

The maintenance of immune equilibrium in the oral cavity, a mucosal surface highly colonized by microorganisms, is a complex process that is largely mediated by the activation and regulated recruitment of leukocytes into gingival tissues. Neutrophils are by far the most abundant leukocyte subset in healthy gingiva and are critical for host defense against oral microorganisms. Circulating neutrophils extravasate from mucogingival blood supply and sub-epithelial capillary loops, and traffic via the junctional epithelium until they reach the gingival sulcus where they form a “defensive wall” against oral biofilms. Neutrophils phagocytose bacteria, release antimicrobial peptides, proteases, oxidants, and inflammatory mediators that protect against bacterial intrusion. Factors that dysregulate neutrophil effector functions, trafficking, or accrual within periodontal tissues significantly enhance susceptibility to periodontal diseases and gingival inflammation. Periodontitis is a dysbiotic chronic inflammatory disease that affects the hard and soft tissues of the periodontium (alveolar bone and gingiva, respectively). Early onset periodontitis and severe gingivitis are highly prevalent in congenital disorders such as leukocyte adhesion deficiency (LAD), Chediak–Higashi syndrome, neutropenias, and chronic granulomatous disease (CGD). However, despite strong clinical association, how neutrophil disorders precipitate into oral pathologies is incompletely understood.1,2
Leucocyte adhesion deficiency Type I (LAD-1) is a rare primary immunodeficiency caused by autosomal recessive mutations in the ITGB2 gene that encodes the CD18 subunit of β2 integrins. The β2 integrin subfamily members consist of heterodimeric receptors formed by dimerization of the CD18β chain with distinct alpha (α) chains. These include LFA1 (CD11a: CD18), complement receptor 3 (CR-3) or Mac1 (CD11b: CD18), complement receptor 4 (CD11c: CD18), and the αDβ2 receptor (CD11d:CD18)). The leukocyte adhesion cascade is a tightly regulated, multi-step process that requires β2 integrins for firm adhesion to the endothelial layer and subsequent leukocyte extravasation. Neutrophils constitutively express high levels of Mac1 and LFA1 that enable binding to endothelial intercellular cell adhesion molecules (ICAM-1 and ICAM-2). Binding is essential for slow rolling and firm adhesion of leukocytes to endothelial cells and the transmigration of neutrophils, into inflamed tissues.3 Loss of β2 integrins results in a failure of neutrophils to emigrate out of blood vessels into peripheral tissues or sites of infection, despite increased numbers in blood. Consequently, LAD-1 patients have significant neutrophilia and present with recurrent life threatening bacterial and fungal infections, with absent neutrophil infiltration and pus. Inflammatory complications of the skin and other mucosal surfaces are also common. Early-onset gingivitis and periodontitis are typical in LAD-1, with generalized alveolar bone recession, acute gingival inflammation, and premature tooth loss. Although severe periodontitis in LAD-1 patients has historically been attributed to compromised neutrophil surveillance, recent studies in both, LAD-1 patients, and relevant mouse models (CD18−/− mice) challenge this notion.2,4 For instance, histiological analysis of gingival tissues from LAD-1 patients showed significant inflammatory infiltrates with a marked absence of neutrophils.2,4 Furthermore, treatment regimens that normally alleviate gingival inflammation in periodontal patients from the general population, such as antibiotic treatment and mechanical removal of oral biofilms, are of limited therapeutic efficacy in LAD-1 patients.4,5
Neutrophil production in the bone marrow and their eventual clearance in the periphery are intricately linked through an essential feedback loop, or “neutrostat.” Stark et al. previously demonstrated that neutrophil numbers in the periphery are maintained by feedback signals that emanate from their phagocytic clearance. Transmigrated neutrophils undergo apoptosis in the peripheral tissues and are ingested by tissue phagocytes in a manner that suppresses the IL-23/IL-17 axis.6 IL-23 is a pro-inflammatory cytokine produced at mucosal surfaces or inflamed sites by tissue Mϕs and dendritic cells, and is essential for the differentiation of Th-17 lymphocytes. Th-17 cells and IL-17 mediate bone resorption by secreting osteoclastogenic cytokines and stimulate granulopoiesis by up-regulating granulocyte colony-stimulating factor (G-CSF) levels. ‘Efferocytosis’ or phagocytic uptake of apoptotic neutrophils blocks IL-23 production and inhibits IL-23 induced IL-17 production, thus regulating neutrophil numbers in the periphery.6 Hajishengallis and colleagues were the first to demonstrate that the paucity of neutrophils in the periodontal tissues of LAD-1 patients and relevant mouse models resulted in the breakdown of efferocytosis mediated inhibition of IL-23/IL-17 axis in oral tissues.5 Gingival tissue specimens from LAD-1 patients had significantly higher numbers of IL-17 producing T cells that correlated with increased IL-23/IL-17 cytokine signatures.5 Consistent with this, blockade of IL-23 in a single LAD-1 patient using ustekinumab, which blocks the p40 subunit common to IL-12 and IL-23, was successful in ameliorating immunopathological complications and gingival inflammation in this patient.7 In their new manuscript, Kajikawa et al.,8 Hajishengallis and colleagues explore the therapeutic efficacy of synthetic agonists that mimic efferocytosis-associated signaling pathways in ameliorating periodontal inflammation.
