

ABSTRACT
An unusual feature of papillomaviruses is that their genomes are packaged into virions along with host histones. Viral minichromosomes were visualized as “beads on a string” by electron microscopy in the 1970s but, to date, little is known about the posttranslational modifications of these histones. To investigate this, we analyzed the histone modifications in HPV16/18 quasivirions, wart-derived bovine papillomavirus (BPV1), and wart-derived human papillomavirus type 1 (HPV1) using quantitative mass spectrometry. The chromatin from all three virion samples had abundant posttranslational modifications (acetylation, methylation, and phosphorylation). These histone modifications were verified by acid urea polyacrylamide electrophoresis and immunoblot analysis. Compared to matched host cell controls, the virion minichromosome was enriched in histone modifications associated with active chromatin and depleted for those commonly found in repressed chromatin. We propose that the viral minichromosome acquires specific histone modifications late in infection that are coupled to the mechanisms of viral replication, late gene expression, and encapsidation. We predict that, in turn, these same modifications benefit early stages of infection by helping to evade detection, promoting localization of the viral chromosome to beneficial regions of the nucleus, and promoting early transcription and replication.
KEYWORDS: HPV, chromatin, epigenetics, histones, papillomavirus
INTRODUCTION
Posttranslational modification of histones regulates many chromatin-associated processes, such as transcription, DNA replication, repair and recombination, and chromatin condensation. The genomes of several DNA viruses are also chromatinized at various stages of the viral life cycle, and this adds an additional layer of regulation to viral transcription and replication (1, 2). In turn, host cell epigenetic processes can modify viral chromatin as part of the intrinsic immune response (3). The small, double-stranded, circular DNA genomes of papillomaviruses are bound by cellular histones to form minichromosomes with chromatin modifications that regulate many aspects of the persistent and productive phases of the viral life cycle (reviewed in reference 1). A relatively novel feature of the papillomaviruses is that the viral genomes are packaged with cellular histones inside the virion; the viral DNA is assembled into about 30 to 32 nucleosomes inside the capsid (4). Although polyomaviruses share this trait (5), other DNA virus families package their genomes either as naked DNA or in complex with other nonhistone DNA-binding proteins or molecules (6).
Papillomaviruses are species-specific and are thought to slowly coevolve with their unique host lineage over millions of years (7). These viruses infect and form reservoirs in dividing stem-cell-like keratinocytes in specific types of stratified epithelia. The productive phase of infection occurs only when these infected cells enter the process of terminal differentiation (7). Viral genomes amplify in cells in a G2-like phase of the cell cycle, and viral particles are produced in the most superficial, differentiated cells destined to be sloughed from the epithelial surface (8, 9). The ∼8 kb papillomavirus genome typically encodes just a few proteins that manipulate cellular processes at each stage of the life cycle, and it is almost certain that they have evolved to exploit chromatin modifications for regulation of their own genomes. These modifications could also promote genome packaging and virion assembly, as well as encapsidation of viral minichromosomes with specific histone modifications that could regulate immediate early viral transcription and replication upon infection of a new host cell.
In 2000, Strahl and Allis predicted that posttranslational modifications of histone tails would modulate chromatin-associated processes (10). The N-terminal tails of all four core histones (H2A, H2B, H3, and H4) can be modified by phosphorylation, methylation, acetylation, ubiquitylation, and sumoylation (11). These modifications can affect the overall structure of chromatin, but also recruit reader proteins with specific domains that recognize these modifications (e.g., bromo-, chromo-, PHD, and Tudor domains) (12). The role of acetylation and methylation of the lysine residues on the N-terminal tails of H3 and H4 are well studied. These modifications generate novel binding sites for chromatin reader proteins and, in addition, acetylation neutralizes the positive charge on lysine residues, resulting in destabilization of the histone-DNA interaction and increased accessibility of the chromatin to other binding factors (13). In general, acetylation is a signature of active chromatin, and the key acetylated residues in H3 are K4, K9, K14, K18, K23, and K27 (14). Methylation can be a mark of active (H3K4) or repressed (H3K9 or K27) chromatin (15). In H4, acetylation of K5, K8, K12, and K16 are signatures of active chromatin (though K16ac can signify either active or repressed chromatin) (16). H4K20 is methylated with mono-, di-, or tri-methylation being associated with different functions (17).
Although it has been known for decades that papillomavirus DNA is packaged in nucleosomes within the virion (4), little is known about the modifications of histones on the packaged genome. Here, we profile the posttranslational modifications on H3 and H4 histones packaged by human papillomavirus type 1 (HPV1), HPV18, and bovine papillomavirus type 1 (BPV1). We show that papillomaviruses contain histones enriched in modifications usually associated with “active” chromatin. These findings provide insight into a relatively unexplored area of papillomavirus biology and may suggest therapeutic epigenetic approaches that could interfere with viral transmission.
RESULTS
The viral minichromosome in the HPV capsid.
To visualize the HPV capsid and encapsidated minichromosome, we developed a structural model of 32 host nucleosomes packaged within an HPV16 capsid. Figure 1A shows the HPV16 capsid derived from cryo-electron microscopy (cryoEM) packed with a mix of H3.1- and H3.3-containing nucleosomes and ∼8,000-bp circular DNA. HPV capsids also contain between 12 and 72 copies of the L2 protein (not shown), most likely positioned under the pentameric L1 capsomeres (18).
FIG 1.

Structural model of HPV16 capsid containing viral minichromosome, histone H3 and H4 tail modifications. (A) Structural model of 13 H3.3 (dark orange) nucleosomes (PDB 5X7X) and 19 H3.1 (light orange) nucleosomes (PDB 3AFA) packed into a sphere approximating the interior of HPV16 capsid (PDB 5KEP) using CellPACK (65) and ePMV (66) in Maxon Cinema 4D. Each nucleosome is wrapped with 146 bp DNA, and the capsid was “packed” with 32 additional 100-bp segments of linker DNA (7,872 bp total). The capsid model does not contain the histone tails, but a single nucleosome is shown to the right with H3 (orange) and H4 (yellow) N-terminal tails (PDB 1KX5). (B) Positions of acetylation and methylation on the N-terminal tails of histone H3 and H4. The histone peptides derived from propionylation and trypsin digestion are indicated. Arginine residues, where cleavage takes place, are indicated in red.
Histone modifications in HPV18 quasivirions.
Initial experiments compared the modifications on histones contained in purified HPV16/18 quasivirions (virions produced using recombinant expression plasmids [19]) to those on histones from 293TT packaging cells to optimize the mass spectrometry workflow (shown in Fig. S1) and to determine whether there was preferential packaging of specific histone modifications. HPV16 quasiviruses were produced with HPV16 capsids containing an HPV18 minicircle genome (supercoiled, recircularized viral DNA generated in bacteria) that was transfected into the 293TT packaging cells. HPV16 capsids were used since they are more stable and infectious than their HPV18 counterparts. Virions were isolated using Benzonase to increase the virion yields necessary for mass spectrometry, but this increases the proportion of cellular chromatin packed in the particles, and we previously determined that 0.1% of particles contained a viral genome (20). Therefore, we also cotransfected plasmids that express the HPV18 E1 and E2 replication proteins to promote replication of the HPV18 genome. This increases packaging of viral genomes by 20-fold (20). Previous studies have also shown that the fine structure of chromatin is more authentic on replicated compared to unreplicated, transfected SV40 DNA (21). Nevertheless, it should be noted that most of the packaged chromatin in quasivirions is derived from host DNA.
Protein isolation and proteomics work flow. Histones were extracted from virions or cellular controls, and lysine residues were chemically derivatized by propionic anhydride. This restricts proteolytic digestion with trypsin to arginine residues. A second round of propionylation of the N termini of the resulting peptides was conducted. The second propionylation reduces the charge state of primary amines and increases the hydrophobicity of the peptide, which allows better retention and separation on-column. Samples were desalted and run through a C18 column before mass spectrometry analysis. Download FIG S1, PDF file, 2.1 MB (2.1MB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Proteins from ∼2.4 × 1012 purified quasivirion particles (1.4 × 109 viral genome equivalents [VGE]) were denatured, separated by SDS-PAGE, and stained with methylene blue. A region of the gel corresponding to the molecular weight of the core histones (H2A, H2B, H3, and H4) was extracted and processed for mass spectrometry. Histones from the 293TT packaging cells were acid extracted and used as “input” controls. The trypsin digestion used for standard mass spectrometry cleaves the N-terminal histone tails into multiple short (1 to 3 residue) peptides that are difficult to retain chromatographically or identify by mass spectrometry. Therefore, histone samples were propionylated both before and after trypsin digestion. This modification restricts digestion at Lys residues and generates cleavage only at Arg residues, resulting in longer peptides. In addition, propionylation reduces the charge state of primary amines and increases the hydrophobicity of the peptide, which allows better retention and separation on-column. Therefore, the histone samples were propionylated prior to trypsin digestion to block cleavage at lysine residues and improve hydrophobicity (22). The peptides were separated by reverse-phase chromatography and injected into the mass spectrometer, where data-independent acquisition (DIA) analysis was performed. The data were analyzed using EpiProfile 2.3 software (23), which calculates the relative abundance of modifications on each histone peptide.
The mass spectrometry (MS) data collected in this study included all histone and histone-like proteins present in the samples. However, we chose to focus on H3 and H4 modifications because these peptides constituted the highest-confidence identifications and because antibodies specific for the modifications we report are widely available. The most common modifications detected were acetylation and methylation of the N-terminal tails of histones H3 (including the variants H3.1 and H3.3) and H4. These peptides, and their sites of acetylation and methylation, are shown in Fig. 1B. The proportion of each modified peptide relative to the total amount of peptide is shown for H3 (Fig. 2A) and H4 (Fig. 2B) and listed in Table S1. The HPV16/18 quasiviruses contained histones with a wide variety and combination of modifications, but for the most part, they were very similar to those of the host 293TT cells. To determine the modification status of each lysine residue in the H3 and H4 tails, the proportion of each modification was summed and is displayed in the pie charts in Fig. 2C. Only the modifications detected on quasivirions are shown, since they were very similar to those of the 293TT host cells. For histone H3, a small amount of K4 was monomethylated (me1), and the majority of K9, K27, and K36 were modified by mono-, di- and tri-methylation. K14, K18, and K23 were all acetylated to various degrees. For histone H4, K5, K8, K12, and K16 showed various degrees of acetylation, while H4 K20 was primarily di-methylated. These modifications are very similar to the epigenetic profile of an average cell. In summary, the HPV16/18 quasiviruses incorporate DNA complexed with histones with the most common modifications, similar in abundance to those of the 293TT packaging cells. Thus, there appears to be no preference for packaging chromatin with specific histone modifications in this system.
FIG 2.

