

Abstract
To better understand the genes with altered expression caused by infection with the novel coronavirus strain SARS-CoV-2 causing COVID-19 infectious disease, a tensor decomposition (TD)-based unsupervised feature extraction (FE) approach was applied to a gene expression profile dataset of the mouse liver and spleen with experimental infection of mouse hepatitis virus, which is regarded as a suitable model of human coronavirus infection. TD-based unsupervised FE selected 134 altered genes, which were enriched in protein-protein interactions with orf1ab, polyprotein, and 3C-like protease that are well known to play critical roles in coronavirus infection, suggesting that these 134 genes can represent the coronavirus infectious process. We then selected compounds targeting the expression of the 134 selected genes based on a public domain database. The identified drug compounds were mainly related to known antiviral drugs, several of which were also included in those previously screened with an in silico method to identify candidate drugs for treating COVID-19.
Keywords: COVID-19, feature extraction, gene expression profile, SARS-CoV-2, tensor decomposition, in silico drug discovery
I. Introduction
THE current pandemic of COVID-19 caused by infection of the new coronavirus strain SARS-CoV-2 is a severe public health problem that must be resolved as soon as possible. To achieve this goal, it is essential to understand the mechanism by which SARS-CoV-2 successfully invades human cells. Although there are many in silico trials for repositioning drugs toward COVID-19 [1]–[3], most of them are to try to find compounds that bind to SARS-CoV-2 proteins with in silico method. On the other hand, we previously identified drug candidate compounds using gene expression profiles of diseases [4], [5]. This strategy can be also applicable to COVID-19. Recently, Pfaender et al. [6] demonstrated that host lymphocyte antigen 6 (LY6E) complex impairs coronavirus fusion and confers immune control of viral disease. The authors also used a transcriptome approach to evaluate the effect of infection of mouse hepatitis virus (MHV), a natural mouse pathogen that causes hepatitis and encephalomyelitis, which is a well-studied model of coronavirus infection. Although they found many pathways that were disturbed after MHV infection, they did not perform a detailed analysis of the genes with altered expression in response to MHV infection.
In this study, we applied tensor decomposition (TD)-based unsupervised feature extraction (FE) [7] to identify genes with altered expression by MHV infection as a model of coronavirus. We further performed functional enrichment on the selected genes to determine their potential associations with coronavirus infection processes, and screened candidate drug compounds targeting these genes. Overall, this work expands TD formalism by exploring the interpretation of six-dimensional tensors in an infectious disease context. Moreover, we demonstrate a novel application of TD to facilitate the drug discovery process, which can offer a valuable resource for researchers to obtain mechanistic insight for identifying effective drugs for infectious diseases such as COVID-19.
The reason why TD was primary employed to be applied to the present data set that we investigated is because the data set was formatted as a six-mode tensor. Since TD is the most famous method to be used to attack the data set formatted as tensor, it is natural to apply TD to it. Although any other methods might be applicable, they will be tried only when TD fails (and as can be seen in the below, it is not the case in this study; TD can work quite well). In addition to this, our proposed approach, TD based unsupervised FE [7], was known to be applicable to wide range of genomics study. Thus, the purpose of this study is to propose a different tensor formulation dealing with such different tensor data representation and to estimate how well the existing method can work to fulfill a new requirement; Using the experiments of mice infected by virus that is related to SARS-CoV-2, but is not SARS-CoV-S itself, identifying critical human genes that play important roles when SARS-CoV-2 infects human lung. The key point is if we can outperform the previous study where SARS-CoV-2 infected human lung cell line [8]. If we can derive the better results than those using MHV infected mouse data set, it is considered a remarkable achievement (and we could do this as can be seen in the below). It is worth noting that our TD employs “samples + genotypes + tissues + treatments + biological replicates + technical replicates” structure that requires a six-axes tensor representation to identify critical human genes and effective drugs for SARS-CoV-2 infection. Reported results via enrichment analysis show the superiority of our TD, attributed to taking into account the relationships among samples, genotypes, tissues, treatments, biological replicates, and technical replicates at once”
II. Materials and Methods
A. Gene Expression Profile Dataset
The gene expression profile was downloaded from the Gene Expression Omnibus (GEO) dataset GSE146074. This dataset comprises the gene expression profiles of the liver and spleen from female mice experimentally infected with MHV or injected with phosphate-buffered saline (PBS) as a control group for comparison. This experiment was performed with mice of two genetic backgrounds, including wild-type (WT) mice and an textit Ly6e-knockout (KO) mutant strain. The number of replicates for each group are listed in Table I. Seventy-two files whose names start with “GSM” (processed file) were used for the analyses.
TABLE I. Number of Biological Replicates. Two Technical Replicates are Available for Each Biological Replicate.
| PBS day 5 | MHV day 3 | MHV day 5 | ||||
|---|---|---|---|---|---|---|
| liver | spleen | liver | spleen | liver | spleen | |
| WT | 3 | 3 | 3 | 3 | 3 | 3 |
| KO | 3 | 3 | 3 | 3 | 3 | 3 |
B. Additional Gene Expression Profile Datasets
In order to validate the suitability of MHV as model SARS-CoV-2 infectious process, two additional gene expression profiles of mouse lung SARS-CoV infectious processes were used (Table II). For GSE33266 and GSE50000, we downloaded two files: GSE33266_series_matrix.txt.gz and GSE50000_series_matrix.txt.gz. Although GSE50000 also includes files for SARS-CoV-MA15, they were omitted for reasons detailed in the Discussion section. For cases with less than five biological replicates, we used some replicates more than once, in order to have five biological replicates for individual cases.
TABLE II. Number of Biological Replicates of SARS-CoV Infection Toward Mouse Lung Gene Expression Profiles.
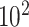
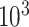
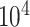
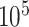
C. TD-Based Unsupervised FE
Although the TD-based unsupervised FE was fully described in the recent book [7], we briefly outline the analysis flow. At first, TD is applied to tensor and singular value vectors are obtained. Individual singular value vectors are attributed to either various experiments or genes. By investigating singular value vectors attributed to experiments, we identify which ones are associated with properties of interest. Then among singular value vectors attributed to genes, those coincident with identified singular value vectors attributed to experiments are selected. Finally, genes having larger contributions toward selected singular value vectors attributed to genes are selected as those associated with the properties of interest.
Fig. 1 shows the flowchart of TD-based unsupervised FE.
Fig. 1.

Flowchart of TD-based unsupervised FE. No dependency: independent of 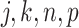 , dependency: dependent upon
, dependency: dependent upon 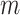 . Singular value vectors with
. Singular value vectors with 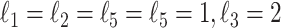 are selected.
are selected.
The gene expression profile dataset was formatted as a tensor, 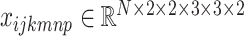 , which represents the expression level of the
, which represents the expression level of the 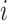 th gene of the
th gene of the 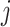 th genotype (
th genotype (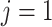 :KO,
:KO, 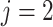 :WT) of the
:WT) of the 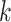 th tissue (
th tissue (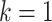 :liver,
:liver, 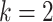 :spleen) of the
:spleen) of the 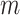 th treatment group (
th treatment group (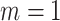 :PBS day 5,
:PBS day 5, 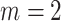 :MHV day 3,
:MHV day 3, 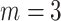 :MHV day 5) for the
:MHV day 5) for the 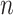 th biological replicate (
th biological replicate (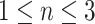 ) and
) and 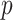 th technical replicate (
th technical replicate (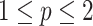 ).
).
The TD is therefore expressed as
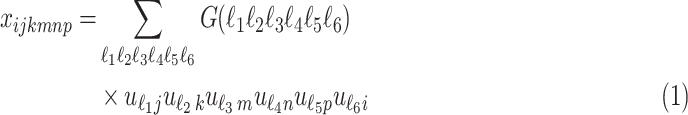 |
where 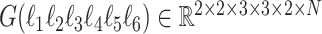 is a core tensor, and
is a core tensor, and 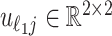 ,
, 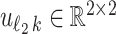 ,
, 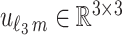 ,
, 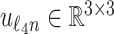 ,
, 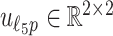 , and
, and 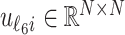 are singular values vectors, which can be obtained via the higher-order singular value decomposition (HOSVD) algorithm [7].
are singular values vectors, which can be obtained via the higher-order singular value decomposition (HOSVD) algorithm [7].
To select 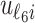 , attributed to selected genes, we need to select
, attributed to selected genes, we need to select 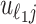 attributed to the genotype,
attributed to the genotype, 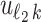 attributed to the tissue,
attributed to the tissue, 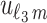 attributed to the treatment,
attributed to the treatment, 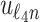 attributed to the biological replicate, and
attributed to the biological replicate, and 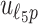 attributed to the technical replicate, associated with desired properties.
attributed to the technical replicate, associated with desired properties.
For this study, we sought to identify genes whose expression is independent of the mouse genotype, tissue type, and replicate. Thus, 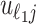 ,
, 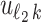 ,
, 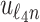 , and
, and 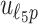 should be independent of
should be independent of 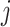 ,
, 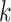 ,
, 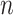 , and
, and 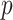 .
.
By contrast, we require 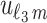 to be dependent on
to be dependent on 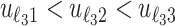 or vice versa. This is because
or vice versa. This is because 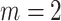 (3 days after MHV infection) must be between
(3 days after MHV infection) must be between 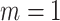 (5 days after PBS injection as the control) and
(5 days after PBS injection as the control) and 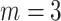 (5 days after MHV infection).
(5 days after MHV infection).
After selecting 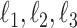 ,
, 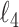 , and
, and 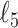 based on the above considerations, we selected
based on the above considerations, we selected 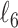 associated with
associated with 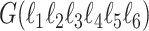 as the largest absolute value, with fixed
as the largest absolute value, with fixed 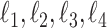 , and
, and 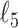 values. Using the selected
values. Using the selected 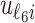 , The
, The  -values,
-values,  s, were attributed to gene expression levels as
s, were attributed to gene expression levels as
 |
where 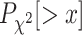 is the cumulative probability distribution of the
is the cumulative probability distribution of the 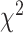 distribution when the argument is larger than
distribution when the argument is larger than 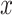 and
and 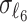 is the standard deviation of
is the standard deviation of 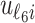 .
.
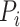 s were adjusted by the Benjamini and Hochberg (BH) criterion [7], and only genes associated with an adjusted
s were adjusted by the Benjamini and Hochberg (BH) criterion [7], and only genes associated with an adjusted 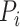 less than 0.01 were selected for further analysis.
less than 0.01 were selected for further analysis.
