

Abstract
Background:
Autophagy plays an important role in causing inflammatory responses initiated by environmental pollutants and respiratory tract infection.
Objective:
We sought to investigate the role of cockroach allergen–induced excessive activation of autophagy in allergic airway inflammation and its underlying molecular mechanisms.
Methods:
Environmental allergen–induced autophagy was investigated in the primary human bronchial epithelial cells (HBECs) and lung tissues of asthmatic mouse model and patients. The role of autophagy in asthma development was examined by using autophagy inhibitor 3-methyladenine in an asthma mouse model. Furthermore, the involvements of reactive oxygen species (ROS) and oxidized Ca2+/calmodulin-dependent protein kinase II (ox-CaMKII) signaling in regulating autophagy during asthma were examined in allergen-treated HBECs and mouse model.
Results:
Cockroach allergen activated autophagy in HBECs and in the lung tissues from asthmatic patients and mice. Autophagy inhibitor 3-methyladenine significantly attenuated airway hyperresponsiveness, TH2-associated lung inflammation, and ROS generation. Mechanistically, we demonstrated a pathological feedforward circuit between cockroach allergen–induced ROS and autophagy that is mediated through CaMKII oxidation. Furthermore, transgenic mice with ROS-resistant CaMKII MM-VVδ showed attenuation of TH2-associated lung inflammation and autophagy. Mitochondrial ox-CaMKII inhibition induced by adenovirus carrying mitochondrial-targeted inhibitor peptide CaMKIIN suppresses cockroach allergen–induced autophagy, mitochondrial dysfunction, mitophagy, and cytokine production in HBECs. Finally, mitochondrial CaMKII inhibition suppressed the expression of one of the key ubiquitin-binding autophagy receptors, optineurin, and its recruitment to fragmented mitochondria. Optineurin knockdown inhibited cockroach allergy–induced mitophagy.
Conclusions:
Our data suggest a previously uncovered axis of allergen-ROS-ox-CaMKII-mitophagy in the development of allergic airway inflammation and asthma.
Keywords: Asthma, cockroach allergen, ROS, CaMKII, mitochondria, autophagy, mitophagy
Graphical Abstract
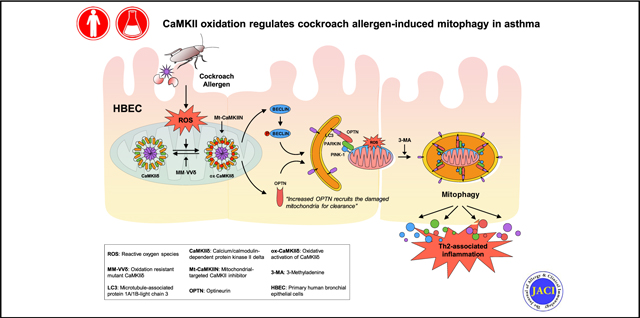
Autophagy is an important homeostatic process by which damaged proteins and organelles are captured by autophagosomes and taken to lysosomes for degradation during cellular stress.1 Studies have demonstrated autophagy participation in inflammatory diseases including asthma.2,3 Increased autophagy has been observed in sputum granulocytes, peripheral blood cells, and peripheral eosinophils from patients with severe asthma.4 Increased autophagy has also been associated with asthma immune mechanisms, extracellular matrix deposition, and airway remodeling.5,6 It has been reported that autophagy can be induced by reactive oxygen species (ROS), which facilitate cellular adaptation and diminish oxidative damage.7 These findings raise the possibility that allergens may induce ROS as signaling molecules to initiate the process of the autophagic cycle. However, the molecular mechanisms underlying the interplay between ROS and autophagy in the development of asthma remain unclear.
The Ca2+/calmodulin-dependent protein kinase II (CaMKII) is a serine/threonine-specific protein kinase that is initially activated by binding calcified calmodulin. Once activated by Ca2+/calmodulin, CaMKII activity can be sustained, even after Ca2+/calmodulin unbinding by oxidation of regulatory domain methionines 281/282 (oxidized Ca2+/calmodulin-dependent protein kinase II [ox-CaMKII]).8 Increased expression of ox-CaMKII has been correlated with the severity of asthma,9 and airway epithelial mitochondrial CaMKII plays an important role in mitochondrial ROS generation and allergic inflammation.10 We have previously demonstrated that oxidant-resistant CaMKII, due to knock in replacement of methionine 281 and 282 with valines on the CaMKIIδ isoform (MM-VVδ), protects against cockroach allergen–induced ROS generation and airway inflammation.11 Recent studies suggested that CaMKII can regulate autophagyby directly phosphorylating Beclin 1,12 a central player in the formation and maturation of autophagosome.13 Beclin 1 also plays a critical role in the initiation of mitophagy by facilitating parkin translocation to mitochondria.14 Mitophagy, also called mitochondrial autophagy, was first introduced by Lemasters15 to describe the selective degradation of mitochondria by autophagy. Mitophagy can specifically target damaged mitochondria for degradation to maintain cellular homeostasis.16 The PTEN-induced putative kinase 1 (PINK1) and Parkin-mediated pathway has been recognized as a key regulatory mechanism of mitophagy.17,18
In the current study, we provided new evidence that excessive autophagy plays a critical role in cockroach allergen–induced airway epithelial ROS generation, cytokine release, and subsequent allergic airway inflammation. By investigating the underlying mechanisms, we revealed a feedforward connection between cockroach allergen–induced ROS and autophagy in airway epithelia cells. Our studies suggest that CaMKII is one of the key signaling pathways downstream to ROS and that ox-CaMKII regulates allergic lung inflammation and autophagy/mitophagy. We further identified optineurin (OPTN) as a critical component in the signaling pathway regulating mitophagy that was controlled by ox-CaMKII. Our data provide insights into the development of allergic airway inflammation in asthma.
METHODS
Animal
All animal experimentations were carried out using 6- to 8-week-old mice. C57BL/6J (wild-type, stock: 000664) mice were purchased from the Jackson Laboratory (Bar Harbor, Maine). ROS-resistant CaMKII-delta (MM-VVδ) mutant mice with C57BL/6 background were provided by Professor Mark Anderson at Johns Hopkins University.11 Animals were maintained underspecific pathogen-free conditions at the animal facility of the Johns Hopkins University School of Medicine. All experiments were carried out with age- and sex-matched controls. The experimental protocol was reviewed and approved by the Institutional Animal Care and Use Committee of the Johns Hopkins University, Baltimore, Md.
Human lung tissues
Dr Kollarik at the Johns Hopkins Asthma Center Histology Laboratory provided paraffin-embedded human airway sections from asthmatic and healthy individuals. The Johns Hopkins Institutional Review Board has approved the study with approval number NA_00046988.
Cockroach allergen–induced mouse model of asthma
Cockroach allergen–induced mouse model of asthma was generated as previously described.19 In some cases, mice were treated with 3-methyladenine (3-MA, 30mg/kg, Sigma-Aldrich, St Louis, Mo) or saline vehicle control by intraperitoneal administration 2 hours before every single challenge.
Airway hyperresponsiveness
Airway responsiveness was measured as previously described.19
Analysis of lung inflammation
Cockroach allergen–induced lung inflammation was determined by the analyses of lung tissue histology and inflammatory cells and cytokines in bronchoalveolar lavage (BAL) fluids as previously described.11,19–21
ELISA
Levels of IL-4, IL-5, IL-13, IL-17, IL-10, TGF-β1, TSLP, IL-33, and IL-25 were measured by ELISA with Ready-Set-Go! ELISA Kits (ThermoFisher) according to the manufacturer’s instructions. Serum levels of allergen specific IgE and IgG1 were also analyzed by ELISA as previously described.19
Immunofluorescence staining
Immunofluorescence staining was performed as previously reported.19 Primary antibodies are provided in Table E1 in this article’s Online Repository at www.jacionline.org. Fluorescent positive cells or positive staining of tissue sections was quantified in at least 4 different hpfs per slide or lung section using Fiji ImageJ v2.00 (National Institutes of Health) and presented as mean fluorescent intensity per square micrometer.
Western blotting
Western blotting was carried out as previously described.11 Primary antibodies are provided in Table E1.
Quantitative RT-PCR
Quantitative RT-PCR was performed as previously reported.19 Primer sequences are provided in Table E2 in this article’s Online Repository at www.jacionline.org.
Assessment of intracellular ROS production
Intracellular ROS were detected using the general and mitochondria-specific oxidative sensitive fluorescent dyes CM-H2DCFDA (ThermoFisher) and MitoSOX Red (ThermoFisher) as previously reported.20
Transfection of GFP-light chain 3 and CaMKIIN into airway epithelial cells
EGFP-light chain 3 (LC3) was a gift from Karla Kirkegaard.22 Approximately 3.0 × 105 human bronchial epithelial cells (HBECs) were seeded onto a 6-well tissue culture plate for 24 hours before transfection with a plasmid expressing EGFP-LC3 or empty vector control using FuGENE 6 Transfection Reagent (Promega) following the manufacturer’s instructions. Similarly, adenovirus containing the cDNA for mitochondrial-targeted GFP (empty vector) or mitochondrial-targeted inhibitor peptide CaMKIIN (Mt-CaMKIIN) (both at 100 MOI) were transfected for 24 hours. Transfection efficiency was assessed by GFP expression using flow cytometry.
Mitochondrial respiration
Mitochondrial respiration was determined by measuring oxygen consumption rates (OCRs) with Seahorse extracellular flux assay kit (Agilent) on a seahorse XF96 Extracellular Flux Analyzer, according to the manufacturer’s protocol. Briefly, HBECs were plated onto wells of a 1% gelatin-coated XF96 plate for 24 hours, and then treated with cockroach extract (CRE) or PBS control for 48 hours. OCRs were measured using the mitochondrial stress test procedure in Dulbecco modified Eagle medium containing 10 mM glucose, 4 mM L-glutamine, and 2 mM sodium pyruvate with pH 7.4. After calibration of the sensor cartridge, the XF96 platewas placed into the seahorse instrument and OCR was measured as p mole per minute per cell. Analysis of mitochondrial function was made using changes in OCR in the presence of 1.0 μM oligomycin (inhibits mitochondrial ATP synthase), 1.0 μM FCCP (uncoupler), and 0.5 μM rotenone and antimycin A (inhibits both complex I and III).
Mitophagy detection
HBECs were transfected with 1 μg of plasmid expressing the mitochondrially targeted Keima (MT-mKeima-Red, addgene)23 using FuGENE 6 Transfection Reagent (Promega) followed by treatment with CaMKIIN or GFP control. The fluorescent protein Keima has an excitation spectrum that changes according to pH. A short wavelength (440 nm) is predominant for excitation in a neutral environment, whereas a long wavelength (586 nm) is predominant in an acidic environment, which occurs when mitochondria are fused with lysosomes. Mitophagy was quantified through the difference in fluorescent wavelengths (586 nm [green]/440 nm [red]) from images obtained using Fiji ImageJ v2.00-rc-69/1.52p (National Institutes of Health). Nuclear staining was carried out using 0.5 μg/mL of Hoechst 33258 (ThermoFisher).
Statistical analysis
All data were analyzed with Graph Pad Prism statistical software program (GraphPad Software, La Jolla, Calif) and are expressed as mean ± SEM. Statistical significance for normally distributed samples was assessed by an independent 2-tailed Student t test or a 1-way ANOVA followed by Turkey post hoc test. A P value of less than .05 was considered statistically significant for all analyses.
RESULTS
Cockroach allergen induces autophagy in human airway epithelial cells and asthmatic lung tissues
To determine whether CRE exposure can elicit autophagy, the expression of autophagy-associated genes was detected in CRE-treated HBECs. Western blot analysis illustrated a dose-dependent induction of autophagy by CE treatment in HBECs, as assessed by the ratio of microtubule-associated protein 1A/1B-LC3 (Fig 1, A and B). Consistently, a dose-dependent increase in the expression of autophagy-related gene (ATG)5 and decrease in p62 were detected in the CRE-treated HBECs. The results were further confirmed by RT-PCR (see Fig E1, A, in this article’s Online Repository at www.jacionline.org). To visualize the induction of autophagy, we transfected a GFP-LC3 construct in HBECs and imaged GFP-LC3 after CRE treatment (20 μg/mL). As shown in Figure 1, C, the autophagy induction by CRE was increased as monitored by the number of LC3 puncta. In line with the finding, cockroach allergen–induced epithelial activation was observed as assessed by the increased levels of TGF-β1, IL-25, and IL-33 in supernatants (Fig 1, D). Furthermore, to determine the role of autophagy in the pathogenesis of asthma, we detected the expression of autophagy markers in the lung tissues of patients with asthma and healthy controls by immunofluorescent staining. As expected, there was a signficant increase in the expression of LC3B and ATG5 in the lung tissues of asthmatic patients compared with healthy controls (Fig 1, E and F). We also examined the expression of Lc3b and Atg5 in the lung tissues of mouse model of asthma.20,24 Consistently, CRE-treated mice showed significantly increased expression of Lc3b in the lung tissues (Fig E1, B and C). The Lc3b was predominantly expressed in the airway epithelium as assessed by the colocalization of Lc3b and epithelial cell marker EpCAM. Similarly, increased expression of Atg5 was observed in the lung tissues of these mice (Fig E1, B and D). Together, these data indicate that cockroach allergen can induce autophagy in primary airway epithelial cells and lung tissues of asthmatic patients and mice.
FIG 1.

