

Abstract
Recruitment of immune cells to a tumor is determined by the complex interplay between cellular and non-cellular components of the tumor microenvironment. Ex vivo platforms that enable identification of key components that promote immune cell recruitment to the tumor could advance the field significantly. Herein, we describe the development of a perfusable multi-cellular tumor-on-a-chip platform involving different cell populations. Cancer cells, monocytes, and endothelial cells were spatially confined within a gelatin hydrogel in a controlled manner by using 3D photopatterning. The migration of the encapsulated endothelial cells against a chemokine gradient created an endothelial layer around the constructs. Employing this platform, we examined the effect of cancer cell-monocyte interaction on T-cell recruitment, where T-cells were dispersed within the perfused media and allowed to infiltrate. The hypoxic environment in the spheroid cultures recruited more T-cells compared to dispersed cancer cells. Moreover, the addition of monocytes to the cancer cells improved T-cell recruitment. The differences in T-cell recruitment were associated with differences in chemokine secretion including chemokines influencing the permeability of the endothelial barrier. This proof-of-concept study shows how integration of microfabrication, microfluidics, and 3D cell culture systems could be used for the development of tumor-on-a-chip platforms involving heterotypic cells and their application in studying recruitment of cells by the tumor-associated microenvironment.
Keywords: Tumor-on-chip, cancer, monocytes, T-cells, immune cell trafficking
Introduction
In recent years, immunotherapies have gained considerable momentum as a potent cancer treatment by mobilizing patients’ immune cells or through transplantation of engineered cells to target and eliminate tumor cells (1-3). CAR T-cell therapy is one such approach where cytotoxic T-cells are armed with Chimeric Antigen Receptors (CAR) that have been shown to efficaciously treat leukemia (1,3-5). Tumor microenvironment which includes both cellular (e.g., immune and stromal cells) and non-cellular cues (e.g., hypoxia and extracellular matrix properties) plays a key role in T-cell infiltration into the tumor site (6-8).
Monocytes are a key cell population of the tumor associated microenvironment which can, depending on their phenotype, either attenuate or support cancer progression (9,10). While M1 macrophages promote the recruitment of cytotoxic T-cells which are shown to eliminate tumor cells, M2 macrophages have been linked to supporting cancer progression through secretion of growth factors such as TGF-β and immune suppressive cytokines like IL-10 (11-13). Moreover, M2 macrophages have been shown to enhance regulatory T-cell trafficking to the tumor environment (9,14-16). In most immunotherapies, the presence of cytotoxic over regulatory T-cells leads to better prognosis (17).
Besides the cellular composition, various extracellular cues like oxygen tension have been shown to alter the function of tumor and tumor-associated cells. Intra-tumoral hypoxia is a common characteristics of the tumor microenvironment and a number of studies have demonstrated the key role played by the hypoxia on tumor progression and prognosis (18). For instance, hypoxia has been shown to promote the secretion of various chemokines from cancer cells and tumor-associated immune cells that are capable of recruiting T-cells as well as monocytes to the tumor environment (19-21). Depending upon the different cell populations present in the tumor microenvironment hypoxia can also influence the function of cytotoxic T-cells (22,23). Thus, hypoxia can have tremendous implications on the recruitment of various cells to the tumor site as well as in determining the outcome of immunotherapies.
As such, a detailed and systematic understanding of the influence of tumor-associated microenvironment on T-cell infiltration is needed. While in vivo studies are invaluable to our understanding of tumor-microenvironment as well as tumor-immune interactions, deconvolution of the interdependency between various factors within the tumor-associated microenvironment is challenging in animal models. Emergence of “tumor-on-chip” technology provides an alternative to study multicellular and tumor-microenvironment interactions in vitro. Tumor-on-a-chip devices have been extensively used to study tumor-stroma interactions, tumor-associated angiogenesis, and recently, tumor-immune interactions (24-27). Herein, we describe the development of a multi-cellular tumor-on-a-chip platform and its potential application to study the effect of tumor microenvironment, especially presence of monocytes and hypoxia, on T-cell recruitment using TALL-104 cells as a model system. We adapted a 3D photopatterning technique to fabricate a bi-layer gelatin methacrylate (GelMA) hydrogel to spatially confine different cell populations (28). Specifically, cancer (breast cancer cells; MCF7 or MDA-MB-231) and immune (THP-1) cells were encapsulated within the inner layer of the bi-layer hydrogel with the outer layer loaded with endothelial cells (HUVECs). The ability of the endothelial cells within the outer layer to migrate towards the chemokine gradient generated by the circulating medium containing serum was used to create an endothelial layer at the periphery of the bi-layer hydrogel constructs (29). Tumor cultures with varying levels of hypoxia were created by using either cancer spheroids (CS) or dispersed cancer cells (DisC); an experimental design adapted based on the mass transfer model described in the results. The constructs were perfused with media containing T-cells (TALL-104) and their context-specific infiltration into the bi-layer constructs was studied as a function of time. Our results demonstrate that cultures involving cancer spheroids (CS) recruited significantly more T-cells compared to groups with dispersed cells (or small cell aggregates) (DisC). Moreover, the presence of monocytes synergistically increased T-cell recruitment into the tumor site.
Materials and Methods
Cell Culture
Human vascular endothelial cells (HUVECs), breast cancer cells (MCF7 and MDA-MB-231), monocytes (THP-1), and T-cells (TALL-104) were obtained from ATCC and cultured according to the manufacture’s protocol. HUVECs were cultured in endothelial medium comprised of 78% Medium 199 (Gibco), 10% fetal bovine serum (FBS) (Gibco), 10% endothelial cell growth medium (Cell Application, Inc.), 1% sodium pyruvate (Gibco), and 1% Penicillin/Streptomycin (P/S). The HUVECs were used below 6 passages. MCF7 and MDA-MB-231 cells were cultured in growth medium containing 89% Dulbecco Modified Eagle’s high glucose medium (DMEM), 10% FBS, and 1% P/S. THP-1 cells were cultured in monocyte medium (79% RPMI-1640 (Gibco) supplemented with 10% FBS, 1% P/S, and 5 μM 2-Mercaptoethanol). TALL-104 cells were cultured in T-cell medium (80% Iscove’s Modified Dulbecco’s Medium (ATCC) supplemented with 20% FBS, 75 units/mL recombinant human IL-2 (Peprotech), 2.5 μg/mL human albumin (Sigma-Aldrich), and 0.5 μg/mL D-Mannitrol (Sigma-Aldrich)). GelMA Hydrogels loaded with HUVECs, MCF7 or MDA-MB-231, and THP-1 were cultured in a mixed medium comprising of 50% endothelial medium and 50% monocyte medium.
Spheroid formation of MCF and MDA-MB-231 cells
MCF7 cells were expanded, trypsinized, and 1 million cells were re-suspended in 4.5 mL of growth medium. To generate the spheroids, MCF7 cell suspension in a 60 mm diameter petri dish was placed on an orbital shaker at 45 rpm and cultured in an incubator at 37 °C and 5% CO2. The cell suspension was cultured for 2 days and spheroids of ~140 μm in diameter were used for the experiments.
For making MDA-MB-231 spheroids, 96 well U-bottom Ultralow attachment plates (CellStar) were coated with 10% BSA overnight. 200 cells were seeded per well and the plate was centrifuged at 2000 rpm for 10 minutes. 3.5% Matrigel (Corning) was added to each well to promote spheroid formation, which is needed as MDA-MB-231 cells lack the ability to form spheroids spontaneously. Spheroids of sizes between 100-150 μm formed in 3-4 days post seeding were used for the experiments.
Preparation of GelMA hydrogel precursor solution
Varying amounts of GelMA were dissolved in PBS to make 7, 8.5, or 10% wt/v solutions to fabricate hydrogels with different stiffnesses (i.e., Young’s modulus). To ensure complete dissolution of GelMA, the solution was incubated at 60°C for 20 minutes. The solution was filter-sterilized by using a syringe filter of 0.22 μm pore size and was kept at 37 °C until use. The corresponding GelMA solution was mixed with 0.01% wt/v ascorbic acid and 2.0 μM LAP prior to photopolymerization.
Characterization of GelMA hydrogels
Cylindrical GelMA hydrogels (5-mm diameter and 4-mm height) of different stiffnesses were created by varying the precursor concentration and exposing the solution to 365 nm wavelength UV light for 5 minutes to polymerize the hydrogel (30). The Young’s moduli of the resulting hydrogels were measured under compressive loading on Instron 3342 Universal Testing machine at a strain rate of 7 mm/min. The linear low strain region of the stress-strain curve was used to estimate the Young’s modulus of the hydrogels.
Fabrication of tumor-on-a-chip device by additive photopatterning
We have previously described the fabrication of a tumor-on-a-chip microfluidic device containing cancer cells encapsulated within a GelMA hydrogel encased by HUVECs layer (29). In brief, the steps involved in the device fabrication can be categorized into: (i) methacrylation of glass, (ii) fabrication of microfluidics device containing polyacrylamide (PAm) hydrogels, and (iii) photopatterning of cell-laden GelMA hydrogels within the device. Supplementary Figure S1 provides a general schematic of the fabrication process.
