

Abstract
Viral fusion glycoproteins catalyze membrane fusion during viral entry. Unlike most enzymes, however, they lack a conventional active site in which formation or scission of a specific covalent bond is catalyzed. Instead, they drive the membrane fusion reaction by cojoining highly regulated changes in conformation to membrane deformation. Despite the challenges in applying inhibitor design approaches to these proteins, recent advances in knowledge of the structures and mechanisms of viral fusogens have enabled the development of small-molecule inhibitors of both class I and class II viral fusion proteins. Here, we review well-validated inhibitors, including their discovery, targets, and mechanism(s) of action, while highlighting mechanistic similarities and differences. Together, these examples make a compelling case for small-molecule inhibitors as tools for probing the mechanisms of viral glycoprotein-mediated fusion and for viral glycoproteins as druggable targets.
Keywords: small molecule, antiviral, viral fusion, inhibitor design, fusion inhibitor
1. INTRODUCTION
Viral glycoproteins (GPs) mediate the initial attachment of the virion to the host cell through interaction with cell surface molecules and catalyze the membrane fusion event that allows the viral genome to be delivered to the interior of the host cell. These functions, which are essential for initiation of the viral infectious cycle, are for some viruses executed by a single GP, but for other viruses, they are separated into two distinct proteins. Viral proteins that mediate membrane fusion during viral entry are known as viral membrane fusion proteins, viral fusion proteins, and viral fusogens. Use of the term fusion in this case is distinct from the more common use of the phrase fusion protein to indicate expression of a protein as a linear sequence with another protein, e.g., green fluorescent protein (GFP) fusion proteins. Although membrane fusion is thermodynamically favorable, the activation barrier is high. Viral fusion proteins lower this energy barrier by coupling membrane fusion to the refolding of their prefusion conformation into the postfusion one (1, 2). Pharmacological interference with these structural changes is an attractive antiviral strategy to prevent the delivery of the viral genome and, hence, to block the infection prior to the onset of viral gene expression and replication. The complex mechanisms regulating the time and place of the fusion reaction reflect the importance of this step in viral infection and its potential as an antiviral target. First, the conformational changes within the viral fusion protein that catalyze membrane fusion are regulated by physiological triggers to ensure delivery of the viral genome to the appropriate compartment of the host cell (1, 2). In some cases, the physiological trigger is the interaction of the viral fusion protein with its attachment receptor and/or coreceptors on the plasma membrane surface. For other viruses, the initial attachment step leads to endocytosis of the virion via clathrin-dependent (3, 4) or caveolin-dependent mechanisms (5–7), and the acidic pH of the endosomal compartment serves as the trigger for changes in the conformation of the viral fusion protein. Proteolytic activation of some viral GPs by cellular proteases during viral assembly, at the cell surface prior to infection or within the endosomal compartment, provides an additional regulatory step to ensure that fusion occurs at the appropriate subcellular location (8, 9). For some viruses, low pH alone can trigger the structural changes necessary for membrane fusion in vitro, whereas during infection the endosomal acidification enables interaction with an intracellular entry receptor that is required for fusion and endosomal escape. One such example is Lassa virus, which undergoes a pH-dependent receptor switch from the primary attachment receptor, the alpha subunit of dystroglycan, to its intracellular receptor, lysosomal-associated membrane protein 1 (10, 11). Another example is Ebola virus, which is internalized by macropinocytosis and, following proteolytic activation of its GP by endosomal cathepsins, engages the endo/lysosomal cholesterol transporter protein Niemann-Pick type C1 (NPC1) prior to fusion and escape to the cytosol (12, 13) (Figure 1).
Figure 1.
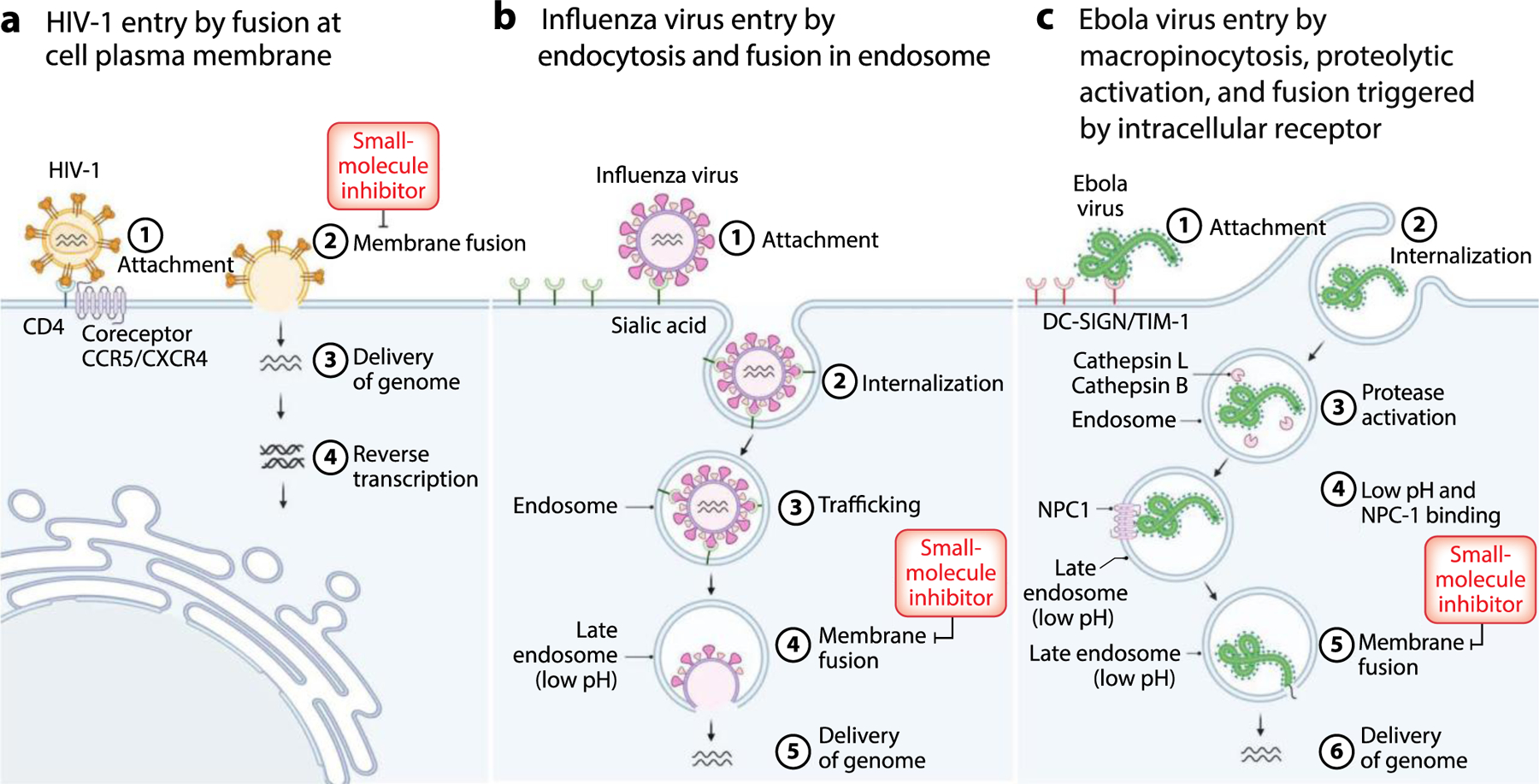
Examples of viral entry pathways. (a) HIV-1 initially binds to the cell via interactions of the receptor-binding subunit gp120 of the viral GP Env with the receptor CD4 on the plasma membrane surface. This interaction induces a conformational change in gp120 that exposes a second receptor-binding domain. Interaction of this site with the coreceptor, CCR5 or CXCR4, triggers structural changes within the fusogenic subunit gp41 that are coupled to fusion of the viral and plasma membranes. (b) Influenza virus initially attaches to sialic acid on the cell surface and is then internalized by clathrin-dependent endocytosis. The reduced pH of the late endosome is the physiological trigger for the structural changes in hemagglutinin leading to fusion of the viral and endosomal membranes. (c) Ebola virus attaches to the host plasma membrane through interaction with surface factors, such as DC-SIGN and TIM-1, and then is internalized by macropinocytosis. The viral GP is proteolytically cleaved by cathepsins B or L in the endosome to form GP1 and GP2. The acidic environment of the late endosome further facilitates the interaction of GP1 with the internal receptor NPC1, which is believed to be the trigger for changes in GP2 structure that are coupled with fusion of the viral and endosomal membranes. Abbreviations: DC-SIGN, dendritic cell-specific intercellular adhesion molecule 3-grabbing nonintegrin; GP, glycoprotein; HIV-1, human immunodeficiency virus type 1; NPC1, Niemann-Pick type C1; TIM-1, T cell immunoglobulin and mucin domain 1. Figure adapted from images created with BioRender.com.
Although contemporary drug discovery efforts have been successful in developing direct-acting antivirals targeting viral enzymes, there are currently no approved small-molecule drugs that target viral GPs. This is because enzymes have active sites that have evolved naturally to bind small molecules and to catalyze formation or scission of distinct covalent bonds. These characteristics enable rational, structure- and mechanism-based inhibitor design and medicinal chemistry. By contrast, viral fusion proteins lack classical active sites and catalyze a process driven by the formation and disruption of multiple noncovalent interactions. Consequently, potential target sites for small molecules that could block fusogenic activity are generally not obvious even in high-resolution structures of viral fusion proteins.
Despite this fundamental challenge, there are increasing examples of small molecules that exert antiviral activity by binding to a viral fusion protein specifically and inhibiting the structural changes that are coupled to membrane fusion during viral entry. These successes have come from a variety of approaches, including (a) high-throughput screens utilizing recombinant proteins and peptides engineered to mimic specific structural changes that accompany membrane fusion; (b) in silico screens utilizing computational docking to identify potential ligands, subsequently validated in biochemical and virological experiments; and (c) unbiased phenotypic screens for compounds with antiviral activity, which have serendipitously yielded compounds that inhibit viral fusion. Careful characterization of the binding sites and mechanisms of these small molecules has led to pharmacological validation of discrete structural targets within viral fusion proteins. This foundational work has been essential for the medicinal chemistry optimization necessary to advance these compounds toward the clinic. Rather than attempting a comprehensive review of all small-molecule fusion protein inhibitors, we focus here on reviewing well-validated inhibitors, how they were discovered, and the experimental data supporting their mechanism(s) and structural target(s) within the viral fusion protein. While medicinal chemistry and structure-activity relationships (SAR) have, in some cases, been critical for inhibitor optimization and played an important role in probing small molecule-protein interactions, we limit the detailed discussion of these results due to the broad focus of this journal and its space limitations.
2. ANTIVIRAL SMALL MOLECULES TARGETING CLASS I VIRAL FUSION PROTEINS
Class I fusion proteins are utilized by diverse viral families, including Retroviridae, Orthomyxoviridae, Paramyxoviridae, Filoviridae, Coronaviridae, and others. The high-resolution crystal structure of the influenza virus hemagglutinin (HA) protein was the first structure of a viral fusion protein (14, 15). Additional high-resolution structures have since been solved, including but not limited to the envelope protein and subunit gp41 of human immunodeficiency virus (HIV) (16, 17), GP and subunit GP2 of Ebola virus (18), the F protein of respiratory syncytial virus (RSV) (19), the F protein of parainfluenza virus (20), and the spike protein of human coronavirus (21, 22). These structures, along with biochemical and biophysical studies, have established our knowledge of the shared characteristics of class I fusion protein structure and function (1). With a largely α-helical secondary structure, class I fusion proteins have an N-terminal hydrophobic fusion peptide (FP), generated by proteolytic cleavage, followed consecutively by N-terminal heptad repeat sequence 1 (HR1) and C-terminal heptad repeat sequence 2 (HR2) and a transmembrane domain (TMD) at the C terminus. The class I fusion proteins are expressed as trimers and require proteolytic activation to form a metastable species competent for fusion. This activation, catalyzed by host proteases, provides a regulatory step that ensures generation of the fusogenic species at the appropriate time and place for productive infection. High-resolution crystal and cryo-electron microscopy structures have shown that both prefusion and postfusion structures of class I fusion proteins are trimers, with the conserved hydrophobic FPs buried at the subunit interface of the prefusion trimer and located at the end of the trimeric hairpin in the postfusion structure, presumably extended into the target membrane (14, 15, 23).
Entry of viruses with class I fusion proteins is initiated as the virion attaches to a receptor on the plasma membrane. The N-terminal subunit [e.g., HA1 of influenza HA and gp120 of HIV type 1 (HIV-1) Env] is generally responsible for this initial attachment step. The structural mechanism leading to membrane fusion catalyzed by class I fusion proteins includes several key steps, including (a) separation of the N-terminal head domains that make the initial contact with the receptor (24); (b) extension of the FP toward the target membrane to form an extended intermediate (often referred to as the prehairpin intermediate) (25, 26); and (c) folding back of the C-terminal HR2 domain against HR1, a zipping up that brings the GP TMD and FP together in a trimeric hairpin or, in some cases, the formation of a postfusion structure with three inner helices and three extended outer peptides. Formation of this parallel trimeric α-helical or extended postfusion structure brings the viral and host membranes together and is the hallmark for all class I fusion proteins. Originally elucidated in the seminal studies of influenza virus HA and HIV-1 Env, this structural mechanism has since been corroborated for other class I viral fusion proteins. The shared features of this model have inspired both mechanistic rationales and experimental approaches for the discovery of small-molecule inhibitors.
2.1. Human Immunodeficiency Virus
HIV-1 enters the host cell by membrane fusion mediated by a class I fusion GP, Env/gp160. Env/gp160 is proteolytically activated by furin or furin-like proteases into gp120 and gp41 subunits, which form the gp120/gp41 trimeric structure that is competent for fusion (17, 27–29) (Figure 2a). Binding of gp120 to the CD4 receptor induces rearrangement of variable loops 1, 2, and 3 (V1–V3) and the formation of a four-stranded β sheet (the bridging sheet) (30, 31), enabling coreceptor binding through interactions of the bridging sheet and V3 with chemokine receptors CCR5 or CXCR4 (30, 32). Binding of gp120 to a coreceptor, in turn, induces further conformational changes that result in the exposure and insertion of the gp41 FP into the target membrane to form the extended intermediate (33). Refolding of the intermediate into the postfusion conformation produces a six-helix bundle, with an inner core and outer layer formed by HR1 and HR2, respectively (16, 17, 34, 35) (Figure 2b). Zipping up of the HR2 and HR1 domains is thought to provide the driving force bringing the transmembrane anchor and the FP of gp41 together, thereby driving fusion of the viral and cellular membranes (Figure 2b). The importance of the HR1-HR2 interaction has been pharmacologically validated by Fuzeon, a Food and Drug Administration (FDA)-approved peptide that prevents this structural change by mimicking HR2 and competing in trans with the cis interaction. A potential approach to pharmacological inhibition of the HR1-HR2 interaction was revealed by structural and biochemical analyses. Peptides N36 and C34, derived from HR1 and HR2, respectively, form a six-helix bundle, in which a cavity formed by N36 hosts hydrophobic residues of C34 (Figure 3). Mutagenesis has demonstrated that interaction of C34 residues with this N36 cavity (also referred to as the inner core cavity or pocket) is important for the stability of the six-helix bundle and for membrane fusion (16, 36). These discoveries led to the hypothesis that a small molecule targeting the cavity would pharmacologically inhibit formation of the six-helix bundle and prevent membrane fusion. (See the sidebar titled Peptides and Small Molecules Have Diverse Mechanisms for Inhibiting Class I Fusion Proteins.) HIV-1 Env-mediated membrane fusion. (a) Schematic diagram of HIV-1 Env including gp120 and gp41 subunits. The locations of the SP, V1–V5, C, protease cleavage site, FP, HR1 and HR2, MPER, TMD, and CT are shown. (b) Schematic of the fusion process.
Figure 2.
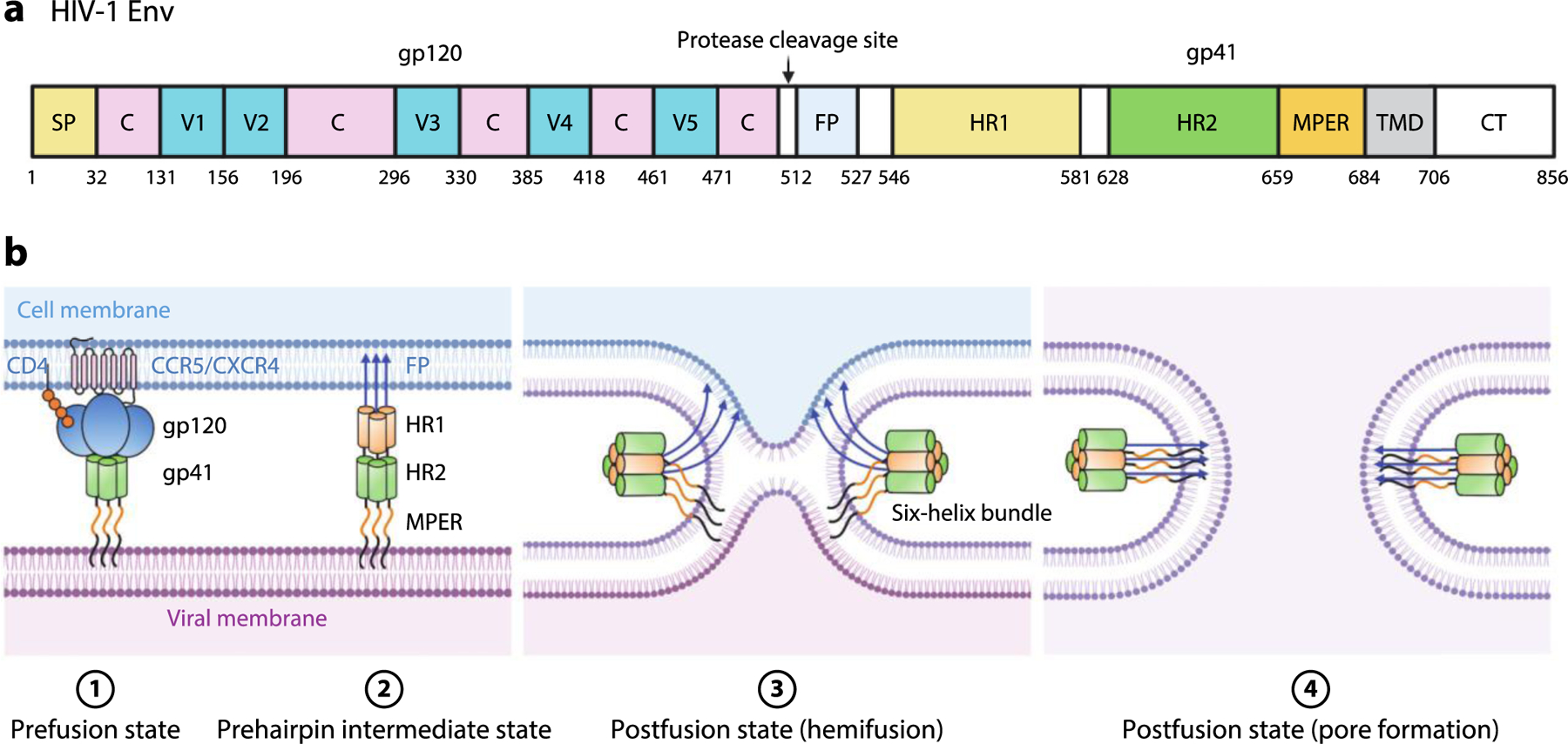
Binding of gp120 to CD4 induces conformational changes that enable interaction with the CCR5 and CXCR4 coreceptors. Interaction with the coreceptors triggers additional changes, including extension of the gp41 FP and its insertion into the host cell membrane to form the extended, prehairpin intermediate and subsequent zipping up of the HR2 domains around the inner HR1 trimeric core to form a six-helix bundle. This brings together the FP and TMD and, therefore, the viral and host membranes, facilitating hemifusion, fusion, and pore formation. Abbreviations: C, conserved region; CT, cytoplasmic tail; FP, fusion peptide; HIV-1, human immunodeficiency virus type 1; HR, heptad repeat; MPER, membrane-proximal external region; SP, signal peptide; TMD, transmembrane domain; V, variable region. Figure adapted from images created with BioRender.com.
