

Abstract
Chronic obstructive pulmonary disease (COPD) is a lung disorder characterized by progressive airflow obstruction associated with inflammation and emphysema, and it is currently one of the leading causes of death worldwide. Recent studies with genetically engineered mice reported that during pulmonary inflammation, basophil-derived interleukin-4 can act on lung-infiltrating monocytes causing aberrant expression of the matrix metalloproteinase-12 (MMP-12). MMP-12 activity in turn causes the destruction of alveolar walls leading to emphysema, making it potentially a valid target for pharmacological intervention. Using NMR- and structure-based optimizations, the current study reports on optimized novel, potent and selective MMP-12 inhibitors with single digit nanomolar affinity in vitro and in vivo efficacy. Using a murine model of elastase-induced emphysema we demonstrated that the most potent agents exhibited a significant decrease in emphysema-like pathology compared to vehicle-treated mice thus suggesting that the reported agents may be potentially translated in novel therapeutics for the treatment of COPD.
Keywords: Drug discovery, MMP-12 inhibitors, COPD, NMR-based drug design, structure-based drug design
Introduction
Chronic obstructive pulmonary disease (COPD) is a lung disease characterized by a progressive and irreversible airflow limitation, and currently ranks among the top leading causes of death worldwide.1 COPD is often associated with an excessive inflammatory response of the lungs to air pollutants, or cigarette smoking. Given the persisting prevalence of cigarette smoking, and the increasing environmental factors and pollutants, the incidence of COPD is expected to continue grow. Moreover COPD has been linked to other sever co-morbidities including cardiovascular disease2 and lung cancer.3 Currently, only a few disease-modifying targeted therapeutics are available for this indication.4–8 Recent studies with a series of genetically engineered mice revealed, however, that the matrix metalloprotease MMP-12 can play a pivotal role in airway inflammation and remodeling.9 Preclinical studies in COPD/emphysema provide experimental support that approaches aimed at blocking MMP-12 could be translated into useful agents for therapeutic intervention. For example, pathological evidence indicates that MMP-12-deficient mice are protected against the development of emphysema induced by cigarette smoke and pollutants.10 However, for these findings to translate in possible therapeutics, novel, effective and selective drug-like MMP-12 agents must be developed. Currently the most potent and selective MMP-12 inhibitor is agent MMP408,11 while previously a dual MMP-9/MMP-12 inhibitor was also reported (e.g. clinical candidate AZD1236).10, 12 We recently reported on the use of the HTS by NMR approach13–15 to derive novel MMP-12 inhibitors.16 Using NMR- and structure-based guided optimizations, this present study reports on a novel series of agents with single-digit nanomolar inhibition against MMP-12. Furthermore, we found that mice exposed to porcine pancreatic elastase but treated with MMP-12 inhibitors (MMP-408, or lead agents compound 25, and 26) exhibited a significant decrease in emphysema-like pathology compared to vehicle-treated mice. Hence, the data demonstrated once again that the HTS by NMR can identify viable hit compounds that can be readily optimized into lead agents for continued drug development. The approach is of general applicability hence in principle can be deployed for the identification of such agents when targeting any metallo-enzymes. Preliminary pharmacological evaluations of compound 25 suggest that the agent could be a suitable candidate for additional efficacy and drug development studies leading eventually to human trials.
Results
NMR- and structure-based approaches to derive novel potent and selective MMP-12 inhibitors.
We previously reported on a novel general approach, termed the HTS by NMR13–15 that allowed the discovery of initial hit agents targeting MMP-12.16 Briefly, the method consisted first in deriving a focused positional scanning (POS) combinatorial library of peptide mimetics (of approximately 100,000 compounds) where each element of the library contained the metal-chelating moiety hydroxamic acid at the C-terminal (Figure 1A).
Figure 1.
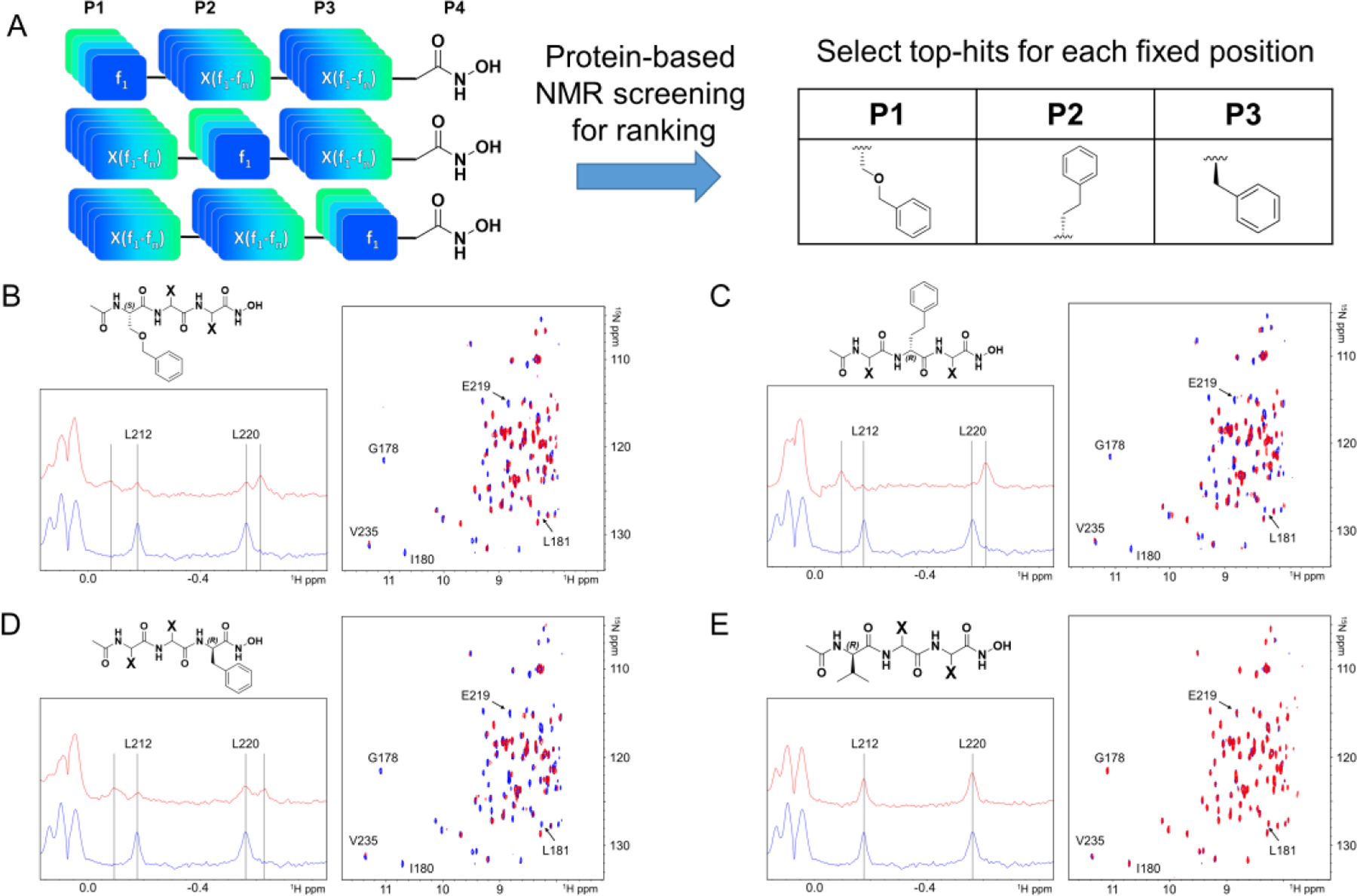
Schematic illustration of the HTS by NMR approach used to derive compound 1. A) From a positional scanning combinatorial library of hydroxamates (46, f1…f46, elements for each of the P1, P2, and P3 positions), NMR-based screening using the catalytic domain of MMP-12 identified P1, P2, and P3 substituents. Rank ordering of the mixtures was accomplished by measuring 1D-1H-aliph, and 2D-[15N, 1H] so-fast HMQC spectra of hMMP-12 (10 μM) collected in the absence (blue) and presence (red) of each mixture (at 500 μM total concentration). Representative spectra for positive mixtures with the indicated fixed position side chains, and a representative negative mixture, are reported in panels B), C), D), and E), respectively.