Methacrylation of glass surfaces:
25 mm x 50 mm rectangular and 15 mm diameter circular glass coverslips were cleaned by using 1.5 M NaOH for 30 minutes and rinsed with DI water. The coverslips were air dried and incubated with 2% (v/v) 3-(trimethoxysilyl) propyl methacrylate solution diluted in 0.5% glacial acetic acid and 99.5% ethanol for 5 minutes. Upon completion of the reaction, the glass surfaces were rinsed with pure ethanol and dried at 60 °C for 30 minutes. The coverslips were used immediately (Fig. S1A).
Fabrication of microfluidic device containing PAm hydrogels:
PAm hydrogels were formed on methacrylated glass surfaces by sandwiching 1.75 μL of the precursor solution (5% wt/v acrylamide, 0.2% wt/v bis-acrylamide, 0.1% wt/v ammonium persulfate, and 0.01% wt/v N,N,N′,N′-tetramethylethylenediamine) between a methacrylated and a non-methacrylated coverslip as described earlier (31). Specifically, the precursor solution was spotted on the center of a methacrylated rectangular coverslip and a non-methacrylated 15 mm diameter coverslip was placed on top of the droplet. This procedure was repeated for the methacrylated circular and the non-methacrylated rectangular coverslips to generate the top layer of the device (Fig. S1A). After 30 minutes of polymerization, the hydrogel sandwiched between the methacrylated and non-methacrylated coverslips were incubated in DI water for 20 minutes and the non-methacrylated coverslips were removed carefully. The hydrogels attached to the methacrylated coverslips were equilibrated overnight in DI water prior to use (Fig. S1A).
The hydrogel tethered coverslip was incorporated into the microfluidic chamber. Towards this, 5 μL of DI water was added to silicon wafer encoded with the flow pattern and the hydrogel tethered coverslip was placed onto the droplet. PDMS solution made from a degassed mixture of Sylgard 184 base and catalyst at a weight ratio of 10:1, was carefully poured onto the silicon wafer until the entire surface was covered. The silicon wafer containing the PAm hydrogel was baked at 60 °C for 2 hours before removing the polymerized PDMS mold with the attached hydrogel (Fig. S1B). The rectangular coverslip and PDMS mold containing the PAm hydrogels were placed in an UV-Ozone chamber under oxygen flow for 5 minutes (Fig. S1C). Care was taken to prevent direct exposure of the PAm hydrogels to deep UV light. After the UV-Ozone treatment, the rectangular coverslip and PDMS mold were bonded together by bringing them into contact while maintaining the alignment between the PAm hydrogels tethered to their surfaces. The microfluidics device was placed in an oven at 60 °C for overnight to complete the bonding (Fig. S1D). The device containing PAm hydrogels was equilibrated in PBS and the flow chamber was UV-sterilized for 45 minutes prior to the cell encapsulation.
Photopatterning of cell-laden GelMA hydrogels within the fluidics device:
To co-culture different cell types, we used an additive photopatterning to encapsulate monocytes, cancer cells, and HUVEC cells within a bi-layer hydrogel (monocytes and cancer cells were encapsulated within the inner layer while the HUVECs were encapsulated within the outer layer) (28). Briefly, 1 mL of monocyte medium containing 5 million THP-1 cells were mixed with 1 mL of growth medium containing MCF7 or MDA-MB-231 cells (CS or DisC). The cell suspension was centrifuged and resuspended in 7, 8.5, or 10% wt/v GelMA solution containing 0.01% wt/v ascorbic acid and 2.0 μM LAP before flowing the solution into the device. The device was placed on a microscope stage mounted with a transparency film containing a 350 μm diameter circular pattern and individual cancer spheroids surrounded by fluorescently labeled THP-1 cells were located. This region was exposed to UV light (365 nm ± 15 nm wavelength) for 20 seconds to form GelMA hydrogels embedded with the selected cells (Fig. S1E). For encapsulation of dispersed cancer cells, locations with cancer cells along with THP-1 cells were identified and photopolymerized. The unreacted solution was removed by flushing with PBS containing 5% P/S. The cell-laden hydrogels were maintained in PBS with 5% P/S for 5 minutes before flowing with a GelMA precursor solution (with 0.01% wt/v ascorbic acid, and 2.0 μM LAP) containing 2 million HUVECs. The device was placed on a transparency film containing a 1000 μm diameter circular pattern and mounted onto a microscope. The GelMA hydrogel containing MCF7 or MDA-MB-231 cells and monocytes were located and positioned to be at the center of the circular pattern prior to exposing the region to UV light for 20 seconds to form the second (i.e. outer) layer embedded with the HUVECs. Unreacted solution was removed with PBS washing. The cell-laden hydrogels within the microfluidics device were perfused with mixed media at a flow rate of 50 μL/hr and maintained in a humidified incubator at 37°C and 5% CO2 (Fig. S1F).
Mass transfer model for hydrogels containing cell aggregates with different sizes
The concentration profile of an arbitrary solute within the cell-laden hydrogel was computed by a 2D diffusion equation in COMSOL Version 4.2. The construct (single layer) was approximated as circle with a diameter of 1000 μm. Within the construct, a large cancer spheroid (140 μm radius) or small cell aggregates (70, 47, or 35 μm radius) were modeled as regions where solute consumption occurs. The total area for a cancer spheroid or small cell aggregates was kept constant at 15,386 μm2 to isolate the effect of spatial distribution of cells on solute availability within the GelMA hydrogel.
The concentration profile for an arbitrary solute within the construct containing cells is governed by the diffusion equation with a convective boundary condition and nutrient consumption (Fig. 3A, Eq. 1).
Figure 3: Mass transfer analyses of hydrogels containing cancer cell aggregates of varying sizes.

(A) Schematic of the mass transfer model illustrating the domain geometry, boundary conditions, and position of the cancer cell aggregates. (B) Profile of diffusion coefficient within a cell aggregate (D;) along its diameter for hydrogels containing different number of cell clusters. For hydrogels with 1, 4, 9, and 16 clusters, the radius of each cluster is 140, 70, 47, and 35 μm, respectively. The x-axis and y-axis denote the locations along the aggregate diameter in microns and the corresponding value of the diffusion coefficient, respectively. Heat map of the diffusion coefficient (C) and normalized concentration of solute at steady state (D) within the GelMA hydrogel containing cancer cell aggregates at specified sizes. The x- and y- axis denotes the spatial positions in microns within the hydrogel. (E) The steady state normalized concentration at the center of the innermost aggregate as a function of ϕ and aggregate size. ϕ is defined as the ratio of diffusion to consumption rate of a solute.
| (Eq. 1) |
D is the diffusion coefficient of an arbitrary solute and is assumed to be spatially variant. The diffusion coefficient within the hydrogel, DG, was approximated to be 10 μm2/s, whereas the coefficient within the cell aggregates, DA, is described by an inverted Gaussian function (32) (Eq. 2).
| (Eq. 2) |
In this equation, xc and yc are the centroids of a cell aggregate and σA is the variance of the inverted Gaussian function. The cell aggregates were assumed to have high and low diffusivity at the periphery and center, respectively, to reflect physiological observations found in tumors (33).
In Equation 1, the consumption rate is described by (kcat/KM)E0C which is the linearized form of the Michaelis-Menten equation where kcat is the catalytic coefficient, KM is the Michaelis constant, and E0 is the enzyme concentration. Here, the catalytic efficiency (kcat/KM) and enzyme concentrations were estimated to be 0.1 μM and 10 mM/min based on values reported in literature (29,34-36). This linear approximation was used by assuming that the nutrients are rapidly consumed such that its concentration is lower than the Michaelis constant. Furthermore, the consumption of nutrients was confined to the regions where the cell aggregates were present and its total area was conserved irrespective of the number of aggregates.
At the boundary of the construct, Equation 1 is governed by a convective boundary condition due to perfusion of media into the device (Eq. 3).
| (Eq. 3) |
| (Eq. 4) |
| (Eq. 5) |
Here, h is the mass transfer coefficient estimated from the laminar flow over a plate, Sc is Schmidt number, and Re is the Reynolds number. Using these equations, the normalized concentration profile for an arbitrary solute at steady state was generated.
Hypoxia detection within tumor-on-chip device
Hypoxia within the cancer spheroids or dispersed cells were detected using ROS-ID Hypoxia/Oxidative stress detection kit (Enzo Life Sciences). After 2 days of culture, MCF7 cell-laden GelMA hydrogels were incubated in 0.5 μM Hypoxia Red Dye (diluted in mixed culture media (50% HUVEC and 50% monocyte media)) for one hour at 37°C and 5% CO2. To remove the unreacted dye, devices were incubated and flushed with PBS for 30 minutes prior to imaging the cells with 561 nm wavelength laser and Texas-red emission filter. Mean fluorescence intensity of the hypoxia red dye was quantified by averaging the dye signal intensity over the total cancer cell area within the inner compartment of the bi-layered hydrogel.