Figure 3.
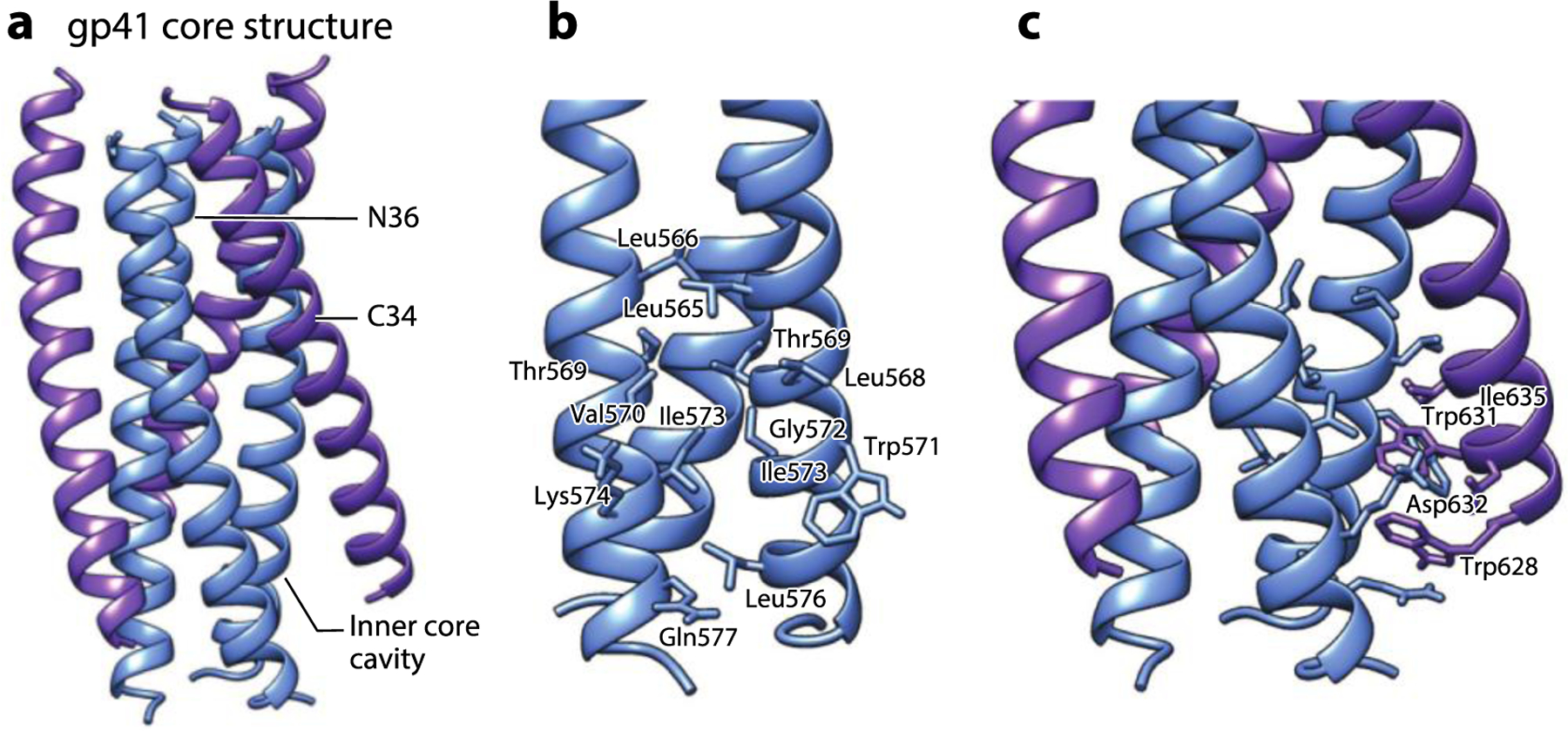
The gp41 core structure formed by HR1- and HR2-derived synthetic peptides. (a) The six-helix bundle structure formed by N36 (blue) and C34 (purple) peptides derived from HR1 and HR2 of gp41, respectively (PDB:1AIK). (b) The zoomed-in view of the hydrophobic cavity at the HR1 core surface formed by residues from the two adjacent N36 helixes. (c) The zoomed-in view of the residues from C34 interacting with the hydrophobic cavity at the HR1 core surface. Abbreviation: HR, heptad repeat. Structural graphics were generated with UCSF Chimera, developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco, with support from NIH P41-GM103311 (166).
PEPTIDES AND SMALL MOLECULES HAVE DIVERSE MECHANISMS FOR INHIBITING CLASS I FUSION PROTEINS.
Biochemical reconstitution experiments in which heptad repeat (HR)-mimicking peptides bind with high affinity and specificity to a recombinant inner core species made up of HR1 peptides provided critical inspiration for inhibiting fusion as an antiviral strategy. T20 (Fuzeon), which corresponds to the HR2 region of human immunodeficiency virus type 1, is the only approved drug that inhibits viral glycoprotein-mediated membrane fusion. Analogous HR2-mimicking peptides have been validated as inhibitors of many other viruses with class I fusion proteins (167), but none have been developed as drugs due to the cost and the poor bioavailability and metabolic stability of peptides. While these HR2-mimicking peptides have been used in assays to identify small molecules that prevent the HR1-HR2 interaction in vitro, the compounds generally have structural mechanisms that differ from T20 and other HR2-derived peptides. Rather than competing directly with HR2 for binding to HR1, the small molecules instead bind in discrete pockets and act by other proposed mechanisms. These include (a) stabilization of the prefusion structure, thereby raising the activation barrier to form the prehairpin intermediate (54–57); (b) destabilization of the prefusion structure, leading to inactivation of the virus due to inappropriate extension of the fusion peptide (58); (c) prevention of six-helix bundle formation (37); and (d) distortion of six-helix bundle structure (59). Notably, these proposed mechanisms may not be mutually exclusive and in many cases cannot be easily distinguished from one another in many experiments—i.e., an inhibitor that prevents formation of the prehairpin intermediate would de facto prevent formation of the postfusion six-helix bundle. It is also important to keep in mind that because the peptides and recombinant proteins used to screen and characterize these inhibitors are experimental models, they may not always accurately mimic the events occurring during authentic infection. While challenging, experiments to validate (or invalidate) proposed mechanisms provide a critical opportunity to test our understanding of glycoprotein structure and fusion mechanisms.
Compound ADS-J1 was initially identified by computational docking of a 20,000-member small-molecule library against the gp41 inner core cavity (PDB:1AIK). ADS-J1 inhibits cell-cell fusion at low micromolar concentrations in vitro (IC50 4.95 μM) and inhibits formation of a six-helix bundle by peptides N36 and C34 (37). Nonetheless, the binding site of ADS-J1 and its mechanism of action were initially disputed due to (a) the selection of resistance mutations mapping to the V3 loop rather than to the inner core cavity on gp41 and (b) experiments suggesting that ADS-J1 instead bound to gp120 and interfered with attachment and/or coreceptor binding (38, 39). Subsequent in vitro experiments showed that ADS-J1 indeed binds to IQN17 (40), a mimic of the gp41 inner core containing a soluble peptide corresponding to the HR1 inner core residues fused to a trimeric coiled coil derived from the GCN4 leucine zipper. In addition, binding of ADS-J1 to IQN17 prevents binding of PIE7 (40, 41), a validated D-amino acid peptide ligand of the inner core cavity on IQN17 (42) (Figure 4). Furthermore, point mutations in the cavity region were shown to confer ADS-J1 resistance (a 30- to greater than 200-fold increase in IC50) to pseudoviruses generated by cotransfection of an HIV-1 Env plasmid and luciferase-expressing Env-deficient HIV-1 vector. While no high-resolution structure has yet been reported for ADS-J1 bound to gp41, a salt bridge between a sulfonic acid of ADS-J1 and Lys574 on N36 was predicted to be a key interaction, and mutation of Lys574 to Asp reduced both binding to ADS-J1 and inhibition of six-helix bundle formation in the IQN17 context (43). Taken together, these experiments provide significant evidence that ADS-J1 interacts specifically with the HR1 cavity region of gp41 and prevents gp41-mediated membrane fusion (42).
Figure 4.
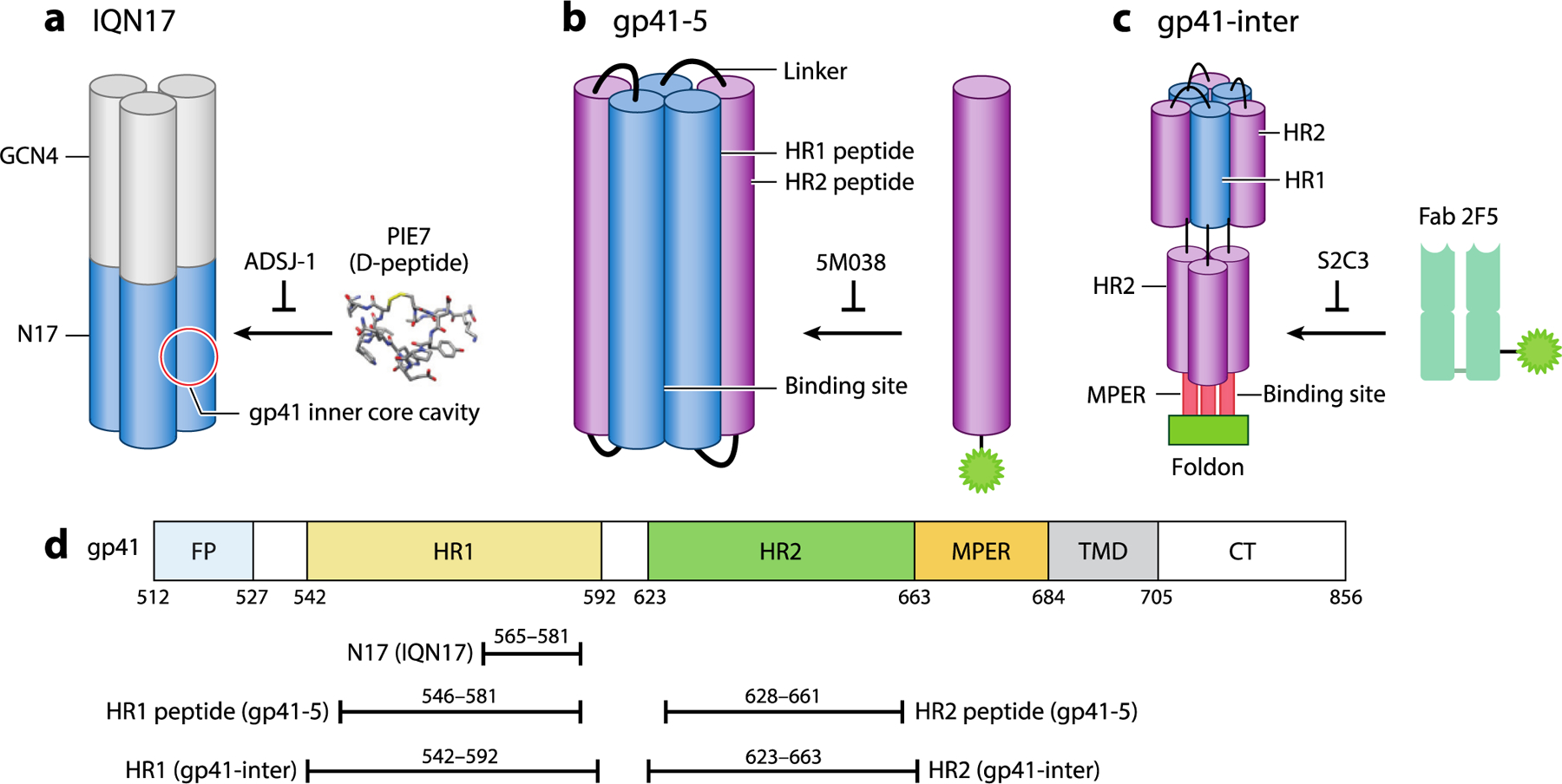
Model peptide and protein systems used in discovery and study of HIV-1 gp41 fusion protein inhibitors. (a) IQN17 mimics the inner core and is derived from a portion of the gp41 HR1 region linked to a trimerization domain derived from GCN4. It was used to validate compounds that prevent binding of PIE7 peptides to the inner core cavity. (40, 41) (b) gp41–5 contains three HR1 peptides and two HR2 peptides derived from gp41 and was used to identify compounds that prevent binding of a fluorescently labeled HR2 peptide (44, 45). (c) gp41-inter mimics the prefusion prehairpin intermediate and was used to identify compounds that prevent binding of Fab 2F5 to the MPER (47, 48). (d) Map of the portions of gp41 present in IQN17, gp41–5, and gp41-inter. Abbreviations: CT, cytoplasmic tail; Fab, fluorescence-labeled antigen-binding fragment; FP, fusion peptide; HIV-1, human immunodeficiency virus type 1; HR, heptad repeat; MPER, membrane-proximal external region; TMD, transmembrane domain. Figure adapted from images created with BioRender.com.
High-throughput screening approaches have also been utilized to identify inhibitors of gp41-mediated membrane fusion. One approach utilized a small protein (gp41–5), which is composed of three HR1 segments forming the inner core and two HR2 segments. Binding of a fluorescently labeled HR2 peptide to gp41–5 in trans was monitored by fluorescence polarization (Figure 4). This approach allowed the identification of small molecules that bind to gp41–5 and prevent its interaction with the HR2 peptide in trans (44, 45). In a high-throughput screen of 34,800 small molecules, compounds 5M038 and 5M041 showed strong inhibition of cell-cell fusion and infection of peripheral blood mononuclear cells with HIV-1 in vitro (IC50 19 μM and 18 μM, respectively) (45). Binding of 5M038 to the HR1 cavity was further supported by nuclear magnetic resonance (NMR) experiments showing (a) the association of 5M038 with an IQN17 containing the residues (Leu57, Trp60, Lys63) corresponding to the inner core cavity of gp41 and (b) competition between 5M038 and a C34 peptide containing the HR2 residues known to bind to the cavity (Trp117, Trp120, Ile124). ELISA-based detection of the six-helix bundle formed by an N36 peptide with a fluorophore-tagged C34 peptide supplied in trans was similarly used to identify compounds NB-2 and NB-64 as gp41 inhibitors (46).
In an alternative approach, the discovery that antibodies targeting the membrane-proximal external region (MPER) are broadly neutralizing was leveraged to screen for small-molecule inhibitors targeting this region (47, 48). A region of approximately 25 highly conserved residues, the MPER lies between the transmembrane region and the extracellular domain of the HIV-1 Env GP (Figure 2) and is the target of broadly neutralizing antibodies (bnAbs) (49–53). To identify small molecules targeting the MPER, Chen and colleagues (47) utilized a protein dubbed gp41-inter, which had been engineered to mimic the prehairpin intermediate of gp41, and a fluorophore-labeled antigen-binding fragment (Fab), 2F5, that recognizes the MPER (Figure 4). A high-throughput screen of 162,102 compounds identified several compounds that prevent binding of 2F5 to gp41-inter. Medicinal chemistry exploration of SARs of a lead compound, dequalinium, led to the development of a derivative, S2C3, that binds to gp41-inter (KD 2 μM) and inhibits cell-cell fusion (IC50 4.4 μM) and HIV-1 infection in vitro (IC50 2.4 μM). Furthermore, S2C3 was shown to block the binding of soluble CD4 to Env by targeting a site distinct from that targeted by gp120-directed antibodies, VRC01 and PG16. Subsequently, the NMR structure of S2C3 with the MPER-TMD revealed that the compound binds in a hydrophobic pocket formed exclusively by the MPER residues. This was corroborated by experiments showing that mutations in this pocket affect susceptibility to inhibition of cell-cell fusion mediated by full-length HIV-1 Env (47). These studies suggested that compounds targeting the MPER pocket inhibit Env-mediated membrane fusion by stabilizing the prefusion conformation and preventing the receptor-induced conformational change to the prehairpin intermediate state. Although a rationale for the initial compound screening was based on competition between compounds and the 2F5 Fab for binding to a model of the prehairpin intermediate state, the NMR study instead suggests that S2C3 reduces the conformational dynamics of the MPER, thereby preventing binding of the 2F5 Fab through an allosteric mechanism rather than through direct competition. This rational screening approach illustrates a valuable complement to the structure- and mechanism-based efforts that targeted the inner core cavity of gp41. Collectively, these examples demonstrate that gp41 has multiple targets for pharmacological inhibition and that judiciously designed screening approaches provide a rational path toward identifying compounds targeting either site. (See the sidebar titled Assays for Characterizing Inhibitors of Viral Glycoproteins.)
