

ABSTRACT
Microorganisms that degrade cellulose utilize extracellular reactions that yield free by-products which can promote interactions with noncellulolytic organisms. We hypothesized that these interactions determine the ecological and physiological traits governing the fate of cellulosic carbon (C) in soil. We performed comparative genomics with genome bins from a shotgun metagenomic-stable isotope probing experiment to characterize the attributes of cellulolytic and noncellulolytic taxa accessing 13C from cellulose. We hypothesized that cellulolytic taxa would exhibit competitive traits that limit access, while noncellulolytic taxa would display greater metabolic dependency, such as signatures of adaptive gene loss. We tested our hypotheses by evaluating genomic traits indicative of competitive exclusion or metabolic dependency, such as antibiotic production, growth rate, surface attachment, biomass degrading potential, and auxotrophy. The most 13C-enriched taxa were cellulolytic Cellvibrio (Gammaproteobacteria) and Chaetomium (Ascomycota), which exhibited a strategy of self-sufficiency (prototrophy), rapid growth, and competitive exclusion via antibiotic production. Auxotrophy was more prevalent in cellulolytic Actinobacteria than in cellulolytic Proteobacteria, demonstrating differences in dependency among cellulose degraders. Noncellulolytic taxa that accessed 13C from cellulose (Planctomycetales, Verrucomicrobia, and Vampirovibrionales) were also more dependent, as indicated by patterns of auxotrophy and 13C labeling (i.e., partial labeling or labeling at later stages). Major 13C-labeled cellulolytic microbes (e.g., Sorangium, Actinomycetales, Rhizobiales, and Caulobacteraceae) possessed adaptations for surface colonization (e.g., gliding motility, hyphae, attachment structures) signifying the importance of surface ecology in decomposing particulate organic matter. Our results demonstrated that access to cellulosic C was accompanied by ecological trade-offs characterized by differing degrees of metabolic dependency and competitive exclusion.
KEYWORDS: decomposition, ecological trade-offs, metagenomics, stable isotope probing, metaproteomics, metabolic dependency, competitive exclusion, surface ecology, soil carbon cycling, ecogenomics
INTRODUCTION
Cellulose is a major structural component of plant biomass and serves as a resource for productive and diverse soil microorganisms (1). Since cellulose is insoluble and highly crystalline, it cannot be transported across cell membranes. Hence, microorganisms rely on extracellular reactions to digest its fibers into soluble by-products for cell metabolism. Due to the structural complexity of lignocellulose, cellulose degradation is facilitated by synergistic interactions between diverse enzymes that differ in specific activity (2, 3). Physiological traits also influence the deconstruction of cellulose fibers, such as those linked to surface colonization like hyphal growth by members of fungi and Actinobacteria (4, 5), gliding motility in Bacteroidetes (6), and the formation of cellulosomes by many anaerobes (7, 8). These circumstances predispose microorganisms involved in cellulose degradation to metabolic, spatial, and ecological interactions. However, due to a reliance on isolation and coculturing, the nature of ecological interactions within cellulose-degrading consortia remain poorly understood, despite evidence of their occurrence in amplicon-based stable-isotope probing (SIP) studies (9–18). Shotgun metagenomics coupled to DNA-SIP now provides the capacity to compare the ecogenomic traits of diverse microorganisms that participate directly in the cellulose economy as it occurs in soil.
The extracellular nature of cellulose degradation creates conditions where the fitness of individuals is contingent on both competition and facilitation. Competition for cellulose and its degradation products impose fitness costs on cellulolytic organisms, promoting antagonistic interactions (19–21). However, facilitation by commensal and mutualistic partners enhances degradation rates to the benefit of cellulolytic organisms (22). Many cellulolytic microbes have close relatives lacking in endoglucanases, suggesting adaptive benefits from the gain or loss of these genes (23). The beneficiaries of community metabolism should be expected to shed energetically costly traits, resulting in adaptive gene loss and evolution of metabolic dependency (24). For example, noncellulolytic bacteria can complement the metabolic functions of cellulolytic bacteria in vitro, through complementary catabolism (25, 26), vitamin biosynthesis (27), amino acid biosynthesis (28), or biosynthesis of other essential metabolites (29). Such metabolic dependency can occur through specific, tightly coupled interactions, such as syntrophic partnerships, or via loosely coupled and nonspecific dependencies, such as a reliance on metabolic by-products or the mortality of community members (30, 31). We expect the anabolic and catabolic by-products of cellulolytic microbes to structure trophic interactions (i.e., the cellulose economy) and the fate of cellulosic carbon in soil.
Shotgun metagenomics and the recovery of metagenome-assembled genomes (MAGs) provide a cultivation-independent means of performing comparative genomics to study the phylogenetic and functional characteristics of microbial communities. This approach has been used to identify ecogenomic traits (32, 33) and to study metabolic dependency in environmental populations (34). By coupling this approach with DNA-SIP, one can distinguish MAGs from organisms that assimilate 13C from labeled substrates by separating and sequencing the 13C-enriched DNA (see Fig. S1 at https://osf.io/tb3n4/). Metagenomic SIP proved effective in resolving traits of cellulolytic and lignolytic populations in forest soil by improving MAG assembly (14). DNA-SIP can be used to estimate the degree of 13C labeling of individual MAGs by measuring the change in buoyant density across the CsCl gradient (35, 36). This gradient-resolved approach offers the capacity to distinguish between highly 13C-enriched DNA, corresponding to taxa with primary access to cellulosic C, less 13C-enriched DNA, corresponding to microbes with peripheral access to cellulosic C, and unenriched DNA from the broader soil community. Information about genomic content coupled to the degree of access to cellulosic C can be used as evidence of ecological trade-offs, such as traits of metabolic dependency (auxotrophy) or antibiotic production, in members of the cellulose economy.
We conducted a metagenomic SIP experiment with 13C-labeled cellulose to test hypotheses about the ecological trade-offs occurring in microbes that access carbon during cellulose degradation. We used comparative genomics to identify features of genome bins grouped by their degree of 13C enrichment (according to gradient-resolved SIP) and competency for cellulose degradation. We expected that cellulolytic microbes would be enriched in secondary metabolite gene clusters (SM), such as those that synthesize antibiotics, to control access to community resources. We further hypothesized that 13C-enriched, noncellulolytic microbes would exhibit signatures of metabolic dependency based on the degree of auxotrophy and/or capacity for degrading microbial necromass (i.e., numbers of genes encoding nucleases, peptidases, chitinases, and other hydrolytic enzymes). We expected early colonizers of cellulose, as identified in a companion study (11), to depend less on the products of community metabolism than late colonists, evidenced by degree of auxotrophy. We also identified traits related to surface colonization, which we anticipated would facilitate interactions on insoluble fibers (37). Results were validated against reference genomes and with metaproteomics to confirm gene expression by target groups. This study aims to evaluate the ecological trade-offs among members of the cellulose economy and to propose a framework for understanding community dynamics responsible for cellulose decomposition in soils.
RESULTS
Overview of metagenome assembly and designation of 13C labeling.
Initially, we performed a DNA-SIP experiment with [13C]cellulose to evaluate the temporal dynamics of decomposition over a 30-day period, profiled using 16S rRNA gene amplicon sequencing as previously described (11). Here, we performed gradient-resolved DNA-SIP using the 13C-labeled DNA at day 30 to discern differences in the degree of 13C incorporation and compare populations based on their relationships to cellulosic C. Deep sequencing was performed on a single gradient-resolved sample to maximize the recovery of genomic information from all members of the cellulose economy to improve our capacity to perform comparative genomics. Comparative genomics was performed in parallel on reference genomes sourced from the NCBI based on similarity to the full-length 16S rRNA gene.
Shotgun metagenomic sequencing of 13C-labeled DNA recovered a total of 1.1 billion reads after quality filtering, estimated by nonpareil (38) to cover 80% of genomic diversity in the DNA pool. Metagenome assembly produced a total of 356,131 contigs greater than 2.5 kb, amounting to a total length of 1.8 Gb (∼230 genomes of 8 Mb). The degree of 13C enrichment was estimated for each contig by comparing the CsCl gradient profile to simulated natural abundance profiles (see Fig. S2 at https://osf.io/tb3n4/). More than half of all contigs were designated either strongly or weakly 13C enriched (total length, 921 Mb), while the remainder were from genomes of abundant soil taxa that lacked evidence of 13C labeling (766 Mb). The occurrence of unlabeled DNA in heavier fractions is common in SIP experiments, primarily reflecting differences in the migration of DNA fragments across the gradient based on G+C content (39). Contigs clustered into coherent sets according to pentanucleotide frequency which grouped by patterns of 13C labeling and taxonomy (Fig. 1a and b), and not average G+C content (Fig. 1c). The random forest model used to predict enrichment status had an overall accuracy of 89.1% with high sensitivity and specificity for both strongly enriched (98.3% and 86.4%, respectively) and unenriched (100% and 93%) contigs (see details in the supplementary methods and Table S1 at https://osf.io/tb3n4/).
FIG 1.

A visual representation of the whole metagenomic assembly reveals the clustering of contigs with respect to taxonomic classification (a), 13C enrichment designation (b), and genome GC content (c). Contigs (>3.5 kb) were clustered by pentanucleotide frequency using the t-SNE multidimension reduction algorithm. This figure reveals the correspondence between taxonomic classification and 13C enrichment status (a versus b) and that enrichment status was not correlated with GC content (b versus c). An interactive html version in the supplementary data package at https://osf.io/tb3n4/ allows for detailed exploration of taxonomic annotations for all contigs.
Phylobin characteristics.
Contigs were grouped into phylobins based on 13C-labeling status and taxonomic classification (rank at the level of order). The large size of many phylobins can be attributed to natural pangenomic diversity (intraspecies) and to the grouping of mixed populations by order (interspecies), as revealed by the analysis of single-copy genes and single nucleotide polymorphisms per single-copy gene (see details in supplementary methods at https://osf.io/tb3n4/). A total of 47, 2, and 46 phylobins greater than 1 Mb were produced from strongly, weakly, and unenriched contig sets, respectively (see Table S2 at https://osf.io/tb3n4/). Of the 95 total phylobins, 38 were deemed high quality (>75% completeness) and were divided into four categories based on enrichment status and cellulolytic potential (inferred from the presence of endoglucanases): strongly 13C enriched and cellulolytic (nstrong = 12), strongly 13C enriched and noncellulolytic (nstrong = 8), weakly 13C enriched and noncellulolytic (nweak = 2), and unenriched (n = 16). Each phylobin represents a genomic-ecological unit, rather than an individual genome, encompassing genomes from at most four to seven genera for enriched and unenriched phylobins, respectively, based on the diversity of assembled full-length 16S rRNA genes (Fig. 2). Both phylobins and representative reference genomes from 13C-enriched cellulolytic taxa were larger (μPhyBin = 24.7 Mb; μRep = 7.1 Mb) than those from 13C-enriched noncellulolytic taxa (μPhyBin = 12.5 Mb; μRep = 5.2 Mb; Wilcoxon test, P ≤ 0.05) and unenriched taxa (μPhyBin = 11.4 Mb; μRep = 5.0 Mb; P < 0.01).
FIG 2.

Members of the cellulose-degrading consortium were defined by their taxonomy and functional capabilities encoded in metagenome-assembled phylobins (PBin) and representative genomes (Rep). Phylobins were categorized by their 13C enrichment and cellulolytic capacity and ranked along the y axis by the relative abundance of SSU rRNA gene fragments recovered (indicated by bar plots). Representative genomes were identified according to similarity of full-length 16S rRNA genes (column 1) and were grouped with their respective phylobins. Representative genomes with less than 97% similarity to a phylobin 16S rRNA gene are shaded in gray. Several phylobins were comprised of genomes from multiple genera, and the size of each (in megabases) and the percentage of peptides assigned to each phylobin are provided. The remaining columns show the presence/absence of genes for endoglucanases or those involved in surface attachment, surface motility (M), and secondary metabolite (SM) production. Boxes are shaded gray if a member of that genus reportedly possesses the ability for attachment or surface motility. Secondary metabolite production is designated if peptides corresponding to antimicrobial gene clusters were detected in the metaproteome. Only the most abundant “unenriched” phylobins are shown (for a full overview, see Table S4 at https://osf.io/tb3n4/).