In the implementation of this approach against hMMP-12, we used a combination of 46 natural and non-natural fn aminoacids to generate 46 mixtures fnXX-CONHOH, 46 mixtures XfnX-CONHOH, and 46 mixtures XXfn-CONHOH, where X represents all the 46 aminoacids.16 Hence, each mixture contained approximately 46 × 46 ~ 2100 compounds (Figure 1A). Subsequently, each mixture was tested against the metalloproteinase hMMP-12 using sensitive protein–NMR screening methods.17 In particular, we rank-ordered the mixtures based on chemical shift changes induced by a given fn element using 1D 1H aliphatic and 2D [15N,1H] so-fast HMQC correlation spectra.13–16, 18–19 The synthesis of the library, and the individual agents, was easily attained by solid phase synthesis and an fmoc-hydroxylamine-2-chlorotrityl resin that after cleavage with 94% of trifluoroacetic acid (TFA) delivered the agents with a C-terminal hydroxamate. Representative 1D 1H aliphatic and 2D [15N,1H] so-fast HMQC correlation spectra for positive top ranking fragments in P1, P2, or P3, along with a negative representative mixture as control, are reported in Figure 1.16 Subsequently, top ranking elements from each position were combined, individual compounds were synthesized encompassing the most active fragments at each position, and tested using NMR and also biochemical assays to more accurately quantify potency and selectivity.16
These studies culminated with the identification of compound 1, that inhibited hMMP-12 with an IC50 value of 29.7 nM (Figure 2A,B).16 Compound 1 did not significantly inhibit MMP-1, MMP-9, MMP-13, and MMP-14 even at 1 μM, while it inhibited appreciably only MMP-3, which is the most closely related metalloproteinase to MMP-12, with ~19% inhibition at 55 nM.16 Given the geometry of the metal chelating moiety, and supported by NMR experimental chemical shift perturbations, a model of the structure of compound 1 in complex with MMP-12 (PDB-ID 5LAB) was generated (Figure 2C,D) that formed the basis for systematic structure-based optimizations of compound 1.
Figure 2.
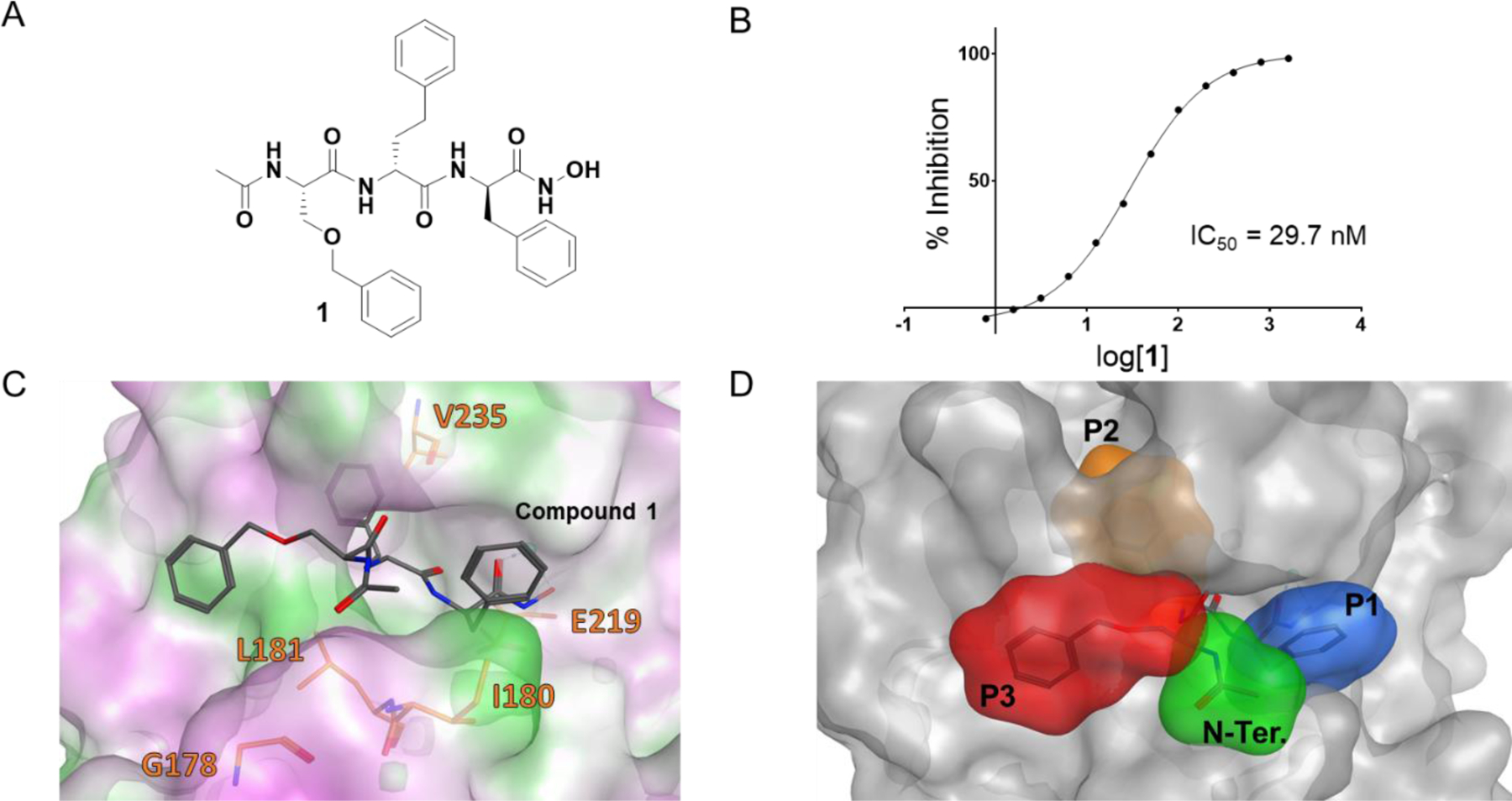
Chemical structure and hMMP-12 inhibition by compound 1. A) Chemical structure of compound 1. B) Dose response inhibition of hMMP-12 by compound 1 indicated an IC50 value of ~ 30 nM. C) Docked geometry of compound 1 obtained with GOLD (Cambridge Crystallographic Data Centre, https://www.ccdc.cam.ac.uk/) and crystal structure of hMMP-12 (PDB-ID 5LAB). The prediction of the docked geometry was largely facilitated by the structural constraints provided by the metal chelating moiety. Surface color scheme: green, lipophilic residues; pink, hydrophilic; whit, neutral. D) As in C) but emphasizing the P1, P2, and P3 substructures being optimized in this present manuscript through SAR studies.
Given the modular nature of the agent, and to reduce the number of possible agents to be synthesized and tested, we opted to systematically fine-tune each substructure independently. Hence, we first probed modifications in the P1 position by introduction of few bioisosteres of the ethoxymethyl benzene present in compound 1 at that position (Table 1). Similarly, replacements of the P2 position D-homo-phenylalanine (Table 2), and the D-phenylalanine in position P3 (Table 3) of compound 1, respectively, were carried out. In each SAR study, rank ordering of the agents was accomplished by NMR chemical shift perturbations followed by enzymatic assays. As reported previously with agents of this series, NMR titrations of test agents with recombinant MMP-12 resulted in slow exchange in the NMR-time scale, in agreement with the nanomolar affinity of the optimized ligands of this series for the target.16 Hence, we used the methyl resonances of residues L212 and I220 that resonate in a spectral region of the 1D 1H NMR spectrum of the protein that is void from overlap with any other resonances from the protein or from ligand hydrogen nuclei (Figure 3). Upon binding of potent ligands, two new resonances appear, shifted by approximately 50 Hz from their position in the unbound state. At a given protein:ligand ratio, the spectrum of hMMP-12 would display the new resonances at an intensity that is proportional to the binding affinity of the test ligand. Hence, we defined a ΔI value as the average ratio between the peak intensities for the methyl resonances of L212 and I220 in the unbound versus the bound form at 1:1 protein/ligand ratio of 10 μM, as a rough measure to rank order the binding affinity of closely related test agents (Figure 3, Tables 1–3). Subsequently, agents that displayed a greater affinity for hMMP-12 compared the parent compound 1 were tested in an enzymatic assay for hMMP-12 inhibition (Tables 1–3).
Table 1:
Initial optimization of compound 1 by modifications at the P1 position.