Quantitative mass spectrometry analysis of modifications on histone H3 and H4 of HPV18 quasiviruses and 293TT control cells. (A) Relative abundance of H3 histone acetylation and methylation as a percentage of total modified and unmodified peptides in HPV16 quasivirions (red) and 293T host cells (blue). Tail peptide sequences TKQTAR, amino acids (aa) 3 to 8; KSTGGKAPR, aa 9 to 17; KQLATKAAR, aa 18 to 40; KSAPATGGVKKPHR, H3.1 aa 27 to 40; KSAPSTGGVKKPHR, H3.3 aa 27 to 40. n = 3. Error = standard deviation (SD). (B) Relative abundance of H4 histone acetylation and methylation as a percentage of total modified and unmodified peptides in HPV16 quasivirions (red) and 293T host cells (blue). Tail peptide sequences: GKGGKGLGKGGAKR, aa 4 to 17; KVLR, aa 20 to 23. n = 3. Error = SD. (C) The proportion of modification on each residue was summed from the raw data for each peptide and averaged among replicates (average, n = 3) from the data in panels A and B and Table S1; raw data are shown in Table S1 for quasivirus versus host.
Histone modifications in HPV16 quasivirions. Relative abundance of posttranslation modifications on histone H3, H4, and H2A in HPV18 quasiviruses and 293TT packaging cells. Data are presented as relative abundance of the given modification as a percentage of total modified and unmodified peptides. The fold change was calculated by dividing the mean quasivirus value by the mean 293TT value for each modification. P value calculated by a two-tailed, equal variance t test. n = 3. Download Table S1, XLSX file, 0.05 MB (48KB, xlsx) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Histone modifications in BPV1 virions extracted from bovine fibropapillomas.
The quasivirion analysis demonstrated that we can successfully quantify histone modifications in both virion and cell samples, and therefore, these methods can be used to compare histone modifications on more biologically relevant wart-derived PV virions with those of their corresponding host cells. Bovine papillomavirus virions were purified from three batches of bovine fibropapilloma tissue previously isolated from cows experimentally infected with bovine papillomavirus 1 (BPV1) (24). Virus was purified on OptiPrep gradients, and each fraction was tested for the presence of BPV L1 protein and histone H3 by immunoblot, BPV1 DNA by quantitative PCR (qPCR), and total protein by Sypro Ruby (Fig. 3). Fractions positive for L1, H3, and viral DNA were pooled. The virion samples were imaged by transmission electron microscopy, showing that they were mostly intact capsids with the expected size and morphology (Fig. 3).
FIG 3.

BPV1 purification and bovine organotypic raft control. (A) A total of 200 μl fractions were collected from the bottom of the BPV OptiPrep gradient. Each fraction was separated by SDS-PAGE and stained for total protein with Sypro Ruby (top) and immunoblotted for BPV L1 (middle) or histone H3 (bottom). Blots are representative of 15 independent experiments. (B) Quantification of immunoblots in panel A. (C) Reverse transcriptase qPCR (RT-qPCR) quantification of BPV1 DNA per fraction (fg). (D) Representative image of transmission electron microscopy of isolated BPV virions. (E) Organotypic raft culture generated from bovine keratinocytes as a cellular control.
Papillomaviruses assemble virions in the differentiated layers of the host epithelium and cause extensive changes in the pathology of the infected tissue; this makes it difficult to match perfectly the relevant host cells for histone extraction. Therefore, we generated three types of bovine keratinocyte cell and tissue samples to serve as controls and also to determine the effect of differentiation on host histone modifications. Primary bovine keratinocytes were cultured as a monolayer under normal proliferative conditions, induced to partially differentiate by the addition of high levels of calcium to the culture medium, or cultured as organotypic skin equivalents (rafts). The keratinocytes in the organotypic raft differentiate into all layers that are present in skin (Fig. 3E and Fig. S2A).
Keratinocyte 3D organotypic raft culture. (A and B) Primary bovine (A) or human (B) keratinocytes were grown in organotypic raft culture, sectioned, and stained with hemotoxylin and eosin. Note: the image in panel A is also shown in Fig. 3. Download FIG S2, PDF file, 2.0 MB (2MB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Histone modifications in wart-derived BPV1 virions and each of the three keratinocyte culture conditions were identified by quantitative mass spectrometry, and the relative abundance of each modification was calculated (Fig. 4). Similar to the HPV quasiviruses, BPV1 capsids contained histones with many of the modifications detected in the cellular controls. However, the proportion of multiple modifications were significantly different between the virus and the three cellular controls (Fig. 4). The data for individual peptides with the greatest change are shown in detail in Fig. S3 for histone H3, and the raw data are shown in Table S2. To determine the individual modification status of each lysine residue in the H3 tail, the proportion of each modification was summed and displayed in the pie charts in Fig. 4B.
FIG 4.

Quantitative mass spectrometry analysis of histone H3 modifications of BPV1 virions and bovine keratinocytes. (A) Relative abundance of the H3 histone acetylation and methylation in histones extracted from BPV1 virions (BPV1, red), proliferating bovine epidermal keratinocytes (BEK grow, dark blue), partially differentiated bovine epidermal keratinocytes (BEK Ca++, blue), or fully differentiated bovine epidermal keratinocytes grown as organotypic skin equivalents (BEK raft, light blue). The peptide sequences are as follows: TKQTAR, aa 3 to 8; KSTGGKAPR, aa 9 to 17; KQLATKAAR, aa 18 to 26; KSAPATGGVKKPHR, H3.1 aa 27 to 40; KSAPSTGGVKKPHR, H3.3 aa 27 to 0. Abundances are calculated as a percentage of total modified and unmodified peptides based on integrated peak areas. n = 3. Error = SD. (B) The proportion of modification of each residue was calculated for BPV1 (average, n = 3) and bovine cell samples (average of all three control cell conditions, n = 9) from the data in panel A and Table S2.
Notable histone H3 modifications in BPV1. Quantitative mass spectrometry analysis of notable and/or significant modifications of histone H3 of BPV1 virions and bovine keratinocytes. Data are presented as the relative abundance of the given modification as a percentage of the total modified and unmodified peptides. Significance was determined by unpaired t test. n.s., P > 0.05; *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; n = 3; error = SD. Download FIG S3, EPS file, 0.5 MB (511.4KB, eps) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Notable histone H4 modifications in BPV1. Quantitative mass spectrometry analysis of notable and/or significant modifications of histone H4 of BPV1 virions and bovine keratinocytes. Data are presented as the relative abundance of the given modification as a percentage of the total modified and unmodified peptides. Significance was determined by unpaired t test. n.s., P > 0.05; *, P < 0.05; **, P < 0.01; ***, P < 0.001 ****, P < 0.0001; n = 3; error = SD. Download FIG S4, EPS file, 0.3 MB (347.6KB, eps) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Histone modifications in BPV1 wart virions. Relative abundance of posttranslation modifications on histone H3 and H4 in BPV1 virions and primary bovine keratinocytes. Cellular controls were collected during normal proliferation (growing), after differentiation in high-calcium medium (Ca++), or after culture in a 3D organotypic raft system (raft). Data are presented as the relative abundance of the given modification as a percentage of total modified and unmodified peptides. The fold change was calculated by dividing the mean BPV value by the mean value of the indicated keratinocyte growth condition for each modification. The P value was calculated by a two-tailed, equal variance t test. n = 3. Download Table S2, XLSX file, 0.1 MB (82.8KB, xlsx) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Histone H3 residue K4 can be acetylated or methylated, but >90% was unmodified in all three cell samples (Fig. 4, Fig. S3, Table S2). In contrast, ∼50% of K4 residues were methylated (mostly K4me1) in the virion particle. H3K4ac was low in all samples but increased by nearly 6-fold in the virions, at ∼0.6% compared to ∼0.1% of the cellular controls. Both acetylation and methylation of H3K4 are usually associated with active chromatin (15). H3 residue K9 can also be acetylated or methylated; acetylation is found in active chromatin, and methylation in repressed chromatin (25). Similar to the activating H3K4 modification, H3K9ac was present, either alone or combinatorially with other modifications, at low levels in all cell samples (∼2%) but was enriched in the virus (∼9%). In contrast, ∼44% H3K9 was methylated (K9me1, K9me2, or K9me3), alone or combinatorially with other modifications, in the virion, but about ∼75% of cellular histones contained this repressive modification. Acetylation of H3K14, also associated with active chromatin, was found in ∼65% of virion H3 peptides compared to ∼47% on average in the cellular samples. Histone H3 peptide 18-26 contains two lysine residues (K18 and K23) that can be acetylated or methylated. Methylation of these residues was extremely low in all samples, but acetylation of both residues was much more abundant in the virion particles. The virions contained H3 peptides with ∼22% K18ac and 54% K23ac, compared to the cell samples that contained, on average, ∼6% and 28%, respectively.
H3 peptide 27-40 differs by a single amino acid, residue 31, between the histone variants H3.1 (Ala) and H3.3 (Ser; Fig. 4), and the key modified residues on these peptides are K27 and K36 (modified as K27ac, K27me1, K27me2, K27me3, and K36me1, K36me2, K36me3). The K27 acetylation mark is associated with active chromatin and di- and tri-methylation with repressed chromatin, while H3K36 methylation is most often found in transcriptionally active chromatin (26, 27). Similar to H3K4, H3K27ac was very low in all samples but more abundant in the virus. Residues K27 and K36 were highly methylated in both H3.1 and H3.3 across all virion and cell samples. K27 was methylated in 82% of H3.1 and 77% of H3.3 peptides in virions compared to 86% of H3.1 and 67% of H3.3 peptides in the cell samples; in all samples, the most abundant peptide form was the single modification H3K27me2. Similarly, K36 was highly methylated in both virus and cells samples: 62% (H3.1) and 78% (H3.3) of residues were methylated in the virion compared to a wider range of 36 to 43% (H3.1) and 43 to 68% (H3.3) across the host cell samples. Overall, HK36 was mostly mono- and di-methylated, and overall, the virion was enriched in these two modified residues (Table S2; BPV versus host).
The proportion of the acetylation and methylation modifications on each lysine residue in the H3 tail is displayed in the pie charts in Fig. 4B for both the BPV1 virions and the bovine cellular controls (the mean of all three cellular controls). These epigenetic signatures highlight the major differences between the BPV1 virions and the cell controls. Overwhelmingly, the virion H3 peptides are enriched in active chromatin modifications (K4me1, K4me2, K4me3, K14ac, K18ac, K23ac, K36me1, K36me2) compared to all three host cell samples (Table S2).
Validation of BPV1 histone H3 modifications.
To validate the mass spectrometry results, we performed immunoblot analysis using antibodies specific to each of the H3 modifications with the greatest enrichment in the BPV1 virions (human and bovine N-terminal histone sequences are identical). As shown in Fig. 5, the levels of H3K4me1, H3K4me3, H3K9ac, H3K14ac, H3K18ac, and H3K23ac were confirmed to be higher in the virions than in cellular controls when the samples were normalized to the level of total histone H3. In agreement with the mass spectrometry data, the BPV virions were strongly depleted for H3K9me3, but not consistently for H3K27me3.
FIG 5.

