münster, Germany) running Zen 2012 Image Analysis Software (Zeiss Inc., Maple Grove, USA) was utilized in capturing the images for the analysis.
2.12. Metabolic profiling
Tissue samples from the peri-ischemic area and from the contralateral side, collected at 1 dpi as described above, were weighed and 80% methanol was added (v/v H2O, LC-MS Ultra CHROMASOLV®, Fluka) in a ratio of 350 μL solvent/100 mg tissue. Extraction was facilitated by breaking the tissue with Teflon coated plastic staff followed with water sonication for 10 min (BRAUSON GWB 2200). Samples were then vortexed and centrifuged (Eppendorf centrifuge 5804 R, 13000 rpm, 5 min at 4 °C). Supernatant was then collected and filtered to HPLC bottles through Acrodisc CR 4 mm (0.45 μm) filter. Quality control samples were made by taking 5 μL of solution from each sample.
The samples were analyzed using a UHPLC-qTOF-MS system (Agilent Technologies, Waldbronn, Karlsruhe, Germany) that consisted of a 1290 LC system, a Jetstream electrospray ionization (ESI) source, and a 6540 UHD accurate-mass qTOF spectrometry as described in Puurunen et al. (2016). Two different chromatographic techniques were used, i.e. reversed phase (RP) and hydrophilic interaction (hilic) chromatography and acquired data in both positive (+) and negative (−) polarity. The sample tray was kept at 4 °C during the analysis. MassHunter Acquisition B.04.00 (Agilent Technologies) software was used for data acquisition. The quality control samples were injected in the beginning of the analysis as well as every 12th injection. The analysis order of the samples was randomized.
The mass spectrometry data processing was performed using MassHunter Profinder B.06.00 (Agilent Technologies, USA). The batch recursive feature extraction function was used to extract ion to molecular features exhibiting isotopic peaks, dimers, and common adducts. Final alignment and quality control of peak spectra were done manually. The data were transferred as compound exchange format files into the Mass Profiler Professional (MPP) software (version 13, Agilent Technologies) for statistical analysis. MS-DIAL ver. 2.52 (Tsugawa et al., 2015) and MassHunter Qualitative Analysis B.07.00 (Agilent Technologies, USA) softwares were used for metabolite identification against MSMS spectra found in public and in-house standard libraries (respectively).
2.13. Statistics
Statistical analyzes were performed using GraphPad Prism software 5.03 (GraphPad Software, LaJolla, CA, USA), with either unpaired two-tailed t-test, one-way ANOVA or two-way ANOVA as appropriate. Cohen’s d effects sizes (mean(cases) − mean(controls)/((SD(cases) + SD (controls))/2) were calculated for the metabolomics data. The statistical test used and n-numbers are indicated in each figure legend. p-values less than 0.05 were considered to be statistically significant. Statistically significant outliers as calculated using GraphPad Prism QuickCalcs were excluded from the datasets. All data are presented as mean ± standard deviation (SD), unless mentioned otherwise.
3. Results
3.1. HX600 is not directly neuroprotective against glutamate exposure in vitro
Since Nurr1 activation has been shown to confer neuroprotection in models of PD and in vitro in neuropathological conditions (Volakakis et al., 2010), we analyzed whether HX600 is neuroprotective in primary cortical neurons exposed to glutamate mimicking ischemia-related excitotoxicity in vitro. Primary cortical neurons were exposed to 100 nM and 1 μM concentrations of HX600 in the presence of glutamate for 24 h, with or without 24 h pretreatment with HX600. Neuronal viability measurement using MTT revealed that HX600 is unable to prevent the direct, glutamate induced neuronal death Fig. 1).
Fig. 1.
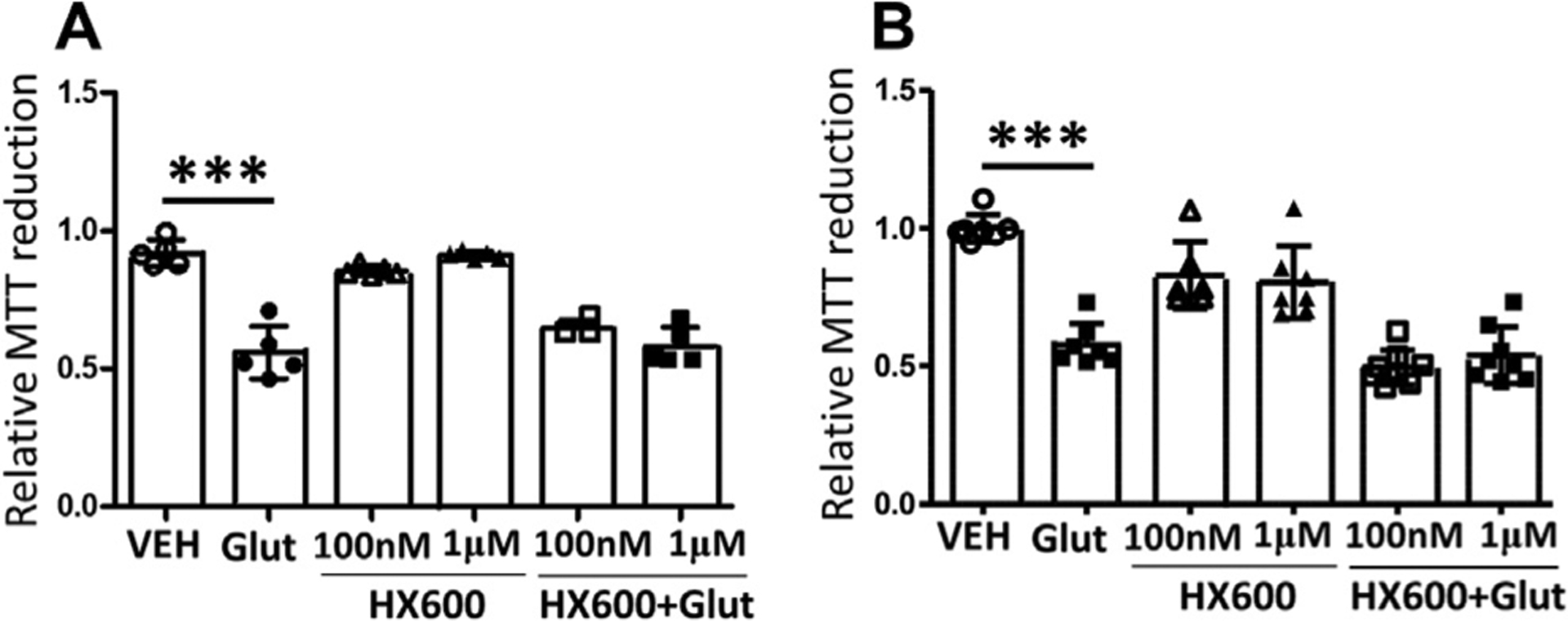
HX600 is not directly neuroprotective against glutamate induced neuronal death. Primary cortical neurons were exposed to 100 nM or 1 μM final concentration of HX600 together with 500 μM glutamate (Glut) for 24 h (A). Alternatively the cells were pretreated with HX600 for 24 h, and thereafter exposed to glutamate for another 24 h while concentration of HX600 was kept unaltered (B). Neuronal viability was measured using MTT assay. HX600 alone did not affect neuronal viability, but neither of the concentrations of HX600 was able to prevent the glutamate induced neuronal death. n = 5 wells in each group. The assay was repeated four times with similar results. 1-way ANOVA with Bonferroni post test, ***p < 0.0001.
3.2. HX600 inhibits the expression of inflammatory mediators in primary microglia and prevents inflammation induced neuronal death
Due to the known ability of Nurr1 to exhibit anti-inflammatory functions, we evaluated whether HX600 alleviates microglial proinflammatory activation. Primary microglia were pretreated with HX600 for 24 h followed by exposure to LPS for 3 h, after which the mRNA levels of inflammatory mediators were measured by qPCR. HX600 significantly reduced the expression of NOS2 (iNOS), Marco, Il1b (IL-1β), Il6 (IL-6) and Mmp9 in LPS treated primary microglia Fig. 2A–E) but failed to alter the expression level of C3 Fig. 2F). To evaluate whether the anti-inflammatory effect of HX600 is able to confer neuroprotection, we used a co-culture model in which primary microglia or BV2 cells are plated on top of primary neurons and exposed to LPS and INF-γ resulting in inflammation induced neuron death. Pretreatment of the neuron – primary microglia co-cultures with HX600 significantly prevented the inflammation induced neuronal death as assessed by MAP-2 immunoreactivity Fig. 3A, B). To confirm that the effect of HX600 is mediated through induction in Nurr1, BV2 cells were transfected with Nr4a2 siRNA. This resulted in 40% knock-down in the expression level of Nurr1 Fig. 3C). Transfection of BV2 cells with Nr4a2 siRNA prevented the HX600 induced protection against inflammation induced neuronal death Fig. 3D), whereas mock transfection of BV2 cells with control siRNA failed to prevent the HX600 effect Fig. 3E).
Fig. 2.
