

Abstract
Summary
ProDy, an integrated application programming interface developed for modelling and analysing protein dynamics, has significantly evolved in recent years in response to the growing data and needs of the computational biology community. We present major developments that led to ProDy 2.0: (i) improved interfacing with databases and parsing new file formats, (ii) SignDy for signature dynamics of protein families, (iii) CryoDy for collective dynamics of supramolecular systems using cryo-EM density maps and (iv) essential site scanning analysis for identifying sites essential to modulating global dynamics.
Availability and implementation
ProDy is open-source and freely available under MIT License from https://github.com/prody/ProDy.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Proteins are dynamic entities. Their structural dynamics is essential to their myriad functions (Bahar et al., 2017). The ProDy application programming interface (API) in Python was introduced in 2011 to provide a unified environment for analyses of protein dynamics and mechanisms which lay the framework for their biological activities (Bakan et al., 2011). The API was upgraded in 2014 by adding a new module, Evol, to enable sequence evolutionary analysis complementing that of structural dynamics (Bakan et al., 2014). The original API featured functions and data structures for spectral mode decomposition and/or normal mode analysis (NMA) based on elastic network models [ENMs, including the Anisotropic Network Model (ANM) (Atilgan et al., 2001) and Gaussian Network Model (GNM) (Bahar et al., 1997)], and principal component analysis (PCA) of experimental structures, allowing users to evaluate and visualize structural dynamics, and make rigorous comparisons of motions derived from experiments and computations. The API has been significantly upgraded since then, and has found wide utility, evidenced by more than 2 million downloads from PyPI and 150 000+ unique website visits.
The current Application Note aims at providing a summary of recent updates. We focus here on three recent modules implemented in ProDy: evaluation of the signature dynamics of protein families (SignDy) (Zhang et al., 2019); characterization of the collective dynamics of supramolecular structures resolved by cryo-EM, using electron density maps as inputs to construct ENMs (Zhang et al., 2020); and essential site scanning analysis (ESSA) (Kaynak et al., 2020); along with general upgrades in ProDy core architecture, yielding a new generation of ProDy, 2.0.
1.1 Inputs and outputs: new file parsers and interfaces
The traditional input for ProDy is a PDB file, either provided by the user or retrieved from the Protein Data Bank (PDB) using an ID or sequence, and the output is structural dynamics. The outputs are various objects, relating to coordinates, sequences and alignments, ensembles and normal modes (Supplementary Fig. S1), as well as various plots facilitated by integration with numeric and scientific Python libraries, NumPy (Harris et al., 2020) and SciPy (Virtanen et al., 2020), and plotting library Matplotlib (Hunter, 2007), and the visualization tool NMWiz as a VMD plug-in (Bakan et al., 2014). Data-handling capabilities of ProDy have been significantly enhanced in version 2.0. For example, development of family-based analysis (in SignDy) led to integration with diverse databases and servers, enabling users to find similar structures upon inputting a single sequence or ID and calculate functional properties for the entire protein family. Other interfaces added include UniProt and QuartataWeb (Li et al., 2020) for drug-target interactions. New parsers include those for the PDBx/mmCIF format (Adams et al., 2019) and cryo-EM maps in MRC2014 format (Cheng et al., 2015) from the EMDataBank (Lawson et al., 2016). A new module, membrANM, was developed for analysing membrane proteins, where the membrane is represented by a disk-shaped elastic network (Fig. 1, lower left) (Lezon and Bahar, 2012), and the force exerted by the membrane network is incorporated into the Hessian of the protein through a system-environment framework (see Supplementary Text).
Fig. 1.