We employed almost the same procedures, apart from different tensor fromats for the two additional gene expression profiles of mouse lung SARS-CoV infectious processes. The tensors formatted and TDs are for GSE33266
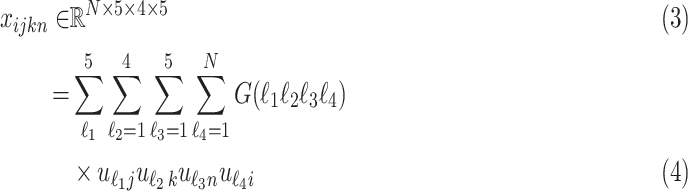 |
which represents the 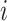 th gene expression profile of
th gene expression profile of 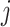 th experiments (
th experiments (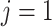 :Mock,
:Mock, 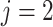 :
: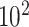 pfu,
pfu,  :
: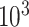 pfu,
pfu,  :
: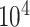 pfu,
pfu, 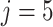 :
: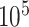 pfu) at the
pfu) at the 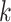 th day after infection (
th day after infection (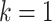 :D1, day 1,
:D1, day 1, 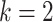 :D2, day 2,
:D2, day 2, 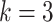 :D4, day 4,
:D4, day 4, 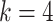 :D7, day 7) of the
:D7, day 7) of the 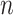 th biological replicate (
th biological replicate (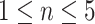 ).
). 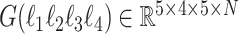 is a core tensor that represents the weight of products of the singular value matrices
is a core tensor that represents the weight of products of the singular value matrices 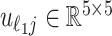 ,
, 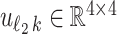 ,
, 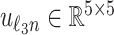 , and
, and 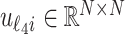 , which are all orthogonal matrices. Those for GSE50000 are
, which are all orthogonal matrices. Those for GSE50000 are
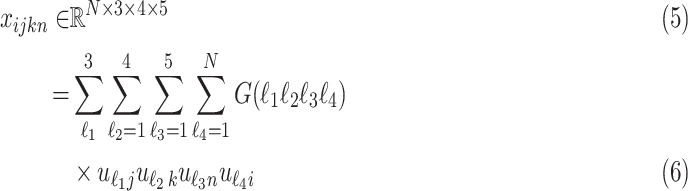 |
which represents the 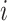 th gene expression profile of the
th gene expression profile of the 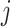 th experiments (
th experiments (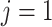 :BatSRBD,
:BatSRBD, 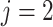 :icSARS,
:icSARS, 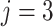 :Mock) at the
:Mock) at the 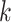 th day after infection (
th day after infection (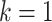 :d1, day 1,
:d1, day 1, 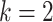 :d2, day 2,
:d2, day 2, 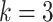 :d4, day 4,
:d4, day 4, 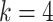 :d7, day 7) of the
:d7, day 7) of the 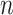 th biological replicate (
th biological replicate (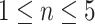 ).
). 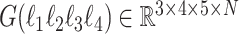 is a core tensor that represents the weight of products of singular value matrices,
is a core tensor that represents the weight of products of singular value matrices, 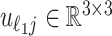 ,
, 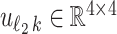 ,
, 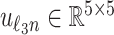 , and
, and 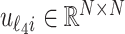 which are all orthogonal matrices.
which are all orthogonal matrices.
The criteria for the selection of singular value vectors were as follows. For GSE33266, 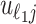 should be a monotonic function of
should be a monotonic function of 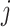 , since it represents the strength of infection,
, since it represents the strength of infection, 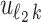 should also be a monotonic function of
should also be a monotonic function of 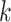 , since it represents time development, and
, since it represents time development, and 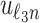 should be constant, since biological replicates should not differ from one another. For GSE50000,
should be constant, since biological replicates should not differ from one another. For GSE50000, 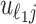 should be distinct between
should be distinct between 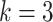 and
and 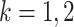 , since it represents the distinction between mock and real infection,
, since it represents the distinction between mock and real infection, 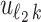 also should be a monotonic function of
also should be a monotonic function of 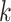 , since it represents time development, and
, since it represents time development, and 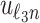 should be constant, since biological replicates should not differ from one another.
should be constant, since biological replicates should not differ from one another.
Downstream procedures for gene selection after identifying singular value vectors are the same as those for MHV. The core tensors, 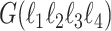 s, were investigated in order to see which
s, were investigated in order to see which 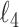 associated with
associated with 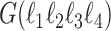 having larger absolute values given
having larger absolute values given 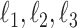 .
. 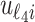 s selected are used for attributing
s selected are used for attributing  -values,
-values, 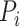 , to genes. Genes associated with adjusted
, to genes. Genes associated with adjusted  -values less than 0.01 were selected.
-values less than 0.01 were selected.
D. Enrichment Analysis
Gene symbols of genes selected by TD-based unsupervised FE with significantly altered expression due to MHV infection were uploaded to Enricher [9] and Metascape [10], which are popular enrichment analysis servers that evaluate the biological properties of genes based on enrichment analysis. There are some explanations about individual categories of Enrichr.
1). Virus-Host PPI P-HIPSTer2020
This is a list of human proteins known to interact with various virus proteins. By comparing uploaded genes with genes in the list, we can estimate the ratio of genes interacting with virus proteins.
2). Virus Perturbations From GEO up/down
From GEO, gene expression profiles of virus infections are retrieved. Then genes associated with altered expression are identified. By comparing uploaded genes with genes in the list, we can estimate the ratio of genes whose expression is altered by virus infection.
3). Drugmatrix
In DrugMatrix data base, gene expression profiles of rat tissues treated with various drugs are recorded. By comparing uploaded genes with genes in the list, we can estimate the ratio of genes whose expression is altered by drug treatment.
4). Drug Perturbations From GEO up/down
From GEO, gene expression profiles of drug treatments are retrieved. Then genes associated with altered expression are identified. By comparing uploaded genes with genes in the list, we can estimate the ratio of genes whose expression is altered by drug treatment.
III. Results
Fig. 2 shows the overview of analyses performed in this study.
Fig. 2.

Overview of analyses.
A. Synthetic Data Sets
In order to demonstrate how effective the tensor is, we employed synthetic data sets composed of multiway and multiclass labels, 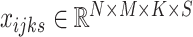 . Here,
. Here, 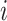 represents
represents 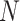 variables, among which partial collections having distinct values between
variables, among which partial collections having distinct values between 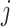 s as well as
s as well as 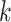 s must be selected, where
s must be selected, where 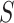 replicates are available for individual combinations of
replicates are available for individual combinations of 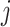 and
and 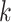 . A typical example is that
. A typical example is that 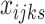 represents the expression of the
represents the expression of the 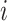 th gene in the
th gene in the 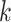 th tissue of patients who belong to the
th tissue of patients who belong to the 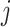 th group, that consists of
th group, that consists of 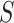 patients. We then need to identify which genes are expressed distinctly in tissue (
patients. We then need to identify which genes are expressed distinctly in tissue (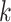 ) -specific as well as patient-group (
) -specific as well as patient-group (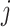 ) specific ways.
) specific ways.
The most popular approach to this problem is linear regression,
 |
where 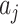 and
and 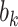 are pre-defined variables that represent some properties of the
are pre-defined variables that represent some properties of the 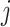 th patient group and
th patient group and 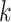 th tissue, respectively.
th tissue, respectively. 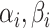 , and
, and 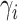 are regression coefficients that are selected such that the discrepancy between both sides of equation is minimized. Then the
are regression coefficients that are selected such that the discrepancy between both sides of equation is minimized. Then the 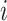 s associated with significant
s associated with significant 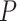 -values are selected as those whose expression levels are different between distinct
-values are selected as those whose expression levels are different between distinct 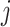 s and
s and 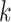 s.
s.
It is not guaranteed that liner regression will correctly represent the dependency of 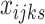 upon
upon 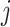 and
and 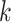 . We generated synthetic data that follows
. We generated synthetic data that follows
 |
where 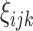 are drawn from
are drawn from 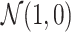 for every combination of
for every combination of 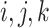 , so that
, so that 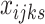 s associated with distinct
s associated with distinct 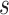 s share the same
s share the same 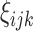 , and
, and 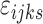 are drawn from
are drawn from 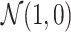 for every combination of
for every combination of 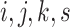 .
. 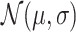 represents a normal distribution that has mean of
represents a normal distribution that has mean of 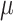 and standard deviation of
and standard deviation of 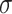 . For
. For 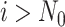 ,
, 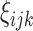 is taken to be zero; this means that
is taken to be zero; this means that 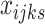 for
for  is simply random variables. The task is to correctly select
is simply random variables. The task is to correctly select  variables associated with the dependence upon
variables associated with the dependence upon 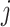 and
and 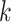 .
.
As an alternative regression analysis to linear regression, we employed
 |
which reflects the multiplicative nature when 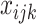 is generated with eq. (8) although it cannot represent
is generated with eq. (8) although it cannot represent 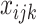 completely since
completely since 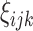 is replaced with
is replaced with 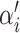 and therefore dependence upon
and therefore dependence upon 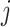 or
or 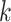 is not assumed.
is not assumed.
In addition to the above two regression analyses, we applied categorical regression, which is equivalent to analysis of variance (ANOVA):
 |
where 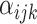 and
and 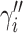 are regression coefficients. Eq. (10) can fully reproduce the
are regression coefficients. Eq. (10) can fully reproduce the 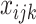 generated by eq. (8), when
generated by eq. (8), when 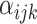 is taken to be
is taken to be 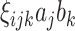 excluding randomness introduced by
excluding randomness introduced by 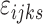 , which can be regarded as residuals.
, which can be regarded as residuals.
Finally, we also applied TD based unsupervised FE to 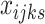 . TD is computed as
. TD is computed as
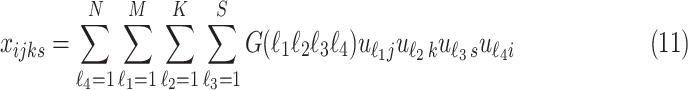 |
Then 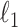 and
and 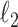 , that have the largest absolute values of the correlation coefficient between
, that have the largest absolute values of the correlation coefficient between 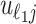 and
and 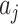 or
or 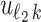 and
and 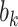 were selected. The top two
were selected. The top two 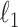 that have the largest absolute
that have the largest absolute 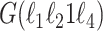 s were selected. The reason why
s were selected. The reason why 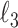 is fixed to be 1 is because
is fixed to be 1 is because 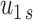 always represent the
always represent the 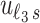 that lacks
that lacks 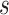 dependency, i.e., is a constant independent of
dependency, i.e., is a constant independent of 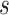 . Since
. Since 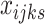 should take the same values between replicates,
should take the same values between replicates, 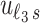 that do not have any
that do not have any 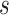 dependence are selected. Then
dependence are selected. Then  -values are attributed to
-values are attributed to  as
as
 |
where summation is taken over two selected 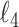 only.
only.
No matter which form of linear regression, eq.(7), eq. (9), categorical regression, eq.(10), or TD, eq.(11), was used to compute 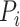 ,
, 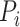 was corrected using the BH criterion [7] and
was corrected using the BH criterion [7] and 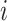 s having corrected
s having corrected 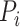 less than threshold
less than threshold 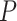 -values were selected as those associated with dependency upon
-values were selected as those associated with dependency upon 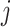 and
and 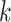 , respectively.