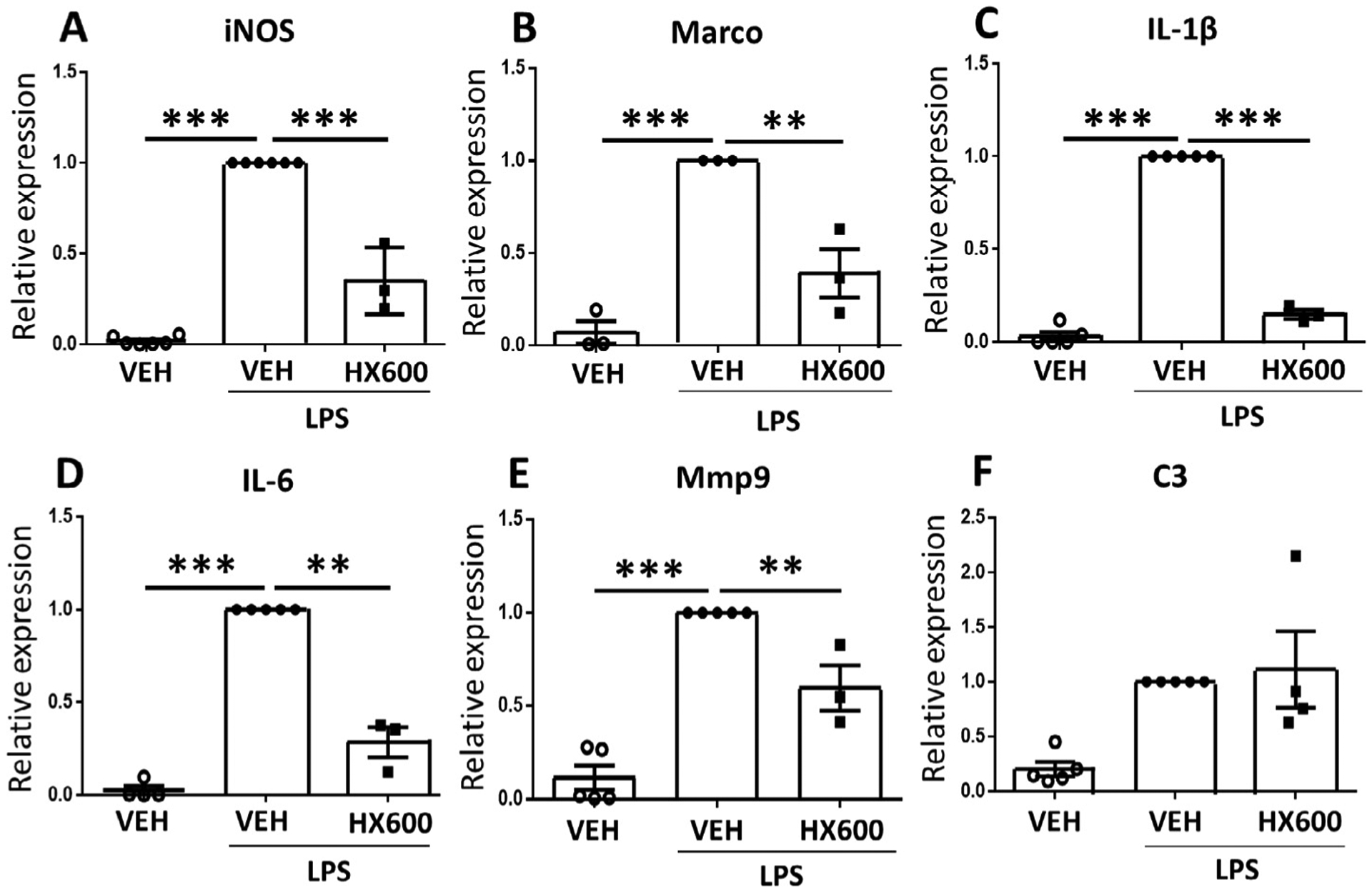
HX600 reduces expression of inflammatory mediators in microglia. Primary microglia were pre-exposed to HX600 for 24 h after which the cells were stimulated with LPS for 3 h. Stimulation of NR4A pathway by HX600 reduced the expression of mRNA levels of NOS2 (iNOS, A) and scavenger receptor Marco (B), Il1b (IL-1β, C), Il6 (IL-6, D), Mmp9 (E) all of which are involved in inflammatory activation of microglia. HX600 failed to alter the expression of C3 (F). Data presented as mean ± SEM, 1-way ANOVA with Bonferroni posthoc test, n = 3–5, **p < 0.01 and ***p < 0.001.
Fig. 3.
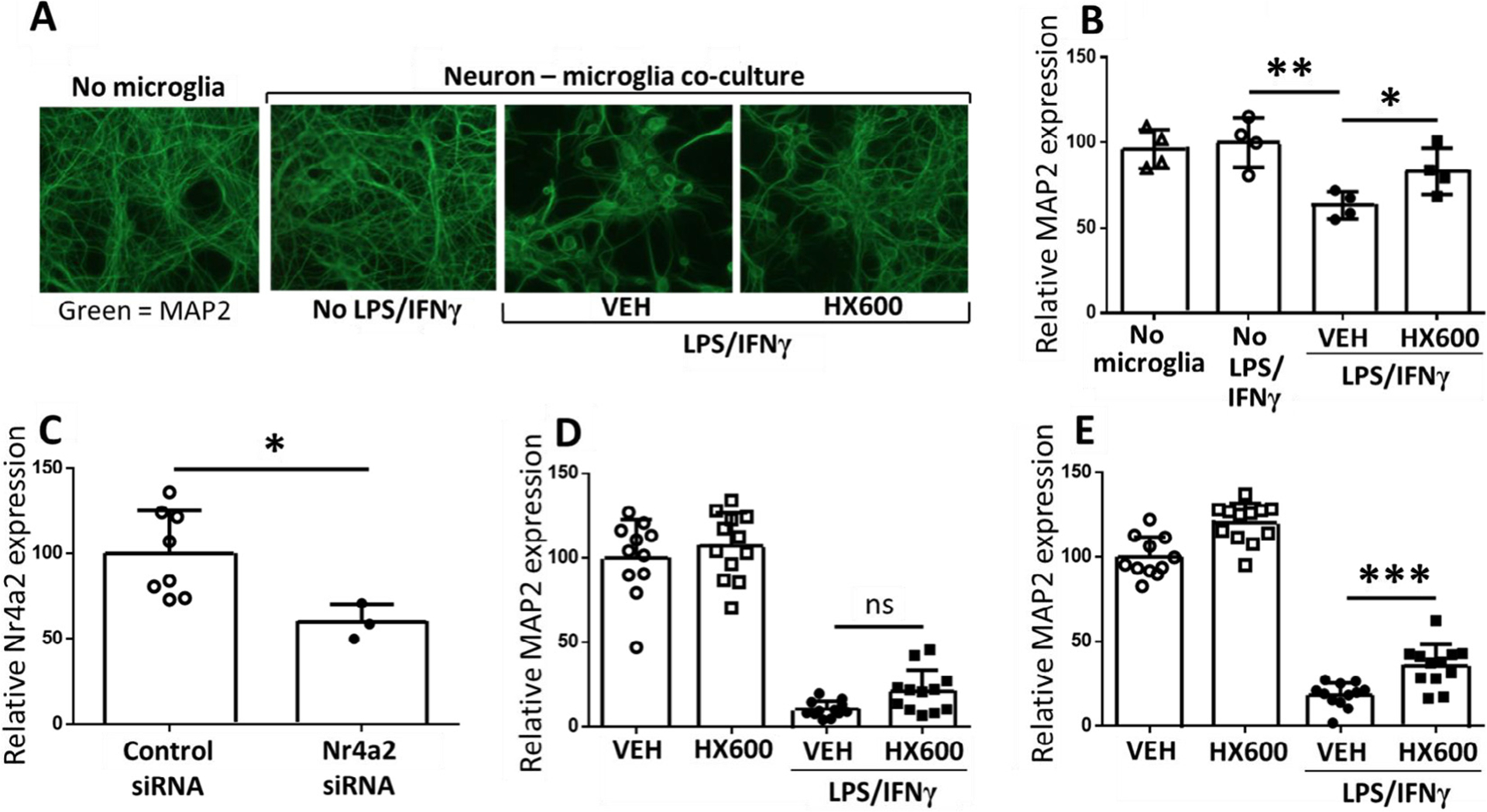
HX600 protects neurons from LPS induced death. Primary microglia or BV2 cells were added on top of primary cortical neurons, and the co-cultures were pre-exposed to HX600 or vehicle for 6 h. After that the cultures received either vehicle or LPS/IFNγ treatment, while HX600 concentration was maintained at 1 μM. HX600 significantly prevented neuronal death. Representative images of the neurons stained with MAP-2 (A) and quantitative analysis (B) from a primary microglia – neuron co-culture. Transfection of BV2 cells with Nurr1 siRNA resulted in 40% knock-down of Nurr1 (C). When the neurons were co-cultured with Nurr1 siRNA transfected BV2 cells, the protective effect of HX600 against the inflammation induced neuronal death was abolished (D). Mock transfection of BV2 cells with control siRNA did not prevent the protection (E). Data presented as mean ± SD, 1-way ANOVA followed by Bonferroni posthoc test (B, D and E) and unpaired two-tailed T-test (C), n = 3–12 wells in each group, each assay repeated three times. *p < 0.05 **p < 0.01 ***p < 0.001.
3.3. HX600 reduces ischemic damage and alleviates motor deficits in permanent ischemia model
To evaluate whether HX600 provides neuroprotection in vivo, HX600 was administered to ischemic mice immediately after ischemia surgery and thereafter every 24 h. Quantification of the lesion volumes at 48 h post stroke revealed that mice treated with HX600 had lesion size 21% smaller than vehicle treated mice Fig. 4A). Fig. 4 B and 4C depict typical examples of the MRI-images of mice treated with vehicle or HX600, respectively. Mice subjected to pMCAo exhibited significantly impaired locomotor activity as measured by Latency-to-move test at 1 dpi; stroke caused the latency time to double when compared to sham-operated mice. These deficits were no longer evident in animals treated with HX600 Fig. 4D).
Fig. 4.
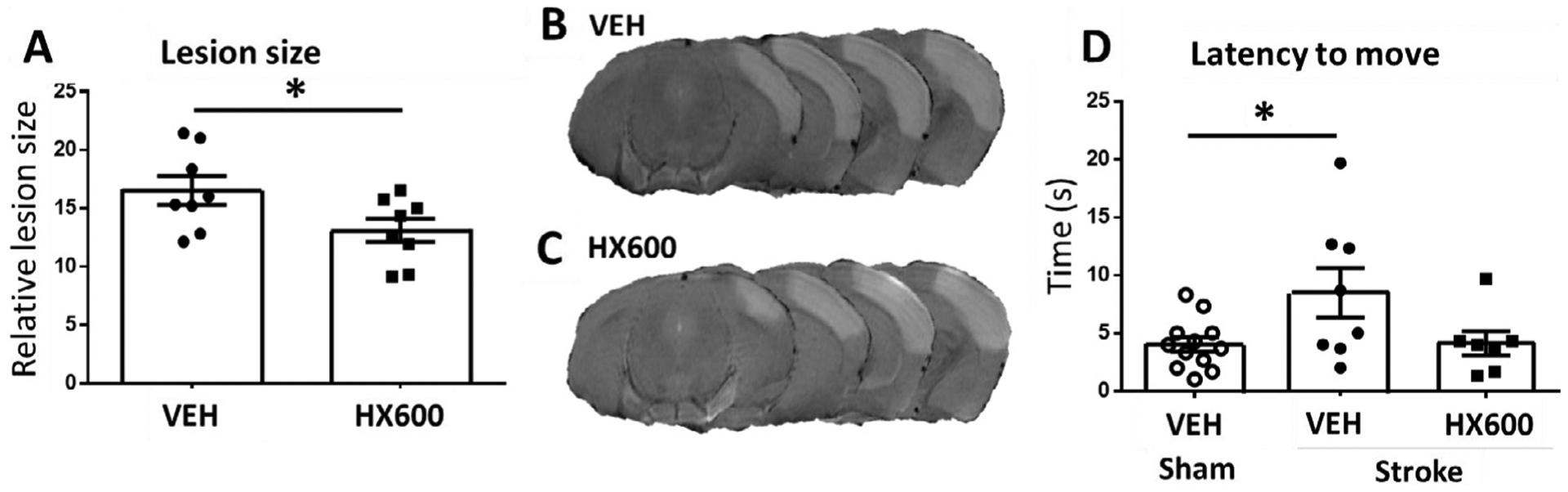
HX600 reduces ischemic damage and improves motor functions in vivo. Lesion size was measured with MRI imaging 48 h after the ischemic insult. Among the HX600 treated mice it was 21% smaller when compared to mice treated with vehicle. Quantitative analysis (A) and representative MRI-images from mice receiving vehicle (B) and HX600 treatment (C). In Latency to move -test ischemia caused clear deterioration of motor function 1 day after the insult (D). HX600 treatment brought the performance back to the level of sham operated mice. Data presented as mean ± SEM, Unpaired two-tailed T-test (A) and 1-way ANOVA followed by Bonferroni posthoc test (D), A: n = 8 in both groups, B: n = 8 in vehicle treated stroke group, 7 in HX600 treated stroke group and 12 in sham group. *p < 0.05.
