

Abstract
Over the past few years, there has been remarkable development in the area of optic neuritis. The discovery of new antibodies has improved our understanding of the pathology of the disease. Antiaquaporin4 antibodies and antimyelin oligodendrocytes antibodies are now considered as distinct entities of optic neuritis with their specific clinical presentation, neuroimaging characteristics, treatment options, and course of the disease. Similarly, there has been a substantial change in the treatment of optic neuritis which was earlier limited to steroids and interferons. The development of new immunosuppressant drugs and monoclonal antibodies has reduced the relapses and improved the prognosis of optic neuritis as well as an associated systemic disease. This review article tends to provide an update on the approach and management of optic neuritis.
Keywords: Myelin oligodendrocytes, multiple sclerosis, neuromyelitis optica, optic neuritis
The landscape of optic neuritis (ON) is rapidly changing with the discovery of new antibodies, advent of latest investigations, and revised diagnostic criteria. In view of these developments, it is vital to revise our approach to the management of ON.[1]
We searched PubMed and Medline for studies published during the last 10 years with the general term “optic neuritis” and specific terms like “typical optic neuritis,” “atypical optic neuritis,” “multiple sclerosis,” “neuromyelitis optic neuritis,” and “myelin oligodendrocyte glycoprotein antibody.” References from identified studies were also reviewed and included if deemed appropriate and important.
Epidemiology
Demyelinating diseases were once considered rare in India with previous Indian studies showing that MS constitutes 0.32% to 1.58% of neurology admissions in hospitals, and a prevalence of approximately 1.33/100,000.[2,3,4,5,6] However, a more recent population-based survey conducted in urban Mangalore has shown a prevalence of 8.3/100,000 for MS and 2.6/100,000 for neuromyelitis optic spectrum diseases (NMOSD).[7] NMOSD is likely to constitute approximately 20% of all demyelinating disorders in India.[8]
This change in epidemiology could be because of greater awareness about the disease, better documentation, and also due to the development of low threshold for MRI among neurologists and ophthalmologists.
ON in India has been reported to be different from the West in terms of clinical features and prognosis. A substantial number of cases from this part of the world have been reported to have atypical presentations and relatively poorer prognosis.[9,10] Recently published data from India suggests that 50% of NMOSD either have aquaporin4 (AQP4) or myelin oligodendrocytes (MOG) antibodies.[11] Similarly, the seropositivity of Anti-MOG+ cases was 20% for AQP4 seronegative cases in the study. The common presentation of anti-AQP4+ was relapsing myelitis and ON with poor outcomes. The clinical picture of MOG in terms of recurrent ON and myelitis with a good response to steroid has been reported to be similar to that of western literature.
Clinical Pattern
Presentation of ON is broadly grouped into typical ON and atypical ON. A typical ON follows a specific clinical pattern and is usually related to a demyelinating lesion which can be isolated or associated with multiple sclerosis (MS). A clinical pattern of ON is considered as atypical ON if it differs from this presentation and has a long list of differentials and includes NMOSD, infectious causes, and autoimmune causes [Table 1].
Table 1.
Etiology of optic neuritis
| Inflammatory | CNS inflammatory disorders | Multiple sclerosis, neuromyelitis optica, spectrum disorders, myelin oligodendrocyte, glycoprotein antibody (MOG) associated disease, autoimmune glial fibrillary acidic protein (GFAP), astrocytopathy |
| Systemic inflammatory disorder | Granulomatous diseases: sarcoidosis and granulomatous polyangiitis Autoimmune diseases: SLE and Sjogren |
|
| Paraneoplastic diseases | CRMP-IgG mediated | |
| Other seronegative | Autoimmune optic neuropathy (AON), chronic relapsing inflammatory optic neuropathy (CRION), relapsing isolated optic neuritis (RION) | |
| Infectious | Viral: VZV, HIV, WNV Bacterial: TB, syphilis, Borrelia, Bartonella, and Toxoplasma |
Each of the etiological causes like MS, NMO, and MOG associated ON may have its own clinical features, prognosis, and recurrence rate. It is imperative to differentiate each etiology from the other because early treatment is crucial for visual function recovery and prevention of relapses.
Typical or demyelinating optic neuritis
Optic neuritis treatment trial (ONTT) is the largest data source that has shaped our understanding of ON. It included 454 patients followed up over a period of 15 years. According to the ONTT, typical ON presents as unilateral, sub-acute, painful loss of vision, seen in young females. The median age of onset is 20–30 years with a female: male ratio of 3:1. Pain in typical ON is classically seen on eye movements and was reported in 90% of cases.[12] The presenting visual acuity in MS-ON was better than 20/200 in around two-thirds of patients with the visual loss progressing over two weeks before becoming stable. Retrobulbar neuritis was seen in two-thirds of patients. ONTT also showed that the visual outcome in MS-ON is same irrespective of the treatment.[13] The visual prognosis in MS-ON is excellent and around 72% of patients maintained visual acuity of 20/20 in both the eyes at 15 years of follow up.[14]
MS is a demyelinating disease of the central nervous system wherein the patient may have relapses or progression of demyelinating episodes depending on the clinical subtype. Around 20% of MS patients have ON as their first episode of demyelination and 50% of MS patients will develop ON during the course of the disease.[15,16] The diagnosis of MS requires dissemination of attacks in space and time either clinically or radiologically and there should be an interval of 30 days between two episodes to consider them as two separate events. A clinical attack is defined as a neurological symptom that is a manifestation of MS and lasts for at least 24 hours.
In 2017, MacDonald criteria for diagnosis of MS were revised.[17] The revision now includes symptomatic and asymptomatic demyelinating lesions. It also includes lesions in the brain stem and spine to consider for dissemination in space. It must be noted that MRI lesions in the optic nerve in a person presenting with ON have not been included in the criteria for diagnosis. Table 2 shows the revised Macdonald criteria adapted from Thompson et al.[17]
Table 2.
Revised 2017 MacDonald criteria for diagnosis of MS
| Clinical Presentation | Additional Data Needed for MS Diagnosis |
|---|---|
| Two or more clinical attacks involving 2 or more clinical locations | None |
| Two or more clinical at same location | Clear history of an attack at additional location |
| Or | |
| additional clinical attack at a different location | |
| Or | |
| involvement of different anatomical sites on MRI | |
| One clinical attack involving 2 different locations | Additional clinical attack disseminated in time |
| Or | |
| Appearance of lesion on MRI on follow up | |
| Or | |
| Presence of oligoclonal band on CSF analysis | |
| One clinical attack at single location | Dissemination in time by |
| Second clinical attack at same location | |
| Or appearance of lesion on follow-up MRI | |
| Or the presence of oligoclonal band on CSF analysis | |
| AND | |
| Dissemination in space by | |
| Clinical attack at a different location | |
| Involvement of different locations on MRI |
Atypical optic neuritis
Clinical features that suggest atypical forms of ON are as follows:
No light perception vision
Optic disc or retinal hemorrhages
Severe optic disc swelling
Macular exudates
Absence of pain
Presence of uveitis
Bilateral visual loss and
Recurrent disease.
Alternative causes of optic neuropathy should be considered in these patients [Table 1].
Neuromyelitis optica
Once considered as a variant of MS, NMOSD is now designated as a separate entity of demyelination, which has a specific clinical pattern. It presents as bilateral or unilateral ON along with simultaneous or sequential transverse myelitis. It is an astrocytopathy characterized by antibodies against aquaporin-4 (AQP4) water channel. AQP4 is a water channel located at the foot processes of astrocytes that helps facilitate water movement across the cell membrane. It is extensively expressed in the brain, spinal cord, and optic nerves and explains the exclusive clinical manifestation of the disease.[18] The antibodies against AQP4 activate the complement system that leads to the damage of astrocytes. Demyelination occurs as a secondary process to the astrocyte injury.[19,20]
Unlike MS, NMOSD is more common in Asian and African population, with high mortality rates reported in the latter.[21,22] It is more common in females as compared to MS, with female: male ratio varying from 3:1–9:1.[23,24] The average age of onset is around 30–40 years which is marginally higher when compared to MS.[25] The classic presentation includes ON, longitudinal extensive transverse myelitis (LETM), and area postrema syndrome (intractable hiccups and vomiting). Longitudinal extensive transverse myelitis (LETM) is a more specific presentation of NMOSD. The concurrent presentation of both ON and LETM is seen in 15%–40% of cases.[26] Visual loss is profound and rapid with two-third of the patients presenting with <20/200 vision.[27] Similarly, the visual prognosis is also poor with 50–60% of patients regaining visual acuity of <20/200. Frequent relapses of both ON and LETM are seen in NMOSD and more common in seropositive patients.[28] The median recurrence rate was 4/patient in the polyphasic group during the follow-up period of 5 years, in an Indian study.[29] Numerous other autoimmune diseases like myasthenia gravis, systemic lupus erythematosus, and Sjogren’s syndrome have also been reported in association with AQP4-ON.[30,31,32] Thus, the manifestation of ON or LETM or any other NMOSD symptom in patients of rheumatological disorders should arouse a suspicion of NMOSD and should be tested early for AQP4-IgG and have an MRI of their brain and spinal cord.