, respectively.
For simplicity, we employed 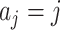 and
and 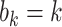 when generating
when generating 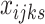 using eq. (8) as well as eq. (9) for regression analysis. Specifically, we chose
using eq. (8) as well as eq. (9) for regression analysis. Specifically, we chose 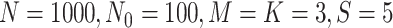 . Generation of
. Generation of 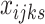 and selection of
and selection of 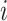 s were repeated 100 times with two distinct threshold
s were repeated 100 times with two distinct threshold 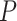 -values, 0.01 or 0.1, for each trial. Table III shows the performance averaged over 100 trials. Although categorical regression, eq. (10), could outperform the other three approaches, since it is able to completely reproduce eq. (8) as shown above, it has one weak point: it cannot explicitly consider the dependence of
-values, 0.01 or 0.1, for each trial. Table III shows the performance averaged over 100 trials. Although categorical regression, eq. (10), could outperform the other three approaches, since it is able to completely reproduce eq. (8) as shown above, it has one weak point: it cannot explicitly consider the dependence of 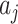 and
and 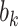 upon
upon 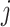 and
and 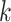 , since
, since 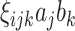 s are estimated as one parameter,
s are estimated as one parameter, 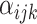 . This limitation might prevent us from selecting
. This limitation might prevent us from selecting 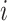 s that are specifically associated with the dependence upon
s that are specifically associated with the dependence upon 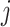 and
and 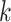 that
that 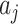 and
and 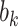 represent. Even if some
represent. Even if some 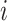 s are selected, it might be because of dependence upon
s are selected, it might be because of dependence upon 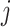 and
and 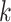 that
that 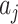 and
and 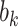 do not represent. There is no way for us to check this point. Taking this problem into account, TD based unsupervised FE, which is the second best approach, is more useful than categorical regression, since TD based unsupervised FE can consider
do not represent. There is no way for us to check this point. Taking this problem into account, TD based unsupervised FE, which is the second best approach, is more useful than categorical regression, since TD based unsupervised FE can consider 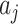 and
and 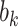 when selecting
when selecting 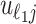 and
and 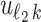 correlated with
correlated with 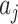 and
and 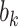 .
.
TABLE III. The Confusion Matrices Obtained Using Synthetic Data (eq.(8), 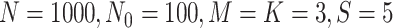 ) and Cpu Time Required for Each Method.
) and Cpu Time Required for Each Method.  s are Threshold
s are Threshold  -Values;
-Values;  Associated With Adjusted
Associated With Adjusted  -Vales Less Than This Threshold Values are Selected.
-Vales Less Than This Threshold Values are Selected.
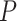
|
0.01 | 0.1 | ||
|---|---|---|---|---|
| TD based unsupervised FE (eq. (11), cpu time 6.5 sec) | ||||
| not selected | selected | not selected | selected | |
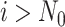
|
900 | 62.2 | 899.9 | 48.44 |
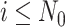
|
0 | 37.8 | 0.1 | 51.56 |
| liner regression (eq. (7), cpu time 65.7 sec) | ||||
| not selected | selected | not selected | selected | |
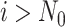
|
899.7 | 75.39 | 894.0 | 58.31 |
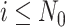
|
0.3 | 24.61 | 6.0 | 41.69 |
| categorical regression (cpu time 70.4 sec) | ||||
| not selected | selected | not selected | selected | |
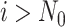
|
897.3 | 0 | — | — |
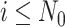
|
2.7 | 100 | — | — |
| eq. (9) (cpu time 84.1 sec) | ||||
| not selected | selected | not selected | selected | |
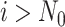
|
900 | 84.55 | 899.3 | 74.0 |
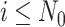
|
0 | 14.55 | 0.7 | 26.0 |
Another advantage of TD based unsupervised FE is the short CPU time required for execution (Table III). The CPU time required for TD based unsupervised FE is approximately one tenth that of other three methods, which must repeat the regression analysis 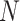 times. This difference might be important when dealing with massive data sets.
times. This difference might be important when dealing with massive data sets.
TD based unsupervised FE can consider the dependence upon 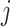 and
and 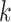 of
of 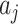 and
and 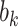 , although other methods cannot because of the random nature of
, although other methods cannot because of the random nature of 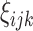 in eq. (8). When an individual
in eq. (8). When an individual 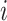 is considered, there are no ways to take into account the dependence upon
is considered, there are no ways to take into account the dependence upon 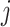 and
and 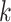 that
that 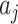 and
and 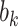 have. However, when
have. However, when 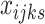 s are averaged over multiple
s are averaged over multiple 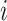 s, there is a possibility that the dependence upon
s, there is a possibility that the dependence upon 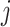 and
and 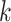 of
of 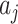 and
and 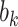 can appear, since the randomness of
can appear, since the randomness of 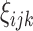 can be smeared out because of averaging. In practice,
can be smeared out because of averaging. In practice, 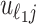 and
and 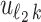 can be such variables that can appear only after averaging, and can represent the dependence upon
can be such variables that can appear only after averaging, and can represent the dependence upon 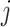 and
and 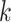 of
of 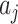 and
and 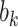 . This is why TD based unsupervised FE can outperform eq. (9), which explicitly considers the multiplicative nature when
. This is why TD based unsupervised FE can outperform eq. (9), which explicitly considers the multiplicative nature when 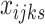 s are generated, and can consider the dependence upon
s are generated, and can consider the dependence upon 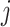 and
and 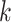 of
of 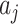 and
and 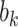 , which categorical regression, eq.(10), cannot.
, which categorical regression, eq.(10), cannot.
B. Selection of Genes
We applied TD based unsupervised FE to the gene expression profiles introduced in the Materials and Methods section. Then other methods were applied to the synthetic data set to form a basis for the comparisons discussed in the Discussions and Conclusions section.
We selected 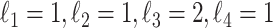 , and
, and 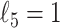 based on the criteria described above (Fig. 3), and
the associated
based on the criteria described above (Fig. 3), and
the associated 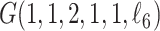 values are listed in Table IV, demonstrating the largest value for
values are listed in Table IV, demonstrating the largest value for 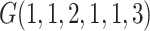 . The associated
. The associated 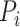 values were computed using
values were computed using 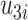 as shown in eq. (2), resulting in selection of 134 genes altered in MHV infection with adjusted P-values less than 0.01 (Table V). Although some mouse-specific genes were included in this list (e.g., genes with symbols starting with “mt”), since there were still several gene symbols that are common between human and mice, we decided to evaluate the potential association of all 134 genes with the infection process of coronavirus.
as shown in eq. (2), resulting in selection of 134 genes altered in MHV infection with adjusted P-values less than 0.01 (Table V). Although some mouse-specific genes were included in this list (e.g., genes with symbols starting with “mt”), since there were still several gene symbols that are common between human and mice, we decided to evaluate the potential association of all 134 genes with the infection process of coronavirus.
Fig. 3.

Singular value vectors obtained by the HOSVD algorithm. U1: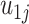 , U2:
, U2: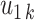 , U3:
, U3: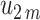 , U4:
, U4: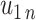 , and U5:
, and U5: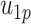 . See Materials and Methods for the meanings of
. See Materials and Methods for the meanings of 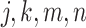 , and
, and 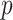 . They are independent of genotypes (j), tissues (k), biological replicates (n) and technical replicates (p), but dependent upon infection (m).
. They are independent of genotypes (j), tissues (k), biological replicates (n) and technical replicates (p), but dependent upon infection (m).
TABLE IV. 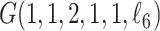 s Computed by the HOSVD Algorithm.
s Computed by the HOSVD Algorithm.
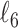
|
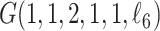
|
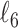
|
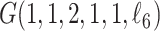
|
|---|---|---|---|
| 1 | -11.846 381 | 6 | 22.375 546 |
| 2 | -28.104 674 | 7 | -41.997 092 |
| 3 | 312.362 569 | 8 | -9.048 416 |
| 4 | -71.001 444 | 9 | 9.212 773 |
| 5 | -189.719 321 | 10 | 3.394 629 |
TABLE V. One Hundred and Thirty Four Genes Selected by TD-Based Unsupervised FE.
| Actb Actg1 Ahsg Alb Ambp Apoa1 Apoa2 Apoc1 Apoe B2m Bst2 C3 Ccnb1ip1 Cd74 Cfb Eef1a1 Eef1g Eef2 Fabp1 Fau Fga Fgb Fgg Fth1 Ftl1 Gapdh Gc Gm10800 Gm2000 Gpx1 H2-Aa H2-D1 H2-K1 H2-T23 Hamp Hba-a2 Hbb-bs Hbb-bt Hist1h3b Hist1h4h Hist2h2aa2 Hp Hpx Hsp90ab1 Hsp90b1 Hspa8 Ifi27l2a Ifitm3 Lars2 Lcn2 Lyz2 mt-Atp6 mt-Atp8 mt-Co1 mt-Co2 mt-Co3 mt-Cytb mt-Nd1 mt-Nd2 mt-Nd3 mt-Nd4 Mt1 Mt2 Myh9 Orm1 Orm2 Pabpc1 Psap Ptma Rack1 Rpl10-ps3 Rpl11 Rpl12 Rpl13 Rpl13a Rpl14 Rpl17 Rpl19 Rpl23a Rpl26 Rpl3 Rpl32 Rpl36 Rpl36a Rpl37a Rpl38 Rpl4 Rpl41 Rpl5 Rpl6 Rpl7 Rpl7a Rpl8 Rplp0 Rplp1 Rplp2 Rps11 Rps12 Rps14 Rps15 Rps17 Rps18 Rps2 Rps21 Rps23 Rps24 Rps27a Rps27rt Rps29 Rps3 Rps3a1 Rps4x Rps5 Rps6 Rps7 Rps8 Rps9 Rpsa S100a8 S100a9 Saa1 Saa2 Serpina1a Serpina1b Serpina1c Serpina1d Serpina3k Tmsb4x Tpt1 Trf Ttr Ubb Ubc Wfdc21 |
In order to see if we could correctly select differentially expressed genes between infected mice and control, we applied t test to expression of 134 genes between infected and control mouse. Then we have found that the null hypothesis that gene expression averaged over selected 134 genes are equal between infected and control mouse was rejected by the 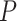 -values of 0.004. Thus we could successfully identify expressed genes between infected mice and control.
-values of 0.004. Thus we could successfully identify expressed genes between infected mice and control.
C. Protein-Protein Interaction With Coronavirus Infection
We first evaluated whether the 134 selected genes could reflect the process of coronavirus infection using the Enrichr server for functional enrichment analysis. Several of the genes were enriched in the category “Virus-Host PPI P-HIPSTer 2020,” which is related to SARS-CoV (Table VI, see the supplementary materials for the full list).
