

ABSTRACT
Bacteriophages (phages) are ubiquitous in nature. These viruses play a number of central roles in microbial ecology and evolution by, for instance, promoting horizontal gene transfer (HGT) among bacterial species. The ability of phages to mediate HGT through transduction has been widely exploited as an experimental tool for the genetic study of bacteria. As such, bacteriophage P1 represents a prototypical generalized transducing phage with a broad host range that has been extensively employed in the genetic manipulation of Escherichia coli and a number of other model bacterial species. Here we demonstrate that P1 is capable of infecting, lysogenizing, and promoting transduction in members of the bacterial genus Sodalis, including the maternally inherited insect endosymbiont Sodalis glossinidius. While establishing new tools for the genetic study of these bacterial species, our results suggest that P1 may be used to deliver DNA to many Gram-negative endosymbionts in their insect host, thereby circumventing a culturing requirement to genetically manipulate these organisms.
IMPORTANCE A large number of economically important insects maintain intimate associations with maternally inherited endosymbiotic bacteria. Due to the inherent nature of these associations, insect endosymbionts cannot be usually isolated in pure culture or genetically manipulated. Here we use a broad-host-range bacteriophage to deliver exogenous DNA to an insect endosymbiont and a closely related free-living species. Our results suggest that broad-host-range bacteriophages can be used to genetically alter insect endosymbionts in their insect host and, as a result, bypass a culturing requirement to genetically alter these bacteria.
KEYWORDS: Sodalis praecaptivus, Sodalis glossinidius, insect endosymbiont, symbiont, transformation, transduction, genetic modification, plasmid transfer, transposition, bacteriophage P1, gene disruption, mutation, paratransgenesis, bacteriophage transduction, symbiosis
INTRODUCTION
Bacteriophages (phages) are the most abundant and diverse biological entities on the planet. With an estimated population size greater than 1 × 1031 (1), these bacterial viruses play essential ecological and evolutionary functions. Phages control the size of bacterial populations and shape the diversity of microbial communities by modulating the abundance of bacterial lineages and promoting, directly and indirectly, the exchange of genetic information among species (2, 3). Historically, phages have played a central role in the development of molecular biology, enabling, for instance, the establishment of DNA as the genetic material of living cells (4). Today, phages are widely used as tools in the study of bacteria. For instance, generalized transducing phages such as P1 allow the rapid transfer of DNA among bacterial strains, greatly facilitating genetic dissection of biological processes (5).
P1 is a temperate bacteriophage capable of alternating between lytic and lysogenic infection. P1 was initially described in studies involving lysogenic strains of Escherichia coli (6). This phage is capable of mediating generalized transduction (7), a property that has fostered its adoption as an important experimental tool for the genetic analysis and manipulation of E. coli (5, 8). Notably, in addition to its habitual E. coli host, P1 can also infect a large number of Gram-negative bacterial species (8–12). This broad host range, along with its well-characterized molecular biology and established experimental procedures, has prompted the use of this phage as an experimental tool for the delivery of DNA to a large number of bacterial species (13–18). Here we establish that P1 is capable of infecting two members of the bacterial genus Sodalis, including Sodalis glossinidius (19, 20).
Sodalis glossinidius is a maternally inherited, Gram-negative bacterial endosymbiont of tsetse flies (Glossina spp.; Diptera: Glossinidae). Similar to other insect endosymbionts, S. glossinidius exists in a stable, chronic association with its insect host and undergoes a predominantly maternal mode of transmission (21–23). Notably, like other insect endosymbionts, this bacterium has undergone an extensive process of genome degeneration as a result of a recent ecological transition from free-living existence to permanent host association (24, 25). Because this process is accompanied by the loss of metabolic capability and stress response pathways (24–29), S. glossinidius has proven refractory to harsh artificial DNA transformation procedures that are commonly employed in model organisms such as Escherichia coli (29). Consequently, this bacterium has remained genetically intractable (30).
In this study, we demonstrate that the bacteriophage P1 is capable of infecting, lysogenizing, and promoting transduction in Sodalis glossinidius, and its free-living close relative, the plant-associated and opportunistic pathogen Sodalis praecaptivus (19, 20). We demonstrate that P1 can be used to mediate generalized transduction of chromosomal and extrachromosomal DNA in S. praecaptivus. We use P1 to transduce autonomous replicating phagemids containing an array of reporter genes and Tn7 transposition systems harboring fluorescent proteins for chromosomal tagging. Finally, we developed a suicide phagemid containing a mariner transposase for random mutagenesis of bacterial strains susceptible to P1 infection. This study establishes a new efficient method for genetic manipulation of Sodalis species (Fig. 1) that can be readily adapted to other Gram-negative bacteria. Furthermore, these results provide a potential means for the genetic modification of bacterial endosymbionts, in their insect host, through the use of P1 as a DNA delivery system.
FIG 1.

Cartoon representation depicting a workflow of the transduction procedure developed for introduction of phagemids in Sodalis species. Following the direction of the arrows, an E. coli P1 lysogen host is transformed with a P1 phagemid. The phagemid is packaged following induction of the P1 prophage, and lysates derived from culture supernatant are used to infect a Sodalis recipient strain. Cells receiving the phagemid are subsequently isolated on plates containing a selective agent.
RESULTS
Bacteriophage P1 infects, lysogenizes, and forms phage particles in Sodalis glossinidius and Sodalis praecaptivus.
P1CMclr-100(ts) is a thermo-inducible P1 variant harboring a chloramphenicol-resistant marker. P1CMclr-100(ts) forms chloramphenicol-resistant lysogens at low temperatures (≤30°C) but produces phage particles at higher temperatures (≥37°C) (31). Consequently, infection of E. coli by P1CMclr-100(ts) yields chloramphenicol-resistant lysogens at 30°C. We took advantage of these P1CMclr-100(ts) properties to test whether S. glossinidius and S. praecaptivus were susceptible to P1 infection. We exposed cultures of these bacteria to increasing concentrations of P1CMclr-100(ts) phage particles and subsequently plated dilutions on solid medium containing chloramphenicol. We established that, similar to the E. coli control (Fig. 2A), exposure to increasing concentrations of P1CMclr-100(ts) particles yielded increasing numbers of chloramphenicol-resistant colonies in both S. glossinidius (Fig. 2B) and S. praecaptivus (Fig. 2C). Importantly, no chloramphenicol-resistant colonies were observed in cultures that were not exposed to P1CMclr-100(ts) particles (Fig. 2B and C).
FIG 2.
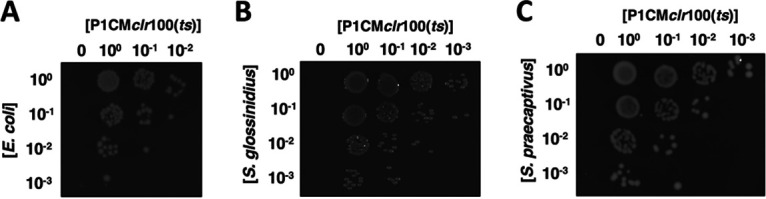
Infection of bacterial strains by phage P1. Lysates derived from an E. coli P1CMclr-100(ts) lysogen (KL463) were used to infect E. coli MG1655 (A), Sodalis glossinidius (B), and Sodalis praecaptivus (C). Plates depict the formation of chloramphenicol-resistant colonies as functions of the concentration of bacteria (vertical axis) and the concentration of P1CMclr-100(ts) lysates (horizontal axis). Note that P1 infection conditions for the strains are different (see Materials and Methods), and images do not reflect efficiency of P1 infection. Images show representative plates of at least three routine experiments.