3.4. HX600 reduces Iba-1, phospho-p38 and TREM-2 immunoreactivities in the ischemic brain
In order to evaluate the impact of HX600 treatment on inflammatory activation in the ischemic brain, series of immunohistochemical stainings were carried out to assess which cell types of the brain are affected by the treatment. HX600 had no effect on CD45 (a marker for microglia/macrophages), GFAP (a marker of astrocytic activation), CD68 (a marker for cells with phagocytic activity) or Ly6G neutrophil immunoreactivities (data not shown), but instead, it markedly reduced the extent of Iba-1 (a marker for activated microglia), phospho-p38 (activated by e.g. inflammatory cytokines) and TREM2 (a marker of phagocytosis) immunoreactivities in the affected area at day 1 post ischemia Fig. 5). To further evaluate the impact of HX600 on microglial activation, we carried out quantitative analysis of microglial morphology which showed that HX600 increased the Iba-1 positive cell perimeter and decreased the Area/Perimeter -ratio Fig. 6).
Fig. 5.

HX600 reduces Iba-1, phospho-p38 and TREM2 immunoreactivities in the ischemic brain. Quantitative analysis of the Iba-1, phospho-p38 and TREM2 immunoreactivities (A–C), and typical representative images of group receiving vehicle (D–F) or HX600 treatment (G–I) at time point 1 day after the insult. Immunoreactivities of Iba-1 and phospho-p38 were analyzed from the peri-ischemic area, and TREM2 at the lesion site. Scale bar 100 μm. Data presented as mean ± SD. Unpaired two-tailed t-test, n = 8 in each group. *p < 0.05 ***p < 0.001.
Fig. 6.
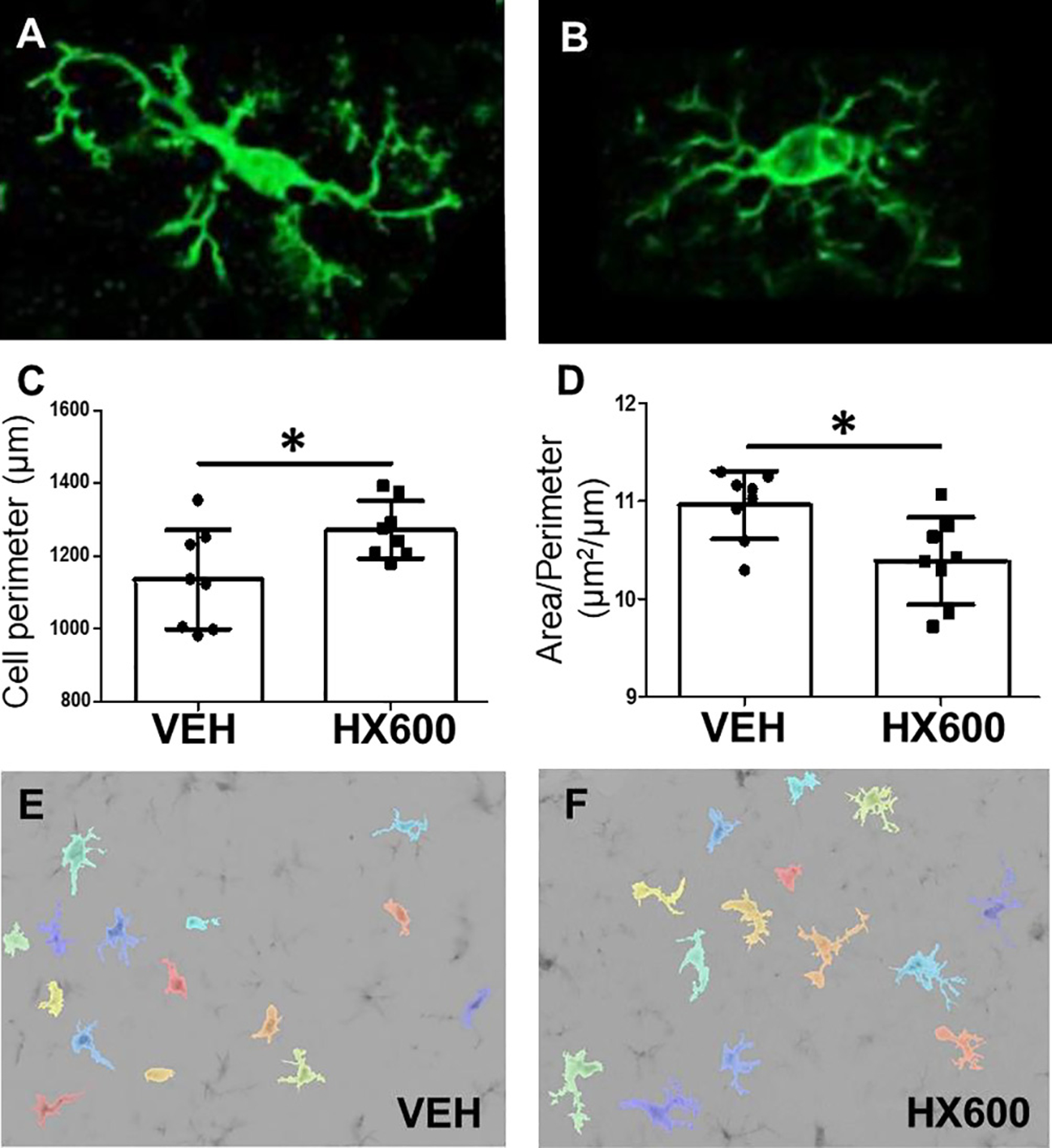
HX600 treatment alters morphology of Iba-1 expressing microglia. Microglia in resting state (A) are characterized by long processes and relatively small soma. When the cells get activated (B), their soma gets larger and processes are shortening, which leads to decreased cell perimeter and increased Area/Perimeter – ratio. Quantitative analysis of cell perimeter (C) and area/perimeter – ratio (D) of Iba-1 positive cells, and representative images of the algorithm used to determine the morphology of microglia in mice treated with vehicle (E) or HX600 (F). Microglia are pseudocolored to aid identification. Data presented as mean ± SD. Unpaired two-tailed t-test, n = 8 in each group. *p < 0.05.
3.5. HX600 treatment normalizes proportions of CD45Hi CD11bLow and Ly6CHi CD45Low cells in the ischemic brain
In order to more specifically evaluate the immune cells of the ischemic brain affected by HX600 treatment, we analyzed proportions of different immune cell types using flow cytometry on freshly isolated cells from the ischemic animals at 3 days post stroke, the time of the peak in leukocyte infiltration. As expected, ischemia induced a significant 59% increase in infiltrating CD45Hi CD11bLow lymphocytes Fig. 7B) and 53% increase in Ly6CHi CD45Low cells, which can be either infiltrating monocytes or activated microglia Fig. 7C), compared to sham operated mice. The levels of these infiltrating cell populations were significantly reduced in HX600 treated mice. Of note is that ischemia induced a small, yet significant loss of resident CD45Low F4/80Hi Ly6CLow microglia, which was prevented by HX600 treatment Fig. 7D).
Fig. 7.