ASSAYS FOR CHARACTERIZING INHIBITORS OF VIRAL GLYCOPROTEINS.
The activity of viral glycoprotein inhibitors can be evaluated in several different assays. Virological assays using live viruses or reporter viruses typically monitor viral or reporter protein expression, viral yield, or virus-induced cytopathic effects to detect inhibition of virus. Because inhibition of any step in the replication cycle can affect these readouts, experiments using these assays commonly restrict the timing and duration of compound exposures to enhance the likelihood that the activity is due to inhibition of viral entry. For example, inhibition of viral infectivity can be monitored by preincubating the small molecule with the virus inoculum and allowing it to be present at the time of infection but absent for the remainder of the experiment. Even when performed under these conditions, these assays cannot distinguish a direct effect on fusion from an effect on other postattachment and postinternalization steps, such as interaction with an intracellular receptor. Assays that monitor glycoprotein-mediated fusion provide a more precise readout on mechanism. Fusogenic activity can be observed by expression of the glycoprotein on the cell surface and the formation of syncytia due to cell-cell fusion upon triggering of the glycoprotein (e.g., exposure to low pH, coreceptor binding, heat). Intracellular fusion of viral particles labeled with lipophilic dyes leads to dequenching when the dye diffuses across the newly fused membrane, a direct readout that can be monitored by fluorescence microscopy. The fusion of authentic virions with liposomes or supported lipid bilayers allows fusion to be monitored under more biochemically defined conditions. In addition, antibodies specific for the postfusion glycoprotein conformation have been used to show that the postfusion structure has not formed, an indirect demonstration that fusion has been prevented.
2.2. Influenza
Influenza virus entry is mediated by the HA trimer (15, 60). Proteolytic cleavage of HA by trypsin-like serine proteases generates HA1 and HA2 subunits, which remain covalently linked by disulfide bridges (Figure 5a). HA1 forms the head domain responsible for receptor binding, whereas HA2 forms the stem domain responsible for membrane fusion during viral entry. Although the sequence of the head region is highly divergent due to immune pressure, the sequence of the stem region is highly conserved and, thus, presents an attractive target for vaccine and antivirals development (61). Viral entry is initiated by interaction of HA1 with sialic acid on the cell surface. Following endocytosis of the virion, acidic pH in the endosomal compartment triggers formation of the extended intermediate, which then progresses to the postfusion form (60) (Figure 5b).
Figure 5.
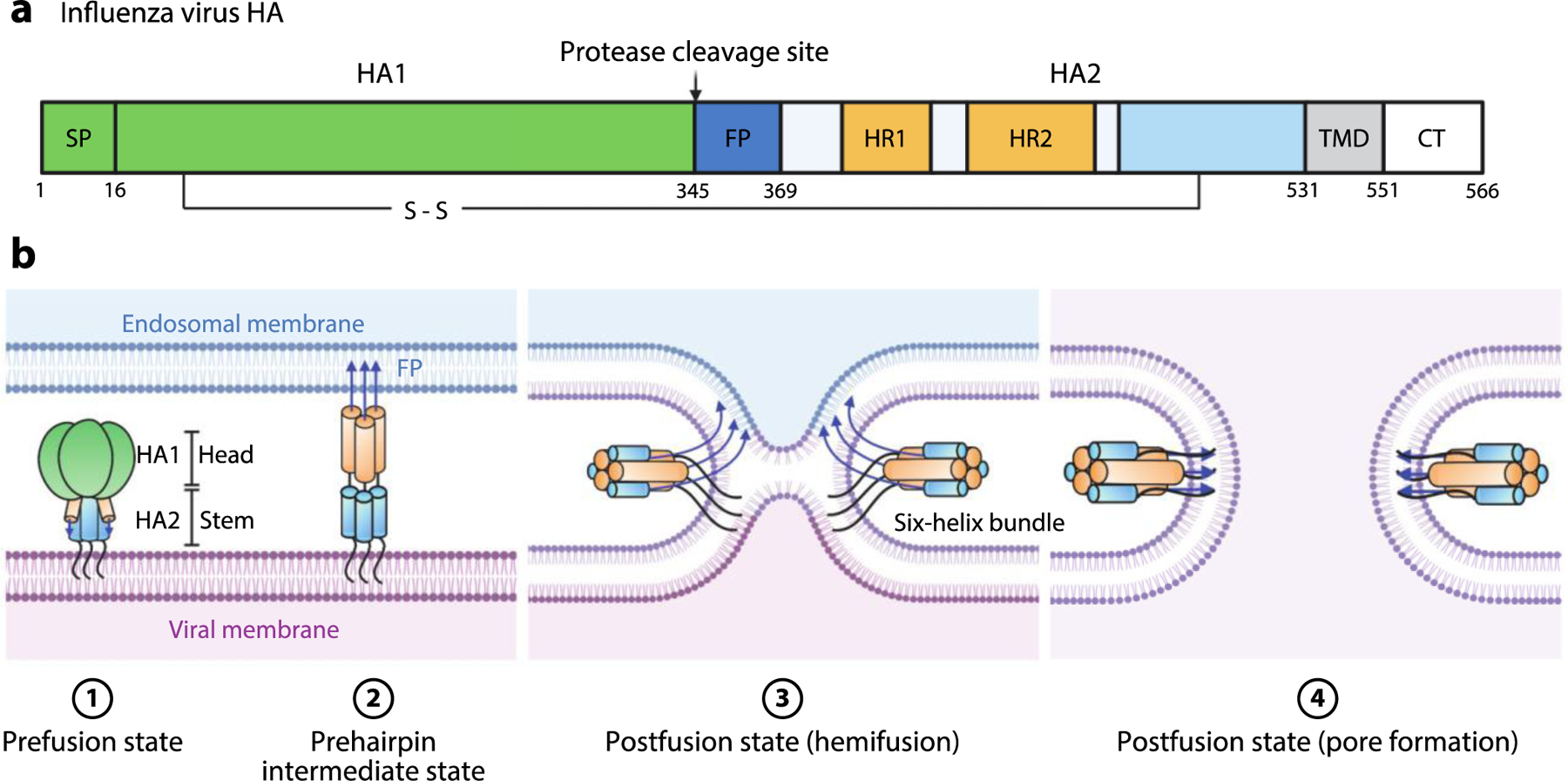
Influenza virus HA-mediated membrane fusion. (a) Schematic diagram of HA. The positions of the SP, HA1, protease cleavage site, HA2, FP, HR1, HR2, disulfide bond, TMD, and CT are shown. (b) Schematic of the fusion process. Viral entry is initiated by binding of HA1 to sialic acid moieties on the plasma membrane. The virion is internalized by endocytosis. The pH of the late endosome is the physiological trigger for extension of the HA2 FP toward the endosomal membrane to produce the extended, prehairpin intermediate. This is followed by refolding of HA2 monomers around the HA1 inner core. Formation of this six-helix bundle drives the viral TMD and FP into proximity, along with the viral and endosomal membranes, thereby facilitating hemifusion and subsequent pore formation upon full fusion of the two membranes. Abbreviations: CT, cytoplasmic tail; FP, fusion peptide; HA, hemagglutinin; HR, heptad repeat; SP, signal peptide; TMD, transmembrane domain. Figure adapted from images created with BioRender.com.
To identify compounds that inhibit HA-mediated fusion, Bodian and colleagues (62) used computational modeling to identify a potential ligand-binding site in the prefusion structure of HA near the FP and then performed an in silico screen to identify potential ligands. Tertiary butylhydroquinone (TBHQ) was discovered to (a) inhibit the pH-induced conformational changes leading to exposure of the FP in vitro, (b) prevent fusion of HA-expressing cells with erythrocyte ghosts, and (c) inhibit influenza virus infection in cell culture (63). Biochemical studies conducted with BHA—the soluble trimeric HA ectodomain produced by bromelain digestion in influenza virus—indicated that TBHQ stabilizes the prefusion conformation of HA—i.e., it raises the activation barrier for viral fusion. Consistent with this, mutations conferring resistance to TBHQ destabilize HA, thereby counteracting the compound’s effect (63). Interestingly, although the computational docking experiments that identified TBHQ as a potential HA inhibitor predicted its binding site to be located at a site proximal to the FP, the high-resolution crystal structure of TBHQ bound to HA demonstrated that it instead binds in a hydrophobic cavity at the interface between protomers of the prefusion HA trimer (54) (Figure 6a–e). (See the sidebar titled Characterization of Inhibitor-Binding Sites.)
Figure 6.
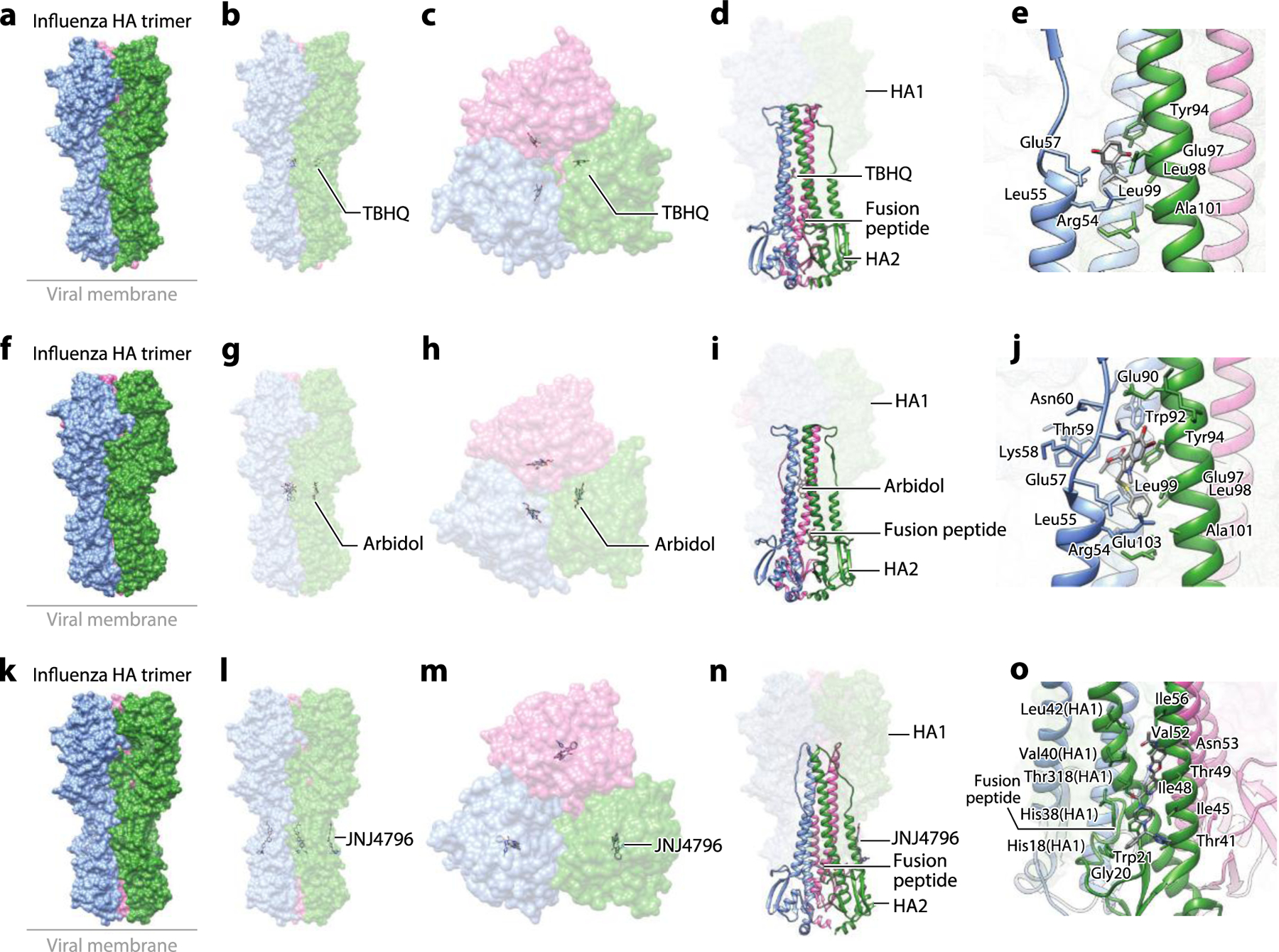
The structure of the influenza HA glycoprotein complexed with small-molecule inhibitors. (a) Structure of the influenza HA trimer complexed with TBHQ (PDB:3EYK). (b) The side view and (c) the top view of the complex structure showing binding of TBHQ in the inner hydrophobic cavity between protomers of the HA trimer. (d) Binding of TBHQ between the adjacent helixes is not proximal to the FP. (e) A zoomed-in view of binding of TBHQ to the hydrophobic cavity. (f) Structure of the influenza HA trimer complexed with arbidol (PDB:5T6S). (g) The side view and (h) the top view of the complex structure showing binding of arbidol in the inner hydrophobic cavity between protomers of the HA trimer. (i) Binding of arbidol between the adjacent helices is not proximal to the FP. (j) A zoomed-in view of arbidol bound in the hydrophobic cavity. (k) Structure of the influenza HA trimer complexed with JNJ4796 (PDB:6CF7). (l) The side view and (m) the top view of the complex structure showing binding of JNJ4796 to the hydrophobic groove at the interface of HA1 and HA2. (n) Binding of JNJ4796 is proximal to the FP. (o) A zoomed-in view of binding of JNJ4796 to the hydrophobic groove at the interface of HA1 and HA2. Abbreviations: FP, fusion peptide; HA, hemagglutinin; TBHQ, tertiary butylhydroquinone. Structural graphics were generated with UCSF Chimera, developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco, with support from NIH P41-GM103311 (166).
CHARACTERIZATION OF INHIBITOR-BINDING SITES.
An equally important part of inhibitor characterization is identification of the inhibitor-binding site on the structure of the target protein. This is critical for understanding mechanisms but also for directing medicinal chemistry optimization of inhibitors. High-resolution cocrystal, cryo-electron microscopy, and nuclear magnetic resonance studies are the gold standards for mapping binding sites and elucidating the protein-small molecule interactions driving antiviral activity and specificity. Chemical cross-linking experiments have also been used to identify inhibitor-binding sites on viral fusion proteins. Lower-resolution, cross-linking results can be enhanced through the use of computational approaches to build binding models, which in turn can be tested via site-directed mutagenesis of the viral fusion protein and medicinal chemistry to modify the inhibitor. While purely computational approaches have been used to identify potential binding sites and to screen for inhibitors due to the relative ease with which they can be implemented, the computational prediction is not always correct. For example, docking experiments that led to the discovery of tertiary butylhydroquinone (TBHQ) as an inhibitor of hemagglutinin-mediated fusion did not correctly identify its binding site. The antiviral activity assays that identified arbidol as an influenza virus inhibitor were agnostic with respect to antiviral target or mechanism. In addition, neither TBHQ- nor arbidol-resistance mutations map to the binding site. This highlights the challenge of identifying and correctly targeting a specific molecular site on viral fusion proteins (as well as other nonenzymatic proteins) and underscores the need to validate the actual binding site, ideally through high-resolution structure determination but also through cross-linking and structure-activity relationship studies that employ both site-directed mutagenesis of the protein and medicinal chemistry on the compounds.
Arbidol is a small-molecule inhibitor of influenza virus that was originally discovered on the basis of its antiviral activity and was subsequently found to target HA and to inhibit its fusion activity (64). The high-resolution crystal structure of arbidol bound to HA shows that it binds in the same hydrophobic cavity between two protomers in the prefusion HA trimer as TBHQ (55) (Figure 6f–j). Through hydrophobic interactions and the formation of inter- and intraprotomer salt bridges, arbidol stabilizes the prefusion conformation of HA and, analogously to TBHQ, raises the energetic barrier for the conformational changes that induce membrane fusion (55). The independent discoveries that TBHQ and arbidol both bind in the interprotomer cavity and act as molecular glue stabilizing HA’s prefusion conformation provide a remarkable validation of the interprotomer cavity as an antiviral target. Although preventing the formation of the postfusion six-helix bundle by competition of HR1 and HR2 interaction may seem to be a more straightforward strategy, the examples of TBHQ and arbidol have demonstrated that pharmacological targeting of the hydrophobic cavity between two protomers in the prefusion HA trimer can effectively block fusion at a much earlier point by stabilizing the prefusion conformation of the GP.
Other small-molecule inhibitors of HA were identified through target-based high-throughput screening. In a screen predating yet analogous to that which identified compounds targeting the MPER region of HIV gp41, AlphaLISA (amplified luminescent proximity homogeneous assay) technology was used to identify compounds that mimic the activity of the bnAb CR626, which recognizes a highly conserved epitope in the HA stem (65). Screening of ~500,000 compounds led to the identification of a class of benzylpiperazines exemplified by JNJ7918, which inhibits the infectivity of several H1N1 strains in cell culture (IC50 1.09 to 12.90 μM) (65). Despite the lack of a high-resolution structure, SAR studies allowed the optimization of JNJ7918 toward the development of JNJ4796, which has 30- to 80-fold increased affinity for the HA stem, 30- to 500-fold improvement in virus neutralization assays, and partial efficacy in murine models of H1N1 infection (65). In mechanism-of-action studies, binding of JNJ4796 to HA was shown to prevent its pH-induced transition to the postfusion form, as evidenced by the loss of the HA1 subunit after reduction of the interchain HA disulfide. Further, binding of JNJ4796 protected prefusion HA from trypsin digestion at low pH, an effect previously reported for CR626 and other stem-targeting neutralizing antibodies that stabilize the prefusion form. High-resolution crystal structures of JNJ4796 bound to two different HA variants (H1N1 A/Solomon Islands/3/2006 and H5N1 A/Vietnam/1203/2004) demonstrated that this compound binds stoichiometrically to the HA prefusion trimer, occupying a highly conserved hydrophobic groove at the interface of HA1 and HA2, which is distinct from the interprotomer cavity targeted by arbidol and TBHQ (Figure 6k–o). As a further validation, residues comprising the JNJ4796-binding site correspond largely to the epitope recognized by CR6261 and other bnAbs, with JNJ4796 making hydrophobic and polar interactions that mimic many of the interactions observed between HA and bnAbs (65). More recently, F0045[S] was discovered using a competitive fluorescence polarization assay that monitored the interactions of a cyclic HR2-mimicking peptide (P7-TAMRA) with the stem region of H1/PR8 HA (66). F0045[S] inhibits the infectivity of several H1N1 viruses and an H5N1 pseudovirus (IC50 1.6 to 22.8 μM). The high-resolution cocrystal structure and biochemical studies demonstrate that F0045[S] acts analogously to JNJ4796 by binding in the hydrophobic groove at the interface of HA1 and HA2, thereby stabilizing the prefusion conformation of HA and preventing the structural changes required for membrane fusion (66).
