

Abstract
Proteasome inhibition with bortezomib has been reported to exert an immunomodulatory action in chronic autoimmune neuropathies. However, bortezomib used for the treatment of multiple myeloma induces a painful toxic polyneuropathy at a higher concentration. Therefore, we addressed this controversial effect and evaluated the neurotoxic and immunomodulatory mode of action of bortezomib in experimental autoimmune neuritis. Bortezomib-induced neuropathy was investigated in Lewis rats using the von Frey hair test, electrophysiological, qPCR and histological analyses of the sciatic nerve as well as dorsal root ganglia outgrowth studies. The immunomodulatory potential of bortezomib was characterized in Lewis rats after experimental autoimmune neuritis induction with P253-78 peptide. Clinical, electrophysiological, histological evaluation, von Frey hair test, flow cytometric and mRNA analyses were used to unravel the underlying mechanisms. We defined the toxic concentration of 0.2 mg/kg bortezomib applied intraperitoneally at Days 0, 4, 8 and 12. This dosage induces a painful toxic neuropathy but preserves axonal regeneration in vitro. Bortezomib at a concentration of 0.05 mg/kg significantly ameliorated experimental autoimmune neuritis symptoms, improved experimental autoimmune neuritis-induced hyperalgesia and nerve conduction studies, and reduced immune cell infiltration. Furthermore, proteasome inhibition induced a transcriptional downregulation of Nfkb in the sciatic nerve, while its inhibitor Ikba (also known as Nfkbia) was upregulated. Histological analyses of bone marrow tissue revealed a compensatory increase of CD138+ plasma cells. Our data suggest that low dose bortezomib (0.05 mg/kg intraperitoneally) has an immunomodulatory effect in the context of experimental autoimmune neuritis through proteasome inhibition and downregulation of nuclear factor 'kappa-light-chain-enhancer' of activated B-cells (NFKB). Higher bortezomib concentrations (0.2 mg/kg intraperitoneally) induce sensory neuropathy; however, the regeneration potential remains unaffected. Our data empathizes that bortezomib may serve as an attractive treatment option for inflammatory neuropathies in lower concentrations.
Keywords: EAN, bortezomib, neuropathies, inflammation, CIDP
Klimas et al. report that low dose bortezomib is immunomodulatory in experimental autoimmune neuritis, through proteasome inhibition and NFKB downregulation. In line with previous clinical trials and case studies, this work underlines the therapeutic potential of low dose bortezomib for autoimmune diseases.
Graphical Abstract
Graphical Abstract.

Introduction
With a prevalence of 0.8–8.9 cases per 100 000, chronic inflammatory demyelinating polyneuropathy (CIDP) is the most common peripheral chronic demyelinating neuropathy.1–3 First-line treatment includes intravenous immunoglobulins, steroids and plasmapheresis.4 Up to 25% of CIDP patients fail to respond to first-line treatment options.5,6 Another 50% experience clinical relapses after treatment, which lead to severe disability.7 Currently available second-line therapies have improved the disease outcome, even though most of them are not immunomodulatory but immunosuppressive. The mechanisms of action of common immunosuppressants used in CIDP are their impact on IL-2 (cyclosporine A), lymphocyte proliferation and survival (azathioprine, cyclosporine A and cyclophosphamide), and B- and T-cell depletion through inhibition of the inosine monophosphate dehydrogenase and thereby de novo purine synthesis (mycophenolate mofetil). Still, a low response rate of up to 38% for patients on second-line agents has been reported.8
Furthermore, humoral factors seem to play a key role in the development of CIDP.6,9 The effect of plasmapheresis on clinical disability and demyelination indicates autoantibodies’ involvement in classical CIDP.10 In addition, it has been shown that the injection of CIDP serum into Lewis rats induces demyelinating neuropathy.11 Subsequently, CD20+ B cell depletion with rituximab leads to clinical improvement of up to 50% of CIDP patients in uncontrolled studies.8 Treatment refractory CIDP patients suffer from an aggressive disease course, posing a particular challenge for neurologists.
Mature circulating, secondary lymphoid organ- and bone-marrow resident plasma cells are not depleted by rituximab. These plasma cells could play a crucial role in these aggressive variants as they are involved in autoantibody production. Proteasome inhibition exerts additional immunomodulatory effects through interaction with the inhibitor of NFKB, a transcription factor promoting autoimmune inflammation.
We have previously reported the successful treatment of 10 patients with aggressive variants of CIDP with bortezomib (BTZ), a reversible proteasome inhibitor. BTZ is known to deplete plasma cells in patients with multiple myeloma.12–14 Its use is, however, restricted mainly due to a bortezomib-induced polyneuropathy (BiPNP)15–17 in the usual oncological schema [1.3 mg/m2 given subcutaneously (s.c.)] on Days 1, 4, 8 and 11, and every 21 days thereafter for an average of four cycles in multiple myeloma. The CIDP patients in our study received one or two cycles of BTZ with 1.3 mg/m2 given s.c. on Days 1, 4, 8 and 11, every 6–12 months, which did not lead to neurotoxic effects.12 To investigate the immunomodulatory and toxic effects of BTZ in different concentrations, we used the model of experimental autoimmune neuritis (EAN) in Lewis rats as a model of autoimmune neuropathies.