TALL-104 cell infiltration
The cell-laden GelMA hydrogels were cultured within the microfluidics device for 4-6 days, which was determined by the endothelial barrier formation. Six million TALL-104 cells were suspended in 100 μL of mixed media, perfused into the device, and incubated at 37 °C and 5% CO2 for 40 minutes. Excess T-cells which were not adhered to the periphery of the bi-layer constructs were removed by washing with PBS. The cell-laden constructs exposed to the T-cells were cultured for 2 days (experimental time used to examine infiltration of the T-cells).
Analysis of TALL-104 cell distribution within cell-laden hydrogels
Fluorescent images of the cell-laden constructs infiltrated with fluorescently labeled TALL-104 cells at DayT0 through DayT2 were analyzed and processed using a custom MATLAB code. In brief, the circular cross section of the hydrogel was partitioned into 7 annular sections of equal spacing as indicated in the inset of Figure 5C. The number of cells within each section was counted and normalized by the total number of cells within the construct. The cell fraction within each section was plotted against the position of the midpoint in each annular region normalized to the radius of the entire construct. To obtain the value for the Distribution of T-cells (DoT), a custom Matlab algorithm was used. The algorithm binarizes the fluorescent images of the TALL cells within the bi-layer hydrogel obtained at DayT 2 and calculates the centroids of individual cell outlines. The squared distance (), defined as the squared distance between the centroid of a particular cell to the center of the bi-layer hydrogel, was calculated for each individual cell. The DoT value was calculated by summing the squared distance for all the cells divided by the total number of TALL cells found attached to or within the bi-layer hydrogel. The above calculations can be mathematically summarized as , where i is the individual cell and n is the total number of cells found attached to or within the bi-layer hydrogel. Here, was used as a product to normalize the slight variations in TALL cell numbers found attached to or within the bi-layer hydrogels under different culture conditions. Larger DoT values indicate that the TALL cells were found at the periphery of the bi-layer hydrogel thus the culture condition did not effectively recruit these cells.
Figure 5: TALL-104 cell infiltration into bi-layer GelMA hydrogels laden with cancer cells (MCF7), monocytes (THP-1), and endothelial cells.

(A, DayT0 Panel) Merged and fluorescence images of T-cells adhered onto the periphery of a bi-layer GelMA hydrogel containing a cancer spheroid, monocytes, and endothelial cells (CS + Mo) immediately after introducing T-cells. (A, DayT 2 Panel) Fluorescence image of the cell-laden construct two days post-infiltration (DayT 2 wherein “C” designates the center of the hydrogel. Monocytes and T-cells are fluorescently labeled by green and red dyes, respectively. Scale bar: 200 μm. (B) Brightfield and fluorescence image of the bi-layer hydrogels with cancer spheroids (CS), dispersed cancer cells and monocytes (DisC + Mo), dispersed cancer cells (DisC), and monocytes (Mo) within the interior of the hydrogel at DayT 2. (C-G) Fraction of TALL-104 cells residing within each annular region, shown in the inset, from DayT 0 to DayT 2 for hydrogels containing CS + Mo, CS, DisC + Mo, DisC, and Mo. The normalized radial position denotes the radial midpoint location in each annulus. The sample sizes for CS+Mo, CS, DisC+Mo, DisC, and Mo are 4, 5, 5, 4, and 5, respectively. (H) Quantification of the Distribution of T-cells (DoT) within the bi-layer hydrogels for different cultures. Lower and higher DoT values indicate the distribution of T-cells towards the center and periphery, respectively.
Flow cytometric characterization of the THP-1 cells
Given the reports that the monocytes can be polarized in the presence of cancer cells, we have characterized the THP-1 cells to identify their alterations based on the culture conditions (37). MCF7 cancer cells (CS or DisC) and THP-1 monocytes cultured for three days were isolated and resuspended in PBS containing Live/Dead fixable Aqua Dead Cell stain (Thermofisher Scientific) at 1:1000 dilution for 10 minutes at room temperature. The conjugation of the Live/Dead dye was quenched by the addition of FACS buffer comprised of 1% BSA and 1 mM EDTA in PBS. The stained cells were washed twice in FACS buffer, centrifuged, and the supernatant was aspirated out. The cells were incubated in Human TruStain FcX (Biolegend) at a dilution of 1:100 in FACS buffer for ten minutes on ice and stained for a battery of markers – anti-human CD45 PE-Cy7 (Clone HI30, Biolegend), anti-human CD68 PE (Clone Y1/82A, Biolegend), anti-human CD86 BV785 (Clone IT2.2, Biolegend), and anti-human CD206 FITC (Clone 15-2, Biolegend). The cells were incubated with the respective antibody, at 1:75 dilution in FACS buffer, on ice for 20 minutes followed by quenching by washing with FACS buffer and analyzed. The THP-1 cells were differentiated into M1 and M2 macrophages following the protocols reported by Genin et al and used as controls (38). For M1 macrophage differentiation, THP-1 cells were incubated for 24 hours in 20 ng/mL IFN-γ (Peprotech) and 10 pg/mL LPS (Sigma Aldrich). For M2 macrophage differentiation, THP-1 cells were exposed to 20 ng/mL IL-4 (Peprotech) and 20 ng/mL IL-13 (Peprotech) for 24 hours.
Chemokine analysis from culture media
MCF7 cancer cells or spheroids were seeded within 12 well plates and cultured in the presence or absence of THP-1 monocytes which were plated into transwell inserts with pore sizes of 0.4 μm. For dispersed cancer cells, a total of 100,000 cells was used and for cancer spheroids, 1/10th of the volume containing cancer spheroids generated from 1 million cancer cells were added to each well. After 3 days of culture, the conditioned media was collected and assayed. To detect the chemokines, Legendplex kit for Human Proinflammatory Chemokine Panel 13-plex (Biolegend) was used according to the manufacturer’s protocol. In brief, the 25 μL of assay buffer, conditioned media, mixed beads, and detection antibodies were added to wells within a V-bottom plate and incubated for 2 hours at room temperature. 25 μL of streptavidin conjugated with phycoerythrin was added to the wells and incubated for an additional 30 minutes. To remove unbound reagents, the V-bottom plate was centrifuged and the supernatant was removed. The beads were resuspended in 200 μL of wash buffer. The quantity of bound analytes was examined through PE intensities using BD LSR Fortessa HTS-2 according to the manufacturer’s instructions.
Statistics
In all experiments, analysis for statistically significant differences between multiple conditions were carried out using one-way ANOVA test with Tukey’s post-hoc test. All bar plots represent the mean value with standard deviation as error bars. Sample sizes are indicated for each experiment within the figure captions for each condition. * and ** indicate statistically significant differences with P < 0.05 and 0.01 between samples designated by • and ➔, respectively. All experiments were independently reproduced at least thrice.
Results
Additive 3D photopatterning for spatial organization and compartmentalization of different cell population towards heterotypic co-cultures
To co-culture different cell types, we have used an additive 3D photopatterning approach, which enabled the formation of cell-laden bi-layer hydrogels. An acellular hydrogel system was used initially to assess the ability of the photopatterning approach to achieve compartmentalized encapsulation of different entities. Towards this, fluorescent particles (0.2 μm) with different emission spectra were encapsulated within an inner hydrogel layer of 350 μm diameter surrounded by an outer hydrogel layer of 650 μm; this results in a bi-layer hydrogel of 1000 μm diameter. This bi-layer hydrogel was gelled between two PAm hydrogels present at the top and bottom of the fluidic device. The X-Z section of the acellular construct showed the inner compartment loaded with red particles surrounded by an outer layer containing green particles (Fig. 1A). Furthermore, the X-Y confocal sections at different Z-positions displayed the PAm-GelMA interface near the top and bottom of the flow chamber (Fig. 1A). The PAm hydrogel layers were incorporated to prevent the migration of cells away from the GelMA hydrogel thereby preserving the spatial organization of the cell-laden hydrogel structures.
Figure 1: Spatially confined co-culture of cancer spheroid, monocytes and endothelial cells.

(A) XZ confocal section of a bi-layer GelMA hydrogel comprised of two concentric cylinders sandwiched by PAm hydrogels at the top and bottom surfaces. The interior and exterior GelMA hydrogels are labeled with red and green fluorescent particles, respectively, while the PAm hydrogels are with magenta fluorescent particles. White arrows and labels Z1, Z2, and Z3 in the XZ section indicate the vertical positions of the XY sections. Vertical Scale bar: 20 μm. Horizontal Scale bar: 50 μm. (B) Brightfield and fluorescence images of monocytes (green), MCF7 cancer spheroid, and endothelial cells embedded in bi-layer GelMA hydrogels immediately after encapsulation (Day 0) and two days in culture (Day 2) with outer layers of varying rigidities, 6.5 and 13 kPa respectively. The boundary of the interior compartment and periphery of the bi-layer hydrogel are marked by red and blue lines, respectively. Scale bar: 200 μm. (C) XZ and XY confocal sections of the interior compartment of a bi-layer hydrogel containing a MCF7 cancer spheroid (green) and THP-1 monocytes (red) cultured for 4 days. The dashed white circle indicates the boundary of the inner gel compartment. Vertical Scale bar: 20 μm. Horizontal Scale bar: 50 μm. The fraction of monocytes retained (D) and change in the cancer spheroid size within the in the inner hydrogel layer (E) as a function of culture time with outer hydrogel layer of varying stiffnesses.