Cockroach allergen induces autophagy in human airway epithelial cells and asthmatic lung tissues. A, Western blot analysis of LC3-I, LC-3II, ATG5, and p62 in HBECs treated with CRE at different concentrations for 4 hours. β-Actin was measured as the loading control. B, Quantitative analysis of Figure 1, A (n = 4). C, Representative immunofluorescence images of HBECs expressing GFP-LC3 after CRE treatment (20 μg/mL), and the average number of puncta per cell was analyzed in the PBS and CRE-treated group (n = 12, number of puncta per cell from 4 different hpfs of 3 independent experiments). D, Quantitative analysis of IL-25, IL-33, and TGF-β1 levels in supernatants of PBS- or CRE-treated HBECs for 4 hours (n = 4–8). E, Representative immunofluorescence images of LC3B and ATG5 expression in lung tissues from asthmatic patients (n = 5) and healthy controls (n = 5). F, Quantitative analysis of Figure 1, E. Data represent mean ± SEM. *P < .05, **P < .01, ***P < .001.
INHIBITION OF AUTOPHAGY SUPPRESSES ALLERGEN-INDUCED AIRWAY HYPERRESPONSIVENESS AND LUNG INFLAMMATION
Next, we investigated the role of autophagy in cockroach allergen–induced airway hyperresponsiveness (AHR) and lung inflammation by using autophagy inhibitor 3-MA in our acute mouse model of asthma following the protocol illustrated in Fig 2, A. 3-MA is a class III group of phosphoinositide 3-kinase inhibitors that has been widely used as an autophagy inhibitor.25 Compared with controls, CRE challenge induced an increased airway resistance, which was attenuated by 3-MA treatment (Fig 2, B). Consistent with our previous findings,20,21 CRE challenge increased airway inflammation compared with PBS treatment. When these CRE-challenged mice were pretreated with 3-MA, the increased lung inflammation was significantly abrogated. Specifically, 3-MA treatment suppressed cockroach allergen–induced peribronchial inflammation (hematoxylin and eosin) and goblet cell hyperplasia (periodic acid-Schiff) (Fig 2, C and D). Moreover, these 3-MA–treated mice showed a marked reduction in the number of total inflammatory cells, particularly eosinophils in the BAL fluids (Fig 2, E). Serum levels of cockroach allergen specific IgE and IgG1 (sIgE and sIgG1) were also reduced in 3-MA–treated mice (Fig 2, F). Furthermore, mice treated with 3-MA showed reduced IL-4, IL-5, IL-13, and TGF-β1, but unchanged IL-17α and IL-10 levels in the BAL fluids (Fig 2, G). These results provide evidence that inhibition of autophagy can prevent cockroach allergen–induced TH2 inflammation. In addition, 3-MA–treated mice showed reduced levels of epithelial-derived cytokines TGF-β1, IL-33, and TSLP in the BAL fluids (see Fig E2, A, in this article’s Online Repository at www.jacionline.org), which were further supported by in vitro analyses of 3-MA–treated CRE-challenged HBECs (Fig E2, B).
FIG 2.