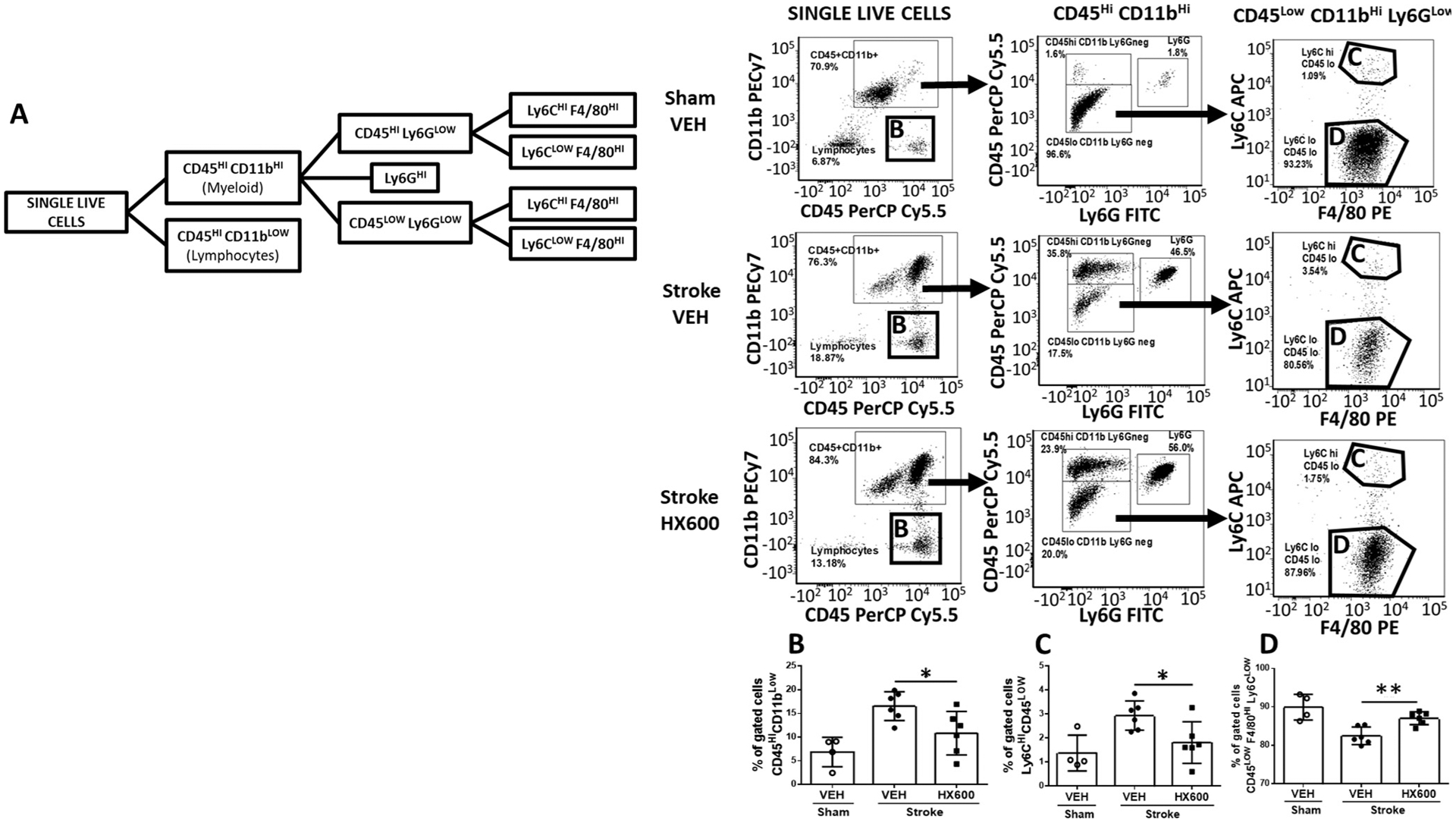
HX600 reduces proportions of lymphocytes and infiltrating macrophages at 3dpi. Gating strategy showing the cell populations analyzed (A). Quantified ratios of CD45Hi CD11bLow infiltrating lymphocytes (B) and Ly6CHi CD45Low infiltrating monocytes or activated microglia (C) and CD45Low F4/80Hi Ly6CLow resident microglia (D). Data presented as mean ± SD. 1-way ANOVA followed by Bonferroni posthoc, n = 6 in stroke groups, n = 4 in sham group. *p < 0.05.
3.6. HX600 treatment normalizes metabolic profile in the peri-ischemic area
To evaluate stroke induced changes in the levels of metabolites as well as those altered by the HX600 treatment, we carried out high-throughput metabolic profiling analysis of the peri-ischemic and the contralateral brain samples (1 dpi) of the HX600 or vehicle treated stroked mice. Results of the analysis are shown in Figs. 8 and 9, and in Supplementary Table 1.
Fig. 8.
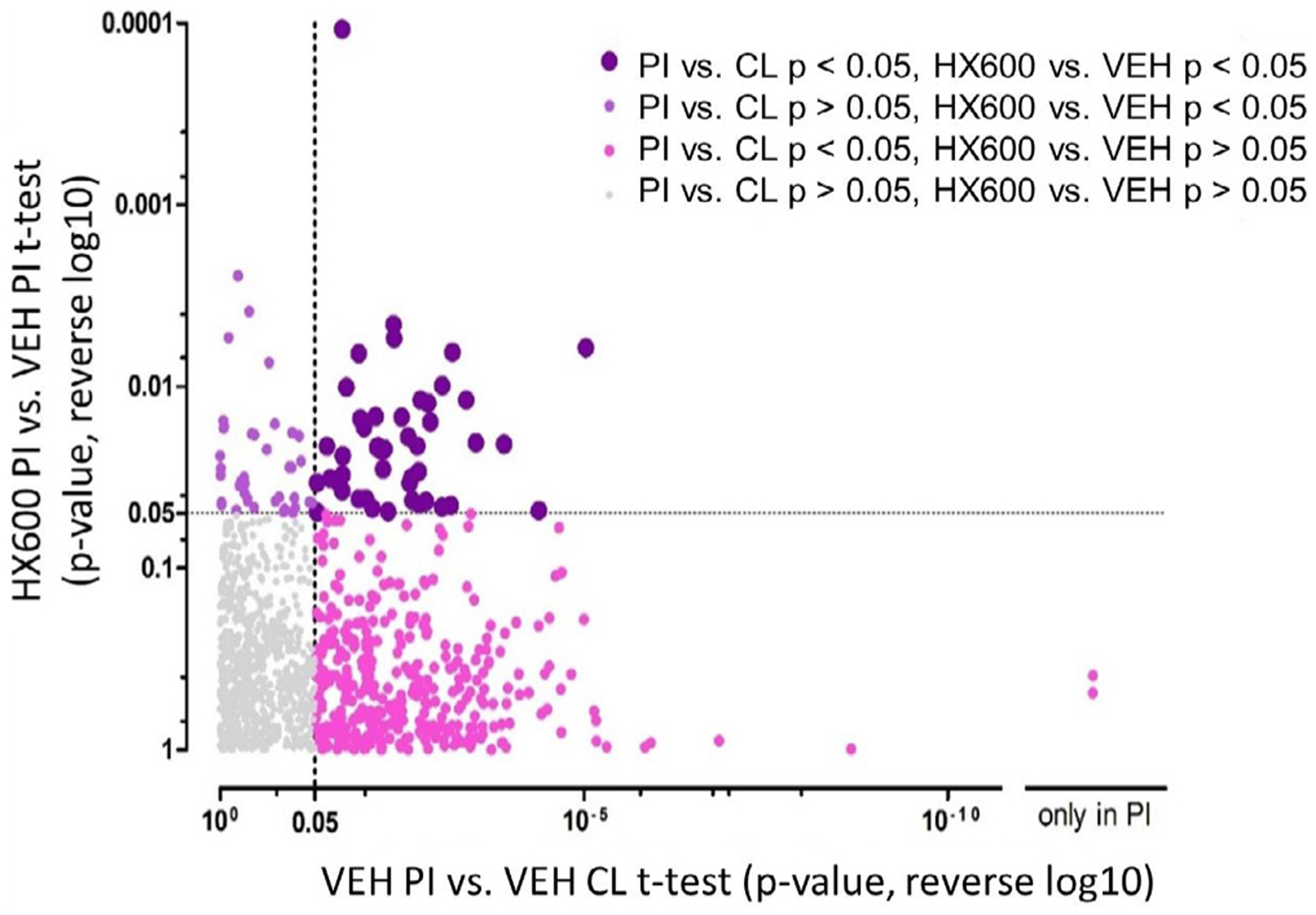
Overview of the metabolite profiling results. 1220 molecular features were detected from the brain samples collected at 1 dpi. When control group brain samples from the ischemic side (PI) were compared to the contralateral side (CL), we observed 414 significantly altered molecular features (x-axis showing reversed log p -values from this comparison). From these, 47 molecular features were significantly different between the ischemic side brain samples from HX600 treated animals and controls (top right quadrant). These molecular features in the top right quadrant were the ones where we observed an effect of both the ischemia and HX600 treatment. Molecular features in the bottom right quadrant were affected by the ischemia but not by the HX600 treatment, and molecular features in the top left quadrant were affected by the HX600 treatment but not by ischemia. CL = contralateral side brain sample, PI = ischemic side brain sample, HX600 = HX600 treated group, VEH = vehicle treated group, p = p-value from t-test. n = 6 mice in each group.
Fig. 9.
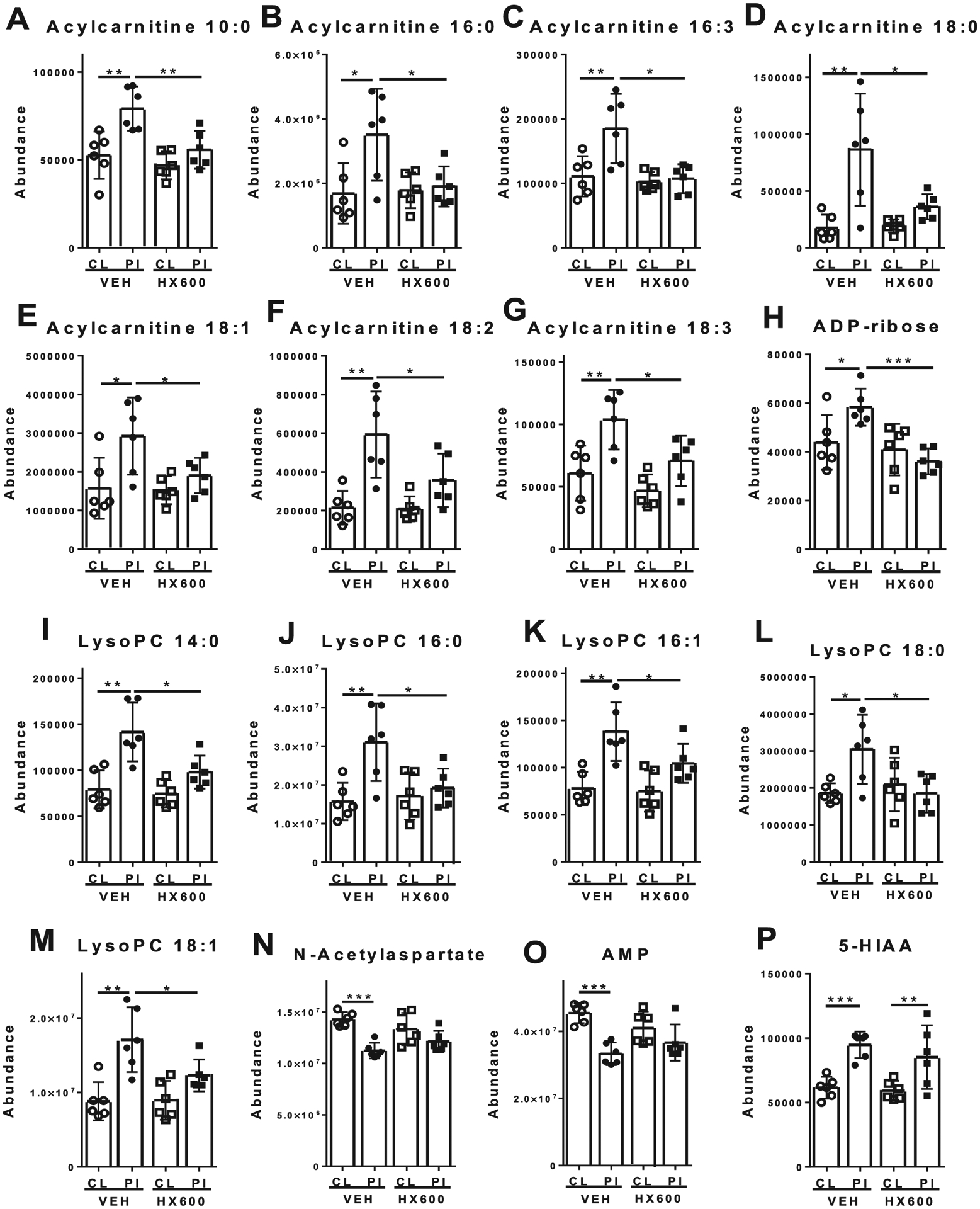
HX600 normalizes levels of acylcarnitines and LysoPCs in the ischemic side of the brain. HX600 treatment normalized the stroke-induced levels of acylcarnitines (A-G), adenosine diphosphate ribose (ADP-ribose, H), and lysophosphatidylcholines (lysoPCs, I-M) in the peri-ischemic area compared to vehicle treated controls. HX600 failed to prevent the stroke-induced alterations in the levels of N-acetylaspartate (NAA; N), adenosine 5′-monophosphate (AMP; O) and 5-hydroxyindoleacetic acid (5-HIAA; P). Data presented as mean ± 95% confidence interval. CL = contralateral side brain sample, PI = ischemic side brain sample, VEH = vehicle treated mice, HX600 = HX600 treated mice. n = 6 mice in each group. *p < 0.05, **p < 0.01, ***p < 0.001.