Tumor-on-a-chip involving heterotypic cells
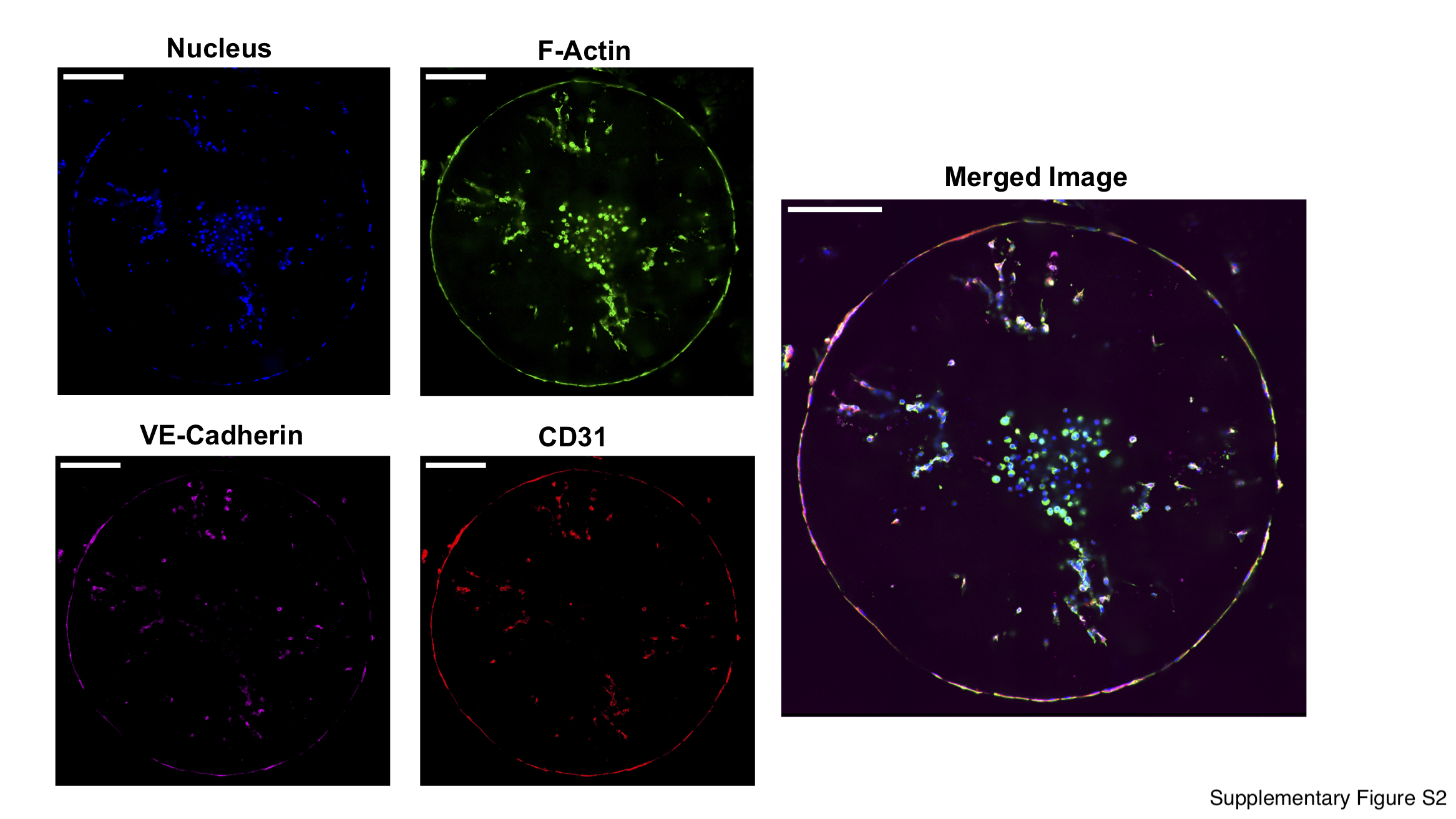
A multi-cellular tumor-on-a-chip co-culture system containing cancer cells (MCF7 or MDA-MB-231), monocytes (THP-1), and endothelial cells (HUVECs) was developed. We first created a tumor-on-a-chip model containing MCF7 tumor spheroids, monocytes, and endothelial cells to investigate the effect of matrix stiffness on the function of the encapsulated cells. We encapsulated cancer spheroids of ~140 μm radius along with monocytes within the inner compartment of the bi-layer hydrogel and endothelial cells within the outer compartment (Fig. 1B). Within two days post-encapsulation, the endothelial cells from the outer layer migrated to the periphery of the construct and self-assembled to form an endothelial layer (Fig. 1B). Characterization of the endothelial layer for F-actin, CD31, VE-Cadherin, and nuclei revealed the cell layer to be confined along the periphery of the constructs, akin to our prior report, as well as a few isolated cells within the hydrogel (Fig. S2). Previously, we have shown that the flow rate of the perfused media with serum can be tuned to induce migration and formation of an endothelial layer at the periphery of cell-laden GelMA hydrogels (29).
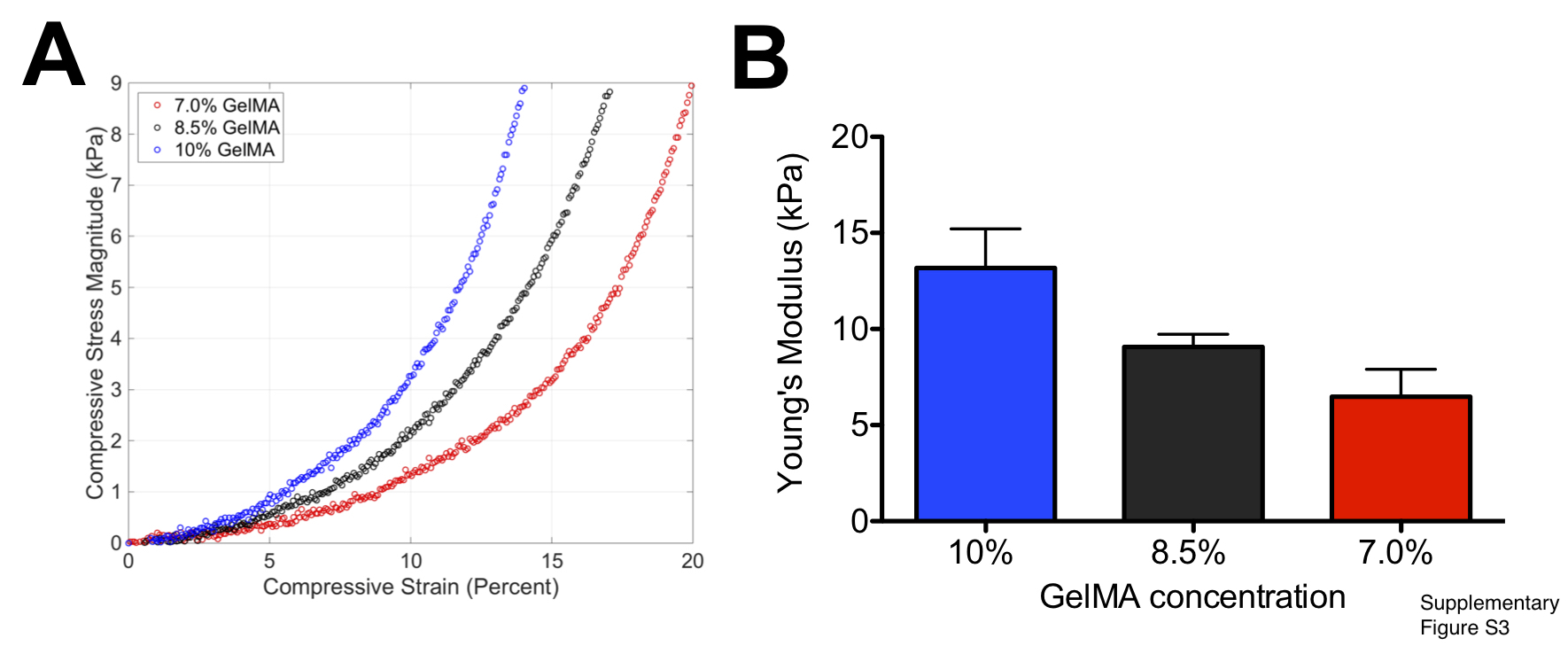
Employing the cell-laden bi-layer hydrogel, we examined the effect of mechanical rigidity of the exterior hydrogel on the encapsulated cells, where the Young’s modulus of the hydrogel was varied through the concentration of the GelMA precursor. Varying the concentration of the GelMA content from 7 to 10% wt/v, hydrogels with Young’s moduli of 6.5, 9, and 13 kPa (Fig. S3A, B) were generated for the outer layer of the bi-layer hydrogel. The Young’s modulus of the inner hydrogel layer was kept constant at 6.5 kPa. This low rigidity range was chosen such that the gels remain sufficiently soft enough to promote cancer cell growth while maintaining the structural integrity of the scaffold for the duration of the culture time. Brightfield images of endothelial cells encapsulated within the GelMA hydrogels showed migration of endothelial cells and formation of an endothelial layer at the periphery after two days of culture irrespective of the stiffness of the outer GelMA layer (Fig. 1B).