Structure of the cellulose economy.
Taxa designated as strongly 13C enriched and cellulolytic (i.e., encoding endoglucanases) represented the greatest proportion of unassembled small subunit (SSU) gene fragments in metagenomes. The most abundant were classified into well-known genera of cellulolytic soil organisms, including Cellvibrio, Herpetosiphon (Chloroflexi), and members of the fungal order Sordariales (predominantly Chaetomium), as well as lesser-known cellulolytic genera, such as Devosia and Sphingomonas (Fig. 2). Peptides from these five taxa were also abundant within the total metaproteome (ntotal = 90,557 peptides; 33,765 unique proteins), occupying the following percentages of total peptides: Rhizobiales, 6.9%; Cellvibrionales, 2.6%; Sphingomonadales, 2.6%; Herpetosiphon, 1.0%; and Sordariales, 0.5%. Endoglucanases were detected in contigs classified into 22 of the 30 genera designated as strongly 13C enriched, consistent with their presence in reference genomes (Fig. 2; see Table S3 at https://osf.io/tb3n4/). The 22 genera designated strongly 13C enriched and cellulolytic comprised 29% of the total SSU rRNA gene fragments. In contrast, the seven genera designated strongly 13C enriched and noncellulolytic (Fig. 2) comprised 2.3% of recovered SSU rRNA reads. The composition of phylobins was in close agreement with prior 16S rRNA gene amplicon data (11) based on gene similarity and taxonomic classification (see Table S3 at https://osf.io/tb3n4/).
A diverse set of endoglucanases was recovered from phylobins, revealing a snapshot of the functional diversity of cellulolytic populations. A total of 430 unique endoglucanase homologs were identified (at a >80% identity threshold) belonging to 40 different carbohydrate-active enzyme (CAZy) families/subfamilies. Eighty-two of these endoglucanases were present within gene clusters that contained a carbohydrate-binding module. A total of 52 peptides in the metaproteome matched endoglucanases; the most abundant was a GH9 from Cellvibrio (Table 1). The second and third most abundant endoglucanases in the metaproteome were encoded by fungi (GH131 and GH7). Overall, most endoglucanases in the metaproteome were encoded by fungi (57%), which was disproportionate to the total relative abundance of fungal peptides (1.2%) in the metaproteome.
TABLE 1.
GH and LPMO gene families encoded in cellulolytic phylobins and detected in metaproteomea

Diverse glycosyl hydrolase (GH) genes and a single family of lytic polysaccharide monooxygenases (LPMO) were identified in cellulolytic phylobins, many of which matched peptides detected in the metaproteome (the number and type of each peptide is indicated). Taxonomic classifications and GH family are provided for the endoglucanase gene fragments found within each phylobin (lowest LCA classified to the order (o_), family (f_), or genus (g_) level). Taxon-specific endoglucanase families are indicated in bold. Five peptides matched endoglucanase genes not belonging to any phylobins. Uncl., unclassified.
Evidence for metabolic dependency and competitive exclusion.
To evaluate potential interactions among 13C-labeled taxa, we assessed the degree of auxotrophy (as an indicator of metabolic dependency) and presence of SM-encoding genes (antibiotic-based competition) in phylobins and their representative genomes. No phylobin or genome was fully prototrophic or auxotrophic for all biosynthetic pathways evaluated (n = 32), with the average phylobin being auxotrophic for 6 amino acids, 1 cofactor, and 2 vitamins and the average representative genome being auxotrophic for 5 amino acids, 1 cofactor, and 1 vitamin. The extent of auxotrophy did not differ significantly between 13C-enriched cellulolytic and 13C-enriched noncellulolytic phylobins or representative genomes (Fig. 3) but did differ among dominant taxa within each group.
FIG 3.

(a) A comparison of the number of complete biosynthetic pathways (prototrophy) revealed that 13C-enriched noncellulolytic (blue) phylobins and representative genomes were slightly less prototrophic than those which were either 13C-enriched cellulolytic (white) or unenriched (red). On average, these differences were not significant (Kruskal-Wallis; phylobins, P = 0.6; representative genomes, P = 0.2), although major populations within each group (Planctomyces versus Cellvibrio) exhibited consistent differences in accordance with hypotheses. (b) Prototrophy was significantly correlated with genome size for phylobins (r = 0.39; P = 0.01) but not representative genomes (r = 0.14; P = 0.28). A ranking of prototrophy in all phylobins and representative genomes is available in Table S4 at https://osf.io/tb3n4/.
The most prototrophic representative genomes were Cellvibrio (31/32 pathways detected; genome size = 5.2 Mb; designated 13C enriched, cellulolytic), Devosia (31/32; 4.2 Mb; 13C enriched, cellulolytic) and Leptothrix (31/32; 4.9 Mb; unenriched, noncellulolytic) (see Table S4 at https://osf.io/tb3n4/). The most auxotrophic representative genomes were Planctomyces (13/32; 3.2 Mb; 13C enriched, noncellulolytic), Nannocystis (16/32; 12.1 Mb; 13C enriched, noncellulolytic), and Vampirovibrio (16/32; 3.0 Mb; 13C enriched, noncellulolytic). These trends were consistent in phylobins, where Cellvibrionales (30/32; ranked 1st in terms of biosynthetic capacity among the 38 phylobins examined) and Rhizobiales (28/32; ranked 3rd) were among the most prototrophic, while Planctomycetales (24/32; 20th), Vampirovibrionales (15/32; 34th), Chthoniobacterales (10/32; 37th; 13C enriched, noncellulolytic), and Chloroflexales (9/32; 38th; 13C enriched, cellulolytic) were among the most auxotrophic. Overall, representative genomes from the phylum Actinobacteria were significantly more auxotrophic than Proteobacteria (μactino = 24.2 versus μproteo = 25.3; Wilcoxon test, P = 0.04) driven largely by Alphaproteobacteria (μalpha = 26.2, P = 0.003; see Fig. S3 at https://osf.io/tb3n4/). This trend was consistent, but not significant, in phylobins (μactino = 23.8 versus μalpha = 26.0). Representative genomes for Actinobacteria and Alphaproteobacteria did not significantly differ in size (μactino = 5.8 Mb versus μalpha = 5.3 Mb; Wilcoxon test, P = 0.46) or completeness (μ = 99.6% in both; P = 0.71).
The number of SM genes encoded in 13C-enriched cellulolytic phylobins (μrep = 14.0; μPhyBin = 40.0) was significantly higher than that in 13C-enriched noncellulolytic (μrep = 5.8; μPhyBin = 12.4; Wilcoxon test, P < 0.01) or unenriched (μrep = 4.8; μPhyBin = 10.0) phylobins. The trend remained after normalization to total phylobin (2.2 read counts per million [rcpm] versus 1.9 rcpm and 1.3 rcpm) or reference genome size (1.6 rcpm versus 1.0 rcpm and 1.0 rcpm). The genomes encoding the greatest number of SMs were Sporocytophaga, several Actinobacteria (Streptomyces, Lentzea, Dactylosporangium, and Kitasatospora) ,and Cellvibrio (see Table S4 at https://osf.io/tb3n4/). Genes encoding type 1 polyketide synthases were consistently more abundant in 13C-enriched cellulolytic taxa than the other two groups (see Fig. S4 at https://osf.io/tb3n4/). Nonribosomal peptide synthetases and bacteriocins were more frequently encoded in both 13C-enriched groups, but only peptides matching cellulolytic taxa were present in the metaproteome (see Fig. S4 at https://osf.io/tb3n4/). The metaproteome was dominated by terpene synthases from Actinobacteria, bacteriocins from Cellvibrio, and nonribosomal peptide synthetases from Sordariales. In contrast to trends in auxotrophy, representative genomes of 13C-enriched cellulolytic Actinobacteria encoded significantly higher numbers of SMs (μ = 20.0; n = 6; P = 0.03) than the cellulolytic Alphaproteobacteria (μ = 5.3; n = 6).
The orders Planctomycetales and Sphingomonadales were represented by phylobins that were weakly 13C enriched, alongside those that were strongly 13C enriched and unenriched (Fig. 2). Only the strongly 13C-enriched Sphingomonadales phylobin encoded endoglucanases and more SMs (predominantly bacteriocins) than the weakly 13C-enriched and unenriched phylobins (2.1 rcpm, 0.4 rcpm, and 1.3 rcpm, respectively). Both 13C-enriched Sphingomonadales phylobins shared the same pattern of auxotrophy (see Fig. S5 at https://osf.io/tb3n4/). No Planctomycetales phylobins encoded endoglucanase nor a substantial number of SMs.
Comparison of cellulolytic and hydrolytic potential.
The functional gene content of representative genomes accounted for substantial variation in enrichment status (Fig. 4). The trend was driven primarily by the relative abundance of glycosyl hydrolases (GH), which were 1.5- to 3-fold higher (after normalization for genome size) in 13C-enriched cellulolytic phylobins and corresponding reference genomes, respectively. This trend was evident in all gene families associated with lignocellulose degradation (GH, carbohydrate-binding modules [CBMs], auxiliary activity enzymes [AA], and polysaccharide lyases [PL]), which collectively explained 63% of the variation in community functional composition along nonparametric multidimensional scaling axis 1 (NMDS1) (Fig. 4a; see Table S5a at https://osf.io/tb3n4/). Genomes were also separated along the secondary axis (NMDS2) based on genome size and peptidase and motility gene content, which explained 16.3, 16%, and 19% of variation, respectively (see Table S5a). The degree of auxotrophy did not correlate with variation in functional gene content in either representative genomes or phylobins (see Table S5b at https://osf.io/tb3n4/). In addition, the relative abundance of biomass-degrading enzymes (e.g., peptidases and nucleases) did not differ with respect to degree of 13C enrichment or cellulolytic capacity, either in phylobins or representative genomes. In contrast, broad differences in functional gene categories were observed between Actinobacteria and Proteobacteria (Fig. 4a).
FIG 4.