The clinical diagnosis of NMOSD is currently based on an international consensus diagnostic criteria published by Wingerchuk et al. 2015.[33] If AQP4 IgG is positive, a patient is considered to have NMO if at least one core clinical criteria is met (ON, longitudinal transverse acute myelitis, area postrema syndrome, acute brainstem syndrome, narcolepsy or diencephalic syndrome, or cerebral syndrome) with the exclusion of alternative diagnoses to explain presentation. If the patient is AQP4 IgG negative, a diagnosis of NMO may be made if two of these core clinical criteria (one of which must be ON, LETM, or area postrema syndrome) with imaging appearance clinically consistent with NMO and without another alternative diagnosis to better explain presentation. These guidelines recommend that the term NMO be changed to NMOSD to be inclusive of patients with NMO features that may not be AQP4 IgG positive or may have presentations outside of the common ON and LETM presentations.
MOG optic neuritis
Although anti-MOG-ON is considered a part of NMOSD, it has its own characteristic clinical and radiological features. MOG antibodies are directed against the glycoprotein located on oligodendrocyte and myelin sheath. Around 20% of NMOSD patients who were seronegative for AQP4-IgG have been found to be positive for MOG antibody.[34] Patients with MOG-IgG are affected in the age range 31–37 years, which is in between AQP4-ON and MS-ON.[35] Interestingly unlike other autoimmune disorders, the F: M ratio is closer to 1:1 in MOG-ON. ON and TM are a common presentation of MOG-IgG disease with 80% of patients presenting with isolated ON.[36] Other neurological manifestations include LETM and acute disseminated encephalomyelitis (ADEM). Visual loss in MOG-ON is often bilateral, severe, and painful. Nearly half of the patients present with disc edema as compared to AQP4-ON and MS-ON. The degree of disc edema in NMOSD, is generally more severe than in cases of MS-ON. Visual prognosis is excellent and many patients gain visual acuity of 20/20. The recurrence of ON in MOG positive cases has been reported to be 50%–80% in various studies.[34,35,36,37] Table 3 shows the differentiating clinical and radiological features of three types of demyelinating diseases.
Table 3.
Differentiating features of the three demyelinating disorders
| Characteristics | MS-ON (Typical) | AQP4-ON | MOG-ON |
|---|---|---|---|
| Gender | Female predominance (60%-75%) | Female predominance (90%) | Equal distribution |
| Laterality | Unilateral | Unilateral or bilateral | Bilateral |
| Visual loss | Mild to moderate | Severe | Moderate to severe |
| Fundus presentation | RBN | RBN > papillitis | Papillitis |
| Myelitis/encephalitis | Rare | LETM is common | ADEM is common |
| Associated diseases | None | Commonly associated with Sjogren’s syndrome, SLE and sarcoidosis | None |
| Visual outcome | Good | Poor | Good |
| Relapse | Common, but less than NMO | Common | Very common |
| MRI of Optic nerve | Retrobulbar, focal lesion involving <50% of length | Retrobulbar and intracranial with chiasmal and optic tract involvement. Longitudinal lesion >50% length is involved | Retrobulbar, longitudinal lesion involving >50% length. Perineural sheath and perineural orbital tissue enhancement |
| MRI Brain | |||
| Area involved | Periventricular, Infratentorial, juxtacortical and subcortical lesions | hypothalamus, thalamus, pituitary, area postrema, brain stem | Supratentorial and cerebellum |
| Type of lesion | Ovid, discrete, arranged perpendicular to ventricles | Confluent, longitudinal and parallel to ventricle | Confluent, longitudinal |
| MRI spine | Short, peripheral and involving <3 vertebrae | Longitudinal, central and involve >3 continuous vertebrae Involves cervical and thoracic spine |
Longitudinal, central, and involve >3 continuous vertebrae Lower spine involving cauda equina |
| CSF | Oligoclonal bands are seen | Pleocytosis with PMN and eosinophils | Pleocytosis may be seen Oligoclonal bands are not seen |
Glial Fibrillary Acidic Protein IgG Astrocytopathy
Patients with GFAP-IgG disease present primarily with neurological manifestations of meningitis or meningoencephalitis. Isolated myelitis is a rare presentation seen in less than 5% of patients.[38,39] The median age of presentation is 44–50 years with no gender predominance. The optic neuropathy presents with bilateral disc edema and mild visual symptoms akin to papilledema.[38,39] Visual acuity is usually normal and mild arcuate defects are seen on visual field examination. They may have mild vitritis with disc edema. Approximately 20% of patients show co-existence of other autoimmune diseases and neoplasm, particularly ovarian teratoma.[38,39] Coexistence of other antibodies like AQP4 IgG is also reported.
Chronic Inflammatory Optic Neuritis
CRION is an inflammatory ON characterized by excellent response to steroids and recurrence on withdrawal or tapering of steroids. Thus, it is also known as steroid-dependent ON. It may involve any age group and has a female preponderance.[40] The disease is often bilateral with simultaneous or sequential involvement of optic nerve. The visual loss is severe and painful. CRION is a diagnosis of exclusion and all other causes of ON; demyelination, autoimmune disease, and infection should be ruled out before making the diagnosis. The diagnostic criteria for CRION include recurrent ON with seronegative for AQP-4 and MOG, response to steroids, and relapse on withdrawal of steroids.[40] Various studies have found out similarities between the clinical features of CRION and MOG-ON and there is a possibility that ON previously labeled as CRION could have been MOG-ON. A few studies have reported MOG IgG seropositivity in CRION phenotype of ON.[41,42]
Optic Neuritis Associated with Autoimmune Diseases
Various systemic autoimmune diseases including Sjogren’s syndrome, sarcoidosis, systemic lupus erythematosus, and antiphospholipid syndrome can manifest as ON during the course of the disease.[43,44,45,46] Optic nerve involvement in these cases could be immune-mediated or due to vasculitis leading to ischemia of the optic nerve. It should be remembered that many of these patients may harbor AQP4 antibodies and ON in these cases may be a manifestation of NMOSD rather than the autoimmune disease itself. Systemic manifestations may help in identifying the etiology. Serological work-up for autoantibodies like ANA, DSDNA, and Rho antibodies should be done to confirm the diagnosis. Patients of systemic autoimmune disease who present with NMOSD kind symptoms should be evaluated for AQP4 antibodies.
Infectious Optic Neuropathy
Multiple viral and bacterial diseases can present as ON. Common infections include syphilis, tuberculosis, Lyme disease, Bartonella, herpetic disease, West Nile virus, and HIV.
One has to be proactive in taking history in such cases. History of preceding fever, exposure to infective organism, and travel to endemic areas should be asked directly. The other features of infective etiology include meningitis, encephalitis, meningoencephalitis, retinitis, vitritis, and infective uveitis. These may help in forming a diagnosis. Complete ophthalmological and systemic examination along with a high index of suspicion is required to diagnose these cases. These patients require extensive laboratory workup and CSF analysis to confirm the diagnosis.
Investigations
Detailed history taking and thorough clinical workup are essential to differentiate ON from other causes of optic neuropathy. Neuroimaging and laboratory workup is a mandatory part of workup to diagnose various etiological causes [Fig. 1].
Figure 1.
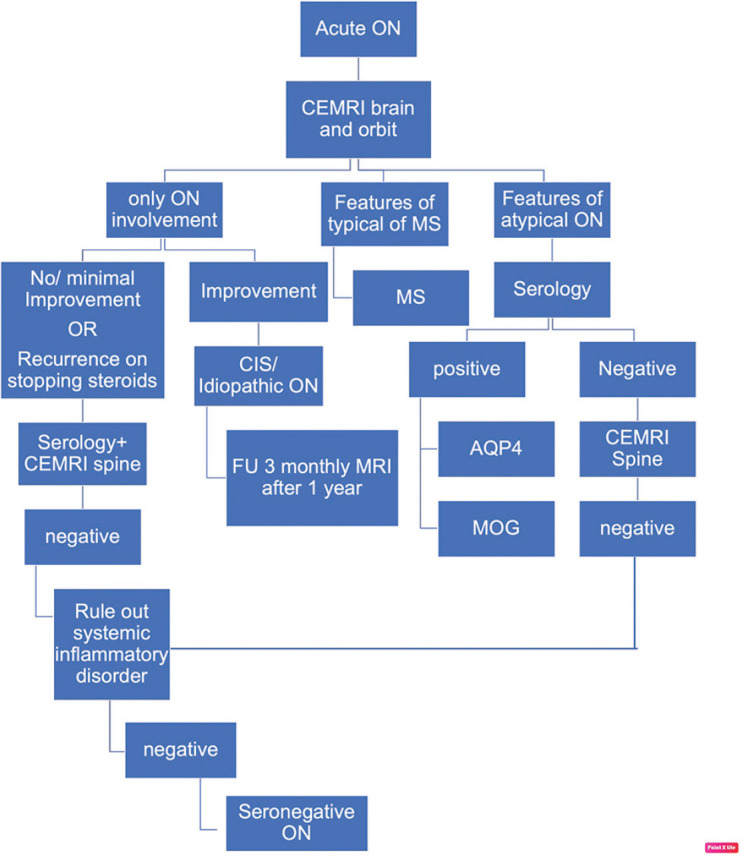
Approach to the diagnosis of acute ON
Neuroimaging
Contrast enhancing magnetic resonance imaging (CEMRI) of the brain, orbits, and spine are indicated for diagnosing the various causes of ON [Table 3], to fulfill the diagnostic criteria for MS and NMO (see above), and prognosticate disease outcomes. It is important to obtain fat-desaturated high-resolution sequences. Fluid attenuated inversion recovery (FLAIR) sequences are particularly helpful for demyelinating lesions.