BPV virions are enriched in histone H3 modifications associated with transcriptional activation and depleted for those associated with transcriptional repression. (A) The left panel shows representative immunoblots of BPV virions and bovine keratinocyte control cells using antibodies against indicated histone H3 modifications. On the right, the immunoblots were stripped and reprobed with antibody against all forms of histone H3 as a loading control. (B) Quantification of immunoblots in panel A. For each band, histone modification signals were normalized to the corresponding bands from the panH3 signal. The resulting levels are represented relative to the signal for BPV1 (K18ac, K4me1, K4me3, K9me3; n = 2) (K9ac, K14ac, K23ac, K27me3; n = 3). Error = SD. Significance was determined by an unpaired t test. n.s., P > 0.05; *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. (C) PanH3 immunoblot against an acid-urea separation of histones from BPV1 virions and bovine cellular controls. The more slowly migrating, upper bands correspond to an increase in acetylation of H3 lysine residues. Recombinant H3 protein (lane 1) indicates the positions of unmodified histone H3. n = 2.
We also performed acid-urea (AU) PAGE to further confirm increased acetylation of histone H3 in the BPV virions. In AU PAGE, proteins are separated on the basis of size and charge. Acetylation of lysines reduces their positive charge, so histones with a higher degree of acetylation will migrate more slowly through the gel. This technique allows us to examine overall protein acetylation levels without the need for specific antibodies against every possible lysine modification. As shown in Fig. 5, most of the signal from the histones extracted from BPV virions was in the more slowly migrating, upper bands, compared to the cellular lanes. This supports the mass spectrometry data and immunoblots and shows that H3 histones packaged in the virions are acetylated to a higher degree than those of the cellular controls.
BPV1 virions are enriched in acetylated histone H4.
Mass spectrometry showed that the majority of H4 lysine residues K5, K8, K12, and K16 were unmodified in the virion and cellular samples. The BPV1 virions were enriched in acetylated K5, K8, and K12, as well as combinatorial acetylation of these lysines compared to each of the cellular controls (Fig. 6 and Fig. S4). In contrast, H4K16ac was depleted in the virions. This was notable since acetylation of K5, K8, and K12 is associated with active chromatin, while the K16ac modification is more complex. No consistent trend was found for the H4K20 methylation, with variation across the cell samples, although a small amount of H4K20ac was present in the virion but not in the cell samples.
FIG 6.
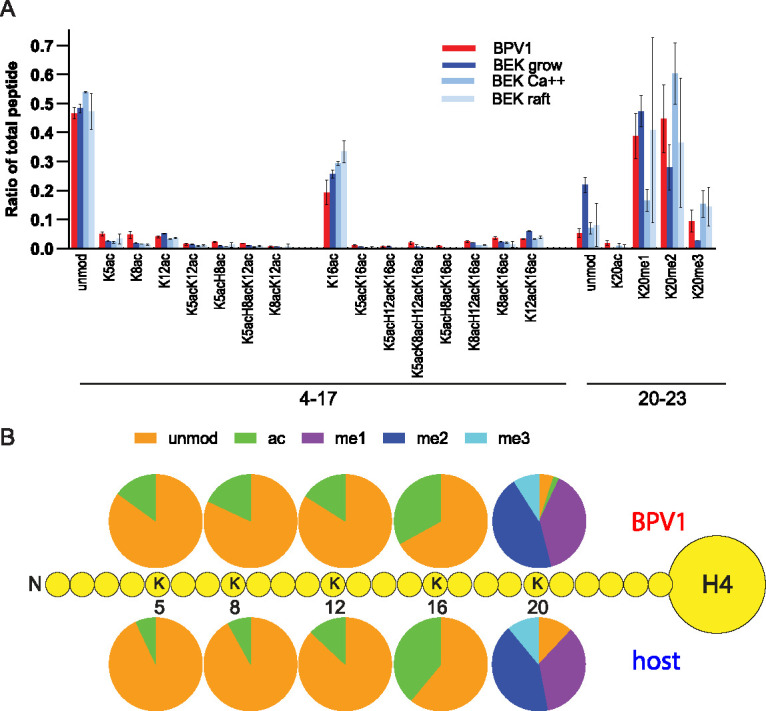
Quantitative mass spectrometry analysis of histone H4 modifications of BPV1 virions and bovine keratinocytes. (A) Relative abundance of the H4 histone acetylation and methylation in histones extracted from BPV1 virions (BPV1), proliferating bovine epidermal keratinocytes (BEK grow), partially differentiated bovine epidermal keratinocytes (BEK Ca++), or fully differentiated bovine epidermal keratinocytes grown as organotypic skin equivalents (BEK raft). The peptide sequences are as follows: GKGGKGLGKGGAKR, aa 4 to 17; KVLR, aa 20 to 23. Abundances are calculated as a percentage of total modified and unmodified peptides based on integrated peak areas. n = 3. Error = SD. (B) The proportion of modification of each residue was calculated for BPV1 (average n = 3) and bovine cell samples (average of all three control cell conditions, n = 9) from the data in panel A and Table S2.
To confirm the enrichment in lysine acetylation of H4 packaged in BPV virions, we performed immunoblots using antibodies specific to H4 acetylated on several different lysines. As shown in Fig. 7, the native BPV virions showed considerably higher levels of acetylation of K5ac, K8ac, and K12ac than those of the control keratinocytes. Additionally, the virion samples had higher signals of tetra-acetylated K5acK8acK12acK16ac. Together, these data suggest that bovine papillomavirus virions contain H4 histones that are more highly acetylated than host keratinocytes.
FIG 7.

BPV virions are enriched in acetylation of histone H4. (A) Representative immunoblots of BPV virions and bovine keratinocyte control cells probed with antibodies against the indicated histone H4 modification. On the right are the same immunoblots, stripped and reprobed with antibody against pan histone H3 to ensure even loading. (B) Quantification of immunoblots in panel A. Histone modification signals were normalized to panH3 signals, and the levels are represented relative to the signal for BPV1. Significance was determined by an unpaired t test. n.s., P > 0.05; *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; n = 2; error = SD.
Histone modifications in HPV1 wart virions. Relative abundance of posttranslation modifications on histone H3 and H4 in HPV1 virions and primary human foreskin keratinocytes. Cellular controls were collected during normal proliferation (growing), after differentiation in high calcium medium (Ca++), or after culture in a 3D organotypic raft system (raft). Data are presented as the relative abundance of the given modification as a percentage of total modified and unmodified peptides. The fold change was calculated by dividing the mean HPV value by the mean value of the indicated keratinocyte growth condition for each modification. The P value was calculated by a two tailed, unequal variance t test. n = 2, HPV1; n = 6, cell samples. Download Table S3, XLSX file, 0.1 MB (80.5KB, xlsx) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Histone modifications in wart-derived HPV1 virions.
To extend these findings to human-specific virus, we were able to isolate sufficient quantities of HPV1 from a palmar wart to perform quantitative mass spectrometry. Primary human foreskin keratinocytes (HFKs) served as control host cells and were cultured in the three conditions described above: proliferating cells, cells partially differentiated with high calcium, and three-dimensional (3D) organotypic rafts (Fig. S2B). Histones were extracted from the HPV1 virions, and cellular controls and were analyzed for histone modifications and variants by quantitative mass spectrometry. As found in the BPV1 virions, the HPV1 virions were enriched in acetylated histone H3, primarily on lysines 14, 18, and 23 (Fig. 8, Fig. S5, Table S3). In all samples, H3K4 was primarily unmodified or monomethylated, and although H3K4me1 was increased in the virion compared to the raft sample, it was similar to the levels in growing and partially differentiated cells. As found in BPV1, the human virions were depleted for H3K9me1 and -me3 and contained a small amount of H3K9ac.
FIG 8.

Quantitative mass spectrometry analysis of modifications on histone H3 of HPV1 virions and primary HFK control cells. (A) Relative abundance of H3 histone acetylation and methylation as a percentage of total modified and unmodified peptides (H3 peptides TKQTAR, aa 3 to 8; KSTGGKAPR, aa 9 to 17; KQLATKAAR, aa 18 to 26; KSAPATGGVKKPHR, H3.1 aa 27 to 40; KSAPSTGGVKKPHR, H3.3 aa 27 to 40) in histones extracted from HPV1 virions (HPV), proliferating human foreskin keratinocytes (growing), partially differentiated human foreskin keratinocytes (Ca++), or fully differentiated human foreskin keratinocytes grown as organotypic skin equivalents (raft). n = 2, HPV1; n = 6, cell samples. Error = SD. (B) The proportion of modification of each residue was calculated for HPV1 (average, n = 2) and human cell samples (average of all three control cell conditions, n = 6) from the data in panel A and Table S3.
Quantitative mass spectrometry analysis of notable and/or significant modifications of histone H3 and H4 of HPV1 virions and human keratinocytes. Data are presented as the relative abundance of the given modification as a percentage of the total modified and unmodified peptides. Significance was determined by unpaired t test. n.s., P > 0.05; *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; n = 3; error = SD. Download FIG S5, EPS file, 0.4 MB (421.2KB, eps) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Histone H4 in the virions was enriched in acetylation on K5, K8, and K12 in comparison to the proliferating and calcium differentiating HFKs (Fig. 9A, Fig. S5, Table S3), but the amounts observed were similar to those of raft-derived histones. There was also less combinatorial acetylation of K5, K8, K12, and K16 in the virions compared to all cell samples, and overall acetylation of H4 K5, K8, K12, and K16 was less in the virion samples than in the average cell sample (Fig. 9B). As was observed for BPV1, virions were enriched for H4K20ac compared to all cellular samples, but there was no consistent pattern of enrichment in methylation patterns on K20.
FIG 9.

Quantitative mass spectrometry analysis of modifications on histone H4 of HPV1 virions and primary HFK control cells. (A) Relative abundance of H4 histone acetylation and methylation as a percentage of total modified and unmodified peptides (H4 peptides GKGGKGLGKGGAKR, aa 4 to 17; KVLR, aa 20 to 23) in histones extracted from HPV1 virions (HPV), proliferating human foreskin keratinocytes (growing), partially differentiated human foreskin keratinocytes (Ca++), or fully differentiated human foreskin keratinocytes grown as organotypic skin equivalents (raft). n = 2, HPV1; n = 6, cell samples. Error = SD. (B) The proportion of modification of each residue was calculated for HPV1 (average, n = 2) and human cell samples (average, n = 6 for each of 3 cell types) from the data in panel A and Table S3.
Together, these data indicate that wart-derived human virions are enriched in histone modifications associated with active chromatin compared to human keratinocyte control cells. Correspondingly, the data indicate that these virions are depleted for transcriptionally repressive histone modifications. This is in agreement with the data from the wart-derived BPV virions.
Histone variant H3.3 is enriched in BPV1 virions.