Supplementary data associated with this article can be found, in the online version, at https://doi.org/10.1016/j.bbi.2018.07.021.
Ischemia was associated with alterations in levels of multiple metabolites compared to the contralateral hemisphere (Supplementary Table 2). These included significantly increased levels of several acylcarnitines (e.g. hexanoylcarnitine, acylcarnitine 18:0, acylcarnitine 18:2, and acylcarnitine 22:6), lysophosphatidylcholines (lysoPCs, e.g. lysoPC 14:0, lysoPC 16:0, lysoPC 18:1, lysoPC 20:4 and lysoPC 22:6), as well as amino acid metabolites (e.g. 5-hydroxyindoleacetic acid [5-HIAA], and trigonelline). On the contrary, ischemia resulted in decrease in several metabolites including amino acids and amino acid metabolites (e.g. N-acetylaspartate [NAA], and glutamate), nucleosides and their metabolites (e.g. adenosine, adenosine 5′-monophosphate [AMP], guanosine, and guanosine 5′-monophosphate [GMP]), and metabolites associated with antioxidant responses (e.g. glutathione and oxidized glutathione). Of note is that when using glutamate exposures to mimic stroke induced excitotoxic insult, it reflects the situation immediately after stroke, whereas these reduced metabolite levels are observed after longer time period when the neurons are already dead.
HX600 treatment normalized the ischemia induced alterations in several metabolites (Fig. 9). Specifically, HX600 prevented the ischemia-induced increase in the levels of acylcarnitine 10:0, acylcarnitine 16:0, acylcarnitine 16:3, acylcarnitine 18:0, acylcarnitine 18:1, acylcarnitine 18:2, and acylcarnitine 18:3 (Fig. 9). Similarly, ischemia induced increase in the levels of lysoPC 14:0, lysoPC 16:0, lysoPC 16:1, lysoPC 18:0, lysoPC 18:1 and ADP-ribose in HX600 treated animals were normalized to the level of the unaffected brain hemisphere and that of vehicle treated controls.
4. Discussion
The nuclear receptor NR4A2/Nurr1 is well known for its ability to confer protection in models of PD. NR4A activation also supports memory enhancement (Hawk et al., 2012) and mediates neuronal survival in neuropathological conditions (Volakakis et al., 2010). Yet, there is very little evidence about the role of NR4A in cerebral stroke. Merino-Zamorano C et al found that Nurr1 induction in endothelial cells after delayed administration of r-tPA contributes to endothelial dysfunction, suggesting that Nurr1 activation may contribute to side-effects of r-tPA treatment (Merino-Zamorano et al., 2015). However, no studies have assessed whether NR4A agonism confers neuroprotection in ischemic stroke.
HX600 has been shown to have pan-agonistic activity on RXRs, and additionally it is able to synergize with retinoic acid receptor (RAR) (Umemiya et al., 1997; Umemiya et al., 1997). In addition, HX600 can activate nuclear receptor NR4A1/Nurr77 (Morita et al., 2005; Ishizawa et al., 2012). Our in vitro data Fig. 3 indicate that the beneficial effects of HX600 may be mediated through Nurr1 receptor activation. However, we cannot rule out the possibility of HX600 activating also other RXR heterodimers and RARs in vivo contributing to the beneficial effects of the treatment.
Here we report for the first time that HX600, an activator for Nurr1/Nurr77, has anti-inflammatory effects both in vitro and in vivo. Although it failed to directly prevent neuronal death against glutamate induced toxicity, it prevents inflammation induced neuronal death and facilitates neuronal survival post stroke. At the brain metabolite level, HX600 treatment normalized the levels of metabolites related to inflammatory and oxidative functions. Considering the known ability of Nurr1 to inhibit inflammatory reactions (Saijo et al., 2009; Ishizawa et al., 2012; Lallier et al., 2016; McMorrow and Murphy, 2011) we propose that targeting Nurr1 provides a novel therapeutic target for ischemic stroke. However, further studies are needed to evaluate whether HX600 can provide protection with delayed administration and whether the protection persist over longer time periods.
HX600 reduced the expression levels of several pro-inflammatory mediators, including NOS2 (iNOS), Marco, Il1b (IL-1β), Il6 (IL-6) and Mmp9 in primary microglia exposed to LPS. All of the analyzed inflammatory mediators are induced in conditions of ischemic stroke (Milne et al., 2005; Niwa et al., 2001; Lambertsen et al., 2012). Marco is a scavenger receptor expressed in microglia and macrophages, and its upregulation has been closely linked to microglial activation and phagocytosis (Granucci et al., 2003) and Mmp9 is a proteinase with the ability to degrade extracellular matrix proteins and in conditions of stroke, cause blood brain barrier (BBB) damage and aggravate further proinflammatory responses (Cauwe et al., 2007; Sandoval and Witt, 2008). iNOS is released by glial cells during inflammation leading to excess amount of nitric oxide which in turn results in tissue damage (Abdel-Salam et al., 2017). iNOS has been shown to be mainly responsible for the inflammation induced neuron death in the microglia-neuron co-culture model (Gresa-Arribas et al., 2012) and thus it is not surprising that HX600 potently prevented the inflammation induced loss of MAP-2 immunoreactivity in this model.
Despite of Nurr1 receptor being abundantly expressed in cortical neurons (Yasuyoshi et al., 2003), HX600 failed to show any benefit in our in vitro -model of excitotoxicity. This may be due to the fact that in pure cortical neuron cultures the neurons are lacking the support they get from the surrounding cells in vivo when such a crisis occurs. Glutamate induced neuronal death is caused by loss of intracellular calcium homeostasis (Vaarmann et al., 2013). Cytosolic calcium concentration is rising shortly after glutamate exposure, goes down again and after that a delayed, more sustained elevation follows, causing a severe disruption to the mitochondrial membrane potential and subsequent neuronal death (Stout and Reynolds, 1999; Abramov and Duchen, 2010). It may be that mere neuronal Nurr1 induction by itself is not enough to save the neurons from a severe insult like this, situation being different in vivo where HX600 affects also the surrounding non-neuronal cells, including microglia and infiltrating immune cell suppressing their activation and subsequently alleviating the stroke induced secondary neuronal damage. Whether HX600 also suppresses the expression of chemotactic factors by brain resident cells is possible, yet beyond the scope of this study.
The anti-inflammatory capacity of HX600 was further outlined by the fact that HX600 treated animals showed reduced levels of Iba-1 and phospho-p38, a serine/threonine protein kinase with important role in microglial activation and inflammatory cytokine production (Bachstetter et al., 2011). The anti-inflammatory effect of HX600 driven Nurr1 activation is likely to be mediated through the previously described capacity of Nurr1 to diminish NFkB activation (Saijo et al., 2009; De Miranda et al., 2015). Indeed, NFkB has been shown to drive the expression of iNOS (Marks-Konczalik et al., 1998; Ozawa et al., 2004). In addition, p38 has been shown to be able to activate NFkB (Madrid et al., 2001) and concomitantly, inhibition of p38 and NFkB signaling is beneficial in ischemic stroke (Koistinaho et al., 2002; Nurmi et al., 2004; Zhao et al., 2017; Rolova et al., 2016).
The anti-inflammatory capacity of HX600 also impacted microglia morphology. Microglial cells in the healthy brain are in ramified shape with long processes constantly inspecting the environment (Nimmerjahn et al., 2005). When microglia get activated, they adopt more amoeboid morphology characterized by shorter processes which are also fewer in number, and larger soma compared to a ramified cell (Perry et al., 2010). These changes affect the area and perimeter of the cell; the rounder the shape gets, the shorter is the perimeter and the larger the area/perimeter -ratio. The microglia morphology analysis showed that Iba-1 positive cells in the peri-ischemic area of HX600 treated mice have larger perimeter and smaller area/perimeter -ratio when compared to vehicle treated mice, being one more attestation towards the anti-inflammatory capacity of HX600.
As infiltrating leucocytes contribute to ischemia induced neuroinflammation, we carried out flow cytometry analysis to investigate how HX600 alters immune cell infiltration, and chose the time point of the peak in macrophage infiltration (Huuskonen et al., 2017). Our data show that HX600 reduced the proportion of infiltrating CD45Hi CD11bLow lymphocytes and Ly6CHi CD45Low infiltrating monocytes (or activated microglia), thus contributing to the reduced pro-inflammatory milieu in the post-ischemic brain. Moreover, HX600 prevented the loss of endogenous microglia suggesting protection of microglia against ischemic insult.