Multiple sclerosis
Optic nerve inflammation is seen as a hyperintense lesion on T2 which enhances with contrast. The involvement of optic nerve in MS is focal and usually limited to the retrobulbar area; focal involvement of intracanalicular and intracranial optic nerve can also be seen [Fig. 2]. The ONTT showed that the presence of demyelinating lesions on baseline brain MRI can predict conversion to clinically defined MS (CDMS) after initial attack of ON. For patients with acute typical ON, the risk of developing clinically definite multiple sclerosis (CDMS) at 15 years, increases from 25% for those who do not have any demyelinating lesions on the baseline MRI to 72% with at least 1 demyelinating lesion on baseline brain MRI.[16] The demyelinating lesions seen on MRI brain are characteristically T2 hyperintense, typically discrete, ovoid, at least 3-mm long, classically located in the periventricular, infratentorial area but also in the juxtracortical and subcortical areas [Fig. 3]. Acute lesions show enhancement with contrast while older lesions are non-enhancing. The spinal cord lesions in MS tend to be peripheral and shorter, i.e., involving <3 vertebrae.
Figure 2.
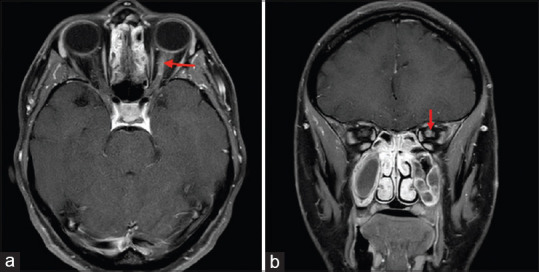
Axial (a) and Coronal (b) T1 weighted contrast and fat saturated MRI sections demonstrate enhancement of the retrobulbar segment of the left optic nerve in a patient with multiple sclerosis
Figure 3.
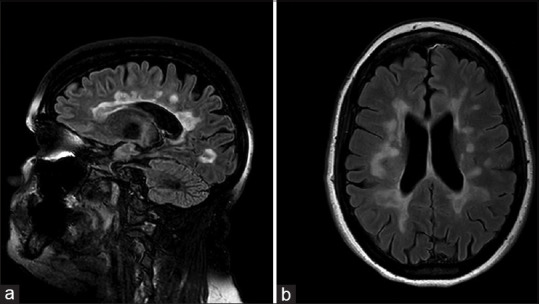
Sagittal (a) and axial (b) T2/FLAIR brain MRI sequences demonstrate focal and confluent foci consistent with demyelinating disease involving corpus callosum and hemispheric white matter in a periventricular location in a patient with multiple sclerosis
Other demyelinating diseases
The neuroimaging in NMOSD has certain characteristic lesions of optic nerve, spine, and brain.[33,47] MRI of the optic nerve in AQP4-ON and MOG- ON shows longitudinal involvement of around 50% of the optic nerve. In AQP4-ON the intracranial part of the optic nerve is more commonly involved and lesion can extend through the optic chiasma and optic tract [Fig. 4]. In contrast, MOG-ON shows the involvement of the retrobulbar part of optic nerve rarely extending in the chiasma. Ramanathan et al.[48] in their study have found out that 64% of AQP4-positive patients had chiasmal involvement compared to only 5% in anti-MOG disease and 15% in MS. The characteristic feature of MOG-ON is perineural enhancement on MRI orbits, with or without fat stranding [Fig. 5], which may be seen in 50% of cases.[35,36] In patients with NMOSD, the MRI spine shows LETM which extends longitudinally for >3 contagious vertebral segments. Lesions are seen in intramedullary area in AQP4 as compared to MS where the lesions are shorter and seen in the periphery. Spinal cord lesions of MOG IgG disease are similar to NMOSD but tend to involve the cauda equina area.
Figure 4.
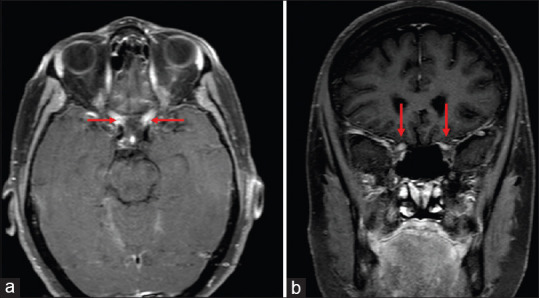
Axial (a) and coronal (b) T1 weighted contrast and fat saturated MRI sections demonstrate edema and enhancement of the both optic nerves from the apex to the optic chiasm in patient with AQP-4 IgG positive NMO disease
Figure 5.
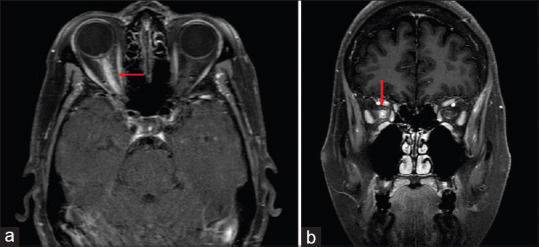
Axial (a) and Coronal (b) T1 weighted contrast and fat saturated MRI sections demonstrate edema and enhancement of the entire prechiasmatic segment of the right optic nerve. There is also enhancement of the optic nerve sheath and adjacent orbital soft tissue in a patient with MOG IgG optic neuropathy
Brain lesions are seen in around 60% of cases of NMOSD and have led to the revision of diagnostic criteria which now includes brainstem lesions as well. The brain lesions in NMOSD are commonly seen around hypothalamus, thalamus, pituitary, and area postrema, which correlates with the increased density of astrocytes in these areas.[49] The brainstem lesions can be seen continuous with the spinal cord lesion. Periventricular lesions are not rare and need to be differentiated from MS lesions. The lesions are confluent, longitudinal and lie parallel to the ventricles. Solitary large tumefactive callosal lesions, “cloud-like” enhancement, radial hemispheric lesions, and posterior reversible encephalopathy syndrome (PRES)-like lesions have also been reported.[44] Brain lesions of MOG-IgG have similar characteristics as those of NMOSD with the commonly involved areas located in supratentorial area and cerebellum.[34]
A distinct pattern of demyelination is seen with GFAP IgG, where lesions are linear with perivascular radial enhancement, extending outward from the ventricles.[38]
In other forms of ON including CRION, brain and spinal MRI are normal. Optic nerve involvement is seen as an enhancement of optic nerve on contrast MRI.
Serology
Serological examination for various antibodies is needed for identifying the variants of NMOSD. Serum testing is generally more sensitive than CSF for APQ4 and MOG antibodies. The sensitivity and specificity of detection of AQP4 and MOG antibodies depend upon the type of assay used for the detection. The assay based on cell line of HEK293 is the most sensitive (77%) while cell-binding assays with fixed cells have a sensitivity of 73%. An ELISA assay has a sensitivity of 60% and GFP-AQP4 fluorescence immune-precipitation assay is sensitive at 53%.[50] Particularly in cases of AQP4-ON, coexisting autoantibodies like antinuclear antibody, anti-double-stranded DNA, anti-Ro, and anti-La may be seen in a substantial number of cases. Serological test to rule out infective causes should be done according to the suspected etiology.
Cerebrospinal fluid analysis
CSF analysis may reveal the presence of oligoclonal bands which are seen in 90% of MS patients and as compared to 20% of patient with NMOSD. CSF analysis in AQP4-ON remarkably shows pleocytosis and increased protein levels. Pleocytosis >50 leukocytes/mL is seen approximately 35% of time in AQP4-ON.[51] CSF analysis is important to rule out infections and shows pleocytosis, high protein, and low glucose level. It is important to do a microbiological examination to rule out infection in general and tuberculosis in particular, considering the fact that infectious causes are more common in our clinical scenario. The presence of GFAP IgG in the CSF of patients with typical clinical presentation and MRI findings will confirm the diagnosis of GFAP astrocytopathy.[38]
Optical coherence tomography
Ophthalmic imaging is not sensitive in differentiating the various causes of ON, but can be used to assess the extent of neuronal damage to the anterior visual pathways.[52,53,54,55,56,57] During the acute stages of ON, the retinal nerve fiber layer (RNFL) may be increased. This is followed by a steady decrease over time accompanied by thinning of the ganglion cell layer. Various studies have shown that ganglion cell layer-inner plexiform layer (GCL-IPL) thinning may be seen earlier than the RNFL thinning and may give an early indication of optic nerve damage.[55,57] Both RNFL and GCL- IPL have been shown to correlate with the visual functions in the affected eye.[52,53,54,55,56,57] These layers can also get affected in the other eye of unilateral cases suggesting a subclinical involvement.[53,56] ON associated with NMOSD has been reported to have more severe RNFL and GCL loss as compared to MS associated ON and MOG disease.[58] The presence of microcystic macular edema in the inner nuclear layer of retina may be associated with poor vision. It is seen more commonly with NMOSD as compared to MS.[58]
Treatment of ON
The treatment of ON can be divided into the management of the acute episode, long-term management for the prevention of relapse, and strategies for neuro-regeneration. An approach to acute and long-term management of ON is provided in Fig. 6.