That these colonies were P1 lysogens, as opposed to recombinants harboring only the P1-derived chloramphenicol-resistant marker, was supported by several lines of evidence. First, the presence of a P1 DNA fragment was detected by polymerase chain reaction (PCR) in both chloramphenicol-resistant S. glossinidius (Fig. 3A and B) and S. praecaptivus clones (Fig. 3C and D), but not in the wild-type strains (Fig. 3A to D, left side). This indicated that chloramphenicol-resistant cells harbor at least part of the P1CMclr-100(ts) genome. Second, lysates prepared from S. glossinidius and S. praecaptivus chloramphenicol-resistant clones, but not their wild-type counterparts, formed plaques in soft agar cultures of E. coli grown at 37°C, a temperature that induces P1CMclr-100(ts) lytic replication (31) (Fig. 3E and F). This indicated that chloramphenicol-resistant S. glossinidius and S. praecaptivus clones can produce phage particles that are lytic to E. coli grown at 37°C. Third, the lytic activity of lysates derived from chloramphenicol-resistant S. praecaptivus cultures propagated at 37°C was 10,000 times higher than those maintained at 30°C (Fig. 3G). This established that higher titers of phage particles were being produced in S. praecaptivus chloramphenicol-resistant clones at a temperature where P1CMclr-100(ts) becomes lytic. Finally, lysates derived from S. glossinidius and S. praecaptivus chloramphenicol-resistant clones, but not their wild-type isogenic counterparts, promoted the formation of chloramphenicol-resistant E. coli cells at 30°C (Fig. 3H). This indicated that the chloramphenicol-resistant marker can be transduced from S. glossinidius and S. praecaptivus back to E. coli.
FIG 3.

Lysogenization and production of infective phage particles by Sodalis glossinidius and Sodalis praecaptivus P1 lysogens. (A) Detection of P1 pacB gene by PCR and agarose gel electrophoresis in S. glossinidius chloramphenicol-resistant clones that emerged following exposure to an E. coli P1CMclr-100(ts) lysogen (KL463). (B) Detection of an S. glossinidius-specific DNA fragment in clones depicted in panel A by PCR and agarose gel electrophoresis. (C) Detection of the P1 pacB gene by PCR and agarose gel electrophoresis in S. praecaptivus chloramphenicol-resistant clones that emerged following exposure to an E. coli P1CMclr-100(ts) lysogen (KL463). (D) Detection of an S. praecaptivus-specific DNA fragment in clones depicted in panel C by PCR and agarose gel electrophoresis. WT, wild type; (−), no DNA control; (+), positive control. (E to G) Formation of phage plaques on soft agar embedded with E. coli MG1655. Soft agar plates were spotted with dilutions of lysates derived from wild-type and S. glossinidius chloramphenicol-resistant (Cmr) P1CMclr-100(ts) lysogen (MP1705) (E), wild-type and S. praecaptivus Cmr P1CMclr-100(ts) lysogen (MP1703) (F), and S. praecaptivus Cmr P1CMclr-100(ts) lysogen (MP1703) grown either at 37°C or 30°C. Plates are representative of the plates of routine experiments. (H) Emergence of Cmr E. coli MG1655 following exposure to lysates derived from wild-type S. glossinidius, S. glossinidius Cmr P1CMclr-100(ts) lysogen (MP1705), wild-type S. praecaptivus, and S. praecaptivus Cmr P1CMclr-100(ts) lysogen (MP1703). The plate is representative of the plates of at least three experiments.
In S. glossinidius, the frequency of chloramphenicol-resistant colonies arising following P1CMclr-100(ts) exposure was similar to those observed for the E. coli control cells, indicating that P1 infection occurs efficiently in this bacterium. In contrast, chloramphenicol-resistant S. praecaptivus colonies emerged at a lower frequency, and higher concentrations of bacterial cells were typically used in P1 infection experiments (see Materials and Methods). Notably, in P1-resistant Salmonella enterica, the efficiency of P1 infection can be drastically increased by mutations in either galU or galE (10). Because these mutations remove the O antigen by truncating the core region of the lipopolysaccharide (LPS) (32, 33), they presumably facilitate access of P1 to its host receptor—conserved structural motifs within the LPS core (8, 10). In particular, while the LPS of S. praecaptivus contains structural components attached to its core region, S. glossinidius is devoid of such structures (see Fig. S1A in the supplemental material). Nonetheless, the lower infectivity of P1 does not appear to be related to the physical occlusion of the P1 receptor by components present in the outer portion of the S. praecaptivus LPS. This is because a mutation in galU results in a truncated LPS in S. praecaptivus but does not affect P1 infectivity (Fig. S1). Hence, unlike S. enterica, this phenotype is not due to the presence of a P1-antagonizing structure(s) in the outer portion of S. praecaptivus LPS. Taken together, these results indicate that phage P1 is capable of infecting and lysogenize in S. glossinidius and S. praecaptivus.
Effect of a galU mutation on LPS composition and P1 infectivity in S. praecaptivus. (A) Schematic representation depicting the inner membrane, peptidoglycan, and outer membrane of a prototypical Gram-negative bacterium. The inset shows a more detailed schematic of structural components of the LPS (left-hand side). Silver-stained sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) of LPS purified from S. praecaptivus galU (CMK36), wild-type S. praecaptivus, and wild-type S. glossinidius (right-hand side). (B) Plates depicting the appearance of chloramphenicol-resistant colonies as functions of the concentration of bacteria (vertical axis) and the concentration of P1CMclr-100(ts) lysates (horizontal axis). Cmr colonies emerge at similar frequencies in wild-type S. praecaptivus (left-hand side plate) and S. praecaptivus galU (CMK36) (right-hand side plate) when cells are exposed to equal amounts of P1CMclr-100(ts) particles. Download FIG S1, TIF file, 2.4 MB (2.5MB, tif) .
Copyright © 2021 Keller et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
P1 generalized transduction in S. praecaptivus.
During the formation of P1 virions, approximately 0.05 to 0.5% of infective phage particles package random DNA fragments derived from the bacterial host (34). These particles can mediate the transfer of bacterial DNA across P1-susceptible strains through generalized transduction. In the laboratory, generalized transduction of DNA can be identified by virtue of genetic markers that are packaged in these phage particles and transferred between bacterial strains. Accordingly, we sought to determine whether P1 could mediate generalized transduction in S. praecaptivus. First, we exposed wild-type S. praecaptivus to phage lysates derived from an S. praecaptivus P1CMclr-100(ts) lysogen harboring the ampicillin-resistant (Ampr) plasmid pSIM6 (35). Following lysate exposure, we were able to retrieve Ampr S. praecaptivus transductants. Importantly, Ampr cells were absent from both phage lysates alone and cultures of wild-type S. praecaptivus that were not exposed to phage (data not shown). In agreement with the notion that these Ampr clones were P1 transductants, diagnostic PCR revealed the presence of a pSIM6 fragment in these cells (Fig. 4A).
FIG 4.