Surprisingly the extent of TREM2 immunoreactivity was reduced in the ischemic lesion of HX600 treated animals. TREM2 is most prominently expressed in the brain by microglia (Colonna, 2003), although the level of expression may vary between different brain areas (Schmid et al., 2002). TREM2 has been suggested to facilitate recovery from brain injuries by promoting phagocytosis and dampening inflammation (Hsieh et al., 2009; Kawabori et al., 2015). However, recent studies indicate that downregulation of TREM2 is unexpectedly mitigating inflammation and microglial activation, and is protective against neuronal damage (Kobayashi et al., 2016; Leyns et al., 2017; Saber et al., 2016; Sieber et al., 2013). One possible mechanism for this seemingly contradictory result might be found from a study conducted by Charles et al (Charles et al., 2008), where bacterial infection was seen to induce TREM2 expression which in turn contributed to reactive oxygen species (ROS) production and aggravation of inflammation (Charles et al., 2008). Since ROS production is heavily involved in inflammation following stroke (Sanderson et al., 2013), it is possible that downregulation of TREM2 expression is affecting inflammation reaction via ROS production similarly as in inflammation induced by bacteria. Of note is that it may take several days until TREM2 expression increases after ischemia; in both permanent (Kawabori et al., 2015) and transient (Sieber et al., 2013) models the peak in TREM2 levels was observed at 7 days after the insult. Since in this study TREM2 immunoreactivity was quantified at the lesion area in early time point, the stained cells were most likely infiltrating macrophages or leukocytes and thus the reduction in TREM2 may reflect the overall reduced number of infiltrating cells.
Ischemic stroke is associated with alterations in several brain metabolites, many of which are associated with inflammatory functions. Only a few papers have described metabolic profiling of brain of rodent stroke models at various time points post stroke (Huang et al., 2018; Feng et al., 2017). Our analysis of the peri-ischemic area and the unaffected contralateral side revealed decreased levels of amino acids and amino acid metabolites (e.g. N-acetyl aspartate [NAA], and glutamate) reflective of the alterations in the excitatory processes in cerebral ischemia. In addition, stroke reduced the levels of nucleosides and their metabolites (e.g. adenosine, adenosine 5′-monophosphate [AMP], guanosine, and guanosine 5′-monophosphate [GMP]), and metabolites associated with antioxidant responses (e.g. glutathione and oxidized glutathione). These data are also in line with that of previously published in a transient ischemia model in mice (Mulder et al., 2016) and in rats (Miura et al., 2010). NAA is considered as a metabolic marker for mitochondria and related to neuronal viability (Céline et al., 2004) and our data are in line with a previous study showing decreased brain levels of NAA and glutamate both at very acute (Yan et al., 2015) as well as at 7 days post stroke in rats (Huang et al., 2018).
Of the stroke induced increases of brain metabolites, HX600 treatment normalized the levels of acylcarnitines, lysoPCs and adenosine diphosphate (ADP) ribose in the ischemic side of the brain compared to vehicle treated controls. Importantly, these molecules can be associated with ischemia induced disruption of mitochondrial function and activation of inflammatory reactions (Jin et al., 2010; Sanderson et al., 2013).
Acylcarnitines are formed from fatty acids by carnitine palmitoyltransferase 1 (CPT1) and serve as an intermediate form, which can be transported by carnitine acylcarnitine translocase (CACT, SLC25A20) into the mitochondria for β-oxidation of fatty acids. Increased levels of acylcarnitines in the peri-ischemic side of the brain may therefore be linked to disrupted mitochondrial function, which HX600 treatment seems to normalize. Importantly, acylcarnitines have been shown to activate proinflammatory signaling pathways and subsequent induction in various proinflammatory mediators, including IL-1β, IL-6 (Rutkowsky et al., 2014), and activation of NFκB pathway (Adams et al., 2009) in macrophages further suggesting the ability of HX600 to diminish inflammation.
LysoPCs are made when one fatty acid group is removed from phosphatidylcholines most often by lipoprotein-associated phospholipase A2 (Lp-PLA2). Under normal conditions, lysophospholipase and LysoPC-acyltransferase then quickly metabolize LysoPCs. However, increased LysoPC and Lp-PLA2 levels have been reported in response to ischemic stroke in animals and humans (Adibhatla et al., 2004; Koizumi et al., 2010; Cucchiara et al., 2009; Tsai et al., 2012); and elevated levels of these molecules have been associated with increased NFkB activation (Masamune et al., 2001; Jiang et al., 2014) LysoPCs increase the recruitment of macrophages and microglia when released from apoptotic cells and adjacent neurons and astrocytes (Lauber et al., 2003; Inose et al., 2015). Thus, increased levels of LysoPCs in the ischemic brain may be associated with increased recruitment of macrophages and microglia, which has been considered to contribute to the ischemic brain injury (Jin et al., 2010) and therefore, HX600 may be able to reduce this recruitment by normalizing the levels of LysoPCs.
ADP-ribose is a building block of polyADP-ribose (PAR) chains, which are formed by enzyme polyADP-ribose polymerase (PARP). PARP is activated by single-strand DNA damage (in combination with other DNA-repairing enzymes). Pharmacological inhibition of this process has been suggested as a treatment option for ischemic brain damage (Strosznajder et al., 2010). Furthermore, ADP-ribose can activate transient receptor potential cation channel M2 (TRPM2) (Fonfria et al., 2004). The physiological role of TRPM2 is still unclear. The TRPM2 gene is highly expressed in the brain, and it has been associated with activation NLRP3 inflammasome (Zhong et al., 2013). Increased ADP-ribose levels in the ischemic side brain samples in the vehicle treated control samples may therefore be associated with increased single-strand DNA damage and inflammation processes, and by reducing ADP-ribose levels, HX600 treatment may reduce these processes.
Taken together, our data show a prominent anti-inflammatory role for HX600 that is driven by Nurr1 activation. Treatment strategies targeting Nurr1 may be beneficial for treatment of conditions involving neuroinflammation including ischemic stroke.
Supplementary Material
Acknowledgements
We thank Ms. Mirka Tikkanen for excellent technical assistance. This study was supported by the Academy of Finland.
References
- Abdel-Salam OME, Youness ER, Mohammed NA, Yassen NN, Khadrawy YA, El-Toukhy SE, et al. , 2017. Nitric oxide synthase inhibitors protect against brain and liver damage caused by acute malathion intoxication. Asian Pac. J. Trop. Med 10 (8), 773–786. [DOI] [PubMed] [Google Scholar]
- Abramov AY, Duchen MR, 2010. Impaired mitochondrial bioenergetics determines glutamate-induced delayed calcium deregulation in neurons. BBA 1800 (3), 297–304. [DOI] [PubMed] [Google Scholar]
- Adams SH, Hoppel CL, Lok KH, Zhao L, Wong SW, Minkler PE, et al. , 2009. Plasma acylcarnitine profiles suggest incomplete long-chain fatty acid beta-oxidation and altered tricarboxylic acid cycle activity in type 2 diabetic African-American women. J. Nutr 139 (6), 1073–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adibhatla RM, Hatcher JF, Dempsey RJ, 2004. Cytidine-5’-diphosphocholine affects CTP-phosphocholine cytidylyltransferase and lyso-phosphatidylcholine after transient brain ischemia. J. Neurosci. Res 76 (3), 390–396. [DOI] [PubMed] [Google Scholar]
- Bachstetter AD, Xing B, de Almeida L, Dimayuga ER, Watterson DM, Van Eldik LJ, 2011. Microglial p38α MAPK is a key regulator of proinflammatory cytokine upregulation induced by toll-like receptor (TLR) ligands or beta-amyloid (Aβ). J. Neuroinflammation 8, 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banati RB, Gehrmann J, Schubert P, Kreutzberg GW, 1993. Cytotoxicity of microglia. Glia 7 (1), 111–118. [DOI] [PubMed] [Google Scholar]
- Barneda-Zahonero B, Servitja J, Badiola N, Miñano-Molina AJ, Fadó R, Saura CA, et al. , 2012. Nurr1 protein is required for N-methyl-D-aspartic acid (NMDA) receptor-mediated neuronal survival. J. Biol. Chem 287 (14), 11351–11362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Béjot Y, Daubail B, Jacquin A, Durier J, Osseby G, Rouaud O, et al. , 2014. May May. Trends in the incidence of ischaemic stroke in young adults between 1985 and 2011: the Dijon Stroke Registry. J. Neurol. Neurosurg. Psychiatr 85 (5), 509–513. [DOI] [PubMed] [Google Scholar]
- Cauwe B, den Steen, Van Philippe, E., Opdenakker G, 2007. The biochemical, biological, and pathological kaleidoscope of cell surface substrates processed by matrix metalloproteinases. Crit. Rev. Biochem. Mol. Biol 42 (3), 113–185. [DOI] [PubMed] [Google Scholar]
- Céline Demougeot, Christine M, Maurice G, Alain B, 2004. N-acetylaspartate: a literature review of animal research on brain ischaemia. J. Neurochem 90 (4), 776–783. [DOI] [PubMed] [Google Scholar]
- Charles Julia F., Humphrey Mary Beth, Zhao Xiaodan, Quarles Ellen, Nakamura Mary C., Aderem Alan, et al. , 2008. The innate immune response to salmonella enterica serovar typhimurium by macrophages is dependent on TREM2-DAP12. Infection Immunity 76 (6), 2439–2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chawla A, Repa JJ, Evans RM, Mangelsdorf DJ, 2001. Nuclear receptors and lipid physiology: opening the X-files. Science 294 (5548), 1866–1870. [DOI] [PubMed] [Google Scholar]
- Colonna M, 2003. TREMs in the immune system and beyond. Nat. Rev. Immunol 3 (6), nri1106. [DOI] [PubMed] [Google Scholar]