Figure 6.
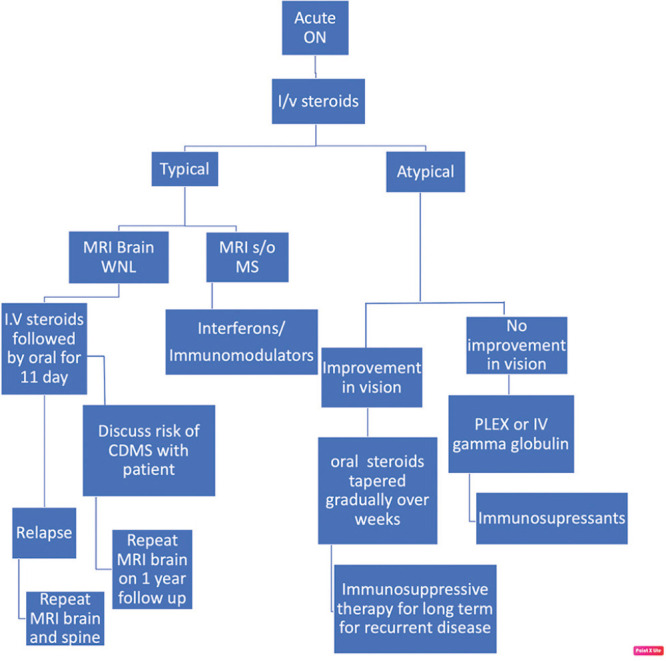
Protocol for management of ON
Treatment of acute episode
Steroids
High-dose intravenous steroids are the treatment of choice for acute ON. The most definitive evidence comes from the ONTT which compared three treatment modalities: intravenous steroids vs oral steroids vs placebo. The results favored intravenous steroids in terms of faster recovery at one month and improved visual functions at 6 months. However, the final visual recovery at 1-year follow-up was similar irrespective of treatment.[59] Intravenous methylprednisolone also delayed the onset of clinically definite multiple sclerosis (CDMS) at 2 years, but no difference was noted thereafter.[13] The recurrence rate at 2-year follow-up was double in the oral steroid group (30%) as compared to the intravenous steroid group (13%) and the placebo group (16%).[13] Thus for a typical ON, intravenous steroids may be administered for faster visual recovery. Oral steroids alone in conventional doses are contraindicated.
A single-blind randomized clinical trial evaluated the efficacy of high dose of oral steroids which is bioequivalent to intravenous steroid. Fifty-five patients received 1250 mg of oral prednisolone which was equally effective to 1000 mg of intravenous methylprednisolone. Improvement in vision was seen at 1 month as well as 6 months.[60] A Cochrane review also compared the efficacy of the two forms of steroid administration and found them to be equally effective.[61]
Studies have also shown that intravenous dexamethasone in a dose of 200 mg/day was equally efficacious as intravenous methylprednisolone 1 g/day, and has the advantage of low cost and less side effects.[62]
A recent Cochrane review evaluating the effects of corticosteroids in typical ON stated that there is no conclusive evidence that the use of either intravenous or oral steroids has benefits in terms of visual recovery, visual field, or contrast sensitivity in MS-ON.[63]
Patients with MOG-ON and CRION show significant visual recovery in response to intravenous steroid treatment (intravenous methylprednisolone 1 g/day for 3–5 days). In these disease conditions, patients are often placed a prolonged, slow oral steroid taper over 6–8 weeks.[36] CRION shows dramatic response with steroid treatment with prompt relief of pain and restoration of normal visual functions.[40] However on steroid withdrawal, the episode of ON tends to relapse often necessitating long-term immunosuppression.
The treatment effect of intravenous steroids is limited for patients with NMOSD and repeated steroid treatment in these patients has not been shown to improve the rate of disease remission.[64] When patients with acute ON are suspected to have NMOSD based on clinical presentation, radiologic features, positive AQP-4 IgG, or poor response to intravenous steroids, immunotherapy (plasma exchange or intravenous immunoglobulin) should be initiated at the earliest.
Acute episodes of ON in an autoimmune disorder like sarcoidosis and sclerosing lupus erythematosus (SLE) are also treated with intravenous steroids. Many of them would require concurrent immunotherapy for more effective results.
Plasmapheresis
Plasmapheresis is indicated for patients with acute ON who fail to respond to standard intravenous steroid treatment, experience recurrent disease, or those with either proven or suspected NMOSD. It is shown to be effective when given as early as possible from the onset of the symptoms.[65,66] The steroid therapy has been shown to be associated with improved Extended Disability Status Scale (EDSS) when given in combination with plasmapheresis in cases of NMOSD.[67]
Intravenous gamma globulin
Evidence is limited for the use of intravenous immunoglobulins (IVIG) in the management of acute ON. IVIG is reserved for the treatment of patients with steroid-refractory MS-ON and MOG-ON.[68,69,70]
Long-term management
Multiple sclerosis
The treatment of MS includes disease-modifying drugs (DMD) such as interferon, immunomodulators like glatiramer acetate (GA) and fingolimod, and monoclonal antibodies like natalizumab.
Various clinical trials like CHAMPS, PRISMS for interferon b-1a, and BENEFIT for interferon b-1b have explored the efficacy of interferons in various doses in delaying the onset of CDMS.[71,72,73,74,75] The results of all these trials showed a decrease in relapse rate and delay in conversion to CDMS for at least 5 years. Interferon beta-1a (Rebif) is available as a subcutaneous injection in 22 mg and 44 mg formulations given 3 times per week. The common side effects include generalized weakness, depression, flu-like symptoms, hypertonia, increased liver enzymes, injection site reactions, leukopenia, and myasthenia.
Glatiramer acetate also known as Copaxone is an immunomodulator used for preventing relapses in RRMS. The drug is given as 40-mg/mL SC injection 3-times-per-week dosage regimen and has shown to reduced mean annualized relapse rates by 34% compared with placebo at 12 months.[76,77]
Dimethyl fumarate (DMF) is an oral immunomodulator indicated for relapsing forms of MS and is shown to have decreased rate of relapses and improve disability.[78,79]
Fingolimod is an immunomodulator that targets the sphingosine-1-phosphate receptor and is specifically used for relapsing-remitting multiple sclerosis (RRMS). It has shown to be highly effective in reducing the relapse rate in RRMS.[80,81] The recommended dosage for fingolimod is 0.5 mg once a day orally. The major side effect is bradycardia; others include macular edema, pulmonary dysfunction, increased risk of infection, and melanoma. Siponimod is a selective sphingosine 1-phosphate (S1P)-1 and -5 receptor modulator that has been recently approved by FDA for SPMS with similar side effects as that of fingolimod.[82] Cladribine, an anti-cancer drug, has also shown efficacy in the treatment of RRMS.[83]
Monoclonal antibodies are the most recent treatment options and are gaining favor for long-term management. They target the CD cells and prevent an antibody-dependent inflammatory reaction. Natalizumab is a monoclonal antibody that binds to the a4 subunit of a4b1 and a4b7 integrins. It is approved for use in relapsing MS.[84,85] Alemtuzumab is also shown to be effective for RRMS while ocrelizumab has been suggested for primary progressive MS as well.[86]
Progressive multifocal leukoencephalopathy (PML) is an important side effect of the drugs used for the treatment of MS, which includes fingolimod, natalizumab, and other monoclonal antibodies and MS patients have become a susceptible population at possible risk of development of PML.[87]
Many of these above-mentioned drugs may not be available in India; and in a resource-limited setting like India, the treatment for a case of MS has to be tailored on an individual basis according to the availability and cost-benefit ratio.
Neuromyelitis optic spectrum disorder
Neuromyelitis optic spectrum disorder (NMOSD) is a debilitating disease with severe relapses and permanent disability. Therefore, it is important to plan long-term immunosuppression to maintain remission and prevent relapses. Various immunosuppressants like azathioprine, methotrexate, and mycophenolate are the commonly used agents. It should also be noted that NMOSD related ON may actually worsen with interferons and differentiation should be made before initiation of the treatment.
Azathioprine is considered as first-line therapy to replace corticosteroids for long-term immunosuppression. It has been found to decrease the relapse rate of NMOSD by 75% and improve the visual acuity and disability scores.[88,89] It is shown to decrease the annual relapse rate when given in a dose of 2.5–3.0 mg/kg per day. As the drug takes time to take effect, it should preferably be started with steroids followed by gradual tapering of steroids. Side-effects include bone-marrow suppression, hypersensitivity reactions, gastrointestinal reactions (nausea and vomiting), liver dysfunction, increased infection risk, and, rarely, pancreatitis. Regular monitoring of liver functions and blood counts are essential.