TABLE VI. SARS-CoV-Related Virus PPI in Enrichr.
| Term | Overlap | P-value | Adjusted P-value |
|---|---|---|---|
| SARS coronavirus excised_polyprotein 1..4369 (gene: orf1ab) | 10/194 |
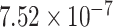
|
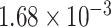
|
| SARS coronavirus P2 full_polyprotein 1..4382 | 10/198 |
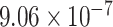
|
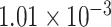
|
| SARS coronavirus nsp7-pp1a/pp1ab (gene: orf1ab) | 4/36 |
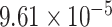
|
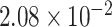
|
| SARS coronavirus 3C-like protease (gene: orf1ab) | 3/19 |
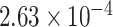
|
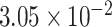
|
These genes were also related to ORF1ab, polyprotein, and 3C-like protease. Interestingly, Woo et al. [11] suggested that ORF1ab, which encodes a replicase polyprotein of CoV-HKU1, is cleaved by papain-like proteases and 3C-like proteinase. Thus, it is reasonable that ORF1ab, polyprotein, and 3C-like protease would be affected during MHV infection VI. Other PPIs detected that are not listed in Table VI (see supplementary materials) were also mainly associated with ORF1ab and polyproteins, suggesting that our strategy has clear capability to elucidate the basic infectious process at the molecular level that is common among various coronaviruses.
D. Virus Perturbation
We next evaluated whether genes with known altered expression by virus perturbation overlapped with the 134 genes selected by our TD-based unsupervised FE approach (Table VII; see the supplementary material for the full list).
TABLE VII. Virus Perturbation in Enricher.
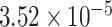
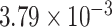
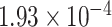
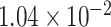
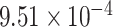
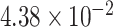
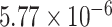
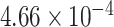
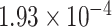
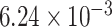
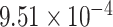
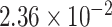
Among these, we detected the overlap of many genes that are pertubated in response to either SARS-CoV or SARS-like bat CoV, which are the genetically closest coronaviruses to the new SARS-CoV-2 strain. This further suggests that our results could have high similarity to the genes perturbated in SARS-CoV-2 infection.
E. TMPRSS2 as a Scavenger Receptor
For further functional enrichment analysis, we uploaded the 134 selected genes to Metascape to identify non-redundant biological terms (Fig. 4). Among the terms identified, “R-HSA-2 173 782: Binding and Uptake of Ligands by Scavenger Receptors” was the third most significantly enriched term. Although it was initially surprising that a scavenger receptor might be related to the response to coronavirus infection, a search of the related literature revealed that the scavenger receptor TMPRSS2 plays a critical role in SARS-CoV-2 infection as well as SARS-CoV infection [12]. Isolation of SARS-CoV-2 was also reported to be enhanced by TMPRSS2-expressing cells [13]. Moreover, TMPRSS2 contains a scavenger receptor domain [14], suggesting that TMPRSS2 activity would be related to detection of scavenger receptor activity. This finding further demonstrates the outstanding capability of our strategy to detect factors related to the SARS-CoV-2 infectious process. Moreover, this analysis suggests that research on the SARS-CoV infection process could be informative for understanding the SARS-CoV-2 infection process when it is not possible to directly investigate SARS-CoV-2 infection.
Fig. 4.

Redundant heatmap of enriched terms generated by uploading the selected 134 genes to Metascape .
F. Drug Discovery
We previously demonstrated that genes selected by TD-based unsupervised FE are useful to screen for drugs that are effective in treating disease or those that may cause adverse effects [15]. Therefore, we used this approach to screen for candidate drugs to treat coronavirus infections based on the individual terms that emerged from the Enrichr analysis.
1). Drug Matrix
In the Enrichr category “DrugMatrix,” the top-ranked drug was related to virus infection (Table VIII; see the supplementary materials for the full list). Most of these viruses are enveloped, single-stranded RNA viruses. Coronaviruses, including SARS-CoV-2, are positive-sense, enveloped, single-stranded RNA viruses, whereas influenza virus is a negative-sense, enveloped, single-stranded RNA virus.
TABLE VIII. Drugs Enriched in the “DrugMatrix” Category in Enrichr. the Full List is Available in the Supplementary Material.
| Term | Overlap | P-value | Adjusted P-value |
|---|---|---|---|
| Primaquine-45 mg/kg in CMC-Rat-Liver-5d-up | 23/315 |
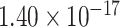
|
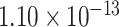
|
| Meloxicam-33 mg/kg in Corn Oil-Rat-Kidney-1d-up | 23/337 |
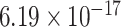
|
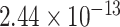
|
| Cytarabine-487 mg/kg in Saline-Rat-Liver-0.25d-up | 22/300 |
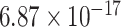
|
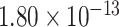
|
| Clotrimazole-60 uM in DMSO-Rat-Primary rat hepatocytes-0.67d-dn | 24/381 |
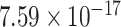
|
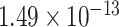
|
| Diclofenac-3.5 mg/kg in Corn Oil-Rat-Liver-5d-dn | 21/269 |
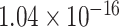
|
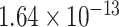
|
| Pyrogallol-1000 mg/kg in Water-Rat-Liver-3d-up | 23/349 |
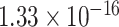
|
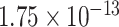
|
| Clindamycin-161 mg/kg in Saline-Rat-Kidney-1d-up | 23/366 |
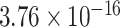
|
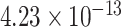
|
| Catechol-195 mg/kg in Saline-Rat-Liver-0.25d-up | 21/290 |
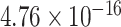
|
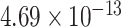
|
| Anisindione-75 mg/kg in CMC-Rat-Liver-5d-up | 21/295 |
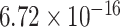
|
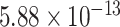
|
| Phenylhydrazine-78 mg/kg in Water-Rat-Liver-3d-up | 22/335 |
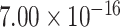
|
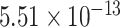
|
| N-Nitrosodiethylamine-1.67 mg/kg in Saline-Rat-Liver-0.25d-up | 23/377 |
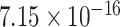
|
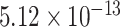
|
| Neomycin-56 mg/kg in Corn Oil-Rat-Liver-1d-dn | 20/259 |
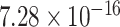
|
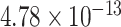
|
Primaquine is known to inhibit the replication of Newcastle disease virus [16], which is in the family of paramyxoviruses that are enveloped, non-segmented, negative-sense single-stranded RNA viruses. Meloxicam is known to have cytotoxic and antiproliferative activity on virus-transformed tumor cells [17], including myelocytomatosis virus and Rous sarcoma virus. Myelocytomatosis virus is a retrovirus, which is an enveloped, negative-sense, single-stranded RNA virus, whereas Rous sarcoma virus is an enveloped, positive-sense, single-stranded RNA virus. Although there are no studies showing that cytarabine is effective against infection of an RNA virus, one report demonstrated that cytarabine can affect DNA virus infection [18]. Pyrogallol was reported to have anti-virus effects on human influenza virus strain A/Udorn/72, avian influenza virus A/swan/Shimane/499/83, herpes simplex virus-1, vesicular stomatitis virus, and retrovirus [19]. As mentioned above, influenza virus is a negative-sense, enveloped, single-stranded RNA virus; herpesvirus is a DNA virus; vesicular stomatitis virus is an enveloped, single-stranded, negative-sense RNA virus; and retroviruses are enveloped, negative-sense, single-stranded RNA viruses. This suggests that a single drug can effectively inhibit a wide range of viruses from DNA viruses to both negative- and positive-sense RNA viruses. The structure-dependent antiviral activity of catechol derivatives in pyroligneous acid against encephalomyocarditis virus was reported, which is a non-enveloped single-stranded RNA virus [20]. To our knowledge, there are no reports that neomycin is effective against RNA viruses; however, one study showed that it could inhibit infection of fibroblasts with human cytomegalovirus [21], which is a DNA virus.
Although not all viruses identified to be related to the 134 genes selected by TD-based unsupervised FE are enveloped, positive-sense, single-stranded RNA viruses similar to SARS-CoV-2, since drugs shown to be effective against other viruses (e.g., DNA viruses) are also often effective against RNA viruses (including pyrogallol that was screened by our strategy), drugs in Table VIII warrant being tested as potential treatments for SARS-CoV-2 infection.
2). Drug Perturbations From GEO
Several promising drug compound candidates were also screened from the GEO “Drug Perturbations from GEO up” and “Drug Perturbations from GEO down” categories, along with available evidence for possible adverse effects (Table IX; see the supplementary material for the full list).
TABLE IX. Drugs Identified in “drug Perturbations From GEO up/down” in Enrichr for the 134 Genes Selected by TD-Based Unsupervised FE.
| Term | Overlap | P-value | Adjusted P-value |
|---|---|---|---|
| Drug Perturbations from GEO up | |||
| coenzyme Q10 5 281 915 mouse GSE15129 sample 3464 | 64/302 |
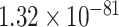
|
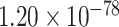
|
| coenzyme Q10 5 281 915 mouse GSE15129 sample 3456 | 63/396 |
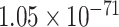
|
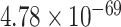
|
| captopril DB01197 mouse GSE19286 sample 2689 | 47/134 |
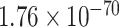
|
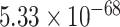
|
| ubiquinol 9 962 735 mouse GSE15129 sample 3463 | 60/346 |
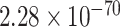
|
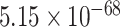
|
| N-METHYLFORMAMIDE 31 254 rat GSE5509 sample 3570 | 56/283 |
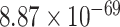
|
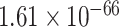
|
| 1-Naphthyl isothiocyanate 11 080 rat GSE5509 sample 3568 | 56/301 |
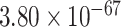
|
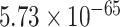
|
| fenretinide 5 288 209 rat GSE3952 sample 3561 | 59/397 |
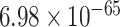
|
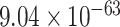
|
| coenzyme Q10 5 281 915 mouse GSE15129 sample 3462 | 50/257 |
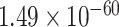
|
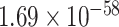
|
| bexarotene DB00307 human GSE6914 sample 2680 | 43/147 |
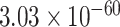
|
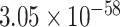
|
| FENRETINIDE 5 288 209 rat GSE3952 sample 3563 | 52/345 |
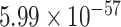
|
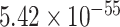
|
| Drug Perturbations from GEO up | |||
| pioglitazone DB01132 rat GSE21329 sample 2841 | 56/321 |
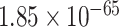
|
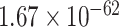
|
| quercetin DB04216 mouse GSE38067 sample 3441 | 59/486 |
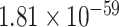
|
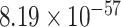
|
| ubiquinol 9 962 735 mouse GSE15129 sample 3461 | 53/349 |
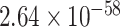
|
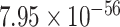
|
| fenretinide 5 288 209 rat GSE3952 sample 3559 | 56/440 |
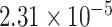 7 7 |
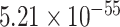
|
| decitabine 451 668 mouse GSE4768 sample 3108 | 45/226 |
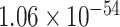
|
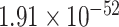
|
| troglitazone DB00197 rat GSE21329 sample 2832 | 50/355 |
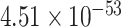
|
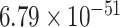
|
| adenosine triphosphate 5957 human GSE30903 sample 3219 | 49/341 |
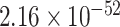
|
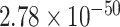
|
| alitretinoin DB00523 rat GSE3952 sample 2673 | 53/483 |
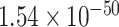
|
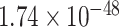
|
| bexarotene 82 146 rat GSE3952 sample 3560 | 48/361 |
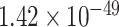
|
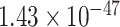
|
| HYPOCHLOROUS ACID 24 341 human GSE11630 sample 3201 | 41/221 |
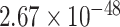
|
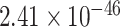
|
| streptozocin DB00428 mouse GSE38067 sample 3439 | 44/287 |
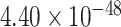
|
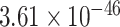
|
| rosiglitazone DB00412 mouse GSE35011 sample 2813 | 44/290 |
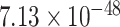
|
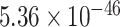
|
| motexafin gadolinium (4 h) DB05428 human GSE2189 sample 3125 | 43/302 |
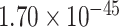
|
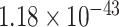
|
Drugs associated with upregulated genes that overlapped with the 134 genes selected by TD-based unsupervised FE are considered to be more likely to cause adverse effects, since they will enhance the expression of genes altered by SARS-CoV infection. Captoprilis is an angiotensin-converting enzyme (ACE) inhibitor, which is known to activate ACE2 that is the receptor that SARS-CoV-2 uses to infect human cells [22], suggesting that this drug might have negative effects for COVID-19 therapy. Coenzyme Q10, which frequently emerged in Table IX, has been reported to accelerate virus infection [23], which could therefore also have negative effects for COVID-19 therapy. Fenretinide is known to effectively inhibit HIV infection [24], and therefore might be a promising drug candidate for SARS-CoV-2 even though it was listed in the “Drug Perturbations from GEO up” category.