Efferocytosis also actively programs resolution of inflammation by the activation of tolerogenic and anti-inflammatory pathways. Defective efferocytosis due to genetic deletion of efferocytic receptors, impaired processing of ingested cells, or microbial manipulation of efferocytic pathways is associated with chronic inflammation, autoimmunity and progressive pathological outcomes.9 Efferosomal processing of an ingested apoptotic cell is a multistep step process. It involves activation of the oxidative burst, formation and acidification of the phagolysosome, and subsequent breakdown of the apoptotic cargo by degradative enzymes.10 Phagolysosomal degradation also generates large amounts of macromolecules such as nucleic acids, lipids, sterols, and fatty acids that need further processing to be metabolically consumed or secreted. Apoptotic cell derived sterols have been shown to activate nuclear receptors such as the liver-X receptor α/β (LXR) and the peroxisome proliferator-activated receptor (PPAR-γ) families. Activation of LXR and PPAR-γ signaling enforces reprograming of efferocytosing Mϕs into phenotypes that promote tissue resolution, by down-regulating pro-inflammatory cytokines, nicotinamide adenine dinucleotide phosphate (NADPH) oxidase activity, and inhibition of inducible nitric oxide synthase (iNOS). This phenotypic transition of Mϕs promotes tissue resolution by the generation of various anti-inflammatory cytokines, lipid mediators, and also up-regulates the expression of efferocytic receptors.9 To restore absent efferocytosis induced anti-inflammatory signals in CD18−/− mice, Kajikawa et al. locally administered synthetic LXR and PPAR agonists. Localized injection of GW3965 (LXR agonist) and GW0742 (PPAR-β/γ) agonists directly into mouse gingiva of CD18−/− mice reduced alveolar bone loss (Figure 1). The combination of these agonist was also efficacious in down-regulating the expression of IL-23/IL-17 and downstream cytokines that modulate neutrophil mobilization (G-CSF) and recruitment into inflamed tissues (CXCL1). Interestingly, the levels of pro-inflammatory cytokines TNF and IL-1β, typically associated with periodontal inflammation and other chronic inflammatory conditions, were not significantly altered on treatment with LXR and PPAR agonists. Instead levels of phosphorylated signal transducer and activator of transcription 3 (STAT-3), an important transcription factor for Th17 development, and IL-17 production were preferentially downregulated. These observations reinstate the importance of IL-23/IL-17 induced inflammatory dysregulation in LAD-1. Thus, Kajikawa et al.8 provide experimental evidence and rationale for the use of LXR and PPAR agonists as therapeutic alternatives to ustekinumab therapy in LAD-1.