The histone variants H3.1 and H3.3 are deposited in chromatin in replication-dependent or replication-independent processes, respectively (28). Mass spectrometry can distinguish between these variants since there is a four-amino acid difference between these variants and so can give us insight into the assembly processes of viral chromatin. In the peptide spanning residues 27 to 40 of histone H3, this results in an alanine to serine difference at residue 31. As measured by mass spectrometry, the ratio of H3.3 to total H3 was 5- to 8-fold higher in BPV1 and 3- to 5-fold higher in the HPV1 virions than the cellular controls (Fig. 10A and B). In comparison, the HPV quasivirions showed no enrichment of H3.3 compared to the control cells (Fig. 10C). There was insufficient material to perform Western blot analysis of H3.3 abundance in HPV1 virions, but to confirm this enrichment in the BPV virions, we performed immunoblot analysis with antibodies specific to H3.3 (Fig. 10D and E). The histones from the BPV virions had much greater levels of H3.3 compared to all three cellular samples (Fig. 10D). To confirm this finding, H3.1 and H3.3 proteins were separated based on hydrophobicity using triton acid urea (TAU) PAGE and immunoblotted with an antibody against all variants of H3 (Fig. 10F and G). This confirmed that about 40% of H3 in the BPV particles was H3.3, very similar to the quantitation by mass spectrometry and much higher than the cellular controls. Therefore, the chromatin in papillomavirus virions is enriched in the histone variant H3.3.
FIG 10.

Wart-derived virions are enriched in the histone H3 variant H3.3. (A) Mass spectrometry analysis of the abundance of H3 variant H3.3 relative to total H3 levels of BPV1 virions and bovine control cells. n = 3 BPV1; n = 6 cell samples; error = SD. (B) Mass spectrometry analysis of the abundance of H3 variant H3.3 relative to total H3 levels of HPV1 virions and primary human control cells. n = 3 BPV1; n = 6 cell samples; error = SD. (C) Mass spectrometry analysis of H3 variant H3.3 in HPV18 quasivirions compared to packaging cells. n = 3. (D) Representative immunoblot of BPV1 and bovine control cells with antibody against H3.3 (top) and all forms of H3 (bottom). n = 3. (E) Quantification of immunoblots as shown in panel D. Significance was determined by unpaired t test. n.s., P > 0.05; *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; n = 3; error = SD. (F) Representative H3 immunoblot (n = 4) from TAU PAGE gel of BPV and control keratinocytes. (G) Quantification of TAU immunoblots as shown in panel F. Significance was determined by paired t test. n.s., P > 0.05; *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; n = 4; error = SD.
DISCUSSION
In this study, we used mass spectrometry to profile the histones of HPV16/18 quasivirions and native BPV1 and HPV1 virions extracted from warts. We demonstrate that the histone profiles of wart-derived virions are distinct from that of the host cell, suggesting that viral chromatin is assembled and/or modified independently from the cellular chromatin. In contrast, the HPV16/18 quasiviruses contained histone modifications similar to those found in the packaging cells, implying that the capsid did not exclude modified chromatin on the basis of space or chemical compatibility. This was not surprising as, due to the promiscuous nature of L1 and L2 capsid assembly, the majority of chromatin packaged by quasiviruses prepared using the Benzonase method is derived from cellular genomic DNA (29). Therefore, most likely, many of the histone modifications we detect in quasiviruses are derived from chromatin containing cellular DNA rather than viral minichromosomes. Nevertheless, these findings provide a good control for our methodology and show that the enhanced histone modifications found in the BPV1 and HPV1 virions are not artifactual. Quasiviruses are a useful model system for early HPV infection, and since quasivirion chromatin mirrors that of the host cells, treatment of packaging cells with small molecule modulators of chromatin-modifying enzymes is a powerful tool to examine the roles of specific modifications in infection studies (20).
The wart-derived HPV1 and BPV1 virions contained histones that were strongly enriched in acetylation of H3 and H4 histone tails. This acetylation could cause the viral minichromosome to be less compact but also provide binding sites for various transcriptional activators. Notably, we have shown that the double bromodomain protein, Brd4, activates early HPV transcription (20), so these modifications could ensure that the viral genome is primed for transcriptional activation by Brd4 immediately after infection. An enrichment of acetylated histones has also been reported in the capsids of the polyomaviruses, SV40, BKV, and mouse polyomavirus (30, 31). We also observed enrichment of H3K4me1, particularly in the bovine virions. H3K4me1 is a mark of cellular enhancer regions, and we have previously shown that H3K4me1 is present throughout the chromatin in HPV replication foci that form in differentiated cells (32). BPV1 particles also showed an enrichment for H3K4me3, a hallmark of actively transcribing genomic regions, and H3K4me2, which is associated with permissive euchromatin. Conversely, H3K9me1, me2, and me3, hallmarks of repressive chromatin, were depleted in the BPV1 and HPV1 virions. The strategy of packaging the viral genome in a chromatin state primed for transcription is likely advantageous for a small virus.
Papillomaviruses also promote various aspects of their life cycle by modulating host epigenetic mechanisms (33), which induces numerous epigenetic changes in host chromatin. Viral proteins bind to and manipulate several host epigenetic modulating enzymes. For example, the E7 protein binds to histone deacetylases (HDACs) and causes a global increase in acetylation (34–36). E7 also reprograms host cells by activating the lysine demethylases KDM6A/B and thus decreasing overall levels of H3K27 trimethylation (35). Both of these trends were observed in the chromatin from wart-derived virions, and it seems likely that viral manipulation of host epigenetic processes also shapes the chromatin in the virion particles.
Mass spectrometry can distinguish between the histone variants H3.1 and H3.3 (37). The H3.1 variant is known to be deposited on DNA in a replication-dependent manner, while H3.3 deposition is associated with transcription and DNA repair (38). We found that H3.3 is enriched several fold in the wart-derived virions compared to the cellular samples. This enrichment could reflect the transcriptionally active nature of the viral DNA before assembly or could reflect the mechanism of DNA replication at late stages of the viral infectious cycle. Papillomaviruses amplify viral DNA in differentiated cells in a G2-like phase of the cell cycle; this is dependent on host DNA damage machinery (39), and likely uses a recombination-directed replication mechanism (40) that does not utilize H3.1. The H3.3 variant is also required for transcription after DNA damage which could promote late viral transcription from amplified DNA templates (41). Several additional modifications enriched in the virions (H3K14ac, H3K18ac, H3K23ac, H4K8ac, H4K12ac, H4K16ac) are linked to DNA damage repair (42, 43). Furthermore, acetyltransferases such as Tip60 aid the loading of DNA repair proteins by relaxing the surrounding chromatin; Tip60 modulates loading of repair proteins and repair of DNA double-strand breaks (44). Thus, an alternative explanation for enrichment of acetylated H3 and H4 could be that these modifications are related to the unique replication mechanism of papillomaviruses at the late stages of infection and may also facilitate the initial replication of the viral genome upon infection of a new host. This is consistent with our understanding of how papillomaviruses hijack specific epigenetic processes for viral replication and transcription throughout the infectious cycle and how they also modulate the host epigenome (1).
Encapsidated viral chromatin confers other advantages to the virus. Long stretches of naked DNA signal the cell that the nucleic acid is foreign and activate the cGAS-STING pathway (45). Trafficking of HPV particles to the nucleus in vesicles helps the virus to evade cGAS/STING sensing (46), but assembly of DNA in chromatin also helps evade detection (45). Our study did not detect histone H1 in virions because we size selected for the smaller canonical histones, but chromatin without the linker histone H1 activates cGAS more efficiently (47). Recent studies have determined the structural mechanism of histone octamer-dependent inhibition of cGAS (48–52) and shown that cGAS interacts with the acidic patch on H2A-H2B heterodimers within adjacent nucleosomes (53); the N-terminal tail of H4 also interacts with this acidic patch to regulate nucleosome stacking and chromatin compaction (47, 54). Therefore, hypothetically, viral minichromosomes with certain histone modifications could more efficiently bypass and evade innate immune surveillance.
The minor capsid protein L2 facilitates genome packaging, but no specific DNA packaging sequence has been identified (55). Could histone modifications on viral chromatin form a packaging signal for viral minichromosomes? On one hand, this seems unlikely because of the permissive nature of chromatin within the quasivirus particles (although this is mostly cellular chromatin), but on the other hand, assembly of authentic papillomavirus particles could take place in concert with specific replication processes and histone modifications in differentiated keratinocytes. The L2 protein localizes to viral replication foci at late stages of the papillomavirus life cycle (56, 57), and in early infection, L2 traffics the viral DNA first to mitotic chromosomes and then to ND10 bodies to establish infection (58, 59). Interaction of L2 with specifically modified viral chromatin could be key to these processes.
In conclusion, we propose that the viral chromatin acquires specific histone modifications late in infection that are coupled to the mechanisms of viral replication, late gene expression, and packaging. We predict that, in turn, these same modifications promote early infection by evading detection, promoting localization of the viral chromosome to beneficial regions of the nucleus, and facilitating early transcription and replication.
MATERIALS AND METHODS
Plasmids.
Plasmids expressing codon-optimized HPV18 E1 and E2 (pMEP9-HPV18E1 and pMEP4-HPV18E2) have been described previously (20). The pShell16 plasmid (Addgene no. 37320) expresses the HPV16 L1 and L2 proteins (60). Minicircle HPV18 genomes were produced in Escherichia coli ZYCY10P3S2T from pMC.BESPX-HPV18 as described previously (67).
Antibodies.
The antibodies used for immunoblots were the following: anti-H3 (Millipore 07-690, 1:5,000), anti-H3K9me3 (Abcam Ab8898, 1:500), anti-H3K27me3 (Abcam Ab6147, 1:200), H3K9ac (Santa Cruz sc-6616, 1:200), anti-H3K14ac (Millipore 07-353, 1:1,000), anti-H3K18ac (Active Motif 39755, 1:1,000), anti-H3K4me (Abcam ab8895, 1:1,000), anti-H3K4me3 (Millipore 05-745, 1:500), anti-H3K27ac (Millipore 07-355, 1:1,000), anti-H3.3 (Abcam ab176840, 1:500), anti-papillomavirus L1 (Millipore MAB837, 1:10,000).
Culture of host cells.
293TT cells were cultured in Dulbecco modified Eagle medium (DMEM)-10% fetal bovine serum (FBS) with 200 μg/ml of hygromycin B (Roche) until 80% confluent. Cells (1 × 106 to 13 × 106 per sample) were collected by trypsinization, washed with phosphate-buffered saline (PBS), and flash-frozen before storage at –80°C.
The human and bovine keratinocytes were as follows: Swiss J2/3T3 murine fibroblast feeders were cultured in DMEM and 10% newborn calf serum (NCS) and lethally irradiated the day before use. Then, 3 × 105 primary human foreskin keratinocytes (HFKs) or bovine epidermal keratinocytes (BEK6) were plated onto the feeder monolayer and incubated for 3 to 4 days at 37°C and 5% CO2 until approximately 70% confluent in Rheinwald-green F-medium (3:1 Ham’s F12/DMEM-high glucose, 5% FBS, 0.4 μg/ml hydrocortisone, 8.4 ng/ml cholera toxin, 10 ng/ml epidermal growth factor [EGF], 24 μg/ml adenine, and 6 μg/ml insulin, 100 units/ml penicillin, 100 μg/ml streptomycin). Feeders were removed with Versene before keratinocytes were collected by trypsinization, pelleted, flash-frozen, and stored at –80°C until protein extraction.
