

ABSTRACT
Severe infections caused by multidrug-resistant Klebsiella pneumoniae sequence type 258 (ST258) highlight the need for new therapeutics with activity against this pathogen. Bacteriophage (phage) therapy is an alternative treatment approach for multidrug-resistant bacterial infections that has shown efficacy in experimental animal models and promise in clinical case reports. In this study, we assessed microbiologic, histopathologic, and survival outcomes following systemic administration of phage in ST258-infected mice. We found that prompt treatment with two phages, either individually or in combination, rescued mice with K. pneumoniae ST258 bacteremia. Among the three treatment groups, mice that received combination phage therapy demonstrated the greatest increase in survival and the lowest frequency of phage resistance among bacteria recovered from mouse blood and tissue. Our findings support the utility of phage therapy as an approach for refractory ST258 infections and underscore the potential of this treatment modality to be enhanced through strategic phage selection.
KEYWORDS: Klebsiella pneumoniae, bacteriophage therapy, bloodstream infections, experimental therapeutics, multidrug resistance
INTRODUCTION
Klebsiella pneumoniae is an encapsulated Gram-negative bacterium that causes a wide range of infections in humans. Pneumonia, urinary tract infections, intra-abdominal infections, including pyogenic liver abscesses, and bloodstream infections with the potential to precipitate sepsis are among the most common types of K. pneumoniae infection (1, 2). These infections often afflict hospitalized patients, particularly those with malignancy, immunocompromise, or respiratory failure requiring mechanical ventilation. Without adequate treatment, K. pneumoniae infections are associated with high rates of morbidity and mortality (3, 4).
Epidemiologic studies have revealed that sequence type 258 (ST258) is the predominant carbapenem-resistant lineage among K. pneumoniae clinical isolates in the United States and in many countries around the world (5–7). Resistance to carbapenems, which in ST258 is primarily conferred by K. pneumoniae carbapenemase (KPC), complicates the management of K. pneumoniae infections and is associated with poorer clinical outcomes (3, 8). Newer drugs that combine a cephalosporin or carbapenem with a potent β-lactamase inhibitor (e.g., ceftazidime-avibactam, meropenem-vaborbactam) have proven efficacious against KPC ST258 and other carbapenem-resistant Enterobacteriaceae (CRE), but resistance to these agents is emerging and expected to increase with continued use (9–11). Hence, novel approaches to treat infections caused by multidrug-resistant ST258 are urgently needed.
Bacteriophages (phages) are viruses that infect and kill bacteria as part of their natural life cycle. Although the concept of using phage as an anti-infective agent has been around for over a century, the number of patients with refractory bacterial infections who have been treated with phage has increased in recent years (12, 13). Positive clinical outcomes have been observed in multiple experimental cases, providing support for the notion that phages can confer therapeutic benefit (14–16).
The prevalence and severity of ST258 infections, in part due to the frequency of carbapenem resistance among ST258 isolates, render infections caused by these bacteria an attractive target for phage therapy. Phages that target multidrug-resistant K. pneumoniae have been administered to at least one patient to date after multiple courses of antibiotics failed to resolve a mixed K. pneumoniae-Acinetobacter baumannii wound infection (16). In that case, the infection cleared and the patient avoided amputation after receiving phages directed against both pathogens (while continuing to receive conventional antibiotic therapy).
More robust evidence demonstrating the therapeutic potential of K. pneumoniae-specific phages has come from animal studies. In animal models of K. pneumoniae pneumonia, liver abscess, and wound infection, phage treatment improved survival outcomes and reduced bacterial burdens of infection (17–21). This led us to question whether phage treatment could provide similar positive effects in the setting of acute ST258 sepsis. To address this knowledge gap, we evaluated the ability of two phages, administered singly and in combination, to rescue mice with ST258 bacteremia.
RESULTS
Mouse model of bacteremia.
Wild-type C57BL/6J mice were injected intraperitoneally (IP) with 106 to 108 CFU of ST258 to determine the optimal dose for phage efficacy testing (Fig. 1). Infection via IP injection of bacteria led to acute systemic disease at the highest doses tested. Weight loss and moribundity that necessitated euthanasia occurred exclusively within the first 48 h of infection, after which time any survivors convalesced for the remainder of the study period. The rapid progression of disease in ST258-infected mice was consistent with the natural history of severe sepsis. Based on these results, an ST258 inoculum of 5 × 107 CFU was used for all subsequent experiments.
FIG 1.
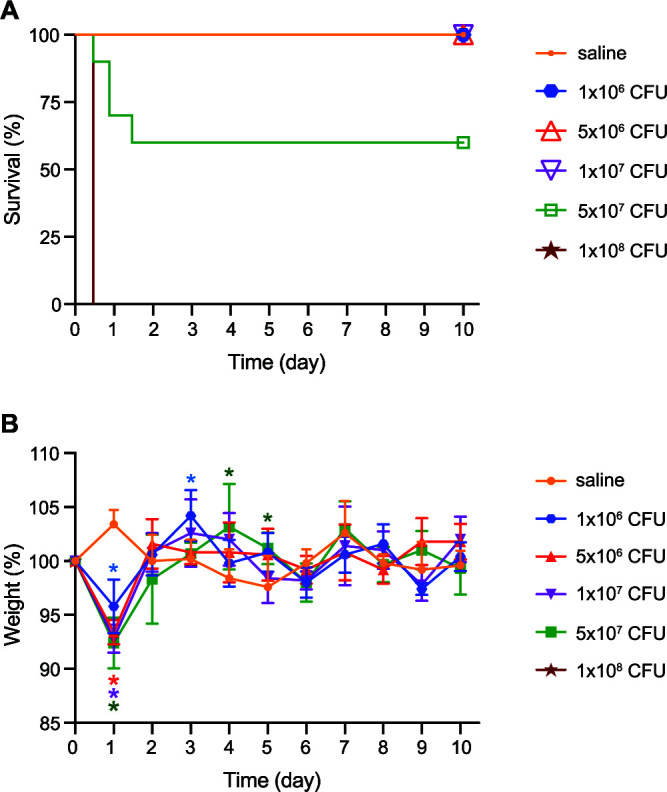
Mouse infection model. (A) Survival of mice after a single IP injection with the indicated inoculum (CFU) of ST258. For groups of mice inoculated with 5 × 107 CFU or 1 × 108 CFU, n = 10. For all other groups, n = 5. (B) The relative daily weight of surviving mice was determined as a percentage of weight on day 0. Data are presented as the mean ± standard deviation. Daily weights were compared among the groups each day. *, P < 0.05 (color-coded to match the CFU key) versus the saline control using a one-way analysis of variance (ANOVA) and Dunnett’s posttest.
Prompt phage treatment rescues ST258-infected mice.