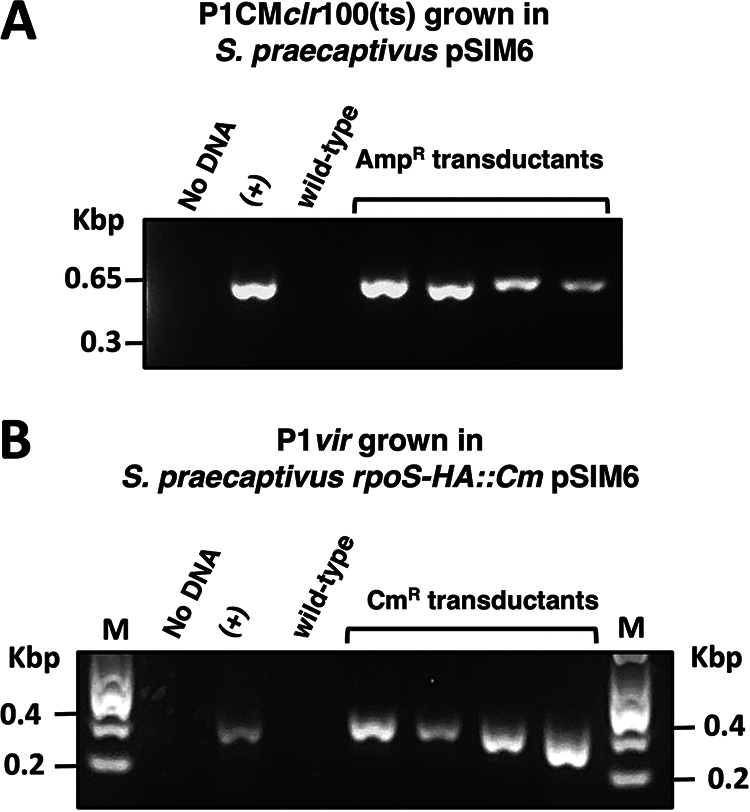
Bacteriophage P1-mediated generalized transduction in S. praecaptivus. (A) Detection of a pSIM6 DNA region by PCR and agarose gel electrophoresis in ampicillin-resistant (Ampr) S. praecaptivus transductants following exposure to lysates derived from S. praecaptivus Cmr P1CMclr-100(ts) (MP1703) harboring Ampr plasmid pSIM6. (B) Detection of the rpoS-HA::Cm chromosomal insertion by PCR and agarose gel electrophoresis in S. praecaptivus transductants following exposure to lysates of P1vir grown in S. praecaptivus rpoS-HA::Cm pSIM6 (MP1522) strain.
Next, we attempted to transduce a chromosomal chloramphenicol-resistant marker (rpoS-HA::Cm) using the P1 lytic strain P1vir. (This P1 strain is widely used as a transducing agent in E. coli due to its inability to lysogenize cells upon infection and the ease with which transducing lysates can be generated [5, 34].) We infected wild-type S. praecaptivus cells with P1vir lysates grown in an S. praecaptivus rpoS-HA::Cm pSIM6 strain. Whereas chloramphenicol-resistant (Cmr) cells emerged from wild-type S. praecaptivus exposed to phage, no Cmr cells were obtained from phage lysates or cultures of naive wild-type S. praecaptivus alone (data not shown). Notably, diagnostic PCR indicated that chloramphenicol-resistant clones were transductants harboring the rpoS-HA::Cm genetic modification (Fig. 4B). Importantly, all of these rpoS-HA::Cm transductants were sensitive to ampicillin, indicating that P1vir mediated the transduction of a discrete portion of the S. praecaptivus genome. Taken together, these results indicate that bacteriophage P1 can be used to mediate generalized transduction in S. praecaptivus.
Introduction of exogenous DNA in S. glossinidius and S. praecaptivus by P1-mediated guided transduction.
Whereas up to 0.5% of P1 particles can contain random fragments of bacterial host DNA (34), the vast majority of virions harbor P1 DNA. This is because the packaging of P1 genome into phage particles is guided by elements encoded within its DNA sequence (36, 37). Particularly, this packaging element can be cloned into plasmids (to produce phagemids) or incorporated into the bacterial chromosome to increase the frequency of P1-mediated transduction of adjacent DNA (17, 38, 39). Indeed, the P1 packaging element can increase the transduction of linked DNA by 1,600-fold above the levels obtained in generalized transduction (38). Hence, this DNA element can be used to increase the number of transducing particles and, consequently, the efficiency of DNA transfer among bacterial strains that are susceptible to P1 infection.
The lack of genetic tools available for the manipulation of Sodalis species, specifically S. glossinidius, prompted us to explore P1 as a plasmid DNA delivery tool for these bacteria. As a proof of principle, we used the aforementioned general technique to transfer a number of P1 phagemids (38) (Fig. 5A) into S. glossinidius. We were able to recover transductants expressing an array of phenotypic traits encoded in the phagemids. These traits included light production (luxCDABE genes), violacein pigment synthesis (vioABCE), β-galactosidase activity (lacZ), or green fluorescence (gfp) (Fig. 5B and C). To expand the tool set available for the modification of Sodalis species, we constructed two phagemids for tagging bacterial chromosomes with fluorescent genes at the Tn7 attachment site (40) and a suicide phagemid encoding a Himar1 transposition system for random mutagenesis (41) (Fig. S2). Following packaging into P1 virions in an E. coli P1CMclr-100(ts) lysogen, these phagemids were efficiently delivered to S. glossinidius and S. praecaptivus (Fig. 5D and E). Together, these results establish that bacteriophage P1 can be used to efficiently deliver replication-competent and suicide vectors into S. glossinidius and S. praecaptivus through a “guided transduction” strategy.
FIG 5.

Transduction of P1 phagemids into Sodalis species. (A) Schematic representation of replication-competent P1 phagemids encoding a number of phenotypic markers (38). (B) Comparison of wild-type (top quadrants) and S. glossinidius transductants (lower quadrants) carrying P1 phagemid BBa_J72114-BBa_J72104 (38). Transductant colonies are purple due to the expression of violacein biosynthetic genes (lower left quadrant), and produce light due to the expression of bioluminescence genes (lower right quadrant). (C) Macrocolonies derived from wild-type S. glossinidius (bottom row) and transductants carrying P1 phagemids BBa_J72114-BBa_J72104 (luxCDABE+ and vioABCE+), BBa_J72114-BBa_J72100 (lacZ+), or BBa_J72113-BBa_J72152 (gfp+) (top row). (D) Transduction of pP1-Tn7-mVenus into S. praecaptivus (left-hand side plate) and pP1-Tn7-mCardinal into S. glossinidius (right-hand side plate). (E) Transduction of phagemid encoding a Himar1 transposition system (pP1-Himar) into S. praecaptivus (left-hand side plate) and S. glossinidius (right-hand side plate).
Schematic representation of P1 phagemids used for tagging the bacterial chromosome at the Tn7 attachment site(s) with genes encoding the fluorescent proteins mCardinal (pP1-Tn7-mCardinal) or mVenus (pP1-Tn7-mVenus) and the suicide P1 phagemid for random mutagenesis with a Himar1 transposition system (pP1-Himar). Download FIG S2, TIF file, 2.6 MB (2.7MB, tif) .
Copyright © 2021 Keller et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
DISCUSSION
In the current study, we demonstrate that bacteriophage P1 can infect, lysogenize, and promote transduction in two species of the genus Sodalis. We show that P1 can mediate generalized transduction in S. praecaptivus (Fig. 4), and we establish that this bacteriophage can be used for the delivery of plasmids and suicide vectors for the genetic manipulation of S. glossinidius and S. praecaptivus (Fig. 5). While these results constitute a significant advance in the development of genetic modification tools to study these bacterial species, they also clear the way for the implementation of P1-based DNA delivery systems to uncultured Sodalis species (42–48) and Gram-negative insect endosymbionts belonging to other genera.
Whereas S. praecaptivus can be genetically engineered with relative ease, the ability of P1 to mediate generalized transduction provides a number of applications for the manipulation of this bacterium. For instance, although S. praecaptivus can be readily modified by recombineering functions of phage λ (λ-Red) (49–51), the use of this technique has two major drawbacks. First, the expression of recombineering functions can be mutagenic (52). This can potentially produce confounding results in subsequent experiments as phenotypes associated with a particular engineered modification may actually result from secondary mutation(s). Second, typical temperature-sensitive plasmids (reppSC101ts ori) harboring recombineering functions cannot be cured from S. praecaptivus by propagating cells at nonpermissive temperatures (≥37°C) in the absence of plasmid selection (our unpublished results). The inability to cure these plasmids can increase the chances of secondary mutations through leaky expression of recombineering functions and hinder the use of plasmids from the same incompatibility group in downstream genetic analyses. Importantly, both of these issues can be overcome by P1-mediated generalized transduction. That is, genomic DNA fragments engineered using λ-Red can be transferred to naive S. praecaptivus cells that lack recombineering plasmids and, therefore, have not been exposed to potential mutagenic events (Fig. 4B).