Differentiated (Ca++) keratinocytes were as follows: cells were grown to confluence, the feeders removed with Versene, and the medium was changed to low-calcium basal medium (Lonza) supplemented with SingleQuots for keratinocytes (bovine pituitary extract, hydrocortisone, and epidermal growth factor) for 24 h. The medium was changed to basal medium supplemented with 1.5 mM CaCl2, and cells cultured for 5 additional days. Cells were collected by trypsinization, pelleted, flash-frozen, and stored at –80°C until protein extraction.
Organotypic raft cultures were as follows: a collagen dermal equivalent was formed by mixing 0.8 ml each of reconstitution buffer (2.2% NaHCO3, 0.05 N NaOH, 200 mM HEPES free acid) and 10× Ham’s F12 medium (Life Technologies) with 7.6 ml rat tail collagen type I (Sigma). Two million NIH 3T3 mouse fibroblasts (ATCC 1658) were suspended in 0.4 ml FBS and mixed with the collagen solution, and 0.75 ml collagen/cell suspension was aliquoted per well of a 12-well tissue culture plate. After 1 h of incubation at 37°C and 5% CO2, the solidified gel was overlaid with 1 ml raft culture medium (3:1 DMEM/F12, 10% fetal calf serum [FCS], 0.4 μg/ml hydrocortisone, 0.01 nM cholera toxin, 5 μg/ml transferrin) and incubated at 37°C for 2 days. Then, 1 × 105 keratinocytes were plated on each collagen disk and cultured in raft culture medium supplemented with 5 ng/ml epidermal growth factor until confluent. The collagen gels were transferred to the liquid-air interface on a metal grid and cultured for 10 days in raft culture medium with 5 ng/ml EGF. To harvest, each raft was bisected and either fixed in 3.7% formaldehyde before paraffin embedding, sectioning, and hematoxylin and eosin staining, or else the epithelial layer was removed and frozen at –80°C for subsequent protein extraction.
Production of HPV16 quasiviruses.
HPV16/18 quasiviruses were produced in 293TT cells as described in reference 61. Briefly, a 75-cm2 flask of 293TT cells was transfected with 19 μg minicircle HPV18 genome, 19 μg pShell16 L1/L2 expression vector, and 6 μg each of pMEP9-HPV18 E1 and pMEP4-HPV18 E2 using Lipofectamine 2000 (Invitrogen). Then, 48 h posttransfection, quasivirus particles were isolated in the presence of Benzonase and purified on an OptiPrep density gradient. Fractions were analyzed for total protein content using Sypro Ruby and for HPV18 DNA using real-time PCR on a QuantStudio 7 PCR system and SYBR green PCR master mix (Roche). A 10-fold dilution series was included to generate a standard curve of cycle threshold versus log10 quantity. PCR was performed in triplicate using forward 5′-CACAATACTATGGCGCGCTTT-3′ and reverse 5′-CCGTGCACAGATCAGGTAGCT-3′ primers, as described previously (62). Fractions that contained L1 and L2 proteins, histones, and HPV18 DNA were selected and pooled. Viral genome equivalents (VGE) and number of virion particles were calculated as previously described (61).
Isolation of bovine papillomavirus virions.
Bovine wart tissue (3 to 5 g) (harvested from BPV1-infected cows by Carl Olson, University of Wisconsin School of Veterinary Medicine; a gift from Carl C. Baker) (24) was minced and vortexed in 10 mM Tris-HCl, pH 7.5, 1 mM MgCl2, and 1% (wt/vol) Brij58 in PBS. Samples were digested with 75 U Benzonase (Millipore) and 50 U Plasmid-Safe (Lucigen) at 37°C for 1 h. Then, 2 mg collagenase H (Sigma) was added, the pH was adjusted to 7 to 7.5, and samples were incubated at 37°C for 15 min followed by 4°C incubation overnight on a rotator. The next day, 0.17 volumes of 5 M NaCl were added, and the tissue was pelleted by centrifugation at 1,000 × g for 5 min. The supernatant was reserved, and the pellet was suspended in salt extraction buffer (10 mM Tris-HCl, pH 7.5, 800 mM NaCl, 1% Brij58, 1% PBS) and sonicated until the solution no longer changed in consistency. Debris was pelleted by centrifugation at 1,000 × g for 5 min, and the supernatant was combined with the previous supernatant. After another clarification step, the supernatant was underlaid with a 1.5-ml cushion of 39% OptiPrep and centrifuged in a Sw32Ti rotor at 30,000 rpm for 2 h at 16°C. The bottom 3 ml was vortexed and centrifuged at 1,000 × g for 10 min at 4°C. The supernatant was separated on an OptiPrep density gradient, and fractions were screened for the presence of BPV1 DNA by qPCR using forward 5′-TTGGTGAGGACAAGCTACAAGTTG-3′ and reverse 5′-TTGGTGAGGACAAGCTACAAGTTG-3′ primers. Fractions that contained L1 and L2 proteins, histone H3 (all identified by immunoblotting), and BPV1 DNA were combined, aliquoted, and stored at –80°C.
Isolation of human papillomavirus virions from wart tissue.
A 0.65-cm-diameter biopsy specimen of a palmar wart was minced in 0.5 ml Dulbecco's PBS (DPBS), 10 mM MgCl2, and 1% Brij58 and digested with 50 U Benzonase and 20 U Plasmid-Safe for 20 min at 37°C. Then, 5 mg collagenase H was added and incubated for 30 min at 37°C and then overnight at 4°C. Next, 180 μl 5 M NaCl was added, and the sample was rocked at 4°C for 2 h. The sample was sonicated, centrifuged at 5,000 × g for 5 min, and the supernatant was transferred to a new tube. The pellet was washed with 0.5 ml DPBS + 0.8 M NaCl before centrifuging was done again. The supernatants were combined, and then virions were purified by OptiPrep ultracentrifugation (as described for quasiviruses). Viral DNA was extracted, analyzed with restriction digest, and cloned into a plasmid vector. Sequencing revealed it to be HPV1.
Mass spectrometry.
Virus histone extraction. BPV1, HPV1, or HPV16/18 quasiviruses (∼1.4 × 109 VGE) were precipitated in 33% trichloroacetic acid (TCA) overnight at 4°C. The precipitate was centrifuged at 20,000 × g for 10 min at 4°C, and the supernatant was removed. The pellet and tube walls were washed with 1 ml cold acetone + 0.1% HCl and centrifuged at 20,000 × g at 4°C for 10 min, and the supernatant was removed. The pellet and tube walls were washed with 1 ml cold acetone and centrifuged at 20,000 × g at 4°C for 10 min, and the supernatant was removed. The pellet was dried overnight in a SpeedVac on low vacuum with no heat. The pellet was dissolved in 60 μl 54 mM Tris-HCl, pH 8, 1× LDS sample buffer (Invitrogen), and 50 mM dithiothreitol (DTT) and heated at 70°C for 10 min. Samples were centrifuged at 16,000 × g for 1 min, and the supernatant was separated on three adjacent wells of a NuPAGE 4 to 12% SDS-PAGE gel (Thermo Fisher) by electrophoresis at 100 V. Proteins were detected with Coomassie G-250 staining (1% Coomassie G-250, 2.55% ortho-phosphoric acid, 10% [wt/vol] ammonium sulfate, 20% ethanol), and bands corresponding to the molecular weight of the core histones (molecular weight, 10 to 20 kDa) were excised from the gel and stored at –80°C.
Gel band processing. Excised gel bands were diced into ∼1-mm cubes and destained with successive washes of liquid chromatography-mass spectrometry (LC-MS)-grade water and 50% acetonitrile until no dye was visible (3 to 4 wash cycles). Gel pieces were fully dehydrated in acetonitrile, rehydrated with a 1:2 (vol/vol) solution of 100 mM ammonium bicarbonate and propionic anhydride (Sigma; catalog no. 240311-60G), and incubated at room temperature for 20 min. The supernatant was removed, and gel pieces were washed with alternating rounds of 50% acetonitrile and 100 mM ammonium bicarbonate until the supernatant pH was ∼8.0. After a second round of propionylation and pH adjustment, gel pieces were dehydrated in 100% acetonitrile and rehydrated in 50 mM ammonium bicarbonate containing 12.5 ng/μl trypsin (Promega; catalog no. V5113). Digestion proceeded overnight at room temperature. Peptides were extracted from the gel with two rounds of 100% acetonitrile dehydration and water rehydration. Extracted peptides were propionylated for two additional rounds with 1:3 (vol/vol) propionic anhydride:2-propanol and adjusted to pH ∼8 with ammonium hydroxide. Fully derivatized samples were desalted over Oligo R3 reverse-phase resin (Thermo Scientific) packed over a C18 Empore base (3M), dried to completion, and resuspended in buffer A (water + 0.1% [vol/vol] formic acid).
Cell histone extraction. Histones were extracted from cell pellets following established protocols (63). Briefly, pellets were resuspended 1:10 (vol/vol) in ice-cold nuclear isolation buffer [15 mM Tris-HCl, pH 7.5, 15 mM NaCl, 60 mM KCl, 5 mM MgCl2, 1 mM CaCl2, 250 mM sucrose, 1 mM DTT, 500 μM 4-(2-aminoethyl)benzenesulfonyl fluoride hydrochloride (AEBSF), 5 nM microcystin, 10 mM sodium butyrate, and 1× HaltTM protease/phosphatase inhibitor cocktail (Fisher)] and pelleted, resuspended 1:10 (vol/vol) in nuclear isolation buffer (NIB) containing 0.3% NP-40 alternative, and incubated on ice for 5 min. The pelleted material was washed twice at 1:10 (vol/vol) in NIB to remove detergent. The nuclear pellet was resuspended 1:5 (vol/vol) in 0.4N H2SO4 and incubated with rotation for 3 h at 4°C. Samples were centrifuged and transferred to clean tubes twice to reduce contamination from adsorbed proteins, adjusted to 33% vol/vol trichloroacetic acid, and incubated at 4°C overnight. Precipitated histones were centrifuged and washed first with acetone + 0.1% HCl and then with neat acetone and dried to completion.
Organotypic raft controls were processed similarly, but with an initial homogenization step in a ground glass pestle to enable effective extraction.
Histone derivatization. After nuclear extraction, histone samples were resuspended in 50 mM ammonium bicarbonate and propionylated for two rounds by addition of 16% (vol/vol) propionic anhydride and 14% (vol/vol) ammonium hydroxide for 10 min at room temperature. Samples were digested with 0.2 μg trypsin at room temperature overnight and then propionylated for another two rounds. Fully derivatized samples were desalted over Oligo R3 reverse-phase resin (Thermo Scientific) packed over a C18 Empore base (3M), dried to completion, and resuspended in buffer A.