We first performed a pilot study to determine conditions optimal for a large-scale phage therapy experiment. Mice infected with ST258 were treated with two lytic phages that had been previously isolated from sewage and characterized in vitro (22). Pharr (P1), a 40.6-kb podophage, and ϕKpNIH-2 (P2), a 49.4-kb siphophage, were injected IP at different times (1, 8, and 24 h after bacterial infection) and at different multiplicities of infection (MOI) (an MOI of 1 or 10) (Fig. 2).
FIG 2.
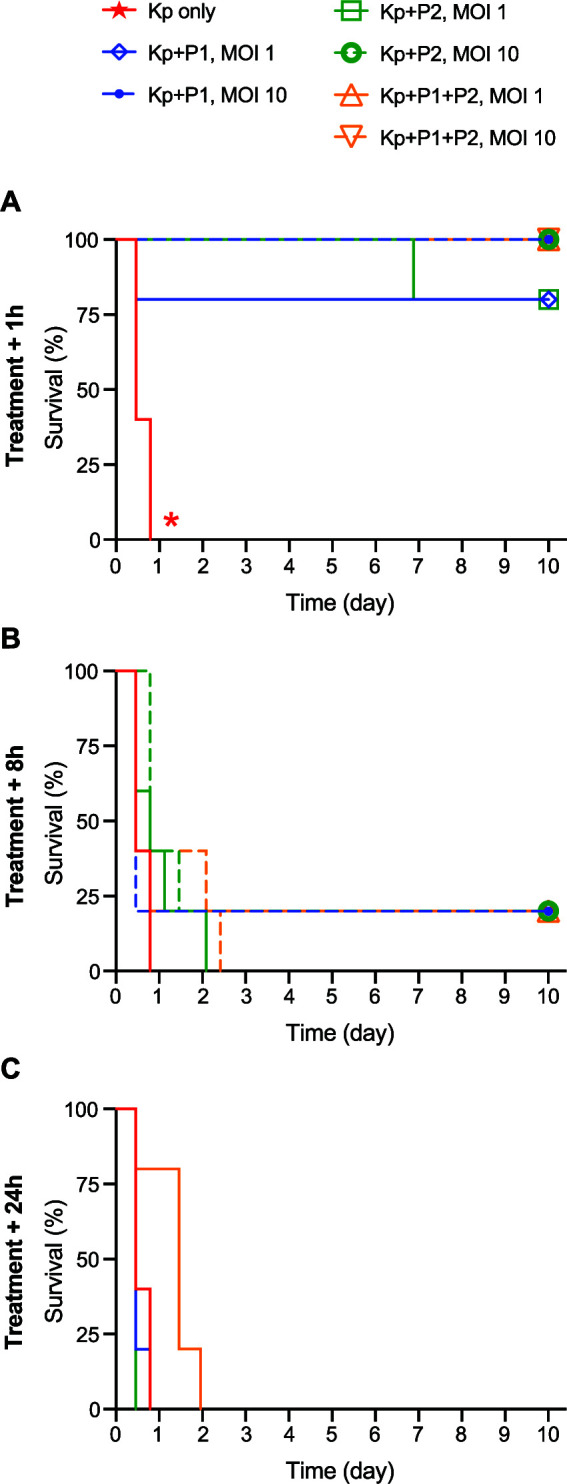
Impact of MOI and timing of phage treatment on survival of infected mice. (A to C) Survival of ST258-infected mice after phage treatment via IP injection at 1 h, 8 h, and 24 h post-bacterial infection as indicated. Colored solid lines (except the red line, which is the saline control group) represent phage MOI 1 treatment groups; dashed lines represent MOI 10 treatment groups. For each group, n = 5. *, P < 0.05 versus all other treatment groups using a log rank (Mantel-Cox) test and Bonferroni correction for multiple comparisons. Kp, K. pneumoniae ST258; P1, Pharr; P2, ϕKpNIH-2.
Overall, results of the pilot study indicated that survival outcomes were strongly influenced by the timing of treatment but not by phage dose. Compared to the saline-treated control group, survival of mice treated with phages P1, P2, or P1 + P2 at 1 h postinfection increased significantly (P < 0.05). By comparison, the ability of phage to rescue mice from severe sepsis diminished dramatically if phage treatment was administered at 8 h or 24 h after bacterial infection (Fig. 2).
We next conducted a definitive, large-scale survival experiment to assess the ability of phage to rescue mice from severe ST258 bacteremia. Wild-type C57BL/6J mice were infected IP with 5 × 107 CFU of ST258 and then treated with P1, P2, or P1 + P2 (each at an MOI of 1) via IP injection 1 h after bacterial infection. All mice in a saline-treated control group progressed quickly to severe disease (Fig. 3, Kp only). In contrast, mice that received treatment with viable phage survived significantly better than those that received saline instead (survival was 93% for ST258-infected mice treated with P1, 80% with P2, and 100% with P1 + P2, versus 0% in the saline control group; P < 0.0001). Although survival among the three treatment groups did not differ statistically, we observed a trend in relative rates of survival following each of the phage treatments (P1 + P2 > P1 > P2) (Fig. 3). Histopathological analyses of liver tissue from these mice 24 h or 48 h after phage treatment support this trend (Table S1 and Fig. S2). The three groups of mice treated with viable phage each survived better than mice treated with heat-killed (HK) phage, whereas there was no significant difference in survival between the HK phage and saline control groups (Fig. 3). Collectively, these data provide support for the idea that phage therapy can moderate the severity of infections caused by multidrug-resistant K. pneumoniae.
FIG 3.
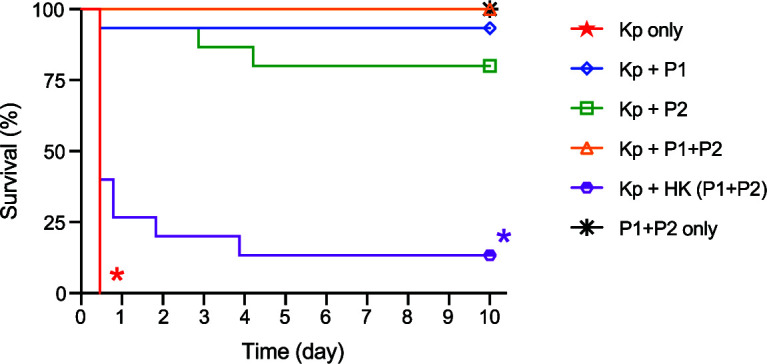
Prompt phage treatment rescues mice with systemic ST258 infection. Survival of mice infected IP with ST258 (5 × 107 CFU) followed by IP treatment with phage (MOI, 1) 1 h later. Equivalent volumes of sterile saline were injected in lieu of phage or bacteria for Kp only and P1 + P2 only groups, respectively. For each group, n = 15. *, P < 0.05 versus all phage treatment groups using a log rank (Mantel-Cox) test and Bonferroni correction for multiple comparisons. The difference between the Kp only and HK phage control groups was not significant. Kp, K. pneumoniae ST258; P1, Pharr; P2, ϕKpNIH-2; HK, heat-killed.