Inhibition of autophagy suppresses allergen-induced lung inflammation. A, Protocol for cockroach allergen–induced mouse model of asthma in the presence or absence of 3-MA during the challenge phase. B, Lung resistance in response to increasing concentrations of methacholine using the forced oscillation technique (FlexiVent, SCIREQ) (n = 6). C, Histological examination of mouse paraffin lung sections stained with hematoxylin and eosin (H&E, upper panel) and periodic acid-Schiff (PAS, lower panel) staining. D, Percentage of PAS+ area among total lung area. E, BAL fluid total and differential (eosinophil, macrophage, neutrophil, and lymphocyte) cell counts as assessed by flow cytometry. F, Serum levels of cockroach allergen–specific IgE and IgG1. G, BAL fluid levels of cytokines. E-G, n = 6–9. Data represent mean ± SEM from 2 independent experiments. *P < .05, **P < .01, ***P < .001.
Feedforward connection between cockroach allergen–induced ROS and autophagy
Recent studies have suggested a cross-talk between ROS and autophagy.7,26 Thus, we asked whether inhibition of autophagy could suppress ROS generation in our mouse model of asthma. Indeed, we found increased ROS expression in the lung tissues of CRE-treated mice (Fig 3, A and B), which was abolished when these mice were pretreated with 3-MA. The results were further supported by in vitro analyses of intracellular and mitochondrial ROS production. Compared with untreated cells, CRE-treated HBECs displayed increased mitochondrial ROS as detected by immunostaining for the colocalization of MitoSOX, a fluorescent mitochondrial ROS reporter dye, with MitoTracker green (Fig 3, C and D). Of note, the increased mitochondrial ROS were significantly inhibited by 3-MA. Similar patterns were observed when intracellular ROS were detected by flow cytometry with CM-H2DCFDA (Fig 3, E and F). These findings suggest that inhibition of autophagy can suppress cockroach allergen–induced ROS generation, particularly mitochondrial ROS.
FIG 3.

Feedforward connection between cockroach allergen–induced ROS and autophagy. A, Representative immunofluorescence images of ROS expression with dihydroethidium (DHE) in lung tissues from cockroach allergen–induced mouse model of asthma treated with or without 3-MA (30 mg/kg, i.p.). B, Quantitative analysis of florescent signal in lung tissue sections (n = 8). C, Representative immunofluorescence images of mitochondrial ROS expression with MitoSOX in cockroach allergen–treated HBECs with or without 3-MA. D, Quantitative analysis of florescent signal in HBECs (n = 8). E, Flow cytometry analysis of intracellular ROS production with CM-H2DCFDA. F, Quantitative analysis of flow cytometry data (n = 5). G, Western blot analysis of LC3-I, LC-3II, ATG5, and p62 expression in CRE-treated HBECs in the presence or absence of either NAC or MitoTEMPO. β-Actin was measured as the loading control. H, Quantitative analysis of Western blots. Data represent mean ± SEM from 2 independent experiments. *P < .05, **P < .01, ***P < .001.
In contrast, we investigated whether ROS can regulate the activation of autophagy by using antioxidant N-acetyl cysteine (NAC) or mitochondrial-targeted antioxidant MitoTEMPO. Compared with media control, CRE treatment led to an increase in the expression of LC3 and ATG5 as detected by Western blot (Fig 3, G and H), which were significantly inhibited by either NAC or MitoTEMPO. In contrast, the CRE-induced decrease in p62 was fully recovered by either NAC or MitoTEMPO treatment. Compared with CRE, no clear autophagy induction was observed for either NAC or MitoTEMPO (see Fig E3, A and B, in this article’s Online Repository at www.jacionline.org). Collectively, these data suggest a feedforward connection between cockroach allergen–induced ROS and autophagy.
Cockroach allergen induces mitochondrial CaMKII oxidation in airway epithelial cells
Our previous studies suggested a critical role of CaMKII oxidation in mast cell activation and the development of asthma.11 Here, we examined whether cockroach allergen can induce CaMKII oxidation in airway epithelial cells. HBECs were treated with CRE at different concentrations for 24 hours, and expression of ox-CaMKII was detected by immunostaining. We found a dose-dependent increase in the expression of ox-CaMKII in HBECs after CRE treatment (see Fig E4, A and B, in this article’s Online Repository at www.jacionline.org). Furthermore, the increased ox-CaMKII was predominantly localized in mitochondria as stained with TOM20, a specific marker for mitochondria. Our results indicate that cockroach allergen can induce CaMKII oxidation, specifically in mitochondria.
ROS-resistant CaMKII prevents allergen-induced autophagy
To better understand the role of ox-CaMKII in asthma, we used an oxidant-resistant CaMKII MM-VVδ (Fig 4, A) to generate a mouse model of asthma.20,24 Histological analysis demonstrated an increased expression of ox-CaMKII in cockroach allergen–induced mouse model of asthma, particularly in the airway epithelial cells, by coimmunostaining of ox-CaMKII and EpCaM (Fig 4, B and C). As expected, ox-CaMKII expression was almost completely absent in the CaMKII MM-VVδ mice. Furthermore, these mice showed protection against CRE-induced peribronchial inflammation (hematoxylin and eosin) and goblet cell hyperplasia (periodic acid-Schiff) (see Fig E5, A and B, in this article’s Online Repository at www.jacionline.org). These CaMKII MM-VVδ mice also showed less total inflammatory cells, particularly eosinophils, in the BAL fluids (Fig E5, C). We next investigated an unexplored link between CaMKII oxidation and autophagy in allergic asthma. Compared with PBS, CRE-treated WT mice showed increased expression of Lc3b in the airways (Fig 4, D and E), which was attenuated in CaMKII MM-VVδ mice. CRE-induced expression of Atg5 in the airways was similarly attenuated in the CaMKII MM-VVδ mice (Fig 4, D and F). In contrast, p62 expression was reduced in CRE-induced wild-type mice, but increased in CaMKII MM-VVδ mice (Fig 4, D and G). These findings were further supported by RT-PCR (Fig E5, D) and Western blot analyses (Fig 4, H and I). Collectively, these data suggest that CaMKII oxidation may regulate cockroach allergen–induced lung inflammation and autophagy.
FIG 4.

ROS-resistant CaMKII prevents allergen-induced autophagy in asthma mouse model. A, Scheme of ROS-resistant CaMKII MM-VVδ (281/282) mice generation. B, Representative immunofluorescence images of ox-CaMKII expression (green) in the airway epithelial cells (EpCAM, red) of CRE-induced mouse model of asthma with wild-type (WT) or CaMKII MM-VVδ (MM-VVδ) mice. C, Quantitative analysis of florescent signal (n = 5–6). D, Representative immunofluorescence images of LC3B, Atg5, and p62 expression (red) in the airway epithelial cells (EpCAM, red) of CRE-induced mouse model of asthma with WT or CaMKII MM-VVδ (MM-VVδ) mice. E-G, Quantitative analysis of florescent signal in Figure 4, D, for LC3 (E), ATG5 (F), and p62 (G) in lung tissues of CRE-induced mouse model of asthma with WT or MM-VVδ mice. n = 6–8 from 2 independent experiments. H, Western blot analysis of LC3-I, LC-3II, ATG5, and p62 in lung tissues of CRE-induced mouse model of asthma with WT or MM-VVδ mice. β-Actin was measured as the loading control. I, Quantitative analysis of Western blots. Data represent mean ± SEM of 2 independent experiments. *P < .05, **P < .01, ***P < .001.
Inhibition of mitochondrial CaMKII suppresses autophagy
To confirm the role of CaMKII in regulating autophagy, we investigated whether inhibiting mitochondrial CaMKII can affect cockroach allergen–induced autophagy. HBECs were infected with adenovirus containing an Mt-CaMKIIN or control adenovirus and confirmed by immunostaining (see Fig E6, A and B, in this article’s Online Repository at www.jacionline.org). We found that CRE exposure increased both LC3 and ATG5, but decreased p62 in HBECs as detected by immunostaining (Fig E6, C and D). However, the increased expression of LC3 and ATG5 and decreased expression of p62 were reversed in HBECs pretransduced with Mt-CaMKIIN. Similar observations were also made by Western blot analysis (Fig 5, A and B). Thus, the data support a view that mitochondrial CaMKII regulates autophagy in bronchial epithelial cells.
FIG 5.