Mycophenolate mofetil is an effective alternative for the patients who are intolerant to azathioprine. There are few studies showing its beneficial role in reducing the relapse rate in NMOSD.[90] It is started in a dose of 500 mg/day, increased in 500 mg steps weekly till a maintenance dose of 2000 mg/day in divided doses. Side effects are hypersensitivity reactions, bone-marrow suppression, gastro-intestinal reactions, liver dysfunction, renal dysfunction, potential risk of lymphoma, and skin malignancy. Monitoring of blood count, kidney and liver function should be done regularly.
Methotrexate is a well-tolerated drug and is used as an alternative to azathioprine in NMOSD cases.[91] It is started with a dose of 7.5 mg weekly with folate supplementation and gradually increased in 2.5 mg steps every week to reach the weekly maintenance dose of 15 mg. The main side effects are bone-marrow suppression, liver toxicity, pulmonary fibrosis, gastrointestinal symptoms, hypersensitivity reactions, and increased infection risk.
Rituximab is one of the most widely used monoclonal antibodies against CD20 that have shown to reduce relapse rate and improve or stabilize the disability in NMOSD.[92,93]
Rituximab is administered as an intravenous infusion in doses of 375 mg/m2/week for 4 weeks or two doses of 1 g each infused 2 weeks apart, and needs to be repeated after 6 months. The main side effects include infusion-related reactions, persistent leukopenia, severe infections, and death.[94]
Eculizumab is the first monoclonal antibody that has been approved by FDA for use in NMOSD.[95]
Myelin oligodendrocyte associated ON
Myelin oligodendrocyte associated ON (MOG-ON) has a good visual prognosis but has more frequent relapses as compared to MS and NMO. The maintenance therapy includes oral steroids slowly tapered over few weeks followed by azathioprine 50–100 mg/day of steroids.[96] It has been reported in a cases series that immunosuppression for at least 3 months after the first attack has been associated with a reduced risk of relapse in these patients.[96] Other immunosuppressants like mycophenolate mofetil, methotrexate, rituximab, and IVIG are also shown to be effective in reducing the relapse rate in MOG-ON.[96]
Glial fibrillary acidic protein
Similar to MOG-ON glial fibrillary acidic protein (GFAP) associated ON also has good prognosis with steroids. For cases with relapse, gradual tapering of steroids and long-term immunosuppressants may be required.[39]
Chronic recurrent inflammatory ON
Chronic recurrent inflammatory ON (CRION) also needs long-term immune suppression to prevent relapses. After treatment of acute attack, the patient is maintained on low-dose corticosteroids. Immunosuppressants like azathioprine, methotrexate, cyclophosphamide, or mycophenolate have been shown to be beneficial for long-term remission.[40]
Optic neuritis associated with systemic inflammatory diseases
Systemic autoimmune diseases need disease-specific therapy. SLE responds better to cyclophosphamide while sarcoidosis can be treated with azathioprine, methotrexate, cyclosporin, mycophenolate, and infliximab. Monoclonal antibodies like rituximab have also been used in cases of refractory sarcoidosis.[97]
Neuroprotective or regeneration therapy
Various agents like erythropoietin, anti-LINGO antibodies, and phenytoin are being explored for neuroprotection and remyelination.[98,99,100]
Conclusion
We conclude that typical ON or MS-ON can be diagnosed with the help of neuroimaging and CSF studies. An atypical ON should be thoroughly investigated since timely diagnosis and prompt treatment is key to prevent relapses and improve the prognosis. The clinical and radiological features of ON may provide a clue to the etiology. Serological investigations and MRI spine help in establishing the etiology and should be obtained when deemed necessary. A stepwise approach is required for the diagnosis and management of an acute episode of ON. A long-term management protocol is a need to prevent further attacks and decrease the disability burden.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
List of Abbreviations
| ADEM | Acute disseminated encephalomyelitis |
|---|---|
| AQP4 | Aquaporin 4 |
| BENEFIT | Betaferon®/Betaseron® in Newly Emerging Multiple Sclerosis for Initial Treatment |
| CDMS | Clinically defined multiple sclerosis |
| CHAMPS | Controlled high-risk Avonex multiple sclerosis trial |
| CRION | Chronic recurrent inflammatory ON |
| CSF | Cerebrospinal fluid |
| DMD | Disease-modifying drugs |
| EDSS | Extended disability status scale |
| GFAP | Glial fibrillary acidic protein IgG astrocytopathy |
| GA | Glatiramer acetate |
| GCL-IPL | Ganglion layer-inner plexiform layer |
| IVIG | Intravenous immunoglobulins |
| LETM | Longitudinal extensive transverse myelitis |
| MOG | Myelin oligodendrocyte glycoprotein |
| MMF | Mycophenolate mofetil |
| MS | Multiple sclerosis |
| NMO | Neuromyelitis optica |
| NMOSD | Neuromyelitis optica spectrum disorder |
| ON | Optic neuritis |
| ONTT | Optic neuritis treatment trial |
| PRISMS | Prevention of relapses and disability by interferon beta-1a subcutaneously in multiple sclerosis |
| PML | Progressive multifocal leukoencephalopathy |
| RRMS | Relapsing-remitting multiple sclerosis |
| RNFL | Retinal nerve fiber layer |
| SPMS | Secondary progressive multiple sclerosis |
| SLE | Sclerosing lupus erythematosus |
References
- 1.Menon V, Saxena R, Misra R, Phuljhele S. Management of optic neuritis. Indian J Ophthalmol. 2011;59:117–22. doi: 10.4103/0301-4738.77020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chopra JS, Radhakrishnan K, Sawhney BB, Pal SR, Banerjee AK. Multiple sclerosis in North-west India. Acta Neurol Scand. 1980;62:312–21. doi: 10.1111/j.1600-0404.1980.tb03042.x. [DOI] [PubMed] [Google Scholar]
- 3.Singhal BS. Multiple sclerosis and related demyelinating disorders in Indian context. Neurol India. 1987;35:1–12. [Google Scholar]
- 4.Bhatia M, Behari M, Ahuja GK. Multiple sclerosis in India:AIIMS experience. J Assoc Physicians India. 1996;44:765–7. [PubMed] [Google Scholar]
- 5.Verma N, Ahuja GK. Spectrum of multiple sclerosis in Delhi region. J Assoc Physicians India. 1982;30:421–2. [PubMed] [Google Scholar]
- 6.Singhal BS. Multiple sclerosis:Indian experience. Ann Acad Med Singapore. 1985;14:32–6. [PubMed] [Google Scholar]
- 7.Pandit L, Kundapur R. Prevalence and patterns of demyelinating central nervous system disorders in urban Mangalore, South India. Mult Scler. 2014;20:1651–3. doi: 10.1177/1352458514521503. [DOI] [PubMed] [Google Scholar]
- 8.Pandit L, Mustafa S, Kunder R, Shetty R, Misri Z, Pai S, et al. Optimizing the management of neuromyelitis optica and spectrum disorders in resource poor settings:Experience from the Mangalore demyelinating disease registry. Ann Indian Acad Neurol. 2013;16:572–6. doi: 10.4103/0972-2327.120474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Saxena R, Phuljhele S, Menon V, Gadaginamath S, Sinha A, Sharma P. Clinical profile and short-term outcomes of optic neuritis patients in India. Indian J Ophthalmol. 2014;62:265–67. doi: 10.4103/0301-4738.121131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pandit L, Shetty R, Misri Z, Bhat S, Amin H, Pai V, et al. Optic neuritis:Experience from a south Indian demyelinating disease registry. Neurol India. 2012;60:470–5. doi: 10.4103/0028-3886.103186. [DOI] [PubMed] [Google Scholar]
- 11.Pandit L, Sato DK, Mustafa S, Takahashi T, D'Cunha A, Malli C, et al. Serological markers associated with neuromyelitis optica spectrum disorders in South India. Ann Indian Acad Neurol. 2016;19:505–9. doi: 10.4103/0972-2327.192389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Optic Neuritis Study Group. The clinical profile of optic neuritis. Experience of the optic neuritis treatment trial. Arch Ophthalmol. 1991;109:1673–8. doi: 10.1001/archopht.1991.01080120057025. [DOI] [PubMed] [Google Scholar]