Histopathology scores Table S1, DOCX file, 0.02 MB (23.9KB, docx) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Quantitation of bacteria and phage in mouse blood and tissue.
To more directly assess the ability of phage to kill ST258 during infection in vivo, blood and tissue samples were collected from ST258-infected mice at 1, 24, and 48 h after phage treatment, and viable bacteria and phage were quantified by plating. Compared with infected mice treated with saline alone or heat-killed (HK) phage, there were significantly fewer bacteria recovered from those treated with P1, P2, or P1 + P2 at all time points tested (Fig. 4A). No viable bacteria (as assessed by CFU) were recovered from the blood of uninfected mice that received phage only, suggesting that all bacteria recovered from infected mice were ST258. Similarly, no phage plaques formed on agar plates containing blood from HK-phage-treated mice, confirming the absence of endogenous, ST258-specific phages in laboratory mouse blood (Fig. 4B).
FIG 4.
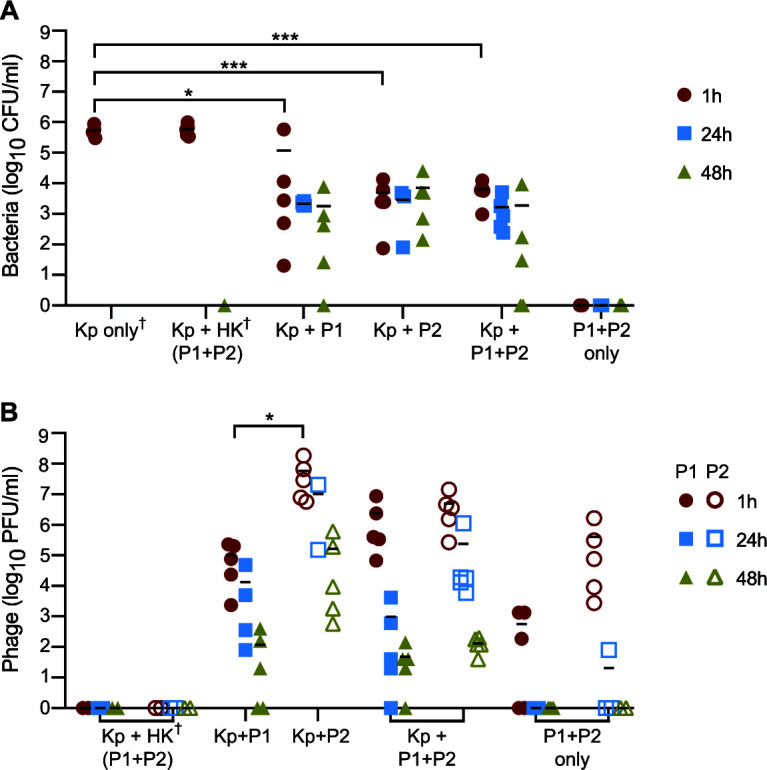
Quantitation of bacteria and phage in mouse blood. (A and B) Bacterial CFU and phage PFU in mouse blood at 1 h, 24 h, and 48 h after phage treatment. Each symbol represents data from a single mouse, up to 5 per treatment group per time point. Bars denote group means. Samples for which no CFU or PFU was recovered are plotted as 0 on the x axis. †, Groups in which blood collection occurred at 12 h instead of 24 h or 48 h due to rapid disease progression. For these groups, bacteria in the blood were estimated to exceed 106 CFU/ml at 12 h but could not be accurately quantified. *, P < 0.05; ***, P < 0.001 as determined using a one-way ANOVA and Tukey’s posttest. Kp, K. pneumoniae ST258; P1, Pharr; P2, ϕKpNIH-2; HK, heat-killed.
Consistent with a sustained reduction in bacteremia after treatment with phage (at 24 h and 48 h), there was a progressive decline in phage titers over time (compare Fig. 4A and B). P2 titers were, on average, higher than those of P1 at all three time points; however, this finding did not correlate with improved mouse survival or lower titers of circulating bacteria in P2-treated animals. Circulating phage titers at 24 h and 48 h were notably higher in infected versus uninfected mice treated with P1 + P2, providing indirect evidence of phage replication in vivo. Both phage and bacteria were also recovered from the kidney, liver, lung, and spleen of infected mice at all three time points (Fig. S1). On average, there were decreased numbers of tissue-associated bacteria in phage-treated mice compared to saline-treated control mice within 24 h after treatment (Fig. S1).
Quantitation of bacteria and phage in tissues. (A to F) Quantification of viable bacteria (A to C) and phage (D to F) in homogenized mouse organs at 1 h, 24 h, and 48 h after phage administration. Data from individual mice, up to 3 per treatment group per time point, are plotted. Bars denote group means. Samples for which no CFU or PFU was recovered are plotted as 0 on the x axis. †, Groups in which tissue collection occurred at 12 h instead of 24 h or 48 h due to rapid disease progression. Data acquired at this earlier time point are plotted. #, Group in which 0/5 mice survived to 48 h. Data were analyzed using a two-way ANOVA and Tukey’s posttest. **, P < 0.01; ****, P < 0.0001. Kp, K. pneumoniae ST258; P1, Pharr; P2, ϕKpNIH-2; HK, heat-killed. Download FIG S1, EPS file, 2.2 MB (2.2MB, eps) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Phage treatment attenuates liver damage in ST258-infected mice. Images represent hematoxylin and eosin (H&E)-stained liver sections from mice infected with ST258 and treated with phage as indicated. Tissue sections were collected during necropsy 48 h post-treatment administration or earlier (12 h) in cases of rapid disease progression (Kp only and Kp + HK P1 + P2 control groups). The original magnifications are ×40 (left column) and ×200 (right column). Representative areas of necrosis are demarcated by a dashed yellow border. Kp, K. pneumoniae ST258; P1, Pharr; P2, ϕKpNIH-2; HK, heat-killed. Download FIG S2, TIF file, 2.5 MB (2.5MB, tif) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Taken together, these data suggest that phage treatment decreases bacterial burden in ST258-infected mice and that this effect correlates with the ability of phage to multiply in infected animals and to prevent mortality.
Development of phage resistance in bacteria recovered from mice.