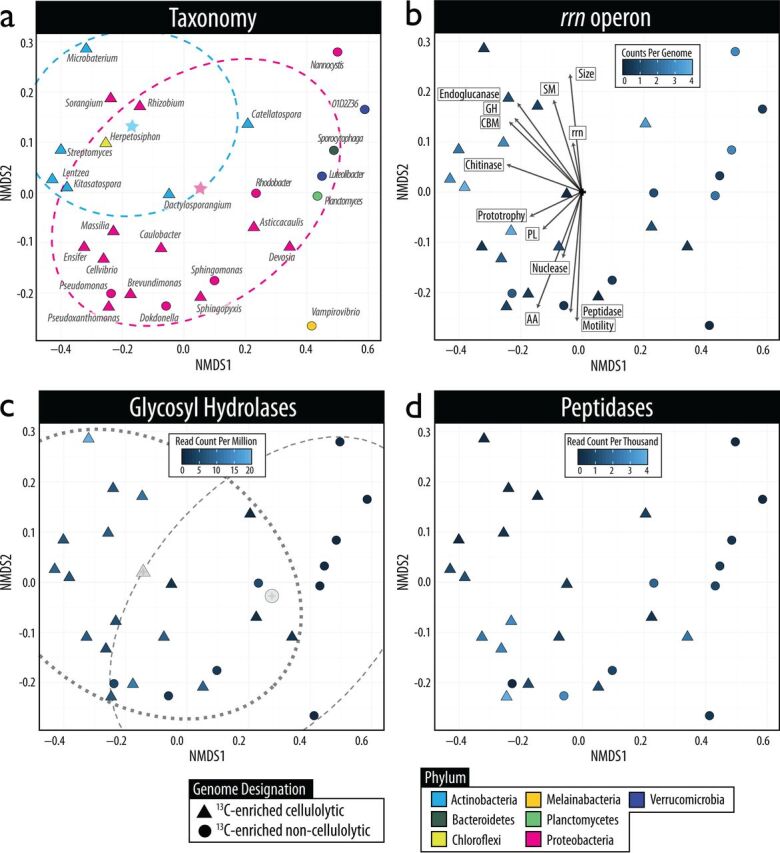
The functional gene contents of representative genomes were compared by NMDS using the Bray-Curtis dissimilarity of gene abundances normalized to genome size. Most variation among representative genomes was attributable to carbohydrate active enzyme content (63% of NMDS1) (see Table S5 at https://osf.io/tb3n4/). Four panels showing the same ordination are colored according to the taxonomic classification at the phylum level (a), the rrn operon copy number (b), the abundance of glycosyl hydrolases (c), and peptidase abundance (d). Genomes formed clusters according to cellulolytic potential (analysis of similarities [ANOSIM], R = 0.498, P < 0.001) with the centroid of each group displayed in panel c. Genomes loosely clustered by phylogenetic differences between Actinobacteria and Proteobacteria, although lacking statistical support (ANOSIM, R = 0.1, P = 0.2), with the centroid (star symbol) for each shown in panel a. In panel b, functional gene data were fitted to the ordination, with the arrow length proportional to the correlation between each variable and ordination axes. Abbreviations: GH, glycosyl hydrolase; SM, secondary metabolite gene cluster; CBM, carbohydrate-binding module; rrn, ribosomal operon; PL, polysaccharide lyase; AA, auxiliary activity; size, genome size.
Temporal dynamics in cellulose economy.
Early and late-stage colonizers of cellulose were identified according to genome-based predictions of growth rate (see Table S4 at https://osf.io/tb3n4/). Taxa designated as 13C enriched and cellulolytic were predicted to have faster generation times based on phylobins (3.0 h) and representative genomes (3.1 h) than 13C-enriched noncellulolytic taxa (5.7 h and 5.6 h, respectively) (Fig. 5a), although these differences were not significant (Kruskal-Wallis, Pphybin = 0.33 and Prep = 0.52). The 13C-enriched noncellulolytic taxa exhibited a bimodal distribution of predicted growth rate (Fig. 5a), and the set of genomes with slower generation times (generation time > 5 h) was significantly more auxotrophic (μslow = 15.8/32 prototrophies) than taxa with faster predicted generation times (<3 h; μfast = 23.8/32; Wilcoxon test, P = 0.05).
FIG 5.

Genome-based predicted generation times of taxa (a) corresponded with temporal patterns in relative abundance in soil microcosms amended with cellulose (b and c), where 13C-enriched cellulolytic phylobins and representative genomes tended to have higher growth rates than noncellulolytic phylobins and their representatives. In panel a, the differences in genome-based predictions of generation time were, on average, not significant (Kruskal-Wallis, Pphybin = 0.33 and Prep = 0.52). However, the most abundant 13C-enriched cellulolytic and noncellulolytic taxa, Cellvibrio and Planctomycetaceae, respectively, exhibited characteristics of faster (b) and slower (c) growth, consistent with expectations of the degree of their metabolic dependency. Several other major taxa, including Devosia, Sphingomonas, and members of the Verrucomicrobia, exhibited similar trends (see Fig. S6 at https://osf.io/tb3n4/). In panels b and c, each panel is divided into two data sets: one corresponding to the relative abundance of bacterial populations in whole DNA extract from soil amended with cellulose, and the other corresponding to the relative abundance in 13C-enriched DNA pools from soils amended with 12C natural abundance or 13C-labeled cellulose. The y axis (read counts per thousand) corresponds to the average relative abundance of heavy CsCl gradient fractions (F2 – F10). A complete ranking of predicted generation times for phylobins and representative genomes is available in Table S4 at https://osf.io/tb3n4/.
The genome-based characterizations of early and late-stage colonizers were consistent with temporal patterns of taxa observed in time course amplicon sequencing data in a companion study (11). The highly prototrophic taxa Cellvibrio and Devosia increased in relative abundance earliest, peaking in 13C enrichment at days 7 to 14 and declining by day 30 (Fig. 5b; see Fig. S6a at https://osf.io/tb3n4/). Chaetomium populations were also early colonizers, showing 13C enrichment by day 7 (see Fig. S7 at https://osf.io/tb3n4/). In contrast, the relative abundance of Actinobacteria was less dynamic, and these organisms tended to become labeled on, or after, day 14. Taxa predicted to be the most slowly growing, and identified as 13C enriched, noncellulolytic, and highly auxotrophic (Planctomyces, Sphingomonas, and members of Verrucomicrobia), began to increase in relative abundance only after day 14 and were maximally 13C enriched on day 30 (Fig. 5c; see Fig. S6c at https://osf.io/tb3n4/).
Surface adhesion and surface motility.
Phylobins from cellulolytic taxa were more likely to encode the capacity for surface adhesion and/or surface motility than other groups, including twitch motility, pilus systems, and fimbriae (see Fig. S8a at https://osf.io/tb3n4/). Surface attachment proteins were abundant in reference genomes from 13C-enriched taxa (both cellulolytic and noncellulolytic) and in phylobins classified as Rhizobiales and Caulobacterales (Fig. S8b). Adhesion proteins used in gliding motility (aglZ and sprB) were present in reference genomes of cellulolytic taxa but absent from phylobins.
DISCUSSION
We performed comparative genomics using metagenomic SIP data to test hypotheses about ecogenomic traits occurring in cellulolytic and noncellulolytic microorganisms participating in the cellulose economy. Taxa identified as 13C enriched and cellulolytic had larger genomes and a greater number of genes encoding CAZymes, secondary metabolites, surface motility, or surface attachment and tended to have faster generation times than 13C-enriched noncellulolytic and unenriched taxa. This evidence supports our hypothesis that the fate of cellulose carbon is mediated by ecological trade-offs between cellulolytic and noncellulolytic taxa. Furthermore, 13C-enriched cellulolytic taxa encoded diverse endoglucanases, representing 39 different subfamilies, but no single taxon encoded more than a third of these enzymes, supporting the importance of synergistic interactions among degraders. Auxotrophy was common among both 13C-labeled cellulolytic and noncellulolytic taxa, indicating that most taxa acquire essential metabolites from other community members. The average phylobin was auxotrophic for 9 of 32 pathways evaluated, although the highest levels of auxotrophy occurred among noncellulolytic 13C-labeled taxa.
The two most prominent 13C-labeled cellulolytic taxa, Cellvibrio and Chaetomium, are commonly abundant in soil from agroecosystems. Both dominated access to cellulosic C in previous SIP cellulose studies of agricultural soils (12, 17) and are favored by tillage (40–43), while Chaetomium populations are early successionists in clear-cut forest soil (18). The most abundant endoglucanase in our metaproteome, a GH9 from Cellvibrio, was homologous to the main cellulase found in worm castings from agricultural soil (NCBI accession no. ACY24809) (44). The predominance of these ruderal taxa, which we found to be fast growing and self-sufficient (i.e., prototrophic), likely reflects the frequent soil disturbances in agroecosystems. Therefore, the ecological attributes of the cellulose economy we observed may be characteristic of managed agricultural lands. The use of sieving to prepare our soil microcosm might have favored disturbance-adapted organisms. However, in a similar SIP-cellulose study, where soils from different ecosystems were subject to the same microcosm conditions, Cellvibrio populations were specific to and dominant in agricultural soil (12). It remains to be seen how the composition and relative importance of the ecological classes we observed differ in less frequently disturbed soils.
Ecological classes of the cellulose economy.
Our results demonstrated that access to cellulosic carbon is mediated by trade-offs related to the capacity to produce carbohydrate-active enzymes and antibiotics, biosynthetic capacity (i.e., prototrophy), growth rate, and adaptation to colonize surfaces. Our results revealed at least three broad ecologically coherent classes which accessed 13C from cellulose: (i) fast-growing, biosynthetically competent cellulolytic taxa (e.g., Cellvibrio, Chaetomium, and Devosia), (ii) slower growing, metabolically dependent (more auxotrophic), cellulolytic taxa (e.g., Actinobacteria and Herpetosiphon), and (iii) slower growing, metabolically dependent (highly auxotrophic), noncellulolytic taxa (e.g., Planctomycetales, Verrucomicrobia, and Vampirovibrionales). Certainly, a wide range of adaptive traits will affect access to cellulosic carbon during decomposition, but these classes provided a useful framework for grouping the major ecogenomic traits we observed. We also propose that these classes may reflect the broader underlying ecological characteristics of microbial consortia involved in decomposing various forms of particulate organic matter in soil.
Independent primary degraders.
Bacteria in the class of independent primary degraders are first to colonize cellulosic materials based on their cellulolytic competency, self-sufficiency, and rapid growth. On average, the phylobins and representative genomes of 13C-enriched cellulolytic taxa were more prototrophic and had lower minimum generation times than their 13C-enriched noncellulolytic counterparts, although these results were statistically insignificant due to the phylogenetic and ecological diversity within groups. Cellvibrio and Devosia were among the most enriched taxa in the [13C]DNA pool (1st and 4th, respectively) and were the two most prototrophic of any genome or phylobin examined. In a companion study, Cellvibrio and Devosia populations peaked earlier than any other 13C-enriched taxa and were in decline as dependent taxa increased in relative abundance (11). Chaetomium exhibited similar early 13C enrichment, occupying upwards of 20% of the [13C]DNA pool by day 7 in a second companion study at the same field site (45). Our method for predicting prototrophy was not validated for eukaryotic genomes; yet, species of Chaetomium are prototrophic—growing on cellulose in minimal medium without the addition of amino acids or cofactors (46). The rapid growth and self-sufficiency of Cellvibrio and Chaetomium were coupled with a strategy of competitive exclusion via the production of antibiotics such as bacteriocin, likely a cellvibriocin (47), and fungicides (48–50). Notably, the early dominance of Cellvibrio and Chaetomium populations and later shift to Actinobacteria were reported in a separate SIP-cellulose study in agricultural soil (17). We expect the competitive nature of these early colonizers and their metabolic by-products to influence the ability of interdependent primary degraders and noncellulolytic taxa to access C derived from cellulose degradation.
Interdependent primary degraders.
Bacteria in the second class, i.e., interdependent primary degraders, primarily Actinobacteria but also Herpetosiphon (Chloroflexi), were cellulolytic but exhibited higher levels of auxotrophy and SM production than early colonists. Populations of Actinobacteria lagged in comparison to Cellvibrio, with the first signs of 13C labeling appearing at day 14 with inconsistent changes in relative abundance over time (11). These trends suggest a greater dependency on external nutrient sources, affected by top down (i.e., mortality-driven) or bottom up (i.e., nutrient limitation because of competition for nutrients) controls, and may also reflect lower growth rates. Actinobacteria encoded and produced the greatest number of SMs and SM peptides. These were primarily terpenoids, which can function in interspecific signaling in soil, potentially facilitating mutualistic interactions upon which Actinobacteria depend (51). The potential benefit of interdependency, as a primary degrader, was apparent in the consistent auxotrophy of Actinobacteria for four of the costliest nonaromatic amino acids to synthesize, namely, the branched-chain acids isoleucine (ranked 1st) and leucine (2nd), the sulfur-containing methionine (3rd), and the amino-group-containing lysine (4th) (52, 53). Auxotrophy for branched-chain amino acids is a signature of genome reduction and dependency in other ubiquitous soil bacteria (54). We hypothesize that the fitness of interdependent primary degraders depends on community interactions, facilitated by their cellulolytic capacity and SM production.