2.3. Ebola Virus
The GP of Ebola virus is a characteristic class I fusion protein consisting of three heterodimers of proteolytically generated GP1 and GP2 subunits (67). Following its initial attachment to the plasma membrane surface, the Ebola virion is internalized by macropinocytosis (68) (Figure 1). Within the endosomal compartment, proteolytic cleavage of GP1 by cathepsins B or L removes the glycan cap and enables the interaction of GP1 with its intracellular receptor, the cholesterol transport protein NPC1 (12, 13, 69, 70). Interaction of NPC1 with the GP1 receptor-binding site is thought to promote the extension of the GP2 fusion loop into the target membrane and folding back of HR2 against HR1 to form the characteristic six-helix bundle associated with membrane fusion (12, 71). Although the fusion loops of most class I fusion proteins are found at the N terminus, the fusion loop of GP2 is found within an internal loop. Phenotypic and computational screens have identified small molecules that bind GP in a way that may inhibit GP-mediated fusion (58, 72, 73). The sequential steps leading to Ebola virus membrane fusion during viral entry have not been fully recapitulated in vitro; consequently, interrogating the effect of small molecules on the structural rearrangements of GP during fusion and testing mechanistic hypotheses have been more challenging for Ebola GP-targeting compounds than analogous inhibitors targeting HIV Env and influenza virus HA.
A screen of FDA- and ex-US-approved drugs and molecular probes identified toremifene and other selective estrogen receptor modulators as inhibitors of a recombinant eGFP-Ebola reporter virus. Toremifene efficiently inhibits eGFP-Ebola virus infection in cell culture (IC50 1 μM) and in a murine model (74). Because it does not affect internalization, cathepsin processing of GP, or endosomal acidification, toremifene was hypothesized to inhibit either trafficking to the late endosome or fusion triggered by GP. The high-resolution cocrystal structure of toremifene bound to GP demonstrated that it binds in a pocket between GP1 and GP2 (58). Toremifene reduces the stability of glycosylated, recombinant GP in thermal shift assays. This destabilization of GP has been proposed but not demonstrated to cause premature triggering of GP2, which could affect the subsequent fusion step. Consistent with this, the antiviral activities of toremifene and other ligands binding to the same pocket correlate with their destabilization of a recombinant GP in the thermal shift assay (58, 75). This effect is notably the opposite of that of arbidol, THBQ, JNJ4796, and F0045[S] on influenza HA. Several other small-molecule inhibitors of GP have been reported to bind in this pocket, based on computational docking corroborated with mutagenesis and/or medicinal chemistry studies (76, 77). Inhibition of GP2 due to small-molecule stabilization of the prefusion conformation—the inhibitory mechanism observed for arbidol and THBQ in influenza HA—has been suggested for compounds identified in a large computational screen (73); however, both the binding site and mechanism of action of these compounds await experimental validation.
2.4. Respiratory Syncytial Virus
RSV has two major GPs on its surface: the attachment GP, G, which is highly variable, and the more conserved fusion GP, F. Although deletion of G attenuates RSV, the virus is still viable, which indicates the F protein is sufficient to mediate the entry process (78, 79). F is proteolytically cleaved into the F1 and F2 subunits that form a metastable prefusion trimer of three F1-F2 heterodimers (80, 81). Although the physiological trigger for F-mediated membrane fusion has not yet been established conclusively, it undergoes conformational changes analogous to those of other class I fusion proteins: the FP inserts into the target membrane to form a prehairpin intermediate, which collapses upon folding of each HR2 helix in an antiparallel fashion against an HR1 segment, to form the six-helix bundle (35). The six-helix bundle formed by peptides N57 and C45—derived from the HR1 and HR2 regions of F, respectively—has been crystallized and provides a high-resolution model for the postfusion structure, including the presence of a potentially druggable hydrophobic cavity, reminiscent of the cavity observed in HIV gp41 (19). The site lies at the carboxy-terminal end of a groove at the surface of the inner coiled coil formed by the N57 peptide (HR1) trimer.
Interest in RSV F inhibitors as antivirals has driven several large phenotypic screens yielding lead compounds that have been the focus of extensive medicinal chemistry optimization. These efforts have developed multiple compounds that (a) potently inhibit laboratory and clinical RSV isolates in cell culture (IC50 and EC50 values sub- to low nanomolar) and in animal models (mouse, cotton rat, and neonatal lamb), (b) inhibit RSV-induced syncytium formation, and (c) cross-link to residue Tyr198 in the HR1 pocket (56, 57, 59, 82–88). Structural and mechanistic studies of these compounds have yielded interesting similarities and potential contrasts with other class I fusion protein inhibitors.
The cocrystal structure of compound TMC-353121 with the postfusion six-helix bundle formed by N52 (HR1) and C39 (HR2) peptides demonstrated that this compound binds in the HR1 pocket, making hydrophobic and electrostatic interactions with both HR1 and HR2 residues (59) (Figure 7a–e). Interestingly, the presence of the C39 (HR2) peptide was required for covalent binding of a photo-cross-linking analog of TMC-353121 in the HR1 pocket of IQN57, a soluble peptide analogous to the gp41 IQN17 protein (Figure 4) containing the hydrophobic pocket of the F protein HR1 fused to a trimeric coiled coil derived from the GCN4 leucine zipper (59). This stands in contrast with compounds targeting the analogous pocket of gp41, which can bind to gp41 IQN17 in the absence of the HR2 peptide (40, 42). The observations that binding of TMC-353121 (a) stabilizes the IQN57-C39 six-helix bundle in thermal shift assays but does not bind to the preformed six-helix bundle and (b) results in a loss of electron density corresponding to the nonhelical, N-terminal portion of the HR2 peptide in the N52 (HR1)-C39 (HR2) six-helix bundle led to the proposal that formation of the more stable, distorted six-helix bundle in the presence TMC-353121 may prevent or mistime the necessary juxtaposition of the viral and host membranes due to interference with the final zipping up of HR2 with HR1 (59) (Figure 7d,e). In presenting this proposed mechanism, however, it was carefully noted that the observed distorted six-helix bundle could represent an artifact of cocrystallization of the compound with separate HR1 and HR2 peptides instead of the full-length RSV F (59).
Figure 7.
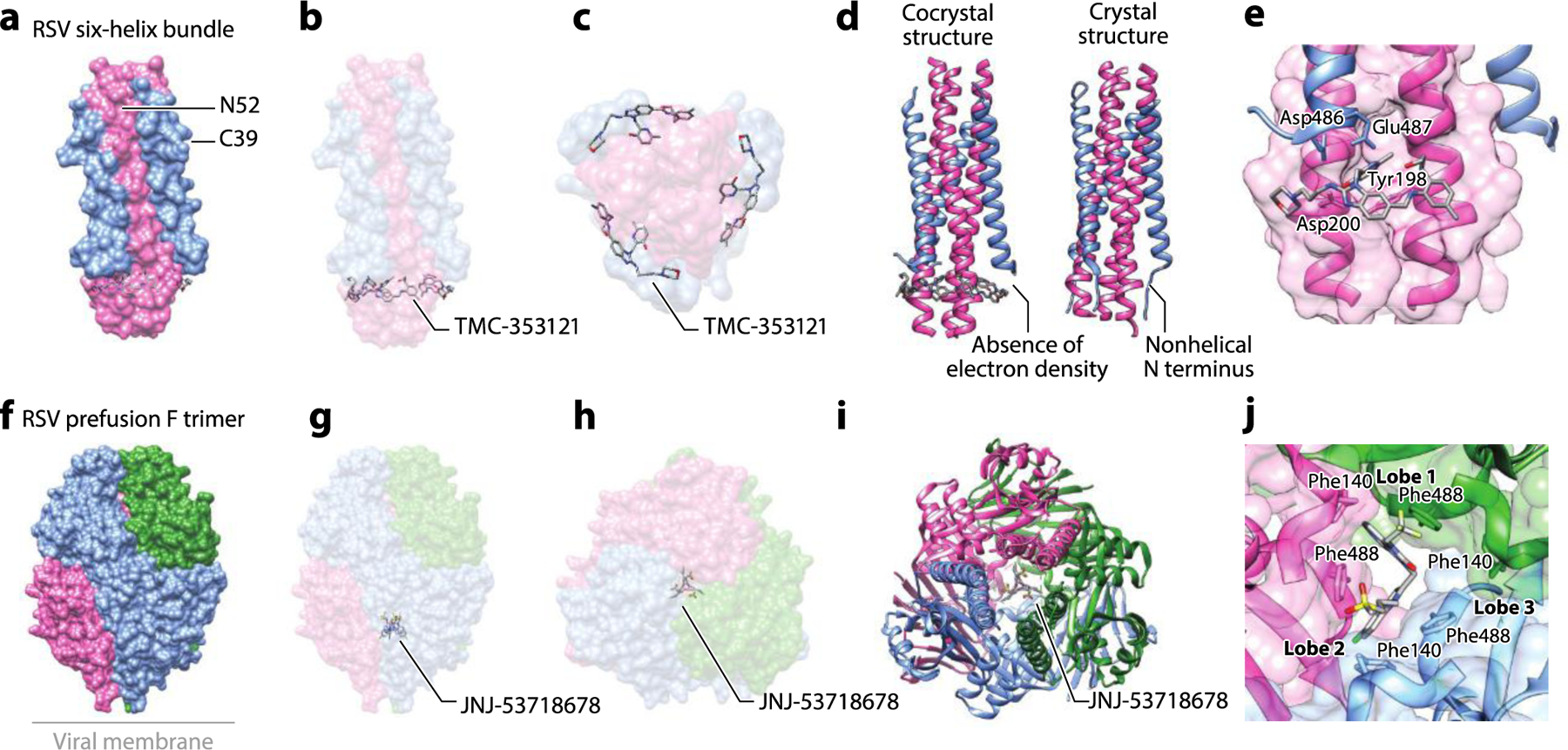
The structure of the RSV F glycoprotein complexed with small-molecule inhibitors. (a) Structure of the RSV six-helix bundle formed by N52 (HR1) and C39 (HR2) peptides with TMC-353121 (PDB:3KPE). (b) The side view and (c) the bottom view of the complex showing binding of TMC-353121 in the HR1 pocket. (d) Binding of TMC-353121 was proposed to distort the six-helix bundle due to absence of electron density at the N-terminal, nonhelical portion of the protein. The crystal structure of the native six-helix bundle (PDB:1G2C) is shown for comparison (19, 59). (e) A zoomed-in view of binding of TMC-353121 to the HR1 pocket. (f) The structure of DS-Cav1, a stabilized RSV prefusion F trimer, with JNJ-53718678 (PDB:5KWW). (g) The side view and (h) the top view of the complex shows JNJ-53718678 in the central cavity. (i) The binding of JNJ-53718678 in the central cavity was shown to stabilize the prefusion F. (j) A zoomed-in view of binding of JNJ-53718678 to two lobes at the interface of the F protein trimer in the central cavity and contacts made to Phe140 and Phe488. Abbreviations: HR, heptad repeat; RSV, respiratory syncytial virus. Structural graphics were generated with UCSF Chimera, developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco, with support from NIH P41-GM103311 (166).
In subsequent structural studies (56, 57) of preclinical F inhibitors targeting the HR1 pocket, including TMC-353121 and JNJ-53718678 (KD 7.4 nM, EC50 RSV infectivity 460 pM, >95% reduction of lung viral titer and RNA with once daily dosing in neonatal lamb model), were studied using DS-Cav1 (89), a recombinant RSV F protein stabilized in the prefusion conformation by an engineered disulfide (S155C-S290C) and mutations S190F-V207L. The structure of JNJ-53718678 with DS-Cav1 (Figure 7f–j) revealed hydrophobic contacts with Phe140 in the FP and HR2 residue Phe488, along with reordering of adjacent residues compared to the unliganded DS-Cav1 structure (57). In differential scanning fluorimetry experiments, these interactions collectively increased the stability of prefusion RSV F and were proposed to raise the barrier for triggering conversion to the postfusion form (57). Consistent with this mechanism, JNJ-53718678 inhibits membrane fusion that occurs upon heat-induced triggering of F expressed on the surface of cells (57). While analogous to the effect of arbidol on influenza HA-mediated fusion, this mechanism differs from the distorted six-helix bundle mechanism that had been proposed for TMC-353121. The hypothesis that TMC-353121 shares a common mechanism with JNJ-53718678, BMS-433771, and other RSV F-targeting compounds is supported by high-resolution crystal structures showing that all of these compounds bind in the same site of the stabilized prefusion F protein (56, 57) (Figure 7f–i). While some of the inhibitors occupy only two of the three equivalent lobes at the interface of the F protein trimer and others all three, all of the compounds have 1:3 inhibitor:F trimer stoichiometry. All five inhibitors form hydrophobic contacts with the aromatic side chains of Phe140 in the FP and Phe488 in HR2 near the viral transmembrane region (56, 57) (Figure 7j). Elucidation of SAR associated with high-affinity binding in isothermal calorimetry experiments and antiviral activity in cell culture revealed π-π and CH-π interactions between heterocyclic groups of the inhibitor with Phe488, Phe140, and Phe137 side chains (57). Additional electrostatic interactions with Asp486 and Glu487 appear to be correlated with the higher potency of inhibition of TMC-353121 and JNJ-2408068 (57). Experiments in which conformation-specific antibodies were used to detect triggering of F by heat demonstrated that binding of the F inhibitors, including TMC-353121, to surface-expressed RSV F prevents triggering of the prefusion protein. These results suggest that all five F-targeting compounds act at an early step in the fusion process rather than at the late stage of zipping up that was originally proposed for TMC-353121 (56, 57). Consistent with this mechanism of action, resistance mutations to TMC-353121 include D486N in the HR2 region but also residues in the FP (F140I, L141W, G143S, V144A) and in the cysteine-rich region of the globular domain between HR1 and HR2 (D392G, K394R, S398L, T400A/I) (59).
From a methodological standpoint, the RSV F inhibitors highlight the inherent challenges in using model peptides and proteins to study viral fusion proteins. Due to their solubility in aqueous solution, these model systems have facilitated both structural and biochemical studies that have been extremely useful in advancing the field. Mechanistic models derived from these model systems, however, benefit greatly when additional experiments can complement the caveats associated with these models, including the absence of significant parts of the viral fusion protein and any associated membranes.
2.5. Inhibitors of Other Class I Fusion Proteins
There have been many additional efforts leading to the discovery of compounds targeting other class I fusion proteins. Important examples include measles virus (90–93), Nipah virus (94), parainfluenza virus (95–98), severe acute respiratory syndrome coronavirus (99), and severe acute respiratory syndrome coronavirus 2 (100). These compounds were discovered and characterized using approaches analogous to those described for the inhibitors of HIV, influenza, and RSV GPs and have been proposed to act by analogous mechanisms including stabilization of the prefusion structure to raise the barrier to membrane, destabilization of prefusion structure to cause premature triggering of the viral fusion protein, and inhibition of six-helix bundle formation. They are included as examples in Supplemental Table 1.
3. ANTIVIRAL SMALL MOLECULES TARGETING CLASS II VIRAL FUSION GLYCOPROTEINS
Class II viral fusion proteins have a structure distinct from their class I counterparts but perform the same function of lowering the energy barrier for membrane fusion. The first members of class II were the E proteins of tick-borne encephalitis virus and other flaviviruses (101–103). The class now also includes the E1 proteins of alphaviruses [Semliki Forest (104, 105), Sindbis (106), chikungunya (107), and rubella (108) viruses] and the Gc proteins of phleboviruses [Rift Valley fever virus, thrombocytopenia syndrome virus, and Uukuniemi virus (109–111)]. In contrast to the class I fusion proteins, which have a largely helical secondary structure, the class II fusion proteins consist predominantly of β strands organized in three domains (I, II, III) that are connected via a membrane-proximal stem region to the C-terminal TMD (112). Domain I (DI) is an eight-stranded β barrel. Domain II (DII), formed from two noncontiguous insertions in DI, is a long, finger-like domain made of twelve β strands and two α helices with the conserved hydrophobic FP at its tip. Domain III (DIII) is an immunoglobulin-like domain that mediates interactions with attachment factors during viral entry and is a major target of host antibody responses (101) (Figure 8a).
Figure. 8.