Inhibition of mitochondrial CaMKII-suppressed autophagy. A, Western blot analysis of LC3-I, LC-3II, ATG5, and p62 in empty-GFP or Mt-CaMKIIN–transfected HBECs with PBS or CRE treatment. β-Actin was measured as the loading control. B, Quantitative analysis of Western blots (n = 3). C, Western blot analysis of p-AMPK, AMPK, p-BECLIN 1, BECLIN 1, p-mTOR, and mTOR expression in empty-GFP or Mt-CaMKIIN–transfected HBECs with PBS or CRE treatment. β-Actin was measured as the loading control. D, Quantitative analysis of Western blots. p-AMPK, Phosphorylation of AMPK; p-MTOR, phosphorylation of mTOR. Data represent mean ± SEM of 2 independent experiments. *P < .05, **P < .01, ***P < .001.
We also explored the further downstream signal molecules that mediate CaMKII-regulated autophagy. CaMKII has been shown to regulate autophagy by directly phosphorylating Beclin 1.12 Consistent with this finding, CRE induced an increase in the phosphorylation of Beclin 1, which was reversed by Mt-CaMKIIN treatment (Fig 5, C and D). Furthermore, we tested several significant signaling molecules (eg, AMP-activated protein kinase [AMPK] and mTOR) that are critical in the initiation of autophagosome.27 Similar to Beclin 1, CRE induced an increase in the phosphorylation of AMPK and a decrease in the phosphorylation of mTOR. Of interest, the CRE-induced increase in the phosphorylation of AMPK and decrease in the phosphorylation of mTOR were reversed by Mt-CaMKIIN. Therefore, CaMKII may play a critical role in cockroach allergen–induced initiation of autophagosome via multiple signaling pathways.
Inhibition of mitochondrial CaMKII protects against allergen-induced mitochondrial respiration
Elevated levels of mitochondrial ROS may lead to mitochondrial dysfunction. We investigated oxygen consumption rate (OCR) in CRE-treated HBECs, an indicator of mitochondrial respiration, as illustrated in Fig 6, A. Basal and maximal respiration, ATP production, and spare respiration capacity were measured in HBECs after 48 hours of CRE treatment (see Fig E7, A, in this article’s Online Repository at www.jacionline.org). Compared with control cells, CRE-challenged cells showed substantially decreased basal respiration rate and ATP production, and increased H+ leak after blocking the proton channel subunit F0 of the ATP synthase with oligomycin (Fig E7, B). Maximal respiration elicited by FCCP, which discharges the pH gradient across the inner mitochondrial membrane, was also substantially lower in CRE-treated HBECs. Similar patterns were observed for the spare respiration capacity. We next investigated the potential effects of ox-CaMKII on cockroach allergen–induced mitochondrial respiration by pretreating HBECs with Mt-CaMKIIN as illustrated in Figure 6, B. Pretreatment of HBECs with Mt-CaMKIIN reversed the CRE-induced reduction in basal and maximal respiration, ATP production, and spare respiration capacity in HBECs (Fig 6, C). Together, these data align with previous studies28 and indicate that inhibition of mitochondrial CaMKII protects against allergen-induced mitochondrial dysfunction in airway epithelial cells.
FIG 6.

Inhibition of mitochondrial CaMKII protects against allergen-induced mitochondrial respiration. A, OCR under basal conditions followed by the sequential measurements after addition of oligomycin, FCCP, rotenone, and antimycin A were determined by Mito Stress test. B, HBECs were transfected with Mt-CaMKIIN and then treated with CRE for OCR measurement. C, Measurement of basal respiration, ATP production, H leak, maximal respiration, and spare respiration capacity in CRE-treated HBECs. n = 7–8. FCCP, Carbonyl cyanide-4-phenylhydrazone; Max, maximum; prod., production; res., respiration. Data represent mean ± SEM of 2 independent experiments. P < .05, **P < .01, ***P < .001.
Inhibition of mitochondrial CaMKII suppresses cockroach allergen–induced mitophagy
Next, we examined the cockroach allergen–induced mitophagy using MT-mKeima-Red,23 which can track the mitochondrial delivery to lysosomes (Fig 7, A). HBECs were transfected with plasmid MT-mKeima-Red and then treated with CRE or PBS. The mitochondria-localized MT-mKeima-Red was displayed in CRE, but not in PBS-treated HBECs (Fig 7, B), indicating an induction of mitophagy by cockroach allergen. We next investigated whether the inhibition of mitochondrial CaMKII can affect cockroach allergen–induced mitophagy. HBECs were transduced with the inhibitory peptide CaMKIIN, then transfected with plasmid MT-mKeima-Red. As expected, Mt-CaMKIIN significantly inhibited CRE-induced mitophagy (Fig 7, C). Furthermore, we examined whether inhibition of mitochondrial CaMKII can affect cockroach allergen–induced epithelial cytokine release. We found higher levels of TGF-β1, IL-25, IL-33, and TSLP in supernatants of CRE-treated HBECs as compared with controls (Fig 7, D), whereas TGF-β1, IL-25, and TSLP, but not IL-33, were significantly reduced in Mt-CaMKIIN–transfected HBECs. Thus, these findings suggest that inhibition of mitochondrial CaMKII suppresses cockroach allergen–induced mitophagy and cytokine release in the airway epithelial cells.
FIG 7.

Inhibition of mitochondrial CaMKII suppressed cockroach allergen–induced mitophagy in airway epithelial cells. A, Scheme of mitophagy detection with MT-mKeima-Red. B, Representative immunofluorescence images of mitophagy with MT-mKeima-Red (red) and mitochondria (Mitotracker, green) in empty-GFP or Mt-CaMKIIN–transfected HBECs with PBS or CRE treatment. C, Quantitative analysis of florescent signals in HBECs (n = 10–18). D, Quantitative analysis of TGF-β1, IL-25, IL-33, and TSLP levels in supernatants in Mock or CaMKIIN-transfected HBECs with PBS or CRE treatment for 4 hours (n = 4). Data represent mean ± SEM. *P < .05, **P < .01, ***P < .001.
Inhibition of mitochondrial CaMKII suppressed mitophagy through controlling OPTN
Finally, we explored the underlying mechanisms by which mitochondrial CaMKII regulates mitophagy. We found that CRE stimulated an increase in the expression of ULK1, PINK1, and PARKIN, the most studied pathways for autophagy. These increased expressions were suppressed in HBECs transfected with Mt-CaMKIIN (Fig 8, A). Furthermore, Western blot analyses showed a similar pattern for the expression of PINK1 and PARKIN (see Fig E8, A and B, in this article’s Online Repository at www.jacionline.org). OPTN, one of the ubiquitin-binding autophagy receptors, was enhanced by CRE treatment, whereas this enhancement was abolished by Mt-CaMKIIN (Fig 8, A). To confirm the involvement of OPTN in cockroach allergen–induced mitophagy, we transfected a GFP-labeled OPTN in HBECs. HBECs treated with CRE showed the recruitment of OPTN-GFP to fragmented mitochondria as assessed by the robust colocalization of OPTN-GFP with MitoTracker (Fig 8, B and C) and OPTN was stably enriched in more than 50% of mitochondria (Fig 8, D). Of note, the recruitment of OPTN-GFP to the fragmented mitochondria was inhibited by Mt-CaMKIIN. Furthermore, we explored the role of OPTN in cockroach allergen–induced mitophagy by OPTN knockdown in HBECs. As expected, knockdown of OPTN attenuated the CRE-induced mitophagy in HBECs (Fig 8, E and F). These results suggest that OPTN is involved in the CaMKII-regulated cockroach allergen–induced mitophagy
FIG 8.