- 13.Beck RW. The optic neuritis treatment trial:Three-year follow-up results. Arch Ophthalmol. 1995;113:136–7. doi: 10.1001/archopht.1995.01100020014004. [DOI] [PubMed] [Google Scholar]
- 14.Optic Neuritis Study Group. Visual function 15 years after optic neuritis:A final follow-up report from the Optic Neuritis Treatment Trial. Ophthalmology. 2008;115:1079–82.e5. doi: 10.1016/j.ophtha.2007.08.004. [DOI] [PubMed] [Google Scholar]
- 15.Miller D, Barkhof F, Montalban X, Thompson A, Filippi M. Clinically isolated syndromes suggestive of multiple sclerosis, part I:Natural history, pathogenesis, diagnosis, and prognosis. Lancet Neurol. 2005;4:281–8. doi: 10.1016/S1474-4422(05)70071-5. [DOI] [PubMed] [Google Scholar]
- 16.Optic Neuritis Study Group. Multiple sclerosis risk after optic neuritis:Final optic neuritis treatment trial follow-up. Arch Neurol. 2008;65:727–32. doi: 10.1001/archneur.65.6.727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thompson AJ, Banwell BL, Barkhof F, Carroll WM, Coetzee T, Comi G, et al. Diagnosis of multiple sclerosis:2017 revisions of the McDonald criteria. Lancet Neurol. 2018;17:162–73. doi: 10.1016/S1474-4422(17)30470-2. [DOI] [PubMed] [Google Scholar]
- 18.Lennon VA, Wingerchuk DM, Kryzer TJ, Pittock SJ, Lucchinetti CF, Fujihara K, et al. A serum autoantibody marker of neuromyelitis optica:Distinction from multiple sclerosis. Lancet. 2004;364:2106–12. doi: 10.1016/S0140-6736(04)17551-X. [DOI] [PubMed] [Google Scholar]
- 19.Hinson SR, Pittock SJ, Lucchinetti CF, Roemer SF, Fryer JP, Kryzer TJ, et al. Pathogenic potential of IgG binding to water channel extracellular domain in neuromyelitis optica. Neurology. 2007;69:2221–31. doi: 10.1212/01.WNL.0000289761.64862.ce. [DOI] [PubMed] [Google Scholar]
- 20.Pittock SJ, Lucchinetti CF. Neuromyelitis optica and the evolving spectrum of autoimmune aquaporin-4 channelopathies:A decade later. Ann N Y Acad Sci. 2016;1366:20–39. doi: 10.1111/nyas.12794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mealy MA, Kessler RA, Rimler Z, Reid A, Totonis L, Cutter G, et al. Mortality in neuromyelitis optica is strongly associated with African ancestry. Neurol Neuroimmunol Neuroinflamm. 2018;5:e468. doi: 10.1212/NXI.0000000000000468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huda S, Whittam D, Bhojak M, Kneen R. Neuromyelitis Optica spectrum disorders. Clin Med. 2019;19:169–76. doi: 10.7861/clinmedicine.19-2-169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Quek AM, McKeon A, Lennon VA, Mandrekar JN, Iorio R, Jiao Y, et al. Effects of age and sex on aquaporin-4 autoimmunity. Arch Neurol. 2012;69:1039–43. doi: 10.1001/archneurol.2012.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bove R, Elsone L, Alvarez E, Borisow N, Cortez MM, Mateen FJ, et al. Female hormonal exposures and neuromyelitis optica symptom onset in a multicentre study. Neurol Neuroimmunol Neuroinflamm. 2017;4:e339. doi: 10.1212/NXI.0000000000000339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pereira WL, Reiche EM, Kallaur AP, Kaimen-Maciel DR. Epidemiological, clinical, and immunological characteristics of neuromyelitis optica:A review. J Neurol Sci. 2015;355:7–17. doi: 10.1016/j.jns.2015.05.034. [DOI] [PubMed] [Google Scholar]
- 26.Pandit L. Neuromyelitis optica spectrum disorders:An update. Ann Indian Acad Neurol. 2015;18(Supp 1):S11–5. doi: 10.4103/0972-2327.164816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hansapinyo L, Vivattanaseth C. Clinical characteristics, treatment outcome and predictive factors in optic neuritis. Open Ophthalmol J. 2018;12:247–55. doi: 10.2174/1874364101812010247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jarius S, Ruprecht K, Wildemann B, Kuempfel T, Ringelstein M, Geis C, et al. Contrasting disease patterns in seropositive and seronegative neuromyelitis optica:A multicentre study of 175 patients. J Neuroinflammation. 2012;9:14. doi: 10.1186/1742-2094-9-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jagtap S, Mandliya A, Sarada C, Nair MD. Neuromyelitis optica and neuromyelitis optica spectrum disorder:Natural history and long-term outcome, an Indian experience. J Neurosci Rural Pract. 2015;6:331–5. doi: 10.4103/0976-3147.158755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pittock SJ, Lennon VA, de Seze J, Vermersch P, Homburger HA, Wingerchuk DM, et al. Neuromyelitis optica and non-organ specific autoimmunity. Arch Neurol. 2008;65:78–83. doi: 10.1001/archneurol.2007.17. [DOI] [PubMed] [Google Scholar]
- 31.Jacob S, Zarei M, Kenton A, Allroggen H. Gluten sensitivity and neuro- myelitis optica:Two case reports. J Neurol Neurosurg Psychiatry. 2005;76:1028–30. doi: 10.1136/jnnp.2004.055491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Leite MI, Coutinho E, Lana-Peixoto M, Apostolos S, Waters P, Sato D, et al. Myasthenia gravis and neuromyelitis optica spectrum disorder:A multicenter study of 16 patients. Neurology. 2012;78:1601–7. doi: 10.1212/WNL.0b013e31825644ff. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wingerchuk DM, Banwell B, Bennett JL, Cabre P, Carroll W, Chitnis T, et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology. 2015;85:177–89. doi: 10.1212/WNL.0000000000001729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Di Pauli F, Berger T. Myelin oligodendrocyte glycoprotein antibody-associated and disorders:Toward a new spectrum of inflammatory demyelinating CNS disorders. Front Immunol. 2018;9:2753. doi: 10.3389/fimmu.2018.02753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Weber MS, Derfuss T, Metz I, Brück W. Defining distinct features of anti-MOG antibody associated central nervous system demyelination. Ther Adv Neurol Disord. 2018;11:1–15. doi: 10.1177/1756286418762083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen JJ, Flanagan EP, Jitprapaikulsan J, López-Chiriboga AS, Fryer JP, Leavitt JA, et al. Myelin oligodendrocyte glycoprotein antibody-positive optic neuritis:Clinical characteristics, radiologic clues, and outcome. Am J Ophthalmol. 2018;195:8–15. doi: 10.1016/j.ajo.2018.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ramanathan S, Mohammad S, Tantsis E, Nguyen TK, Merheb V, Fung VS, et al. Clinical course, therapeutic and responses and outcomes in relapsing MOG antibody-associated demyelination. J Neurol Neurosurg Psychiatry. 2018;89:127–37. doi: 10.1136/jnnp-2017-316880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Flanagan EP, Hinson SR, Lennon VA, Fang B, Aksamit AJ, Morris PP, et al. Glial fibrillary acidic protein immunoglobulin G as biomarker of autoimmune astrocytopathy:Analysis of 102 patients. Ann Neurol. 2017;81:298–309. doi: 10.1002/ana.24881. [DOI] [PubMed] [Google Scholar]
- 39.Kunchok A, Zekeridou A, McKeon A. Autoimmune glial fibrillary acidic protein astrocytopathy. Curr Opin Neurol. 2019;32:452–8. doi: 10.1097/WCO.0000000000000676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Petzold A, Plant GT. Chronic relapsing inflammatory optic neuropathy:A systematic review of 122 cases reported. J Neurol. 2014;261:17–26. doi: 10.1007/s00415-013-6957-4. [DOI] [PubMed] [Google Scholar]
- 41.Lee HJ, Kim B, Waters P, Woodhall M, Irani S, Ahn S, et al. Chronic relapsing inflammatory optic neuropathy (CRION):A manifestation of myelin oligodendrocyte glycoprotein antibodies. J Neuroinflammation. 2018;15:302. doi: 10.1186/s12974-018-1335-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chalmoukou K, Alexopoulos H, Akrivou S, Stathopoulos P, Reindl M, Dalakas M. Anti-MOG antibodies are frequently associated with steroid-sensitive recurrent optic neuritis. Neurol Neuroimmunol Neuroinflamm. 2015;2:e131. doi: 10.1212/NXI.0000000000000131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.de Andrade FA, Guimarães Moreira Balbi G, Bortoloti de Azevedo LG, Provenzano SáG, Vieira de Moraes Junior H, Mendes Klumb E, et al. Neuro-ophthalmologic manifestations in systemic lupus erythematosus. Lupus. 2017;26:522–8. doi: 10.1177/0961203316683265. [DOI] [PubMed] [Google Scholar]