Mass spectrometry acquisition. All solvents used in the analysis of MS samples were LC-MS grade. All peptides were separated over a 75 μm inner diameter silica capillary column packed in-house with Repro-Sil Pur C18-AQ 3 μm resin.
HPV16 quasivirion samples and associated 293TT controls were analyzed with an Easy-nLC system (Thermo Fisher) running 0.1% (vol/vol) formic acid (buffer A) and 80% (vol/vol) acetonitrile with 0.1% (vol/vol) formic acid (buffer B), coupled to an Orbitrap Fusion Tribrid mass spectrometer. Peptides were eluted with a gradient of 4 to 38% buffer B over 45 min. Full MS scans from 300 to 1,100 m/z were analyzed in the Orbitrap device at 60,000 full-width half maximum (FWHM) resolution and 2 × 105 automatic gain control (AGC) target value for 100 ms maximum injection time (MIT). MS2 analyses were performed in data-independent acquisition (DIA) mode with 50 m/z isolation windows. Spectra were acquired in the ion trap operating in rapid mode. High-energy collisional dissociation (HCD) fragmentation was applied at 28%, with a stepped collision energy of ±5%. The AGC target was set to 1 × 104, and MIT, to 50 ms.
BPV virion samples and BEK6 controls were analyzed with an Easy-nLC system (Thermo Fisher) running 0.1% (vol/vol) formic acid (buffer A) and 80% (vol/vol) acetonitrile with 0.1% (vol/vol) formic acid (buffer B), coupled to an Orbitrap Fusion Tribrid mass spectrometer. Peptides were eluted with a gradient of 5 to 34% buffer B over 45 min. Full MS scans from 300 to 1,100 m/z were analyzed in the Orbitrap at 60,000 FWHM resolution and 4 × 105 AGC target value, for 50 ms MIT. MS2 analyses were performed in DIA mode with 50 m/z isolation windows. Spectra were acquired in the ion trap operating in normal mode. HCD fragmentation was applied at 30%. The AGC target was set to 1 × 104, and MIT, to 50 ms.
HPV1 virion sample and HFK22 controls were analyzed in technical duplicate with an Easy-nLC system running 0.1% (vol/vol) formic acid (buffer A) and 80% (vol/vol) acetonitrile with 0.1% (vol/vol) formic acid (buffer B) coupled to a Q-Exactive HF-X Orbitrap mass spectrometer. Peptides were eluted with a stepped gradient of 0 to 5% buffer B over 2 min and 5 to 30% buffer B over 49 min. Full MS scans from 295 to 1,100 m/z were acquired in the Orbitrap at 120,000 FWHM resolution and 3 × 106 AGC target value, for 50 ms MIT. MS2 analyses were performed in DIA mode with 24 m/z isolation windows, with an AGC target of 1 × 106 and 30 ms MIT. HCD fragmentation was applied at 27%, with a stepped collision energy of ±5%.
Data analysis.
MS raw data were searched using EpiProfile v2.3 with a 10-ppm mass tolerance. Retention times were manually validated in Skyline (64), and any mis-assigned peaks were corrected in EpiProfile. The relative ratio for each modification state was calculated as the area under the extracted ion chromatographic peak relative to the summed peak areas of all modification states detected for that peptide sequence. H3.3 total protein abundance was calculated as the summed peak areas of all unmodified and modified forms of H3.3 peptide 27-40 relative to the summed peak areas of all unmodified and modified forms of peptide 27-40 for both H3 and H3.3. P values were calculated using a two-tailed, equal variance t test for quasivirions and BPV1 and two-tailed, unequal variance t test for HPV1.
Acid urea gel electrophoresis and triton acid urea (TAU) gel electrophoresis and immunoblotting.
Acid extraction of cellular histones. Frozen cell pellets or rafts were thawed, and histones were extracted in 750 μl 0.4N H2SO4 for 4 h at 4°C on a rotator. Lysates were clarified by centrifugation at 16,000 × g at 4°C for 10 min, and 100% TCA was added dropwise to the supernatant to a final concentration of 33%. After overnight incubation on ice, samples were centrifuged at 16,000 × g at 4°C for 10 min, the supernatant was discarded, and the pellet was washed in 100 μl ice-cold acetone before centrifugation for 5 min at 16,000 × g at 4°C. The supernatant was removed, and the acetone wash was repeated. Pellets were air dried, dissolved in 100 μl H2O, and aliquoted and stored at –80°C until use.
Preparation of virion proteins for acid urea (AU). BPV1 virions were precipitated by adding TCA dropwise to a final concentration of 33% and incubating overnight at 4°C. Samples were centrifuged at 16,000 × g at 4°C for 10 min, the supernatant was discarded, and the pellet was washed with 100 μl ice-cold acetone before centrifugation for 5 min at 16,000 × g at 4°C. The supernatant was removed, and the acetone wash was repeated. Pellets were air dried, dissolved in 100 μl H2O, and aliquoted and stored at –80°C until use.
Acid urea gels. A separating gel (15% acrylamide, 0.1% bis-acrylamide, 6 M urea, 5% acetic acid, 0.06% tetramethylethylenediamine, 0.14% ammonium persulfate) overlaid with a stacking gel without wells (6% acrylamide, 0.04% bis-acrylamide, 6 M urea, 0.06% tetramethylethylenediamine, 0.14% ammonium persulfate) was polymerized in 18 by 16-cm glass gel casting plates. Then, 500 μl AU sample buffer (6 M urea, 5% acetic acid, 0.02% pyronin Y, 12.5 mg/ml protamine sulfate) was loaded on top of stacking gel, and gels were electrophoresed at 300 V overnight in 5% acetic acid (AU running buffer). Protein samples were dried in a SpeedVac (low vacuum, no heat) and dissolved in 20 μl AU sample buffer + 50 mM DTT. Next, 0.5 μg histone H3.1 (New England Biolabs [NEB] catalog no. M2503S) and H3.3 (NEB; catalog no. M2507S) were used as controls, and 10 μg cytochrome c was used as a visual marker of protein migration during electrophoresis. Because of the fragile nature of the stacking gel, samples were loaded into the space between the teeth of the comb and separated by electrophoresis at 200 V (TAU) or 400 V (AU) until the cytochrome c marker had just run off the bottom of the gel.
Triton-acid urea gels. A separating gel (15% acrylamide, 0.1% bis-acrylamide, 6 M urea, 5% acetic acid, 0.37% Triton X-100, 0.06% tetramethylethylenediamine, 0.14% ammonium persulfate) was polymerized in plastic cassettes (13.3 by 8.7 cm) with 12+2 well comb (no stacking gel). Protein samples were prepared as described for acid urea gels. Gels were electrophoresed in 5% acetic acid (vol/vol) running buffer at 200 V until the cytochrome c ran just off the bottom of the gel. Transfer conditions were as described for acid urea gels.
Immunoblot transfer. Proteins were transferred to 0.45-μm polyvinylidene difluoride (PVDF) membrane in AU transfer buffer (0.7% acetic acid) at 500 mA for 20 min (proteins migrate toward the anode). Immediately after transfer, immunoblots were performed as described below.
Sodium dodecyl sulfate polyacrylamide gel electrophoresis and immunodetection.
Cell protein extraction. Growing and differentiated cellular samples were cultured as described. Keratinocyte monolayers were rinsed with ice-cold PBS and lysed in 1 ml SDS lysis buffer (1% [wt/vol] SDS, 10 mM Tris-HCl, pH 8, 1 mM EDTA, pH 8) heated to 95°C. Lysates were sonicated using a Bioruptor and heated to 95°C, and debris was pelleted by centrifugation at 16,100 × g for 5 min. The supernatant was stored in aliquots at –80°C. Frozen organotypic rafts were thawed, suspended in 200 μl SDS lysis buffer, and heated to 95°C. Samples were sonicated using a Bioruptor and homogenized with ReadyPrep protein minigrinders (Bio-Rad). Debris was removed by centrifugation at 20,000 × g at 4°C, and supernatant was stored in aliquots at –80°C. A Pierce BCA protein assay kit (Thermo Fischer) was used to determine protein concentrations.
Sample preparation and electrophoresis. Equivalent amounts of virus preparations were combined with LDS sample buffer to 1×, and DTT, to 50 mM. Cellular protein lysates were thawed, and LDS sample buffer (Thermo Fisher) and DTT were added to final concentrations of 1× and 50 mM, respectively. Samples were heated at 70°C for 10 min before loading. Proteins were separated on 1.0 mm NuPAGE 12% Bis-Tris protein gels (Thermo Fischer) in 1 liter MES [2-(N-morpholino)ethanesulfonic acid] running buffer at 150 V.
Protein transfer. Proteins were transferred to 0.45-μm PVDF membrane (Millipore) in 1× NuPAGE transfer buffer (Thermo Fisher), 10% methanol, at 60 V for 3 h or at 20 V overnight.
Immunodetection. For immunoblots probing for histone modifications, protein samples could not be loaded based on equal protein amounts. Therefore, a series of dilutions of each viral and cellular protein preparation was first compared until the signal for total H3 protein was in a comparable range. The optimized amounts of viral preparation and cellular extracts were analyzed by immunoblotting for histone modifications. Membranes were stained with Ponceau S (Sigma) to ensure uniform loading and transfer and blocked in 5% skim milk in TBST (Tris-buffered saline/0.1% Tween) (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.1% Tween) for 1 h. Primary antibodies were diluted in 5% milk/TBST and incubated overnight at 4°C. Horseradish peroxidase conjugated secondary antibodies (Invitrogen) were used at a dilution of 1:10,000. Proteins were detected with SuperSignal West Dura extended duration substrate (Thermo Fisher), and chemiluminescent signals were captured using a G:Box (Syngene) and quantitated using Syngene GeneTools software. The blots were stripped after imaging with One Minute Plus stripping buffer (GM Biosciences) and reprobed with the pan-histone H3 antibody to check for even loading. The histone modification signals were normalized to the total histone H3 signals, and the levels were calculated relative to the normalized signal for BPV1. P values were calculated by two-tailed, equal variance t tests.
ACKNOWLEDGMENTS
This work was supported by the Intramural Research Programs of the National Institute of Allergy and Infectious Diseases (AM) and the National Cancer Institute (CB) of the National Institutes of Health. This work was also supported through NIH R01-AI145266 and R01-CA097093 (M.D.W.) and R01-AI118891 and P01-CA196539 (B.A.G.).
Footnotes
Citation Porter SS, Liddle JC, Browne K, Pastrana DV, Garcia BA, Buck CB, Weitzman MD, McBride AA. 2021. Histone modifications in papillomavirus virion minichromosomes. mBio 12:e03274-20. https://doi.org/10.1128/mBio.03274-20.
Contributor Information
Alison A. McBride, Email: amcbride@nih.gov.
Thomas Shenk, Princeton University.