FIGURE 1: LXR/RXR activation suppresses IL-23/IL-17 mediated gingival inflammation and alveolar bone loss:
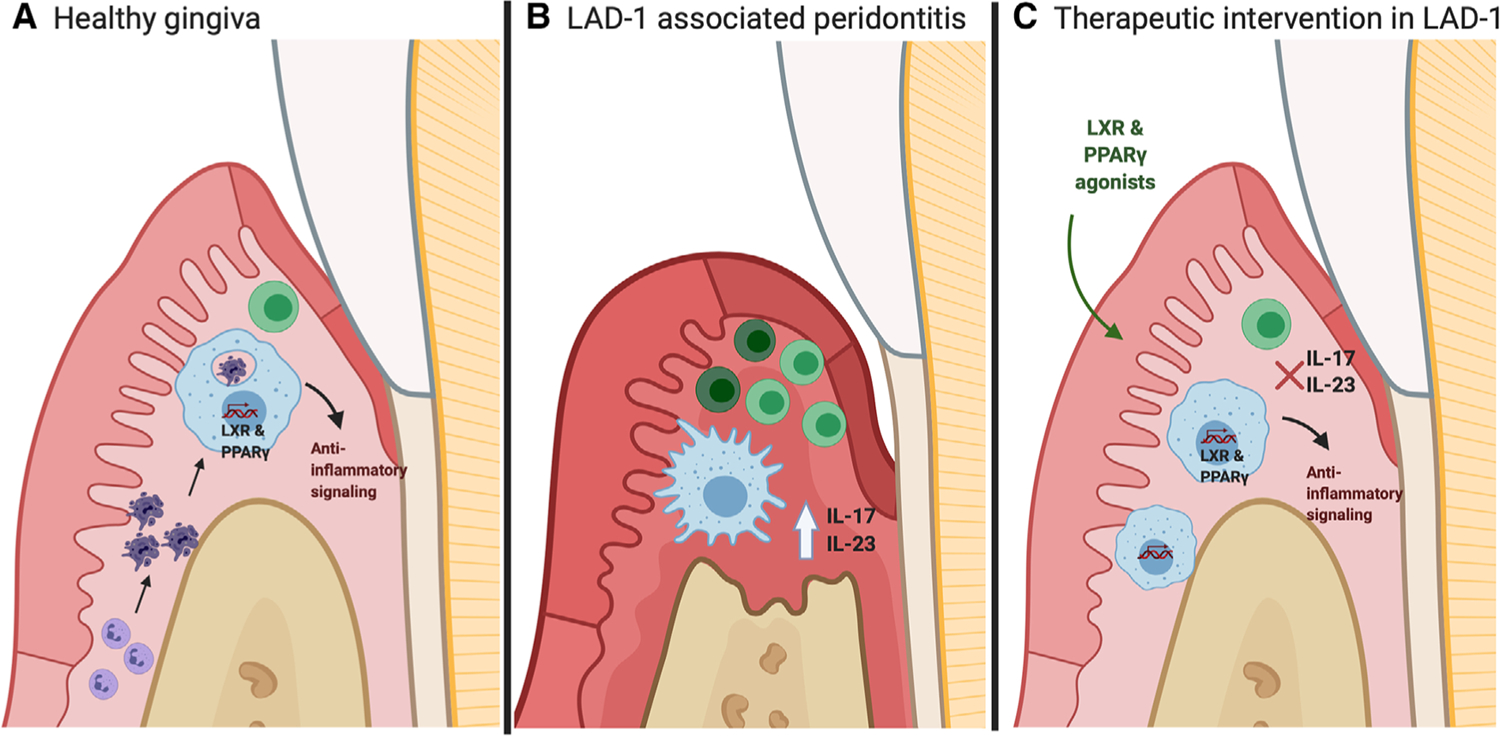
(A) Efferocytosis of apoptotic neutrophils by tissue Mϕs/dendritic cells activates LXR/RXR dependent anti-inflammatory mediators, suppressing IL-23 levels in the gingiva. (B) Genetic deletion of CD18 results in dramatic loss of neutrophil transmigration into gingival tissues, disrupting efferocytosis induced feedback inhibition of the IL-23/IL-17 axis. (C) Intra-gingival administration of LXR and PPAR agonists induces the expression of LXR/RXR dependent genes, consequently suppressing IL-23/IL-17 mediated alveolar bone loss and gingival inflammation
REFERENCES
- 1.Zeng MY, Miralda I, Armstrong CL, Uriarte SM, Bagaitkar J. The roles of NADPH oxidase in modulating neutrophil effector responses. Mol Oral Microbiol. 2019;34(2):27–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dinauer MC. Inflammatory consequences of inherited disorders affecting neutrophil function. Blood. 2019;133(20):2130–2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol. 2013;13(3):59–75. [DOI] [PubMed] [Google Scholar]
- 4.Deas DE, Mackey SA, McDonnell HT. Systemic disease and periodontitis: manifestations of neutrophil dysfunction. Periodontol 2000. 2003;32:82–104. [DOI] [PubMed] [Google Scholar]
- 5.Moutsopoulos NM, Konkel J, Sarmadi M, et al. Defective neutrophil recruitment in leukocyte adhesion deficiency type I disease causes local IL-17-driven inflammatory bone loss. Sci Transl Med. 2014;6(229):229ra40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stark MA, Huo Y, Burcin TL, et al. Phagocytosis of apoptotic neutrophils regulates granulopoiesis via IL-23 and IL-17. Immunity. 2005;22(3):285–294. [DOI] [PubMed] [Google Scholar]
- 7.Moutsopoulos NM, Zerbe CS, Wild T, et al. Interleukin-12 and interleukin-23 blockade in leukocyte adhesion deficiency type 1. N Engl J Med. 2017;376(12):1141–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kajikawa T, Wang B, Li X, et al. Frontline science: activation of metabolic nuclear receptors restores periodontal tissue homeostasis in mice with leukocyte adhesion deficiency-1. J Leukoc Biol. 2020;108:1501–1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Doran AC, Yurdagul A Jr, Tabas I. Efferocytosis in health and disease. Nat Rev Immunol. 2020;20:254–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bagaitkar J, Huang J, Zeng MJ, et al. NADPH oxidase activation regulates apoptotic neutrophil clearance by murine macrophages. Blood. 2018;131(21):2367–2378. [DOI] [PMC free article] [PubMed] [Google Scholar]