- 44.Marie I, Hervé F, Borg JY, Levesque H. Retrobulbar optic neuritis revealing primary anti-phospholipid antibody syndrome. Scand J Rheumatol. 2007;36:156–7. doi: 10.1080/03009740701218832. [DOI] [PubMed] [Google Scholar]
- 45.Phuljhele S, Pujari A, Obedulla H, Saxena R, Sharma P. Atypical presentation of primary Sjögren's syndrome as optic neuritis. Can J Ophthalmol. 2019;54:e30–2. doi: 10.1016/j.jcjo.2018.04.013. [DOI] [PubMed] [Google Scholar]
- 46.Goel J, Anadure R, Gupta S, Wilson V, Saxena R, Sahu S, et al. A Study of the clinical profile, radiologic features, and therapeutic outcomes in neurosarcoidosis from two tertiary care centers in Southern India. Neurol India. 2020;68:609–16. doi: 10.4103/0028-3886.288976. [DOI] [PubMed] [Google Scholar]
- 47.Geraldes R, Ciccarelli O, Barkhof F, De Stefano N, Enzinger C, Filippi M, et al. The current role of MRI in differentiating multiple sclerosis from its imaging mimics. Nat Rev Neurol. 2018;14:199–213. doi: 10.1038/nrneurol.2018.14. [DOI] [PubMed] [Google Scholar]
- 48.Ramanathan S, Prelog K, Barnes EH, Tantsis EM, Reddel SW, Henderson AP, et al. Radiological differentiation of optic neuritis with myelin oligodendrocyte glycoprotein antibodies, aquaporin-4 antibodies, and multiple sclerosis. Mult Scler. 2016;22:470–82. doi: 10.1177/1352458515593406. [DOI] [PubMed] [Google Scholar]
- 49.Pittock SJ, Weinshenker BG, Lucchinetti CF, Wingerchuk DM, Corboy JR, Lennon VA. Neuromyelitis optica brain lesions localized at sites of high aquaporin 4 expression. Arch. Neurol. 2006;63:964–8. doi: 10.1001/archneur.63.7.964. [DOI] [PubMed] [Google Scholar]
- 50.Jacob A, McKeon A, Nakashima I, Sato DK, Elsone L, Fujihara K, et al. Current concept neuromyelitis optica (NMO) and NMO spectrum disorders. J Neurol Neurosurg Psychiatry. 2013;84:922–30. doi: 10.1136/jnnp-2012-302310. [DOI] [PubMed] [Google Scholar]
- 51.Takano R, Misu T, Takahashi T, Sato S, Fujihara K, Itoyama Y. Astrocytic damage is far more severe than demyelination in NMO:A clinical CSF biomarker study. Neurology. 2010;75:208–16. doi: 10.1212/WNL.0b013e3181e2414b. [DOI] [PubMed] [Google Scholar]
- 52.Petzold A, de Boer JF, Schippling S, Vermersch P, Kardon R, Green A, et al. Optical coherence tomography in multiple sclerosis:A systematic review and meta-analysis. Lancet Neurol. 2010;9:921–32. doi: 10.1016/S1474-4422(10)70168-X. [DOI] [PubMed] [Google Scholar]
- 53.Saxena R, Bandyopadhyay G, Singh D, Singh S, Sharma P, Menon V. Evaluation of changes in retinal nerve fiber layer thickness and visual functions in cases of optic neuritis and multiple sclerosis. Indian J Ophthalmol. 2013;61:562–6. doi: 10.4103/0301-4738.121071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tan O, Chopra V, Lu AT, Schuman JS, Ishikawa H, Wollstein G, et al. Detection of macular ganglion cell loss in glaucoma by fourier-domain optical coherence tomography. Ophthalmology. 2009;116:2305–140. doi: 10.1016/j.ophtha.2009.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pillay G, Ganger A, Singh D, Bhatia R, Sharma P, Menon V, et al. Retinal nerve fiber layer and ganglion cell layer changes on optical coherence tomography in early multiple sclerosis and optic neuritis cases. Indian J Ophthalmol. 2018;66:114–9. doi: 10.4103/ijo.IJO_539_17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Costello F. The afferent visual pathway:Designing a structural-functional paradigm of multiple sclerosis. ISRN Neurol. 2013;2013:134858. doi: 10.1155/2013/134858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Saidha S, Syc SB, Durbin MK, Eckstein C, Oakley JD, Meyer SA, et al. Visual dysfunction in multiple sclerosis correlates better with optical coherence tomography derived estimates of macular ganglion cell layer thickness than peripapillary retinal nerve fiber layer thickness. Mult Scler. 2011;17:1449–63. doi: 10.1177/1352458511418630. [DOI] [PubMed] [Google Scholar]
- 58.Bennett JL, de Seze J, Lana-Peixoto M, Palace J, Waldman A, Schippling S, et al. Neuromyelitis optica and multiple sclerosis:Seeing differences through optical coherence tomography. Mult Scler. 2015;21:678–88. doi: 10.1177/1352458514567216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Beck RW, Cleary PA. Optic neuritis treatment trial. One-year follow-up results. Arch Ophthalmol. 1993;111:773–5. doi: 10.1001/archopht.1993.01090060061023. [DOI] [PubMed] [Google Scholar]
- 60.Morrow SA, Fraser JA, Day C, Bowman D, Rosehart H, Kremenchutzky M, et al. Effect of treating acute optic neuritis with bioequivalent oral vs intravenous corticosteroids- A randomized clinical trial. JAMA Neurol. 2018;75:690–6. doi: 10.1001/jamaneurol.2018.0024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Burton JM, O'Connor PW, Hohol M, Beyene J. Oral versus intravenous steroids for treatment of relapses in multiple sclerosis. Cochrane Database Syst Rev. 2009;8:CD006921. doi: 10.1002/14651858.CD006921.pub2. [DOI] [PubMed] [Google Scholar]
- 62.Menon V, Mehrotra A, Saxena R, Jaffery NF. Comparative evaluation of megadose methylprednisolone with dexamethasone for treatment of primary typical optic neuritis. Indian J Ophthalmol. 2007;55:355–9. doi: 10.4103/0301-4738.33821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gal RL, Vedula SS, Beck R. Corticosteroids for treating optic neuritis. Cochrane Database Syst Rev. 2015;2015:CD001430. doi: 10.1002/14651858.CD001430.pub4. doi:10.1002/14651858.CD001430.pub4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kleiter I, Gahlen A, Borisow N, Fischer K, Wernecke KD, Wegner B, et al. Neuromyelitis optica:Evaluation of 871 attacks and 1,153 treatment courses. Ann Neurol. 2016;79:206–16. doi: 10.1002/ana.24554. [DOI] [PubMed] [Google Scholar]
- 65.Srisupa-Olan T, Siritho S, Kittisares K, Jitprapaikulsan J, Sathukitchai C, Prayoonwiwat N. Beneficial effect of plasma exchange in acute attack of neuromyelitis optica spectrum disorders. Mult Scler Relat Disord. 2018;20:115–21. doi: 10.1016/j.msard.2018.01.010. [DOI] [PubMed] [Google Scholar]
- 66.Bonnan M, Valentino R, Debeugny S, Merle H, Fergé JL, Mehdaoui H, et al. Short delay to initiate plasma exchange is the strongest predictor of outcome in severe attacks of NMO spectrum disorders. J Neurol Neurosurg Psychiatry. 2018;89:346–51. doi: 10.1136/jnnp-2017-316286. [DOI] [PubMed] [Google Scholar]
- 67.Abboud H, Petrak A, Mealy M, Sasidharan S, Siddique L, Levy M. Treatment of acute relapses in neuromyelitis optica:Steroids alone versus steroids plus plasma exchange. Mult Scler. 2016;22:185–92. doi: 10.1177/1352458515581438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tselis A, Perumal J, Caon C, Hreha S, Ching W, Din M, et al. Treatment of corticosteroid refractory optic neuritis in multiple sclerosis patients with intravenous immunoglobulin. Eur J Neurol. 2008;15:1163–7. doi: 10.1111/j.1468-1331.2008.02258.x. [DOI] [PubMed] [Google Scholar]
- 69.Achiron A, Kishner I, Sarova-Pinhas I, Raz H, Faibel M, Stern Y, et al. Intravenous immunoglobulin treatment following the first demyelinating event suggestive of multiple sclerosis:A randomized, double-blind, placebo-controlled trial. Arch Neurol. 2004;61:1515–20. doi: 10.1001/archneur.61.10.1515. [DOI] [PubMed] [Google Scholar]
- 70.Dos Passos GR, Oliveira LM, da Costa BK, Apostolos-Pereira SL, Callegaro D, Fujihara K, et al. MOG-IgG-associated optic neuritis, encephalitis, and myelitis:Lessons learned from neuromyelitis optica spectrum disorder. Front Neurol. 2018;9:217. doi: 10.3389/fneur.2018.00217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jacobs LD, Beck RW, Simon JH, Kinkel RP, Brownscheidle CM, Murray TJ, et al. Intramuscular interferon beta-1a therapy initiated during a first demyelinating event in multiple sclerosis. CHAMPS Study Group. N Engl J Med. 2000;343:898–904. doi: 10.1056/NEJM200009283431301. [DOI] [PubMed] [Google Scholar]