Secondary consumers: mutualists, opportunists, and parasites.
The third class, that of secondary consumers, was defined by characteristics of their dependency on the metabolism of primary degraders, indicating that these taxa are secondary consumers of cellulosic carbon. Members of this group all possessed several signatures of metabolic dependency, which included high levels of auxotrophy, the lack of necessary genes for cellulose degradation, and late-stage and low levels of 13C enrichment. The most prominent secondary consumers were members of Planctomycetales, Vampirovibrionales, and Verrucomicrobia (Luteolibacter, “Candidatus Xiphinematobacter,” and 01D2Z36). These taxa all reached maximal relative abundance only after independent primary degraders had become enriched (Cellvibrio, Devosia, and Chaetomium) and remained abundant even after their decline. This pattern suggests a dependence on products of community metabolism either through cometabolism, the consumption of metabolic by-products, or the consumption of macromolecules released during the turnover of microbial biomass. The consequence of these dependencies (i.e., mutualistic versus antagonistic, etc.) could not be determined from our data, but previous research indicates that these taxa encompass a range of symbiotic relationships, including mutualism, opportunism, and parasitism (MOP).
Planctomyces are commonly found to colonize the surfaces of marine algae and to metabolize polysaccharides but not cellulose (55–57). They purportedly assimilate oligosaccharides into their cells, indicating the ability to scavenge higher-molecular-weight degradation by-products (13, 58–60). The capacity of Planctomyces to attach to surfaces with holdfast and their distinct tolerance to a range of antibiotics would advantage an opportunistic lifestyle, particularly among antibiotic-producing primary degraders (61–63). The cultured representatives for the two other highly auxotrophic 13C-enriched noncellulolytic taxa are obligate symbionts, namely, Vampirovibrio and “Candidatus Xiphinematobacter.” The former are algal parasites that encode a range of GHs (64) but lack endoglucanases, and the latter can be endobionts of nematodes and are abundant in forest litter, cellulose-degrading consortia, or in association with Basidiomycota (65–68). “Candidatus Xiphinematobacter” and Planctomyces have been previously identified as part of the cellulose economy in agricultural soil using SIP, with the former hypothesized to be a late-stage, mutualistic cellulosic C consumer (10, 15).
Differentiating between an opportunistic or mutualistic relationship among primary degraders and noncellulolytic taxa presents a challenge where lignocellulose is being decomposed. The degradation of other plant biomass by noncellulolytic taxa can benefit primary degraders, supported by the fact that secondary consumers possess the capacity to degrade other carbohydrate polymers, like Vampirovibrio and Planctomyces (57, 64). One set of phylobins provided evidence for what could be considered opportunistic “cheating” (24). Phylobins from Sphingomonadales differed in terms of weak and strong 13C enrichment yet shared the same pattern of auxotrophy. The strongly enriched phylobins encoded several endoglucanases and bacteriocins, while the equally sized, weakly enriched phylobins lacked these capabilities. While we cannot rule out some form of mutualism, the weakly 13C-enriched phylobin did not encode any unique CAZyme families associated with hemicellulose or pectin degradation. These data suggest that the strongly labeled cellulolytic strain is degrading [13C]cellulose extracellularly and the weakly 13C-enriched strain can access degradation products as well as other sources of unlabeled carbon present in soil. The capacity of Sphingomonas to degrade cellulose through the activity of extracellular enzymes has been reported (69, 70).
Role of surface ecology.
Several major populations of microbes that accessed 13C from cellulose were capable of surface adherence and/or surface motility. Genes encoding surface attachment were present in phylobins, or have been previously reported (Fig. 2), in Rhizobiaceae (Ensifer/Sinorhizobium, Rhizobium, and Agrobacterium), Hyphomicrobiaceae (Devosia), Sphingomonadaceae (Sphingomonas), and Caulobacteraceae (Asticcacaulis, Brevundimonas, and Caulobacter), as well as in Pseudoxanthomonas and Planctomycetaceae (Planctomyces and Rhodopirellula) (71–73). Each of these genera, except for those in Planctomycetaceae, are represented by isolates capable of degrading cellulose (74–81). For these organisms, attachment would provide preferential access to the extracellular by-products of cellulose degradation. This phenomenon is exemplified by the abundance of sugar transporters located on the stalk used by Caulobacter to adhere to surfaces (82, 83). Attachment may also facilitate cooperation to crowd out competitors from accessing resources, as observed in the social behavior of Caulobacter during xylan degradation (84) or in the coordination of extracellular degradative processes by the surface-gliding bacteria Herpetosiphon and Sorangium (85, 86). Social interactions and cell surface density are critical determinants of the rate and efficiency of decomposition of particulate carbon (22). Thus, the dynamics of surface attachment have ramifications for microbial ecology and evolution, as observed in the rumen (87, 88) and aquatic niches (22, 31, 89), which have not yet been studied in soil or in relation to biogeochemical cycling. For example, our observations suggest that the universal priming effect induced by cellulose (90) might result from promoting the growth of surface-adapted taxa which can subsequently gain access to insoluble, less bioavailable C pools, such as particulate and surface-associated organic matter (91).
Diversity at the subgenus level in the cellulose economy.
Deep shotgun metagenomics provided a comprehensive set of genomes from taxa present in the cellulose economy but was ineffective at resolving the genomes of closely related species. Phylobins were comprised of large pangenomes, which limited our ability to test for adaptive gene loss among closely related species, known to be important in the evolution of metabolic dependencies (24, 92). The recovery of large single-genus phylobins for Myxococcales (Sorangium), Cellvibrionales (Cellvibrio), Planctomycetales (Planctomyces), and Micrococcales (Microbacterium) provided evidence of sizeable pangenomic genetic diversity, which might reflect niche partitioning among close relatives. However, the degree of 13C enrichment within these single-genus phylobins did not differ, except for Planctomycetales and Sphingomonadales (i.e., “weak” versus “strong” phylobins). We conclude that few differences in the capacity to access cellulosic carbon was evident among closely related populations.
Conclusions.
The taxonomic composition of 13C-labeled populations was consistent with past SIP-cellulose experiments, represented by populations of Sordariales (Chaetomiaceae), Actinobacteria (Microbacteriaceae, Streptomycetaceae, and Micrococcaceae), Alphaproteobacteria (Rhizobiaceae, Caulobacteraceae, and Sphingomonadaceae), Deltaproteobacteria (Polyangiaceae), Gammaproteobacteria (Cellvibrionaceae), Bacteroidetes (Cytophagaceae and Sphingobacteriaceae), Planctomyces (Planctomycetaceae and Pirellulaceae), and Verrucomicrobia (Verrucomicrobiaceae, Opitutaceae, and Chthoniobacteraceae), although missing Betaproteobacteria (9–18). Yet, for the first time, comparative genomics was used to reveal the ecogenomic traits of these taxa and to deepen understanding of the ecological strategies they employ to gain access to cellulosic C. We identified self-sufficient cellulolytic bacteria and fungi (e.g., Cellvibrio and Chaetomium) that sought to restrict access via competitive exclusion and other more interdependent cellulolytic bacteria (e.g., Actinobacteria and Herpetosiphon), whose fitness depended on the metabolic by-products of the community. A third class of noncellulolytic taxa that accessed cellulosic C (e.g., Planctomycetes, Vampirovibrio, Verrucomicrobia) were characterized by dependency on community resources that encompassed mutualistic, opportunistic, and parasitic interactions, which have not yet been fully described due to challenges in cultivability (64). Overall, our findings attest to the existence of an economy underscored by ecological interactions and novel physiological adaptations that impact the degradation of cellulose in soil. These findings improve our capacity to interpret and quantify the effect of community structure and function on decomposition and carbon cycling.
MATERIALS AND METHODS
Sample description and recovery of 13C-enriched DNA.
DNA-SIP was performed using an agricultural soil incubated with 13C-labeled cellulose for 30 days to capture the labeling of primary and secondary populations, as performed previously (11). In brief, a microcosm was prepared with 10 g of soil from a tilled agricultural field under organic management in Penn Yan, New York, as previously described (93). The soil was sieved (2 mm), homogenized, wetted to and maintained at 50% water holding capacity, corresponding to 70% water filled pore space (94), and preincubated for 2 weeks prior to initiation of the experiment. After the initial soil respiration subsided, an amendment designed to approximate the composition of plant biomass was added at a total of 2.9 mg C g−1 soil (dry weight). By weight, the mixture was comprised of 38% 13C-labeled bacterial cellulose (99 atom% 13C), 23% lignin alkali, 20% xylose, 3% arabinose, 1% galactose, 1% glucose, 0.5% mannose, 10.6% amino acids, and 2.9% Murashige Skoog basal salt mixture (11). Bacterial cellulose was harvested from Gluconoacetobacter xylinus grown on 99 atom% [13C]glucose as the sole carbon source as previously described (11). After incubation, extracted DNA was subjected to CsCl density gradient centrifugation and fractionated into 35 100-μl aliquots. Shotgun metagenomes were prepared from eight gradient fractions, starting at a buoyant density (BD) of 1.749 g ml−1 (F6) and continuing to a BD of 1.717 g ml−1 (F13). A schematic overview of the methods used in this study is presented in Fig. S1 at https://osf.io/tb3n4/. The 16S rRNA gene and ITS1 region amplicon data from this DNA-SIP experiment (11) and a companion study (45) are available at the NCBI under BioProject no. PRJNA317227 and PRJNA589050, respectively.
DNA and peptide sequencing.
Shotgun metagenomes were generated by multiplexing DNA from each gradient fraction using the Nextera XT library preparation kit and then sequenced using three lanes of the Illumina HiSeq 2500 (150 bp, paired end). A subsequent round of sequencing was performed on each gradient fraction using a single lane of MiSeq (250 bp, paired end) using a library prepared with the Illumina Nextera XT DNA library prep kit (product number FC-131-1024; Illumina). The raw sequencing data are archived in the European Nucleotide Archive (BioProject no. PRJEB23737). A full description of protein extraction, purification, digestion, mass spectroscopy, and peptide annotation is available in the supplementary methods at https://osf.io/tb3n4/. In brief, protein was extracted from 5 g of soil with the NoviPure soil protein kit (Qiagen), initially separated and massed using a Waters nano-Acquity M-class dual pumping UPLC system (Milford, MA) and a Q-Exactive HF mass spectrometer (Thermo Scientific, San Jose, CA). Twenty-four fractions were subsequently submitted for liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis using an LTQ Orbitrap Velos mass spectrometer (ThermoFisher, Waltham, MA). Peptides were identified from LC-MS/MS data using predicted protein sequences from the metagenome and filtered with a false discovery rate cutoff of 1%.
Assembly and classification of SSU RNA genes.
Partial 16S and 18S rRNA gene fragments were identified in unassembled reads to estimate relative abundances. Fragments were identified using Infernal (v. 1.1.2) (95) and assigned taxonomy using the mothur implementation of the RDP Classifier (96, 97) with the Silva database (silva.nr_v128) as the reference (98). Full-length 16S or 18S rRNA genes were assembled using MATAM (99), also using the Silva database. We manually recovered a full-length 16S rRNA gene for Vampirovibrio, which was prevalent in SSU fragments, but not assembled by MATAM (see details in the supplementary methods at https://osf.io/tb3n4/).
Shotgun metagenome assembly.