- Cucchiara BL, Messe SR, Sansing L, MacKenzie L, Taylor RA, Pacelli J, et al. , 2009. Lipoprotein-associated phospholipase A2 and C-reactive protein for risk-stratification of patients with TIA. Stroke 40 (7), 2332–2336. [DOI] [PubMed] [Google Scholar]
- De Miranda BR, Popichak KA, Hammond SL, Jorgensen BA, Phillips AT, Safe S, et al. , 2015. The Nurr1 activator 1,1-Bis(3’-Indolyl)-1-(p-Chlorophenyl)methane blocks inflammatory gene expression in BV-2 microglial cells by inhibiting nuclear factor κB. Mol. Pharmacol 87 (6), 1021–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhungana H, Malm T, Denes A, Valonen P, Wojciechowski S, Magga J, et al. , 2013. Aging aggravates ischemic stroke-induced brain damage in mice with chronic peripheral infection. Aging Cell 12 (5), 842–850. [DOI] [PubMed] [Google Scholar]
- Erdö F, Trapp T, Mies G, Hossmann K, 2004. Immunohistochemical analysis of protein expression after middle cerebral artery occlusion in mice. Acta Neuropathol. 107 (2), 127–136. [DOI] [PubMed] [Google Scholar]
- Evans MRB, White P, Cowley P, Werring DJ, 2017. Revolution in acute ischaemic stroke care: a practical guide to mechanical thrombectomy. Pract. Neurol 17 (4), 252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng S, Aa N, Geng J, Huang J, Sun R, Ge C, et al. , 2017. Pharmacokinetic and metabolomic analyses of the neuroprotective effects of salvianolic acid A in a rat ischemic stroke model. Acta Pharmacol. Sin 38 (11), 1435–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonfria E, Marshall ICB, Benham CD, Boyfield I, Brown JD, Hill K, et al. , 2004. TRPM2 channel opening in response to oxidative stress is dependent on activation of poly(ADP-ribose) polymerase. Br. J. Pharmacol 143 (1), 186–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GBD 2015 Mortality and Causes of Death Collaborators. Global, regional, and national life expectancy, all-cause mortality, and cause-specific mortality for 249 causes of death, 1980–2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet (2016) 388 (10053) 1459–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granucci F, Petralia F, Urbano M, Citterio S, Di Tota F, Santambrogio L, et al. , 2003. The scavenger receptor MARCO mediates cytoskeleton rearrangements in dendritic cells and microglia. Blood 102 (8), 2940–2947. [DOI] [PubMed] [Google Scholar]
- Gresa-Arribas Núria, Viéitez Cristina, Dentesano Guido, Serratosa Joan, Saura Josep, Solà Carme, 2012. Modelling neuroinflammation in vitro: a tool to test the potential neuroprotective effect of anti-inflammatory agents. PLoS One 7 (9), e45227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond SL, Popichak KA, Li X, Hunt LG, Richman EH, Damale P, et al. , 2018. The Nurr1 ligand,1,1-bis(3’-indolyl)-1-(p-chlorophenyl)methane, modulates glial reactivity and is neuroprotective in MPTP-induced parkinsonism. J. Pharmacol. Exp. Ther 06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond SL, Safe S, 2015. Tjalkens RB A novel synthetic activator of Nurr1 induces dopaminergic gene expression and protects against 6-hydroxydopamine neurotoxicity in vitro. Neurosci. Lett 607, 83–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawk JD, Bookout AL, Poplawski SG, Bridi M, Rao AJ, Sulewski ME, et al. , 2012. NR4A nuclear receptors support memory enhancement by histone deacetylase inhibitors. J. Clin. Invest 122 (10), 3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honkaniemi J, Sharp FR, 1996. Global ischemia induces immediate-early genes encoding zinc finger transcription factors. J. Cereb. Blood Flow Metab 16 (4), 557–565. [DOI] [PubMed] [Google Scholar]
- Honkaniemi J, States BA, Weinstein PR, Espinoza J, Sharp FR, 1997. Expression of zinc finger immediate early genes in rat brain after permanent middle cerebral artery occlusion. J. Cereb. Blood Flow Metab 17 (6), 636–646. [DOI] [PubMed] [Google Scholar]
- Hsieh CL, Koike M, Spusta S, Niemi E, Yenari M, Nakamura MC, et al. , 2009. A role for TREM2 ligands in the phagocytosis of apoptotic neuronal cells by microglia. J. Neurochem 109 (4), 1144–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Q, Li C, Xia N, Zhao L, Wang D, Yang Y, et al. , 2018. Neurochemical changes in unilateral cerebral hemisphere during the subacute stage of focal cerebral ischemia-reperfusion in rats: an ex vivo 1H magnetic resonance spectroscopy study. Brain Res. 1684, 67–74. [DOI] [PubMed] [Google Scholar]
- Huuskonen MT, Tuo Q, Loppi S, Dhungana H, Korhonen P, McInnes LE, et al. , 2017. The copper bis(thiosemicarbazone) complex CuII(atsm) is protective against cerebral ischemia through modulation of the inflammatory milieu. Neurotherapeutics 14 (2), 519–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inose Y, Kato Y, Kitagawa K, Uchiyama S, Shibata N, 2015. Activated microglia in ischemic stroke penumbra upregulate MCP-1 and CCR2 expression in response to lysophosphatidylcholine derived from adjacent neurons and astrocytes. Neuropathology 35 (3), 209–223. [DOI] [PubMed] [Google Scholar]
- Ishizawa M, Kagechika H, Makishima M, 2012. NR4A nuclear receptors mediate carnitine palmitoyltransferase 1A gene expression by the rexinoid HX600. Biochem. Biophys. Res. Commun 418 (4), 780–785. [DOI] [PubMed] [Google Scholar]
- Jiang Z, Kokalari B, Redai IG, Hanman N, Macphee CH, Haczku A. Lack of lipoprotein associated phospholipase A2 (Lp-PLA2) enhanced phagocytosis and IL-10 expression and decreased NF-kB activation In CD206+ M2 macrophages in gene deficient mice. In: B21. AIRWAY HOST DEFENSE American Thoracic Society; 2014. p. A2485. [Google Scholar]
- Jin R, Yang G, Li G, 2010. Inflammatory mechanisms in ischemic stroke: role of inflammatory cells. J. Leukoc. Biol 87 (5), 779–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadkhodaei B, Alvarsson A, Schintu N, Ramsköld D, Volakakis N, Joodmardi E, et al. , 2013. Transcription factor Nurr1 maintains fiber integrity and nuclear-encoded mitochondrial gene expression in dopamine neurons. PNAS 110 (6), 2360–2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawabori M, Kacimi R, Kauppinen T, Calosing C, Kim JY, Hsieh CL, et al. , 2015. Triggering receptor expressed on myeloid cells 2 (TREM2) deficiency attenuates phagocytic activities of microglia and exacerbates ischemic damage in experimental stroke. J. Neurosci 35 (8), 3384–3396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawabori M, Kacimi R, Kauppinen T, Calosing C, Kim JY, Hsieh CL, et al. , 2015. Triggering receptor expressed on myeloid cells 2 (TREM2) deficiency attenuates phagocytic activities of microglia and exacerbates ischemic damage in experimental stroke. J. Neurosci 35 (8), 3384–3396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kissela BM, Khoury JC, Alwell K, Moomaw CJ, Woo D, Adeoye O, et al. , 2012. Age at stroke. Neurology 79 (17), 1781–1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi M, Konishi H, Sayo A, Takai T, Kiyama H, 2016. TREM2/DAP12 signal elicits proinflammatory response in microglia and exacerbates neuropathic pain. J. Neurosci 36 (43), 11138–11150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koistinaho M, Kettunen MI, Goldsteins G, Keinanen R, Salminen A, Ort M, et al. , 2002. Amyloid precursor protein transgenic mice that harbor diffuse A deposits but do not form plaques show increased ischemic vulnerability: role of inflammation. Proc. Nat. Acad. Sci 99 (3), 1610–1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koizumi S, Yamamoto S, Hayasaka T, Konishi Y, Yamaguchi-Okada M, Goto-Inoue N, et al. , 2010. Imaging mass spectrometry revealed the production of lyso-phosphatidylcholine in the injured ischemic rat brain. Neuroscience 168 (1), 219–225. [DOI] [PubMed] [Google Scholar]
- Lallier SW, Graf AE, Waidyarante GR, Rogers LK, 2016. Nurr1 expression is modified by inflammation in microglia. NeuroReport 27 (15), 1120–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambertsen KL, Biber K, Finsen B, 2012. Inflammatory cytokines in experimental and human stroke. J. Cereb. Blood Flow Metab 32 (9), 1677–1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauber K, Bohn E, Kröber SM, Xiao Y, Blumenthal SG, Lindemann RK, et al. , 2003. Apoptotic cells induce migration of phagocytes via caspase-3-mediated release of a lipid attraction signal. Cell 113 (6), 717–730. [DOI] [PubMed] [Google Scholar]
- Leyns CEG, Ulrich JD, Finn MB, Stewart FR, Koscal LJ, Serrano JR, et al. , 2017. TREM2 deficiency attenuates neuroinflammation and protects against neuro-degeneration in a mouse model of tauopathy. PNAS 114 (43), 11524–11529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubjuhn J, Gastens A, von Wilpert G, Bargiotas P, Herrmann O, Murikinati S, et al. , 2009. Functional testing in a mouse stroke model induced by occlusion of the distal middle cerebral artery. J. Neurosci. Methods 184 (1), 95–103. [DOI] [PubMed] [Google Scholar]
- Madrid LV, Mayo MW, Reuther JY, Baldwin AS, 2001. Akt stimulates the transactivation potential of the RelA/p65 Subunit of NF-kappa B through utilization of the Ikappa B kinase and activation of the mitogen-activated protein kinase p38. J Biol Chem. 276 (22), 18934–18940. [DOI] [PubMed] [Google Scholar]
- Malm T, Mariani M, Donovan LJ, Neilson L, Landreth GE, 2015. Activation of the nuclear receptor PPAR delta is neuroprotective in a transgenic mouse model of Alzheimer’s disease through inhibition of inflammation. J. Neuroinflammation 12 (1), 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks-Konczalik J, Chu SC, Moss J, 1998. Cytokine-mediated transcriptional induction of the human inducible nitric oxide synthase gene requires both activator protein 1 and nuclear factor kappaB-binding sites. J. Biol. Chem 273 (35), 22201–22208. [DOI] [PubMed] [Google Scholar]
- Masamune A, Sakai Y, Yoshida M, Satoh A, Satoh K, Shimosegawa T, 2001. Lysophosphatidylcholine activates transcription factor NF-kappaB and AP-1 in AR42J cells. Dig. Dis. Sci 46 (9), 1871–1881. [DOI] [PubMed] [Google Scholar]