We next assessed whether bacteria recovered from the blood and tissue of phage-treated mice had developed resistance to phage. ST258 colonies recovered from mouse blood and homogenized tissue samples were picked and cross-streaked with phages P1 and P2. Isolates were classified as being phage-sensitive (bacterial clearing in the region of overlap between bacterial and phage streaks), partially phage-resistant (bacterial growth inhibition in the region of overlap), or completely phage-resistant (bacterial confluence in the region of overlap). Overall, we detected a high frequency of colonies with at least some degree of phage resistance (Fig. 5). Partially resistant bacteria were especially prevalent, representing more than 90% (64/70) of colonies with discernible resistance. There were, however, fewer phage-resistant ST258 recovered 24 h and 48 h following treatment with the combination of phages compared to that with either phage alone. These data suggest that development of phage resistance is a potential caveat of the phage therapy approach, a problem addressed at least in part by using a combination of phages for treatment. Further work is needed to better understand the mechanisms by which phage resistance develops in vivo and the extent to which phage resistance alters clinical outcomes.
FIG 5.

Development of bacterial resistance to phage in blood and tissue. (A to D) Percentage of bacterial isolates recovered from blood (A and C) or tissue (B and D) that demonstrated complete or partial resistance to phage P1 (A and B) or P2 (C and D). Up to five bacterial isolates per mouse were tested for phage resistance using a cross-streak assay, equating to a maximum of 25 isolates (A and C) or 15 isolates (B and D) represented per bar. Data are presented as the mean ± standard deviation. Kp, K. pneumoniae ST258; P1, Pharr; P2, ϕKpNIH-2.
Bacterial and phage viability in mouse and human blood ex vivo.
To test whether increased survival of ST258-infected mice after phage treatment was linked directly to phage-mediated killing of bacteria, we evaluated the ability of phage (P1, P2, and P1 + P2) to kill ST258 in heparinized murine blood and murine serum (Fig. 6A and B). Compared to untreated control assays or those containing HK phage, survival of ST258 in mouse blood and serum decreased significantly following incubation with phage added singly or in combination (Fig. 6A and B). These data provide support to the idea that phage-mediated lysis of ST258 contributed to—or was largely responsible for—the observed rescue of mice infected with ST258 (Fig. 3).
FIG 6.

Bacteria and phage survival in whole blood and serum. (A and C) (Left) Percentage bacterial recovery after 1 h of incubation with phage at an MOI of 1 or sterile saline (Kp only) in heparinized murine (A) and human (C) whole blood. (Right) Percentage phage recovery after 1 h of incubation in heparinized murine (A) and human (C) whole blood (no bacteria added). (B and D) In vitro assay from panels A and C performed in murine (B) and human (D) serum. Data are presented as the mean ± standard deviation. P values were determined using a one-way ANOVA and Dunnett’s posttest (left side, panels A and B) or by unpaired two-tailed t test (right side, black and white bars). *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. Kp, K. pneumoniae ST258; P1, Pharr; P2, ϕKpNIH-2; HK, heat-killed.
The mouse survival data and assays with mouse blood were used to assess the potential role of phage in the treatment of ST258 bacteremia. As a first step toward providing an estimation of this therapeutic potential in humans, we tested the ability of these phages to kill ST258 in heparinized human blood and human serum (Fig. 6C and D). In contrast to the high rate of bacterial survival observed in murine blood (Fig. 6A), the ST258 strain used for these studies was killed efficiently in human blood (ST258 survival was <50% under all conditions tested) (Fig. 6C). These results are consistent with previous studies that show limited survival of some ST258 clinical isolates in human blood (23). Addition of phage failed to decrease bacterial survival further, an outcome that was observed in human blood as well as in human serum (Fig. 6D). Depletion of immunoglobulin G (IgG) restored the bactericidal effect of P1 and P1 + P2 (but not P2) in human serum, indicating that host antibody may diminish the activity of select phages in vivo (Fig. S3B and C). Further studies are needed to provide a more complete understanding of the interaction of phage with components of human blood.
Bacterial and phage survival in treated human serum and LB medium. (A to C) (Left) Percentage bacterial recovery after 1 h of incubation with phage at an MOI of 1 or sterile saline (Kp only) in LB medium (A), heat-inactivated human serum (B), and IgG-depleted human serum (C). Mean bacterial survival was compared between each phage group and the control (Kp only) group. For panels A and C, **, P < 0.01 and ****, P < 0.0001 versus the Kp control assay using a one-way ANOVA and Dunnett’s posttest. (Right) Percentage phage PFU recovery after 1 h of incubation in LB medium (A), heat-inactivated human serum (B), and IgG-depleted human serum (C) (no bacteria added). Data are presented as the mean ± standard deviation. Kp, K. pneumoniae ST258; P1, Pharr; P2, ϕKpNIH-2; HK, heat-killed. Download FIG S3, EPS file, 1.7 MB (1.7MB, eps) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
DISCUSSION
A dearth of treatment options for infections caused by multidrug-resistant bacteria has led to renewed interest in the therapeutic potential of phage. Despite increasing numbers of experimental phage therapy cases, controlled data to justify and guide clinical phage use remains limited. In this study, we demonstrate that prompt, systemic phage administration rescues mice with K. pneumoniae ST258 bacteremia. We present evidence of lower bacterial burdens in blood and tissues of phage-treated mice, with the caveat that CFU quantitation may have been impacted by phage lysis occurring in samples after extraction. We also report trends in survival differences between mice receiving 3 distinct phage treatments.
Not all phage therapy cases have culminated in positive clinical outcomes, a point which has sparked speculation as to whether phage selection had been suboptimal in those cases or if circumstances were unfavorable for other reasons (24, 25). Phages are known to exhibit a wide range of diversity, yet the specific structural and functional characteristics of a phage that enhance its therapeutic efficacy remain undefined (26). Considering that P2 was associated with higher titers in mouse blood and tissue as well as lower rates of resistance among recovered bacteria, it is notable that survival was higher among mice treated with P1. The shorter latent period of P1 compared with P2 (16 min versus 24 min) may account in part for the enhanced survival of P1-treated mice (22). The time to first dose of effective antibiotics has been shown to be a critical determinant of outcome in severe sepsis (27). Analogously, a short time to lysis may be an independent predictor of phage efficacy in cases of Gram-negative bacteremia.
As exemplified by the ST258-specific phages in this study, predicting relative therapeutic efficacy among two or more phages is challenging given the array of phage-, bacterium-, and host-specific factors likely to be involved. Traditionally, clinically deployed phages have been selected on the basis of their lytic activity in laboratory culture (28, 29). This one-dimensional valuation neglects a plethora of physiologic variables, including those related to host immunity. To determine whether in vitro phage screening is more informative when performed in biological fluids, we evaluated survival of ST258 and phage in murine and human whole blood and serum.
Compared to LB media, murine whole blood provided a much closer approximation of the bactericidal effect of phage in vivo. However, the phages tested here had no impact on ST258 survival in human blood and human serum. We observed that P1’s viability in human serum increased significantly after depletion of IgG and correlated with greater bactericidal effect of this phage in vitro (Fig. S3). These results indicate that human blood components can interfere with phage activity and potentially impair phage efficacy in vivo. Additional work is needed to identify features that predispose phages like P1 to high rates of neutralization in the blood. Conceivably this knowledge could inform phage selection, dosing, and engineering and thereby enhance its therapeutic effect.