REFERENCES
- 1.Burley M, Roberts S, Parish JL. 2020. Epigenetic regulation of human papillomavirus transcription in the productive virus life cycle. Semin Immunopathol 42:159–171. doi: 10.1007/s00281-019-00773-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Knipe DM, Cliffe A. 2008. Chromatin control of herpes simplex virus lytic and latent infection. Nat Rev Microbiol 6:211–221. doi: 10.1038/nrmicro1794. [DOI] [PubMed] [Google Scholar]
- 3.Knipe DM. 2015. Nuclear sensing of viral DNA, epigenetic regulation of herpes simplex virus infection, and innate immunity. Virology 479-480:153–159. doi: 10.1016/j.virol.2015.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Favre M, Breitburd F, Croissant O, Orth G. 1977. Chromatin-like structures obtained after alkaline disruption of bovine and human papillomaviruses. J Virol 21:1205–1209. doi: 10.1128/JVI.21.3.1205-1209.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Germond J, Hirt B, Oudet P, Gross-Bellark M, Chambon P. 1975. Folding of the DNA double helix in chromatin-like structures from simian virus 40. Proc Natl Acad Sci U S A 72:1843–1847. doi: 10.1073/pnas.72.5.1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Teif VB, Bohinc K. 2011. Condensed DNA: condensing the concepts. Prog Biophys Mol Biol 105:208–222. doi: 10.1016/j.pbiomolbio.2010.07.002. [DOI] [PubMed] [Google Scholar]
- 7.Kranjec C, Doorbar J. 2016. Human papillomavirus infection and induction of neoplasia: a matter of fitness. Curr Opin Virol 20:129–136. doi: 10.1016/j.coviro.2016.08.011. [DOI] [PubMed] [Google Scholar]
- 8.Nakahara T, Peh WL, Doorbar J, Lee D, Lambert PF. 2005. Human papillomavirus type 16 E1^E4 contributes to multiple facets of the papillomavirus life cycle. J Virol 79:13150–13165. doi: 10.1128/JVI.79.20.13150-13165.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Banerjee NS, Wang HK, Broker TR, Chow LT. 2011. Human papillomavirus (HPV) E7 induces prolonged G2 following S phase reentry in differentiated human keratinocytes. J Biol Chem 286:15473–15482. doi: 10.1074/jbc.M110.197574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Strahl BD, Allis CD. 2000. The language of covalent histone modifications. Nature 403:41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 11.Arnaudo AM, Garcia BA. 2013. Proteomic characterization of novel histone post-translational modifications. Epigenetics Chromatin 6:24. doi: 10.1186/1756-8935-6-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bannister AJ, Kouzarides T. 2011. Regulation of chromatin by histone modifications. Cell Res 21:381–395. doi: 10.1038/cr.2011.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang T, Cooper S, Brockdorff N. 2015. The interplay of histone modifications: writers that read. EMBO Rep 16:1467–1481. doi: 10.15252/embr.201540945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liang G, Lin JCY, Wei V, Yoo C, Cheng JC, Nguyen CT, Weisenberger DJ, Egger G, Takai D, Gonzales FA, Jones PA. 2004. Distinct localization of histone H3 acetylation and H3-K4 methylation to the transcription start sites in the human genome. Proc Natl Acad Sci U S A 101:7357–7362. doi: 10.1073/pnas.0401866101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ruthenburg AJ, Allis CD, Wysocka J. 2007. Methylation of lysine 4 on histone H3: intricacy of writing and reading a single epigenetic mark. Mol Cell 25:15–30. doi: 10.1016/j.molcel.2006.12.014. [DOI] [PubMed] [Google Scholar]
- 16.Dion MF, Altschuler SJ, Wu LF, Rando OJ. 2005. Genomic characterization reveals a simple histone H4 acetylation code. Proc Natl Acad Sci U S A 102:5501–5506. doi: 10.1073/pnas.0500136102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Balakrishnan L, Milavetz B. 2010. Decoding the histone H4 lysine 20 methylation mark. Crit Rev Biochem Mol Biol 45:440–452. doi: 10.3109/10409238.2010.504700. [DOI] [PubMed] [Google Scholar]
- 18.Buck CB, Cheng N, Thompson CD, Lowy DR, Steven AC, Schiller JT, Trus BL. 2008. Arrangement of L2 within the papillomavirus capsid. J Virol 82:5190–5197. doi: 10.1128/JVI.02726-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pyeon D, Lambert PF, Ahlquist P. 2005. Production of infectious human papillomavirus independently of viral replication and epithelial cell differentiation. Proc Natl Acad Sci U S A 102:9311–9316. doi: 10.1073/pnas.0504020102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McKinney CC, Kim MJ, Chen D, McBride AA. 2016. Brd4 activates early viral transcription upon human papillomavirus 18 infection of primary keratinocytes. mBio 7:e01644-16. doi: 10.1128/mBio.01644-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cereghini S, Yaniv M. 1984. Assembly of transfected DNA into chromatin: structural changes in the origin-promoter-enhancer region upon replication. EMBO J 3:1243–1253. doi: 10.1002/j.1460-2075.1984.tb01959.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Garcia BA, Mollah S, Ueberheide BM, Busby SA, Muratore TL, Shabanowitz J, Hunt DF. 2007. Chemical derivatization of histones for facilitated analysis by mass spectrometry. Nat Protoc 2:933–938. doi: 10.1038/nprot.2007.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yuan ZF, Sidoli S, Marchione DM, Simithy J, Janssen KA, Szurgot MR, Garcia BA. 2018. EpiProfile 2.0: a computational platform for processing epi-proteomics mass spectrometry data. J Proteome Res 17:2533–2541. doi: 10.1021/acs.jproteome.8b00133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baker CC, Howley PM. 1987. Differential promoter utilization by the bovine papillomavirus in transformed cells and productively infected wart tissues. EMBO J 6:1027–1035. doi: 10.1002/j.1460-2075.1987.tb04855.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Black JC, Whetstine JR. 2011. Chromatin landscape: methylation beyond transcription. Epigenetics 6:9–15. doi: 10.4161/epi.6.1.13331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kizer KO, Phatnani HP, Shibata Y, Hall H, Greenleaf AL, Strahl BD. 2005. A novel domain in Set2 mediates RNA polymerase II interaction and couples histone H3 K36 methylation with transcript elongation. Mol Cell Biol 25:3305–3316. doi: 10.1128/MCB.25.8.3305-3316.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wiles ET, Selker EU. 2017. H3K27 methylation: a promiscuous repressive chromatin mark. Curr Opin Genet Dev 43:31–37. doi: 10.1016/j.gde.2016.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tagami H, Ray-Gallet D, Almouzni G, Nakatani Y. 2004. Histone H3.1 and H3.3 complexes mediate nucleosome assembly pathways dependent or independent of DNA synthesis. Cell 116:51–61. doi: 10.1016/s0092-8674(03)01064-x. [DOI] [PubMed] [Google Scholar]
- 29.Buck CB, Thompson CD, Pang YY, Lowy DR, Schiller JT. 2005. Maturation of papillomavirus capsids. J Virol 79:2839–2846. doi: 10.1128/JVI.79.5.2839-2846.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schaffhausen BS, Benjamin TL. 1976. Deficiency in histone acetylation in nontransforming host range mutants of polyoma virus. Proc Natl Acad Sci U S A 73:1092–1096. doi: 10.1073/pnas.73.4.1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fang CY, Shen CH, Wang M, Chen PL, Chan MW, Hsu PH, Chang D. 2015. Global profiling of histone modifications in the polyomavirus BK virion minichromosome. Virology 483:1–12. doi: 10.1016/j.virol.2015.04.009. [DOI] [PubMed] [Google Scholar]
- 32.Sakakibara N, Chen D, Jang MK, Kang DW, Luecke HF, Wu SY, Chiang CM, McBride AA. 2013. Brd4 is displaced from HPV replication factories as they expand and amplify viral DNA. PLoS Pathog 9:e1003777. doi: 10.1371/journal.ppat.1003777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mac M, Moody CA. 2020. Epigenetic regulation of the human papillomavirus life cycle. Pathogens 9:483. doi: 10.3390/pathogens9060483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang B, Laribee RN, Klemsz MJ, Roman A. 2004. Human papillomavirus type 16 E7 protein increases acetylation of histone H3 in human foreskin keratinocytes. Virology 329:189–198. doi: 10.1016/j.virol.2004.08.009. [DOI] [PubMed] [Google Scholar]
- 35.McLaughlin-Drubin ME, Crum CP, Munger K. 2011. Human papillomavirus E7 oncoprotein induces KDM6A and KDM6B histone demethylase expression and causes epigenetic reprogramming. Proc Natl Acad Sci U S A 108:2130–2135. doi: 10.1073/pnas.1009933108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Longworth MS, Laimins LA. 2004. The binding of histone deacetylases and the integrity of zinc finger-like motifs of the E7 protein are essential for the life cycle of human papillomavirus type 31. J Virol 78:3533–3541. doi: 10.1128/jvi.78.7.3533-3541.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.El Kennani S, Crespo M, Govin J, Pflieger D. 2018. Proteomic analysis of histone variants and their PTMs: strategies and pitfalls. Proteomes 6:29. doi: 10.3390/proteomes6030029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shi L, Wen H, Shi X. 2017. The histone variant H3.3 in transcriptional regulation and human disease. J Mol Biol 429:1934–1945. doi: 10.1016/j.jmb.2016.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moody CA, Laimins LA. 2009. Human papillomaviruses activate the ATM DNA damage pathway for viral genome amplification upon differentiation. PLoS Pathog 5:e1000605. doi: 10.1371/journal.ppat.1000605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sakakibara N, Chen D, McBride AA. 2013. Papillomaviruses use recombination-dependent replication to vegetatively amplify their genomes in differentiated cells. PLoS Pathog 9:e1003321. doi: 10.1371/journal.ppat.1003321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Adam S, Polo SE, Almouzni GJC. 2013. Transcription recovery after DNA damage requires chromatin priming by the H3. 3 histone chaperone HIRA. Cell 155:94–106. doi: 10.1016/j.cell.2013.08.029. [DOI] [PubMed] [Google Scholar]
- 42.Dyer N, Young L, Ott S. 2016. Artifacts in the data of Hu et al. Nat Genet 48:2–4. doi: 10.1038/ng.3392. [DOI] [PubMed] [Google Scholar]
- 43.Tamburini BA, Tyler JK. 2005. Localized histone acetylation and deacetylation triggered by the homologous recombination pathway of double-strand DNA repair. Mol Cell Biol 25:4903–4913. doi: 10.1128/MCB.25.12.4903-4913.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Murr R, Loizou JI, Yang YG, Cuenin C, Li H, Wang ZQ, Herceg Z. 2006. Histone acetylation by Trrap-Tip60 modulates loading of repair proteins and repair of DNA double-strand breaks. Nat Cell Biol 8:91–99. doi: 10.1038/ncb1343. [DOI] [PubMed] [Google Scholar]
