

Summary
Motivation
Routine infectious disease surveillance is increasingly based on large-scale whole-genome sequencing databases. Real-time surveillance would benefit from immediate assignments of each genome assembly to hierarchical population structures. Here we present pHierCC, a pipeline that defines a scalable clustering scheme, HierCC, based on core genome multi-locus typing that allows incremental, static, multi-level cluster assignments of genomes. We also present HCCeval, which identifies optimal thresholds for assigning genomes to cohesive HierCC clusters. HierCC was implemented in EnteroBase in 2018 and has since genotyped >530 000 genomes from Salmonella, Escherichia/Shigella, Streptococcus, Clostridioides, Vibrio and Yersinia.
Availability and implementation
https://enterobase.warwick.ac.uk/ and Source code and instructions: https://github.com/zheminzhou/pHierCC
Supplementary information
Supplementary data are available at Bioinformatics online.
1. Introduction
Following its introduction in 2011 (Mellmann et al., 2011), core genome multi-locus sequence typing (cgMLST) was widely adopted as a scalable, portable and easily communicable genotyping solution for the genome-based, routine surveillance of bacterial pathogens (Jolley et al., 2012; Jones et al., 2019; Moura et al., 2016). In a cgMLST scheme, bacterial genomes are assigned to sequence types (STs) consisting of 1000s of integers, which each represents a distinct sequence variant (allele) of a soft core gene (Zhou et al., 2020). A pairwise comparison of allelic differences between STs approximates the genetic distance between genomes and can be used for downstream phylogenetic analyses (Zhou et al., 2018). However, STs are arbitrary constructs, and natural bacterial populations can each encompass multiple, related STs.
Several single-level clustering schemes have been applied to cgMLST schemes to extract single-level clusters (SCs) from hierarchical clustering. Such SCs were equated with sub-lineages in Listeria (Moura et al., 2016) or single source outbreaks of Salmonella serovar Enteritidis (Coipan et al., 2020). However, because SC schemes identify only one clustering level, they ignore the wide spectrum of genetic diversities and the existence of multiple hierarchical clusters (HCs) of natural populations.
Unlike MLST schemes, multiple multi-level clustering schemes for bacterial pathogens exist that are based on core genomic single nucleotide polymorphisms (SNPs). For example, SnapperDB assigns Salmonella genomes to so-called SNP addresses, consisting of seven hierarchical single-linkage clusters based on SNP distances (Dallman et al., 2018). Similarly, genomes of Yersinia pestis or Salmonella Typhi are assigned to one of multiple levels of sub-lineages based on their placement in a phylogeny (Morelli et al., 2010; Wong et al., 2016). However, SNP-based approaches are restricted to relatively uniform clades because, unlike cgMLST schemes that can extend to the genus level (Zhou et al., 2020), the SNP calls and phylogenetic reconstructions become less reliable at higher levels of intra-genus diversity.
Here we present pHierCC, a pipeline that defines a scalable HierCC scheme that assigns bacterial genomes in real-time to multi-level clusters spanning a wide spectrum of genetic diversities. We also present HCCeval, which identifies optimal levels from the HierCC scheme, and yields multi-level HCs that likely represent hierarchical natural bacterial populations up to the species level.
2. pHierCC workflow
pHierCC firstly calculates a minimum spanning tree (Fig. 1) based on a distance metric that minimizes the topological distortion due to missing genes (Supplementary Text). The resulting tree is used to assign every genome to clusters in multiple HCs. Subsequent assignments by pHierCC are performed in ‘production mode’. In production mode, new genomes that are equidistant to multiple clusters are automatically assigned to the oldest cluster (lowest cluster designation) in order to ensure the long-term stability of nomenclature designations (Supplementary Text).
Fig. 1.
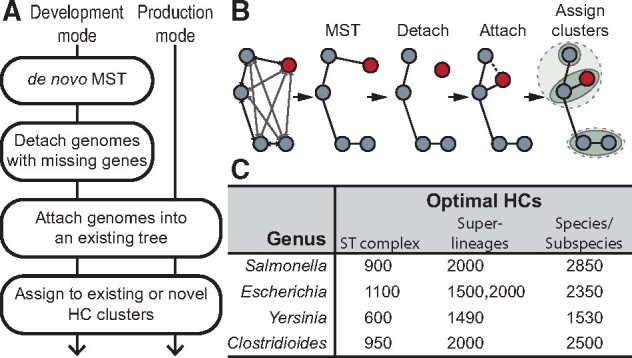
(A) The workflows of pHierCC in development or production mode. (B) Cartoon of the workflow in development mode. The node in red indicates a genome that carries numerous missing genes. (C) The optimal HC levels identified by HCCeval in four EnteroBase databases
3. Evaluation of optimal HCs with HCCeval
We developed an additional tool, HCCeval, to identify the optimal HCs that distinguish between natural, genetically discrete populations within a given set of STs and their corresponding HierCC assignments. HCCeval compares both normalized mutual information (NMI) (Kvalseth, 2017) and Silhouette scores (Rousseeuw, 1987) at each HC level with clustering at other levels as a measure of their relative stability (Supplementary Fig. S1). NMI measures the similarity of clustering by a given pair of hierCC levels as a harmonic mean of homogeneity and completeness between the two. HCCeval provides a heat map of these NMI scores in which sets of continuous HC levels that identify highly similar clusters (NMI ≥0.9) form visually recognizable blocks of stable HC clusters that do not change dramatically at slightly different thresholds of allelic differences. Silhouette evaluates cluster cohesiveness by comparing the internal pairwise similarities of each genome in a cluster with its similarities to genomes from other clusters. The greatest silhouette score for each stable NMI block is likely to correspond to an HC level that is optimal for identifying natural populations.