Inhibition of mitochondrial CaMKII suppressed mitophagy through controlling OPTN. A, Quantitative PCR analysis of ULK1, PINK1, PARKIN, and OPTN in empty-GFP or Mt-CaMKIIN–transfected HBECs with PBS or CRE treatment (n = 6). B, Representative immunofluorescence images of HBECs expressing OPTN-GFP after CRE treatment in the presence or absence of Mt-CaMKIIN transfection. C and D, Number of OPTN foci (Fig 8, C) and percentage (Fig 8, D) of OPTN-MitoTracker were quantified in the PBS and CRE-treated group. n = 10–18. E, Representative images of mitophagy with MT-mKeima-Red (red) and mitochondria (Mitotracker, green) in lentivirus-shRNA-Mock or lentivirus-shRNA-OPTN–transfected HBECs with PBS or CRE treatment. F, Quantitative analysis of florescent signals in HBECs (n = 16). Data represent mean ± SEM from 2 independent experiments. *P < .05, **P < .01, ***P < .001.
DISCUSSION
In the present study, we examined whether cockroach allergen, one of the major triggers for developing asthma, can induce autophagy in the airway epithelial cells and explored the role of autophagy in cockroach allergen–induced airway inflammation and its underlying mechanisms.
Our in vivo results demonstrated that autophagy was increased in the lung tissues of cockroach allergen–treated mice, particularly in the airway epithelium of these mice. Further in vitro analysis in the human airway epithelial cells revealed that treatment with cockroach allergen led to increased autophagy as indicated by the increased LC3 and ATG5 and decreased p62 expression. Importantly, by visualizing autophagosome formation in real time in live cells using GFP-LC3, a mammalian expression vector containing the human LC3 gene fused at its 5′ end to the GFP gene, we found a time-dependent increase in the number of autophagosomes in the airway epithelial cells exposed to cockroach allergen. These results demonstrated that cockroach allergen could directly stimulate autophagy in airway epithelial cells. Although we mainly focused on airway epithelial cells, which are the first line of defense against environmental pollutants and allergens, cockroach allergen could also induce autophagy in other structural and immune cells. Our results support previous findings on the association of autophagy with asthma and provide further evidence that environmental allergens can induce autophagy, which may contribute to epithelial cell cytokine release and subsequent airway inflammation and asthma.
Despite the increased autophagy in asthma, the exact role of autophagy in asthma remains still poorly understood. Our findings indicate that inhibition of autophagy using 3-MA, a widely used autophagy inhibitor that can block the formation of autophagosome, significantly attenuated AHR and lung eosinophilic inflammation and levels of TH2 cytokines. These results suggest that blockage of autophagy may prevent cockroach allergen–induced AHR and TH2-associated inflammation. Recent studies showed very similar results—that treatment with 3-MA suppressed ovalbumin-induced eosinophilia, lung inflammation, oxidative stress, mitochondrial damage, and formation of eosinophil extracellular traps in asthma.29 Furthermore, inhibition of autophagy attenuated the ovalbumin-induced airway epithelial fibrosis30 and HDM-induced airway inflammation, AHR, and features of airway fibrosis and remodeling.5 These findings implicated that autophagy could exacerbate asthma. However, autophagy may also have a protective mechanism given that autophagy is a key homeostatic process in which cytosolic components are degraded and recycled through lysosomes. For example, autophagy-deficient mice (atg5−/− and atg7−/−) develop spontaneous sterile lung inflammation,31 and deficiency of cell-specific autophagy (eg, CD11C-Cre:Atg5fl/fl and ER-Cre:Atg7fl/fl) results in increased airway inflammation and airway hyperreactivity.32–34 The reasons for the dual roles of autophagy in asthma remain unclear. It is postulated that autophagy may have a protective role in maintaining homeostasis at baseline or excessive autophagy during acute infection may become detrimental in a prolonged exposure to environmental pollutants/allergens or inflammation.35 Furthermore, autophagy may play distinct roles in different cell types, thereby leading to different phenotypic changes.
Increased ROS have been shown to cause airway and lung damage and be associated with severity of asthma.36–38 We have previously shown that environmental allergens can induce ROS, which are primary mediators to induce airway epithelial cytokine release (eg, TSLP and IL-33).20,39 Similarly, ROS have been considered as a key checkpoint responsible for IL-33 release from damaged airway epithelium.40 Another important role of ROS is to activate autophagy,7 and that persistent ROS overproduction results in the dysregulation of autophagy, leading to airway epithelial cell damage, inflammatory cytokine release, and exacerbation of allergic inflammation in asthma. Thus, an attractive hypothesis would be that ROS, as signaling molecules, might contribute to epithelial barrier dysfunction, cytokine release, and allergic airway inflammation through activating autophagy. Indeed, we found that cockroach allergen–induced autophagy can be blocked by either antioxidant NAC or mitochondrial-targeted antioxidant MitoTEMPO, suggesting that ROS, particularly mitochondrial ROS, may be critical in cockroach allergen–induced autophagy. In contrast, autophagy can also regulate ROS generation. Autophagy is generally thought to eliminate intracellular sources of ROS; however, prolonged activation of autophagy or impaired autophagy under a disease state may promote ROS production.41 Indeed, we found that inhibition of autophagy suppressed cockroach allergen–induced ROS generation in a mouse model of asthma and in the primary airway epithelial cells. Together, these studies suggest a pathological feedforward connection between ROS and autophagy in airway epithelium that may exacerbate cytokine release and allergic airway inflammation in asthma.
Our study also provides more detailed molecular mechanisms underlying ROS-autophagy interplay. It was suggested that ROS can induce autophagy by directly oxidizing autophagy-related proteins.26 CaMKII has been suggested to be one of the key signaling pathways downstream to ROS and can be activated to be oxidized CaMKII.8,42 We have previously suggested a strong association for ox-CaMKII with asthma.9,11 Our current studies demonstrated a novel role for ox-CaMKII in cockroach allergen–induced autophagy. First, cockroach allergen can induce the expression of ox-CaMKII in HBECs. Next, the increased ox-CaMKII was significantly abrogated by NAC or MitoTEMPO, indicating that cockroach allergen could induce ox-CaMKII though ROS generation and mitochondrial ROS. Furthermore, ox-CaMKII may serve as a key signaling molecule that contributes to cockroach allergen–induced lung inflammation and autophagy. Especially, CaMKII MM-VVδ mice showed reduced lung inflammation and autophagy in comparison to wild-type controls. This raises the possibility that the reduced inflammation may be due to the reduced autophagy in CaMKII MM-VVδ mice and further suggests that ox-CaMKII may be critical in cockroach allergen–induced autophagy and subsequently airway inflammation and asthma.
To identify the underlying mechanisms by which CaMKII regulates autophagy, we tested several key signaling molecules (eg, Beclin 1,12 AMPK,43–45 and mTOR43,46) for autophagy activation and determined whether CaMKII is involved in controlling their expression. Of these signaling molecules, Beclin 1 was directly phosphorylated by CaMKII to promote K63-linked ubiquitination of Beclin 1 and activation of autophagy.12 We found that inhibition of CaMKII could attenuate cockroach allergen–induced phosphorylation of Beclin 1, supporting that CaMKII may play a role in cockroach allergen–induced autophagy through phosphorylation of Beclin 1. Similar effects were observed for the phosphorylation of AMPK and mTOR. In addition, CaMKII may regulate autophagy through other signaling molecules. For example, several transcription factors (eg, p53 and Forkhead box O347 and transcription factor EB48) and histone modifications (histone H4K16 acetylation, H3K9 dimethylation, and H3K27 trimethylation)49 have drawn attention in regulating autophagy.
Mitochondria are critical in maintaining cellular homeostasis, and the clearance of damaged or depolarized mitochondria is through mitophagy.17,50 Mitophagy is a selective degradation of mitochondria by autophagy.15 It often happens to the damaged mitochondria following the exposure to environmental pollutants/allergens or stress. Defective removal of damaged mitochondria can induce hyperactivation of inflammatory signaling pathways, leading to chronic inflammation. Both enhanced and impaired mitophagy have been associated with chronic obstructive pulmonary disease and lung fibrosis.51,52 Our current studies provide evidence that cockroach allergen can lead to mitochondrial dysfunction with decreased basal and maximal respiration and ATP turnover rate, and that inhibition of mitochondrial CaMKII can reverse this process. Furthermore, we demonstrated that cockroach allergen can induce mitophagy, and the epithelial mitochondria-targeted inhibition of CaMKII significantly suppressed cockroach allergen–induced mitophagy. Collectively, these data suggest that mitochondrial CaMKII may contribute to the cockroach allergen-mitochondrial dysfunction and mitophagy.
Pink1- and Parkin-mediated mitochondrial autophagy is currently the most well-established molecular mechanism for the regulation of mitophagy in mammalian cells.52,53 Active PINK1 accumulates on the outer mitochondrial membrane to recruit Parkin to damaged mitochondria, resulting in the phosphorylation and activation of Parkin. The activated Parkin triggers the polyubiquitination of several outer mitochondrial membrane proteins, which interact directly with LC3 to regulate mitochondrial elimination. Studies have also suggested other proteins that are involved in the molecular regulation of mitophagy, such as Nip3-like protein X, BCL2 interacting protein 3, and Fun14 domain-containing protein 1.16,53,54 In this study, we suggest one of the novel mechanisms that CaMKII modulates mitophagy through regulating OPTN, a ubiquitin-binding autophagy receptor. OPN can bind both ubiquitin and LC3 to target ubiquitinated substrates to newly forming autophagosomes for the autophagic clearance of damaged mitochondria.1 Our studies demonstrated that cockroach allergen can induce the recruitment of OPTN to fragmented mitochondria, and OPTN knockdown protects against cockroach allergy–induced mitophagy. Thus, one of the potential mechanisms could be that CaMKII regulates mitophagy through regulating OPTN. However, future studies are needed to explore the mechanisms for ox-CaMKII–regulated OPTN and mitophagy.
Conclusions
We have provided evidence for the first time that cockroach allergen induces autophagy in airway epithelial cells. Inhibition of autophagy suppresses cockroach allergen–induced AHR, airway epithelial cytokine release, and TH2-associated lung inflammation. Most importantly, we found that there is a feedforward loop between cockroach allergen–induced ROS generation and autophagy, and identified CaMKII as a linkage molecule that is activated by ROS and lead to downstream regulation of autophagy/mitophagy. We further revealed one of the novel mechanisms that CaMKII modulates mitophagy through regulating OPTN. These results suggest an allergen-ROS-ox-CaMKII-mitophagy axis in the development of allergic airway inflammation, and CaMKII may be an attractive therapeutic target for asthma.
METHODS
Cockroach allergen–induced mouse model of asthma
Mice were sensitized by intratracheal inhalation of 20 μg CRE (Stallergenes Greer, Inc, Cambridge, Mass) in 50 μL of PBS on days 0 to 4. Mice were then challenged on days 10 to 13. Control mice received PBS during the sensitization and challenge phases. On day 14, mice were sacrificed, BAL fluid was harvested for total and differential counts of lavage cells and ELISA measurements, and lung tissues were dissected for histological analyses. Blood was taken to screen for serum antibodies against CREs. In some cases, mice were treated with 3-MA (30 mg/kg, Sigma-Aldrich, St Louis, Mo) or saline vehicle control by intraperitoneal administration 2 hours before every single challenge.
Analysis of lung inflammation
For the histological assessment of lung inflammation, lung tissues were fixed in 4% paraformaldehyde and embedded in paraffin. Lung sections (5 μM) were prepared from these lung tissues and stained with hematoxylin and eosin and periodic acid-Schiff. Images were obtained using a NIKON ECLIPSETi-U microscope equipped with DS-Fi2 camera (NIKON). For analysis of BAL fluids, total cells in the bronchial lavage fluids were counted using the Countess II Automated Cell Counter (ThermoFisher, Waltham, Mass), and cellular differential percentages were determined by means characterized by flow cytometry on a FACScalibur flow cytometer (BD Biosciences, San Jose, Calif), and analyzed using FlowJo v10.4 (Treestar, Woodburn, Ore). Eosinophils are characterized as SSChi, SiglecF+ (clone 1RNM44N, ThermoFisher), and Mac-3− (clone M3/84, BD Biosciences) cells, alveolar macrophages as SSChi, SiglecF+, and Mac-3+, granulocytes as SSChi and Gr-1+ (clone RB6–8C5, BioLegend, San Diego, Calif), and lymphocytes as FSClo/SSClo and CD3+ (clone 145–2C11, BioLegend).
Quantitative RT-PCR
The purity of RNA was tested by measuring the ratio of absorbance at 260 nm over 280 nm. For 1RT-PCR, 1 μg RNA was reverse transcribed into complementary DNA using the SuperScript First-Strand Synthesis System (Invitrogen), andthen quantitative RT-PCR was performed with SYBR GreenMaster Mix (Qiagen, Hilden, Germany) on an ABI Prism 7300 detection system. Relative expression was calculated by using the 2−ΔΔCT method as described by Livak and Schmittgen.E2 The levels of mRNA were normalized to the internal control gene.
Assessment of intracellular ROS production
Normal HBECs (CRL-4051, ATCC, Old Town Manassas, Va) were loaded with 2 μM of CM-H2DCFDA and treated with 3-MA or vehicle control 1 hour before CRE treatment. CM-H2DCFDA (excitation/emission: 492–495/517–527 nm) fluorescent intensity was assessed using a FACScalibur flow cytometer (BD Biosciences). In a separate experiment, CRE (20 or 50 μg/mL) was used to treat HBECs loaded with the antioxidant NAC (1 or 10 μM), MitoTEMPO (10 or 100 μM), or vehicle controls 1 hour before allergen treatment. For mitochondrial ROS measurement, HBECs were cultured onto 35-mm glass-bottom dish (Mattek, Ashland, Mass), loaded with 10 μM of MitoSOX Red and 50 nM of MitoTracker Green (ThermoFisher), and treated with 3-MA or vehicle control 1 hour before CRE treatment (50 μg/mL) in F-12K media at 37°C in a 5% CO2 atmosphere. Cells were imaged with a NIKON ECLIPSE Ti-U microscope equipped with PCO Edge LT monochrome camera. Florescent signal was quantified using Fiji ImageJ v2.00-rc-69/1.52p (National Institutes of Health).
OPTN knockdown
HBECs were treated with Mission shRNA (Sigma-Aldrich, TRCN0000083745) lentivirus particles expressing OPTN shRNA at various multiplicity of infection (at 1, 5, 10, and 20). Controls cells received lentivirus mock treatments. Cells were selected with puromycin in complete media for 7 continuous days. Selected clones were expanded, and OPTN expression was confirmed using quantitative PCR. Mitophagy was assessed in 3 different lentivirus particles expressing OPTN shRNA and mock-treated HBE clones using MT-mKeima-Red 24 hours after CRE or PBS treatment.
Extended Data
FIG E1.