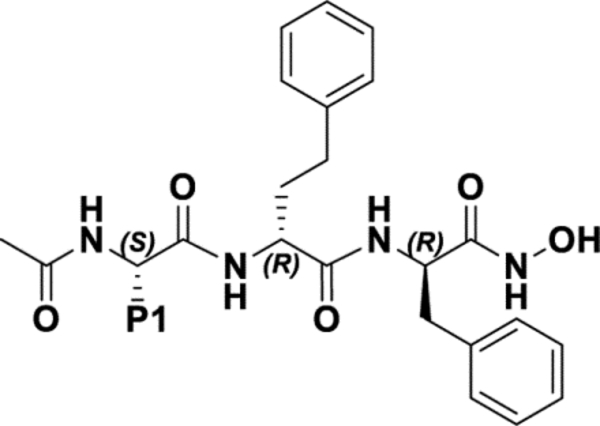
| |||
|---|---|---|---|
| Cmpd | P1 | NMR rankinga | IC50b [nM] |
| 1 |

|
+++ | 26.52 ± 0.03 (n = 8) |
| 2 |

|
++++ | 19.5 ± 0.3 (n = 2) |
| 3 |

|
++ | 34.9 ± 2.8 (n = 2) |
| 4 |

|
+ | 138.2 ± 2.3 (n = 2) |
| 5 |
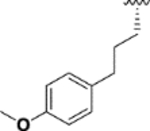
|
++++ | 20.5 ± 0.3 (n = 2) |
NMR rank ordering was obtained by following the side chain methyl resonances of residues L212 and I220 in the 1D 1H NMR spectra of MMP-12 (10 μM) collected in absence and in presence of 10 μM of test agents. ΔI is the ratio of the peak intensities of L212 and I220 in the bound versus the unbound form at 1:1 ratio (10 μM). ++++, ΔI > 1 and greater than effect induced by compound 1; +++, ΔI > 1; ++, ΔI ~ 1; +, ΔI < 1 (see Figure 3C and supplementary Figure S2).
IC50 values were obtained as described in the experimental section.
Table 2:
Initial optimization of compound 1 by modifications at the P2 position.

| |||
|---|---|---|---|
| Cmpd | P2 | NMR rankinga | IC50b [nM] |
| 1 |

|
+++ | 26.52 ± 0.03 (n = 8) |
| 6 |
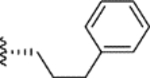
|
+ | N.D. |
| 7 |

|
inactive | N.D. |
| 8 |
|
inactive | N.D |
| 9 |
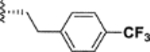
|
++ | 16.7 ± 0.1 (n = 2) |
| 10 |

|
++++ | 16.3 ± 0.1 (n = 2) |
NMR rank ordering was obtained by following the side chain cross peaks of residues L212 and I220 in 1D 1H NMR spectra of MMP-12 (10 μM) collected in absence and in presence of 10 μM of test agents. ΔI is the ratio of the peak intensities of L212 and I220 in the bound versus the unbound form at 1:1 ratio (10 μM). ++++, ΔI > 1 and greater than the effect induced by compound 1; +++, ΔI > 1; ++, ΔI ~ 1; +, ΔI < 1 (see Figure 3C and supplementary Figure S2).
IC50 values were obtained as described in the experimental section.
Table 3:
Initial optimization of compound 1 by modifications at the P3 position.
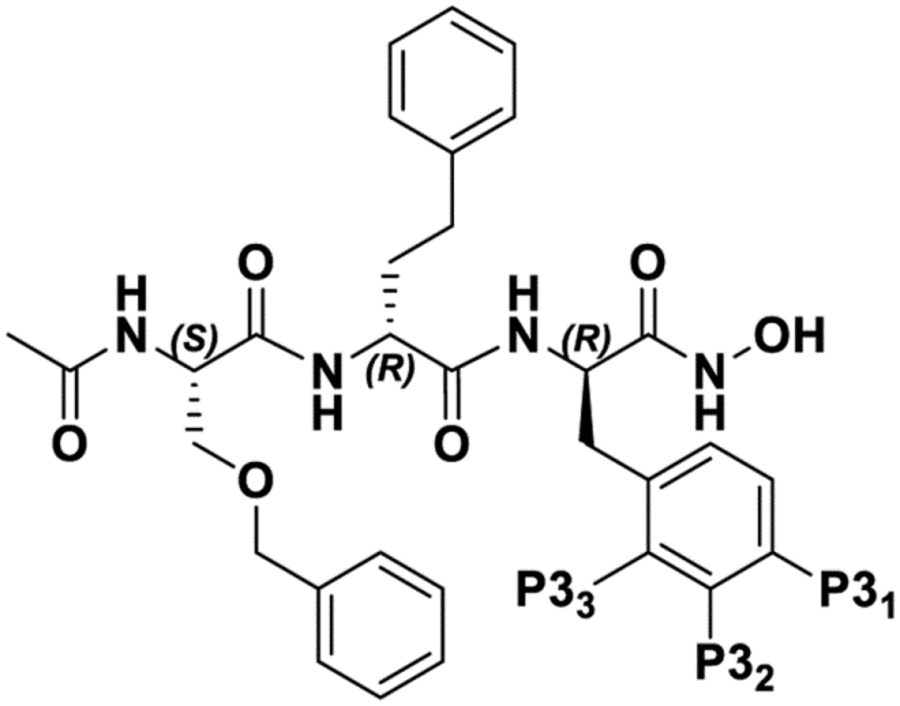
| |||||
|---|---|---|---|---|---|
| Cmpd | P31 | P32 | P33 | NMR rankinga | IC50b [nM] |
| 1 | −H | −H | −H | +++ | 26.52 ± 0.03 (n = 8) |
| 11 | −OH | −H | −H | +++ | 42.1 ± 0.4 (n = 2) |
| 12 | −OSO2F | −H | −H | + | N.D. |
| 13 | −F | −H | −H | +++ | 42.0 ± 0.1 (n = 2) |
| 14 | −CF3 | −H | −H | ++ | 57.1 ±1.8 (n = 2) |
| 15 | −OCOCH3 | −H | −H | ++ | 36.1 ±1.2 (n = 2) |
| 16 | −Cl | −H | −H | +++ | 35.1 ± 0.3 (n = 2) |
| 17 | −CH3 | −H | −H | ++++ | 20.9 ± 0.2 (n = 2) |
| 18 | −H | −F | −H | ++++ | 30.2 ±1.8 (n = 2) |
| 19 | −H | −Cl | −H | +++ | 47.2 ± 0.7 (n = 2) |
| 20 | −H | CH3 | −H | +++ | 35.5 ± 0.4 (n = 2) |
| 21 | −H | −H | −F | ++++ | 27.8 ± 0.9 (n = 2) |
| 22 | −H | −H | −Cl | + | N.D. |
| 23 | −H | −H | −CH3 | + | N.D. |
| 24 | −F | −F | −F | ++ | 75.9 ± 5.3 (n = 2) |
NMR rank ordering was obtained by following the side chain cross peaks of residues L212 and I220 in 1D 1H NMR spectra of MMP-12 (10 μM) collected in absence and in presence of 10 μM of test agents. ΔI is the ratio of the peak intensities of L212 and I220 in the bound versus the unbound form at 1:1 ratio (10 μM). ++++, ΔI > 1 and greater than effect induced by compound 1; +++, ΔI > 1; ++, ΔI ~ 1; +, ΔI < 1 (see Figure 3C and supplementary Figure S2).
IC50 values were obtained as described in the experimental section.
Figure 3. Summary of optimizations of the P1-P3 substituents on the backbone of compound 1.

A) Chemical substructures that appeared more active than reference compound 1 via the NMR binding assay, and by the hMMP-12 inhibition in vitro enzymatic assay (Tables 1–3). B) 1D 1H aliphatic portion of the spectra of MMP-12 catalytic domain (10 μM) recorded in absence (blue), and in presence (red) of compound 1 (10 μM). ΔI for the selected resonances was derived as the ratio of the intensities of the bound versus apo form (IBND/IAPO), used for ranking purposes. C) 1D 1H aliphatic portion of the spectra of MMP-12 catalytic domain (10 μM) measured in the absence and the presence of agents 1, 2, 5, 9, 10, and 17 (each at 10 μM). Spectra were recorded on a 700 MHz Bruker instrument equipped with a TCI cryo-probe.