In contrast to the drugs in the above list, those associated with downregulated genes that overlapped with the 134 genes selected by TD-based unsupervised FE are considered to be able to effectively suppress SARS-CoV-2 infection, since they will inhibit the expression of genes altered by SARS-CoV infection. Pioglitazone was also included in the list of candidate compounds for SARS-CoV-2 screened by an in silico method [25]. Quercetin was reported to inhibit the cell entry of SARS-CoV-2 [26], and was also included in the list of candidate compounds for SARS-CoV-2 screened by an in silico method [27]. Fenretinide was also included in the drugs identified as effective compounds in the “Drug perturbations from GEO up” category as described above. Decitabine is one of the drugs used in HIV combination therapy [28]. Troglitazone impedes the oligomerization of sodium taurocholate co-transporting polypeptide and entry of hepatitis B virus into hepatocytes [29], which is a partially double-stranded DNA virus. Finally, motexafin gadolinium was reported to selectively induce apoptosis in HIV-1-infected CD4+ T helper cells [30].
Based on these observations, our strategy appears to be useful to identify potential drug compounds for SARS-CoV-2.
G. Comparison With in Silico Drug Discovery
Finally, we compared the drugs screened out using our approach from the “Drug perturbations from GEO up/down” lists with those screened from two in silico drug discovery studies [25], [27]
1). Comparison With Wu et al. [25]
We found multiple hits, which are summarized in Table X.The main drugs identified included doxycycline, ascorbic acid, isotretinoin, pioglitazone, cortisone, and tibolone.
TABLE X. List of in Silico Screened drugs [25] Whose Target Genes Were Also Enriched in the 134 Genes Selected by TD-Based Unsupervised FE.
| Term | Overlap | P-value | Adjusted P-value |
|---|---|---|---|
| Drug Perturbations from GEO up | |||
| doxycycline DB00254 mouse GSE29848 sample 3209 | 32/267 | 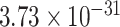 |
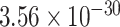 |
| doxycycline DB00254 human GSE2624 sample 3074 | 28/175 | 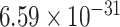 |
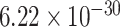 |
| doxycycline DB00254 human GSE2624 sample 3077 | 27/209 | 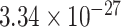 |
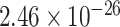 |
| doxycycline DB00254 human GSE2624 sample 3076 | 25/272 | 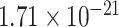 |
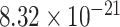 |
| doxycycline DB00254 mouse GSE29848 sample 3207 | 22/225 | 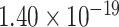 |
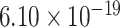 |
| doxycycline DB00254 mouse GSE29848 sample 3208 | 12/291 | 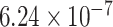 |
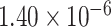 |
| ascorbic acid 54 670 067 human GSE11919 sample 3190 | 8/313 | 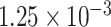 |
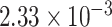 |
| isotretinoin DB00982 human GSE10432 sample 2772 | 8/308 | 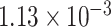 |
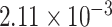 |
| pioglitazone DB01132 rat GSE21329 sample 2843 | 48/400 | 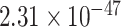 |
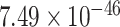 |
| pioglitazone DB01132 rat GSE21329 sample 2842 | 42/349 | 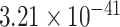 |
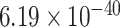 |
| pioglitazone DB01132 rat GSE21329 sample 2842 | 42/349 | 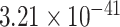 |
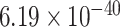 |
| pioglitazone DB01132 rat GSE20219 sample 2794 | 13/292 | 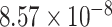 |
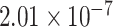 |
| pioglitazone 4829 mouse GSE1458 sample 2587 | 11/318 | 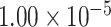 |
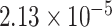 |
| pioglitazone DB01132 mouse GSE32536 sample 2797 | 7/307 | 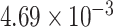 |
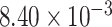 |
| pioglitazone DB01132 rat GSE20219 sample 2795 | 7/330 | 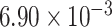 |
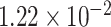 |
| hydrocortisone DB00741 human GSE7890 sample 2751 | 40/305 | 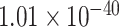 |
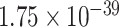 |
| tibolone 444 008 human GSE12446 sample 3204 | 15/287 | 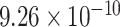 |
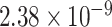 |
| Drug Perturbations from GEO down | |||
| doxycycline DB00254 human GSE2624 sample 3075 | 20/358 | 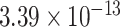 |
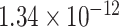 |
| doxycycline DB00254 mouse GSE29848 sample 3208 | 17/309 | 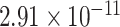 |
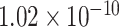 |
| doxycycline DB00254 human GSE2624 sample 3077 | 18/391 | 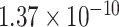 |
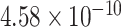 |
| doxycycline DB00254 mouse GSE29848 sample 3207 | 17/375 | 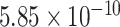 |
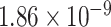 |
| doxycycline DB00254 human GSE2624 sample 3074 | 17/425 | 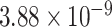 |
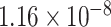 |
| doxycycline DB00254 mouse GSE29848 sample 3209 | 9/333 | 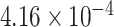 |
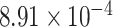 |
| ascorbic acid 54 670 067 human GSE11919 sample 3190 | 19/287 | 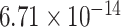 |
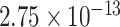 |
| Ascorbic acid 54 670 067 mouse GSE37676 sample 3132 | 15/306 | 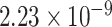 |
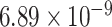 |
| pioglitazone DB01132 rat GSE21329 sample 2841 | 56/321 | 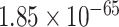 |
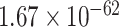 |
| pioglitazone 4829 mouse GSE1458 sample 2587 | 28/282 | 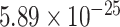 |
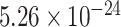 |
| pioglitazone DB01132 rat GSE20219 sample 2794 | 25/308 | 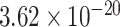 |
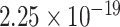 |
| pioglitazone DB01132 rat GSE20219 sample 2795 | 21/270 | 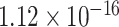 |
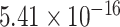 |
| pioglitazone DB01132 human GSE8157 sample 2796 | 12/269 | 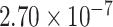 |
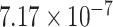 |
| tibolone 444 008 human GSE12446 sample 3204 | 35/313 | 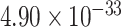 |
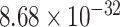 |
Wu et al. [25] identified 29 potential PLpro inhibitors, 27 potential 3CLpro inhibitors, and 20 potential RdRp inhibitors from the ZINC drug database, and identified 13 potential PLpro inhibitors, 26 potential 3Clpro inhibitors, and 20 Potential RdRp inhibitors from their in-house natural product database. Doxycycline was among both the potential PLpro and 3CLpro inhibitors; ascorbic acid and isotretinoin were among the potential PLpro inhibitors; pioglitazone was among the potential 3CLpro inhibitors; and cortisone and tibolone were included in the potential RdRp inhibitors from the ZINC drug database. These multiple hits also further support the suitability of our strategy.
2). Comparison With Ubani et al. [27]
Ubani et al. [27] screened a library of 22 phytochemicals with antiviral activity obtained from the PubChem database for activity against the spike envelope glycoprotein and main protease of SARS-CoV-2. Among these, we found only one hit that overlapped with our screened out drugs, which was quercetin (Table XI).
TABLE XI. List of in Silico Screened drugs [27] Whose Target Genes are Also Enriched in the 134 Genes Selected by TD Based Unsupervised FE.