We compared characteristics of the phages tested here to those of other phages that have shown efficacy in animal models of K. pneumoniae infection to determine whether specific features correlated strongly with treatment success (17–21). No strong correlation was apparent, although at present, the number of relevant phages for comparison is limited and the variability in experimental conditions under which these phages have been applied as treatment is wide. More informative insights may be derived from a larger, more homogeneous data set.
Our observation that treatment with a two-phage cocktail was associated with greater survival of ST258-infected mice than treatment with either phage individually supports the use of combination phage therapy (as opposed to monophage therapy) as a first-line approach. Consistent with this idea, we recovered phage-resistant bacteria from mice in all phage-treated groups, with a trend toward lower recovery in combination phage-treated mice. However, there are confounding factors (e.g., accelerated rate of decay in phage titer for certain premixed phage cocktails) that make it challenging to predict whether a particular phage cocktail will confer a net therapeutic advantage (30). Further investigation is needed in order to assess the impact of individual phages in a given phage cocktail.
Overall, our results demonstrate that phage therapy is effective for treatment of systemic ST258 infection in a mouse model. Substantial work remains to be done before it can be determined how readily these results might be translated into a therapeutic for humans (31, 32). A deeper understanding of the direct and indirect factors that shape phage-bacteria interactions in vivo will be critical to both predict and maximize the efficacy of clinical phage therapy in the future.
MATERIALS AND METHODS
Study approval and ethics statement.
All animal studies and procedures were approved by the Animal Care and Use Committee at Rocky Mountain Laboratories (RML), National Institute of Allergy and Infectious Diseases (NIAID), under RML protocol 2018-016. Human heparinized whole blood and serum were obtained from healthy individuals in accordance with a protocol (01-I-N055) approved by the Institutional Review Board for Human Subjects, NIAID. Informed consent was obtained from each donor.
Bacterial strains and phages.
MKP103, a derivative of the highly drug-resistant K. pneumoniae ST258 clinical isolate KPNIH1, was used to establish infection in mice (33). MKP103 was generated by Colin Manoil and colleagues at the University of Washington following targeted knockout of the carbapenemase gene blaKPC-3 in the pKpQIL plasmid of KPNIH1 (34). Experiments were performed with MKP103, not KPNIH1, given the increased antibiotic susceptibility of the former. Pharr (P1) and ϕKpNIH-2 (P2) are lytic phages capable of infecting MKP103. P1 and P2 were isolated from sewage as previously described (22).
Preparation of bacteria for injection.
On the day of mouse infection, MKP103 was grown to early exponential growth phase (optical density at 600 nm [OD600], 0.75) in Luria-Bertani (LB) broth before being pelleted by centrifugation and washed two times in sterile injection-grade saline. Bacteria were resuspended in the requisite volume of sterile saline to yield the desired inoculum (CFU) in a 100-μl injection volume.
Preparation of phage for injection.
Phages were amplified in broth cultures of exponential-phase MKP103. Phage lysates were extracted with 1:10 (vol/vol) chloroform and centrifuged at 6,500 × g for 10 min. The supernatant was passed through a 0.45-μm filter before undergoing centrifugation at 12,000 × g for 10 h at 4°C. Phage pellets were resuspended in minimal volumes of gelatin-free SM buffer (10 mM MgSO4, 50 mM Tris-HCl pH 7.5, 100 mM NaCl) and applied to a cesium chloride density gradient as described previously (35). After ultracentrifugation in a Beckman Coulter type 70.1 Ti rotor at 139,000 × g for 4 h at 4°C, the phage band was extracted and transferred to a Thermo Scientific Slide-A-Lyzer dialysis cassette (10K MWCO). Phages were dialyzed against high-salt SM buffer (1 M NaCl) for 12 h and then against unmodified SM buffer (100 mM NaCl) two times for 4 h each time. Purified phage preparations were diluted in sterile SM buffer to achieve the desired concentrations for injection.
Endotoxin measurement.
The endotoxin content of each purified phage preparation was quantified with a limulus amebocyte lysate (LAL) kinetic turbidimetric assay (Charles River Endosafe KTA2). Endotoxin concentrations below 2,000 endotoxin units (EU) per 100 μl were deemed safe for injection in accordance with published data (36).
Mouse bacteremia model.
Female C57BL/6J mice aged 16 to 17 weeks were purchased from The Jackson Laboratory. Mice were group-housed and allowed food and water ad libitum. Mice were acclimated for at least 5 days before the start of an experiment. On the day of experiment, mice were anesthetized and subsequently injected IP with 100 μl bacterial suspension. Phage treatment groups received equivalent volumes of phage suspension or saline (untreated control) on the contralateral side at predetermined time points. After infection, mice were checked regularly for signs of disease. Mice that met criteria for euthanasia were euthanized immediately in accordance with the approved ACUC protocol (2018-016), as indicated above.
Mouse treatment.
Purified P1, P2, and P1 + P2 phage preparations were concentration-adjusted to produce multiplicities of infection (MOI) approximately equal to 1 or 10 in a fixed injection volume. For each phage or phage combination, a single 100-μl IP injection of phage at an MOI 1 or 10 was administered at 1 h, 8 h, or 24 h after bacterial infection. For a given MOI, the total PFU remained constant. Therefore, mice in the MOI 1 treatment group received either 5 × 107 PFU of P1 or P2, respectively, or 2.5 × 107 PFU of P1 plus 2.5 × 107 PFU of P2 in combination.
Murine blood and tissue collection.
Mice were sacrificed at 1 h, 24 h, and 48 h post-phage treatment. Intracardiac whole blood was collected from each mouse (up to 5 mice per group). Select organs (bilateral lungs, liver, spleen, and right kidney) were harvested from up to 3 mice per group, combined with 1 ml sterile phosphate-buffered saline (PBS), and homogenized with a rotor-stator tissue homogenizer (Omni, Inc.). Heparinized whole blood (5 U heparin/ml blood) and tissue homogenates were plated on Klebsiella-selective media (Klebsiella Chromoselect selective agar base; Sigma, no. 90925 and Klebsiella selective supplement; Sigma, no. 15821) and LB agar for enumeration of colony counts.
Histopathology.