These studies were aimed at the identification of optimized substituents for each of the 3 positons of the backbone of compound 1 (Figure 3). In particular, substituents that were more hydrophobic than the benzyl-ester in P1 resulted more active (compounds 2 and 5) while more rigid analogues were significantly less potent that compound 1 (Table 1). Commercially available amino-acids carrying isosters of the P2 position side chain resulted in agents listed in Table 2, that when tested by the NMR and enzymatic assays resulted less potent than compound 1, with the exception of compounds 9 and 10, that introduced a p-CF3 or p-methoxyl group in the P2 side chain, respectively. However, under the same experimental conditions, compound 10 appeared more active in the NMR-binding assay, hence was selected as the top ranking for the P2 position. Replacement of the D-Phe in position P3 was also probed by synthesizing and testing the agents listed in Table 3. Substitutions in the para position with a methyl group (compound 17), and in the meta (compound 18) or ortho (compound 21) position with a fluorine atom, increased potency of the agents relative to compound 1. However, unfortunately di- or tri-substituted D-Phe such as p-methyl,o-F-D-Phe, p-methyl,m-F-D-Phe, or p-methyl,o-F,m-F-D-Phe were not commercially available. Hence, the side chain of compound 17 was selected as top ranking for the P3 position.
Therefore, these studies ultimately led to the synthesis of compounds 25 and 26 that encompass the optimal commercially available P1, P2, and P3 substituents (Figure 3). The morpholino group was introduced at the N-terminal of these agents to increase their solubility. Enzyme activity inhibition assay using the SensoLyte® 520 hMMP-12 Fluorimetric Assay Kit (Anaspec) revealed that compounds 25 and 26 were competitive inhibitors for hMMP-12 with remarkable IC50 values of ~ 4 nM (Figure 4A, B). As controls, we also tested the pan-MMP inhibitor GM6001, and the reportedly potent and selective MMP-12 inhibitor compound MMP408.11 When tested side by side, and using the exact same assay protocol and kit (SensoLyte® 520 MMP-12 Fluorimetric Assay Kit) control agents pan-MMP inhibitor GM6001, and MMP-12 selective inhibitor MMP408 displayed IC50 values of ~2.5 nM, and ~19 nM, respectively (Figure 4D, E). Hence, while the IC50 value obtained for GM6001 is in close agreement with that reported in literature for this non-selective MMP inhibitor, in our assay agent MMP408 resulted significantly less potent than previously reported.11 While absolute IC50 values depend on the assay parameters, the relative potency between compounds 25, 26, and MMP408, tested under the same experimental conditions, reveals that our agents are > 5 times more potent than MMP408.
Figure 4.

Chemical structures and comparative dose response values for optimized MMP-12 inhibitors. A) Chemical structure of compound 25 and relative dose response curve for inhibitor of MMP-12. B) Chemical structure of compound 26 and relative dose response curve for inhibitor of MMP-12. C) Overlay of [15N, 1H] sofast HMQC spectra of MMP-12 catalytic domain (10 μM) recorded in absence (blue) or in presence of compound 25 (red) or compound 26 (green), each at 10 μM. D) Chemical structure of the pan-MMP inhibitor GM6001 and relative dose response curve for inhibition of MMP-12. E) Chemical structure of compound MMP408 and relative dose response curve for inhibition of MMP-12.
Similar to compound 1, compounds 25 and 26 did not display significant inhibition of MMP-1, MMP-9, MMP-13, and MMP-14, while appreciable inhibition of the closely related MMP-3 was observed with both agents and with GM6001 (Figure 5).
Figure 5.
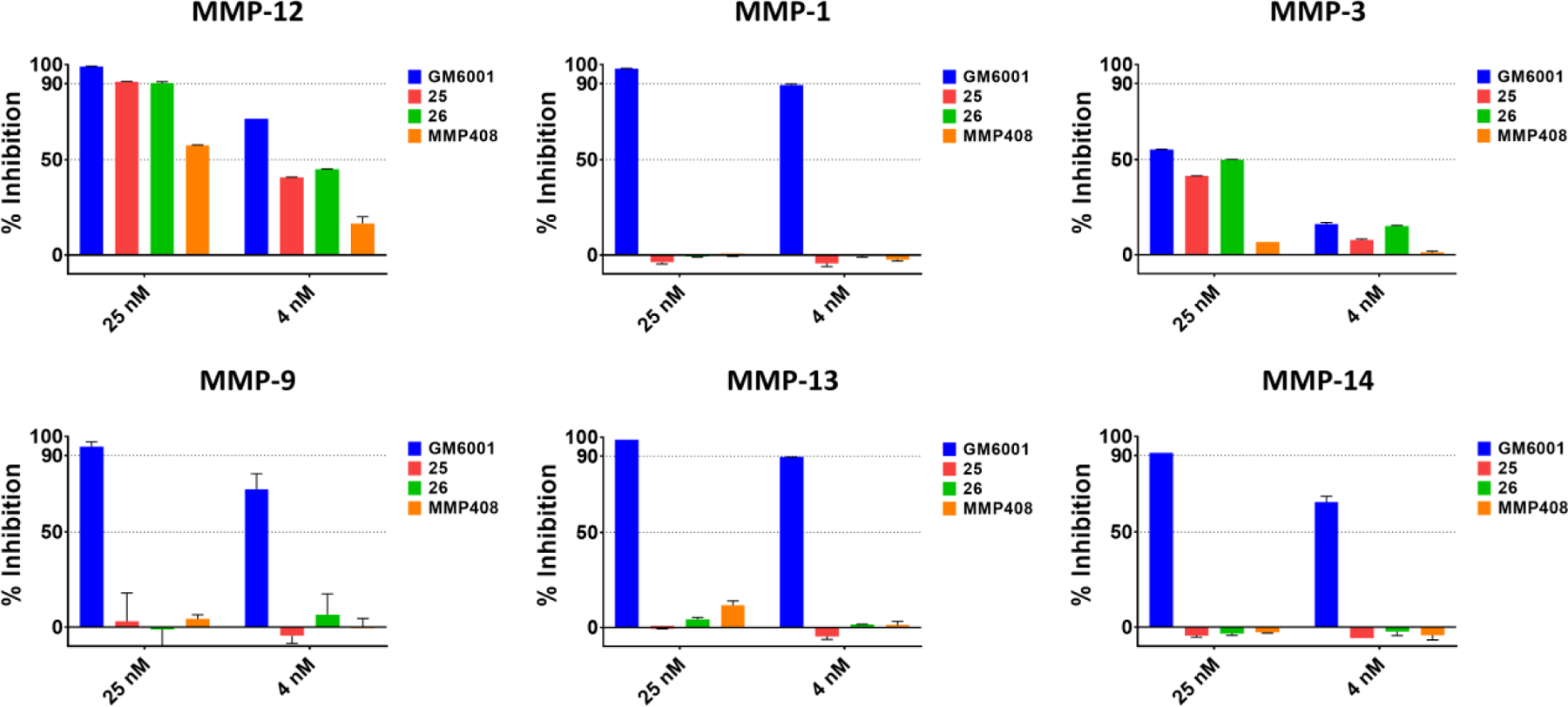
Selectivity assays for compounds 25, 26, GM6001, and MMP408. For each indicated enzyme, agents were tested at 25 nM or at 4 nM and percent inhibition was measured using the catalytic domains of the respective enzyme and the SensoLyte® 520 Fluorimetric Assay Kit (Anaspec).
Anticipating their use in in vivo efficacy studies, we further conducted a preliminary pharmacokinetic study with compound 25 (Figure 6A). When administered at 30 mg/kg intraperitoneally, we observed peak plasma drug concentration reaching ~ 400 ng/ml corresponding to ~ 580 nM, hence > 20 times above the in vitro determined IC90 value. Intriguingly, the chemical structures of compound 25 and 26 resemble the FDA-approved proteasome inhibitor carfilzomib (Figure 6A),20–25 with the notable difference that the metal chelating hydroxamate in our agents is a reactive epoxide in carfilzomib, that is necessary to irreversibly inactivate the proteasome. Hence, we can speculate that our agents, presenting a similar chemical structure but lacking the potential chemical liability of the epoxide present in carfilzomib, could also be useful as potential therapeutic agents. In summary, these SAR studies on hit compound 1, originally selected out of 100,000 molecules within the combinatorial library and the HTS by NMR approach,16 resulted in viable drug-like lead agents 25 and 26.