| Term | Overlap | P-value | Adjusted P-value |
|---|---|---|---|
| Drug Perturbations from GEO up | |||
| quercetin DB04216 mouse GSE38141 sample 3435 | 33/280 |
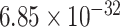
|
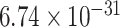
|
| quercetin DB04216 mouse GSE38136 sample 3438 | 31/254 |
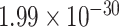
|
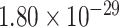
|
| quercetin DB04216 mouse GSE38136 sample 3437 | 37/472 |
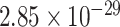
|
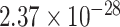
|
| quercetin 5 280 343 rat GSE7479 sample 3409 | 33/394 |
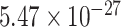
|
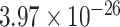
|
| quercetin DB04216 mouse GSE38136 sample 3436 | 30/297 |
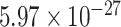
|
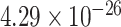
|
| quercetin DB04216 mouse GSE38067 sample 3440 | 26/227 |
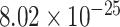
|
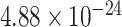
|
| quercetin 5 280 343 human GSE7259 sample 3416 | 29/327 |
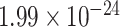
|
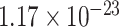
|
| quercetin DB04216 mouse GSE38067 sample 3441 | 20/114 |
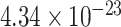
|
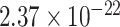
|
| quercetin 5 280 343 human GSE13899 sample 3182 | 16/307 |
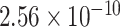
|
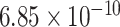
|
| quercetin DB04216 mouse GSE4262 sample 3428 | 14/360 |
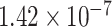
|
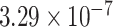
|
| quercetin DB04216 mouse GSE4262 sample 3429 | 9/229 |
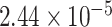
|
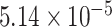
|
| quercetin 5 280 343 human GSE7259 sample 3415 | 9/336 |
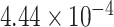
|
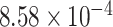
|
| quercetin DB04216 mouse GSE4262 sample 3433 | 8/323 |
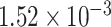
|
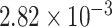
|
| quercetin DB04216 mouse GSE4262 sample 3434 | 8/324 |
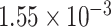
|
|
| quercetin DB04216 mouse GSE4262 sample 3431 | 7/252 |
|
|
| quercetin DB04216 mouse GSE4262 sample 3427 | 8/360 |
|
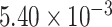
|
| quercetin DB04216 mouse GSE4262 sample 3432 | 5/254 |
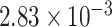
|
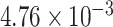
|
| Drug Perturbations from GEO down | |||
| quercetin DB04216 mouse GSE38067 sample 3441 | 59/486 |
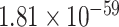
|
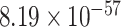
|
| quercetin DB04216 mouse GSE38136 sample 3437 | 26/128 |
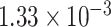 1 1 |
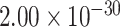
|
| quercetin 5 280 343 human GSE7259 sample 3415 | 29/264 |
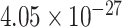
|
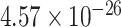
|
| quercetin DB04216 mouse GSE38136 sample 3436 | 30/303 |
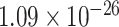
|
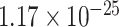
|
| quercetin 5 280 343 human GSE13899 sample 3182 | 26/293 |
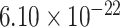
|
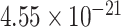
|
| quercetin DB04216 mouse GSE38067 sample 3440 | 28/373 |
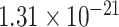
|
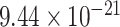
|
| quercetin DB04216 mouse GSE38136 sample 3438 | 27/346 |
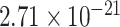
|
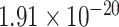
|
| quercetin DB04216 mouse GSE38141 sample 3435 | 22/320 |
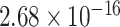
|
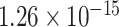
|
| quercetin 5 280 343 human GSE7259 sample 3416 | 18/273 |
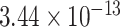
|
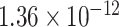
|
| quercetin 5 280 343 rat GSE7479 sample 3409 | 14/206 |
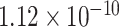
|
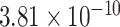
|
| quercetin DB04216 mouse GSE4262 sample 3431 | 11/348 |
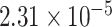
|
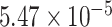
|
| quercetin DB04216 mouse GSE4262 sample 3427 | 5/240 |
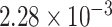
|
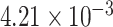
|
IV. Discussion and Conclusion
In this paper, we present a novel evaluation method to identify drugs that could be used to effectively treat COVID-19. We applied a TD-based unsupervised FE method to select genes with altered expression caused by MHV infection in mice. Although the dataset analyzed for this study was not based on SARS-CoV-2 infection, the 134 genes selected by TD-based unsupervised FE can still be considered useful for gaining a better understanding of the infectious mechanism of SARS-CoV-2 for several reasons. First, the 134 genes selected were enriched in general RNA virus proteins that play important roles during infectious processes. This suggests that the infectious mechanism represented by the 134 genes in the mouse model is also applicable to SARS-CoV-2 infection. In fact, these genes were also enriched in processes related to scavenger receptor activity, which might reflect the critical role of TMPRSS2 activity in SARS-CoV-2 replication, suggesting a potential therapeutic target.
Following these achievements, we tried to identify potential drug candidate compounds that could influence the 134 selected genes. Among these, we screened out several candidate compounds that are known antiviral drugs, including those that were screened out as drug candidate compounds for SARS-CoV-2 using in silico methods.
The question arises whether MHV is a suitable model system for SARS-CoV-2 infection, since there are more datasets available for SARS-CoV infection in mouse lung. In order to evaluate the suitability of MHV as a model of the SARS-CoV-2 infectious process, we also performed additional analyses using two SARS-CoV infectious processes in mouse lung (see Materials and Methods). As can be seen in Figs. 5 and 6, 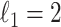 was selected as singular value vector
was selected as singular value vector 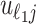 with monotonic dependence upon
with monotonic dependence upon 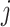 ,
, 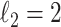 was selected as a singular value vector
was selected as a singular value vector 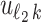 with monotonic dependence upon
with monotonic dependence upon 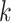 , and
, and 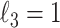 was selected as a singular value vector
was selected as a singular value vector 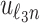 with constant values regardless of
with constant values regardless of 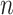 for GSE33266, while
for GSE33266, while 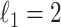 was selected as a singular value vector
was selected as a singular value vector 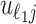 with distinct values between mock (
with distinct values between mock (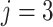 ) and infectious samples (
) and infectious samples (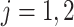 ),
), 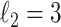 was selected as a singular value vector
was selected as a singular value vector 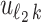 with monotonic dependence upon
with monotonic dependence upon 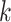 , and
, and 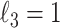 was selected as a singular value vector
was selected as a singular value vector 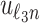 with constant values regardless of
with constant values regardless of 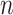 for GSE50000. Then
for GSE50000. Then 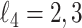 and
and 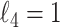 were selected for GSE33266 and GSE50000, respectively, as those associated with the larger absolute values of
were selected for GSE33266 and GSE50000, respectively, as those associated with the larger absolute values of 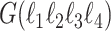 given
given 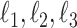 .
.  -values,
-values,  , were attributed to gene
, were attributed to gene 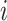 using a cumulative
using a cumulative 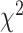 distribution,
distribution, 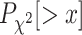 , as described in the Materials and Methods; 569 gene symbols associated with selected genes were selected for GSE33266 and 475 were selected for GSE50000 (see Supplementary Material).
Fig. 7 shows the Venn diagram of these two sets of genes and 134 genes selected by TD based unsupervised FE using GSE146074. Although there are some overlaps, the majority of genes are not shared among these three gene sets.
, as described in the Materials and Methods; 569 gene symbols associated with selected genes were selected for GSE33266 and 475 were selected for GSE50000 (see Supplementary Material).
Fig. 7 shows the Venn diagram of these two sets of genes and 134 genes selected by TD based unsupervised FE using GSE146074. Although there are some overlaps, the majority of genes are not shared among these three gene sets.
Fig. 5.

Singular value vectors obtained by the HOSVD algorithm applied to GSE33266 (Table II). U1: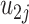 , U2:
, U2: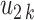 , and U3:
, and U3: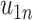 . See Materials and Methods for the meanings of
. See Materials and Methods for the meanings of 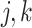 and
and 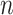 .
.
Fig. 6.

Singular value vectors obtained by the HOSVD algorithm applied to GSE50000 (Table II). U1: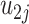 , U2:
, U2: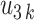 , and U3:
, and U3: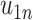 . See Materials and Methods for the meanings of
. See Materials and Methods for the meanings of 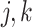 and
and 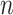 .
.
Fig. 7.

Venn diagrams of 134 genes selected by TD bases unsupervised FE using GSE146074, 569 genes selected by TD bases unsupervised FE using GSE33266, and 475 genes selected by TD bases unsupervised FE using GSE50000.
These two sets of genes were uploaded to Enrichr and checked for significant overlap with coronavirus PPI genes (Table XII).Two sets of selected genes significantly overlap with genes that are believed to interact with SARS-CoV proteins, in spite of the fact that these two gene sets are not highly coincident with the 134 genes selected by the TD based unsupervised FE using GSE146074 (Fig. 7). TD based unsupervised FE therefore has the ability to predict PPI using gene expression profiles, no matter which data sets among GSE146074, GSE33266, and GSE50000 are used. These findings also suggest that the results shown in Table VI are unlikely to be accidental, but provide evidence that the genes selected by TD based unsupervised FE are those interacting with SARS-CoV proteins during infectious processes.
TABLE XII. SARS-CoV-Related Virus PPI in Enrichr.
| Term | Overlap | P-value | Adjusted P-value |
|---|---|---|---|
| GSE33266 | |||
| SARS coronavirus excised_polyprotein 1..4369 (gene: orf1ab) | 16/194 |
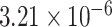
|
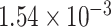
|
| SARS coronavirus P2 full_polyprotein 1..4382 | 16/198 |
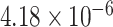
|
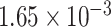
|
| SARS coronavirus nsp9-pp1a/pp1ab (gene: orf1ab) | 5/13 |
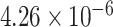
|
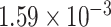
|
| SARS coronavirus 3C-like proteinase (gene: orf1ab) | 5/19 |
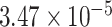
|
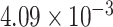
|
| SARS coronavirus nsp8-pp1a/pp1ab (gene: orf1ab) | 7/45 |
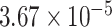
|
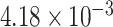
|
| SARS coronavirus 2-O-ribose methyltransferase (2-o-MT) (gene: orf1ab) | 4/11 |
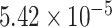
|
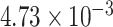
|
| SARS coronavirus nsp7-pp1a/pp1ab (gene: orf1ab) | 6/36 |
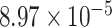
|
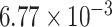
|
| SARS coronavirus endoRNAse (gene: orf1ab) | 3/6 |
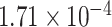
|
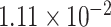
|
| SARS coronavirus nsp4-pp1a/pp1ab (gene: orf1ab) | 4/16 |
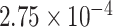
|
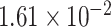
|
| SARS coronavirus formerly known as growth-factor-like protein (gene: orf1ab) | 4/17 |
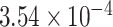
|
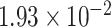
|
| SARS coronavirus Tor2 replicase 1AB | 9/108 |
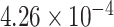
|
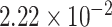
|
| SARS coronavirus P2 full_polyprotein 1..7073 | 9/109 |
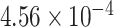
|
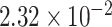
|
| SARS coronavirus RNA-dependent RNA polymerase (gene: orf1ab) | 3/9 |
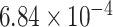
|
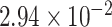
|
| SARS coronavirus leader protein (gene: orf1ab) | 4/20 |
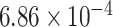
|
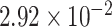
|
| SARS coronavirus nsp3-pp1a/pp1ab (gene: orf1ab) | 9/118 |
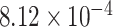
|
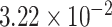
|
| GSE50000 | |||
| SARS coronavirus 3C-like proteinase (gene: orf1ab) | 4/19 |
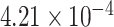
|
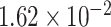
|
| SARS coronavirus hypothetical protein sars7a | 5/38 |
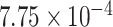
|
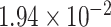
|
| SARS coronavirus P2 hypothetical protein sars7a | 5/38 |
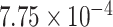
|
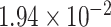
|
| SARS coronavirus Tor2 Orf8 | 5/38 |
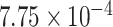
|
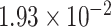
|
| SARS coronavirus 2-O-ribose methyltransferase (2-o-MT) (gene: orf1ab) | 3/11 |
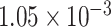
|
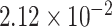
|
| SARS coronavirus nsp9-pp1a/pp1ab (gene: orf1ab) | 3/13 |
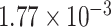
|
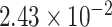
|
| SARS coronavirus nsp13-pp1ab (ZD, NTPase/HEL; RNA (gene: orf1ab) | 3/14 |
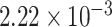
|
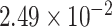
|
| SARS coronavirus P2 spike glycoprotein precursor | 6/71 |
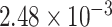
|
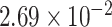
|
| SARS coronavirus E2 glycoprotein precursor (gene: S) | 6/72 |
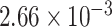
|
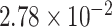
|
| SARS coronavirus Tor2 spike glycoprotein | 6/72 |
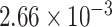
|
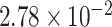
|
| SARS coronavirus nsp4-pp1a/pp1ab (gene: orf1ab) | 3/16 |
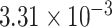
|
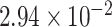
|
| SARS coronavirus nsp3-pp1a/pp1ab (gene: orf1ab) | 7/118 |
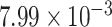
|
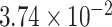
|
Another possible concern is that the studies are not based upon direct investigation of SARS-CoV-2, but are based upon a closely related virus. This limitation suggests that these results might not be applicable to SARS-CoV-2. In order to address this point, we compared the 134 genes (Table V) with genes reported to interact with SARS-CoV-2 proteins [31] (Table XIII).One hundred and thirty-four genes significantly overlap with human genes reported to interact with SARS-CoV-2 proteins. TD based unsupervised FE therefore appears to have the ability to predict PPI, even when only gene expression profiles from a related virus are available. Especially, it is remarkable that the present study could outperform the inference in the previous study [8] (asterisked ones in Table XIII) where SARS-CoV-2 infected human lung cell lines are investigated. This is possibly because of the superiority of in vivo study toward in vitro study in spite of the usage of different species. Our proposed method, TD based unsupervised FE, can make use of the data set taken from different species infected by related but not exactly same virus whereas other methods could not (see below). Since it is unrealistic to intentionally infect human subject SARS-CoV-2, it is important to have the methodology that can make use of data set taken from other species than human.