In contrast, the establishment of P1-mediated transduction provides a considerable advancement in our ability to genetically manipulate S. glossinidius. This is because S. glossinidius is recalcitrant to DNA transformation by standard techniques such as heat shock and electroporation (29, 30, 53). Whereas we have recently developed a method for DNA transfer to S. glossinidius via conjugation (30), P1-mediated transduction provides an alternative, simpler method for the introduction of exogenous DNA into this bacterium. Altered chromosomal fragments, replication-competent plasmids carrying an array of functions, and suicide vectors engineered for allelic replacement or containing transposition systems can be quickly transduced into S. glossinidius in a simple protocol. Given the large DNA packaging capability of P1 (up to 100 kbp) (5), this bacteriophage can be efficiently used for a variety of applications, including the delivery of bacterial artificial chromosomes (bacterial artificial chromosome [BAC] vectors) or large plasmids encoding multiple genome editing CRISPR systems (54) that are not easily transferred by conjugation (30). Additionally, the P1 packing sequence can be incorporated into DNA fragments used in insertional mutagenesis (39), enabling rapid and efficient combination of mutations via P1 “guided transduction.” This approach can greatly facilitate the implementation of several analyses (e.g., complementation and epistasis) to identify and dissect genetic components and pathways governing bacterial behaviors and interactions with eukaryotic hosts.
Beyond the genus Sodalis, the results highlighted in this study have potential broad implications for the genetic modification of uncultured Gram-negative insect endosymbionts. In Gram-negative bacteria, the LPS is the major structural constituent of the outer leaflet of the outer membrane. The LPS is composed of a highly conserved lipid A “anchor,” a conserved core polysaccharide and, sometimes, a hypervariable outer component designated O antigen (see Fig. S1A in the supplemental material) (76). Bacteriophage P1 has a broad host range, in part, because it recognizes, as its host receptor, structural features of the conserved LPS core (8). In addition to E. coli and several species of the Gammaproteobacteria, P1 has been shown to be capable of infecting various members within the Alpha, Beta-, and Deltaproteobacteria, and even bacterial species residing outside the Proteobacteria phylum such as Flavobacterium sp. strain M64 (8–12).
Notably, a large number of economically important insect species—including several disease vectors of animals and plants—harbor maternally inherited, Gram-negative bacterial endosymbionts (24, 25). However, unlike S. glossinidius and a handful of other species, the vast majority of these endosymbionts have not been isolated in pure culture (14, 19, 55–59). This is because these bacteria undergo a process of genome degeneration and size reduction during the course of long-term evolution and specialization within their eukaryotic insect hosts. This process leads to the loss of many physiological functions that are required for replication outside the host (24, 25, 27, 29). Notably, classical protocols of bacterial genetics require the manipulation of large numbers of cells and the subsequent isolation of rare genetic events as bacterial colonies on selective agar plates. Consequently, the implementation of genetics for the study of insect endosymbionts has remained scarce and limited to species that can grow in axenic culture, form colonies on agar plates, and are receptive to exogenous DNA (29, 30).
The ability of bacteriophage P1 to deliver DNA to a large number of bacterial species suggests a clear method in which the requirement for culturing may be bypassed. That is, similar to viral vectors that are commonly utilized in gene therapy in mammalians (60), P1 could be used to deliver DNA to bacterial endosymbionts inside their insect hosts. Specifically, insects could be microinjected (23, 27, 61–63) with P1 virions packaged with recombinant DNA. The establishment of successful P1 infections and subsequent enrichment of transductant endsymbionts to near-homogeneous or clonal populations could be attained by making use of phenotypic markers (Fig. 5) and implementing antibiotic selection regiments in insects (64–69). Whereas this approach would preferentially target recently acquired endosymbionts by virtue of their ability to exist intracellularly and extracellularly in various insect tissues (21–23, 25), ancient obligate intracellular endosymbionts could also be subjected to infection and genetic modification via P1, if they transiently exit host cells (70).
The results presented in this study pave the way for the development of tractable genetic systems for S. glossinidius and, potentially, a myriad of Gram-negative bacterial endosymbionts of insects. While this may empower the use of genetics to study these obscure bacteria, it also has clear translational applications. P1-mediated DNA delivery into insect endosymbionts may allow the engineering of bacterial traits aimed at modifying aspects of insect ecology (29, 30), mitigating their burden on economic activities and human health.
MATERIALS AND METHODS
Microbial strains, phages, plasmids, and growth conditions.
Microbial strains, phages, and plasmids used in this study are presented in Table S1 in the supplemental material. Unless indicated, all Escherichia coli strains were propagated at 30, 37, or 42°C in Luria-Bertani (LB) broth or agar (1.5% [wt/vol]). Sodalis glossinidius was grown at 27°C in brain heart infusion broth supplemented with 10 mM MgCl2 (BHI) or on brain heart infusion agar (1.2% [wt/vol]) supplemented with 10 mM MgCl2 (BHI agar). S. glossinidius was also propagated on BHI agar plates supplemented with 10% defibrinated horse blood (BHIB). Sodalis praecaptivus was grown at 30, 39, or 42°C in Luria-Bertani broth or agar (1.5% [wt/vol]) lacking sodium chloride. For experiments involving P1 infection or generation of lysates, the growth medium was supplemented with CaCl2 and MgCl2 to a final concentration of 10 mM, respectively. Growth of S. glossinidius on BHIB agar plates was carried out under microaerophilic conditions, which was achieved either using BD GasPak EZ Campy Gas Generating sachets or a gas mixture (5% oxygen and 95% CO2). For all strains, growth in liquid medium was carried out in shaking water bath incubators with aeration (250 rpm). When required, medium was supplemented with ampicillin (100 μg/ml), chloramphenicol (20 μg/ml for E. coli or S. praecaptivus and 10 μg/ml for S. glossinidius), kanamycin (50 μg/ml for E. coli and 25 μg/ml for S. glossinidius or S. praecaptivus). Arabinose was used at a concentration of 0.5 or 1% (wt/vol); 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal) was used at a concentration of 100 μg/ml.
Microbial strains, phages, and plasmids used in this study. Download Table S1, XLSX file, 0.01 MB (15.9KB, xlsx) .
Copyright © 2021 Keller et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Lipopolysaccharide extraction and detection.
Extraction of lipopolysaccharide (LPS) from S. glossinidius and S. praecaptivus cultures was carried out as described previously (71). Extracted samples were separated in a NuPAGE 10% Bis-Tris gel in NuPAGE MES SDS Running (ThermoFisher Scientific). LPS in gels were stained with ProteoSilver silver stain kit (Sigma-Aldrich).
Construction of phagemid pP1-Tn7-mCardinal.
Oligonucleotide sequences used in this study are presented in Table S2. Phusion high-fidelity DNA polymerase (New England BioLabs) was used in PCRs with primers 469 and 470 and plasmid BBa_J72113-BBa_J72152 (38) as the template. The PCR product was ligated into pMRE-Tn7-163 (72), previously digested with SbfI, using NEBuilder HiFi DNA Assembly (New England BioLabs). The integrity of the construct was verified by DNA sequencing and the ability to be efficiently transduced by P1 particles.