4. Implementation in EnteroBase
In 2018, EnteroBase calculated an initial set of HierCC HCs in development mode for representative genomes from Salmonella, Escherichia, Yersinia and Clostridioides. These representatives consisted of one genome per ribosomal MLST type, and their HCs were evaluated by HCCeval. Visual inspection of the results identified three to four stable blocks of HCs for each genus (Supplementary Fig. S1 and Frentrup et al., 2019). The highest HC blocks (HC1530–HC2850 depending on genus) distinguish subspecies or species (Fig. 1C) (http://enterobase.readthedocs.io/en/latest/HierCC_lookup.html). The lowest blocks (HC600–HC1100) identify ST complexes or comparable populations defined by 7-gene MLST schemes (Frentrup et al., 2019; Zhou et al., 2020). Additionally, EnteroBase publishes HC assignments for HC0, HC2, HC5, HC10, HC20, HC50, HC100, HC200 and HC400 even though these do not form blocks in the NMI comparisons. In 2020, HierCC schemes for Vibrio and Streptococcus were added, but their final evaluations are still in progress.
Experience has indicated that some infectious outbreaks (European Centre for Disease Prevention and Control and European Food and Safety Authority, 2020; Jones et al., 2019) or recent local transmissions (Frentrup et al., 2019; Zhou et al., 2020) are each associated with a cluster defined by one of these low diversity HC levels. However, our current experience indicates that clusters of genetically closely associated genomes may not necessarily represent traditional transmission chains because several have now been identified that have spread globally and continued to exist over decades. We therefore recommend that the final definition of a transmission chain continue to be based on epidemiological criteria in addition to genetic similarities. We also recommend that genetic analyses should be performed over a range of HCs in order to ensure that populations are truly distinct.
5. Conclusions
In this article, we introduce pHierCC, a pipeline that defines HierCC, a scalable, fine-grained, incremental clustering scheme for bacterial genomes based on their cgMLST allelic profiles. HierCC was integrated into EnteroBase in 2018, and pHierCC has currently assigned >530 000 genomes from Salmonella, Escherichia/Shigella, Streptococcus, Clostridioides, Vibrio and Yersinia into 12–13 multi-level clusters ranging from sub-clonal variation to species. pHierCC is also available as a stand-alone package that can be used for any bacterial genus with a cgMLST scheme and where large numbers of genomes are available for the initial assignments.
Supplementary Material
Acknowledgements
We gratefully acknowledge feedback for Salmonella HierCC scheme from Francois-Xavier Weill and María Pardos de la Gándara, Institut Pasteur, and the maintenance of EnteroBase by Khaled Mohamed.
Funding
This project was supported by the Wellcome Trust (202792/Z/16/Z).
Conflict of Interest: We have no conflict of interest to declare.
Contributor Information
Zhemin Zhou, Warwick Medical School, University of Warwick, Coventry CV4 7AL, UK.
Jane Charlesworth, Warwick Medical School, University of Warwick, Coventry CV4 7AL, UK.
Mark Achtman, Warwick Medical School, University of Warwick, Coventry CV4 7AL, UK.
References
- Coipan C.E. et al. (2020) Concordance of SNP- and allele-based typing workflows in the context of a large-scale international Salmonella Enteritidis outbreak investigation. Microb. Genom. 6:e000318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dallman T. et al. (2018) SnapperDB: a database solution for routine sequencing analysis of bacterial isolates. Bioinformatics, 34, 3028–3029. [DOI] [PubMed] [Google Scholar]
- European Centre for Disease Prevention and Control and European Food and Safety Authority. (2020) Multi-country outbreak of Salmonella Typhimurium and S. Anatum infections linked to Brazil nuts. EFSA Supporting Publications, October 21, 2020, 17, 1944E.
- Frentrup M. et al. (2019) A publicly accessible database for Clostridioides difficile genome sequences supports tracing of transmission chains and epidemics. Microb. Genom., 6, mgen000410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jolley K.A. et al. (2012) Resolution of a meningococcal disease outbreak from whole-genome sequence data with rapid web-based analysis methods. J. Clin. Microbiol., 50, 3046–3053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones G. et al. (2019) Outbreak of Salmonella enterica serotype Poona in infants linked to persistent Salmonella contamination in an infant formula manufacturing facility, France, August 2018 to February 2019. Euro. Surveill., 24, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kvalseth T.O. (2017) On normalized mutual information: measure derivations and properties. Entropy, 19, 631. [Google Scholar]
- Mellmann A. et al. (2011) Prospective genomic characterization of the German enterohemorrhagic Escherichia coli O104:H4 outbreak by rapid next generation sequencing technology. PLoS One, 6, e22751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morelli G. et al. (2010) Yersinia pestis genome sequencing identifies patterns of global phylogenetic diversity. Nat. Genet., 42, 1140–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moura A. et al. (2016) Whole genome-based population biology and epidemiological surveillance of Listeria monocytogenes. Nat. Microbiol., 2, 16185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousseeuw P.J. (1987) Silhouettes: a graphical aid to the interpretation and validation of cluster analysis. J. Comput. Appl. Math., 20, 53–65. [Google Scholar]
- Wong V.K. et al. ; International Typhoid Consortium. (2016) An extended genotyping framework for Salmonella enterica serovar Typhi, the cause of human typhoid. Nat. Commun., 7, 12827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z. et al. (2018) GrapeTree: visualization of core genomic relationships among 100,000 bacterial pathogens. Genome Res., 28, 1395–1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z. et al. ; the Agama Study Group. (2020) The EnteroBase user's guide, with case studies on Salmonella transmissions, Yersinia pestis phylogeny, and Escherichia core genomic diversity. Genome Res., 30, 138–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.