Metagenomes for each gradient fraction were composited and assembled using an iterative process to maximize assembly quality (see Fig. S1 and details in the supplementary methods at https://osf.io/tb3n4/). In brief, an initial de novo assembly was performed using megahit (v1.1.1-2-g02102e1) (100). Contigs shorter than 2,500 bp were discarded (∼7% of total). Contigs were then classified by the lowest common ancestor (LCA) algorithm implemented by MEGAN (v. 6) (101) based on DIAMOND BLASTX searches (102) against the NCBI nr database (downloaded 3 February 2017). To improve assembly, two additional assemblies were performed on read sets with reduced sequence diversity. This reduction was achieved by segregating unassembled, quality-processed reads by mapping to (i) the LCA taxonomy of the initial assembly, at the rank of order, and (ii) publicly available genomes represented in the full-length 16S rRNA gene library (see Table S6 at https://osf.io/tb3n4/). All assemblies were then merged using MeGAMerge (103) with the latest version of MUMMer (104) (v.4beta) designed for large data sets. Merging improved the assembly statistics as determined by QUAST (105), increasing N50 from 4,407 to 5,419 (see Table S7 at https://osf.io/tb3n4/).
Designating 13C enrichment of contigs with gradient-resolved SIP.
The relative abundance of every contig across the density gradient (a gradient profile) was determined by calculating average read depth using “jgi summarize bam contig depths” from MetaBAT (106) (v. 2.12.1). The gradient profile of each contig was also simulated with natural abundance of 13C (∼1.1 atom% C) to control for variation in GC content by using methods outlined previously (39, 107). A random forest regression model was used to assign a categorical degree of 13C enrichment for each contig, namely, “strongly” and “weakly” enriched, “unenriched,” and “bimodal” (i.e., local maxima in both heavy and light portions of the gradient), and “undetermined” (see examples in Fig. S2 at https://osf.io/tb3n4/). The following features were used to build the model: the number of local maxima and minima (and the fraction in which they occurred) and the average read depth in each fraction for observed and simulated gradient profiles. Data from 600 manually curated contigs were used to train the model, which was implemented in the R package caret (108). Model validation was performed on 20% of the training set (see R code in the supplementary data at https://osf.io/tb3n4/).
Genome binning.
Common tools for reconstructing MAGs, based on kmer frequency and covariance (in our case across the CsCl gradient), were prone to cross-contamination (see supplementary methods at https://osf.io/tb3n4/). In addition, MAGs constructed using standard practices failed to recover genomes from taxa known to be abundant in the metagenome and 13C labeled, including Chaetomium, Vampirovibrio, and members of Verrucomicrobia and Chloroflexales. Given these limitations, we opted to define a genomic unit based on 13C enrichment and LCA classification of contigs, which we term a “phylobin.” Phylobins consisted of contig sets divided by 13C enrichment status (i.e., strong, weak. and unenriched) and by the taxonomic rank at the level of order (e.g., strongly enriched Cellvibrionales). We justify this approach accordingly: (i) DNA-SIP selectively enriched for a relatively narrow subset of taxa within a given order, and (ii) phylogenetically related organisms with similar enrichment status are likely to share similar genomic and ecological traits. There is no universally appropriate taxonomic rank or phylogenic depth for grouping organisms, since functional traits are conserved at various phylogenetic depths (109). We chose the rank of order as the cutoff because LCA often fails to accurately classify to the species level taxa that are poorly represented in the NCBI nr database. Hence, aggregating at the rank of order decreases the risk of losing genomic information. Prior research has shown that aggregating microbiome data by taxonomic order produced the greatest discriminating power of relevant soil microbial processes (110). The loss of resolution of individual genomes was compensated for by performing all analyses in parallel on reference genomes chosen based on the similarity of full-length SSU rRNA genes recovered in our study (μsimilarity = 98%, n = 89) or, in some cases, by the only available representative genome for that genus or clade (n = 38) (see Table S6 at https://osf.io/tb3n4/).
Functional gene annotation.
Functional genes were annotated using curated databases relating to genes for motility, adhesion, secondary metabolite (SM) biosynthetic gene clusters, and catabolic enzymes for biomass and cellulose. Fungal genomes were annotated only for SM cellulolytic enzymes. SMs were annotated using the default settings of antiSMASH for bacteria or fungi (v. 4.1.0) (111). Genes involved in cellulolytic activity, namely, those encoding glycosyl hydrolases (GH), endoglucanases (specific GH families), carbohydrate-binding modules (CBMs), polysaccharide lyases (PL), and auxiliary activity enzymes (AA), were annotated using DIAMOND BLASTX searches against a local version of the CAZy database, which includes bacterial and fungal genes (112) (downloaded 20 December 2017). The following endoglucanase-containing glycosyl hydrolase and lytic polysaccharide monooxygenase families were deemed to confer cellulolytic ability: GH5(1), GH5(2), GH5(4), GH5(5), GH5(7), GH5(8), GH5(9), GH5(11), GH5(12), GH5(13), GH5(15), GH5(16), GH5(18), GH5(19), GH5(22), GH5(23), GH5(24), GH5(25), GH5(26), GH5(27), GH5(28), GH5(29), GH5(30), GH5(31), GH5(36), GH5(37), GH5(38), GH5(39), GH5(40), GH5(41), GH5(43), GH5(44), GH5(45), GH5(46), GH5(47), GH5(48), GH5(49), GH5(50), GH5(51), GH5(53), GH6, GH7, GH8, GH9, GH12, GH44, GH45, GH48, GH51, GH61 (now AA9), GH74, GH124, GH131, and AA10. Chitinases were represented by CAZy families GH18 and GH19. Genes encoding nucleases, adhesion (curli and holdfast proteins), and motility were annotated using DIAMOND BLASTX searches against a local version of the NCBI COG database (113) (downloaded 1 May 2018) and, in the case of motility, mapped to KEGG biosynthetic pathways for synthesizing complete motility apparatus (see Table S8 at https://osf.io/tb3n4/). Genes encoding peptidases were annotated using DIAMOND BLASTX searches against a local version of the MEROPS database (114) (downloaded 1 July 2018). The capacity for gliding motility was assessed using canonical genes from three model organisms: the focal adhesion protein in Myxococcus xanthus (AglZ) (115), the SprB and RemA adhesins in Flavobacterium johnsoniae (116, 117), and Gli349 and Gli521 in Mycoplasma mobile (118, 119). Additional adhesion gene families were annotated using compilations of well-characterized proteins, including unipolar polysaccharide synthesis proteins (upp) (120) and tight adherence proteins (tad) (121). All annotations were based on a sequence identity cutoff of ≥60% across 90% of the full-length gene.
Auxotrophies were determined for each representative genome and phylobin based on genome-enabled metabolic models (GEMs) in KBase (122), according to Henry et al. (123). Briefly, flux balance analysis was performed on GEMs under two growth conditions: on a rich medium containing all potential biomass precursors and on a minimal medium containing only C and essential nutrients. The number of critical enzyme-catalyzed reactions was calculated for each GEM according to the following criteria: (i) the reaction was not involved in central C metabolism, (ii) the reaction was essential and carried flux only under minimal (i.e., not under rich) medium conditions, and (iii) the flux of the reaction was coupled to the production of an essential compound. A genome was considered auxotrophic for a compound if the number of its critical reactions for its biosynthesis was below a compound-specific threshold or if the number of gap-filled critical reactions exceeded a compound-specific threshold. Thresholds were set based on auxotrophy profiles from a dozen well-characterized bacteria in the Bacteroidetes, Firmicutes, Alphaproteobacteria, and Gammaproteobacteria.
Statistical analyses.
Statistics were performed in R v.3.4.2 (124) with the following packages: reshape2, ggplot2, plyr (125–127), Hmisc (128), and phyloseq (129). Nonparametric multidimensional scaling (NMDS) was performed using metaMDS from the R package vegan (130). The relative amount of variation in the primary and secondary NMDS axes explained by functional traits was calculated using the R package relaimpo (131). Pairwise multiple comparisons based on the Kruskal-Wallis test (kruskalmc) were performed using the R package pgirmess (132). Minimum generation times were predicted for all phylobins and representative genomes using growthpred (v.1.07) (133) based on codon usage bias using ribosomal genes (identified by COG ID) as the set of highly expressed genes. All analyses can be reproduced using R scripts, and data are available in the supplementary data package at https://osf.io/tb3n4/.
Data availability.
All analyses can be reproduced using R scripts, and data are available in the supplementary data package at https://osf.io/tb3n4/. Supplementary data, figures, tables, and methods are hosted at the Open Science Framework at https://osf.io/tb3n4/. The shotgun metagenomes (BioProject no. PRJEB23737), 16S rRNA (no. PRJNA317227), and ITS (no. PRJNA589050) amplicon libraries associated with this study are available through the NCBI.
ACKNOWLEDGMENTS
This work was supported by the U.S. Department of Energy, Office of Biological & Environmental Research Genomic Science Program, under award numbers DE-SC0016364 and DE-SC0004486.
We declare that we have no competing interests.
R.C.W. performed all data analysis, research, and writing. C.P.R., performed metagenomic sequencing, commented on the manuscript, and provided the basic code for producing Fig. 1. P.W. performed predictions of prototrophy/auxotrophy. M.L. performed the metaproteomics. D.H.B. guided the formulation of research questions and made significant design and writing contributions.
Footnotes
Citation Wilhelm RC, Pepe-Ranney C, Weisenhorn P, Lipton M, Buckley DH. 2021. Competitive exclusion and metabolic dependency among microorganisms structure the cellulose economy of an agricultural soil. mBio 12:e03099-20. https://doi.org/10.1128/mBio.03099-20.