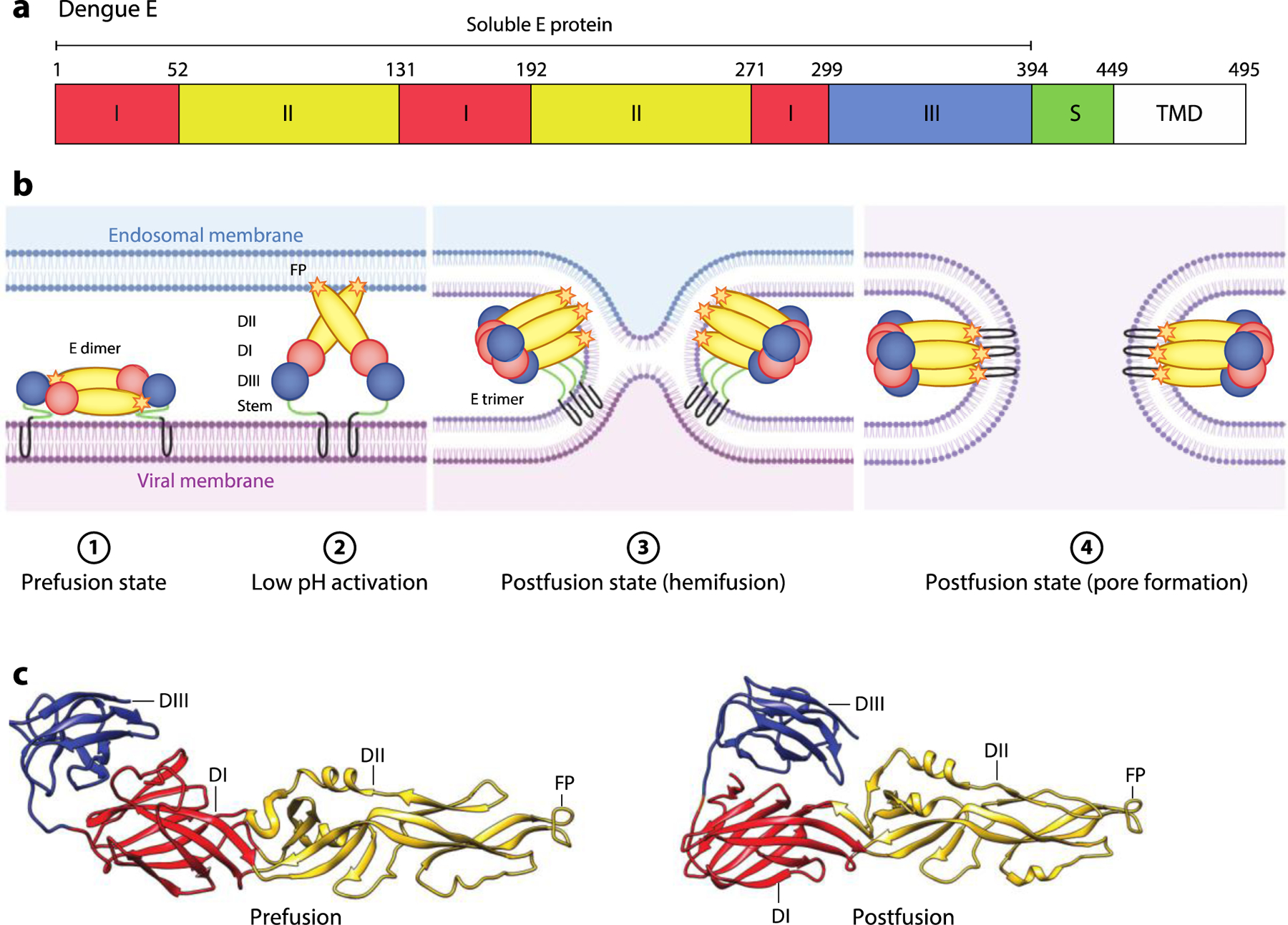
Flavivirus E-mediated membrane fusion. (a) Schematic diagram of dengue E. The locations of DI, DII, DIII, stem region S region, TMD, and soluble E protein (residues 1–394) are shown. (b) Schematic of the E-mediated fusion process. In the prefusion state, the mature E dimer is anchored on the viral membrane. Exposure to low pH in the endosome triggers dissociation of the E dimer and extension of the FP toward the endosomal membrane. Rearrangement of E as a trimeric species is followed by movement of DIII toward DI. Zipping up of this extended intermediate through new interactions of the S region with DII pulls the viral TMD and FP into proximity, and with them, the viral and endosomal membranes. This facilitates hemifusion and subsequent formation of a fusion pore. (c) Pre- (PDB:1OAN) and postfusion (PDB:1OK8) structures of dengue E protein. The DI, DII, DIII, and FP are indicated. Abbreviations: D, domain; FP, fusion peptide; S, stem; TMD, transmembrane domain. Figure adapted from images created with BioRender.com.
Unlike class I fusion proteins, which are trimeric in both pre- and postfusion conformations but undergo extensive refolding during membrane fusion, the flavivirus E protein undergoes large changes in oligomerization and domain orientation while maintaining similar secondary structure during viral maturation and cell entry. On immature virions, E exists as a trimer of heterodimers with its viral chaperone, pre-membrane (prM) (113). This association protects the FP from premature triggering as the new virion traffics through the Golgi (114, 115). prM is cleaved by furin or furin-like proteases to produce pr and M proteins in the trans-Golgi network (114, 115). While M remains associated with the viral membrane, pr dissociates from the FP upon encountering the neutral pH of the extracellular milieu (114, 115) (Figure 9). This event coincides with reorganization of E as the fusion-competent dimer on mature, infectious virions. Membrane fusion mediated by class II fusion proteins is associated with dissociation of the prefusion dimer to monomers that reorganize as trimers. In the alphaviruses, the attachment and membrane fusion functions are performed by separate proteins, E2 and E1, respectively. The fusion loop of E1 is buried beneath E2 in the prefusion conformation (105). E2 is expressed in precursor form as pE2, which is cleaved by furin-like proteases into E2 and E3 proteins (116), with E3 acting analogously to the flavivirus pr protein (117, 118).
Figure 9.
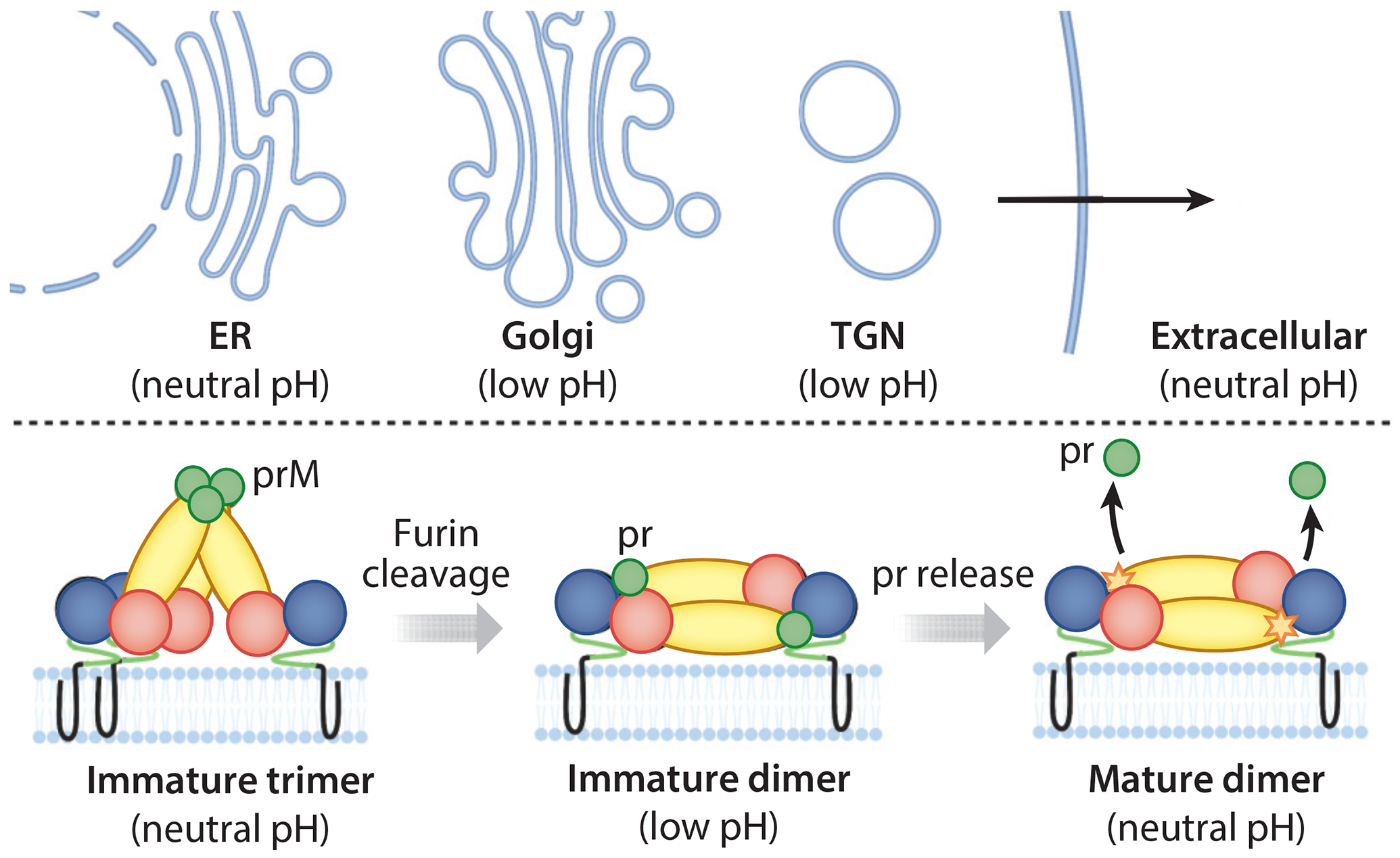
Processing of the flavivirus E protein during virion maturation. Immature virions are assembled at neutral pH in the ER. prM-E heterodimers organize as trimers on the surface of immature virions. As the immature virions traffic in the TGN, prM is cleaved by furin-like proteases into pr and M (not visible), and the E proteins reorganize as dimers. The pr peptide remains associated with the fusion peptide of E until the virion reaches the neutral pH of the extracellular space. Abbreviations: ER, endoplasmic reticulum; prM, pre-membrane; TGN, trans-Golgi network. Figure adapted from images created with BioRender.com.
Viral entry mediated by class II fusion proteins is initiated by attachment to the host cell through interactions of either DIII of E (flaviviruses) or the E2 protein (alphaviruses) with factors on the plasma membrane surface. This triggers internalization of the virion through clathrin-dependent endocytosis. Acidic pH in the endosomal compartment is a physiological trigger for reorganization of the prefusion dimer to postfusion trimer. A structural mechanism for class II fusion proteins has been developed based on differences in the pre- and postfusion conformations of the soluble envelope proteins (minus the stem region and membrane domain) and biochemical experiments (104, 119, 120). The key features of the mechanism (Figure 8b,c) include (a) rotation of DII about the hinge region between DI and DII, which results in extension of the FP away from the virion surface and enables formation of trimer contacts; (b) insertion of the fusion loop into the target membrane, which induces trimerization; and (c) folding of the stem region and membrane anchor back toward the fusion loop in a zipping up reaction that drives fusion of the viral and target membranes (104, 112, 119, 120). Consistent with this model, mutations at the interface between DI and DII alter the pH threshold for fusion (121), and the addition of soluble DIII or synthetic peptides corresponding to the stem region inhibits the fusion reaction in vitro (122, 123).
3.1. Dengue Virus
A potential mechanism for pharmacological inhibition of the dengue virus E protein was serendipitously discovered when a soluble prefusion E dimer (sE2) comprised by DI, DII, and DIII of dengue virus 2 (DENV2) cocrystallized with n-octyl-β-D-glucoside (βOG), a detergent present at millimolar concentrations in the crystallization buffer (102) (Figure 10a). The high-resolution structure of this complex revealed that the detergent molecule docks in a hydrophobic pocket located between DI and DII. Notably, the kl loop (residues 270–279), which lies in the hinge region between DI and DII, forms a lid at the top of the pocket in the unliganded structure. In the βOG-bound structure, the hairpin shifts upward, opening the pocket (Figure 10b). This observation suggested that small molecules targeting this pocket might block E-mediated fusion by interfering with the conformational changes required for this process, in particular by hindering the change in angle between DI and DII required to transition to the postfusion conformation of the protein or by stabilizing an early intermediate state that occurs prior to dimer dissociation (102, 120). This hypothesis was also supported by prior research demonstrating that mutations in this hinge region, including many residues corresponding to the βOG pocket, affect the pH threshold for fusion (124, 125) (Figure 10c).
Figure 10.
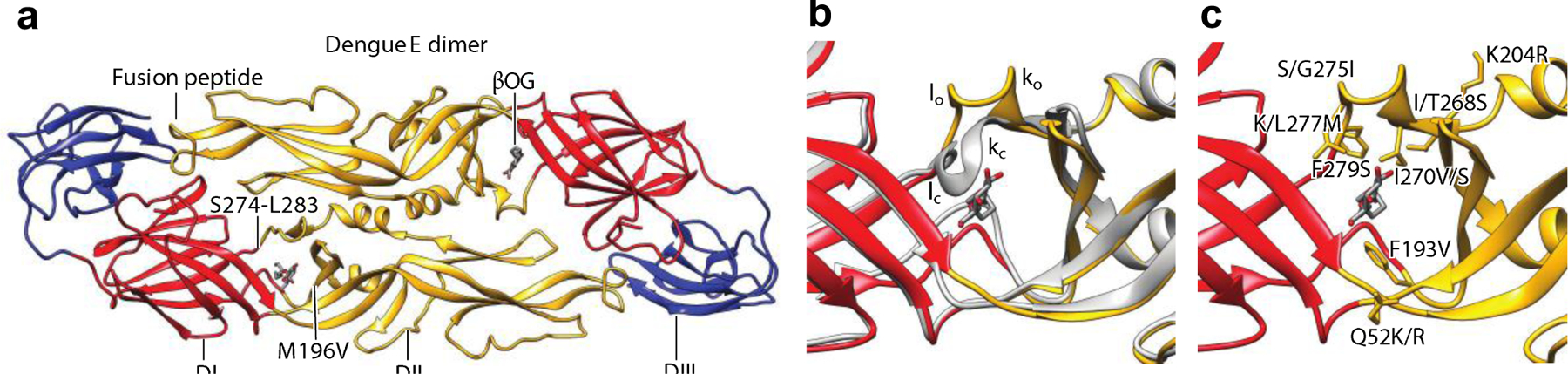
Cocrystal structure of a DENV2 sE2 with βOG. (a) Structure of a soluble prefusion dimer containing DI, DII, and DIII of DENV2 with βOG bound in the hydrophobic pocket at the interface of DI and DII (PDB:1OKE). (b) The zoomed-in view of the βOG-binding pocket (PDB:1OKE) superposed on the unliganded structure (gray; PDB:1OAN), indicating the change in the kl hairpin. (c) The zoomed-in view of the location of mutations that alter the threshold pH for fusion. Abbreviations: βOG, n-octyl-β-D-glucoside; D, domain; DENV2, dengue virus 2; sE2, soluble prefusion E dimer. Structural graphics were generated with UCSF Chimera, developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco, with support from NIH P41-GM103311 (166).
Several dengue E inhibitors have been identified through virtual screening to identify potential ligands of the βOG pocket (126–130). The first of these studies identified compound P02, which inhibits a luciferase reporter pseudovirus derived from the related yellow fever virus in cell culture (IC50 13 μM) (130). Computational docking studies suggested that P02 makes close contact with several residues in the βOG pocket, including a hydrophobic contact with Leu207 (130). Saturation transfer difference NMR spectroscopy using sE2 protein showed loss of specific βOG resonances in the presence of P02 (130). This indicated that P02 causes displacement of βOG but could not address whether this occurs directly or allosterically, and inhibition of fusion was not demonstrated. In addition, P02 may have more than one mode of action based on its activity against a subgenomic yellow fever virus replicon, a model system used to study viral RNA replication that lacks the E protein entirely (IC50 13 μM pseudovirus, 17 μM replicon). Subsequent E inhibitors discovered in virtual screens have been shown to inhibit dengue virus entry, to coelute with soluble prefusion E protein of dengue virus, and, in some cases, to inhibit E-mediated cell-cell fusion (126–130). While these inhibitors are predicted to bind in the βOG pocket, no high-resolution structural data, biochemical cross-linking experiments, or gain/loss-of-inhibition established with resistance mutations or medicinal chemistry have yet been published for these compounds.
A phenotypic screen using immunofluorescence-based detection of dengue-infected cells identified GNF-2, an allosteric inhibitor of BCR-Abl kinase (131), as an inhibitor of DENV2 (EC90 15 μM) (132). Mode-of-action studies revealed that GNF-2 inhibits a post-entry step of the infectious cycle mediated by cellular Abl kinases; however, an additional effect on viral entry was not recapitulated with other Abl kinase inhibitors (132). The effect on viral entry was mapped to inhibition of E, as evidenced by interaction of a biotinylated analog of GNF-2 with dengue virions and sE2. Medicinal chemistry established distinct SAR for the antiviral activities mediated by E versus Abl kinases, leading to related 4,6-disubstituted pyrimidines (1-100-1) and 2,4-diamino pyrimidines (2-12-2, 7-148-6) that inhibit DENV2 infectivity with single-digit micromolar EC90 values. These compounds prevent E-mediated membrane fusion in an assay in which fusion of virions with trypsin-encapsulating liposomes leads to mixing of the inner contents of the virion and liposomes and digestion of the viral core protein on the interior side of the viral membrane (133). Direct evidence for binding in the βOG pocket was provided by cross-linking of an analog of compound 1-100-1 to a region between residues Ser274 and Leu283 in the βOG pocket of sE2 (133). Bolstering this, a resistance mutation, Met196Val, selected during serial passage of DENV2 in the presence of 7–148-6 was mapped to the βOG pocket. A panel of mutations (Phe193Leu, Met196Val, Gln200Glu/Ala, Gln271Glu/Ala, Met272Ser, Phe279Ser) in the βOG pocket was further used to map loss of binding of 1–100-1, 2–12-2, 7–148-6, and GNF-2 to sE2 (133) and to show that structurally related inhibitors have similar patterns of sensitivity to different mutations. Together, these studies provide the best current evidence for the βOG pocket as a druggable target.
A different approach to the discovery of dengue E inhibitors was enabled by the demonstration that the zipping up of the stem peptide along the outside of the postfusion E trimer can be modeled with a synthetic peptide corresponding to the membrane-proximal stem region (stem peptide) and a soluble postfusion E trimer of DENV2 (sE3) (134). This model formed the basis of a competitive fluorescence polarization assay used to identify compounds that prevent the interaction of the stem region with sE3 (134). High-throughput screening of ~30,000 compounds followed by secondary screening for inhibition of DENV2 infectivity led to the identification of cyanohydrazone 1662G07 (sE3-stem IC50 ~15 μM, DENV2 infectivity EC90 16.9 μM). Medicinal chemistry established SAR for inhibition of DENV2 infectivity and led to the development of compound 3-110-22, which inhibits DENV2 infectivity with EC90 0.75 μM and prevents E-mediated fusion with trypsin-encapsulating liposomes in vitro (134). Closer study, however, revealed that 3-110-22 has no effect on binding of the stem peptide to sE3, suggesting that its mechanism differs from that of 1662G07 (134). 3-110-22 was later shown to bind with KD 0.6 μM to sE2 in biolayer interferometry experiments, and loss of binding was observed when specific mutations were made in the βOG pocket (133). The binding site of 1662G07 has not been mapped, and whether its antiviral activity is due to direct competition with the sE3-stem peptide interaction was not pursued further. Additional medicinal chemistry to reduce nonspecific, pan-assay interference activities of 3-110-22 and to improve its microsome stability resulted in compound JBJ-01-162-04 (DENV2 infectivity EC90 1.5 μM), which was shown to inhibit DENV2 infection in a murine model (135). Finally, a competitive amplified luminescent proximity homogeneous assay screen (AlphaScreen) using DENV2 sE2 and a biotinylated derivative of GNF-2 was used for target-based high-throughput screening. This led to the identification of several additional lead compounds (S4105, K786–9739, C200–5340, G199–0398, C200–9144, S7337, S1633, C066–4182) that inhibit DENV2 infectivity with EC90 values in the low micromolar range, block E-mediated fusion of virions with synthetic liposomes, and exhibit sensitivity to mutations in the βOG pocket (136). Validation of the assay through correlation of AlphaScreen and antiviral activities suggests that the target-based assay provides a reliable screening tool for identification of ligands of the βOG pocket.