Oligonucleotide sequences used in this study. Download Table S2, XLSX file, 0.01 MB (15.4KB, xlsx) .
Copyright © 2021 Keller et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Construction of phagemid pP1-Tn7-mVenus.
Oligonucleotide sequences used in this study are presented in Table S2. Phusion high-fidelity DNA polymerase (New England BioLabs) was used in PCRs with primers 469 and 470 and plasmid BBa_J72113-BBa_J72152 (38) as the template. The PCR product was ligated into pMRE-Tn7-166 (72), previously digested with SbfI, using NEBuilder HiFi DNA Assembly (New England BioLabs). The integrity of the construct was verified by DNA sequencing and the ability to be efficiently transduced by P1 particles.
Construction of phagemid pP1-Himar.
Oligonucleotide sequences used in this study are presented in Table S2. Phusion high-fidelity DNA polymerase (New England BioLabs) was used in PCRs with primers 475 and 476 and plasmid BBa_J72113-BBa_J72152 (38) as the template. The PCR product was ligated into pMarC9-R6k (73), previously digested with EcoRI and HindIII, using NEBuilder HiFi DNA Assembly (New England BioLabs). The integrity of the construct was verified by DNA sequencing and the ability to be efficiently transduced by P1 particles.
Recombineering procedure for S. praecaptivus.
Oligonucleotide sequences used in this study are presented in Table S2. An S. praecaptivus strain harboring plasmid pSIM6 (35) was grown overnight in LB broth supplemented with 100 μg/ml of ampicillin at 30°C and 250 rpm. Cells were diluted (1:100) in 30 ml of the same medium and grown to an optical density at 600 nm (OD600) between 0.45 and 0.5. The culture flask was then grown in a water bath at 42°C and 250 rpm for 25 min. Cells were immediately transferred to a 50-ml conical tube, collected by centrifugation (7,000 rpm for 2.5 min at 4°C), and resuspended in 40 ml of ice-cold deionized H2O (dH2O). Cells were collected again by centrifugation, and this washing procedure was repeated a second time. Finally, cells were resuspended in 150 μl of ice-cold dH2O. Homologous recombination was obtained by electroporating 70 μl of cell suspension with 10 μl of purified PCR products generated with primers 251 and 252 (galU::Kn) or primers 84 and 85 (rpoS-HA::Cm) and plasmids pKD4 and pKD3 (74) as the templates, respectively.
Preparation of phage lysates derived from EMG16 P1 lysogens.
Lysates from E. coli EMG16 harboring selected phagemids were prepared following arabinose induction as described previously (38).
Preparation of stocks of P1vir phage lysates.
Stocks of P1vir phage lysates were prepared by infecting E. coli MG1655 (75), as described previously (5).
Preparation of phage lysates derived from P1CMclr-100(ts) lysogens.
E. coli P1CMclr-100(ts) lysogens were grown overnight at 30°C. Cultures were diluted (1:100 [vol/vol]) into fresh medium and grown to an OD600 value of 0.3 to 0.4. Subsequently, cultures were shifted to 42°C and propagated until extensive cell lysis (3 to 4 h). At times, cultures were allowed to grow at 42°C for 16 h prior to the preparation of lysates. Partially lysed cells were disrupted by vortexing the cultures following the addition of chloroform (1 volume of chloroform per 100 volumes of culture). Cell debris was removed by centrifugation (12 min, 4,000 rpm, room temperature), and the supernatant was passed through a 0.22-μm polyethersulfone membrane filter. S. praecaptivus P1CMclr-100(ts) lysogens were prepared as described above, except that cells were grown for 2 h at 42°C and 16 h at 37°C prior to lysate preparation. S. glossinidius P1CMclr-100(ts) lysogens were grown in BHI to an OD600 of 0.4. Cultures were heat shocked at 37°C for 2 h. The cultures were treated with chloroform (1 volume of chloroform per 100 volumes of culture) and processed as described for E. coli and S. praecaptivus.
Infection by P1vir, P1CMclr-100(ts), and P1 transducing particles.
Following overnight growth in LB, E. coli cultures were diluted in fresh LB supplemented with 10 mM CaCl2 and MgCl2 to an OD600 of 1. One-milliliter aliquots of these cell solutions were incubated for 30 min at 30°C in the presence or absence of various concentrations of P1 lysate. Cells were subsequently collected by centrifugation (1 min, 13,000 rpm, room temperature), and the supernatants were replaced by 1 ml of LB containing 5 mM sodium citrate. Cells were grown for 1 h at 30°C and 250 rpm prior to plating. S. praecaptivus cells grown overnight in LB were collected by centrifugation (1 min, 13,000 rpm, room temperature) and resuspended in fresh LB supplemented with 10 mM CaCl2 and MgCl2. One milliliter of these resuspended solutions was incubated for 30 min at 30°C in the presence or absence of various concentrations of P1 lysate. Cells were collected by centrifugation (1 min, 13,000 rpm, room temperature), and the supernatants were replaced by 1 ml of LB containing 5 mM sodium citrate. Cells were plated following 1 h of growth at 30°C and 250 rpm. S. glossinidius cells were grown in BHI for 3 to 5 days to an OD600 of ≈0.5. Cells were collected by centrifugation and concentrated to an OD600 of 1. One milliliter of concentrated cultures was incubated for 60 min at 30°C in the presence or absence of various concentrations of P1 lysate. Cells were collected by centrifugation (1 min, 13,000 rpm, room temperature), and the supernatants were replaced by 10 ml of BHI. Cultures were incubated overnight at 27°C overnight with shaking prior to plating.
Curing of pP1-Tn7 phagemids.
Transduction of pP1-Tn7 phagemids into S. glossinidius and S. praecaptivus was initially selected on plates containing ampicillin (Fig. 5D; see also Fig. S2 in the supplemental material). Because episomes harboring reppSC101ts origins of replication are not easily cured from S. praecaptivus (see Discussion), the curing of phagemids was performed only in S. glossinidius. The strategy used to identify S. glossinidius clones lacking phagemids was similar to the one adopted elsewhere (53). Briefly, to identify S. glossinidius clones that contained the chloramphenicol-resistant marker at the Tn7 attachment site and had lost the ampicillin-resistant plasmid, cultures were propagated in BHI containing chloramphenicol and 1% arabinose. After four passages, cells were diluted and plated. Single colonies were screened for sensitivity to ampicillin. Transposon insertion at the Tn7 attachment site was verified by PCR with primers 1018 and 1019.
Image acquisition, analysis, and manipulation.
DNA agarose gel electrophoresis and bacterial colonies, with the exception of S. glossinidius macrocolonies, were detected using an Amersham Imager 680 (GE Healthcare). S. glossinidius macrocolonies expressing green fluorescent protein (GFP) were detected using a dark reader (Clare Chemical Research) and documented with an iPhone. When oversaturated, the intensity of signals in images were adjusted across the entire images using Preview (Apple).
ACKNOWLEDGMENTS
We thank Serap Aksoy (Yale University) for kindly providing us with a culture of Sodalis glossinidius and Hubert Salvail (Yale University) for assistance obtaining an E. coli P1CMclr-100(ts) lysogen.
M.H.P. is supported by grant AI148774 from the National Institutes of Health and start-up funds from The Pennsylvania State University College of Medicine.
We declare that we have no conflicts of interest.
Contributor Information
Mauricio H. Pontes, Email: mpontes@pennstatehealth.psu.edu.