REFERENCES
- 1.Wohl DL, Arora S, Gladstone JR. 2004. Functional redundancy supports biodiversity and ecosystem function in a closed and constant environment. Ecology 85:1534–1540. doi: 10.1890/03-3050. [DOI] [Google Scholar]
- 2.Van Dyk JS, Pletschke BI. 2012. A review of lignocellulose bioconversion using enzymatic hydrolysis and synergistic cooperation between enzymes—factors affecting enzymes, conversion and synergy. Biotechnol Adv 30:1458–1480. doi: 10.1016/j.biotechadv.2012.03.002. [DOI] [PubMed] [Google Scholar]
- 3.Shoseyov O, Shani Z, Levy I. 2006. Carbohydrate binding modules: biochemical properties and novel applications. Microbiol Mol Biol Rev 70:283–295. doi: 10.1128/MMBR.00028-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lynd LR, Weimer PJ, Van Zyl WH, Pretorius IS. 2002. Microbial cellulose utilization: fundamentals and biotechnology. Microbiol Mol Biol Rev 66:506–577. doi: 10.1128/mmbr.66.3.506-577.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schwarze FWMR. 2007. Wood decay under the microscope. Fungal Biol Rev 21:133–170. doi: 10.1016/j.fbr.2007.09.001. [DOI] [Google Scholar]
- 6.Berg B, von Hofsten B, Pettersson G. 1972. Electronmicroscopic observations on the degradation of cellulose fibres by Cellvibrio fulvus and Sporocytophaga myxococcoides. J Appl Bacteriol 35:215–219. doi: 10.1111/j.1365-2672.1972.tb03692.x. [DOI] [PubMed] [Google Scholar]
- 7.Bayer EA, Kenig R, Lamed R. 1983. Adherence of Clostridium thermocellum to cellulose. J Bacteriol 156:818–827. doi: 10.1128/JB.156.2.818-827.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Doi RH, Kosugi A. 2004. Cellulosomes: plant-cell-wall-degrading enzyme complexes. Nat Rev Microbiol 2:541–551. doi: 10.1038/nrmicro925. [DOI] [PubMed] [Google Scholar]
- 9.Eichorst SA, Kuske CR. 2012. Identification of cellulose-responsive bacterial and fungal communities in geographically and edaphically different soils by using stable isotope probing. Appl Environ Microbiol 78:2316–2327. doi: 10.1128/AEM.07313-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schellenberger S, Kolb S, Drake HL. 2010. Metabolic responses of novel cellulolytic and saccharolytic agricultural soil bacteria to oxygen. Environ Microbiol 12:845–861. doi: 10.1111/j.1462-2920.2009.02128.x. [DOI] [PubMed] [Google Scholar]
- 11.Pepe-Ranney C, Campbell AN, Koechli CN, Berthrong S, Buckley DH. 2016. Unearthing the ecology of soil microorganisms using a high resolution DNA-SIP approach to explore cellulose and xylose metabolism in soil. Front Microbiol 7:703. doi: 10.3389/fmicb.2016.00703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Verastegui Y, Cheng J, Engel K, Kolczynski D, Mortimer S, Lavigne J, Montalibet J, Romantsov T, Hall M, McConkey BJ, Rose DR, Tomashek JJ, Scott BR, Charles TC, Neufeld JD. 2014. Multisubstrate isotope labeling and metagenomic analysis of active soil bacterial communities. mBio 5:e01157-14. doi: 10.1128/mBio.01157-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang X, Sharp CE, Jones GM, Grasby SE, Brady AL, Dunfield PF. 2015. Stable-isotope probing identifies uncultured Planctomycetes as primary degraders of a complex heteropolysaccharide in soil. Appl Environ Microbiol 81:4607–4615. doi: 10.1128/AEM.00055-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wilhelm RC, Singh R, Eltis LD, Mohn WW. 2019. Bacterial contributions to delignification and lignocellulose degradation in forest soils with metagenomic and quantitative stable isotope probing. ISME J 13:413–429. doi: 10.1038/s41396-018-0279-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Haichar FEZ, Achouak W, Christen R, Heulin T, Marol C, Marais MF, Mougel C, Ranjard L, Balesdent J, Berge O. 2007. Identification of cellulolytic bacteria in soil by stable isotope probing. Environ Microbiol 9:625–634. doi: 10.1111/j.1462-2920.2006.01182.x. [DOI] [PubMed] [Google Scholar]
- 16.Štursová M, Žifčáková L, Leigh MB, Burgess R, Baldrian P. 2012. Cellulose utilization in forest litter and soil: identification of bacterial and fungal decomposers. FEMS Microbiol Ecol 80:735–746. doi: 10.1111/j.1574-6941.2012.01343.x. [DOI] [PubMed] [Google Scholar]
- 17.Kramer S, Dibbern D, Moll J, Huenninghaus M, Koller R, Krueger D, Marhan S, Urich T, Wubet T, Bonkowski M, Buscot F, Lueders T, Kandeler E. 2016. Resource partitioning between bacteria, fungi, and protists in the detritusphere of an agricultural soil. Front Microbiol 7:1524. doi: 10.3389/fmicb.2016.01524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wilhelm RC, Cardenas E, Leung HTC, Szeitz A, Jensen LD, Mohn WW. 2017. Long-term enrichment of stress-tolerant cellulolytic soil populations following timber harvesting evidenced by multi-omic stable isotope probing. Front Microbiol 8:537. doi: 10.3389/fmicb.2017.00537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Romaní AM, Fischer H, Mille-Lindblom C, Tranvik LJ. 2006. Interactions of bacteria and fungi on decomposing litter: differential extracellular enzyme activities. Ecology 87:2559–2569. doi: 10.1890/0012-9658(2006)87[2559:IOBAFO]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 20.Folman LB, Klein Gunnewiek PJA, Boddy L, De Boer W. 2008. Impact of white-rot fungi on numbers and community composition of bacteria colonizing beech wood from forest soil. FEMS Microbiol Ecol 63:181–191. doi: 10.1111/j.1574-6941.2007.00425.x. [DOI] [PubMed] [Google Scholar]
- 21.Book AJ, Lewin GR, Mcdonald BR, Takasuka TE, Fox G, Currie CR. 2016. Evolution of high cellulolytic activity in symbiotic Streptomyces through selection of expanded gene content and coordinated gene expression. PLoS Biol 14:e1002475. doi: 10.1371/journal.pbio.1002475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ebrahimi A, Schwartzman J, Cordero OX. 2019. Cooperation and spatial self-organization determine rate and efficiency of particulate organic matter degradation in marine bacteria. Proc Natl Acad Sci U S A 116:23309–23316. doi: 10.1073/pnas.1908512116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Berlemont R, Martiny AC. 2013. Phylogenetic distribution of potential cellulases in bacteria. Appl Environ Microbiol 79:1545–1554. doi: 10.1128/AEM.03305-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Morris JJ, Lenski RE, Zinser ER. 2012. The Black Queen Hypothesis: evolution of dependencies through adaptative gene loss. mBio 3:e00036-12. doi: 10.1128/mBio.00036-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ng TK, Zeikus JG. 1981. Ethanol production by thermophilic bacteria: fermentation of cellulosic substrates by cocultures of Clostridium thermocellum and Clostridium thermohydrosulfuricum. Appl Environ Microbiol 41:1337–1343. doi: 10.1128/AEM.41.6.1337-1343.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jiao Y, Navid A, Stewart BJ, Mckinlay JB, Thelen MP, Pett-Ridge J. 2012. Syntrophic metabolism of a co-culture containing Clostridium cellulolyticum and Rhodopseudomonas palustris for hydrogen production. Int J Hydrogen Energy 37:11719–11726. doi: 10.1016/j.ijhydene.2012.05.100. [DOI] [Google Scholar]
- 27.Cavedon K, Canale-Parola E. 1992. Physiological interactions between a mesophilic cellulolytic Clostridium and a non-cellulolytic bacterium. FEMS Microbiol Lett 86:237–245. doi: 10.1111/j.1574-6968.1992.tb04815.x. [DOI] [Google Scholar]
- 28.Mori Y. 1990. Characterization of symbiotic coculture of Clostridium thermohydrosulfuricum YM3 and Clostridium thermocellum YM4. Appl Environ Microbiol 56:37–42. doi: 10.1128/AEM.56.1.37-42.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kato S, Haruta S, Cui ZJ, Ishii M, Igarashi Y. 2004. Effective cellulose degradation by a mixed-culture system composed of a cellulolytic Clostridium and aerobic non-cellulolytic bacteria. FEMS Microbiol Ecol 51:133–142. doi: 10.1016/j.femsec.2004.07.015. [DOI] [PubMed] [Google Scholar]
- 30.D'Souza G, Shitut S, Preussger D, Yousif G, Waschina S, Kost C. 2018. Ecology and evolution of metabolic cross-feeding interactions in bacteria. Nat Prod Rep 35:455–488. doi: 10.1039/c8np00009c. [DOI] [PubMed] [Google Scholar]
- 31.Enke TN, Datta MS, Schwartzman J, Cermak N, Schmitz D, Barrere J, Pascual-García A, Cordero OX. 2019. Modular assembly of polysaccharide-degrading marine microbial communities. Curr Biol 29:1528–1535.e6. doi: 10.1016/j.cub.2019.03.047. [DOI] [PubMed] [Google Scholar]
- 32.He S, Stevens SLR, Chan LK, Bertilsson S, Glavina del Rio T, Tringe SG, Malmstrom RR, McMahon KD. 2017. Ecophysiology of freshwater Verrucomicrobia inferred from metagenome-assembled genomes. mSphere 2:e00277-17. doi: 10.1128/mSphere.00277-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee STM, Kahn SA, Delmont TO, Shaiber A, Esen ÖC, Hubert NA, Morrison HG, Antonopoulos DA, Rubin DT, Eren AM. 2017. Tracking microbial colonization in fecal microbiota transplantation experiments via genome-resolved metagenomics. Microbiome 5:50. doi: 10.1186/s40168-017-0270-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Garcia SL, Buck M, McMahon KD, Grossart HP, Eiler A, Warnecke F. 2015. Auxotrophy and intrapopulation complementary in the interactome of a cultivated freshwater model community. Mol Ecol 24:4449–4459. doi: 10.1111/mec.13319. [DOI] [PubMed] [Google Scholar]
- 35.Youngblut ND, Barnett SE, Buckley DH. 2018. HTSSIP: an R package for analysis of high throughput sequencing data from nucleic acid stable isotope probing (SIP) experiments. PLoS One 13:e0189616. doi: 10.1371/journal.pone.0189616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Barnett SE, Buckley DH. 2020. Simulating metagenomic stable isotope probing datasets with MetaSIPSim. BMC Bioinformatics 21:37. doi: 10.1186/s12859-020-3372-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nilsson T, Björdal C. 2008. The use of kapok fibres for enrichment cultures of lignocellulose-degrading bacteria. Int Biodeterior Biodegrad 61:11–16. doi: 10.1016/j.ibiod.2007.06.009. [DOI] [Google Scholar]
- 38.Rodriguez-R LM, Konstantinidis KT. 2014. Nonpareil: a redundancy-based approach to assess the level of coverage in metagenomic datasets. Bioinformatics 30:629–635. doi: 10.1093/bioinformatics/btt584. [DOI] [PubMed] [Google Scholar]
- 39.Youngblut ND, Buckley DH. 2014. Intra-genomic variation in G + C content and its implications for DNA stable isotope probing. Environ Microbiol Rep 6:767–775. doi: 10.1111/1758-2229.12201. [DOI] [PubMed] [Google Scholar]
- 40.Koechli CN. 2016. Land management affects microbial community composition and function in carbon cycling. Ph.D. thesis. Cornell University, Ithaca, NY. [Google Scholar]