Figure 6. In vivo pharmacokinetics and docked structure of compound 25.

A) In vivo PK studies with compound 25 after i.p. administration (30 mg/kg) in Balb-C mice. Compound 25 plasma concentration was measured by LC/MS at the indicated time points after administration of the agent (formulation: 10% DMSO, 40% PEG400, and 10% (2-hydroxypropyl)-Beta-cyclodextrin in PBS; Cmax = 400 ng/ml; tmax = 2 hr; t1/2 ~ 5h). The chemical structure of compound 25 is displayed side by side with that of the FDA approved proteasome inhibitor carfilzomib. The chemical resemblance of the structures of compound 25 (and 26) with that of this FDA approved drug provides some confidence on the translational potential of our MMP-12 targeting agents. B) Docked geometry of compound 25 in complex with MMP-12. The docked structure is largely supported by the geometry of the metal chelating moiety that was used as constraint in GOLD (Cambridge Crystallographic Data Centre, https://www.ccdc.cam.ac.uk/) and the crystal structure of MMP-12 (PDB-ID 5LAB). Surface color scheme: green, lipophilic regions; pink, hydrophilic regions; white, neutral regions.
Efficacy of agents 25 and 26 in a murine model of elastase-induced emphysema
Recent studies have identified a role for MMP-12 in promoting the emphysematous lung tissue destruction associated with chronic obstructive pulmonary disease.9–10 To test the efficacy of compound 25 and compound 26 in an in vivo model system, we chose to investigate the impacts of pharmacological MMP-12 inhibition in a well-established murine model of elastase-induced emphysema. In this model, mice instilled with elastase exhibit early markers of inflammation and injury within hours to days following elastase treatment. While initial inflammatory cell influx and heightened airway cytokine release wane in the first week following elastase instillation, lung injury following elastase treatment evolves over several weeks, with dramatic tissue destruction evident within 3–4 weeks following elastase challenge. Using this model, we assessed the impacts of MMP-12 inhibition on the development of tissue destruction at 21 days following elastase treatment. As expected based on our chosen (21-day) time point, there was not a significant difference in total BALF cellularity/influx amongst all groups (Figure 7A), although differential cell analysis did identify a significant impact of elastase treatment on lymphocytes influx, as shown in Figure 7.
Figure 7. Effects of MMP-12 inhibition on lung lavage cellularity at 21 days post-PPE instillation.
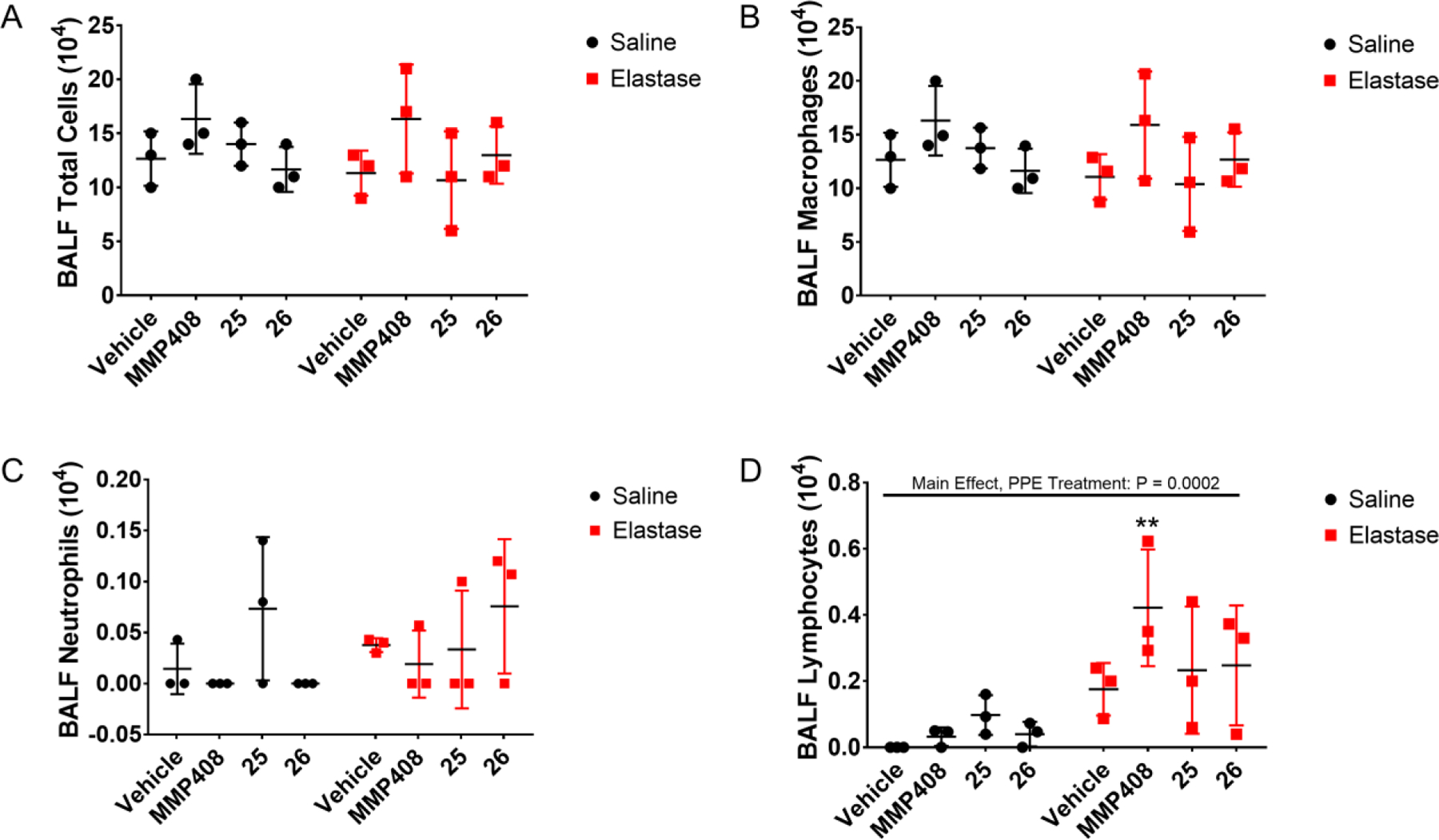
Mice were challenged with a single intranasal instillation of porcine pancreatic elastase, then treated daily for 7 days with vehicle control or MMP-12 inhibitors MMP408, compound 25, or compound 26 via intraperitoneal inoculation beginning 2 days following elastase treatment. Bronchoalveolar lavage was performed and fluid was assessed for total cells, and differential cell counts performed to enumerate individual cell populations. (A) Total BALF cells; (B) BALF macrophages; (C) BALF neutrophils; (D) BALF lymphocytes. ** P < 0.01 versus Vehicle + Saline control, based on two-way ANOVA with Tukey post-hoc comparisons. Significant two-way ANOVA main effects indicated at top of graph.
To assess for tissue destruction in saline- or elastase-treated animals, we performed mean linear intercept (MLI) analysis. Based on this analysis, we identified significant lung tissue destruction in murine lungs exposed to porcine pancreatic elastase (PPE) and treated with the vehicle control, compared to murine lungs exposed to the saline control and treated with the vehicle control (Figure 8). Notably, however, mice exposed to PPE but treated with MMP-12 inhibitors (MMP-408, compound 25, or compound 26) exhibited a significant decrease in emphysema-like pathology compared to PPE + vehicle-treated mice, with MLI measurements for mice treated with PPE + MMP-12 inhibitors exhibiting no significant differences compared to saline (no PPE)-treated mice (Figure 8).
Figure 8. Effects of MMP-12 inhibition on lung tissue destruction at 21 days post-PPE instillation.

Mice were challenged with a single intranasal instillation of porcine pancreatic elastase, then treated daily for 7 days with vehicle control or MMP-12 inhibitors MMP408, compound 25, or compound 26 via intraperitoneal inoculation beginning 2 days following elastase treatment. At day 21, lungs were inflated in formalin at 20 cm pressure, and tissue were paraffin-embedded, sectioned, and stained with hematoxylin and eosin. Mean linear intercept analyses were performed to assess the impact of MMP-12 inhibition on PPE-induced lung tissue destruction as measured by increased mean linear intercept. ** P < 0.01 versus Vehicle + Saline control, based on two-way ANOVA with Tukey post-hoc comparisons. Significant two-way ANOVA main effects indicated at top of graph.