Barbara J. Campbell, Clemson University
REFERENCES
- 1.Hendrix RW, Smith MC, Burns RN, Ford ME, Hatfull GF. 1999. Evolutionary relationships among diverse bacteriophages and prophages: all the world's a phage. Proc Natl Acad Sci U S A 96:2192–2197. doi: 10.1073/pnas.96.5.2192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Touchon M, Moura de Sousa JA, Rocha EP. 2017. Embracing the enemy: the diversification of microbial gene repertoires by phage-mediated horizontal gene transfer. Curr Opin Microbiol 38:66–73. doi: 10.1016/j.mib.2017.04.010. [DOI] [PubMed] [Google Scholar]
- 3.Soucy SM, Huang J, Gogarten JP. 2015. Horizontal gene transfer: building the web of life. Nat Rev Genet 16:472–482. doi: 10.1038/nrg3962. [DOI] [PubMed] [Google Scholar]
- 4.Hershey AD, Chase M. 1952. Independent functions of viral protein and nucleic acid in growth of bacteriophage. J Gen Physiol 36:39–56. doi: 10.1085/jgp.36.1.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thomason LC, Costantino N, Court DL. 2007. E. coli genome manipulation by P1 transduction. Curr Protoc Mol Biol Chapter 1:Unit 1.17. doi: 10.1002/0471142727.mb0117s79. [DOI] [PubMed] [Google Scholar]
- 6.Bertani G. 1951. Studies on lysogenesis. I. The mode of phage liberation by lysogenic Escherichia coli. J Bacteriol 62:293–300. doi: 10.1128/JB.62.3.293-300.1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lennox ES. 1955. Transduction of linked genetic characters of the host by bacteriophage P1. Virology 1:190–206. doi: 10.1016/0042-6822(55)90016-7. [DOI] [PubMed] [Google Scholar]
- 8.Yarmolinsky MB, Sternberg N. 1988. Bacteriophage P1, p 291–438. In Calendar R (ed), The bacteriophages, vol 1. Plenum Press, New York, NY. [Google Scholar]
- 9.Goldberg RB, Bender RA, Streicher SL. 1974. Direct selection for P1-sensitive mutants of enteric bacteria. J Bacteriol 118:810–814. doi: 10.1128/JB.118.3.810-814.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ornellas EP, Stocker BA. 1974. Relation of lipopolysaccharide character to P1 sensitivity in Salmonella typhimurium. Virology 60:491–502. doi: 10.1016/0042-6822(74)90343-2. [DOI] [PubMed] [Google Scholar]
- 11.Kaiser D, Dworkin M. 1975. Gene transfer to myxobacterium by Escherichia coli phage P1. Science 187:653–654. doi: 10.1126/science.803710. [DOI] [PubMed] [Google Scholar]
- 12.Murooka Y, Harada T. 1979. Expansion of the host range of coliphage P1 and gene transfer from enteric bacteria to other Gram-negative bacteria. Appl Environ Microbiol 38:754–757. doi: 10.1128/AEM.38.4.754-757.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Streicher S, Gurney E, Valentine RC. 1971. Transduction of the nitrogen-fixation genes in Klebsiella pneumoniae. Proc Natl Acad Sci U S A 68:1174–1177. doi: 10.1073/pnas.68.6.1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.O'Connor KA, Zusman DR. 1983. Coliphage P1-mediated transduction of cloned DNA from Escherichia coli to Myxococcus xanthus: use for complementation and recombinational analyses. J Bacteriol 155:317–329. doi: 10.1128/JB.155.1.317-329.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Downard JS. 1988. Tn5-mediated transposition of plasmid DNA after transduction to Myxococcus xanthus. J Bacteriol 170:4939–4941. doi: 10.1128/jb.170.10.4939-4941.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wolf-Watz H, Portnoy DA, Bölin I, Falkow S. 1985. Transfer of the virulence plasmid of Yersinia pestis to Yersinia pseudotuberculosis. Infect Immun 48:241–253. doi: 10.1128/IAI.48.1.241-243.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Westwater C, Schofield DA, Schmidt MG, Norris JS, Dolan JW. 2002. Development of a P1 phagemid system for the delivery of DNA into Gram-negative bacteria. Microbiology (Reading) 148:943–950. doi: 10.1099/00221287-148-4-943. [DOI] [PubMed] [Google Scholar]
- 18.Butela K, Lawrence JG. 2012. Genetic manipulation of pathogenicity loci in non-Typhimurium Salmonella. J Microbiol Methods 91:477–482. doi: 10.1016/j.mimet.2012.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dale C, Maudlin I. 1999. Sodalis gen. nov. and Sodalis glossinidius sp. nov., a microaerophilic secondary endosymbiont of the tsetse fly Glossina morsitans morsitans. Int J Syst Bacteriol 49:267–275. doi: 10.1099/00207713-49-1-267. [DOI] [PubMed] [Google Scholar]
- 20.Chari A, Oakeson KF, Enomoto S, Jackson DG, Fisher MA, Dale C. 2015. Phenotypic characterization of Sodalis praecaptivus sp. nov., a close non-insect-associated member of the Sodalis-allied lineage of insect endosymbionts. Int J Syst Evol Microbiol 65:1400–1405. doi: 10.1099/ijs.0.000091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aksoy S, Chen X, Hypsa V. 1997. Phylogeny and potential transmission routes of midgut-associated endosymbionts of tsetse (Diptera: Glossinidae). Insect Mol Biol 6:183–190. doi: 10.1111/j.1365-2583.1997.tb00086.x. [DOI] [PubMed] [Google Scholar]
- 22.Cheng Q, Aksoy S. 1999. Tissue tropism, transmission and expression of foreign genes in vivo in midgut symbionts of tsetse flies. Insect Mol Biol 8:125–132. doi: 10.1046/j.1365-2583.1999.810125.x. [DOI] [PubMed] [Google Scholar]
- 23.De Vooght L, Caljon G, Van Hees J, Van Den Abbeele J. 2015. Paternal transmission of a secondary symbiont during mating in the viviparous tsetse fly. Mol Biol Evol 32:1977–1980. doi: 10.1093/molbev/msv077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McCutcheon JP, Boyd BM, Dale C. 2019. The life of an insect endosymbiont from the cradle to the grave. Curr Biol 29:R485–R495. doi: 10.1016/j.cub.2019.03.032. [DOI] [PubMed] [Google Scholar]
- 25.Moran NA, McCutcheon JP, Nakabachi A. 2008. Genomics and evolution of heritable bacterial symbionts. Annu Rev Genet 42:165–190. doi: 10.1146/annurev.genet.41.110306.130119. [DOI] [PubMed] [Google Scholar]
- 26.Toh H, Weiss BL, Perkin SA, Yamashita A, Oshima K, Hattori M, Aksoy S. 2006. Massive genome erosion and functional adaptations provide insights into the symbiotic lifestyle of Sodalis glossinidius in the tsetse host. Genome Res 16:149–156. doi: 10.1101/gr.4106106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pontes MH, Smith KL, De Vooght L, Van Den Abbeele J, Dale C. 2011. Attenuation of the sensing capabilities of PhoQ in transition to obligate insect-bacterial association. PLoS Genet 7:e1002349. doi: 10.1371/journal.pgen.1002349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Clayton AL, Oakeson KF, Gutin M, Pontes A, Dunn DM, von Niederhausern AC, Weiss RB, Fisher M, Dale C. 2012. A novel human-infection-derived bacterium provides insights into the evolutionary origins of mutualistic insect-bacterial symbioses. PLoS Genet 8:e1002990. doi: 10.1371/journal.pgen.1002990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pontes MH, Dale C. 2006. Culture and manipulation of insect facultative symbionts. Trends Microbiol 14:406–412. doi: 10.1016/j.tim.2006.07.004. [DOI] [PubMed] [Google Scholar]