- 45.Zierhut C, Yamaguchi N, Paredes M, Luo JD, Carroll T, Funabiki H. 2019. The cytoplasmic DNA sensor cGAS promotes mitotic cell death. Cell 178:302–315.e23. doi: 10.1016/j.cell.2019.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Uhlorn BL, Jackson R, Bratton SM, Li S, Van Doorslaer K, Campos SK. 2020. Vesicular trafficking permits evasion of cGAS/STING surveillance during initial human papillomavirus infection. PLoS Pathog 16:e1009028. doi: 10.1371/journal.ppat.1009028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Uggenti C, Lepelley A, Depp M, Badrock AP, Rodero MP, El-Daher M-T, Rice GI, Dhir S, Wheeler AP, Dhir A, Albawardi W, Frémond M-L, Seabra L, Doig J, Blair N, Martin-Niclos MJ, Della Mina E, Rubio-Roldán A, García-Pérez JL, Sproul D, Rehwinkel J, Hertzog J, Boland-Auge A, Olaso R, Deleuze J-F, Baruteau J, Brochard K, Buckley J, Cavallera V, Cereda C, De Waele LMH, Dobbie A, Doummar D, Elmslie F, Koch-Hogrebe M, Kumar R, Lamb K, Livingston JH, Majumdar A, Lorenço CM, Orcesi S, Peudenier S, Rostasy K, Salmon CA, Scott C, Tonduti D, Touati G, Valente M, van der Linden H, Van Esch H, et al. 2020. cGAS-mediated induction of type I interferon due to inborn errors of histone pre-mRNA processing. Nat Genet 52:1364–1372. doi: 10.1038/s41588-020-00737-3. [DOI] [PubMed] [Google Scholar]
- 48.Zierhut C, Funabiki H. 2020. Regulation and consequences of cGAS activation by self-DNA. Trends Cell Biol 30:594–605. doi: 10.1016/j.tcb.2020.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Boyer JA, Spangler CJ, Strauss JD, Cesmat AP, Liu P, McGinty RK, Zhang Q. 2020. Structural basis of nucleosome-dependent cGAS inhibition. Science 370:450–454. doi: 10.1126/science.abd0609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhao B, Xu P, Rowlett CM, Jing T, Shinde O, Lei Y, West AP, Liu WR, Li P. 2020. The molecular basis of tight nuclear tethering and inactivation of cGAS. Nature 587:673–677. doi: 10.1038/s41586-020-2749-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pathare GR, Decout A, Gluck S, Cavadini S, Makasheva K, Hovius R, Kempf G, Weiss J, Kozicka Z, Guey B, Melenec P, Fierz B, Thoma NH, Ablasser A. 2020. Structural mechanism of cGAS inhibition by the nucleosome. Nature 587:668–672. doi: 10.1038/s41586-020-2750-6. [DOI] [PubMed] [Google Scholar]
- 52.Michalski S, de Oliveira Mann CC, Stafford CA, Witte G, Bartho J, Lammens K, Hornung V, Hopfner KP. 2020. Structural basis for sequestration and autoinhibition of cGAS by chromatin. Nature 587:678–682. doi: 10.1038/s41586-020-2748-0. [DOI] [PubMed] [Google Scholar]
- 53.Kujirai T, Zierhut C, Takizawa Y, Kim R, Negishi L, Uruma N, Hirai S, Funabiki H, Kurumizaka H. 2020. Structural basis for the inhibition of cGAS by nucleosomes. Science 370:455–458. doi: 10.1126/science.abd0237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chen Q, Yang R, Korolev N, Liu CF, Nordenskiold L. 2017. Regulation of nucleosome stacking and chromatin compaction by the histone H4 N-terminal tail-H2A acidic patch interaction. J Mol Biol 429:2075–2092. doi: 10.1016/j.jmb.2017.03.016. [DOI] [PubMed] [Google Scholar]
- 55.Stauffer Y, Raj K, Masternak K, Beard P. 1998. Infectious human papillomavirus type 18 pseudovirions. J Mol Biol 283:529–536. doi: 10.1006/jmbi.1998.2113. [DOI] [PubMed] [Google Scholar]
- 56.Day PM, Roden RB, Lowy DR, Schiller JT. 1998. The papillomavirus minor capsid protein, L2, induces localization of the major capsid protein, L1, and the viral transcription/replication protein, E2, to PML oncogenic domains. J Virol 72:142–150. doi: 10.1128/JVI.72.1.142-150.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Swindle CS, Zou N, Van Tine BA, Shaw GM, Engler JA, Chow LT. 1999. Human papillomavirus DNA replication compartments in a transient DNA replication system. J Virol 73:1001–1009. doi: 10.1128/JVI.73.2.1001-1009.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Day PM, Baker CC, Lowy DR, Schiller JT. 2004. Establishment of papillomavirus infection is enhanced by promyelocytic leukemia protein (PML) expression. Proc Natl Acad Sci U S A 101:14252–14257. doi: 10.1073/pnas.0404229101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.DiGiuseppe S, Keiffer TR, Bienkowska-Haba M, Luszczek W, Guion LG, Muller M, Sapp M. 2015. Topography of the human papillomavirus minor capsid protein L2 during vesicular trafficking of infectious entry. J Virol 89:10442–10452. doi: 10.1128/JVI.01588-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Buck CB, Pastrana DV, Lowy DR, Schiller JT. 2005. Generation of HPV pseudovirions using transfection and their use in neutralization assays. Methods Mol Med 119:445–462. doi: 10.1385/1-59259-982-6:445. [DOI] [PubMed] [Google Scholar]
- 61.Porter SS, McBride AA. 2020. Human papillomavirus production quasivirus and infection of primary human keratinocytes. Curr Protoc Microbiol 57:e101. doi: 10.1002/cpmc.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Stepp WH, Meyers JM, McBride AA. 2013. Sp100 provides intrinsic immunity against human papillomavirus infection. mBio 4:e00845-13–e00813. doi: 10.1128/mBio.00845-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kulej K, Avgousti DC, Sidoli S, Herrmann C, Della Fera AN, Kim ET, Garcia BA, Weitzman MD. 2017. Time-resolved global and chromatin proteomics during herpes simplex virus (HSV-1) infection. Mol Cell Proteomics 16:S92–S107. doi: 10.1074/mcp.M116.065987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.MacLean B, Tomazela DM, Shulman N, Chambers M, Finney GL, Frewen B, Kern R, Tabb DL, Liebler DC, MacCoss MJ. 2010. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26:966–968. doi: 10.1093/bioinformatics/btq054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Johnson GT, Autin L, Al-Alusi M, Goodsell DS, Sanner MF, Olson AJ. 2015. cellPACK: a virtual mesoscope to model and visualize structural systems biology. Nat Methods 12:85–91. doi: 10.1038/nmeth.3204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Johnson GT, Autin L, Goodsell DS, Sanner MF, Olson AJ. 2011. ePMV embeds molecular modeling into professional animation software environments. Structure 19:293–303. doi: 10.1016/j.str.2010.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Henno L, Tombak E-M, Geimanen J, Orav M, Ustav E, Ustav M. 2017. Analysis of human papillomavirus genome replication using two- and three- dimensional agarose gel electrophoresis. Curr Protoc Microbiol 45:14B.10.1–14B.10.37. doi: 10.1002/cpmc.28. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Protein isolation and proteomics work flow. Histones were extracted from virions or cellular controls, and lysine residues were chemically derivatized by propionic anhydride. This restricts proteolytic digestion with trypsin to arginine residues. A second round of propionylation of the N termini of the resulting peptides was conducted. The second propionylation reduces the charge state of primary amines and increases the hydrophobicity of the peptide, which allows better retention and separation on-column. Samples were desalted and run through a C18 column before mass spectrometry analysis. Download FIG S1, PDF file, 2.1 MB (2.1MB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Histone modifications in HPV16 quasivirions. Relative abundance of posttranslation modifications on histone H3, H4, and H2A in HPV18 quasiviruses and 293TT packaging cells. Data are presented as relative abundance of the given modification as a percentage of total modified and unmodified peptides. The fold change was calculated by dividing the mean quasivirus value by the mean 293TT value for each modification. P value calculated by a two-tailed, equal variance t test. n = 3. Download Table S1, XLSX file, 0.05 MB (48KB, xlsx) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Keratinocyte 3D organotypic raft culture. (A and B) Primary bovine (A) or human (B) keratinocytes were grown in organotypic raft culture, sectioned, and stained with hemotoxylin and eosin. Note: the image in panel A is also shown in Fig. 3. Download FIG S2, PDF file, 2.0 MB (2MB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Notable histone H3 modifications in BPV1. Quantitative mass spectrometry analysis of notable and/or significant modifications of histone H3 of BPV1 virions and bovine keratinocytes. Data are presented as the relative abundance of the given modification as a percentage of the total modified and unmodified peptides. Significance was determined by unpaired t test. n.s., P > 0.05; *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; n = 3; error = SD. Download FIG S3, EPS file, 0.5 MB (511.4KB, eps) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Notable histone H4 modifications in BPV1. Quantitative mass spectrometry analysis of notable and/or significant modifications of histone H4 of BPV1 virions and bovine keratinocytes. Data are presented as the relative abundance of the given modification as a percentage of the total modified and unmodified peptides. Significance was determined by unpaired t test. n.s., P > 0.05; *, P < 0.05; **, P < 0.01; ***, P < 0.001 ****, P < 0.0001; n = 3; error = SD. Download FIG S4, EPS file, 0.3 MB (347.6KB, eps) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Histone modifications in BPV1 wart virions. Relative abundance of posttranslation modifications on histone H3 and H4 in BPV1 virions and primary bovine keratinocytes. Cellular controls were collected during normal proliferation (growing), after differentiation in high-calcium medium (Ca++), or after culture in a 3D organotypic raft system (raft). Data are presented as the relative abundance of the given modification as a percentage of total modified and unmodified peptides. The fold change was calculated by dividing the mean BPV value by the mean value of the indicated keratinocyte growth condition for each modification. The P value was calculated by a two-tailed, equal variance t test. n = 3. Download Table S2, XLSX file, 0.1 MB (82.8KB, xlsx) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Histone modifications in HPV1 wart virions. Relative abundance of posttranslation modifications on histone H3 and H4 in HPV1 virions and primary human foreskin keratinocytes. Cellular controls were collected during normal proliferation (growing), after differentiation in high calcium medium (Ca++), or after culture in a 3D organotypic raft system (raft). Data are presented as the relative abundance of the given modification as a percentage of total modified and unmodified peptides. The fold change was calculated by dividing the mean HPV value by the mean value of the indicated keratinocyte growth condition for each modification. The P value was calculated by a two tailed, unequal variance t test. n = 2, HPV1; n = 6, cell samples. Download Table S3, XLSX file, 0.1 MB (80.5KB, xlsx) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Quantitative mass spectrometry analysis of notable and/or significant modifications of histone H3 and H4 of HPV1 virions and human keratinocytes. Data are presented as the relative abundance of the given modification as a percentage of the total modified and unmodified peptides. Significance was determined by unpaired t test. n.s., P > 0.05; *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; n = 3; error = SD. Download FIG S5, EPS file, 0.4 MB (421.2KB, eps) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.