Discussion and conclusions
The search for possible therapeutic applications of potent and selective metallo-enzyme inhibitors has remained very active in the past two decades. Small molecules with potentially favorable pharmacological properties have been developed,26 including the design of allosteric inhibitors27 or the deployment of novel metal chelating groups.28–32 MMP-12 is involved in the inflammatory response in chronic obstructive pulmonary disease (COPD) in mice,10 while in humans with asthma and COPD, MMP-12 aberrant activation is associated with disease severity.9 In a recent phase II trial, the dual MMP-12/MMP-9 inhibitor AZD1236 was tested in a randomized short trial (6 weeks) on moderate to severe COPD, and it showed an acceptable safety profile, although the therapeutic efficacy could not be demonstrated given the limited duration of the study.12 In addition, FP-025 (Forsee Pharmaceutical) reported on an ongoing phase II trial of their MMP-12 inhibitor, to assess its efficacy on allergen-induced airway inflammation in mild eosinophilic house dust mite allergic asthma (https://clinicaltrials.gov/ct2/show/NCT03858686). Another more recently reportedly potent and selective MMP-12 inhibitor is agent MMP408.11 However, when tested side by side with compounds 25, or 26, MMP408 exhibited significantly less potency than our agents that in turn are as potent as the pan-MMP inhibitor GM6001, yet displayed a greater selectivity for MMP-12. Following a preliminary pharmacokinetic study, compound 25 was administered in mice via intraperitoneal injections and displayed a favorable drug plasma levels and half-life. Interestingly, the chemical structures of optimized agents 25 and 26 structurally resemble the FDA-approved proteasome inhibitor carfilzomib (Figure 5), suggesting potentially a drug-like nature of the agents. Using a well-established animal model of elastase-induced emphysema, we found that mice exposed to porcine pancreatic elastase were protected by treatment with MMP-12 inhibitors (MMP-408, compound 25, or compound 26) from developing emphysema-like pathology compared to untreated mice, indicating that our agents have significant therapeutic potential. This study once again emphasizes the effectiveness of the HTS by NMR approach in deriving novel, potent and selective agents, particularly when an anchoring moiety can be incorporated in the positional scanning combinatorial library, as we have recently reported.13, 15–17, 33 The present SAR studies also further suggest that possible further optimizations of 25 or 26 could include o- and/or m-fluorination of the D-Phe in position P3.
In summary, we are confident that the identified agents represent innovative, potent and effective MMP-12 inhibitors, hence worthy of continued drug development for their potential translation in novel therapeutics for the treatment of MMP-12-mediated airway inflammatory conditions and potential other co-morbidities associated with COPD.
Experimental Section
General Chemistry.
All common solvent and reagents were obtained by commercial sources. NMR spectra were recorded on Bruker Avance III 700 MHz and these were used both for quality control and to verify the concentration of the stock solutions used for dose response measurements and in vivo studies. An Agilent LC-TOF instrument was used to obtain high-resolution mass spectral data. Purification of all agents was obtained using RP-HPLC on a JASCO preparative system equipped with a PDA detector. The instrument is also equipped with a fraction collector controlled by a ChromNAV system (JASCO). For all agents, a Luna C18 10μ 10 × 250mm (Phenomenex) column was used to purify agents to > 95% purity. For intermediate reagents that were not commercially available, RP-chromatography purification was performed using a CombiFlash (Teledyne ISCO). GM6001 was obtained from Enzo Life science. MMP408 was obtained from EMD Millipore Corp.
Peptide Synthesis
Peptides were synthesized by using standard solid-phase synthesis protocols, using an fmoc-hydroxylamine-2-chlorotrityl resin that introduces the hydroxamic acid at the C-terminus of the peptides, after cleavage. For each coupling reaction, 3 eq. of Fmoc-AA, 3 eq. of HATU, 3 eq. of OximaPure, and 5 eq. of DIPEA in 1 ml of DMF were used. The coupling reaction was allowed to proceed for 1 h. Fmoc deprotection was performed by treating the resin-bound peptide with 20% piperidine in DMF twice. Peptides were cleaved from Rink amide resin with a cleavage cocktail containing TFA/TIS/water (94:3:3) for 3 h. The cleaving solution was filtered from the resin, evaporated under reduced pressure and the peptides precipitated in Et2O, centrifuged and dried in high vacuum. The crude peptide was purified by preparative RP-HPLC using a Luna C18 column (Phenomenex) and water/acetonitrile gradient (30% to 70%) containing 0.1% TFA. The final compounds were characterized by HRMS.
For the synthesis of fmoc-amino acids, 1 eq. of the unprotected amino acid and Na2CO3 were dissolved in THF/H2O (1:1) and cooled to 0°C. 1.1 eq. of Fmoc Chloride was dissolved in THF and added dropwise to the mixture. The reaction was stirred for 2 h at 0°C. The organic solvent was evaporated under reduced pressure and the pH lowered to 0 using concentrated HCl. The aqueous phase was extracted 3 times with AcOEt and the collected organic phase were dried with Na2SO4, filtered and evaporated. The resulting crude was purified using a CombiFlash Rf (Teledyne ISCO) using cyclohexane/Ethyl Acetate (10% to 100%). Fmoc protection was required for 4-methoxy-D-homophenylalanine for the synthesis of both compounds 25, and 26; and for (S)-2-Amino-5-(4-methoxyphenyl)pentanoic acid for the synthesis of compound 26. Detailed experimental procedures and analytical data for compounds 25 and 26 are provided as supplementary information.
Protein expression and purification
The catalytic domain (Gly106-Gly263) of human macrophage metalloelastase (hMMP-12), was expressed by cloning the gene into a pET21 vector (Novagen) using NdeI and BamHI as restriction enzymes and then transfected into E. coli strain BL21 Codon Plus cells. Expression of uniformly 15N-labeled hMMP-12 in M9 minimal media containing 15 mM (15NH4)2SO4was induced with 0.5 mM IPTG at 37 °C for 4 h. Then protein forms inclusion bodies that were isolated and solubilized in a solution of 8 M urea (in 20 mM Tris–HCl, pH 8). The protein was purified in two steps including a first size-exclusion chromatography (Pharmacia HiLoad Superdex 75 16/60) in 6 M urea (in 50 mM sodium acetate). A second cation exchange purification step was carried using a Mono-S column (Pharmacia) and a sodium chloride linear gradient (from 0 to 500 mM). Protein refolding was accomplished by a multiple dialyses into decreasing concentrations of urea (from 4 M up to 2 M; 50 mM Tris–HCl, pH 7.2, 10 mM CaCl2, 0.1 mM ZnCl2, 300 mM NaCl). Further dialysis steps were performed exchanging the protein into the final buffer containing 20 mM Tris–HCl (pH 7.2), 10 mM CaCl2, 0.1 mM ZnCl2, 300 mM NaCl. The final buffer also contained 200 mM of acetohydroxamic acid (AHA), a weak MMP-12 inhibitor, to prevent the self-proteolysis. Recombinant MMP-1 (Cat. # AS-55575-1), MMP-3 (Cat. # 72006), MMP-9 (Cat. # AS-55576-1), MMP-13 (Cat. # AS-72257), and MMP-14 (Cat. # AS-72068) were obtained from AnaSpec.
Nuclear Magnetic Resonance
Solution Nuclear Magnetic Resonance (NMR) experiments were conducted on a 700 MHz Bruker Avance III spectrometer equipped with a TCI cryoprobe. Each samples contained 10 μM of 15N-labeled hMMP-12 catalytic domain in absence or in presence of 10 μM of each compounds, in 20 mM Tris-HCl pH 7.2, 300 mM NaCl, 200 mM acetohydroxamic acid (AHA), 10 mM CaCl2, and 0.1 mM ZnCl2 with 1% final d6-DMSO. For each sample 2D [15N, 1H] so-fast HMQC, and 1D 1H-aliph experiments were acquired. For ranking purpose were measured the ratio between the intensity of the bound peaks (IBND), and the intensity of the apo peaks (IAPO), for the aliphatic residues L212 and I220, that we called ΔI. For a value of ΔI > 1 and producing an effect greater than what observed with compound 1 we gave a rank of ++++, for a value of ΔI > 1 we gave a rank of +++, for a value of ΔI ~ 1 we gave a rank of ++, and for a value of ΔI < 1 we gave a rank of +.