- 41.Degrune F, Theodorakopoulos N, Colinet G, Hiel MP, Bodson B, Taminiau B, Daube G, Vandenbol M, Hartmann M. 2017. Temporal dynamics of soil microbial communities below the seedbed under two contrasting tillage regimes. Front Microbiol 8:1127. doi: 10.3389/fmicb.2017.01127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sharma-Poudyal D, Schlatter D, Yin C, Hulbert S, Paulitz T. 2017. Long-term no-till: a major driver of fungal communities in dryland wheat cropping systems. PLoS One 12:e0184611. doi: 10.1371/journal.pone.0184611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Orgiazzi A, Lumini E, Nilsson RH, Girlanda M, Vizzini A, Bonfante P, Bianciotto V. 2012. Unravelling soil fungal communities from different Mediterranean land-use backgrounds. PLoS One 7:e34847. doi: 10.1371/journal.pone.0034847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Beloqui A, Nechitaylo TY, López-Cortés N, Ghazi A, Guazzaroni ME, Polaina J, Strittmatter AW, Reva O, Waliczek A, Yakimov MM, Golyshina OV, Ferrer M, Golyshin PN, Lo N, Polaina J, Strittmatter AW, Reva O, Waliczek A, Yakimov MM, Golyshina OV, Ferrer M, Golyshin PN. 2010. Diversity of glycosyl hydrolases from cellulose-depleting communities enriched from casts of two earthworm species. Appl Environ Microbiol 76:5934–5946. doi: 10.1128/AEM.00902-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Koechli C, Campbell AN, Pepe-Ranney C, Buckley DH. 2019. Assessing fungal contributions to cellulose degradation in soil by using high-throughput stable isotope probing. Soil Biol Biochem 130:150–158. doi: 10.1016/j.soilbio.2018.12.013. [DOI] [Google Scholar]
- 46.Vasil'chenko LG, Khromonygina VV, Karapetyan KN, Vasilenko OV, Rabinovich ML. 2005. Cellobiose dehydrogenase formation by filamentous fungus Chaetomium sp. INBI 2-26(−). J Biotechnol 119:44–59. doi: 10.1016/j.jbiotec.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 47.Halliwell G, Sweet C. 1973. Bacteriocinogenic activity in the genus Cellvibrio. J Gen Microbiol 77:363–369. doi: 10.1099/00221287-77-2-363. [DOI] [Google Scholar]
- 48.Tathan S, Sibounnavong P, Sibounnavong P, Soytong K, To-Anun C. 2012. Biological metabolites from Chaetomium spp to inhibit Drechslera oryzae causing leaf spot of rice. Int J Agric Technol 8:1691–1701. [Google Scholar]
- 49.Hung PM, Wattanachai P, Kasem S, Poeaim S. 2015. Efficacy of Chaetomium species as biological control agents against Phytophthora nicotianae root rot in citrus. Mycobiology 43:288–296. doi: 10.5941/MYCO.2015.43.3.288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu L, Huang X, Zhao J, Zhang J, Cai Z. 2019. Characterizing the key agents in a disease-suppressed soil managed by reductive soil disinfestation. Appl Environ Microbiol 85:e02992-18. doi: 10.1128/AEM.02992-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schulz-Bohm K, Martín-Sánchez L, Garbeva P. 2017. Microbial volatiles: small molecules with an important role in intra- and inter-kingdom interactions. Front Microbiol 8:2484. doi: 10.3389/fmicb.2017.02484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wagner A. 2005. Energy constraints on the evolution of gene expression. Mol Biol Evol 22:1365–1374. doi: 10.1093/molbev/msi126. [DOI] [PubMed] [Google Scholar]
- 53.Barton MD, Delneri D, Oliver SG, Rattray M, Bergman CM. 2010. Evolutionary systems biology of amino acid biosynthetic cost in yeast. PLoS One 5:e11935. doi: 10.1371/journal.pone.0011935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brewer TE, Handley KM, Carini P, Gilbert JA, Fierer N. 2016. Genome reduction in an abundant and ubiquitous soil bacterium ‘Candidatus Udaeobacter copiosus.’ Nat Microbiol 2:16198. doi: 10.1038/nmicrobiol.2016.198. [DOI] [PubMed] [Google Scholar]
- 55.Jeske O, Jogler M, Petersen J, Sikorski J, Jogler C. 2013. From genome mining to phenotypic microarrays: Planctomycetes as source for novel bioactive molecules. Antonie Van Leeuwenhoek 104:551–567. doi: 10.1007/s10482-013-0007-1. [DOI] [PubMed] [Google Scholar]
- 56.Ward LM, Hemp J, Pace LA, Fischer WW. 2015. Draft genome sequence of Herpetosiphon geysericola GC-42, a nonphototrophic member of the Chloroflexi class Chloroflexia. Genome Announc 3:e01352-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lage OM, Bondoso J. 2014. Planctomycetes and macroalgae, a striking association. Front Microbiol 5:267. doi: 10.3389/fmicb.2014.00267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wiegand S, Jogler M, Jogler C. 2018. On the maverick Planctomycetes. FEMS Microbiol Rev 42:739–760. doi: 10.1093/femsre/fuy029. [DOI] [PubMed] [Google Scholar]
- 59.Erbilgin O, McDonald KL, Kerfeld CA. 2014. Characterization of a planctomycetal organelle: a novel bacterial microcompartment for the aerobic degradation of plant saccharides. Appl Environ Microbiol 80:2193–2205. doi: 10.1128/AEM.03887-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Reintjes G, Arnosti C, Fuchs B, Amann R. 2019. Selfish, sharing and scavenging bacteria in the Atlantic Ocean: a biogeographical study of bacterial substrate utilisation. ISME J 13:1119–1132. doi: 10.1038/s41396-018-0326-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cayrou C, Raoult D, Drancourt M. 2010. Broad-spectrum antibiotic resistance of Planctomycetes organisms determined by Etest. J Antimicrob Chemother 65:2119–2122. doi: 10.1093/jac/dkq290. [DOI] [PubMed] [Google Scholar]
- 62.Ward NL, Staley JT, Schmidt JM. 2015. Planctomyces, p 1–23. In Bergey’s manual of systematics of archaea and bacteria. John Wiley & Sons, New York, NY. [Google Scholar]
- 63.Schlesner H. 1994. The development of media suitable for the microorganisms morphologically resembling Planctomyces spp., Pirellula spp., and other Planctomycetales from various aquatic habitats using dilute media. Syst Appl Microbiol 17:135–145. doi: 10.1016/S0723-2020(11)80042-1. [DOI] [Google Scholar]
- 64.Soo RM, Woodcroft BJ, Parks DH, Tyson GW, Hugenholtz P. 2015. Back from the dead; the curious tale of the predatory cyanobacterium Vampirovibrio chlorellavorus. PeerJ 3:e968. doi: 10.7717/peerj.968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gui H, Purahong W, Hyde KD, Xu J, Mortimer PE. 2017. The arbuscular mycorrhizal fungus Funneliformis mosseae alters bacterial communities in subtropical forest soils during litter decomposition. Front Microbiol 8:1120. doi: 10.3389/fmicb.2017.01120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wei Y, Zhou H, Zhang J, Zhang L, Geng A, Liu F, Zhao G, Wang S, Zhou Z, Yan X. 2015. Insight into dominant cellulolytic bacteria from two biogas digesters and their glycoside hydrolase genes. PLoS One 10:e0129921. doi: 10.1371/journal.pone.0129921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Vandekerckhove TTM, Willems A, Gillis M, Coomans A. 2000. Occurrence of novel verrucomicrobial species, endosymbiotic and associated with parthenogenesis in Xiphinema americanum-group species (Nematoda, Longidoridae). Int J Syst Evol Microbiol 50:2197–2205. doi: 10.1099/00207713-50-6-2197. [DOI] [PubMed] [Google Scholar]
- 68.Warmink JA, Nazir R, Van Elsas JD. 2009. Universal and species-specific bacterial ‘fungiphiles’ in the mycospheres of different basidiomycetous fungi. Environ Microbiol 11:300–312. doi: 10.1111/j.1462-2920.2008.01767.x. [DOI] [PubMed] [Google Scholar]
- 69.Li D, Feng L, Liu K, Cheng Y, Hou N, Li C. 2016. Optimization of cold-active CMCase production by psychrotrophic Sphingomonas sp. FLX-7 from the cold region of China. Cellulose 23:1335–1347. doi: 10.1007/s10570-016-0859-4. [DOI] [Google Scholar]
- 70.Zhu L, Si M, Li C, Xin K, Chen C, Shi X, Huang R, Zhao L, Shen X, Zhang L. 2015. Sphingomonas gei sp. nov., isolated from roots of Geum aleppicum. Int J Syst Evol Microbiol 65:1160–1166. doi: 10.1099/ijs.0.000074. [DOI] [PubMed] [Google Scholar]
- 71.Rosenberg E (ed). 2014. The prokaryotes: Alphaproteobacteria and Betaproteobacteria, 4th ed. Springer, Berlin, Germany. [Google Scholar]
- 72.Chen H, Jogler M, Rohde M, Klenk HP, Busse HJ, Tindall BJ, Spröer C, Overmann J. 2012. Reclassification and emended description of Caulobacter leidyi as Sphingomonas leidyi comb. nov., and emendation of the genus Sphingomonas. Int J Syst Evol Microbiol 62:2835–2843. doi: 10.1099/ijs.0.039636-0. [DOI] [PubMed] [Google Scholar]
- 73.Kumar M, Revathi K, Khanna S. 2015. Biodegradation of cellulosic and lignocellulosic waste by Pseudoxanthomonas sp R-28. Carbohydr Polym 134:761–766. doi: 10.1016/j.carbpol.2015.08.072. [DOI] [PubMed] [Google Scholar]
- 74.Michaud P, Belaich A, Courtois B, Courtois J. 2002. Cloning, sequencing and overexpression of a Sinorhizobium meliloti M5N1CS carboxymethyl-cellulase gene. Appl Microbiol Biotechnol 58:767–771. doi: 10.1007/s00253-002-0953-4. [DOI] [PubMed] [Google Scholar]
- 75.Yoo S-H, Weon H-Y, Kim B-Y, Hong S-B, Kwon S-W, Cho Y-H, Go S-J, Stackebrandt E. 2006. Devosia soli sp. nov., isolated from greenhouse soil in Korea. Int J Syst Evol Microbiol 56:2689–2692. doi: 10.1099/ijs.0.64214-0. [DOI] [PubMed] [Google Scholar]
- 76.Kim S, Gong G, Park TH, Um Y. 2013. Asticcacaulis solisilvae sp. nov., isolated from forest soil. Int J Syst Evol Microbiol 63:3829–3834. doi: 10.1099/ijs.0.047423-0. [DOI] [PubMed] [Google Scholar]
- 77.Song N, Cai HY, Yan ZS, Jiang HL. 2013. Cellulose degradation by one mesophilic strain Caulobacter sp. FMC1 under both aerobic and anaerobic conditions. Bioresour Technol 131:281–287. doi: 10.1016/j.biortech.2013.01.003. [DOI] [PubMed] [Google Scholar]
- 78.Robledo M, Rivera L, Jiménez-Zurdo JI, Rivas R, Dazzo F, Velázquez E, Martínez-Molina E, Hirsch AM, Mateos PF. 2012. Role of Rhizobium endoglucanase CelC2 in cellulose biosynthesis and biofilm formation on plant roots and abiotic surfaces. Microb Cell Fact 11:125. doi: 10.1186/1475-2859-11-125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Matthysse AG, Thomas DL, White AR. 1995. Mechanism of cellulose synthesis in Agrobacterium tumefaciens. J Bacteriol 177:1076–1081. doi: 10.1128/jb.177.4.1076-1081.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hu X, Yu J, Wang C, Chen H. 2014. Cellulolytic bacteria associated with the gut of Dendroctonus armandi larvae (Coleoptera: Curculionidae: Scolytinae). Forests 5:455–465. doi: 10.3390/f5030455. [DOI] [Google Scholar]
- 81.Talia P, Sede SM, Campos E, Rorig M, Principi D, Tosto D, Hopp HE, Grasso D, Cataldi A. 2012. Biodiversity characterization of cellulolytic bacteria present on native Chaco soil by comparison of ribosomal RNA genes. Res Microbiol 163:221–232. doi: 10.1016/j.resmic.2011.12.001. [DOI] [PubMed] [Google Scholar]
- 82.Ireland MME, Karty JA, Quardokus EM, Reilly JP, Brun YV. 2002. Proteomic analysis of the Caulobacter crescentus stalk indicates competence for nutrient uptake. Mol Microbiol 45:1029–1041. doi: 10.1046/j.1365-2958.2002.03071.x. [DOI] [PubMed] [Google Scholar]
- 83.Blanvillain S, Meyer D, Boulanger A, Lautier M, Guynet C, Denancé N, Vasse J, Lauber E, Arlat M. 2007. Plant carbohydrate scavenging through TonB-dependent receptors: a feature shared by phytopathogenic and aquatic bacteria. PLoS One 2:e224. doi: 10.1371/journal.pone.0000224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.D’Souza G, Ackermann M. 2018. Growing in spatial collectives helps Caulobacter crescentus cells to utilise complex plant derived polysaccharides. 17th Conference of the International Society of Microbial Ecology, Leipzig, Germany.