Expression of autophagy in cockroach allergen (CRE)-treated HBECs and mouse model of asthma. A, HBECs were treated with different dosages of CRE (0–50 μg/mL) for 4 hours. Expression of LC3A, LC3B, ATG5, and p62 was measured by quantitative PCR (n = 3). B, Representative immunofluorescence images of Lc3b and Atg5 expression in lung tissues from cockroach allergen–induced mouse model of asthma. C and D, Relative expression analysis of Lc3b (Fig E2, C) and Atg5 (Fig E1, D). n = 12. Data represent mean ± SEM. *P < .05, **P < .01, ***P < .001.
FIG E2.

Inhibition of autophagy suppresses epithelial cytokine release in cockroach allergen–treated HBECs and mouse model of asthma. A, Levels of TGF-β1, IL-25, IL-33, and TSLP in the BAL fluids of CRE-induced mouse model of asthma in the presence or absence of 3-MA (n = 8). B, HBECs were treated with CRE (50 μg/mL) in the presence or absence of 3-MA for 16 hours. Levels of TGF-β1, IL-25, IL-33, and TSLP were measured by ELISA (n = 8). Data represent mean ± SEM. *P < .05, **P < .01, ***P < .001.
FIG E3.

No clear autophagy was induced by either NAC or MitoTEMPO. A, Representative Western blot analyses of LC3A/B-I, LC3A/B-II, ATG5, and p62 expression in media, NAC, MitoTEMPO (MT), and cockroach allergen–treated HBECs. β-Actin was used as a loading control. B, Quantification of Western blots in Figure E3, A.
FIG E4.

Cockroach allergen induces CaMKII oxidation in airway epithelial cells. A, Representative immunofluorescence images of ox-CaMKII expression (red) in mitochondria (TOM20, green) in HBECs with different doses of CRE. B, Relative expression analysis of florescent signal (n = 8). Data represent mean ± SEM. *P < .05, ***P < .001.
FIG E5.

ROS-resistant CaMKII prevents allergen-induced lung inflammation. A, Histological examination of mouse paraffin lung sections stained with hematoxylin and eosin (H&E, upper panel) and periodic acid-Schiff (PAS, lower panel) staining. B, Percentage of PAS+ cell area. C, BAL fluid total and eosinophil cell counts as assessed by flow cytometry (n = 5). D, Expression of Lc3a, Lc3b, Atg5, and p62 in the lung tissues of WT and CaMKII MM-VV mice was measured by quantitative PCR (n = 4–6). Data represent mean ± SEM. *P < .05, **P < .01, ***P < .001.
FIG E6.

Inhibition of mitochondrial CaMKII suppressed autophagy. A, Adenovirus containing the cDNA for mitochondrial-targeted GFP (empty vector) or CaMKIIN. B, HBECs were transfected with or without Mt-CaMKIIN and confirmed by immunostaining. C, Representative immunofluorescence images of LC3B, ATG5, and p62 expression (red) in empty-GFP or Mt-CaMKIIN–transfected HBECs with PBS or cockroach allergen treatment. D, Quantitative analysis of florescent signals (n = 6). Data represent mean ± SEM. *P < .05, **P < .01, ***P < .001.
FIG E7.

Cockroach allergen induces mitochondrial respiration. A, HBECs were treated with CRE for 48 hours and then used for OCR measurement. B, Basal respiration, ATP production, H leak, maximal respiration, and spare respiration capacity were measured in CRE-treated HBECs (n = 7–8). Max, Maximum; Prod., production; Res., respiration. Data represent mean ± SEM. *P < .05, **P < .01, ***P < .001.
FIG E8.