Enzymatic assays
The enzymatic assays to profile the inhibitory effects of all the compounds on hMMP-12, and against a panel of 5 closely related MMPs (MMP-1, MMP-3, MMP-9, MMP-13, and MMP-14) were performed using the SensoLyte® 520 Fluorimetric Assay Kit (AnaSpec) for each MMPs. Assay kit for MMP-12 Cat. # AS-71157, MMP-1 Cat. # AS-71150, MMP-3 Cat. # AS-71152, MMP-9 Cat. # AS-71155, MMP-13 Cat. # AS-71156, and MMP-14 Cat. # AS-72025. The assay was performed according to their protocols. Briefly, 10 ng of each MMPs were incubated on a black flat-bottom 96-well plate (Cat. # 9502867) at RT for 15 minutes in absence or in presence of different concentration of each compound in a total volume of 50 μL. After 15 minutes, 50 μL of each substrate solution were added. The reagents were mixed together by shaking the plate gently for 30 sec. Immediately after the fluorescence was measured using the VICTOR X5 microplate reader (PerkinElmer) every 3 min for 60 min. For SAR purpose, IC50 values for agents in Table 1, 2, and 3, were extrapolated from % Inhibition of MMP-12 in presence of 25 nM of each agent. The IC50 values were calculated from dose-response curves using GraphPad Prism 7.
Molecular modeling
Compound 1, and 25 were docked using Gold (Cambridge Crystallographic Data Center; www.ccdc.cam.ac.uk) and Protein Data Bank entry 5LAB. The docking preparation for both protein and ligands were performed using MOE 2019.0101 (Chemical Computing Group). The surface representations were prepared using MOE 2019.0101 (Chemical Computing Group).
Pharmacokinetics studies
For these studies, compound 25 was administered i.p. (30 mg/kg) to 5 Balb-C mice. Retrorbital bleeding was used to collect blood samples at times 30 min, 1h, 2h, 4h, 8h, and 24h and the samples analyzed for compound 25 plasma concentration via extraction followed by LC/MS and compared to a standard calibration curve prepared with purified agent. Compound 25 was soluble in 10% DMSO, 40% PEG400, and 10% (2-hydroxypropyl)-Beta-cyclodextrin in PBS. Mice weight varied between 22 g and 26 g, each receiving approximately 200 μL (weight adjusted to administer 30 mg/Kg of compound 25) of formulated agent. The experiments were conducted at the University of California San Diego in vivo pharmacology core facility, according to a UCSD Institutional Animal Care and Use Committee (IACUC) approved protocol.
Murine Model
C57BL/6 mice were purchased from Jackson Laboratories (Bar Harbor, ME) and maintained in a pathogen-free vivarium at room temperature with 12-hour light/dark cycles. All mice were 6–8 weeks and both male and female mice were used for the experiment. All animal-use and euthanasia protocols were approved by the UC Riverside Institutional Animal Care and Use Committee (IACUC).
In Vivo Porcine Pancreatic Elastase Exposure
C57BL/6 mice were given a single intranasal instillation with 50 μl of saline or 0.9 U porcine pancreas elastase (Sigma-Aldrich, St. Louis, MO) diluted in phosphate buffered saline (1X PBS). Two days following the instillation, mice were treated with 200 μl of vehicle control, MMP-408, compound 25, or compound 26 via intraperitoneal injection. Mice were continually given treatment once a day for 7 consecutive days. Twenty-one days following the initial intranasal instillation, mice were sacrificed. Bronchoalveolar lavage fluid (BALF) was collected by making an incision in the trachea and inserting a cannula with syringe (BD Biosciences, CA), then washing with 1 mL of cold PBS three times. BALF was centrifuged at 1200 RPM for 5 minutes, then cell pellets were combined and resuspended in 200 μl of PBS to be used in cell counts and cell differential analysis. The lungs were isolated, slowly filled with 1 mL of 10% formalin then hung in formalin overnight for fixation. The following day, lungs were moved into 70% ethanol and stored at 4°C until paraffin-embedding.
Cell Counts and Cell Differentials
Cell pellets were resuspended in 200 μl of PBS, then 10 μl of the solution was put onto a hemocytometer for total cell counts. For cell differentials, 150 μl of the suspended cell solution was put on a microscope slide using a cytospin. Using a Revolve light microscope (La Jolla, CA), a total of 300 cells were counted per slide, and were differentiated by cell type. The number of each cell type identified was then divided by 300 to get the percentage of each cell type amongst the total cell population, then that percentage was multiplied by the total cell count to represent the total number of each cell type in the BALF.
Mean Linear Intercept (MLI) Analysis
Mouse lungs were fixed in formalin and paraffin-embedded, then sectioned and stained in hematoxylin and eosin by the University of California Irvine Department of Pathology Experimental Tissue Resource Core Facility. The lungs were imaged on a Revolve light microscope (La Jolla, CA) at 10x magnification. MLI measurements were calculated using an established indirect method on ImageJ software. Briefly, alveolar walls/intersections were counted along a line of known length, then length was divided by the number of intersections to calculate MLI. This process was repeated for a total of five MLI measurements per image, with there being four images per mouse lung. The average MLI measurement for each lung was calculated and statistically analyzed using GraphPad Prism software (San Diego, CA).
Supplementary Material
Acknowledgements
Financial support was obtained in part by the NIH grants NS107479, CA168517, CA242620 (to MP), R00ES025819 (to TMN), and by a City of Hope/UCR (CUBRI) research grant (to MP). MP holds the Daniel Hays Chair in Cancer Research at the School of Medicine at UCR. The authors declare no conflict of interest. Our agents can be distributed in small amounts (1-5 mg) for research purposes upon request and signing of a standard material transfer agreement.
Abbreviations used
- COPD
Chronic obstructive pulmonary disease
- MLI
Mean Linear Intercept
- MMP
Matrix metalloproteinase
- PPE
porcine pancreatic elastase
Footnotes
Supporting Information: The Supporting Information is available free of charge on the ACS Publications website. Figure S1 reports analytical data on compounds; Figure S2, reports NMR spectral data used to rank order agents in Tables 1, 2, and 3; HRMS data for agents is also provided. Coordinates for the docked geometries reported in the manuscript are also included. Molecular Formula Strings file is also provided.