At predetermined time points (or upon reaching endpoint criteria), animals were euthanized and tissues were collected. Collected tissues were fixed in 10% neutral buffered formalin (Cancer Diagnostics, Durham, NC) for a minimum of 7 days. Tissues were placed in cassettes and processed with a Sakura VIP-6 Tissue Tek (Sakura Finetek, USA) on a 12-h automated schedule using a graded series of ethanol, xylene, and Paraplast Extra. Embedded tissues were sectioned at 5 μm and dried overnight at 42°C prior to staining with hematoxylin and eosin. Images were obtained using an Olympus BX51 microscope and an Olympus DP74 camera (Olympus Corporation). Image brightness and contrast were adjusted evenly using Adobe Photoshop, and figures were created using Adobe Illustrator CC 2020 (Adobe, San Jose, CA).
Cross-streak assay assessing phage resistance.
Small volumes (20 μl) of high-titer, cesium-purified phage preparations were streaked onto Terrific Broth (TB) plates and allowed to absorb. Individual colonies appearing after overnight incubation of mouse blood and tissue homogenate plate cultures were picked and streaked perpendicular to the dried phage streak. Cross-streaked plates were incubated overnight and assessed the following day for bacterial growth or clearance in the area of intersection.
In vitro bacterial and phage survival in blood and serum.
Bacterial survival in whole blood and serum was measured after 1 h of incubation according to previously described protocols with minor modifications (23, 37). Briefly, exponential-phase ST258 and purified phage at an MOI of ∼1 were added to heparinized human blood (5 U heparin/ml blood), freshly separated normal human serum, or thawed IgG-depleted human serum (Cell Sciences) for a total volume of 600 μl (∼1 × 106 CFU/ml blood) in 1.7-ml tubes. For assays with mouse blood, ST258 and phage were added to heparinized mouse blood pooled from 5 adult C57BL/6J mice (5 U heparin/ml blood) or thawed mouse serum from a commercial source (Innovative Research, Inc.) for a total volume of 300 μl (∼1 × 106 CFU/ml blood) in 1.7-ml tubes. In some experiments, mouse or human serum was inactivated by heating at 56°C for 30 min prior to use. Assay mixtures were rotated slowly at 37°C for 1 h, at which time bacteria and phage were plated for enumeration the next day. Bacteria were quantified by colony counts, and phage, by spot titer. Percentage survival was calculated with the equation (CFUt1/CFUt0) × 100, where CFUt0 is the number of CFU in the starting inoculum at time zero and CFUt1 is the number of CFU recovered at 1 h.
Statistical analysis.
Data were analyzed using GraphPad Prism 8.2.0 as indicated in the figure legends. Pairwise comparisons of mouse survival (between groups) were determined using a log rank (Mantel-Cox) test combined with a Bonferroni correction for multiple comparisons (to reduce type 1 error), and 0.05 was used as the initial significance threshold before applying the Bonferroni correction.
ACKNOWLEDGMENTS
We gratefully acknowledge the staff of the Rocky Mountain Veterinary Branch for their expert animal care assistance.
This work was supported by the Intramural Research Program of the National Institutes of Health, National Institute of Allergy and Infectious Diseases, and the National Cancer Institute, Center for Cancer Research.
Footnotes
This article is a direct contribution from Frank R. DeLeo, a Fellow of the American Academy of Microbiology, who arranged for and secured reviews by Anne-Catrin Uhlemann, Columbia University, and Biswajit Biswas, Naval Medical Research Center.
Citation Hesse S, Malachowa N, Porter AR, Freedman B, Kobayashi SD, Gardner DJ, Scott DP, Adhya S, DeLeo FR. 2021. Bacteriophage treatment rescues mice infected with multidrug-resistant Klebsiella pneumoniae ST258. mBio 12:e00034-21. https://doi.org/10.1128/mBio.00034-21.
Contributor Information
Shayla Hesse, Email: shayla.hesse@nih.gov.
Paul Keim, Northern Arizona University.
REFERENCES
- 1.Podschun R, Ullmann U. 1998. Klebsiella spp. as nosocomial pathogens: epidemiology, taxonomy, typing methods, and pathogenicity factors. Clin Microbiol Rev 11:589–603. doi: 10.1128/CMR.11.4.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tsay RW, Siu LK, Fung CP, Chang FY. 2002. Characteristics of bacteremia between community-acquired and nosocomial Klebsiella pneumoniae infection: risk factor for mortality and the impact of capsular serotypes as a herald for community-acquired infection. Arch Intern Med 162:1021–1027. doi: 10.1001/archinte.162.9.1021. [DOI] [PubMed] [Google Scholar]
- 3.Xu L, Sun X, Ma X. 2017. Systematic review and meta-analysis of mortality of patients infected with carbapenem-resistant Klebsiella pneumoniae. Ann Clin Microbiol Antimicrob 16:18. doi: 10.1186/s12941-017-0191-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ramos-Castaneda JA, Ruano-Ravina A, Barbosa-Lorenzo R, Paillier-Gonzalez JE, Saldana-Campos JC, Salinas DF, Lemos-Luengas EV. 2018. Mortality due to KPC carbapenemase-producing Klebsiella pneumoniae infections: systematic review and meta-analysis: mortality due to KPC Klebsiella pneumoniae infections. J Infect 76:438–448. doi: 10.1016/j.jinf.2018.02.007. [DOI] [PubMed] [Google Scholar]
- 5.Pitout JD, Nordmann P, Poirel L. 2015. Carbapenemase-producing Klebsiella pneumoniae, a key pathogen set for global nosocomial dominance. Antimicrob Agents Chemother 59:5873–5884. doi: 10.1128/AAC.01019-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.David S, Reuter S, Harris SR, Glasner C, Feltwell T, Argimon S, Abudahab K, Goater R, Giani T, Errico G, Aspbury M, Sjunnebo S, Eu SWG, Group ES, Feil EJ, Rossolini GM, Aanensen DM, Grundmann H, EuSCAPE Working Group, ESGEM Study Group . 2019. Epidemic of carbapenem-resistant Klebsiella pneumoniae in Europe is driven by nosocomial spread. Nat Microbiol 4:1919–1929. doi: 10.1038/s41564-019-0492-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van Duin D, Arias CA, Komarow L, Chen L, Hanson BM, Weston G, Cober E, Garner OB, Jacob JT, Satlin MJ, Fries BC, Garcia-Diaz J, Doi Y, Dhar S, Kaye KS, Earley M, Hujer AM, Hujer KM, Domitrovic TN, Shropshire WC, Dinh A, Manca C, Luterbach CL, Wang M, Paterson DL, Banerjee R, Patel R, Evans S, Hill C, Arias R, Chambers HF, Fowler VG, Kreiswirth BN, Bonomo RA, Multi-Drug Resistant Organism Network Investigators . 2020. Molecular and clinical epidemiology of carbapenem-resistant Enterobacterales in the USA (CRACKLE-2): a prospective cohort study. Lancet Infect Dis 20:731–741. doi: 10.1016/S1473-3099(19)30755-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bonomo RA, Burd EM, Conly J, Limbago BM, Poirel L, Segre JA, Westblade LF. 2018. Carbapenemase-producing organisms: a global scourge. Clin Infect Dis 66:1290–1297. doi: 10.1093/cid/cix893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shields RK, Nguyen MH, Press EG, Chen L, Kreiswirth BN, Clancy CJ. 2017. Emergence of ceftazidime-avibactam resistance and restoration of carbapenem susceptibility in Klebsiella pneumoniae carbapenemase-producing K pneumoniae: a case report and review of literature. Open Forum Infect Dis 4:ofx101. doi: 10.1093/ofid/ofx101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shields RK, Potoski BA, Haidar G, Hao B, Doi Y, Chen L, Press EG, Kreiswirth BN, Clancy CJ, Nguyen MH. 2016. Clinical outcomes, drug toxicity, and emergence of ceftazidime-avibactam resistance among patients treated for carbapenem-resistant Enterobacteriaceae infections. Clin Infect Dis 63:1615–1618. doi: 10.1093/cid/ciw636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sun D, Rubio-Aparicio D, Nelson K, Dudley MN, Lomovskaya O. 2017. Meropenem-vaborbactam resistance selection, resistance prevention, and molecular mechanisms in mutants of KPC-producing Klebsiella pneumoniae. Antimicrob Agents Chemother 61. doi: 10.1128/AAC.01694-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Organization World Health. 2015. Global action plan on antimicrobial resistance. https://www.who.int/antimicrobial-resistance/publications/global-action-plan/en/.