Similarly, irrespective of the stiffness of the outer layer, a substantial number of monocytes remained within the interior layer of the bi-layer hydrogel throughout culture time (Fig. 1B). Furthermore, confocal sections after 4 days of culture showed monocytes (red) at the periphery of the cancer spheroid (green), which were confined within the inner layer of the bi-layer hydrogel (Fig. 1C). Using these confocal images, a 3-D image of the cancer spheroid and monocytes was constructed (Supplementary Movie 1). Quantification of the number of monocytes suggests ~80% of the cells remained in the inner compartment of the bi-layer construct irrespective of the stiffness of the outer hydrogel layer (Fig. 1D). Although the stiffness of the outer layer did not have any significant effect on the endothelial cells and monocytes, it affected the growth of the cancer spheroids. Analyses following 4 days showed that bi-layer constructs where the exterior layers were of low rigidity (6.5 kPa) promoted the growth of cancer spheroids compared to those with higher stiffnesses (Fig. 1E). Hence, for all subsequent studies, GelMA hydrogels with a Young’s modulus of 6.5 kPa were used to create both the inner and outer layers of the bi-layer hydrogel. We have also compared the bi-layer hydrogels against that of a corresponding monolayer hydrogel gelled between two PAm hydrogels at the top and bottom surfaces of the fluidic chamber (Fig. S4A). When a single layer hydrogel was used, monocytes along with the endothelial cells migrated to the periphery of the construct and intravasated into the perfusing media irrespective of the presence of a cancer spheroid (Fig. S4B, C). This is evident by the decrease in monocyte and endothelial cell number in the interior of the hydrogel at 1 and 2 days post-encapsulation. Moreover, a higher density of cells was observed near the periphery of the hydrogel compared to its center (Fig. S4D, E). Thus, a bi-layer structure is needed to ensure the confinement of respective cells and their spatial distribution.
To create a tumor model with densely packed but dispersed cells, a mixture of cancer cells and fluorescently labeled monocytes was encapsulated within the inner compartment of the bi-layer hydrogel. Confocal sections along the XY and XZ planes show a homogeneous distribution of cancer cells (green) and monocytes (red) within the inner compartment (Fig. 2A); a reconstructed 3-dimensional image of the co-culture system is shown in Supplementary Movie 2 and a magnified image of the dispersed cancer cells within the hydrogel construct is shown in Supplementary Figure S5. Time course brightfield and fluorescent images of the encapsulated cells indicate that monocytes remained within the inner compartment among the cancer cells even after 4 days of culture (Fig. 2B). The endothelial cells encapsulated within the outer compartment migrated towards the periphery and assembled to form an endothelial layer (Fig. 2B). Additionally, we assessed the retention of cancer cells and monocytes within the GelMA hydrogel by using fluorescently labeled cells. Throughout culture time, the majority of monocytes and cancer cells were found to remain within the inner compartment of the bi-layer hydrogel (Fig. 2C). In the case of dispersed cancer cells, they were found to form small aggregates of 2-5 cells by Day 4 post-encapsulation (Fig. S5). Besides the growth pattern, the viability of the encapsulated cells was confirmed by live/dead staining, which showed ~84. 2 ± 5.6 % cells to be alive four days post-encapsulation.
Figure 2: Co-culture involving dispersed cancer cells (MCF7), monocytes (THP-1), and endothelial cells.

(A) Confocal sections of dispersed cancer cells (green) and monocytes (red) in the interior of the bi-layer GelMA hydrogel at Day 0. XZ section along with XY section at vertical positions (Z1-Z3) show homogeneously distributed cancer cells and monocytes. The dashed white circle indicates the boundary of the inner gel compartment. Vertical scale bar: 20 μm. Horizontal scale bar: 50 μm. (B) Time course brightfield and fluorescent images of cell-laden bi-layer GelMA hydrogel. The interior hydrogel contains a mixture of cancer cells and monocytes (green) while the exterior contains endothelial cells. The boundaries of the interior and exterior hydrogels are lined with red and blue circles, respectively. Scale bar: 200 μm. (C) Quantification of normalized monocyte and cancer cell numbers within the GelMA hydrogel as a function of culture time.
Solute gradient within different tumor-on-a-chip cultures
We used a mass transfer model to determine whether the two encapsulations (spheroid vs. dispersed cells) lead to different nutrient gradients within the cell mass and the effect of cell aggregate size on the gradient (Fig. 3A). Towards this, we assessed how the concentration of an arbitrary solute at the center of the construct differs between a spheroid (140 μm radius) and small cell aggregates (70, 47, and 35 μm radii). The spatial profile of the diffusion coefficient within the cell aggregates, DA, for different sizes of cell aggregates is plotted in Figure 3B and the corresponding 2D heatmap is illustrated in Figure 3C. The normalized concentration profile for an arbitrary solute at steady state is shown in Fig. 3D. The results indicate that decreasing cell aggregate size from 140 to 35 μm in diameter leads to increased nutrient availability at the interior of the construct. To generalize these findings towards more solutes, the ϕ parameter, defined as the ratio of the diffusion rate, DA/Aaag, to consumption rate, (kcat/KM)E0, within a cancer cell aggregate was used. Aagg denotes the area of an aggregate obtained from multiplying its perimeter by the height of the chamber. To account for solutes of varying diffusivity and consumption rate, we varied ϕ from 1 to 100 and estimated the solute concentration at the center for all cultures. The results showed size-dependent solute concentration at the center of the construct where lower concentration of solute was found in hydrogels with large spheroids compared to those with small cell aggregates (Fig. 3E). These findings along with the fact that the total area of cell aggregates was maintained constant irrespective of their number suggest that aggregate packing influences the gradient generation more than the number of aggregates. This further implies that spheroids will encounter a significantly higher oxygen gradient compared to dispersed cells even with higher cell numbers.
Cancer spheroid and dispersed cancer cells experience differential hypoxia
Results from the numerical analyses were verified by examining the concentration of oxygen within the different cultures. Varying extent of hypoxia within the bi-layer constructs containing cancer spheroid (CS), dispersed cancer cells (DisC), cancer spheroid with monocytes (CS + Mo), and dispersed cancer cells with monocytes (DisC + Mo) was examined by using a hypoxia detecting fluorescent dye. The brightfield and fluorescent images of cell-laden hydrogels are shown in Figure 4A and 4B. Monocytes were labeled with a green fluorescent dye while the intensity of the red dye corresponds to hypoxia. The fluorescent intensity plot in the direction of the white arrow in Figure 4B show higher fluorescence signal corresponding to higher levels of hypoxia in cultures containing tumor spheroids (Fig. 4C). These observations are quantified in Figure 4D by calculating the mean fluorescent intensities of the hypoxia dye within various culture conditions.
Figure 4: Culture condition-dependent hypoxia in GelMA hydrogels containing cancer cells (MCF7) and monocytes (THP-1) after 2 days of culture.

(A) Brightfield images of GelMA hydrogels containing a cancer spheroid (CS), dispersed cancer cells (DisC), cancer spheroid with monocytes (CS + Mo), and dispersed cancer cells with monocytes (DisC + Mo). (B) Fluorescent image of hypoxia detection dye within encapsulated cells. Increased intensity of red fluorescence denotes more hypoxia experienced by the cells. Monocytes within the hydrogels are labeled with green fluorescent dye. (C) Line profile of red fluorescence intensity along the midline of the fluorescent images, which is indicated by the white dashed line in the fluorescent image containing CS. (D) Mean fluorescence intensity within different cultures. Scale bar: 200 μm.