3.2. Other Flaviviruses
Cocrystallization of βOG or other small molecules with other flavivirus E proteins has not been reported even after deliberate efforts to obtain cocrystals with βOG (103, 137–139). Despite this, alignment of residues in the βOG pocket suggests at least partial conservation of the inhibitor-binding site. Consistent with this, validated inhibitors of the DENV2 βOG pocket were shown to have varying levels of activity against Zika virus (ZIKV), West Nile virus, and Japanese encephalitis virus (133, 135). As an initial step in discovery of inhibitors of other flavivirus E proteins, recombinant prefusion Zika E protein and a 3-110-22-based probe were used in an analogous competitive AlphaScreen to screen ~27,000 compounds (140). Secondary assays for inhibition of ZIKV infectivity confirmed seven lead compounds that inhibit ZIKV infectivity with single-digit EC90 values and that inhibit fusion of Zika virions with liposomes (140). While these results are consistent with inhibition of Zika E through pharmacological targeting of the pocket analogous to the βOG pocket on dengue E, this conclusion awaits direct evidence in the form of high-resolution structures, cross-linking, and/or resistance, mutagenesis, and medicinal chemistry SAR studies.
3.3. Hepatitis C Virus
Hepatitis C virus (HCV) has two surface GPs, E1 and E2, which form a heterodimer essential for viral entry (141). Secondary structure predictions suggested that both E1 and E2 have homology to other class II fusion GPs; however, the currently available structural data reveal significant differences. The E2 protein has an N-terminal domain with three hypervariable regions (HVR1, HVR2, HVR3) and a C-terminal transmembrane helix. Two high-resolution crystal structures of the core domain of E2—corresponding to an N-terminal deletion protein that begins at HVR2— in complex with different antibody fragments revealed an Ig-like β sandwich made up of two antiparallel β sheets, each stabilized by disulfide bonds (142, 143) (Figure 11). Although they share a similar Ig fold, the compact globular shape of E2 is apparently unlike other class II fusion proteins. The N-terminal domain of E1 (residues 192–271) was crystallized as a homohexamer, with each monomer having a hairpin-helix-sheet structure, in which the N-terminal hairpin and the helix and three-stranded β sheet form two distinct dimer interfaces with adjacent monomers (144) (Figure 11). The resulting homodimer is a six-stranded sheet with greater structural similarity to phosphatidylcholine transfer protein than to known class II fusion proteins (144). This discovery has raised the suggestion that the HCV GPs represent a unique class of fusion proteins (144, 145). Further studies are required to determine if this is the case because the model proteins crystallized lack significant portions of E1 and E2 as well as the viral membrane; moreover, the fragment of E1 was crystallized under low pH buffer conditions. Consequently, the current structures may differ from the physiologically relevant prefusion structures of E1 and E2 in ways that are not yet known.
Figure 11.
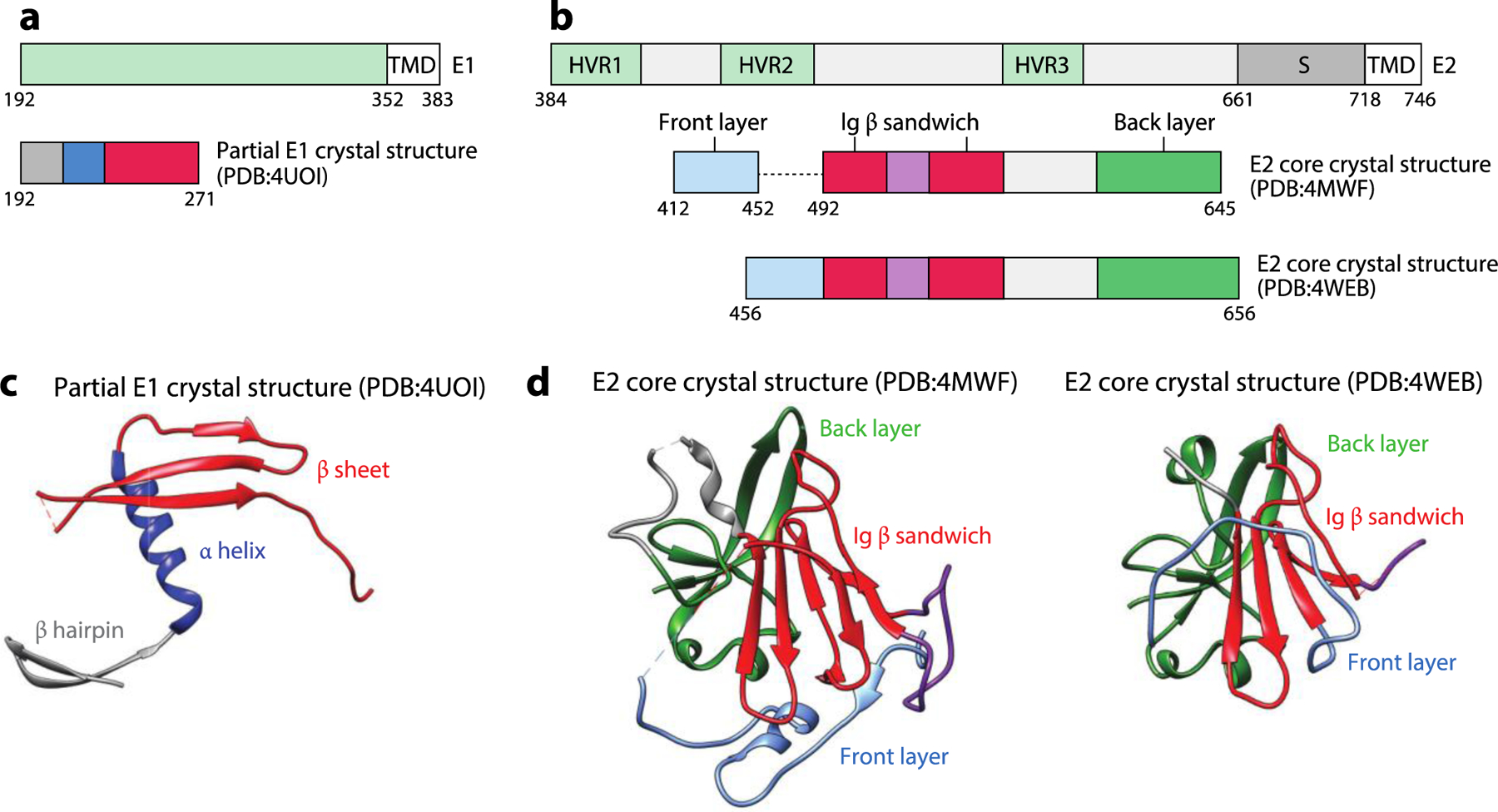
The HCV E1 and E2 proteins. (a) The schematic diagrams of E1 and the portion of E1 present in the crystallized construct. (b) The schematic diagram of E2 and the portion of E2 present in the crystallized construct. (c) The crystal structure of partial E1 consisting of the N-terminal hairpin, the helix, and a three-stranded β sheet (PDB:4UOI). (d) The crystal structures of the E2 core consist of the front layer, Ig β sandwich, and back layer (PDB:4MWF and PDB:4WEB). Abbreviations: HCV, hepatitis C virus; HVR, hypervariable region; S, stem; TMD, transmembrane domain. Structural graphics were generated with UCSF Chimera, developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco, with support from NIHP41-GM103311 (166).
HCV entry follows a complex path beginning with the interaction of E2 with the low-density lipoprotein receptor and glycosaminoglycans or other host factors on the plasma membrane surface. This is followed by coordinated, sequential interactions with receptor-like scavenger receptor class B type I, CD81, CLDN1, and occludin that lead to the uptake of HCV into the endosome and membrane fusion induced by low pH (146–152). The aforementioned structures and a wealth of genetic and biochemical studies have provided insight into E2’s function in attachment and binding of coreceptors and identified a putative FP in E1 (146, 153–157). Despite these discoveries, elucidation of HCV’s fusion mechanism has been challenging due to the lack of high-resolution structures containing both E1 and E2 in pre- and postfusion forms, the physiological association of HCV virions with lipoproteins, the complexity of the HCV entry mechanism, and the absence of an experimental model that biochemically recapitulates E1/E2-mediated fusion.
The lack of mechanistic information about HCV-mediated fusion has not impeded the discovery of small-molecule inhibitors of HCV fusion. Phenotypic assays using a Cre-Lox-based reporter system to detect HCV infection of cells based on Gaussia luciferase reporter activity enabled discovery of small molecules that act at any step in the viral replication cycle (158). Multiple compounds affecting an early step in the replication cycle were identified from a ~350,000-member library (159). A subsequent medicinal chemistry effort to optimize the antiviral activity and biopharmaceutical and pharmacokinetic properties of a set of related aryloxazoles led to the development of compound 18a (fluoxazolevir), which inhibits multiple HCV genotypes in cell culture (EC50 values 0.008–2.007 μM) (160, 161). Most impressively, tests of 18a against HCV genotypes 1b, 2a, and 3 in the humanized chimeric mouse model of infection demonstrated 1–2 log decreases in viral titer as well as activity against multidrug-resistant HCV mutants. Also, 18a exhibited evidence of synergy with daclatasvir, an approved drug that interferes with HCV RNA replication through its interaction with the viral nonstructural protein 5A, suggesting that 18a (a) acts a step distinct from daclatasvir and (b) has potential for use in combination with daclatasvir.
Chlorcyclizine (CCZ), an antihistamine that was identified in an analogous phenotypic screen (EC50 0.022 μM), has been shown to inhibit a late stage of viral entry and to reduce HCV genotype 1b and 2a titers by 2- and 1.5-log, respectively, in the albumin–urokinase plasminogen activator/severe combined immunodeficient chimeric mouse model (162, 163). Although inhibition of E1/E2-mediated membrane fusion could not be assayed directly for 18a or CCZ, time-of-addition experiments and experiments in which viral attachment, internalization, and fusion were synchronized through control of temperature and endosomal pH are consistent with effects on fusion. In photo-cross-linking experiments, derivatives of 18a and CCZ were shown to cross-link to E1when incubated with an E1/E2 protein (residues 192–746) expressed recombinantly in a lentivirus expression system (161) or when added directly to HCV-infected cells in culture (162), respectively. While the location of 18a’s cross-links to E1 were not mapped, a diazirine-conjugated CCZ analog was shown to covalently modify E1 residues 214, 215, and 221 (162). Mutations that confer resistance without loss of viral fitness map to the predicted fusion loop for both 18a (Ala274Ser, Ile374Thr, Asp382Glu, and Val414Ala) (161) and CCZ (Met267Val, Ala274Thr, Leu286Ile, Gln289His, Phe291Leu, Met267Val/Phe291Leu, and Leu286Ile/Phe291Leu) (162). The location of these mutations suggests that 18a and CCZ may directly hinder function of the predicted fusion loop; however, the possibility that these mutations act by a more general mechanism (e.g., by destabilizing a prefusion conformation, analogously to the mutations conferring TBHQ-resistance to influenza virus and F inhibitor-resistance to RSV) cannot currently be excluded. Molecular modeling based on the crystal structures of HCV E2 and partial E1 predicts binding of CCZ in a hydrophobic pocket formed by the fusion loop within the ectodomain of E1 (162); however, the orientation of the compound in this binding model is only partially consistent with the location of the cross-linked residues. While further biochemical and structural studies are needed to confirm this model and to assess whether 18a binds in the same pocket, the activity of these compounds both in vitro and in vivo provides ample motivation for these efforts.
4.4. THE FUTURE OF VIRAL FUSION PROTEIN INHIBITORS: OUTSTANDING QUESTIONS
The development of small molecules targeting viral fusion proteins as antiviral drugs has lagged behind the development of other classes of direct-acting antivirals, most notably those targeting viral enzymes. This is partly due to the inherent challenges of applying rational, structure-based drug discovery and optimization approaches to this target class. A second challenge has been the diversity of viral fusion proteins and their structural tolerance of mutations to escape immune pressure. This raises legitimate concerns that a limited spectrum of activity and poor resistance profile would render viral fusion protein inhibitors of limited use as therapeutics. Despite these challenges, the significant efforts reviewed here demonstrate that viral fusion proteins are druggable targets and that viral fusion protein inhibitors can exert potent and specific antiviral activity both in vitro and in vivo. This is, perhaps, unsurprising considering that, while the surface epitopes of viral envelopes are diverse, conservation of the fusion machinery targeted by these compounds resembles the conservation of enzyme active sites. With such inhibitors now in hand, the field is poised to both advance antivirals toward the clinic (as in the case of the RSV inhibitors) and deploy these compounds as tools to better understand fusion mechanisms. In advancing this exciting area of antivirals discovery, obvious areas for growth would be the extension of these efforts to other viral fusion proteins—in particular, class III viral fusion proteins because none have been published at the time this review was prepared. Beyond this, several important questions beg attention.
One such question regards the stoichiometry required for inhibition. How many functionally redundant copies of the fusion protein on the virion surface must be inhibitor bound to prevent fusion? Clearly, saturating all copies of the fusion protein on the virion surface would be difficult. Fortunately, the potency of some fusion protein inhibitors and their affinity for recombinant fusion protein suggest that saturated binding is not necessary for antiviral activity. This was more directly analyzed in single-particle experiments monitoring hemifusion of dengue virus-like particles (VLPs) labeled with a self-quenching lipophilic dye with a supported lipid bilayer (164). The inclusion of a fluorophore-conjugated analog of the E inhibitor 3-110-22 showed that hemifusion was blocked once ~20% of the copies of E on the VLP were inhibitor bound (164). Since this represents a single example, analyses of other inhibitor series and viruses are warranted to determine how the inhibitory threshold is affected by the location of the binding site or specific interactions of the small molecule with the fusion protein. Alternatively, the inhibitory threshold could be an inherent property of a particular virus that is maintained across inhibitors.
Related to this and pertinent to drug resistance is whether mutations can affect this inhibitory threshold and reduce drug susceptibility independently of their effects on affinity of drug binding. If so, this would present a novel resistance mechanism specific to this antiviral class. Identifying binding modes and sites that are prone to this type of resistance would inform drug development efforts and could provide unique opportunities to probe the cooperativity of fusion proteins on the virion surface. A second aspect of antiviral resistance worth exploring is whether viral fusion proteins are dominant drug targets. This concept refers to the fact that a drug-resistant mutant genome initially exists in the infected cell within a population of largely wild-type, drug-susceptible genomes. Kirkegaard and colleagues (165) have elegantly shown that viral capsid proteins are dominant drug targets. When a resistance mutation is introduced into the population, progeny virions contain both drug-resistant and drug-susceptible forms of the capsid protein. Outgrowth of the drug-resistant mutant is prevented by the drug susceptibility of the wild-type proteins within the newly synthesized virion. In this example, the oligomeric structure and function of the viral capsid protein are critical for the dominance of drug susceptibility. Viral fusion proteins may follow this paradigm; consequently, examining this phenomenon and identification of inhibitor characteristics (e.g., binding site and binding mode, inter-versus intraprotomer contacts) that confer a dominant phenotype may be useful in optimizing the resistance profile of this class of antivirals.
Finally, the effects of viral fusion protein inhibitors on virion morphogenesis warrant exploration. Most fusion protein inhibitor studies have focused almost exclusively on inhibition of viral entry. Despite this, specific, coordinated structural changes occur during virion morphogenesis. Moreover, reverse genetic studies have demonstrated that even single amino acid substitutions in viral fusion proteins can completely block production of viral particles. This suggests that targeting fusion proteins during virion morphogenesis presents another opportunity for antiviral intervention. Agents that have this dual mode of action may have superior potencies as well as more advantageous resistance profiles due to the high likelihood that single suppressor mutations that rescue fusion will be unable to rescue morphogenesis, and vice versa. Examining whether existing viral fusion protein inhibitors affect late stages of the viral replication cycle is a logical first step in this line of investigation. This can be supplemented by the development of assays to detect compounds that interfere with the maturation of viral fusion proteins and the production of infectious particles. Identifying single sites within viral fusion proteins that can mediate pharmacological inhibition of both virion fusion and morphogenesis could confer distinct advantages.
There is a large unmet need for antivirals to combat existing as well as emerging viral pathogens, and agents that have broad-spectrum activity against related viruses and that have high barriers to resistance are sorely needed. Small-molecule inhibitors of viral fusion proteins represent an attractive opportunity for drug development. They lack known homologs in humans and have conserved structures and functions in catalyzing membrane fusion during viral entry. The small molecules and discovery strategies discussed here provide the foundation for developing direct-acting antivirals and for examining new concepts—including inhibitory thresholds, drug dominance, and dual modes of action—to develop solutions for the current challenge.
Supplementary Material
ACKNOWLEDGMENTS
The authors gratefully acknowledge support from NIH/NIAID Award AI146152. Structural graphics in this review were generated with UCSF Chimera, developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco, with support from NIH P41-GM103311.