- 85.Lee N, Reichenbach H. 2006. The genus Herpetosiphon. In Dworkin M, Falkow S, Rosenberg E, Schleifer K-H, Stackebrandt E (ed), The prokaryotes, 3rd ed. Springer, Berlin, Germany. [Google Scholar]
- 86.Schneiker S, Perlova O, Kaiser O, Gerth K, Alici A, Altmeyer MO, Bartels D, Bekel T, Beyer S, Bode E, Bode HB, Bolten CJ, Choudhuri JV, Doss S, Elnakady YA, Frank B, Gaigalat L, Goesmann A, Groeger C, Gross F, Jelsbak L, Jelsbak L, Kalinowski J, Kegler C, Knauber T, Konietzny S, Kopp M, Krause L, Krug D, Linke B, Mahmud T, Martinez-Arias R, McHardy AC, Merai M, Meyer F, Mormann S, Muñoz-Dorado J, Perez J, Pradella S, Rachid S, Raddatz G, Rosenau F, Rückert C, Sasse F, Scharfe M, Schuster SC, Suen G, Treuner-Lange A, Velicer GJ, Vorhölter F-J, et al. 2007. Complete genome sequence of the myxobacterium Sorangium cellulosum. Nat Biotechnol 25:1281–1289. doi: 10.1038/nbt1354. [DOI] [PubMed] [Google Scholar]
- 87.Shi Y, Weimer PJ. 1997. Competition for cellobiose among three predominant ruminal cellulolytic bacteria under substrate-excess and substrate-limited conditions. Appl Environ Microbiol 63:743–748. doi: 10.1128/AEM.63.2.743-748.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Mosoni P, Fonty G, Gouet P. 1997. Competition between ruminal cellulolytic bacteria for adhesion to cellulose. Curr Microbiol 35:44–47. doi: 10.1007/s002849900209. [DOI] [PubMed] [Google Scholar]
- 89.Wucher BR, Bartlett TM, Hoyos M, Papenfort K, Persat A, Nadell CD. 2019. Vibrio cholerae filamentation promotes chitin surface attachment at the expense of competition in biofilms. Proc Natl Acad Sci U S A 116:14216–14221. doi: 10.1073/pnas.1819016116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Perveen N, Barot S, Maire V, Cotrufo MF, Shahzad T, Blagodatskaya E, Stewart CE, Ding W, Siddiq MR, Dimassi B, Mary B, Fontaine S. 2019. Universality of priming effect: an analysis using thirty five soils with contrasted properties sampled from five continents. Soil Biol Biochem 134:162–171. doi: 10.1016/j.soilbio.2019.03.027. [DOI] [Google Scholar]
- 91.Lehmann J, Kleber M. 2015. The contentious nature of soil organic matter. Nature 528:60–68. doi: 10.1038/nature16069. [DOI] [PubMed] [Google Scholar]
- 92.Cordero OX, Polz MF. 2014. Explaining microbial genomic diversity in light of evolutionary ecology. Nat Rev Microbiol 12:263–273. doi: 10.1038/nrmicro3218. [DOI] [PubMed] [Google Scholar]
- 93.Berthrong ST, Buckley DH, Drinkwater LE. 2013. Agricultural management and labile carbon additions affect soil microbial community structure and interact with carbon and nitrogen cycling. Microb Ecol 66:158–170. doi: 10.1007/s00248-013-0225-0. [DOI] [PubMed] [Google Scholar]
- 94.Campbell A. 2015. Illuminating microbial contributions to soil carbon cycling. Cornell University, Ithaca, NY. [Google Scholar]
- 95.Nawrocki EP, Eddy SR. 2013. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29:2933–2935. doi: 10.1093/bioinformatics/btt509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wang Q, Garrity GM, Tiedje JM, Cole JR. 2007. Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73:5261–5267. doi: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Van Horn DJ, Weber CF. 2009. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75:7537–7541. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Pruesse E, Quast C, Knittel K, Fuchs BM, Ludwig W, Peplies J, Glöckner FO. 2007. SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res 35:7188–7196. doi: 10.1093/nar/gkm864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Pericard P, Dufresne Y, Couderc L, Blanquart S, Touzet H. 2018. MATAM: reconstruction of phylogenetic marker genes from short sequencing reads in metagenomes. Bioinformatics 34:585–591. doi: 10.1093/bioinformatics/btx644. [DOI] [PubMed] [Google Scholar]
- 100.Li D, Liu C-M, Luo R, Sadakane K, Lam T-W. 2015. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31:1674–1676. doi: 10.1093/bioinformatics/btv033. [DOI] [PubMed] [Google Scholar]
- 101.Huson DH, Beier S, Flade I, Górska A, El-Hadidi M, Mitra S, Ruscheweyh HJ, Tappu R. 2016. MEGAN community edition—interactive exploration and analysis of large-scale microbiome sequencing data. PLoS Comput Biol 12:e1004957. doi: 10.1371/journal.pcbi.1004957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Buchfink B, Xie C, Huson DH. 2015. Fast and sensitive protein alignment using DIAMOND. Nat Methods 12:59–60. doi: 10.1038/nmeth.3176. [DOI] [PubMed] [Google Scholar]
- 103.Scholz M, Lo CC, Chain PSG. 2015. Improved assemblies using a source-agnostic pipeline for metagenomic assembly by merging (MeGAMerge) of contigs. Sci Rep 4:6480. doi: 10.1038/srep06480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kurtz S, Phillippy A, Delcher A, Smoot M, Shumway M, Antonescu C, Salzberg S. 2004. Versatile and open software for comparing large genomes. Genome Biol 5:R12. doi: 10.1186/gb-2004-5-2-r12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Gurevich A, Saveliev V, Vyahhi N, Tesler G. 2013. QUAST: quality assessment tool for genome assemblies. Bioinformatics 29:1072–1075. doi: 10.1093/bioinformatics/btt086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kang DD, Froula J, Egan R, Wang Z. 2015. MetaBAT, an efficient tool for accurately reconstructing single genomes from complex microbial communities. PeerJ 3:e1165. doi: 10.7717/peerj.1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Barnett SE, Buckley DH. 2019. Simulating metagenomic stable isotope probing datasets with MetaSIPSim. bioRxiv doi: 10.1101/735787. [DOI] [PMC free article] [PubMed]
- 108.Kuhn M. 2011. caret: classification and regression training. R package. R Foundation for Statistical Computing, Vienna, Austria. [Google Scholar]
- 109.Martiny AC, Treseder K, Pusch G. 2013. Phylogenetic conservatism of functional traits in microorganisms. ISME J 7:830–838. doi: 10.1038/ismej.2012.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Chang H, Haudenshield JS, Bowen CR, Allen R, Hartman GL. 2017. Metagenome-wide association study and machine learning prediction of bulk soil microbiome and crop productivity. Front Microbiol 8:519. doi: 10.3389/fmicb.2017.00519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Blin K, Medema MH, Kottmann R, Lee SY, Weber T. 2017. The antiSMASH database, a comprehensive database of microbial secondary metabolite biosynthetic gene clusters. Nucleic Acids Res 45:D555–D559. doi: 10.1093/nar/gkw960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Cantarel BI, Coutinho PM, Rancurel C, Bernard T, Lombard V, Henrissat B. 2009. The Carbohydrate-Active EnZymes database (CAZy): an expert resource for glycogenomics. Nucleic Acids Res 37:D233–D238. doi: 10.1093/nar/gkn663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Tatusov RL, Galperin MY, Natale DA, Koonin EV. 2000. The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res 28:33–36. doi: 10.1093/nar/28.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Rawlings ND, Barrett AJ, Bateman A. 2012. MEROPS: the database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res 40:D343–D350. doi: 10.1093/nar/gkr987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Islam ST, Mignot T. 2015. The mysterious nature of bacterial surface (gliding) motility: a focal adhesion-based mechanism in Myxococcus xanthus. Semin Cell Dev Biol 46:143–154. doi: 10.1016/j.semcdb.2015.10.033. [DOI] [PubMed] [Google Scholar]
- 116.Shrivastava A, Rhodes RG, Pochiraju S, Nakane D, McBride MJ. 2012. Flavobacterium johnsoniae RemA is a mobile cell surface lectin involved in gliding. J Bacteriol 194:3678–3688. doi: 10.1128/JB.00588-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Nakane D, Sato K, Wada H, McBride MJ, Nakayama K. 2013. Helical flow of surface protein required for bacterial gliding motility. Proc Natl Acad Sci U S A 110:11145–11150. doi: 10.1073/pnas.1219753110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Uenoyama A, Kusumoto A, Miyata M. 2004. Identification of a 349-kilodalton protein (Gli349) responsible for cytadherence and glass binding during gliding of Mycoplasma mobile. J Bacteriol 186:1537–1545. doi: 10.1128/jb.186.5.1537-1545.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Seto S, Uenoyama A, Miyata M. 2005. Identification of a 521-kilodalton protein (Gli521) involved in force generation or force transmission for Mycoplasma mobile gliding. J Bacteriol 187:3502–3510. doi: 10.1128/JB.187.10.3502-3510.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Fritts RK, Lasarre B, Stoner AM, Posto AL, McKinlay JB. 2017. A Rhizobiales-specific unipolar polysaccharide adhesin contributes to Rhodopseudomonas palustris biofilm formation across diverse photoheterotrophic conditions. Appl Environ Microbiol 83:1–14. doi: 10.1128/AEM.03035-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kachlany SC, Planet PJ, Desalle R, Fine DH, Figurski DH. 2001. Genes for tight adherence of Actinobacillus actinomycetemcomitans: from plaque to plague to pond scum. Trends Microbiol 9:429–437. doi: 10.1016/s0966-842x(01)02161-8. [DOI] [PubMed] [Google Scholar]
- 122.Arkin AP, Cottingham RW, Henry CS, Harris NL, Stevens RL, Maslov S, Dehal P, Ware D, Perez F, Canon S, Sneddon MW, Henderson ML, Riehl WJ, Murphy-Olson D, Chan SY, Kamimura RT, Kumari S, Drake MM, Brettin TS, Glass EM, Chivian D, Gunter D, Weston DJ, Allen BH, Baumohl J, Best AA, Bowen B, Brenner SE, Bun CC, Chandonia J-M, Chia J-M, Colasanti R, Conrad N, Davis JJ, Davison BH, DeJongh M, Devoid S, Dietrich E, Dubchak I, Edirisinghe JN, Fang G, Faria JP, Frybarger PM, Gerlach W, Gerstein M, Greiner A, Gurtowski J, Haun HL, He F, Jain R, Joachimiak MP, Keegan KP, Kondo S, et al. 2018. KBase: the United States Department of Energy Systems Biology Knowledgebase. Nat Biotechnol 36:566–569. doi: 10.1038/nbt.4163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Henry CS, Dejongh M, Best AA, Frybarger PM, Linsay B, Stevens RL. 2010. High-throughput generation, optimization and analysis of genome-scale metabolic models. Nat Biotechnol 28:977–974. doi: 10.1038/nbt.1672. [DOI] [PubMed] [Google Scholar]
- 124.R Core Team. 2017. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. [Google Scholar]
- 125.Wickham H. 2016. ggplot2: elegant graphics for data analysis. Springer International Publishing. [Google Scholar]
- 126.Wickham H. 2007. Reshaping data with the reshape package. J Stat Soft 21:1–20. [Google Scholar]
- 127.Wickham H. 2009. The split-apply-combine strategy for data analysis. J Stat Soft 40:1–29. [Google Scholar]
- 128.Harrell FE, Jr. 2015. Hmisc: Harrell miscellaneous. R Package. R Foundation for Statistical Computing, Vienna, Austria. [Google Scholar]
- 129.McMurdie PJ, Holmes S. 2013. Phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 8:e61217. doi: 10.1371/journal.pone.0061217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Oksanen J, Blanchet F, Kindt R, Legendre P, Minchin PR, O’Hara RB, Simpson G, Solymos M, Stevens HH, Szoecs E, Wagener H. 2015. Vegan: community ecology package. R Package. R Foundation for Statistical Computing, Vienna, Austria. [Google Scholar]
- 131.Groemping U. 2006. Relative importance for linear regression in R: the package relaimpo. J Stat Soft 17:1–27. [Google Scholar]
- 132.Giraudoux P. 2018. pgirmess: spatial analysis and data mining for field ecologists. 1.6.9. https://cran.r-project.org/web/packages/pgirmess/index.html.
- 133.Vieira-Silva S, Rocha EPC. 2010. The systemic imprint of growth and its uses in ecological (meta)genomics. PLoS Genet 6:e1000808. doi: 10.1371/journal.pgen.1000808. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All analyses can be reproduced using R scripts, and data are available in the supplementary data package at https://osf.io/tb3n4/. Supplementary data, figures, tables, and methods are hosted at the Open Science Framework at https://osf.io/tb3n4/. The shotgun metagenomes (BioProject no. PRJEB23737), 16S rRNA (no. PRJNA317227), and ITS (no. PRJNA589050) amplicon libraries associated with this study are available through the NCBI.