The authors declare no competing financial interest
References
- 1.Maselli DJ; Bhatt SP; Anzueto A; Bowler RP; DeMeo DL; Diaz AA; Dransfield MT; Fawzy A; Foreman MG; Hanania NA; Hersh CP; Kim V; Kinney GL; Putcha N; Wan ES; Wells JM; Westney GE; Young KA; Silverman EK; Han MK; Make BJ, Clinical Epidemiology of COPD: Insights From 10 Years of the COPDGene Study. Chest 2019, 156 (2), 228–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Andre S; Conde B; Fragoso E; Boleo-Tome JP; Areias V; Cardoso J; Cronica, G. D.-G. d. I. n. D. P. O., COPD and Cardiovascular Disease. Pulmonology 2019, 25 (3), 168–176. [DOI] [PubMed] [Google Scholar]
- 3.Caramori G; Ruggeri P; Mumby S; Ieni A; Lo Bello F; Chimankar V; Donovan C; Ando F; Nucera F; Coppolino I; Tuccari G; Hansbro PM; Adcock IM, Molecular links between COPD and lung cancer: new targets for drug discovery? Expert Opin Ther Targets 2019, 23 (6), 539–553. [DOI] [PubMed] [Google Scholar]
- 4.Pires N; Pinto P; Marcal N; Ferreira AJ; Rodrigues C; Barbara C; Disease, G. D. I. G. o. C. O. P., Pharmacological treatment of COPD - New evidence. Pulmonology 2019, 25 (2), 90–96. [DOI] [PubMed] [Google Scholar]
- 5.Lopez-Campos JL; Quintana Gallego E; Carrasco Hernandez L, Status of and strategies for improving adherence to COPD treatment. Int J Chron Obstruct Pulmon Dis 2019, 14, 1503–1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kisialiou A; Prinzi G; Lamonaca P; Cardaci V; Tomino C; Fini M; Bonassi S; Russo P, Pharmacological Management of Chronic Obstructive Lung Disease (COPD). Evidence from a Real-World Perspective - Part 2. Curr Med Chem 2019, 26 (10), 1734–1745. [DOI] [PubMed] [Google Scholar]
- 7.Franssen FM; Alter P; Bar N; Benedikter BJ; Iurato S; Maier D; Maxheim M; Roessler FK; Spruit MA; Vogelmeier CF; Wouters EF; Schmeck B, Personalized medicine for patients with COPD: where are we? Int J Chron Obstruct Pulmon Dis 2019, 14, 1465–1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cazzola M; Rogliani P; Stolz D; Matera MG, Pharmacological treatment and current controversies in COPD. F1000Res 2019, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Morita H; Nagai R, MMP12, lung function, and COPD in high-risk populations. N Engl J Med 2010, 362 (13), 1241; author reply 1242. [PubMed] [Google Scholar]
- 10.Le Quement C; Guenon I; Gillon JY; Valenca S; Cayron-Elizondo V; Lagente V; Boichot E, The selective MMP-12 inhibitor, AS111793 reduces airway inflammation in mice exposed to cigarette smoke. Br J Pharmacol 2008, 154 (6), 1206–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li W; Li J; Wu Y; Wu J; Hotchandani R; Cunningham K; McFadyen I; Bard J; Morgan P; Schlerman F; Xu X; Tam S; Goldman SJ; Williams C; Sypek J; Mansour TS, A selective matrix metalloprotease 12 inhibitor for potential treatment of chronic obstructive pulmonary disease (COPD): discovery of (S)-2-(8-(methoxycarbonylamino)dibenzo[b,d]furan-3-sulfonamido)-3-methylbutanoic acid (MMP408). J Med Chem 2009, 52 (7), 1799–1802. [DOI] [PubMed] [Google Scholar]
- 12.Magnussen H; Watz H; Kirsten A; Wang M; Wray H; Samuelsson V; Mo J; Kay R, Safety and tolerability of an oral MMP-9 and −12 inhibitor, AZD1236, in patients with moderate-to-severe COPD: a randomised controlled 6-week trial. Pulm Pharmacol Ther 2011, 24 (5), 563–570. [DOI] [PubMed] [Google Scholar]
- 13.Wu B; Barile E; De SK; Wei J; Purves A; Pellecchia M, High-Throughput Screening by Nuclear Magnetic Resonance (HTS by NMR) for the Identification of PPIs Antagonists. Curr Top Med Chem 2015, 15 (20), 2032–2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu B; Zhang Z; Noberini R; Barile E; Giulianotti M; Pinilla C; Houghten RA; Pasquale EB; Pellecchia M, HTS by NMR of combinatorial libraries: a fragment-based approach to ligand discovery. Chem Biol 2013, 20 (1), 19–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bottini A; Wu B; Barile E; De SK; Leone M; Pellecchia M, High-Throughput Screening (HTS) by NMR Guided Identification of Novel Agents Targeting the Protein Docking Domain of YopH. ChemMedChem 2016, 11 (8), 919–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baggio C; Cerofolini L; Fragai M; Luchinat C; Pellecchia M, HTS by NMR for the Identification of Potent and Selective Inhibitors of Metalloenzymes. ACS Med Chem Lett 2018, 9 (2), 137–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu B; Zhang Z; Noberini R; Barile E; Giulianotti M; Pinilla C; Houghten RA; Pasquale EB; Pellecchia M, HTS by NMR of combinatorial libraries: a fragment-based approach to ligand discovery. Chem Biol 2013, 20 (1), 19–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Barile E; Pellecchia M, NMR-based approaches for the identification and optimization of inhibitors of protein-protein interactions. Chem Rev 2014, 114 (9), 4749–4763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang JW; Zhang Z; Wu B; Cellitti JF; Zhang X; Dahl R; Shiau CW; Welsh K; Emdadi A; Stebbins JL; Reed JC; Pellecchia M, Fragment-based design of small molecule X-linked inhibitor of apoptosis protein inhibitors. J Med Chem 2008, 51 (22), 7111–7118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kuhn DJ; Chen Q; Voorhees PM; Strader JS; Shenk KD; Sun CM; Demo SD; Bennett MK; van Leeuwen FW; Chanan-Khan AA; Orlowski RZ, Potent activity of carfilzomib, a novel, irreversible inhibitor of the ubiquitin-proteasome pathway, against preclinical models of multiple myeloma. Blood 2007, 110 (9), 3281–3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang Z; Yang J; Kirk C; Fang Y; Alsina M; Badros A; Papadopoulos K; Wong A; Woo T; Bomba D; Li J; Infante JR, Clinical pharmacokinetics, metabolism, and drug-drug interaction of carfilzomib. Drug Metab Dispos 2013, 41 (1), 230–237. [DOI] [PubMed] [Google Scholar]
- 22.Herndon TM; Deisseroth A; Kaminskas E; Kane RC; Koti KM; Rothmann MD; Habtemariam B; Bullock J; Bray JD; Hawes J; Palmby TR; Jee J; Adams W; Mahayni H; Brown J; Dorantes A; Sridhara R; Farrell AT; Pazdur R, U.s. Food and Drug Administration approval: carfilzomib for the treatment of multiple myeloma. Clin Cancer Res 2013, 19 (17), 4559–4563. [DOI] [PubMed] [Google Scholar]
- 23.Yang J; Wang Z; Fang Y; Jiang J; Zhao F; Wong H; Bennett MK; Molineaux CJ; Kirk CJ, Pharmacokinetics, pharmacodynamics, metabolism, distribution, and excretion of carfilzomib in rats. Drug Metab Dispos 2011, 39 (10), 1873–1882. [DOI] [PubMed] [Google Scholar]
- 24.Khan ML; Stewart AK, Carfilzomib: a novel second-generation proteasome inhibitor. Future Oncol 2011, 7 (5), 607–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.O’Connor OA; Stewart AK; Vallone M; Molineaux CJ; Kunkel LA; Gerecitano JF; Orlowski RZ, A phase 1 dose escalation study of the safety and pharmacokinetics of the novel proteasome inhibitor carfilzomib (PR-171) in patients with hematologic malignancies. Clin Cancer Res 2009, 15 (22), 7085–7091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Amar S; Fields GB, Potential clinical implications of recent matrix metalloproteinase inhibitor design strategies. Expert Rev Proteomics 2015, 12 (5), 445–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Remacle AG; Golubkov VS; Shiryaev SA; Dahl R; Stebbins JL; Chernov AV; Cheltsov AV; Pellecchia M; Strongin AY, Novel MT1-MMP small-molecule inhibitors based on insights into hemopexin domain function in tumor growth. Cancer Res 2012, 72 (9), 2339–2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Agrawal A; Johnson SL; Jacobsen JA; Miller MT; Chen LH; Pellecchia M; Cohen SM, Chelator fragment libraries for targeting metalloproteinases. ChemMedChem 2010, 5 (2), 195–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Johnson S; Barile E; Farina B; Purves A; Wei J; Chen LH; Shiryaev S; Zhang Z; Rodionova I; Agrawal A; Cohen SM; Osterman A; Strongin A; Pellecchia M, Targeting metalloproteins by fragment-based lead discovery. Chem Biol Drug Des 2011, 78 (2), 211–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Martin DP; Blachly PG; Marts AR; Woodruff TM; de Oliveira CA; McCammon JA; Tierney DL; Cohen SM, ‘Unconventional’ coordination chemistry by metal chelating fragments in a metalloprotein active site. J Am Chem Soc 2014, 136 (14), 5400–5406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fullagar JL; Garner AL; Struss AK; Day JA; Martin DP; Yu J; Cai X; Janda KD; Cohen SM, Antagonism of a zinc metalloprotease using a unique metal-chelating scaffold: tropolones as inhibitors of P. aeruginosa elastase. Chem Commun (Camb) 2013, 49 (31), 3197–3199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tanakit A; Rouffet M; Martin DP; Cohen SM, Investigating chelating sulfonamides and their use in metalloproteinase inhibitors. Dalton Trans 2012, 41 (21), 6507–6515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Baggio C; Udompholkul P; Barile E; Pellecchia M, Enthalpy-Based Screening of Focused Combinatorial Libraries for the Identification of Potent and Selective Ligands. ACS Chem Biol 2017, 12 (12), 2981–2989. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.