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Harrison SC. 2015. Viral membrane fusion. Virology 479:498–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Earp LJ, Delos SE, Park HE, White JM. 2004. The many mechanisms of viral membrane fusion proteins. In Membrane Trafficking in Viral Replication, ed. Marsh M, pp. 25–66. Berlin: Springer [Google Scholar]
- 3.Acosta EG, Castilla V, Damonte EB. 2008. Functional entry of dengue virus into Aedes albopictus mosquito cells is dependent on clathrin-mediated endocytosis. J. Gen. Virol 89:474–84 [DOI] [PubMed] [Google Scholar]
- 4.Blanchard E, Belouzard S, Goueslain L, Wakita T, Dubuisson J, et al. 2006. Hepatitis C virus entry depends on clathrin-mediated endocytosis. J. Virol 80:6964–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Empig CJ, Goldsmith MA. 2002. Association of the caveola vesicular system with cellular entry by filoviruses. J. Virol 76:5266–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhu Y-Z, Xu Q-Q, Wu D-G, Ren H, Zhao P, et al. 2012. Japanese encephalitis virus enters rat neuroblastoma cells via a pH-dependent, dynamin and caveola-mediated endocytosis pathway. J. Virol 86:13407–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Macovei A, Radulescu C, Lazar C, Petrescu S, Durantel D, et al. 2010. Hepatitis B virus requires intact caveolin-1 function for productive infection in HepaRG cells. J. Virol 84:243–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Millet JK, Whittaker GR. 2015. Host cell proteases: critical determinants of coronavirus tropism and pathogenesis. Virus Res 202:120–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schornberg K, Matsuyama S, Kabsch K, Delos S, Bouton A, White J. 2006. Role of endosomal cathepsins in entry mediated by the Ebola virus glycoprotein. J. Virol 80:4174–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hulseberg CE, Fénéant L, Szymańska KM, White JM. 2018. Lamp1 increases the efficiency of Lassa virus infection by promoting fusion in less acidic endosomal compartments. mBio 9:e01818–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jae LT, Raaben M, Herbert AS, Kuehne AI, Wirchnianski AS, et al. 2014. Lassa virus entry requires a trigger-induced receptor switch. Science 344:1506–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carette JE, Raaben M, Wong AC, Herbert AS, Obernosterer G, et al. 2011. Ebola virus entry requires the cholesterol transporter Niemann–Pick C1. Nature 477:340–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang H, Shi Y, Song J, Qi J, Lu G, et al. 2016. Ebola viral glycoprotein bound to its endosomal receptor Niemann-Pick C1. Cell 164:258–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wilson IA, Skehel JJ, Wiley DC. 1981. Structure of the haemagglutinin membrane glycoprotein of influenza virus at 3 Å resolution. Nature 289:366–73 [DOI] [PubMed] [Google Scholar]
- 15.Bullough PA, Hughson FM, Skehel JJ, Wiley DC. 1994. Structure of influenza haemagglutinin at the pH of membrane fusion. Nature 371:37–43 [DOI] [PubMed] [Google Scholar]
- 16.Chan DC, Fass D, Berger JM, Kim PS. 1997. Core structure of gp41 from the HIV envelope glycoprotein. Cell 89:263–73 [DOI] [PubMed] [Google Scholar]
- 17.Weissenhorn W, Dessen A, Harrison S, Skehel J, Wiley D. 1997. Atomic structure of the ectodomain from HIV-1 gp41. Nature 387:426–30 [DOI] [PubMed] [Google Scholar]
- 18.Weissenhorn W, Carfí A, Lee K-H, Skehel JJ, Wiley DC. 1998. Crystal structure of the Ebola virus membrane fusion subunit, GP2, from the envelope glycoprotein ectodomain. Mol. Cell 2:605–16 [DOI] [PubMed] [Google Scholar]
- 19.Zhao X, Singh M, Malashkevich VN, Kim PS. 2000. Structural characterization of the human respiratory syncytial virus fusion protein core. PNAS 97:14172–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yin H-S, Wen X, Paterson RG, Lamb RA, Jardetzky TS. 2006. Structure of the parainfluenza virus 5 F protein in its metastable, prefusion conformation. Nature 439:38–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kirchdoerfer RN, Cottrell CA, Wang N, Pallesen J, Yassine HM, et al. 2016. Pre-fusion structure of a human coronavirus spike protein. Nature 531:118–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wrapp D, Wang N, Corbett KS, Goldsmith JA, Hsieh C-L, et al. 2020. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 367:1260–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Böttcher C, Ludwig K, Herrmann A, van Heel M, Stark H. 1999. Structure of influenza haemagglutinin at neutral and at fusogenic pH by electron cryo-microscopy. FEBS Lett 463:255–59 [DOI] [PubMed] [Google Scholar]
- 24.Daniels RS, Downie JC, Hay AJ, Knossow M, Skehel JJ, et al. 1985. Fusion mutants of the influenza virus hemagglutinin glycoprotein. Cell 40:431–39 [DOI] [PubMed] [Google Scholar]
- 25.Carr CM, Kim PS. 1993. A spring-loaded mechanism for the conformational change of influenza hemagglutinin. Cell 73:823–32 [DOI] [PubMed] [Google Scholar]
- 26.Tsurudome M, Glück R, Graf R, Falchetto R, Schaller U, Brunner J. 1992. Lipid interactions of the hemagglutinin HA2 NH2-terminal segment during influenza virus-induced membrane fusion. J. Biol. Chem 267:20225–32 [PubMed] [Google Scholar]
- 27.Hallenberger S, Bosch V, Angliker H, Shaw E, Klenk H-D, Garten W. 1992. Inhibition of furin-mediated cleavage activation of HIV-1 glycoprotein gpl60. Nature 360:358–61 [DOI] [PubMed] [Google Scholar]
- 28.Lu M, Blacklow SC, Kim PS. 1995. A trimeric structural domain of the HIV-1 transmembrane glycoprotein. Nat. Struct. Biol 2:1075–82 [DOI] [PubMed] [Google Scholar]
- 29.Liu J, Bartesaghi A, Borgnia MJ, Sapiro G, Subramaniam S. 2008. Molecular architecture of native HIV-1 gp120 trimers. Nature 455:109–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kwong PD, Wyatt R, Robinson J, Sweet RW, Sodroski J, Hendrickson WA. 1998. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature 393:648–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen B, Vogan EM, Gong H, Skehel JJ, Wiley DC, Harrison SC. 2005. Structure of an unliganded simian immunodeficiency virus gp120 core. Nature 433:834–41 [DOI] [PubMed] [Google Scholar]
- 32.Speck RF, Wehrly K, Platt EJ, Atchison RE, Charo IF, et al. 1997. Selective employment of chemokine receptors as human immunodeficiency virus type 1 coreceptors determined by individual amino acids within the envelope V3 loop. J. Virol 71:7136–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chan DC, Kim PS. 1998. HIV entry and its inhibition. Cell 93:681–84 [DOI] [PubMed] [Google Scholar]
- 34.Tan K, Liu J, Wang J, Shen S, Lu M. 1997. Atomic structure of a thermostable subdomain of HIV-1 gp41. PNAS 94:12303–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Colman PM, Lawrence MC. 2003. The structural biology of type I viral membrane fusion. Nat. Rev. Mol. Cell Biol 4:309–19 [DOI] [PubMed] [Google Scholar]
- 36.Chan DC, Chutkowski CT, Kim PS. 1998. Evidence that a prominent cavity in the coiled coil of HIV type 1 gp41 is an attractive drug target. PNAS 95:15613–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Debnath AK, Radigan L, Jiang S. 1999. Structure-based identification of small molecule antiviral compounds targeted to the gp41 core structure of the human immunodeficiency virus type 1. J. Med. Chem 42:3203–9 [DOI] [PubMed] [Google Scholar]
- 38.Armand-Ugón M, Clotet-Codina I, Tintori C, Manetti F, Clotet B, et al. 2005. The anti-HIV activity of ADS-J1 targets the HIV-1 gp120. Virology 343:141–49 [DOI] [PubMed] [Google Scholar]
- 39.González-Ortega E, Mena M-P, Permanyer M, Ballana E, Clotet B, Esté JA. 2010. ADS-J1 inhibits HIV-1 entry by interacting with gp120 and does not block fusion-active gp41 core formation. Antimicrob. Agents Chemother 54:4487–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Eckert DM, Malashkevich VN, Hong LH, Carr PA, Kim PS. 1999. Inhibiting HIV-1 entry: discovery of D-peptide inhibitors that target the gp41 coiled-coil pocket. Cell 99:103–15 [DOI] [PubMed] [Google Scholar]
- 41.Welch BD, VanDemark AP, Heroux A, Hill CP, Kay MS. 2007. Potent D-peptide inhibitors of HIV-1 entry. PNAS 104:16828–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang H, Qi Z, Guo A, Mao Q, Lu H, et al. 2009. ADS-J1 inhibits human immunodeficiency virus type 1 entry by interacting with the gp41 pocket region and blocking fusion-active gp41 core formation. Antimicrob. Agents Chemother 53:4987–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yu F, Lu L, Liu Q, Yu X, Wang L, et al. 2014. ADS-J1 inhibits HIV-1 infection and membrane fusion by targeting the highly conserved pocket in the gp41 NHR-trimer. Biochim. Biophys. Acta Biomembr 1838:1296–305 [DOI] [PubMed] [Google Scholar]
- 44.Root MJ, Kay MS, Kim PS. 2001. Protein design of an HIV-1 entry inhibitor. Science 291:884–88 [DOI] [PubMed] [Google Scholar]
- 45.Frey G, Rits-Volloch S, Zhang X-Q, Schooley RT, Chen B, Harrison SC. 2006. Small molecules that bind the inner core of gp41 and inhibit HIV envelope-mediated fusion. PNAS 103:13938–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jiang S, Lu H, Liu S, Zhao Q, He Y, Debnath AK. 2004. N-substituted pyrrole derivatives as novel human immunodeficiency virus type 1 entry inhibitors that interfere with the gp41 six-helix bundle formation and block virus fusion. Antimicrob. Agents Chemother 48:4349–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xiao T, Frey G, Fu Q, Lavine CL, Scott DA, et al. 2020. HIV-1 fusion inhibitors targeting the membrane-proximal external region of Env spikes. Nat. Chem. Biol 16:529–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fu Q, Shaik MM, Cai Y, Ghantous F, Piai A, et al. 2018. Structure of the membrane proximal external region of HIV-1 envelope glycoprotein. PNAS 115:E8892–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Salzwedel K, West JT, Hunter E. 1999. A conserved tryptophan-rich motif in the membrane-proximal region of the human immunodeficiency virus type 1 gp41 ectodomain is important for Env-mediated fusion and virus infectivity. J. Virol 73:2469–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Montero M, van Houten NE, Wang X, Scott JK. 2008. The membrane-proximal external region of the human immunodeficiency virus type 1 envelope: dominant site of antibody neutralization and target for vaccine design. Microbiol. Mol. Biol. Rev 72:54–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Muster T, Steindl F, Purtscher M, Trkola A, Klima A, et al. 1993. A conserved neutralizing epitope on gp41 of human immunodeficiency virus type 1. J. Virol 67:6642–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stiegler G, Kunert R, Purtscher M, Wolbank S, Voglauer R, et al. 2001. A potent cross-clade neutralizing human monoclonal antibody against a novel epitope on gp41 of human immunodeficiency virus type 1. AIDS Res. Hum. Retrovir 17:1757–65 [DOI] [PubMed] [Google Scholar]
- 53.Huang J, Ofek G, Laub L, Louder MK, Doria-Rose NA, et al. 2012. Broad and potent neutralization of HIV-1 by a gp41-specific human antibody. Nature 491:406–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Russell RJ, Kerry PS, Stevens DJ, Steinhauer DA, Martin SR, et al. 2008. Structure of influenza hemagglutinin in complex with an inhibitor of membrane fusion. PNAS 105:17736–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kadam RU, Wilson IA. 2017. Structural basis of influenza virus fusion inhibition by the antiviral drug Arbidol. PNAS 114:206–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Battles MB, Langedijk JP, Furmanova-Hollenstein P, Chaiwatpongsakorn S, Costello HM, et al. 2016. Molecular mechanism of respiratory syncytial virus fusion inhibitors. Nat. Chem. Biol 12:87–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Roymans D, Alnajjar SS, Battles MB, Sitthicharoenchai P, Furmanova-Hollenstein P, et al. 2017. Therapeutic efficacy of a respiratory syncytial virus fusion inhibitor. Nat. Commun 8:167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhao Y, Ren J, Harlos K, Jones DM, Zeltina A, et al. 2016. Toremifene interacts with and destabilizes the Ebola virus glycoprotein. Nature 535:169–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Roymans D, De Bondt HL, Arnoult E, Geluykens P, Gevers T, et al. 2010. Binding of a potent small-molecule inhibitor of six-helix bundle formation requires interactions with both heptad-repeats of the RSV fusion protein. PNAS 107:308–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Skehel JJ, Wiley DC. 2000. Receptor binding and membrane fusion in virus entry: the influenza hemagglutinin. Annu. Rev. Biochem 69:531–69 [DOI] [PubMed] [Google Scholar]
- 61.Chen GL, Subbarao K. 2009. Attacking the flu: Neutralizing antibodies may lead to ‘universal’ vaccine. Nat. Med 15:1251–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bodian DL, Yamasaki RB, Buswell RL, Stearns JF, White JM, Kuntz ID. 1993. Inhibition of the fusion-inducing conformational change of influenza hemagglutinin by benzoquinones and hydroquinones. Biochemistry 32:2967–78 [DOI] [PubMed] [Google Scholar]
- 63.Hoffman LR, Kuntz ID, White JM. 1997. Structure-based identification of an inducer of the low-pH conformational change in the influenza virus hemagglutinin: irreversible inhibition of infectivity. J. Virol 71:8808–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Leneva IA, Russell RJ, Boriskin YS, Hay AJ. 2009. Characteristics of arbidol-resistant mutants of influenza virus: implications for the mechanism of anti-influenza action of arbidol. Antiv. Res 81:132–40 [DOI] [PubMed] [Google Scholar]
- 65.van Dongen MJP, Kadam RU, Juraszek J, Lawson E, Brandenburg B, et al. 2019. A small-molecule fusion inhibitor of influenza virus is orally active in mice. Science 363:eaar6221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yao Y, Kadam RU, Lee C-CD, Woehl JL, Wu NC, et al. 2020. An influenza A hemagglutinin small-molecule fusion inhibitor identified by a new high-throughput fluorescence polarization screen. PNAS 117:18431–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Volchkov VE, Feldmann H, Volchkova VA, Klenk H-D. 1998. Processing of the Ebola virus glycoprotein by the proprotein convertase furin. PNAS 95:5762–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mulherkar N, Raaben M, de la Torre JC, Whelan SP, Chandran K. 2011. The Ebola virus glycoprotein mediates entry via a non-classical dynamin-dependent macropinocytic pathway. Virology 419:72–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chandran K, Sullivan NJ, Felbor U, Whelan SP, Cunningham JM. 2005. Endosomal proteolysis of the Ebola virus glycoprotein is necessary for infection. Science 308:1643–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bornholdt ZA, Ndungo E, Fusco ML, Bale S, Flyak AI, et al. 2016. Host-primed Ebola virus GP exposes a hydrophobic NPC1 receptor-binding pocket, revealing a target for broadly neutralizing antibodies. mBio 7:e02154–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Harrison SC. 2008. Viral membrane fusion. Nat. Struct. Mol. Biol 15:690–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Basu A, Li B, Mills DM, Panchal RG, Cardinale SC, et al. 2011. Identification of a small-molecule entry inhibitor for filoviruses. J. Virol 85:3106–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Singleton CD, Humby MS, Yi HA, Rizzo RC, Jacobs A. 2019. Identification of Ebola virus inhibitors targeting GP2 using principles of molecular mimicry. J. Virol 93:e00676–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Johansen LM, Brannan JM, Delos SE, Shoemaker CJ, Stossel A, et al. 2013. FDA-approved selective estrogen receptor modulators inhibit Ebola virus infection. Sci. Transl. Med 5:190ra79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ren J, Zhao Y, Fry EE, Stuart DI. 2018. Target identification and mode of action of four chemically divergent drugs against Ebolavirus infection. J. Med. Chem 61:724–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gaisina IN, Peet NP, Wong L, Schafer AM, Cheng H, et al. 2020. Discovery and structural optimization of 4-(aminomethyl) benzamides as potent entry inhibitors of Ebola and Marburg virus infections. J. Med. Chem 63:7211–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cui Q, Cheng H, Xiong R, Zhang G, Du R, et al. 2018. Identification of diaryl-quinoline compounds as entry inhibitors of Ebola virus. Viruses 10:678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Karron RA, Buonagurio DA, Georgiu AF, Whitehead SS, Adamus JE, et al. 1997. Respiratory syncytial virus (RSV) SH and G proteins are not essential for viral replication in vitro: clinical evaluation and molecular characterization of a cold-passaged, attenuated RSV subgroup B mutant. PNAS 94:13961–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Schlender J, Zimmer G, Herrler G, Conzelmann K-K. 2003. Respiratory syncytial virus (RSV) fusion protein subunit F2, not attachment protein G, determines the specificity of RSV infection. J. Virol 77:4609–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.McLellan JS, Ray WC, Peeples ME. 2013. Structure and function of respiratory syncytial virus surface glycoproteins. In Challenges and Opportunities for Respiratory Syncytial Virus Vaccines, ed. Anderson LJ, Graham BS, pp. 83–104. Berlin: Springer; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yin H-S, Wen X, Paterson RG, Lamb RA, Jardetzky TS. 2006. Structure of the parainfluenza virus 5 F protein in its metastable, prefusion conformation. Nature 439:38–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cianci C, Yu K-L, Combrink K, Sin N, Pearce B, et al. 2004. Orally active fusion inhibitor of respiratory syncytial virus. Antimicrob. Agents Chemother 48:413–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Andries K, Moeremans M, Gevers T, Willebrords R, Sommen C, et al. 2003. Substituted benzimidazoles with nanomolar activity against respiratory syncytial virus. Antiv. Res 60:209–19 [DOI] [PubMed] [Google Scholar]
- 84.Douglas JL, Panis ML, Ho E, Lin K-Y, Krawczyk SH, et al. 2003. Inhibition of respiratory syncytial virus fusion by the small molecule VP-14637 via specific interactions with F protein. J. Virol 77:5054–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bonfanti J-F, Meyer C, Doublet F, Fortin J, Muller P, et al. 2008. Selection of a respiratory syncytial virus fusion inhibitor clinical candidate. 2. Discovery of a morpholinopropylaminobenzimidazole derivative (TMC353121). J. Med. Chem 51:875–96 [DOI] [PubMed] [Google Scholar]