Monocytes and cancer spheroids synergistically recruit T-cells into the cell-laden GelMA hydrogel
We next determined the effect of culture condition-dependent T-cell recruitment to different cultures— CS + Mo, CS, DisC + Mo, DisC, and Mo. The TALL-104 cells were allowed to extravasate from the surrounding media into cultures and their infiltration to the cell-laden hydrogels was examined as a function of time. The brightfield images of the cultures containing a cancer spheroid and monocytes immediately after T-cell infiltration assay (DayT 0) showed the cancer spheroid at the center of the construct surrounded by numerous single monocytes and an endothelial layer at the periphery of the construct (Fig. 5A). Fluorescent images at DayT 0 indicate the presence of T-cells, labeled red, at the hydrogel periphery and monocytes, labeled green, near the center of the GelMA hydrogel (Fig. 5A, DayT 0 Panel). Fluorescence imaging of the constructs following two days of infiltration (DayT 2) reveals high distribution of T-cells (red) throughout the interior of the hydrogel (Fig. 5A, DayT 2 Panel). The corresponding magnified fluorescent image of the center of the hydrogel (designated by “C”) showed a large number of T-cells near the cancer spheroid and amongst the monocytes (green) (Fig. 5A, DayT 2 Panel). Brightfield and fluorescent images of T-cells within different hydrogel cultures at DayT 2 reveal differential recruitment of T-cells (Fig. 5B). The differential recruitment of T-cells into various cultures was quantified by examining the fraction of T-cells at various normalized radial positions within the circular XY section of the hydrogel at DayT 0, DayT 1, and DayT 2. Here, normalized radial position (R0) of 0 and 1 indicates the center and the edge of the constructs, respectively. Our analysis indicates that cultures containing cancer spheroids show a higher fraction of T-cells at the normalized radial positions lower than 0.56 at DayT 1 and DayT 2 compared to cultures containing dispersed cancer cells (Fig. 5C-F). These radial positions are adjacent to the inner compartment of the bi-layer hydrogel. This indicates that cancer spheroids are better at recruiting T-cells compared to those with dispersed cells or small cell aggregates (DisC and DisC + Mo). Furthermore, the inclusion of monocytes leads to higher T-cell presence at lower normalized positions at DayT 1 and DayT 2 (Fig. 5C-F), suggesting that the presence of monocytes promotes T-cell infiltration. A corresponding plot for each day of infiltration for different culture conditions is given in Fig. S6A-C. In contrast, cultures containing only monocytes failed to exhibit any significant T-cell recruitment as the majority of T-cells remained at the periphery of the construct at two days of culture (maximum experimental time used to examine the cell infiltration) (Fig. 5G). To concisely summarize the recruitment of T-cells into each construct, the distribution of T-cells (DoT) within the constructs at DayT 2 was calculated. Large and small values of DoT indicate that the peripheral and central location of T-cells, respectively. Using this analysis, the DoT values for constructs with different cell types from lowest to highest are CS + Mo < CS < DisC + Mo < DisC < Mo (Fig. 5H). Interestingly, cultures without the endothelial layer at the periphery showed a lack of T-cell extravasation.
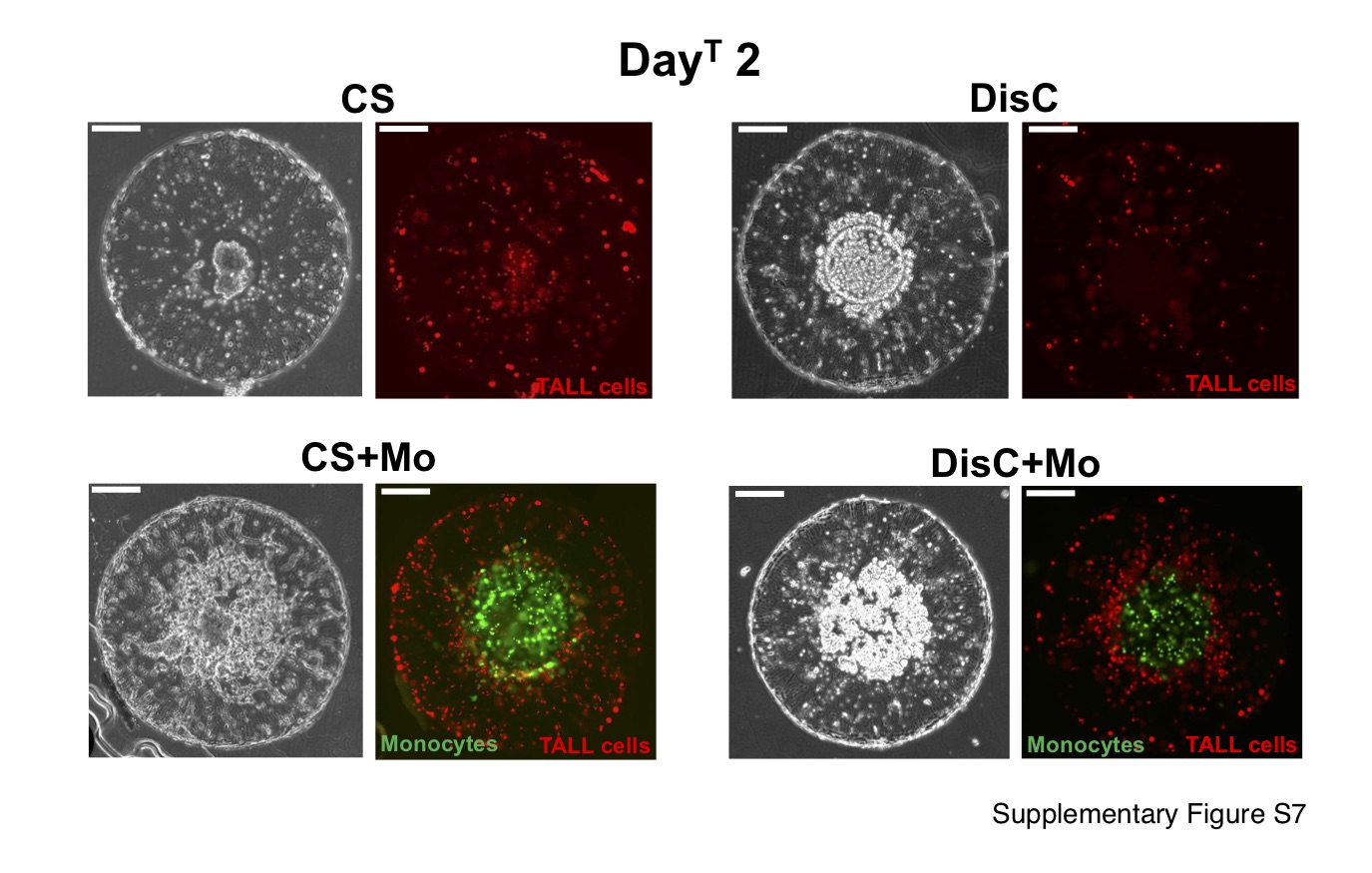
The findings from the MCF7 cells were further confirmed by using MDA-MB-231 cells. Similar to MCF cells, cultures containing MDA-MB-231 cells recruited the TALL cells albeit with subtle differences (Fig. S7). While the presence of monocytes promoted T cell infiltration, unlike MCF7 cells, MDA-MB-231 cells induced deeper infiltration of TALL cells into the hydrogel. Quantification and spatial distribution of the recruited TALL cells within the MDA-MB-231 cultures was not performed due to the differences between the DisC and CS cultures. As mentioned in methods, the addition of Matrigel was used to promote spheroid formation among the MDA-MB-231 cells, which was not used in the DisC cultures because addition of Matrigel to DisC could interfere with the monocytes.
Culture condition dependent phenotypic changes of monocytes
Given the significant differences in T-cell recruitment in the presence and absence of cancer cells, we determined the potential phenotypic changes, if any, that the monocytes undergo during co-culture. Within in vivo tumor models, recruitment of T-cells to the cancer microenvironment indicates an inflammatory response that is triggered and propagated by innate immune cells such as M1 macrophages (39). Thus, we examined whether THP-1 monocytes differentiated into macrophages (CD68hi) and polarized into M1 (CD68hi, CD206int, CD86hi) phenotype during co-culture with MCF7 cancer cells (38,40). After three days of co-culture, monocytes cultured with DisC showed only a minor increase in CD68 expression while CD205 and CD86 expressions remained unchanged compared to a THP-1 monoculture (negative control) (Fig. 6A, B). Similarly, monocytes cultured with CS also showed only minor changes compared to negative control although CD205 and CD86 expression were higher than that of monocytes in DisC+Mo (Fig. 6A, B). However, the change in CD68, CD206, and CD86 expression on THP-1 cells in both DisC+Mo and CS+Mo cultures were significantly lower than our positive control where THP-1 monocytes were partially differentiated to M1 or M2 macrophage by treating with a cocktail containing LPS/ IFN-γ and IL-4/IL-13, respectively (Fig. 6A, B). Together the data indicate that the THP-1 cells are significantly different from M1 or M2 phenotypes and the presence of cancer cells did not significantly change their polarization.
Figure 6: Culture condition dependent chemokine secretion and changes in THP-1 phenotypes.

(A) FACS analysis of THP-1 monocytes from different cultures showing representatives intensity histograms for CD68, CD80, and CD206. M1 and M2 within the legends indicate THP-1 cells differentiated into either M1 or M2 macrophages. (B) Mean fluorescence intensity (MFI) of CD68, CD80, and CD206 normalized to MFI of these markers of THP-1 monoculture. (C) Chemokines of different cultures containing MCF7 cells and THP-1 monocytes.