- McMorrow JP, Murphy EP, 2011. Inflammation: a role for NR4A orphan nuclear receptors? Biochem. Soc. Trans 39 (2), 688–693. [DOI] [PubMed] [Google Scholar]
- Merino-Zamorano C, Hernández-Guillamon M, Jullienne A, Le Béhot A, Bardou I, Parés M, et al. , 2015. NURR1 involvement in recombinant tissue-type plasminogen activator treatment complications after ischemic stroke. Stroke 46 (2), 477–484. [DOI] [PubMed] [Google Scholar]
- Milne SA, McGregor AL, McCulloch J, Sharkey J, 2005. Increased expression of macrophage receptor with collagenous structure (MARCO) in mouse cortex following middle cerebral artery occlusion. Neurosci. Lett 383 (1–2), 58–62. [DOI] [PubMed] [Google Scholar]
- Miura D, Fujimura Y, Yamato M, Hyodo F, Utsumi H, Tachibana H, et al. , 2010. Ultrahighly sensitive in situ metabolomic imaging for visualizing spatiotemporal metabolic behaviors. Anal Chem. 82 (23), 9789–9796. [DOI] [PubMed] [Google Scholar]
- Morita K, Kawana K, Sodeyama M, Shimomura I, Kagechika H, Makishima M, 2005. Selective allosteric ligand activation of the retinoid X receptor heterodimers of NGFI-B and Nurr1. Biochem Pharmacol. 71 (1–2), 98–107. [DOI] [PubMed] [Google Scholar]
- Mulder IA, Clara E, Wermer MJ, Mathias H, Tolner EA, den Maagdenberg AM, et al. , 2016. Funnel-freezing versus heat-stabilization for the visualization of metabolites by mass spectrometry imaging in a mouse stroke model. Proteomics 16 (11–12), 1652–1659. [DOI] [PubMed] [Google Scholar]
- Nimmerjahn A, Kirchhoff F, Helmchen F, 2005. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308 (5726), 1314–1318. [DOI] [PubMed] [Google Scholar]
- Niwa M, Inao S, Takayasu M, Kawai T, Kajita Y, Nihashi T, et al. , 2001. Time course of expression of three nitric oxide synthase isoforms after transient middle cerebral artery occlusion in rats. Neurol. Med. Chir. (Tokyo) 41 (2), 73. [DOI] [PubMed] [Google Scholar]
- Nurmi A, Lindsberg PJ, Koistinaho M, Zhang W, Juettler E, Karjalainen-Lindsberg M, et al. , 2004. Nuclear factor-kappaB contributes to infarction after permanent focal ischemia. Stroke 35 (4), 987–991. [DOI] [PubMed] [Google Scholar]
- Ozawa N, Shichiri M, Iwashina M, Fukai N, Yoshimoto T, Hirata Y, 2004. Laminar shear stress up-regulates inducible nitric oxide synthase in the endothelium. Hypertens Res. 27 (2), 93–99. [DOI] [PubMed] [Google Scholar]
- Pearen MA, Muscat GEO, 2010. Minireview: nuclear hormone receptor 4A signaling: implications for metabolic disease. Mol. Endocrinol 24 (10), 1891–1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry VH, Nicoll JAR, Holmes C, 2010. Microglia in neurodegenerative disease. Nat. Rev. Neurol 6 (4), 193–201. [DOI] [PubMed] [Google Scholar]
- Puurunen J, Tiira K, Lehtonen M, Hanhineva K, Lohi H, 2016. Non-targeted metabolite profiling reveals changes in oxidative stress, tryptophan and lipid metabolisms in fearful dogs. Behav. Brain Funct 12 (1), 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quain DA, Parsons MW, Loudfoot AR, Spratt NJ, Evans MK, Russell ML, et al. , 2008. Improving access to acute stroke therapies: a controlled trial of organised prehospital and emergency care. Med. J. Aust 189 (8), 429–433. [DOI] [PubMed] [Google Scholar]
- Raivich G, Bohatschek M, Kloss CU, Werner A, Jones LL, Kreutzberg GW, 1999. Neuroglial activation repertoire in the injured brain: graded response, molecular mechanisms and cues to physiological function. Brain Res. Brain Res. Rev 30 (1), 77–105. [DOI] [PubMed] [Google Scholar]
- Rolova T, Dhungana H, Korhonen P, Valonen P, Kolosowska N, Konttinen H, et al. , 2016. August Aug. Deletion of nuclear factor kappa B p50 subunit decreases inflammatory response and mildly protects neurons from transient forebrain ischemia-induced damage. Aging Dis. 7 (4), 450–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutkowsky JM, Knotts TA, Ono-Moore KD, McCoin CS, Huang S, Schneider D, et al. , 2014. Acylcarnitines activate proinflammatory signaling pathways. Am. J. Physiol. Endocrinol. Metab 306 (12), 1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saber M, Kokiko-Cochran O, Puntambekar SS, Lathia JD, Lamb BT, 2016. Triggering receptor expressed on myeloid cells 2 deficiency alters acute macrophage distribution and improves recovery after traumatic brain injury. J. Neurotrauma 15,;34(2), 423–435. [DOI] [PubMed] [Google Scholar]
- Saijo K, Winner B, Carson CT, Collier JG, Boyer L, Rosenfeld MG, et al. , 2009. A Nurr1/CoREST pathway in microglia and astrocytes protects dopaminergic neurons from inflammation-induced death. Cell 137 (1), 47–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanderson TH, Reynolds CA, Kumar R, Przyklenk K, Hüttemann M, 2013. Molecular mechanisms of ischemia-reperfusion injury in brain: pivotal role of the mitochondrial membrane potential in reactive oxygen species generation. Mol. Neurobiol 47 (1), 9–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandoval KE, Witt KA, 2008. Blood-brain barrier tight junction permeability and ischemic stroke. Neurobiol. Dis 32 (2), 200–219. [DOI] [PubMed] [Google Scholar]
- Schmid CD, Sautkulis LN, Danielson PE, Cooper J, Hasel KW, Hilbush BS, et al. , 2002. December Dec. Heterogeneous expression of the triggering receptor expressed on myeloid cells-2 on adult murine microglia. J. Neurochem 83 (6), 1309–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekiya T, Kashiwagi I, Yoshida R, Fukaya T, Morita R, Kimura A, et al. , 2013. Nr4a receptors are essential for thymic regulatory T cell development and immune homeostasis. Nat. Immunol 14, 230. [DOI] [PubMed] [Google Scholar]
- Shuaib A, Xu Wang C, Yang T, Noor R, 2002. December Dec. Effects of nonpeptide V(1) vasopressin receptor antagonist SR-49059 on infarction volume and recovery of function in a focal embolic stroke model. Stroke 33 (12), 3033–3037. [DOI] [PubMed] [Google Scholar]
- Sieber Matthias W., Jaenisch Nadine, Brehm Martin, Guenther Madlen, Linnartz-Gerlach Bettina, Neumann Harald, et al. , 2013. Attenuated inflammatory response in triggering receptor expressed on myeloid cells 2 (TREM2) knock-out mice following stroke. PLoS One 8 (1), e52982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skerrett R, Malm T, Landreth G, 2014. Nuclear receptors in neurodegenerative diseases. Neurobiol. Dis 72 Pt A, 104–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stout AK, Reynolds IJ, 1999. High-affinity calcium indicators underestimate increases in intracellular calcium concentrations associated with excitotoxic glutamate stimulations. Neuroscience 89 (1), 91–100. [DOI] [PubMed] [Google Scholar]
- Stroke Facts|cdc.gov [Internet]; 2017. [updated −09–06T08:10:23Z; cited Sep 15, 2017]. Available from: https://www.cdc.gov/stroke/facts.htm.
- Strosznajder RP, Czubowicz K, Jesko H, Strosznajder JB, 2010. Poly(ADP-ribose) metabolism in brain and its role in ischemia pathology. Mol. Neurobiol 41 (2–3), 187–196. [DOI] [PubMed] [Google Scholar]
- Tsai T, Chen Y, Lin H, Liu C, Chang H, Lu C, et al. , 2012. Link between lipoprotein-associated phospholipase A2 gene expression of peripheral-blood mononuclear cells and prognostic outcome after acute ischemic stroke. J. Atheroscler. Thromb 19 (6), 523–531. [DOI] [PubMed] [Google Scholar]
- Tsugawa H, Cajka T, Kind T, Ma Y, Higgins B, Ikeda K, et al. , 2015. MS-DIAL: data-independent MS/MS deconvolution for comprehensive metabolome analysis. Nat. Methods 12 (6), 523–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umemiya H, Kagechika H, Fukasawa H, Kawachi E, Ebisawa M, Hashimoto Y, et al. , 1997. Action mechanism of retinoid-synergistic dibenzodiazepines. Biochem. Biophys. Res. Commun 233 (1), 121–125. [DOI] [PubMed] [Google Scholar]
- Umemiya H, Fukasawa H, Ebisawa M, Eyrolles L, Kawachi E, Eisenmann G, et al. , 1997. Regulation of retinoidal actions by diazepinylbenzoic acids. Retinoid synergists which activate the RXR-RAR heterodimers. J. Med. Chem 40 (26), 4222–4234. [DOI] [PubMed] [Google Scholar]
- Vaarmann A, Kovac S, Holmström KM, Gandhi S, Abramov AY, 2013. Dopamine protects neurons against glutamate-induced excitotoxicity. Cell Death Dis. 4 (1), e455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volakakis Nikolaos, Kadkhodaei Banafsheh, Joodmardi Eliza, Wallis Karin, Panman Lia, Silvaggi Jessica, et al. , 2010. NR4A orphan nuclear receptors as mediators of CREB-dependent neuroprotection. Proc. Nat. Acad. Sci. U.S.A 107 (27), 12317–12322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volakakis N, Kadkhodaei B, Joodmardi E, Wallis K, Panman L, Silvaggi J, et al. , 2010. NR4A orphan nuclear receptors as mediators of CREB-dependent neuroprotection. Proc. Natl. Acad. Sci. U.S.A 107 (27), 12317–12322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Q, Castillo SO, Nikodem VM, 1996. Distribution of messenger RNAs for the orphan nuclear receptors Nurr1 and Nur77 (NGFI-B) in adult rat brain using in situ hybridization. Neuroscience 75 (1), 221–230. [DOI] [PubMed] [Google Scholar]
- Yan G, Dai Z, Xuan Y, Wu R, 2015. Early metabolic changes following ischemia onset in rats: an in vivo diffusion-weighted imaging and 1H-magnetic resonance spectroscopy study at 7.0 T. Mol. Med. Rep 11 (6), 4109–4114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuyoshi A, Mami I, Takeshi K, Sachiyo I, Akira O, 2003. Organization and development of corticocortical associative neurons expressing the orphan nuclear receptor Nurr1. J. Comp. Neurol 466 (2), 180–196. [DOI] [PubMed] [Google Scholar]
- Zhao H, Chen Z, Xie L, Liu G, 2017. Suppression of TLR4/NF-κB signaling pathway improves cerebral ischemia-reperfusion injury in rats. Mol. Neurobiol [DOI] [PubMed] [Google Scholar]
- Zhong Z, Zhai Y, Liang S, Mori Y, Han R, Sutterwala FS, et al. , 2013. TRPM2 links oxidative stress to NLRP3 inflammasome activation. Nat. Commun 4, 1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.