- 13.CDC. 2019. Biggest threats and data: 2019 AR threats report. https://www.cdc.gov/DrugResistance/Biggest-Threats.html.
- 14.Schooley RT, Biswas B, Gill JJ, Hernandez-Morales A, Lancaster J, Lessor L, Barr JJ, Reed SL, Rohwer F, Benler S, Segall AM, Taplitz R, Smith DM, Kerr K, Kumaraswamy M, Nizet V, Lin L, McCauley MD, Strathdee SA, Benson CA, Pope RK, Leroux BM, Picel AC, Mateczun AJ, Cilwa KE, Regeimbal JM, Estrella LA, Wolfe DM, Henry MS, Quinones J, Salka S, Bishop-Lilly KA, Young R, Hamilton T. 2017. Development and use of personalized bacteriophage-based therapeutic cocktails to treat a patient with a disseminated resistant Acinetobacter baumannii Infection. Antimicrob Agents Chemother 61:e00954-17. doi: 10.1128/AAC.00954-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dedrick RM, Guerrero-Bustamante CA, Garlena RA, Russell DA, Ford K, Harris K, Gilmour KC, Soothill J, Jacobs-Sera D, Schooley RT, Hatfull GF, Spencer H. 2019. Engineered bacteriophages for treatment of a patient with a disseminated drug-resistant Mycobacterium abscessus. Nat Med 25:730–733. doi: 10.1038/s41591-019-0437-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nir-Paz R, Gelman D, Khouri A, Sisson BM, Fackler J, Alkalay-Oren S, Khalifa L, Rimon A, Yerushalmy O, Bader R, Amit S, Coppenhagen-Glazer S, Henry M, Quinones J, Malagon F, Biswas B, Moses AE, Merril G, Schooley RT, Brownstein MJ, Weil YA, Hazan R. 2019. Successful treatment of antibiotic-resistant, poly-microbial bone infection with bacteriophages and antibiotics combination. Clin Infect Dis 69:2015–2018. doi: 10.1093/cid/ciz222. [DOI] [PubMed] [Google Scholar]
- 17.Cao F, Wang X, Wang L, Li Z, Che J, Wang L, Li X, Cao Z, Zhang J, Jin L, Xu Y. 2015. Evaluation of the efficacy of a bacteriophage in the treatment of pneumonia induced by multidrug resistance Klebsiella pneumoniae in mice. Biomed Res Int 2015:752930. doi: 10.1155/2015/752930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chhibber S, Kaur S, Kumari S. 2008. Therapeutic potential of bacteriophage in treating Klebsiella pneumoniae B5055-mediated lobar pneumonia in mice. J Med Microbiol 57:1508–1513. doi: 10.1099/jmm.0.2008/002873-0. [DOI] [PubMed] [Google Scholar]
- 19.Hung CH, Kuo CF, Wang CH, Wu CM, Tsao N. 2011. Experimental phage therapy in treating Klebsiella pneumoniae-mediated liver abscesses and bacteremia in mice. Antimicrob Agents Chemother 55:1358–1365. doi: 10.1128/AAC.01123-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kumari S, Harjai K, Chhibber S. 2010. Evidence to support the therapeutic potential of bacteriophage Kpn5 in burn wound infection caused by Klebsiella pneumoniae in BALB/c mice. J Microbiol Biotechnol 20:935–941. doi: 10.4014/jmb.0909.09010. [DOI] [PubMed] [Google Scholar]
- 21.Chadha P, Katare OP, Chhibber S. 2016. In vivo efficacy of single phage versus phage cocktail in resolving burn wound infection in BALB/c mice. Microb Pathog 99:68–77. doi: 10.1016/j.micpath.2016.08.001. [DOI] [PubMed] [Google Scholar]
- 22.Hesse S, Rajaure M, Wall E, Johnson J, Bliskovsky V, Gottesman S, Adhya S. 2020. Phage resistance in multidrug-resistant Klebsiella pneumoniae ST258 evolves via diverse mutations that culminate in impaired adsorption. mBio 11:e02530-19. doi: 10.1128/mBio.02530-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.DeLeo FR, Kobayashi SD, Porter AR, Freedman B, Dorward DW, Chen L, Kreiswirth BN. 2017. Survival of carbapenem-resistant Klebsiella pneumoniae sequence type 258 in human blood. Antimicrob Agents Chemother 61:e02533-16. doi: 10.1128/AAC.02533-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.LaVergne S, Hamilton T, Biswas B, Kumaraswamy M, Schooley RT, Wooten D. 2018. Phage therapy for a multidrug-resistant Acinetobacter baumannii craniectomy site infection. Open Forum Infect Dis 5:ofy064. doi: 10.1093/ofid/ofy064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aslam S, Courtwright AM, Koval C, Lehman SM, Morales S, Furr CL, Rosas F, Brownstein MJ, Fackler JR, Sisson BM, Biswas B, Henry M, Luu T, Bivens BN, Hamilton T, Duplessis C, Logan C, Law N, Yung G, Turowski J, Anesi J, Strathdee SA, Schooley RT. 2019. Early clinical experience of bacteriophage therapy in 3 lung transplant recipients. Am J Transplant 19:2631–2639. doi: 10.1111/ajt.15503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Henry M, Lavigne R, Debarbieux L. 2013. Predicting in vivo efficacy of therapeutic bacteriophages used to treat pulmonary infections. Antimicrob Agents Chemother 57:5961–5968. doi: 10.1128/AAC.01596-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rhodes A, Evans LE, Alhazzani W, Levy MM, Antonelli M, Ferrer R, Kumar A, Sevransky JE, Sprung CL, Nunnally ME, Rochwerg B, Rubenfeld GD, Angus DC, Annane D, Beale RJ, Bellinghan GJ, Bernard GR, Chiche J-D, Coopersmith C, De Backer DP, French CJ, Fujishima S, Gerlach H, Hidalgo JL, Hollenberg SM, Jones AE, Karnad DR, Kleinpell RM, Koh Y, Lisboa TC, Machado FR, Marini JJ, Marshall JC, Mazuski JE, McIntyre LA, McLean AS, Mehta S, Moreno RP, Myburgh J, Navalesi P, Nishida O, Osborn TM, Perner A, Plunkett CM, Ranieri M, Schorr CA, Seckel MA, Seymour CW, Shieh L, Shukri KA, et al. 2017. Surviving Sepsis Campaign: international guidelines for management of sepsis and septic shock: 2016. Crit Care Med 45:486–552. doi: 10.1097/CCM.0000000000002255. [DOI] [PubMed] [Google Scholar]