Culture condition dependent differential chemokine secretion
Since the phenotypic analysis did not show any difference, we next determined the changes in chemokine secretion in different culture conditions. TALL-104 cells are shown to express chemokine receptors CCR3, CCR5, and CCR6 which can be activated by one or a combination of CCL4, CCL5, CCL11, and CCL20 (41). Analyses of proinflammatory chemokine panel suggest low levels of CCL4, CCL11, and CCL20 in cultures containing only monocytes (Fig. 6C). On the contrary, monocytes in combination with cancer cells were found to have significantly higher levels of chemokine concentrations in the culture media, except for CCL20, albeit with some differences. Compared to CS groups which showed upregulation of CCL4, CCL5 and CCL11 in the presence of monocytes, DisC groups only showed upregulation of CCL5 (Fig. 6C). Besides these cytokines, we have also examined CXCL8, which is shown to destabilize cell-cell junctions within the endothelium and contribute to “leaky” vasculature (42,43). Not only did the presence of cancer cells showed higher levels of CXCL8 in the co-cultures, the concentration of CXCL-8 was found to be the highest among all the cytokines detected (Fig. 6C).
Discussion
We have created a heterotypic co-culture system containing breast cancer cells, monocytes, and endothelial cells within a microfluidics device to examine T-cell infiltration. Towards this, we have used an additive photopatterning technique to encapsulate and position different cell types within a bi-layer hydrogel. A mixture of monocytes and cancer cells (either as spheroids (CS) or dispersed cells (DisC) with approximately similar or higher cell density to that of spheroids) was confined within the inner layer of the hydrogel with endothelial cells in the outer layer. Similar to prior findings, the endothelial cells in the exterior layer migrated to the periphery of the hydrogel in response to a morphogen gradient and formed an endothelial layer (29). This layer facilitated adhesion of TALL cells prior to their subsequent infiltration.
Examining the effect of Young’s modulus of hydrogel on cell functions identified hydrogels with lower values (6.5kPa) compared to hydrogels with higher Young’s modulus to be optimal for the growth of the encapsulated MCF7 spheroids. This finding concurs with prior reports, which showed preferential growth of MCF7 spheroids within alginate hydrogels with lower rigidities (44). Moreover, recent studies have implicated that the solid stresses experienced by cancer spheroids encapsulated within matrices could inhibit their growth (45). It is conceivable that rigid matrices will impart higher solid stresses to the growing spheroids compared to softer matrices thus supporting our observed trend of higher proliferation within GelMA hydrogels with 6.5 kPa compared to those with higher stiffnesses.
TALL-104 cells were used as a model cell type for activated T-cells. This cell line is generated from a patient with acute lymphoblastic leukemia and have been immortalized through successive passages (46). Moreover, TALL-104 cells exhibit cytotoxic activity towards cancer cell lines in a MHC un-restricted manner although their function and viability are highly dependent on the presence of IL-2 (46). In comparison to primary human T-cells activated with anti-CD3 antibody, IL-2, and IFN-γ, TALL-104 cells were found to express similar chemokine receptors such as CCR5 and CCR6, in addition to CCR3 (41).
As evident from Fig 5 and Fig. S7, the TALL cells extravasated into the constructs irrespective of the cancer cell line (MCF7 or MDA-MB-231) used, and the presence of monocytes promoted the extravasation. To investigate a possible mechanism that underlies the differential recruitment of TALL-104 cells under various culture conditions, we examined for the phenotypic alterations that the monocytes undergo in culture as well as for the presence of different chemokines. Although the monocytes in co-cultures maintained its phenotype close to their native state, there were significant differences in the chemokines with the co-cultures showing higher levels of CCL4, CCL5, CCL11, and CCL20. These chemokines are shown to trigger various receptors, CCR3, CCR4, and CCR5, on TALL cells (41). Concomitant with the synergistic recruitment of TALL-104 cells in the presence of monocytes, higher levels of CCL5 were detected in DisC+Mo and CS+Mo compared to DisC and CS, respectively. This finding is also supported by prior studies which reported increased CCL5 expression in THP-1 monocytes when co-cultured with cancer cell lines such as AGS, a gastric adenocarcinoma cell line (47). Interestingly, cultures with CS+Mo recruited the highest number of TALL-104 cells and secreted the highest concentration of CCL4, CCL5, and CCL11 compared to all other conditions. The higher levels of CCL5 and CCL20 in CS+Mo and CS compared to DisC+Mo and DisC could be associated with hypoxia (19-21,47,48). In addition to these chemokines, CXCL8, a chemokine that activates CXCR1 and CXCR2 both of which are absent on TALL-104 cells, was also found in the co-cultures. In contrast to directly recruiting TALL cells, CXCL8 can potentially influence the recruitment by acting on endothelial monolayer at the periphery of the bi-layer hydrogel. Indeed, this chemokine has been found to induce the loss of tight junction proteins and gap formations between endothelial cells through cytoskeletal reorganization thus increasing the endothelial barrier permeability (42,43). The high levels of CXCL8 and its correlation to the degree of TALL-104 cell recruitment in different culture conditions suggest this chemokine might be playing a crucial role in T-cell recruitment by promoting endothelial permeability (i.e., leaky endothelium). A direct comparison between the two cancer cell lines was not carried out due to their intrinsic differences such as invasiveness, which could influence hydrogel degradation and thereby the infiltration of the extravasating cells (49). There were also differences in experimental protocols used for spheroid formation as mentioned in methods and results.
It is important to note that the ex vivo co-culture system described herein does not entirely recapitulate the tumor microenvironment which is comprised of different cell types such as T-regulatory cells, stromal cells like fibroblasts, and innate immune cells (17). Incorporation of these key cell populations into the platform is needed to understand the role of tumor associated microenvironment on cytotoxic T-cell recruitment. For example, presence of alternatively activated macrophages in the tumor microenvironment has been shown to limit the recruitment of cytotoxic T-cells (9). Similarly, the physicochemical properties of the tumor ECM can also contribute to the T-cell recruitment. By designing biomaterials with tumor-specific physicochemical cues, one could envision incorporating insoluble cues of the tumor microenvironment into this ex vivo platform. Identifying the factors that contribute to T-cell recruitment without the incorporation of these key components of the tumor microenvironment may not provide an accurate insight and hence recapitulation of more accurate tumor microenvironment is needed. Nevertheless, this system presents a proof-of-concept approach to modulate the cellular composition and the cells’ spatial organization within an in vitro tumor model and illustrates its impact on studying recruitment of cells such as T-cells. Additional cell populations can be incorporated into the platform either by including them along with other cells or can be spatially confined through photopatterning. Such ex vivo platforms with the ability to tune the cellular and non-cellular components of the tumor microenvironment provides a powerful tool to not only understand the fundamental basis of how the cancer microenvironment is sculpted but also to develop translational approaches, particularly for immunotherapies.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Statement of Significance.
The study describes how tumor-on-chip platforms could be designed to create a heterogenous mix of cells and non-cellular components to study the effect of the tumor microenvironment on immune cell recruitment.
Acknowledgements
AA acknowledges the support from ARCS foundation and the Ruth L. Kirschstein National Research Service Award NIH/NHLBI T32 HL 105373.
Footnotes
Conflicts: There are no conflicts of interests to declare.