- 30.Kendra CG, Keller CM, Bruna RE, Pontes MH. 2020. Conjugal DNA transfer in the maternally inherited symbiont of tsetse flies Sodalis glossinidius. mSphere 5:e00864-20. doi: 10.1128/mSphere.00864-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rosner JL. 1972. Formation, induction, and curing of bacteriophage P1 lysogens. Virology 48:679–689. doi: 10.1016/0042-6822(72)90152-3. [DOI] [PubMed] [Google Scholar]
- 32.Wilkinson RG, Stocker BA. 1968. Genetics and cultural properties of mutants of Salmonella typhimurium lacking glucosyl or galactosyl lipopolysaccharide transferases. Nature 217:955–957. doi: 10.1038/217955a0. [DOI] [PubMed] [Google Scholar]
- 33.Osborn MJ. 1968. Biochemical characterization of mutants of Salmonella typhimurium lacking glucosyl or galactosyl lipopolysaccharide transferases. Nature 217:957–960. doi: 10.1038/217957a0. [DOI] [PubMed] [Google Scholar]
- 34.Ikeda H, Tomizawa JI. 1965. Transducing fragments in generalized transduction by phage P1. I. Molecular origin of the fragments. J Mol Biol 14:85–109. doi: 10.1016/s0022-2836(65)80232-7. [DOI] [PubMed] [Google Scholar]
- 35.Datta S, Costantino N, Court DL. 2006. A set of recombineering plasmids for gram-negative bacteria. Gene 379:109–115. doi: 10.1016/j.gene.2006.04.018. [DOI] [PubMed] [Google Scholar]
- 36.Sternberg N, Coulby J. 1987. Recognition and cleavage of the bacteriophage P1 packaging site (pac). I. Differential processing of the cleaved ends in vivo. J Mol Biol 194:453–468. doi: 10.1016/0022-2836(87)90674-7. [DOI] [PubMed] [Google Scholar]
- 37.Sternberg N, Coulby J. 1987. Recognition and cleavage of the bacteriophage P1 packaging site (pac). II. Functional limits of pac and location of pac cleavage termini. J Mol Biol 194:469–479. doi: 10.1016/0022-2836(87)90675-9. [DOI] [PubMed] [Google Scholar]
- 38.Kittleson JT, DeLoache W, Cheng HY, Anderson JC. 2012. Scalable plasmid transfer using engineered P1-based phagemids. ACS Synth Biol 1:583–589. doi: 10.1021/sb300054p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huang H, Masters M. 2014. Bacteriophage P1 pac sites inserted into the chromosome greatly increase packaging and transduction of Escherichia coli genomic DNA. Virology 468-470:274–282. doi: 10.1016/j.virol.2014.07.029. [DOI] [PubMed] [Google Scholar]
- 40.Peters JE, Craig NL. 2001. Tn7: smarter than we thought. Nat Rev Mol Cell Biol 2:806–814. doi: 10.1038/35099006. [DOI] [PubMed] [Google Scholar]
- 41.Rubin EJ, Akerley BJ, Novik VN, Lampe DJ, Husson RN, Mekalanos JJ. 1999. In vivo transposition of mariner-based elements in enteric bacteria and mycobacteria. Proc Natl Acad Sci U S A 96:1645–1650. doi: 10.1073/pnas.96.4.1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Heddi A, Charles H, Khatchadourian C, Bonnot G, Nardon P. 1998. Molecular characterization of the principal symbiotic bacteria of the weevil Sitophilus oryzae: a peculiar G+C content of an endocytobiotic DNA. J Mol Evol 47:52–61. doi: 10.1007/pl00006362. [DOI] [PubMed] [Google Scholar]
- 43.Fukatsu T, Koga R, Smith WA, Tanaka K, Nikoh N, Sasaki-Fukatsu K, Yoshizawa K, Dale C, Clayton DH. 2007. Bacterial endosymbiont of the slender pigeon louse, Columbicola columbae, allied to endosymbionts of grain weevils and tsetse flies. Appl Environ Microbiol 73:6660–6668. doi: 10.1128/AEM.01131-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nováková E, Hypsa V. 2007. A new Sodalis lineage from bloodsucking fly Craterina melbae (Diptera, Hippoboscoidea) originated independently of the tsetse flies symbiont Sodalis glossinidius. FEMS Microbiol Lett 269:131–135. doi: 10.1111/j.1574-6968.2006.00620.x. [DOI] [PubMed] [Google Scholar]
- 45.Chrudimský T, Husník F, Nováková E, Hypša V. 2012. Candidatus Sodalis melophagi sp. nov.: phylogenetically independent comparative model to the tsetse fly symbiont Sodalis glossinidius. PLoS One 7:e40354. doi: 10.1371/journal.pone.0040354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Smith WA, Oakeson KF, Johnson KP, Reed DL, Carter T, Smith KL, Koga R, Fukatsu T, Clayton DH, Dale C. 2013. Phylogenetic analysis of symbionts in feather-feeding lice of the genus Columbicola: evidence for repeated symbiont replacements. BMC Evol Biol 13:109. doi: 10.1186/1471-2148-13-109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Santos-Garcia D, Silva FJ, Morin S, Dettner K, Kuechler SM. 2017. The all-rounder Sodalis: a new bacteriome-associated endosymbiont of the lygaeoid bug Henestaris halophilus (Heteroptera: Henestarinae) and a critical examination of its evolution. Genome Biol Evol 9:2893–2910. doi: 10.1093/gbe/evx202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Šochová E, Husník F, Nováková E, Halajian A, Hypša V. 2017. Arsenophonus and Sodalis replacements shape evolution of symbiosis in louse flies. PeerJ 5:e4099. doi: 10.7717/peerj.4099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Clayton AL, Enomoto S, Su Y, Dale C. 2017. The regulation of antimicrobial peptide resistance in the transition to insect symbiosis. Mol Microbiol 103:958–972. doi: 10.1111/mmi.13598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Enomoto S, Chari A, Clayton AL, Dale C. 2017. Quorum sensing attenuates virulence in Sodalis praecaptivus. Cell Host Microbe 21:629–636.e5. doi: 10.1016/j.chom.2017.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Thomason LC, Sawitzke JA, Li X, Costantino N, Court DL. 2014. Recombineering: genetic engineering in bacteria using homologous recombination. Curr Protoc Mol Biol 106:1.16.1–1.16.39. doi: 10.1002/0471142727.mb0116s106. [DOI] [PubMed] [Google Scholar]
- 52.Murphy KC, Campellone KG. 2003. Lambda Red-mediated recombinogenic engineering of enterohemorrhagic and enteropathogenic E. coli. BMC Mol Biol 4:11. doi: 10.1186/1471-2199-4-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pontes MH, Dale C. 2011. Lambda red-mediated genetic modification of the insect endosymbiont Sodalis glossinidius. Appl Environ Microbiol 77:1918–1920. doi: 10.1128/AEM.02166-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Adiego-Pérez B, Randazzo P, Daran JM, Verwaal R, Roubos JA, Daran-Lapujade P, van der Oost J. 2019. Multiplex genome editing of microorganisms using CRISPR-Cas. FEMS Microbiol Lett 366:fnz086. doi: 10.1093/femsle/fnz086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hypsa V, Dale C. 1997. In vitro culture and phylogenetic analysis of "Candidatus Arsenophonus triatominarum," an intracellular bacterium from the triatomine bug, Triatoma infestans. Int J Syst Bacteriol 47:1140–1144. doi: 10.1099/00207713-47-4-1140. [DOI] [PubMed] [Google Scholar]
- 56.Sabri A, Leroy P, Haubruge E, Hance T, Frère I, Destain J, Thonart P. 2011. Isolation, pure culture and characterization of Serratia symbiotica sp. nov., the R-type of secondary endosymbiont of the black bean aphid Aphis fabae. Int J Syst Evol Microbiol 61:2081–2088. doi: 10.1099/ijs.0.024133-0. [DOI] [PubMed] [Google Scholar]