- 72.Kinkel RP, Kollman C, O'Connor P, Murray TJ, Simon J, Arnold D, et al. IM interferon beta-1a delays definite multiple sclerosis 5 years after a first demyelinating event. Neurology. 2006;66:678–84. doi: 10.1212/01.wnl.0000200778.65597.ae. [DOI] [PubMed] [Google Scholar]
- 73.Comi G, Filippi M, Barkhof F, Durelli L, Edan G, Fernández O, et al. Effect of early interferon treatment on conversion to definite multiple sclerosis:A randomised study. Lancet Lond Engl. 2001;357:1576–82. doi: 10.1016/s0140-6736(00)04725-5. [DOI] [PubMed] [Google Scholar]
- 74.Ebers GC. Randomised double-blind placebo-controlled study of interferon b-1a in relapsing/remitting multiple sclerosis. Lancet. 1998;352:1498–504. [PubMed] [Google Scholar]
- 75.Kappos L, Freedman MS, Polman CH, Edan G, Hartung H-P, Miller DH, et al. Effect of early versus delayed interferon beta-1b treatment on disability after a first clinical event suggestive of multiple sclerosis:A 3-year follow-up analysis of the BENEFIT study. Lancet Lond Engl. 2007;370:389–97. doi: 10.1016/S0140-6736(07)61194-5. [DOI] [PubMed] [Google Scholar]
- 76.Comi G, Martinelli V, Rodegher M, Moiola L, Bajenaru O, Carra A, et al. Effect of glatiramer acetate on conversion to clinically definite multiple sclerosis in patients with clinically isolated syndrome (PreCISe study):A randomised, double-blind, placebo-controlled trial. Lancet Lond Engl. 2009;374:1503–11. doi: 10.1016/S0140-6736(09)61259-9. [DOI] [PubMed] [Google Scholar]
- 77.Khan O, Rieckmann P, Boyko A, Selmaj K, Zivadinov R. Three times weekly glatiramer acetate in relapsing-remitting multiple sclerosis. Ann Neurol. 2013;73:705–13. doi: 10.1002/ana.23938. [DOI] [PubMed] [Google Scholar]
- 78.Gold R, Kappos L, Arnold DL, Bar-Or A, Giovannoni G, Selmaj K, et al. Placebo-controlled phase 3 study of oral BG-12 for relapsing multiple sclerosis. N Engl J Med. 2012;367:1098–107. doi: 10.1056/NEJMoa1114287. [DOI] [PubMed] [Google Scholar]
- 79.Fox RJ, Miller DH, Phillips JT, Hutchinson M, Havrdova E, Kita M, et al. Placebo-controlled phase 3 study of oral BG-12 or glatiramer in multiple sclerosis. N Engl J Med. 2012;367:1087–97. doi: 10.1056/NEJMoa1206328. [DOI] [PubMed] [Google Scholar]
- 80.Cohen JA, Tenenbaum N, Bhatt A, Zhang Y, Kappos L. Extended treatment with fingolimod for relapsing multiple sclerosis:The 14-year LONGTERMS study results. Ther Adv Neurol Disord. 2019;12:17562, 9878324. doi: 10.1177/1756286419878324. doi:10.1177/1756286419878324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Derfuss T, Sastre-Garriga J, Montalban X, Rodegher M, Wuerfel J, Gaetano L, et al. The ACROSS study:Long-term efficacy of fingolimod in patients with relapsing-remitting multiple sclerosis. Mult Scler J Exp Transl Clin. 2020;6:2055217320907951. doi: 10.1177/2055217320907951. doi:10.1177/2055217320907951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kappos L, Bar-Or A, Cree BAC, Fox RJ, Giovannoni G, Gold R, et al. Siponimod versus placebo in secondary progressive multiple sclerosis (EXPAND):A double-blind, randomised, phase 3 study. Lancet. 2018;391:1263–73. doi: 10.1016/S0140-6736(18)30475-6. [DOI] [PubMed] [Google Scholar]
- 83.Giovannoni G, Soelberg Sorensen P, Cook S, Rammohan K, Rieckmann P, Comi G, et al. Safety and efficacy of cladribine tablets in patients with relapsing-remitting multiple sclerosis:Results from the randomized extension trial of the CLARITY study. Mult Scler. 2018;24:1594–604. doi: 10.1177/1352458517727603. [DOI] [PubMed] [Google Scholar]
- 84.Polman CH, O'Connor PW, Havrdova E, Hutchinson M, Kappos L, Miller DH, et al. A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med. 2006;354:899–910. doi: 10.1056/NEJMoa044397. [DOI] [PubMed] [Google Scholar]
- 85.Clerico M, Artusi CA, Liberto AD, Rolla S, Bardina V, Barbero P, et al. Natalizumab in multiple sclerosis:Long-term management. Int J Mol Sci. 2017;18:940. doi: 10.3390/ijms18050940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ghezzi A. European and American guidelines for multiple sclerosis treatment. Neurol Ther. 2018;7:189–94. doi: 10.1007/s40120-018-0112-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Anton R, Haas M, Arlett P, Weise M, Balabanov P, Mazzaglia G, et al. Drug-induced progressive multifocal leukoencephalopathy in multiple sclerosis:European regulators'perspective. Clin Pharmacol Ther. 2017;102:283–9. doi: 10.1002/cpt.604. [DOI] [PubMed] [Google Scholar]
- 88.Costanzi C, Matiello M, Lucchinetti CF, Weinshenker BG, Pittock SJ, Mandrekar J, et al. Azathioprine:Tolerability, efficacy, and predictors of benefit in neuromyelitis optica. Neurology. 2011;77:659–66. doi: 10.1212/WNL.0b013e31822a2780. [DOI] [PubMed] [Google Scholar]
- 89.Bichuetti DB, Lobato de Oliveira EM, Oliveira DM, Amorin de SN, Gabbai AA. Neuromyelitis optica treatment:Analysis of 36 patients. Arch Neurol. 2010;67:1131–6. doi: 10.1001/archneurol.2010.203. [DOI] [PubMed] [Google Scholar]
- 90.Jacob A, Matiello M, Weinshenker BG, Wingerchuk DM, Lucchinetti C, Shuster E, et al. Treatment of neuromyelitis optica with mycophenolate mofetil:Retrospective analysis of 24 patients. Arch Neurol. 2009;66:1128–33. doi: 10.1001/archneurol.2009.175. [DOI] [PubMed] [Google Scholar]
- 91.Kitley J, Elsone L, George J, Waters P, Woodhall M, Vincent A, et al. Methotrexate is an alternative to azathioprine in neuromyelitis optica spectrum disorders with aquaporin-4 antibodies. J Neurol Neurosurg Psychiatry. 2013;84:918–21. doi: 10.1136/jnnp-2012-304774. [DOI] [PubMed] [Google Scholar]
- 92.Bedi GS, Brown AD, Delgado SR, Usmani N, Lam BL, Sheremata WA. Impact of rituximab on relapse rate and disability in neuromyelitis optica. Mult Scler. 2011;17:1225–30. doi: 10.1177/1352458511404586. [DOI] [PubMed] [Google Scholar]
- 93.Etemadifar M, Salari M, Mirmosayyeb O, Serati M, Nikkhah R, Askari M, et al. Efficacy and safety of rituximab in neuromyelitis optica:Review of evidence. J Res Med Sci. 2017;22:18. doi: 10.4103/1735-1995.200275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Damato V, Evoli A, Iorio R. Efficacy and safety of rituximab therapy in neuromyelitis optica spectrum disorders:A systematic review and meta-analysis. JAMA Neurol. 2016;73:1342–8. doi: 10.1001/jamaneurol.2016.1637. [DOI] [PubMed] [Google Scholar]
- 95.Pittock SJ, Berthele A, Fujihara K, Kim HJ, Levy M, Palace J, et al. Eculizumab in aquaporin-4-positive neuromyelitis optica spectrum disorder. N Engl J Med. 2019;381:614–25. doi: 10.1056/NEJMoa1900866. [DOI] [PubMed] [Google Scholar]
- 96.Chun BY, Cestari DM. Myelin oligodendrocyte glycoprotein-IgG-associated optic neuritis. Curr Opin Ophthalmol. 2018;29:508–13. doi: 10.1097/ICU.0000000000000520. [DOI] [PubMed] [Google Scholar]
- 97.Cinetto F, Compagno N, Scarpa R, Malipiero G, Agostini C. Rituximab in refractory sarcoidosis:A single centre experience. Clin Mol Allergy. 2015;13:19. doi: 10.1186/s12948-015-0025-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sühs KW, Hein K, Sättler MB, Görlitz A, Ciupka C, Scholz K, et al. A randomized, double-blind, phase 2 study of erythropoietin in optic neuritis. Ann Neurol. 2012;72:199–210. doi: 10.1002/ana.23573. [DOI] [PubMed] [Google Scholar]
- 99.Cadavid D, Balcer L, Galetta S, Aktas O, Ziemssen T, Vanopdenbosch L, et al. Safety and efficacy of opicinumab in acute optic neuritis (RENEW):A randomised, placebo-controlled, phase 2 trial. The Lancet. Neurology. 2017;16:189–99. doi: 10.1016/S1474-4422(16)30377-5. [DOI] [PubMed] [Google Scholar]
- 100.Raftopoulos R, Hickman SJ, Toosy A, Sharrack B, Mallik S, Paling D, et al. Phenytoin for neuroprotection in patients with acute optic neuritis:A randomised, placebo-controlled, phase 2 trial. Lancet Neurol. 2016;15:259–69. doi: 10.1016/S1474-4422(16)00004-1. [DOI] [PubMed] [Google Scholar]