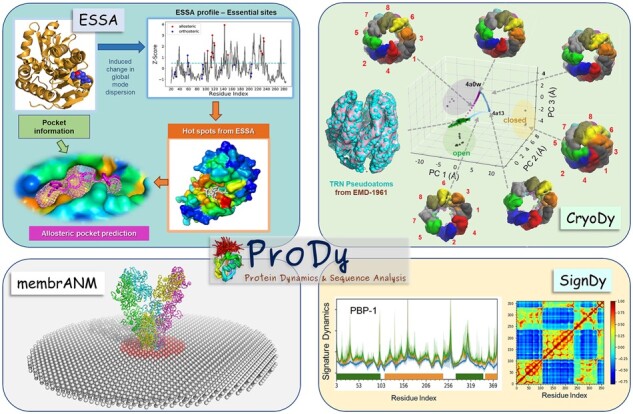
Illustration of four new modules implemented in ProDy 2.0. Results are presented (starting from the top left, clockwise) for: ESSA [applied to glutamate racemase (PDB: 2JFN), and β-lactamase (PDB: 1PZO)]; CryoDy (exploring the conformational space accessible to the mammalian chaperonin CCT/TRiC); SignDy (signature residue-fluctuations-profile and cross-correlations for PBP-1 domain family); and membrANM [constructed for a glutamate receptor (PDB: 3KG2)]
1.2 SignDy: signature dynamics of protein families
The SignDy module enables comparative analysis of the equilibrium dynamics of structural homologs and evaluation of their signature dynamics that often reflect their shared functional mechanisms (Zhang et al., 2019). The method is applicable to structural homologs that may share little sequence identity and/or exhibit functional diversity, as illustrated in Supplementary Figure S2 for 116 CATH superfamilies and the family of the periplasmic binding protein 1 (PBP-1) domains. The module evaluates the generic features shared by family members as well as specific features of subfamilies. This is made possible by (i) interfaces to various structural classification databases and servers for finding structure homologues (family members) given one input structure or ID; (ii) improved protein structure alignment protocols, including CEAlign (Shindyalov and Bourne, 1998) and automated chain matching procedures; (iii) optimal matching of normal modes accessible to family members; and (iv) comparative analyses using metrics such as covariance or mode-mode overlap. Residue fluctuation profiles and cross-correlations averaged over family members (Fig. 1, lower right) define the signature dynamics, and deviations from the means describe their differentiation among family members. SignDy permits generation of dendrograms to cluster family members by their dynamics.
1.3 CryoDy: dynamics of Cryo-EM resolved structures
CryoDy (Zhang et al., 2020) is designed to characterize the structural dynamics of cryo-EM resolved structures. It uses the topology-representing-network (TRN) algorithm to map electron densities associated with multiple residues to pseudo-atoms (ENM nodes), thus enabling efficient ENM-NMA and the use of low-resolution maps. The pipeline provides information on structural and dynamic properties, including allosteric signal propagation paths based on existing ProDy tools, and sampling of conformational landscapes through a new implementation of the adaptive ANM method, which works for both pseudo-atomic and atomic models (see Fig. 1, upper right). Its integration in ProDy permits a wealth of ENM-based analyses, in contrast to the powerful but more specialized tools in Scipion (de la Rosa-Trevin et al., 2016).
1.4 ESSA: essential site scanning analysis
ESSA (Kaynak et al., 2020) identifies essential residues, defined as those whose perturbation makes the highest impact (usually a shift to higher frequency) on the global modes intrinsically accessible to the system, being involved in biological activities (active or allosteric sites) or mechanical responses (hinges) (Supplementary Fig. S3a–c). ESSA identifies these residues by evaluating the effect of increased crowding near each residue on the frequency dispersion of ENM modes. The change in global mode dispersion is measured by z-scores, which represent the mean shift in the frequency of the softest modes after pairwise matching between the original and perturbed models. ESSA integrates information on pocket geometry and local hydrophobic density data (Song et al., 2017) from Fpocket (Le Guilloux et al., 2009 ) to provide an automated protocol for detecting allosteric pockets (Fig. 1, upper left and Supplementary Fig. S3d).
2 Conclusion
Over the years, ProDy has been closing the gap in protein dynamics evaluations between theory and experiments. By virtue of its modular, object-oriented design and integration with scientific computing libraries, ProDy lends itself to easy development, scalability and reproducibility. The features presented here extend its capabilities to analyse supramolecular systems resolved at low resolution (CryoDy), assess the conservation and differentiation of structural dynamics (SignDy), and identify essential sites that may impact the functional dynamics upon ligand binding (ESSA).
Supplementary Material
Acknowledgements
J.M.K. thanks the Molecular Sciences Software Institute (MolSSI) and particularly Dr Andrew Abi-Mansour for guidance during his fellowship.
Funding
This work was supported by the National Institutes of Health [P41GM103712 to I.B.] and the Molecular Sciences Software Institute [COVID-19 Seed Software Fellowship to J.K.].
Conflict of Interest: none declared.
Contributor Information
She Zhang, Department of Computational and Systems Biology, School of Medicine, University of Pittsburgh, Pittsburgh, PA 15213, USA.
James M Krieger, Department of Computational and Systems Biology, School of Medicine, University of Pittsburgh, Pittsburgh, PA 15213, USA.