- 57.Dale C, Beeton M, Harbison C, Jones T, Pontes M. 2006. Isolation, pure culture, and characterization of "Candidatus Arsenophonus arthropodicus," an intracellular secondary endosymbiont from the hippoboscid louse fly Pseudolynchia canariensis. Appl Environ Microbiol 72:2997–3004. doi: 10.1128/AEM.72.4.2997-3004.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Brandt JW, Chevignon G, Oliver KM, Strand MR. 2017. Culture of an aphid heritable symbiont demonstrates its direct role in defence against parasitoids. Proc Biol Soc 284:20171925. doi: 10.1098/rspb.2017.1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Masson F, Calderon Copete S, Schüpfer F, Garcia-Arraez G, Lemaitre B. 2018. In vitro culture of the insect endosymbiont Spiroplasma poulsonii highlights bacterial genes Iinvolved in host-symbiont interaction. mBio 9:e00024-18. doi: 10.1128/mBio.00024-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kay MA, Glorioso JC, Naldini L. 2001. Viral vectors for gene therapy: the art of turning infectious agents into vehicles of therapeutics. Nat Med 7:33–40. doi: 10.1038/83324. [DOI] [PubMed] [Google Scholar]
- 61.Boyle L, O'Neill SL, Robertson HM, Karr TL. 1993. Interspecific and intraspecific horizontal transfer of Wolbachia in Drosophila. Science 260:1796–1799. doi: 10.1126/science.8511587. [DOI] [PubMed] [Google Scholar]
- 62.Oliver KM, Russell JA, Moran NA, Hunter MS. 2003. Facultative bacterial symbionts in aphids confer resistance to parasitic wasps. Proc Natl Acad Sci U S A 100:1803–1807. doi: 10.1073/pnas.0335320100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Doremus MR, Smith AH, Kim KL, Holder AJ, Russell JA, Oliver KM. 2018. Breakdown of a defensive symbiosis, but not endogenous defences, at elevated temperatures. Mol Ecol 27:2138–2151. doi: 10.1111/mec.14399. [DOI] [PubMed] [Google Scholar]
- 64.Wilkinson TL. 1998. The elimination of intracellular microorganisms from insects: an analysis of antibiotic-treatment in the pea aphid (Acyrthosiphon pisum). Comp Biochem Physiol Ser 119:871–881. doi: 10.1016/S1095-6433(98)00013-0. [DOI] [Google Scholar]
- 65.Dale C, Welburn SC. 2001. The endosymbionts of tsetse flies: manipulating host-parasite interactions. Int J Parasitol 31:628–631. doi: 10.1016/s0020-7519(01)00151-5. [DOI] [PubMed] [Google Scholar]
- 66.Dobson SL, Rattanadechakul W. 2001. A novel technique for removing Wolbachia infections from Aedes albopictus (Diptera: Culicidae). J Med Entomol 38:844–849. doi: 10.1603/0022-2585-38.6.844. [DOI] [PubMed] [Google Scholar]
- 67.Koga R, Tsuchida T, Sakurai M, Fukatsu T. 2007. Selective elimination of aphid endosymbionts: effects of antibiotic dose and host genotype, and fitness consequences. FEMS Microbiol Ecol 60:229–239. doi: 10.1111/j.1574-6941.2007.00284.x. [DOI] [PubMed] [Google Scholar]
- 68.Pais R, Lohs C, Wu Y, Wang J, Aksoy S. 2008. The obligate mutualist Wigglesworthia glossinidia influences reproduction, digestion, and immunity processes of its host, the tsetse fly. Appl Environ Microbiol 74:5965–5974. doi: 10.1128/AEM.00741-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.McLean AH, van Asch M, Ferrari J, Godfray HC. 2011. Effects of bacterial secondary symbionts on host plant use in pea aphids. Proc Biol Sci 278:760–766. doi: 10.1098/rspb.2010.1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zaidman-Rémy A, Vigneron A, Weiss BL, Heddi A. 2018. What can a weevil teach a fly, and reciprocally? Interaction of host immune systems with endosymbionts in Glossina and Sitophilus. BMC Microbiol 18:150. doi: 10.1186/s12866-018-1278-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Davis MR, Jr, Goldberg JB. 2012. Purification and visualization of lipopolysaccharide from Gram-negative bacteria by hot aqueous-phenol extraction. J Vis Exp 2012:3916. doi: 10.3791/3916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Schlechter RO, Jun H, Bernach M, Oso S, Boyd E, Muñoz-Lintz DA, Dobson RCJ, Remus DM, Remus-Emsermann MNP. 2018. Chromatic bacteria − a broad host-range plasmid and chromosomal insertion toolbox for fluorescent protein expression in bacteria. Front Microbiol 9:3052. doi: 10.3389/fmicb.2018.03052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lee HH, Ostrov N, Wong BG, Gold MA, Khalil AS, Church GM. 2019. Functional genomics of the rapidly replicating bacterium Vibrio natriegens by CRISPRi. Nat Microbiol 4:1105–1113. doi: 10.1038/s41564-019-0423-8. [DOI] [PubMed] [Google Scholar]
- 74.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Blattner FR, Plunkett G, III, Bloch CA, Perna NT, Burland V, Riley M, Collado-Vides J, Glasner JD, Rode CK, Mayhew GF, Gregor J, Davis NW, Kirkpatrick HA, Goeden MA, Rose DJ, Mau B, Shao Y. 1997. The complete genome sequence of Escherichia coli K-12. Science 277:1453–1462. doi: 10.1126/science.277.5331.1453. [DOI] [PubMed] [Google Scholar]
- 76.Silipo A, Molinaro A. 2010. The diversity of the core oligosaccharide in lipopolysaccharides. Subcell Biochem 53:69–99. doi: 10.1007/978-90-481-9078-2_4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Effect of a galU mutation on LPS composition and P1 infectivity in S. praecaptivus. (A) Schematic representation depicting the inner membrane, peptidoglycan, and outer membrane of a prototypical Gram-negative bacterium. The inset shows a more detailed schematic of structural components of the LPS (left-hand side). Silver-stained sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) of LPS purified from S. praecaptivus galU (CMK36), wild-type S. praecaptivus, and wild-type S. glossinidius (right-hand side). (B) Plates depicting the appearance of chloramphenicol-resistant colonies as functions of the concentration of bacteria (vertical axis) and the concentration of P1CMclr-100(ts) lysates (horizontal axis). Cmr colonies emerge at similar frequencies in wild-type S. praecaptivus (left-hand side plate) and S. praecaptivus galU (CMK36) (right-hand side plate) when cells are exposed to equal amounts of P1CMclr-100(ts) particles. Download FIG S1, TIF file, 2.4 MB (2.5MB, tif) .
Copyright © 2021 Keller et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Schematic representation of P1 phagemids used for tagging the bacterial chromosome at the Tn7 attachment site(s) with genes encoding the fluorescent proteins mCardinal (pP1-Tn7-mCardinal) or mVenus (pP1-Tn7-mVenus) and the suicide P1 phagemid for random mutagenesis with a Himar1 transposition system (pP1-Himar). Download FIG S2, TIF file, 2.6 MB (2.7MB, tif) .
Copyright © 2021 Keller et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Microbial strains, phages, and plasmids used in this study. Download Table S1, XLSX file, 0.01 MB (15.9KB, xlsx) .
Copyright © 2021 Keller et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Oligonucleotide sequences used in this study. Download Table S2, XLSX file, 0.01 MB (15.4KB, xlsx) .
Copyright © 2021 Keller et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.