TABLE XIII. Coincidence Between 134 Genes and Human Genes Reported to Interact With SARS-CoV-2 Proteins [31]. Asterisked Ones are the Cases That Outperformed the Inference in Previous Study [8] That Investigated Gene Expression Profiles of Human Lung Cell Lines Infected by SARS-CoV-2.
| SARS-CoV-2 proteins | P values | Odds Ratio |
|---|---|---|
| SARS-CoV2 E |
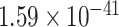 (*) (*) |
20.2 (*) |
| SARS-CoV2 M |
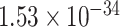 (*) (*) |
13.7 (*) |
| SARS-CoV2 N |
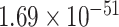 (*) (*) |
31.8 (*) |
| SARS-CoV2 nsp1 |
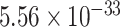 (*) (*) |
19.8 (*) |
| SARS-CoV2 nsp10 |
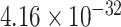 (*) (*) |
23.0 (*) |
| SARS-CoV2 nsp11 |
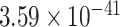 (*) (*) |
19.1 (*) |
| SARS-CoV2 nsp12 |
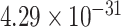 (*) (*) |
18.3 (*) |
| SARS-CoV2 nsp13 |
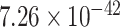 (*) (*) |
18.6 (*) |
| SARS-CoV2 nsp14 |
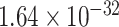 (*) (*) |
22.3 (*) |
| SARS-CoV2 nsp15 |
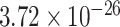 (*) (*) |
16.4 (*) |
| SARS-CoV2 nsp2 |
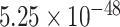 (*) (*) |
22.7 (*) |
| SARS-CoV2 nsp4 |
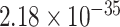 (*) (*) |
16.0 (*) |
| SARS-CoV2 nsp5 |
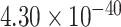 (*) (*) |
25.5 (*) |
| SARS-CoV2 nsp5_C145 A |
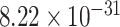 (*) (*) |
25.1 (*) |
| SARS-CoV2 nsp6 |
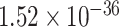 (*) (*) |
16.0 (*) |
| SARS-CoV2 nsp7 |
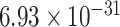 (*) (*) |
14.7 (*) |
| SARS-CoV2 nsp8 |
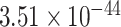 (*) (*) |
19.6 (*) |
| SARS-CoV2 nsp9 |
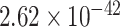 (*) (*) |
23.3 (*) |
| SARS-CoV2 orf10 |
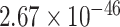 (*) (*) |
22.3 (*) |
| SARS-CoV2 orf3a |
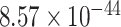 (*) (*) |
19.8 (*) |
| SARS-CoV2 orf3b |
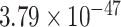 (*) (*) |
24.8 (*) |
| SARS-CoV2 orf6 |
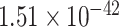 (*) (*) |
21.9 (*) |
| SARS-CoV2 orf7a |
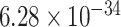 (*) (*) |
15.3 (*) |
| SARS-CoV2 orf8 |
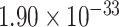 (*) (*) |
14.0 (*) |
| SARS-CoV2 orf9b |
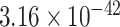 (*) (*) |
22.3 (*) |
| SARS-CoV2 orf9c |
 (*) (*) |
13.9 (*) |
Finally, we compared identified drugs (“Drug Pert GEO up/down” and “DrugMarix“ in the Supplementary Material) with those reported as possible drugs against SARS-CoV-2 [32]. Among the 142 drugs identified by Zhou et al. [32], as many as 25 drugs were found to significantly affect 134 genes in at least one experiment within either DrugMatrix, or GEO, using Enrichr with adjusted 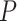 -values of less than 0.05 (Table XIV). Thus, our suggestions for drug repositioning are also supported.
-values of less than 0.05 (Table XIV). Thus, our suggestions for drug repositioning are also supported.
TABLE XIV. Number of Experiments Associated With Adjusted 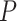 -Values in Various Enrichr Categories for the Drugs Identified in Another study [32].
-Values in Various Enrichr Categories for the Drugs Identified in Another study [32].
| GEO up | GEO down | DrugMatrix | |
|---|---|---|---|
| Methotrexate | 2 | 1 | 35 |
| Fluorouracil | 4 | 5 | 3 |
| Testosterone | 1 | 12 | |
| Stanolone | 2 | 1 | |
| Menadione | 1 | ||
| Hydrocortisone | 1 | 20 | |
| Mestranol | 6 | ||
| Hexestrol | 4 | ||
| Mercaptopurine | 15 | ||
| Paroxetine | 8 | ||
| Vinblastine | 16 | ||
| Phenylbutazone | 3 | ||
| Naloxone | 6 | ||
| Hydralazine | 11 | ||
| Vinorelbine | 10 | ||
| Carvedilol | 16 | ||
| Colchicine | 12 | ||
| Amitriptyline | 12 | ||
| Epinephrine | 12 | ||
| Dactinomycin | 6 | ||
| Melatonin | 8 | ||
| Methyltestosterone | 6 | ||
| Omeprazole | 19 | ||
| Oxymetholone | 6 | ||
| Progesterone | 20 |
Since it is unlikely that this level of agreement is purely accidental, the drugs identified in the present study can be useful candidates for further evaluation for COVID-19 therapy. This work therefore provides a foundation for further research pertaining to utilizing advanced learning concepts to analyze COVID-19 infectious disease.
Final concerns to be addressed might be the comparison with methods other than TD based unsupervised FE applied to synthetic data sets. When considering the synthetic data set, categorical regression, eq. (10), outperformed TD based unsupervised FE. Although eq. (9) cannot be better than TD based unsupervised FE (Table III), its performance is still comparable. If these two more easily understood methods are better than or comparable to TD based unsupervised FE, TD based unsupervised FE, which is more difficult to interpret, is useless. In order to check this point, we applied categorical regression and eq. (9), which were modified as
 |
and
 |
where 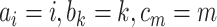 , to the present set (GSE146074). Then
, to the present set (GSE146074). Then 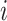 s associated with adjusted
s associated with adjusted 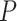 -values less than 0.01 were selected. There are too many genes that passed this screening to evaluate: 25 609 and 20 217, respectively. This observation suggests that these two methods lack the ability to screen for a limited number of genes that are likely to interact with SARS-CoV-2 proteins, since only a limited number of human genes will interact with SARS-CoV-2 proteins (
-values less than 0.01 were selected. There are too many genes that passed this screening to evaluate: 25 609 and 20 217, respectively. This observation suggests that these two methods lack the ability to screen for a limited number of genes that are likely to interact with SARS-CoV-2 proteins, since only a limited number of human genes will interact with SARS-CoV-2 proteins (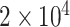 are as many as all human protein coding genes). Although this finding is enough reason to reject the use of these two methods in favor of TD based unsupervised FE if only the top ranked genes are selected, it might be possible to identify a limited number of genes that significantly overlap with genes reported to interact with SARS-CoV-2 proteins. We selected the 134 top ranked genes using the
are as many as all human protein coding genes). Although this finding is enough reason to reject the use of these two methods in favor of TD based unsupervised FE if only the top ranked genes are selected, it might be possible to identify a limited number of genes that significantly overlap with genes reported to interact with SARS-CoV-2 proteins. We selected the 134 top ranked genes using the 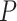 -values produced by these two methods and uploaded them to Enrichr. There were no SARS-CoV-2 proteins that significantly interact with these 134 genes. Since a significant interaction with SARS-CoV-2 proteins is the primary requirement for genes to be used to screen candidate drug compounds, these two methods appear to be inadequate for the present purpose.
-values produced by these two methods and uploaded them to Enrichr. There were no SARS-CoV-2 proteins that significantly interact with these 134 genes. Since a significant interaction with SARS-CoV-2 proteins is the primary requirement for genes to be used to screen candidate drug compounds, these two methods appear to be inadequate for the present purpose.
If we employ more complicated and sophisticated methods to select genes, it might be possible to identify a limited number of genes that significantly interact with SARS-CoV-2 proteins. However, TD based unsupervised FE is simple and rapid (see the CPU time in Table III) enough to achieve the present purpose, and does so with acceptable accuracy.
Finally, we would like to discuss why we did not use SARS-CoV-MA15 data. This is simply because we could not get any significant overlaps with human proteins supposed to be interact with SARS-CoV proteins. This is possibly because SARS-CoV-MA15 is believed to be adapted to mouse infection processes while we checked human proteins. Thus, even if organism used is not human but mouse, MHV is suitable model for human SARS-CoV-2 infection and our method has ability to distinguish between MHV and SARS-CoV-15 where the former remains an effective model of human SARS-CoV-2 infectious process as Pfaender et al. [6] correctly assumed while the latter is not.
One might wonder why we have employed one specific algorithm, HOSVD, among those developed to apply TD to tenors. Although it was fully described in the recently published book [7], we outline it here very briefly.
-
•
Although CP decomposition [7] is more popular implementation, it cannot give us suitable solution when the number of features are much larger than that of samples as the problems dealt in this study because of its heavily dependence upon initial values [7].
-
•
There are other implementations to derive Tucker decomposition than HOSVD, other methods cannot give us suitable solution because other methods require the optimization within the limited number of given singular value vectors. Since HOSVD does not perform optimization and singular value vectors can be obtained independent of the number of singular value vectors to be computed, HOSVD does not destroy important singular value vectors with small contribution; since the number of genes considers is as small as a few hundreds, which is less than 1 % of total number of genes
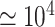 , we cannot ignore singular value vectors with small contributions that might reflect the property of a few hundred genes.