Yan Zhang, Department of Computational and Systems Biology, School of Medicine, University of Pittsburgh, Pittsburgh, PA 15213, USA.
Cihan Kaya, Department of Computational and Systems Biology, School of Medicine, University of Pittsburgh, Pittsburgh, PA 15213, USA.
Burak Kaynak, Department of Computational and Systems Biology, School of Medicine, University of Pittsburgh, Pittsburgh, PA 15213, USA.
Karolina Mikulska-Ruminska, Department of Computational and Systems Biology, School of Medicine, University of Pittsburgh, Pittsburgh, PA 15213, USA.
Pemra Doruker, Department of Computational and Systems Biology, School of Medicine, University of Pittsburgh, Pittsburgh, PA 15213, USA.
Hongchun Li, Department of Computational and Systems Biology, School of Medicine, University of Pittsburgh, Pittsburgh, PA 15213, USA.
Ivet Bahar, Department of Computational and Systems Biology, School of Medicine, University of Pittsburgh, Pittsburgh, PA 15213, USA.
References
- Adams P.D. et al. (2019) Announcing mandatory submission of PDBx/mmCIF format files for crystallographic depositions to the Protein Data Bank (PDB). Acta Crystallogr. D Struct. Biol., 75, 451–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atilgan A.R. et al. (2001) Anisotropy of fluctuation dynamics of proteins with an elastic network model. Biophys. J., 80, 505–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahar I. et al. (1997) Direct evaluation of thermal fluctuations in proteins using a single-parameter harmonic potential. Folding Des., 2, 173–181. [DOI] [PubMed] [Google Scholar]
- Bahar I. et al. (2017) Protein Actions: Principles and Modeling. Garland Science, Abingdon. [Google Scholar]
- Bakan A. et al. (2014) Evol and ProDy for bridging protein sequence evolution and structural dynamics. Bioinformatics, 30, 2681–2683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakan A. et al. (2011) ProDy: protein dynamics inferred from theory and experiments. Bioinformatics, 27, 1575–1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng A. et al. (2015) MRC2014: extensions to the MRC format header for electron cryo-microscopy and tomography. J. Struct. Biol., 192, 146–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Rosa-Trevin J.M. et al. (2016) Scipion: a software framework toward integration, reproducibility and validation in 3D electron microscopy. J. Struct. Biol., 195, 93–99. [DOI] [PubMed] [Google Scholar]
- Harris C.R. et al. (2020) Array programming with NumPy. Nature, 585, 357–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter J.D. (2007) Matplotlib: a 2D graphics environment. Comput. Sci. Eng., 9, 90–95. [Google Scholar]
- Kaynak B.T. et al. (2020) Essential site scanning analysis: a new approach for detecting sites that modulate the dispersion of protein global motions. Comput. Struct. Biotechnol. J., 18, 1577–1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson C.L. et al. (2016) EMDataBank unified data resource for 3DEM. Nucleic Acids Res., 44, D396–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Guilloux V. et al. (2009) Fpocket: an open source platform for ligand pocket detection. BMC Bioinformatics, 10, 168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lezon T.R., Bahar I. (2012) Constraints imposed by the membrane selectively guide the alternating access dynamics of the glutamate transporter GltPh. Biophys. J., 102, 1331–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. et al. (2020) QuartataWeb: integrated chemical-protein-pathway mapping for polypharmacology and chemogenomics. Bioinformatics, 36, 3935–3937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- , Shindyalov I.N., Bourne P.E. (1998) Protein structure alignment by incremental combinatorial extension (CE) of the optimal path. Protein Eng., 11, 739–747. [DOI] [PubMed] [Google Scholar]
- Song K. et al. (2017) Improved method for the identification and validation of allosteric sites. J. Chem. Inf. Model., 57, 2358–2363. [DOI] [PubMed] [Google Scholar]
- Virtanen P. et al. ; SciPy 1.0 Contributors. (2020) SciPy 1.0: fundamental algorithms for scientific computing in Python. Nat. Methods, 17, 261–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S. et al. (2019) Shared signature dynamics tempered by local fluctuations enables fold adaptability and specificity. Mol. Biol. Evol., 36, 2053–2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y. et al. (2020) State-dependent sequential allostery exhibited by chaperonin TRiC/CCT revealed by network analysis of Cryo-EM maps. Prog. Biophys. Mol. Biol., 160, 104–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.