- 86.Mackman RL, Sangi M, Sperandio D, Parrish JP, Eisenberg E, et al. 2015. Discovery of an oral respiratory syncytial virus (RSV) fusion inhibitor (GS-5806) and clinical proof of concept in a human RSV challenge study. J. Med. Chem 58:1630–43 [DOI] [PubMed] [Google Scholar]
- 87.Bonfanti J-F, Doublet F, Fortin J, Lacrampe J, Guillemont J, et al. 2007. Selection of a respiratory syncytial virus fusion inhibitor clinical candidate, part 1: improving the pharmacokinetic profile using the structure–property relationship. J. Med. Chem 50:4572–84 [DOI] [PubMed] [Google Scholar]
- 88.Cianci C, Langley DR, Dischino DD, Sun Y, Yu K-L, et al. 2004. Targeting a binding pocket within the trimer-of-hairpins: small-molecule inhibition of viral fusion. PNAS 101:15046–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.McLellan JS, Chen M, Joyce MG, Sastry M, Stewart-Jones GBE, et al. 2013. Structure-based design of a fusion glycoprotein vaccine for respiratory syncytial virus. Science 342:592–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sun A, Prussia A, Zhan W, Murray EE, Doyle J, et al. 2006. Nonpeptide inhibitors of measles virus entry. J. Med. Chem 49:5080–92 [DOI] [PubMed] [Google Scholar]
- 91.Plemper RK, Doyle J, Sun A, Prussia A, Cheng L-T, et al. 2005. Design of a small-molecule entry inhibitor with activity against primary measles virus strains. Antimicrob. Agents Chemother 49:3755–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hashiguchi T, Fukuda Y, Matsuoka R, Kuroda D, Kubota M, et al. 2018. Structures of the prefusion form of measles virus fusion protein in complex with inhibitors. PNAS 115:2496–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Plemper RK, Erlandson KJ, Lakdawala AS, Sun A, Prussia A, et al. 2004. A target site for template-based design of measles virus entry inhibitors. PNAS 101:5628–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Niedermeier S, Singethan K, Rohrer SG, Matz M, Kossner M, et al. 2009. A small-molecule inhibitor of Nipah virus envelope protein-mediated membrane fusion. J. Med. Chem 52:4257–65 [DOI] [PubMed] [Google Scholar]
- 95.Marcink T, Yariv E, Rybkina K, Más V, Bovier F, et al. 2020. Hijacking the fusion complex of human parainfluenza virus as an antiviral strategy. mBio 11:e03203–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Farzan SF, Palermo LM, Yokoyama CC, Orefice G, Fornabaio M, et al. 2011. Premature activation of the paramyxovirus fusion protein before target cell attachment with corruption of the viral fusion machinery. J. Biol. Chem 286:37945–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Porotto M, Murrell M, Greengard O, Moscona A. 2003. Triggering of human parainfluenza virus 3 fusion protein (F) by the hemagglutinin-neuraminidase (HN) protein: an HN mutation diminishes the rate of F activation and fusion. J. Virol 77:3647–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Bottom-Tanzer SF, Rybkina K, Bell JN, Alabi CA, Mathieu C, et al. 2019. Inhibiting human parainfluenza virus infection by preactivating the cell entry mechanism. mBio 10:e02900–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Adedeji AO, Severson W, Jonsson C, Singh K, Weiss SR, Sarafianos SG. 2013. Novel inhibitors of severe acute respiratory syndrome coronavirus entry that act by three distinct mechanisms. J. Virol 87:8017–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Chen CZ, Xu M, Pradhan M, Gorshkov K, Petersen JD, et al. 2020. Identifying SARS-CoV-2 entry inhibitors through drug repurposing screens of SARS-S and MERS-S pseudotyped particles. ACS Pharmacol. Transl. Sci 3:1165–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Rey FA, Heinz FX, Mandl C, Kunz C, Harrison SC. 1995. The envelope glycoprotein from tick-borne encephalitis virus at 2 Å resolution. Nature 375:291–98 [DOI] [PubMed] [Google Scholar]
- 102.Modis Y, Ogata S, Clements D, Harrison SC. 2003. A ligand-binding pocket in the dengue virus envelope glycoprotein. PNAS 100:6986–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Nybakken GE, Nelson CA, Chen BR, Diamond MS, Fremont DH. 2006. Crystal structure of the West Nile virus envelope glycoprotein. J. Virol 80:11467–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Gibbons DL, Vaney M-C, Roussel A, Vigouroux A, Reilly B, et al. 2004. Conformational change and protein–protein interactions of the fusion protein of Semliki Forest virus. Nature 427:320–25 [DOI] [PubMed] [Google Scholar]
- 105.Lescar J, Roussel A, Wien MW, Navaza J, Fuller SD, et al. 2001. The fusion glycoprotein shell of Semliki Forest virus: an icosahedral assembly primed for fusogenic activation at endosomal pH. Cell 105:137–48 [DOI] [PubMed] [Google Scholar]
- 106.Li L, Jose J, Xiang Y, Kuhn RJ, Rossmann MG. 2010. Structural changes of envelope proteins during alphavirus fusion. Nature 468:705–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Voss JE, Vaney M-C, Duquerroy S, Vonrhein C, Girard-Blanc C, et al. 2010. Glycoprotein organization of Chikungunya virus particles revealed by X-ray crystallography. Nature 468:709–12 [DOI] [PubMed] [Google Scholar]
- 108.DuBois RM, Vaney M-C, Tortorici MA, Al Kurdi R, Barba-Spaeth G, et al. 2013. Functional and evolutionary insight from the crystal structure of rubella virus protein E1. Nature 493:552–56 [DOI] [PubMed] [Google Scholar]
- 109.Dessau M, Modis Y. 2013. Crystal structure of glycoprotein C from Rift Valley fever virus. PNAS 110:1696–701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Halldorsson S, Behrens A-J, Harlos K, Huiskonen JT, Elliott RM, et al. 2016. Structure of a phleboviral envelope glycoprotein reveals a consolidated model of membrane fusion. PNAS 113:7154–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Överby AK, Pettersson RF, Grünewald K, Huiskonen JT. 2008. Insights into bunyavirus architecture from electron cryotomography of Uukuniemi virus. PNAS 105:2375–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kuhn RJ, Zhang W, Rossmann MG, Pletnev SV, Corver J, et al. 2002. Structure of dengue virus: implications for flavivirus organization, maturation, and fusion. Cell 108:717–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Zhang Y, Corver J, Chipman PR, Zhang W, Pletnev SV, et al. 2003. Structures of immature flavivirus particles. EMBO J 22:2604–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Yu IM, Zhang W, Holdaway HA, Li L, Kostyuchenko VA, et al. 2008. Structure of the immature dengue virus at low pH primes proteolytic maturation. Science 319:1834–37 [DOI] [PubMed] [Google Scholar]
- 115.Li L, Lok S-M, Yu IM, Zhang Y, Kuhn RJ, et al. 2008. The flavivirus precursor membrane-envelope protein complex: structure and maturation. Science 319:1830–34 [DOI] [PubMed] [Google Scholar]
- 116.De Curtis I, Simons K. 1988. Dissection of Semliki Forest virus glycoprotein delivery from the trans-Golgi network to the cell surface in permeabilized BHK cells. PNAS 85:8052–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Uchime O, Fields W, Kielian M. 2013. The role of E3 in pH protection during alphavirus assembly and exit. J. Virol 87:10255–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Yap ML, Klose T, Urakami A, Hasan SS, Akahata W, Rossmann MG. 2017. Structural studies of Chikungunya virus maturation. PNAS 114:13703–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Bressanelli S, Stiasny K, Allison SL, Stura EA, Duquerroy S, et al. 2004. Structure of a flavivirus envelope glycoprotein in its low-pH-induced membrane fusion conformation. EMBO J 23:728–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Modis Y, Ogata S, Clements D, Harrison SC. 2004. Structure of the dengue virus envelope protein after membrane fusion. Nature 427:313–19 [DOI] [PubMed] [Google Scholar]
- 121.Cecilia D, Gould EA. 1991. Nucleotide changes responsible for loss of neuroinvasiveness in Japanese encephalitis virus neutralization-resistant mutants. Virology 181:70–77 [DOI] [PubMed] [Google Scholar]
- 122.Liao M, Kielian M. 2005. Domain III from class II fusion proteins functions as a dominant-negative inhibitor of virus membrane fusion. J. Cell Biol 171:111–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Schmidt AG, Yang PL, Harrison SC. 2010. Peptide inhibitors of flavivirus entry derived from the E protein stem. J. Virol 84:12549–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Monath TP, Arroyo J, Levenbook I, Zhang Z-X, Catalan J, et al. 2002. Single mutation in the flavivirus envelope protein hinge region increases neurovirulence for mice and monkeys but decreases viscerotropism for monkeys: relevance to development and safety testing of live, attenuated vaccines. J. Virol 76:1932–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Lee E, Weir RC, Dalgarno L. 1997. Changes in the dengue virus major envelope protein on passaging and their localization on the three-dimensional structure of the protein. Virology 232:281–90 [DOI] [PubMed] [Google Scholar]
- 126.Kampmann T, Yennamalli R, Campbell P, Stoermer MJ, Fairlie DP, et al. 2009. In silico screening of small molecule libraries using the dengue virus envelope E protein has identified compounds with antiviral activity against multiple flaviviruses. Antiv. Res 84:234–41 [DOI] [PubMed] [Google Scholar]
- 127.Poh MK, Yip A, Zhang S, Priestle JP, Ma NL, et al. 2009. A small molecule fusion inhibitor of dengue virus. Antiv. Res 84:260–66 [DOI] [PubMed] [Google Scholar]
- 128.Wang Q-Y, Patel SJ, Vangrevelinghe E, Xu HY, Rao R, et al. 2009. A small-molecule dengue virus entry inhibitor. Antimicrob. Agents Chemother 53:1823–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Yennamalli R, Subbarao N, Kampmann T, McGeary RP, Young PR, Kobe B. 2009. Identification of novel target sites and an inhibitor of the dengue virus E protein. J. Comput.-Aided Mol. Des 23:333–41 [DOI] [PubMed] [Google Scholar]
- 130.Zhou Z, Khaliq M, Suk J-E, Patkar C, Li L, et al. 2008. Antiviral compounds discovered by virtual screening of small-molecule libraries against dengue virus E protein. ACS Chem. Biol 3:765–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Adrián FJ, Ding Q, Sim T, Velentza A, Sloan C, et al. 2006. Allosteric inhibitors of Bcr-abl–dependent cell proliferation. Nat. Chem. Biol 2:95–102 [DOI] [PubMed] [Google Scholar]
- 132.Clark MJ, Miduturu C, Schmidt AG, Zhu X, Pitts JD, et al. 2016. GNF-2 inhibits dengue virus by targeting Abl kinases and the viral E protein. Cell Chem. Biol 23:443–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.de Wispelaere M, Lian W, Potisopon S, Li P-C, Jang J, et al. 2018. Inhibition of flaviviruses by targeting a conserved pocket on the viral envelope protein. Cell Chem. Biol 25:1006–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Schmidt AG, Yang PL, Harrison SC. 2010. Peptide inhibitors of dengue-virus entry target a late-stage fusion intermediate. PLOS Pathog 6:e1000851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Li P-C, Jang J, Hsia C-Y, Groomes PV, Lian W, et al. 2019. Small molecules targeting the flavivirus E protein with broad-spectrum activity and antiviral efficacy in vivo. ACS Infect. Dis 5:460–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Lian W, Jang J, Potisopon S, Li P-C, Rahmeh A, et al. 2018. Discovery of immunologically inspired small molecules that target the viral envelope protein. ACS Infect. Dis 4:1395–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Zhang Y, Zhang W, Ogata S, Clements D, Strauss JH, et al. 2004. Conformational changes of the flavivirus E glycoprotein. Structure 12:1607–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Modis Y, Ogata S, Clements D, Harrison SC. 2005. Variable surface epitopes in the crystal structure of dengue virus type 3 envelope glycoprotein. J. Virol 79:1223–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Kanai R, Kar K, Anthony K, Gould LH, Ledizet M, et al. 2006. Crystal structure of West Nile virus envelope glycoprotein reveals viral surface epitopes. J. Virol 80:11000–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Pitts J, Hsia C-Y, Lian W, Wang J, Pfeil M-P, et al. 2019. Identification of small molecule inhibitors targeting the Zika virus envelope protein. Antiv. Res 164:147–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Lindenbach BD, Rice CM. 2013. The ins and outs of hepatitis C virus entry and assembly. Nat. Rev. Microbiol 11:688–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Kong L, Giang E, Nieusma T, Kadam RU, Cogburn KE, et al. 2013. Hepatitis C virus E2 envelope glycoprotein core structure. Science 342:1090–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Khan AG, Whidby J, Miller MT, Scarborough H, Zatorski AV, et al. 2014. Structure of the core ectodomain of the hepatitis C virus envelope glycoprotein 2. Nature 509:381–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.El Omari K, Iourin O, Kadlec J, Sutton G, Harlos K, et al. 2014. Unexpected structure for the N-terminal domain of hepatitis C virus envelope glycoprotein E1. Nat. Commun 5:4874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.El Omari K, Iourin O, Harlos K, Grimes JM, Stuart DI. 2013. Structure of a pestivirus envelope glycoprotein E2 clarifies its role in cell entry. Cell Rep 3:30–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Scarselli E, Ansuini H, Cerino R, Roccasecca RM, Acali S, et al. 2002. The human scavenger receptor class B type I is a novel candidate receptor for the hepatitis C virus. EMBO J 21:5017–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Pileri P, Uematsu Y, Campagnoli S, Galli G, Falugi F, et al. 1998. Binding of hepatitis C virus to CD81. Science 282:938–41 [DOI] [PubMed] [Google Scholar]
- 148.Agnello V, Ábel G, Elfahal M, Knight GB, Zhang Q-X. 1999. Hepatitis C virus and other flaviviridae viruses enter cells via low density lipoprotein receptor. PNAS 96:12766–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Germi R, Crance JM, Garin D, Guimet J, Lortat-Jacob H, et al. 2002. Cellular glycosaminoglycans and low density lipoprotein receptor are involved in hepatitis C virus adsorption. J. Med. Virol 68:206–15 [DOI] [PubMed] [Google Scholar]
- 150.Farquhar MJ, Hu K, Harris HJ, Davis C, Brimacombe CL, et al. 2012. Hepatitis C virus induces CD81 and claudin-1 endocytosis. J. Virol 86:4305–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Evans MJ, von Hahn T, Tscherne DM, Syder AJ, Panis M, et al. 2007. Claudin-1 is a hepatitis C virus co-receptor required for a late step in entry. Nature 446:801–5 [DOI] [PubMed] [Google Scholar]
- 152.Ploss A, Evans MJ, Gaysinskaya VA, Panis M, You H, et al. 2009. Human occludin is a hepatitis C virus entry factor required for infection of mouse cells. Nature 457:882–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Garry RF, Dash S. 2003. Proteomics computational analyses suggest that hepatitis C virus E1 and pestivirus E2 envelope glycoproteins are truncated class II fusion proteins. Virology 307:255–65 [DOI] [PubMed] [Google Scholar]
- 154.Perin PM, Haid S, Brown RJP, Doerrbecker J, Schulze K, et al. 2016. Flunarizine prevents hepatitis C virus membrane fusion in a genotype-dependent manner by targeting the potential fusion peptide within E1. Hepatology 63:49–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Li H-F, Huang C-H, Ai L-S, Chuang C-K, Chen SSL. 2009. Mutagenesis of the fusion peptide-like domain of hepatitis C virus E1 glycoprotein: involvement in cell fusion and virus entry. J. Biomed. Sci 16:89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Lavillette D, Pécheur E-I, Donot P, Fresquet J, Molle J, et al. 2007. Characterization of fusion determinants points to the involvement of three discrete regions of both E1 and E2 glycoproteins in the membrane fusion process of hepatitis C virus. J. Virol 81:8752–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Petracca R, Falugi F, Galli G, Norais N, Rosa D, et al. 2000. Structure-function analysis of hepatitis C virus envelope-CD81 binding. J. Virol 74:4824–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Hu Z, Lan K-H, He S, Swaroop M, Hu X, et al. 2014. Novel cell-based hepatitis C virus infection assay for quantitative high-throughput screening of anti-hepatitis C virus compounds. Antimicrob. Agents Chemother 58:995–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Hu Z, Hu X, He S, Yim HJ, Xiao J, et al. 2015. Identification of novel anti-hepatitis C virus agents by a quantitative high throughput screen in a cell-based infection assay. Antiv. Res 124:20–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.He S, Li K, Lin B, Hu Z, Xiao J, et al. 2017. Development of an aryloxazole class of hepatitis C virus inhibitors targeting the entry stage of the viral replication cycle. J. Med. Chem 60:6364–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Ma CD, Imamura M, Talley DC, Rolt A, Xu X, et al. 2020. Fluoxazolevir inhibits hepatitis C virus infection in humanized chimeric mice by blocking viral membrane fusion. Nat. Microbiol 5:1532–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Hu Z, Rolt A, Hu X, Ma CD, Le DJ, et al. 2020. Chlorcyclizine inhibits viral fusion of hepatitis C virus entry by directly targeting HCV envelope glycoprotein 1. Cell Chem. Biol 27:780–92.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.He S, Lin B, Chu V, Hu Z, Hu X, et al. 2015. Repurposing of the antihistamine chlorcyclizine and related compounds for treatment of hepatitis C virus infection. Sci. Transl. Med 7:282ra49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Chao LH, Jang J, Johnson A, Nguyen A, Gray NS, et al. 2018. How small-molecule inhibitors of dengue-virus infection interfere with viral membrane fusion. eLife 7:e36461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Tanner EJ, Liu H, Oberste MS, Pallansch M, Collett MS, Kirkegaard K. 2014. Dominant drug targets suppress the emergence of antiviral resistance. eLife 3:e03830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, et al. 2004. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem 25:1605–12 [DOI] [PubMed] [Google Scholar]
- 167.Pessi A 2015. Cholesterol-conjugated peptide antivirals: a path to a rapid response to emerging viral diseases. J. Peptide Sci 21:379–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.