- 28.Henry M, Biswas B, Vincent L, Mokashi V, Schuch R, Bishop-Lilly KA, Sozhamannan S. 2012. Development of a high throughput assay for indirectly measuring phage growth using the OmniLog(TM) system. Bacteriophage 2:159–167. doi: 10.4161/bact.21440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gill JJ, Hyman P. 2010. Phage choice, isolation, and preparation for phage therapy. Curr Pharm Biotechnol 11:2–14. doi: 10.2174/138920110790725311. [DOI] [PubMed] [Google Scholar]
- 30.Bretaudeau L, Tremblais K, Aubrit F, Meichenin M, Arnaud I. 2020. Good manufacturing practice (GMP) compliance for phage therapy medicinal products. Front Microbiol 11:1161. doi: 10.3389/fmicb.2020.01161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Duplessis CA, Biswas B. 2020. A review of topical phage therapy for chronically infected wounds and preparations for a randomized adaptive clinical trial evaluating topical phage therapy in chronically infected diabetic foot ulcers. Antibiotics 9:377. doi: 10.3390/antibiotics9070377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Aslam S, Lampley E, Wooten D, Karris M, Benson C, Strathdee S, Schooley RT. 2020. Lessons learned from the first 10 consecutive cases of intravenous bacteriophage therapy to treat multidrug-resistant bacterial infections at a single center in the United States. Open Forum Infect Dis 7:ofaa389. doi: 10.1093/ofid/ofaa389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Snitkin ES, Zelazny AM, Thomas PJ, Stock F, Henderson DK, Palmore TN, Segre JA, Group NCSP . 2012. Tracking a hospital outbreak of carbapenem-resistant Klebsiella pneumoniae with whole-genome sequencing. Sci Transl Med 4:148ra116. doi: 10.1126/scitranslmed.3004129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ramage B, Erolin R, Held K, Gasper J, Weiss E, Brittnacher M, Gallagher L, Manoil C. 2017. Comprehensive arrayed transposon mutant library of Klebsiella pneumoniae outbreak strain KPNIH1. J Bacteriol 199:e00352-17. doi: 10.1128/JB.00352-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Haase-Pettingell C. 2000. CsCl step gradient to purify phage. http://web.mit.edu/king-lab/www/cookbook/cscl_grad_phage.htm. Accessed September 2017.
- 36.Purswani MU, Eckert SJ, Arora HK, Noel GJ. 2002. Effect of ciprofloxacin on lethal and sublethal challenge with endotoxin and on early cytokine responses in a murine in vivo model. J Antimicrob Chemother 50:51–58. doi: 10.1093/jac/dkf091. [DOI] [PubMed] [Google Scholar]
- 37.Eraso JM, Olsen RJ, Beres SB, Kachroo P, Porter AR, Nasser W, Bernard PE, DeLeo FR, Musser JM. 2016. Genomic landscape of intrahost variation in group A Streptococcus: repeated and abundant mutational inactivation of the fabT gene encoding a regulator of fatty acid synthesis. Infect Immun 84:3268–3281. doi: 10.1128/IAI.00608-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Histopathology scores Table S1, DOCX file, 0.02 MB (23.9KB, docx) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Quantitation of bacteria and phage in tissues. (A to F) Quantification of viable bacteria (A to C) and phage (D to F) in homogenized mouse organs at 1 h, 24 h, and 48 h after phage administration. Data from individual mice, up to 3 per treatment group per time point, are plotted. Bars denote group means. Samples for which no CFU or PFU was recovered are plotted as 0 on the x axis. †, Groups in which tissue collection occurred at 12 h instead of 24 h or 48 h due to rapid disease progression. Data acquired at this earlier time point are plotted. #, Group in which 0/5 mice survived to 48 h. Data were analyzed using a two-way ANOVA and Tukey’s posttest. **, P < 0.01; ****, P < 0.0001. Kp, K. pneumoniae ST258; P1, Pharr; P2, ϕKpNIH-2; HK, heat-killed. Download FIG S1, EPS file, 2.2 MB (2.2MB, eps) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Phage treatment attenuates liver damage in ST258-infected mice. Images represent hematoxylin and eosin (H&E)-stained liver sections from mice infected with ST258 and treated with phage as indicated. Tissue sections were collected during necropsy 48 h post-treatment administration or earlier (12 h) in cases of rapid disease progression (Kp only and Kp + HK P1 + P2 control groups). The original magnifications are ×40 (left column) and ×200 (right column). Representative areas of necrosis are demarcated by a dashed yellow border. Kp, K. pneumoniae ST258; P1, Pharr; P2, ϕKpNIH-2; HK, heat-killed. Download FIG S2, TIF file, 2.5 MB (2.5MB, tif) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Bacterial and phage survival in treated human serum and LB medium. (A to C) (Left) Percentage bacterial recovery after 1 h of incubation with phage at an MOI of 1 or sterile saline (Kp only) in LB medium (A), heat-inactivated human serum (B), and IgG-depleted human serum (C). Mean bacterial survival was compared between each phage group and the control (Kp only) group. For panels A and C, **, P < 0.01 and ****, P < 0.0001 versus the Kp control assay using a one-way ANOVA and Dunnett’s posttest. (Right) Percentage phage PFU recovery after 1 h of incubation in LB medium (A), heat-inactivated human serum (B), and IgG-depleted human serum (C) (no bacteria added). Data are presented as the mean ± standard deviation. Kp, K. pneumoniae ST258; P1, Pharr; P2, ϕKpNIH-2; HK, heat-killed. Download FIG S3, EPS file, 1.7 MB (1.7MB, eps) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.