, we cannot ignore singular value vectors with small contributions that might reflect the property of a few hundred genes. -
•
Not all TD cannot give us weight to evaluate the relations between singular value vectors attributed to distinct instances. Although HOSVD can give us
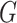 that can evaluate coincidence between
that can evaluate coincidence between 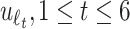 , for example tensor train decomposition [7] cannot give us something that corresponds to
, for example tensor train decomposition [7] cannot give us something that corresponds to 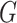 . Thus we cannot know which singular value vectors attributed to gene should be used for selecting genes based upon the evaluation other singular value vectors attributed to, e.g., samples.
. Thus we cannot know which singular value vectors attributed to gene should be used for selecting genes based upon the evaluation other singular value vectors attributed to, e.g., samples.
Because of these reasons, we have specifically employed HOSVD.
In the book [7], we carried out decompositions of data modeled as three-mode tensor and five-mode tensor in addition to others, which were different from this study as we consider a decomposition of different application and data modeled as six-mode tensor 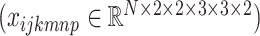 .
.
Biographies

Y-H. Taguchi received a B.S. and Ph.D. degrees in physics from the Tokyo Institute of Technology, Tokyo, Japan. He is currently a Full Professor with the Department of Physics, Chuo University, Japan. His researchs have been published in leading journals, such as the Physical Review Letters, Bioinformatics, and Scientific Reports. His research interests include bioinformatics, machine learning, and nonlinear physics. He is also an Editorial Board Member of Frontiers in Genetics:RNA, PloS ONE, BMC Medical Genomics, Medicine (Lippincott Williams & Wilkins journal), BMC Research Notes, non-coding RNA (MDPI), and IPSJ Transaction on Bioinformatics.

Turki Turki received the B.S. degree in computer science from King Abdulaziz University, Jeddah, Saudi Arabia, the M.S. degree in computer science from NYU Tandon School of Engineering, New York, NY, USA, and the Ph.D. degree in computer science from the New Jersey Institute of Technology, Newark, NJ, USA. He is currently an Assistant Professor with the Department of Computer Science, King Abdulaziz University. His research interests include artificial intelligence, machine learning, deep learning, data mining, data science, big data analytics, and bioinformatics. His research has been accepted and published in journals, such as Frontiers in Genetics, BMC Genomics, BMC Systems Biology, Expert Systems with Applications, Computers in Biology and Medicine, and Current Pharmaceutical Design. He was the recipient of several distinction awards from the Deanship of Scientific Research at King Abdulaziz University. He is supported by King Abdulaziz University and is currently working on several biomedicine related projects. He was on the program committees of several international conferences. He is an Editorial Board Member of Sustainable Computing: Informatics and Systems and Computers in Biology and Medicine.
Funding Statement
This work was supported in part by KAKENHI under Grants 19H05270, 20H04848, and 20K12067 and in part by the Deanship of Scientific Research (DSR) at King Abdulaziz University, Jeddah, under Grant KEP-8-611-38.
Contributor Information
Y-H. Taguchi, Email: tag@granular.com.
Turki Turki, Email: tturki@kau.edu.sa.
References
- [1].Cavasotto C. N. and Filippo J. I. Di, “In silico drug repurposing for COVID-19: Targeting SARS-CoV-2 proteins through docking and consensus ranking,” Mol. Inform., vol. 40, no. 1 2021, Art. no. 2000115. [DOI] [PubMed] [Google Scholar]
- [2].Elmezayen A. D., Al-Obaidi A., Şhin A. T., and Yelekçi K., “Drug repurposing for coronavirus (COVID-19): In silico screening of known drugs against coronavirus 3CL hydrolase and protease enzymes,” J. Biomol. Struct. Dyn., pp. 1–13, 2020. [Online]. Available: https://doi.org/10.1080/07391102.2020.1758791 [DOI] [PMC free article] [PubMed]
- [3].Kalamatianos K., “Drug repurposing for coronavirus (COVID-19): In silico screening of known drugs against the SARS-CoV-2 spike protein bound to angiotensin converting enzyme 2 (ACE2) (6M0J),” Aug. 2020. [Online]. Available: https://doi.org/10.26434/chemrxiv.12857678.v1 [DOI] [PMC free article] [PubMed]
- [4].Taguchi Y. H., “Identification of candidate drugs using tensor-decomposition-based unsupervised feature extraction in integrated analysis of gene expression between diseases and drug matrix datasets,” Sci. Rep., vol. 7, no. 1, pp. 1-16, Oct. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Taguchi Y. H., “Drug candidate identification based on gene expression of treated cells using tensor decomposition-based unsupervised feature extraction for large-scale data,” BMC Bioinf., vol. 19, no. S13, pp. 27–42, Feb. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Pfaender S. et al. , “LY6E impairs coronavirus fusion and confers immune control of viral disease,” Nature Microbiol., vol. 5, no. 11, pp. 1330–1339, Jul. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Taguchi Y. H., Unsupervised Feature Extraction Applied to Bioinformatics: PCA and TD Based Approach. Switzerland: Springer International, 2020. [Google Scholar]
- [8].Taguchi Y.-H. and Turki T., “A new advanced in silico drug discovery method for novel coronavirus with tensor decomposition-based unsupervised feature extraction,” PLos One, vol. 15, no. 9, pp. 1–16, Sep. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Kuleshov M. V. et al. , “Enrichr: A comprehensive gene set enrichment analysis web server 2016 update,” Nucleic Acids Res., vol. 44, no. W1, pp. W90–W97, May 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Zhou Y. et al. , “Metascape provides a biologist-oriented resource for the analysis of systems-level datasets,” Nature Commun., vol. 10, no. 1, pp. 1–10, Apr. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Woo P. C., Huang Y., Lau S. K., H.-W. Tsoi, and Yuen K.-Y., “In silico analysis of ORF1ab in coronavirus HKU1 genome reveals a unique putative cleavage site of coronavirus HKU1 3C-like protease,” Microbiol. Immunol., vol. 49, no. 10, pp. 899–908, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Hoffmann M. et al. , “SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor,” Cell, vol. 181, no. 2, pp. 271–280.e8, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Matsuyama S. et al. , “Enhanced isolation of SARS-CoV-2 by TMPRSS2-expressing cells,” Proc. Nat. Acad. Sci., vol. 117, no. 13, pp. 7001–7003, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Bugge T. H., Antalis T. M., and Wu Q., “Type II transmembrane serine proteases,” J. Biol. Chem., vol. 284, no. 35, pp. 23177–23181, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Taguchi Y. H. and Turki T., “Neurological disorder drug discovery from gene expression with tensor decomposition,” Curr. Pharmaceut. Des., vol. 25, no. 43, pp. 4589–4599, 2019. [DOI] [PubMed] [Google Scholar]
- [16].Burdick J. R. and Durand D. P., “Primaquine diphosphate: Inhibition of newcastle disease virus replication,” Antimicrobial Agents Chemother., vol. 6, no. 4, pp. 460–464, 1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Culita1 D. C. et al. , “Evaluation of cytotoxic and antiproliferative activity of Co(II), Ni(II), Cu(II) and Zn(II) complexes with Meloxicam on virus - transformed tumor cells daniela,” Revista De Chimie, vol. 63, no. 4, pp. 384–389, 2012. [Google Scholar]
- [18].Renis H. E., “Antiviral activity of cytarabine in herpesvirusinfected rats,” Antimicrobial Agents Chemother., vol. 4, no. 4, pp. 439–444, 1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Ueda K., Kawabata R., Irie T., Nakai Y., Tohya Y., and Sakaguchi T., “Inactivation of pathogenic viruses by plant-derived tannins: Strong effects of extracts from persimmon (diospyros kaki) on a broad range of viruses,” PLos One, vol. 8, no. 1, pp. 1–10, Jan. 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Li R. et al. , “Structure-dependent antiviral activity of catechol derivatives in pyroligneous acid against the encephalomycarditis virus,” RSC Adv., vol. 8, pp. 35888–35896, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Lobert P. E., Hober D., Delannoy A. S., and Wattré P., “Evidence that neomycin inhibits human cytomegalovirus infection of fibroblasts,” Arch. Virol., vol. 141, no. 8, pp. 1453–1462, Aug. 1996. [DOI] [PubMed] [Google Scholar]
- [22].Abuohashish H. M., Ahmed M. M., Sabry D., Khattab M. M., and Al-Rejaie S. S., “ACE-2/Ang1-7/Mas cascade mediates ace inhibitor, captopril, protective effects in estrogen-deficient osteoporotic rats,” Biomed. Pharmacother., vol. 92, pp. 58–68, 2017. [DOI] [PubMed] [Google Scholar]
- [23].Cheng W. et al. , “Coenzyme Q plays opposing roles on bacteria/fungi and viruses in drosophila innate immunity,” Int. J. Immunogenet., vol. 38, no. 4, pp. 331–337, 2011. [DOI] [PubMed] [Google Scholar]
- [24].Finnegan C. M. and Blumenthal R., “Fenretinide inhibits HIV infection by promoting viral endocytosis,” Antiviral Res., vol. 69, no. 2, pp. 116–123, 2006. [DOI] [PubMed] [Google Scholar]
- [25].Wu C. et al. , “Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods,” Acta Pharm. Sinica B., vol. 10, no. 5, pp. 766–788, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Yi L. et al. , “Small molecules blocking the entry of severe acute respiratory syndrome coronavirus into host cells,” J. Virol., vol. 78, no. 20, pp. 11334–11339, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Ubani A. et al. , “Molecular docking analysis of some phytochemicals on two SARS-CoV-2 targets,” Biorxiv, 2020. [Online]. Available: https://www.biorxiv.org/content/early/2020/04/01/2020.03.31.017657
- [28].Clouser C. L., Patterson S. E., and Mansky L. M., “Exploiting drug repositioning for discovery of a novel HIV combination therapy,” J. Virol., vol. 84, no. 18, pp. 9301–9309, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Fukano K. et al. , “Troglitazone impedes the oligomerization of sodium taurocholate cotransporting polypeptide and entry of hepatitis B virus into hepatocytes,” Front. Microbiol., vol. 9, 2019, Art. no. 3257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Perez O. D., Nolan G. P., Magda D., Miller R. A., Herzenberg L. A., and Herzenberg L. A., “Motexafin gadolinium (Gd-Tex) selectively induces apoptosis in HIV-1 infected CD4+ T helper cells,” Proc. Nat. Acad. Sci., vol. 99, no. 4, pp. 2270–2274, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Gordon D. E. et al. , “A SARS-CoV-2-human protein-protein interaction map reveals drug targets and potential drug-repurposing,” Nature, vol. 583, pp. 459–468, Apr. 2020. https://doi.org/10.1038/s41586-020-2286-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Zhou Y., Hou Y., Shen J., Huang Y., Martin W., and Cheng F., “Network-based drug repurposing for novel coronavirus 2019-nCoV/SARS-CoV-2,” Cell Discov., vol. 6, no. 1, pp. 1–18, Mar. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]