References
- 1.Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric Antigen Receptor T Cells for Sustained Remissions in Leukemia. New Engl J Med 2014;371:1507–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lee DW, Barrett DM, Mackall C, Orentas R, Grupp SA. The Future Is Now: Chimeric Antigen Receptors as New Targeted Therapies for Childhood Cancer. Clin Cancer Res 2012;18:2780–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kochenderfer JN, Rosenberg SA. Treating B-cell cancer with T cells expressing anti-CD19 chimeric antigen receptors. Nat Rev Clin Oncol 2013;10:267–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gettinger SN, Horn L, Gandhi L, Spigel DR, Antonia SJ, Rizvi NA, et al. Overall Survival and Long-Term Safety of Nivolumab (Anti-Programmed Death 1 Antibody, BMS-936558, ONO-4538) in Patients With Previously Treated Advanced Non-Small-Cell Lung Cancer. J Clin Oncol 2015;33:2004–U32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, Activity, and Immune Correlates of Anti-PD-1 Antibody in Cancer. New Engl J Med 2012;366:2443–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Newick KM E; Albelda SM Chimeric antigen receptor T-cell therapy for solid tumors. Molecular Therapy-Oncolytics 2016;3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Craddock JA, Lu A, Bear A, Pule M, Brenner MK, Rooney CM, et al. Enhanced Tumor Trafficking of GD2 Chimeric Antigen Receptor T Cells by Expression of the Chemokine Receptor CCR2b. J Immunother 2010;33:780–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Harlin H, Meng Y, Peterson AC, Zha YY, Tretiakova M, Slingluff C, et al. Chemokine Expression in Melanoma Metastases Associated with CD8(+) T-Cell Recruitment. Cancer Res 2009;69:3077–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Noy R, Pollard JW. Tumor-Associated Macrophages: From Mechanisms to Therapy (vol 41, pg 49, 2014). Immunity 2014;41:866- [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shiao SLG AP; Rugo HS; Coussens LM Immune microenvironments in solid tumors: new targets for therapy. Genes and Development 2011;25:2559–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Burkholder B, Huang RY, Burgess R, Luo SH, Jones VS, Zhang WJ, et al. Tumor-induced perturbations of cytokines and immune cell networks. Bba-Rev Cancer 2014;1845:182–201 [DOI] [PubMed] [Google Scholar]
- 12.DeNardo D, Andreu P, Coussens LM. Interactions between lymphocytes and myeloid cells regulate pro- versus anti-tumor immunity. Cancer Metast Rev 2010;29:309–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Braumuller H, Wieder T, Brenner E, Assmann S, Hahn M, Alkhaled M, et al. T-helper-1-cell cytokines drive cancer into senescence. Nature 2013;494:361–5 [DOI] [PubMed] [Google Scholar]
- 14.Schlecker E, Stojanovic A, Eisen C, Quack C, Falk CS, Umansky V, et al. Tumor-Infiltrating Monocytic Myeloid-Derived Suppressor Cells Mediate CCR5-Dependent Recruitment of Regulatory T Cells Favoring Tumor Growth. J Immunol 2012;189:5602–11 [DOI] [PubMed] [Google Scholar]
- 15.Liou GY, Doppler H, Necela B, Krishna M, Crawford HC, Raimondo M, et al. Macrophage-secreted cytokines drive pancreatic acinar-to-ductal metaplasia through NF-kappa B and MMPs. J Cell Biol 2013;202:563–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol 2010;11:889–96 [DOI] [PubMed] [Google Scholar]
- 17.Fridman WH, Zitvogel L, Sautes-Fridman C, Kroemer G. The immune contexture in cancer prognosis and treatment. Nat Rev Clin Oncol 2017;14:717–34 [DOI] [PubMed] [Google Scholar]
- 18.Schito L, Semenza GL. Hypoxia-Inducible Factors: Master Regulators of Cancer Progression. Trends Cancer 2016;2:758–70 [DOI] [PubMed] [Google Scholar]
- 19.Lin SS, Wan SY, Sun L, Hu JL, Fang DD, Zhao RP, et al. Chemokine C-C motif receptor 5 and C-C motif ligand 5 promote cancer cell migration under hypoxia. Cancer Sci 2012;103:904–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ye LY, Chen W, Bai XL, Xu XY, Zhang Q, Xia XF, et al. Hypoxia-Induced Epithelial-to-Mesenchymal Transition in Hepatocellular Carcinoma Induces an Immunosuppressive Tumor Microenvironment to Promote Metastasis. Cancer Res 2016;76:818–30 [DOI] [PubMed] [Google Scholar]
- 21.Nagarsheth N, Wicha MS, Zou WP. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nature Reviews Immunology 2017;17:559–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Millrud CR, Bergenfelz C, Leandersson K. On the origin of myeloid-derived suppressor cells. Oncotarget 2017;8:3649–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Noman MZ, Hasmim M, Messai Y, Terry S, Kieda C, Janji B, et al. Hypoxia: a key player in antitumor immune response. A Review in the Theme: Cellular Responses to Hypoxia. Am J Physiol-Cell Ph 2015;309:C569–C79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shirure VS, Bi Y, Curtis MB, Lezia A, Goedegebuure MM, Goedegebuure SP, et al. Tumor-on-a-chip platform to investigate progression and drug sensitivity in cell lines and patient-derived organoids. Lab Chip 2018;18:3687–702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen MB, Hajal C, Benjamin DC, Yu C, Azizgolshani H, Hynes RO, et al. Inflamed neutrophils sequestered at entrapped tumor cells via chemotactic confinement promote tumor cell extravasation. Proc Natl Acad Sci U S A 2018;115:7022–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Osaki T, Sivathanu V, Kamm RD. Vascularized microfluidic organ-chips for drug screening, disease models and tissue engineering. Curr Opin Biotechnol 2018;52:116–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kumar V, Varghese S. Ex Vivo Tumor-on-a-Chip Platforms to Study Intercellular Interactions within the Tumor Microenvironment. Adv Healthc Mater 2018:e1801198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Davey SK, Aung A, Agrawal G, Lim HL, Kar M, Varghese S. Embedded 3D Photopatterning of Hydrogels with Diverse and Complex Architectures for Tissue Engineering and Disease Models. Tissue Eng Part C-Me 2015;21:1188–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aung AT J; Lim HL; Varghese S Chemotaxis-driven assembly of endothelial barrier in a tumor-on-a-chip platform. Lab Chip 2016;16:1886–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lin S, Sangaj N, Razafiarison T, Zhang C, Varghese S. Influence of physical properties of biomaterials on cellular behavior. Pharm Res 2011;28:1422–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Aung A, Bhullar IS, Theprungsirikul J, Davey SK, Lim HL, Chiu YJ, et al. 3D cardiac mu tissues within a microfluidic device with real-time contractile stress readout. Lab Chip 2016;16:153–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kaemmerer E, Melchels FPW, Holzapfel BM, Meckel T, Hutmacher DW, Loessner D. Gelatine methacrylamide-based hydrogels: An alternative three-dimensional cancer cell culture system. Acta Biomater 2014;10:2551–62 [DOI] [PubMed] [Google Scholar]
- 33.Namazi H, Kulish VV, Wong A, Nazeri S. Mathematical Based Calculation of Drug Penetration Depth in Solid Tumors. Biomed Res Int 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sun ZJ, Murry DJ, Sanghani SP, Davis WI, Kedishvili NY, Zou Q, et al. Methylphenidate is stereoselectively hydrolyzed by human carboxylesterase CES1A1. J Pharmacol Exp Ther 2004;310:469–76 [DOI] [PubMed] [Google Scholar]
- 35.Di Carlo D, Aghdam N, Lee LP. Single-cell enzyme concentrations, kinetics, and inhibition analysis using high-density hydrodynamic cell isolation arrays. Anal Chem 2006;78:4925–30 [DOI] [PubMed] [Google Scholar]
- 36.Aung A, Gupta G, Majid G, Varghese S. Osteoarthritic chondrocyte-secreted morphogens induce chondrogenic differentiation of human mesenchymal stem cells. Arthritis Rheum 2011;63:148–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stewart DA, Yang Y, Makowski L, Troester MA. Basal-like breast cancer cells induce phenotypic and genomic changes in macrophages. Mol Cancer Res 2012;10:727–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Genin M, Clement F, Fattaccioli A, Raes M, Michiels C. M1 and M2 macrophages derived from THP-1 cells differentially modulate the response of cancer cells to etoposide. Bmc Cancer 2015;15:577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Poh AR, Ernst M. Targeting Macrophages in Cancer: From Bench to Bedside. Front Oncol 2018;8:49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Raggi F, Pelassa S, Pierobon D, Penco F, Gattorno M, Novelli F, et al. Regulation of Human Macrophage M1-M2 Polarization Balance by Hypoxia and the Triggering Receptor Expressed on Myeloid Cells-1. Front Immunol 2017;8:1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schmidt TL. Cytokine-induced killer cell trafficking to tumors: A chemokine-directed migration. Stanford University, Doctor of Philosophy Thesis 2010 [Google Scholar]
- 42.Schraufstatter IU, Chung J, Burger M. IL-8 activates endothelial cell CXCR1 and CXCR2 through Rho and Rac signaling pathways. Am J Physiol Lung Cell Mol Physiol 2001;280:L1094–103 [DOI] [PubMed] [Google Scholar]
- 43.Yu H, Huang X, Ma Y, Gao M, Wang O, Gao T, et al. Interleukin-8 regulates endothelial permeability by down-regulation of tight junction but not dependent on integrins induced focal adhesions. Int J Biol Sci 2013;9:966–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cavo M, Fato M, Penuela L, Beltrame F, Raiteri R, Scaglione S. Microenvironment complexity and matrix stiffness regulate breast cancer cell activity in a 3D in vitro model. Sci Rep 2016;6:35367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kalli M, Stylianopoulos T. Defining the Role of Solid Stress and Matrix Stiffness in Cancer Cell Proliferation and Metastasis. Front Oncol 2018;8:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cesano AS D Two unique human leukemic T-cell lines endowed with a stable cytotoxic function and a different spectrum of target reactivity analysis and modulation of their lytic mechanisms. In Vitro Cell Developmental Biology 1992;28A:648–56 [DOI] [PubMed] [Google Scholar]
- 47.Ding H, Zhao L, Dai S, Li L, Wang F, Shan B. CCL5 secreted by tumor associated macrophages may be a new target in treatment of gastric cancer. Biomed Pharmacother 2016;77:142–9 [DOI] [PubMed] [Google Scholar]
- 48.Al-Soudi A, Kaaij MH, Tas SW. Endothelial cells: From innocent bystanders to active participants in immune responses. Autoimmun Rev 2017;16:951–62 [DOI] [PubMed] [Google Scholar]
- 49.Kam Y, Guess C, Estrada L, Weidow B, Quaranta V. A novel circular invasion assay mimics in vivo invasive behavior of cancer cell lines and distinguishes single-cell motility in vitro. Bmc Cancer 2008;8:198. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.