Inhibition of mitochondrial CaMKII suppressed mitophagy pathway PINK1/Parkin. A, Western blot analysis of PINK1 and PARKIN expression in Ad5-Mock or Ad-CaMKIIN–transfected HBECs with PBS or CRE treatment. β-Actin was measured as the loading control. B, Relative expression analysis of Figure E8, A (n = 4). Data represent mean ± SEM. *P < .05, **P < .01.
TABLE E1.
Antibodies used for WB and immunostaining
| Target | Specie | Clone | Assay (dilution) | Company |
|---|---|---|---|---|
| AMPK | Rabbit | D5A2 | ● WB (1:500) | Cell Signaling |
| ATG5 | Rabbit | NB110-53818 | ● WB (1:500) ● IF (1:300) |
NOVUSBIO |
| BECLIN-1 | Rabbit | 3738 | ● WB (1:500) | Cell Signaling |
| Ep-CAM | Mouse | VU1D9 | ● IF (1:500) | Cell Signaling |
| GAPDH | Rabbit | 14C10 | ● WB (1:1000) | Cell Signaling |
| GFP | Rat | FM264G | ● IF (1:500) | BioLegend |
| LC3A/B | Rabbit | NB100-2220 | ● WB (1:1000) ● IF (1:200) |
NOVUSBIO |
| mTOR | Rabbit | 7C10 | ● WB (1:500) | Cell Signaling |
| ox-CAMKII | Rabbit | 07-1387 | ● WB (1:500) ● IF (1:100) |
EMD Millipore |
| p-AMPK | Rabbit | D4D6D | ● WB (1:500) | Cell Signaling |
| p-BECLIN-1 | Rabbit | D9A5G | ● WB (1:500) | Cell Signaling |
| p-mTOR | Rabbit | D9C2 | ● WB (1:500) | Cell Signaling |
| p62 | Rabbit | NBP1-48320 | ● WB (1:2000) | NOVUSBIO |
| PARKIN | Mouse | 4211 | ● WB (1:1000) | Cell Signaling |
| PINK1 | Rabbit | 6946 | ● WB (1:500) | Cell Signaling |
| TOM20 | Mouse | F-10 | ● IF (1:200) | Santa Cruz Biotechnology |
| β-Actiin | Mouse | 2F1-1 | ● WB (1:1000) | BioLegend |
GAPDH, Glyceraldehyde 3-phosphate dehydrogenase; p-AMPK, phosphorylation of AMPK; p-MTOR, phosphorylation of mTOR; WB, Western blot.
TABLE E2.
Primers used for RT-PCR assay
| Gene | NCBI GeneID | Primer bank ID | Amplicon size | Sequence |
|---|---|---|---|---|
| ATG5 | 9474 | 92859692c1 | 133 | ● Fwd: 5′-AAAGATGTGCTTCGAGATGTGT |
| ● Rev: 5′-CACTTTGTCAGTTACCAACGTCA | ||||
| GAPDH | 2597 | 378404907c1 | 197 | ● Fwd: 5′-GGAGCGAGATCCCTCCAAAAT |
| ● Rev: 5′-GGCTGTTGTCATACTTCTCATGG | ||||
| GUSB | 2990 | 268834191c1 | 127 | ● Fwd: 5′-GTCTGCGGCATTTTGTCGG |
| ● Rev: 5′-CACACGATGGCATAGGAATGG | ||||
| LC3A | 84557 | 377652329c1 | 127 | ● Fwd: 5′-AACATGAGCGAGTTGGTCAAG |
| ● Rev: 5′-GCTCGTAGATGTCCGCGAT | ||||
| LC3B | 81631 | 12383056a1 | 167 | ● Fwd: 5′-GATGTCCGACTTATTCGAGAGC |
| ● Rev: 5′-TTGAGCTGTAAGCGCCTTCTA | ||||
| OPTN | 10133 | 56549110c1 | 124 | ● Fwd: 5′-CCAAACCTGGACACGTTTACC |
| ● Rev: 5′-CCTCAAATCTCCCTTTCATGGC | ||||
| p62 | 8878 | 214830450c1 | 92 | ● Fwd: 5′-GCACCCCAATGTGATCTGC |
| ● Rev: 5′-CGCTACACAAGTCGTAGTCTGG | ||||
| PARKIN | 5071 | 169790970c1 | 129 | ● Fwd: 5′-GTGTTTGTCAGGTTCAACTCCA |
| ● Rev: 5′-GAAAATCACACGCAACTGGTC | ||||
| PINK1 | 65018 | 112382374c1 | 114 | ● Fwd: 5′-GCCTCATCGAGGAAAAACAGG |
| ● Rev: 5′-GTCTCGTGTCCAACGGGTC | ||||
| ULK1 | 8408 | 225637564c2 | 118 | ● Fwd: 5′-AGCACGATTTGGAGGTCGC |
| ● Rev: 5′-GCCACGATGTTTTCATGTTTCA |
Fwd, Forward; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; Rev, reverse.
Key messages.
Cockroach allergen can induce autophagy and ROS in airway epithelial cells.
There is a feedforward connection between cockroach allergen–induced ROS and autophagy.
Excessive activation of autophagy contributes to AHR, ROS generation, and TH2-associated lung inflammation.
ROS-activated CaMKII plays a role in downstream regulation of autophagy/mitophagy.
Acknowledgments
We thank Dr Mark Anderson for providing CaMKII MM-VVδ mice and Mt-CaMKIIN. We also thank Dr Kollarik for providing paraffin-embedded human airway sections from asthmatic and healthy individuals.
The research was supported by grants from the US National Institutes of Health (grant nos. R56 AI143668, 2R56ES021739, R21 AI137547, and R01AI141642 to P.G.).
Abbreviations used
- AHR
Airway hyperresponsiveness
- AMPK
AMP-activated protein kinase
- ATG
Autophagy-related gene
- BAL
Bronchoalveolar lavage
- CaMKII
Ca2+/calmodulin-dependent protein kinase II
- CRE
Cockroach extract
- HBEC
Human bronchial epithelial cell
- LC3
Light chain 3
- Mt-CaMKIIN
Mitochondrial-targeted inhibitor peptide CaMKIIN
- NAC
N-acetyl cysteine
- MT-mKeima-Red
Mitochondrially targeted Keima-Red
- OCR
Oxygen consumption rate
- OPTN
Optineurin
- ox-CaMKII
Oxidized Ca2+/calmodulin-dependent protein kinase II
- PINK1
PTEN-induced putative protein kinase 1
- ROS
Reactive oxygen species
- TSLP
Thymic stromal lymphopoietin
- 3-MA
3-methyladenine
Footnotes
Disclosure of potential conflict of interest: The authors declare that they have no relevant conflicts of interest.
REFERENCES
- 1.Sachdeva K, Do DC, Zhang Y, Hu X, Chen J, Gao P. Environmental exposures and asthma development: autophagy, mitophagy, and cellular senescence. Front Immunol 2019;10:2787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lee J, Kim HS. The role of autophagy in eosinophilic airway inflammation. Immune Netw 2019;19:e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zeki AA, Yeganeh B, Kenyon NJ, Post M, Ghavami S. Autophagy in airway diseases: a new frontier in human asthma? Allergy 2016;71:5–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ban GY, Pham DL, Trinh TH, Lee SI, Suh DH, Yang EM, et al. Autophagy mechanisms in sputum and peripheral blood cells of patients with severe asthma: a new therapeutic target. Clin Exp Allergy 2016;46:48–59. [DOI] [PubMed] [Google Scholar]
- 5.McAlinden KD, Deshpande DA, Ghavami S, Xenaki D, Sohal SS, Oliver BG, et al. Autophagy activation in asthma airways remodeling. Am J Respir Cell Mol Biol 2019;60:541–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xia F, Deng C, Jiang Y, Qu Y, Deng J, Cai Z, et al. IL4 (interleukin 4) induces autophagy in B cells leading to exacerbated asthma. Autophagy 2018;14:450–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kaushal GP, Chandrashekar K, Juncos LA. Molecular interactions between reactive oxygen species and autophagy in kidney disease. Int J Mol Sci 2019;20:3791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Erickson JR, Joiner ML, Guan X, Kutschke W, Yang J, Oddis CV, et al. A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell 2008;133:462–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sanders PN, Koval OM, Jaffer OA, Prasad AM, Businga TR, Scott JA, et al. CaMKII is essential for the proasthmatic effects of oxidation. Sci Transl Med 2013;5:195ra97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sebag SC, Koval OM, Paschke JD, Winters CJ, Jaffer OA, Dworski R, et al. Mitochondrial CaMKII inhibition in airway epithelium protects against allergic asthma. JCI Insight 2017;2:e88297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Qu J, Do DC, Zhou Y, Luczak E, Mitzner W, Anderson ME, et al. Oxidized CaMKII promotes asthma through the activation of mast cells. JCI Insight 2017;2: e90139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li X, Wu XQ, Deng R, Li DD, Tang J, Chen WD, et al. CaMKII-mediated Beclin 1 phosphorylation regulates autophagy that promotes degradation of Id and neuroblastoma cell differentiation. Nat Commun 2017;8:1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kang R, Zeh HJ, Lotze MT, Tang D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ 2011;18:571–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Choubey V, Cagalinec M, Liiv J, Safiulina D, Hickey MA, Kuum M, et al. BECN1 is involved in the initiation of mitophagy: it facilitates PARK2 translocation to mitochondria. Autophagy 2014;10:1105–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lemasters JJ. Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res 2005;8:3–5. [DOI] [PubMed] [Google Scholar]
- 16.Palikaras K, Lionaki E, Tavernarakis N. Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat Cell Biol 2018;20:1013–22. [DOI] [PubMed] [Google Scholar]
- 17.Matsuda N, Tanaka K. Uncovering the roles of PINK1 and parkin in mitophagy. Autophagy 2010;6:952–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Koyano F, Okatsu K, Kosako H, Tamura Y, Go E, Kimura M, et al. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 2014;510:162–6. [DOI] [PubMed] [Google Scholar]
- 19.Do DC, Mu J, Ke X, Sachdeva K, Qin Z, Wan M, et al. miR-511–3p protects against cockroach allergen-induced lung inflammation by antagonizing CCL2. JCI Insight 2019;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Qiu L, Zhang Y, Do DC, Ke X, Zhang S, Lambert K, et al. miR-155 modulates cockroach allergen- and oxidative stress-induced cyclooxygenase-2 in asthma. J Immunol 2018;201:916–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ke X, Do DC, Li C, Zhao Y, Kollarik M, Fu Q, et al. Ras homolog family member A/Rho-associated protein kinase 1 signaling modulates lineage commitment of mesenchymal stem cells in asthmatic patients through lymphoid enhancer-binding factor 1. J Allergy Clin Immunol 2019;143:1560–74.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jackson WT, Giddings TH Jr, Taylor MP, Mulinyawe S, Rabinovitch M, Kopito RR, et al. Subversion of cellular autophagosomal machinery by RNA viruses. PLoS Biol 2005;3:e156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vargas JNS, Wang C, Bunker E, Hao L, Maric D, Schiavo G, et al. Spatiotemporal control of ULK1 activation by NDP52 and TBK1 during selective autophagy. Mol Cell 2019;74:347–62.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu T, Zhou Y, Qiu L, Do DC, Zhao Y, Cui Z, et al. Aryl hydrocarbon receptor protects lungs from cockroach allergen-induced inflammation by modulating mesenchymal stem cells. J Immunol 2015;195:5539–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wu YT, Tan HL, Shui G, Bauvy C, Huang Q, Wenk MR, et al. Dual role of 3-methyladenine in modulation of autophagy via different temporal patterns of inhibition on class I and III phosphoinositide 3-kinase. J Biol Chem 2010;285: 10850–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dickinson JD, Sweeter JM, Warren KJ, Ahmad IM, De Deken X, Zimmerman MC, et al. Autophagy regulates DUOX1 localization and superoxide production in airway epithelial cells during chronic IL-13 stimulation. Redox Biol 2018;14: 272–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ghosh R, Pattison JS. Macroautophagy and chaperone-mediated autophagy in heart failure: the known and the unknown. Oxid Med Cell Longev 2018;2018: 8602041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Roy SJ, Koval OM, Sebag SC, Ait-Aissa K, Allen BG, Spitz DR, et al. Inhibition of CaMKII in mitochondria preserves endothelial barrier function after irradiation. Free Radic Biol Med 2020;146:287–98. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 29.Silveira JS, Antunes GL, Kaiber DB, da Costa MS, Ferreira FS, Marques EP, et al. Autophagy induces eosinophil extracellular traps formation and allergic airway inflammation in a murine asthma model. J Cell Physiol 2019;235:267–80. [DOI] [PubMed] [Google Scholar]
- 30.Cho IH, Choi YJ, Gong JH, Shin D, Kang MK, Kang YH. Astragalin inhibits autophagy-associated airway epithelial fibrosis. Respir Res 2015;16:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Abdel Fattah E, Bhattacharya A, Herron A, Safdar Z, Eissa NT. Critical role for IL-18 in spontaneous lung inflammation caused by autophagy deficiency. J Immunol 2015;194:5407–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Suzuki Y, Maazi H, Sankaranarayanan I, Lam J, Khoo B, Soroosh P, et al. Lack of autophagy induces steroid-resistant airway inflammation. J Allergy Clin Immunol 2016;137:1382–9.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Inoue D, Kubo H, Taguchi K, Suzuki T, Komatsu M, Motohashi H, et al. Inducible disruption of autophagy in the lung causes airway hyper-responsiveness. Biochem Biophys Res Commun 2011;405:13–8. [DOI] [PubMed] [Google Scholar]
- 34.Pu Q, Gan C, Li R, Li Y, Tan S, Li X, et al. Atg7 deficiency intensifies inflammasome activation and pyroptosis in Pseudomonas sepsis. J Immunol 2017;198:3205–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Racanelli AC, Kikkers SA, Choi AMK, Cloonan SM. Autophagy and inflammation in chronic respiratory disease. Autophagy 2018;14:221–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee IT, Yang CM. Role of NADPH oxidase/ROS in pro-inflammatory mediators-induced airway and pulmonary diseases. Biochem Pharmacol 2012;84:581–90. [DOI] [PubMed] [Google Scholar]
- 37.Poon A, Eidelman D, Laprise C, Hamid Q. ATG5, autophagy and lung function in asthma. Autophagy 2012;8:694–5. [DOI] [PubMed] [Google Scholar]
- 38.Rogers LK, Cismowski MJ. Oxidative stress in the lung—the essential paradox. Curr Opin Toxicol 2018;7:37–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang E, Liu X, Tu W, Do DC, Yu H, Yang L, et al. Benzo(a)pyrene facilitates dermatophagoides group 1 (Der f 1)-induced epithelial cytokine release through aryl hydrocarbon receptor in asthma. Allergy 2019;74:1675–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Uchida M, Anderson EL, Squillace DL, Patil N, Maniak PJ, Iijima K, et al. Oxidative stress serves as a key checkpoint for IL-33 release by airway epithelium. Allergy 2017;72:1521–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wible DJ, Bratton SB. Reciprocity in ROS and autophagic signaling. Curr Opin Toxicol 2018;7:28–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Luczak ED, Anderson ME. CaMKII oxidative activation and the pathogenesis of cardiac disease. J Mol Cell Cardiol 2014;73:112–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 2011;13: 132–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim J, Kim YC, Fang C, Russell RC, Kim JH, Fan W, et al. Differential regulation of distinct Vps34 complexes by AMPK in nutrient stress and autophagy. Cell 2013; 152:290–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jang JE, Eom JI, Jeung HK, Cheong JW, Lee JY, Kim JS, et al. AMPK-ULK1-mediated autophagy confers resistance to BET inhibitor JQ1 in acute myeloid leukemia stem cells. Clin Cancer Res 2017;23:2781–94. [DOI] [PubMed] [Google Scholar]
- 46.Glick D, Barth S, Macleod KF. Autophagy: cellular and molecular mechanisms. J Pathol 2010;221:3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhao J, Brault JJ, Schild A, Cao P, Sandri M, Schiaffino S, et al. FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell Metab 2007;6:472–83. [DOI] [PubMed] [Google Scholar]
- 48.Napolitano G, Ballabio A. TFEB at a glance. J Cell Sci 2016;129:2475–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Baek SH, Kim KI. Epigenetic control of autophagy: nuclear events gain more attention. Mol Cell 2017;65:781–5. [DOI] [PubMed] [Google Scholar]
- 50.Gkikas I, Palikaras K, Tavernarakis N. The role of mitophagy in innate immunity. Front Immunol 2018;9:1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bueno M, Lai YC, Romero Y, Brands J, St Croix CM, Kamga C, et al. PINK1 deficiency impairs mitochondrial homeostasis and promotes lung fibrosis. J Clin Invest 2015;125:521–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tsubouchi K, Araya J, Kuwano K. PINK1-PARK2-mediated mitophagy in COPD and IPF pathogeneses. Inflamm Regen 2018;38:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, et al. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015; 524:309–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Saito T, Sadoshima J. Molecular mechanisms of mitochondrial autophagy/mitophagy in the heart. Circ Res 2015;116:1477–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
REFERENCES
- E1.Qu J, Do DC, Zhou Y, Luczak E, Mitzner W, Anderson ME, et al. Oxidized CaMKII promotes asthma through the activation of mast cells. JCI Insight 2017;2: e90139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- E2.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 2001;25: 402–8. [DOI] [PubMed] [Google Scholar]