Materials and methods
Animals, in vivo treatment with BTZ and experimental design
All experiments were carried out in accordance with the European Communities Council Directive of 22 September 2010 (2010/63/EEC) for the care of laboratory animals and with local government authorization (Landesamt für Natur, Umwelt und Verbraucherschutz North Rhine-Westphalia; Az.: 81–02.04.2018.A043). A total of 58 6- to 8-week-old female Lewis rats were purchased from Charles River (Sulzfeld, Germany) and kept under standardized conditions in our local animal facility (Medical Faculty, Ruhr-University Bochum) in pathogen-free cages with food and water available ad libitum. Animals were in the weight range of 140–160 g at arrival and could acclimatize for at least 1 week. At experimental onset, animals were weighed repeatedly and randomly divided into four animals per condition. BTZ was diluted in 5% dimethyl sulphoxide (DMSO). Two experimental setups were used to describe the effects of BTZ.
Toxic setting with three conditions in healthy animals
Animals were treated with 0.2 (n = 4) and 0.05 (n = 4) mg/kg BTZ or with 5% DMSO (n = 4) intraperitoneally (i.p.) at Days 0, 4, 8 and 12 and were sacrificed on Day 21. This setting was performed twice with 0.2 mg/kg BTZ. A total of 26 rats were used, 20 for in vivo studies and 6 additional for an in vitro dorsal root ganglia (DRG) outgrowth study. Prior investigations in healthy Sprague Dawley rats described 0.2 mg/kg BTZ as toxic, while 0.1 mg/kg did not cause a BiPNP.18
Therapeutic setting with three conditions in EAN
Animals were treated with 0.1 (n = 4) and 0.05 mg/kg BTZ (n = 4) or with 5% DMSO (n = 4) i.p. at Days 9 (day of first clinical symptoms), 13, 17 and 21 post immunization (p.i.) and were sacrificed on Day 23 p.i. This setting was performed three times with 0.05 mg/kg BTZ and twice with 0.1 mg/kg BTZ. A total of 32 rats were used.
Induction of EAN and assessment of the clinical score
In the therapeutic setting, EAN was induced with neuritogenic P2 peptide, corresponding to the amino acids 53–78 of rat myelin P2 protein, synthesized by Dr Rudolf Volkmer from Charité University-Hospital Berlin, Germany. P2 peptide (250 µg) was emulsified in complete Freund’s adjuvants containing 1 mg/ml Mycobacterium tuberculosis H37RA (Difco) and injected s.c. into the tail base under anaesthesia with xylazine and ketamine (CP-Pharma). Disease onset was daily assessed by a blinded investigator using the following EAN score system: 0 = normal; 1 = less lively; 2 = impaired righting/limb tail; 3 = absent righting; 4 = ataxic gait/abnormal position; 5 = mild paraparesis; 6 = moderate paraparesis; 7 = severe paraplegia; 8 = tetraparesis; 9 = moribund; 10 = death.19 With scores ≥7, the animals were euthanized from animal welfare measures.
Von Frey hair test
To detect mechanical allodynia/nociception, the 50% paw withdrawal threshold was assessed with the von Frey hair test (vF) in both settings. Monofilaments were purchased at BioSebLab. Tests were performed in a plastic box with a customized plexiglass platform of 3 mm thickness and with 1.5 mm diameter holes in a grid pattern. The platforms’ surface was obscured using sandblast to prevent disturbance of rats by the investigator’s hand movement.20 Animals were allowed to habituate for 30 min in the test chamber. Monofilaments with a range of 0.4–28 weight force (G) were applied consecutively on the mid-plantar skin of the left and right hind paw. Each hind paw was tested at least six times with a minimum of 6 s rest in between. The 50% paw withdrawal threshold was calculated according to a modified up-and-down method.21 To assess treatment impact on allodynia/hyperalgesia, tests were performed at Days 0 and 6 p.i. and 1 day prior BTZ administration at Days 10, 14, 18 and 22 p.i. In the toxic setting, the tests were performed at Days 0, 2, 4, 6, 14, 16 and 18.
Nerve conduction studies
At experimental termination at Day 21 in toxic setting and Day 23 p.i. in therapeutic setting, nerve conduction studies (NCS) evaluating motor nerve conduction velocity (MNCV) and F-wave latency were performed by a blinded investigator. For strain, gender, age and weight, see ‘Animals, in vivo treatment with BTZ and experimental design’ section. Rats were successively anaesthetized as with 10 mg/kg Xylazine (Xylavet, CP-Pharma) and 50 mg/kg Ketamine (Ketamine, CP-Pharma) through i.p. injection. Temperature differences were minimized by conducting the study as soon as the anaesthesia took effect and by stabilizing the animals’ body temperature with a heating lamp. Paired needle electrodes for stimulation were inserted into the right sciatic notch (hip, proximal) and into the right malleolus (distal) to assess the NCV with a fully digital recording Keypoint apparatus (Dante, Skovlunde, Denmark). Stimulations with supramaximal rectangular pulses of 0.05 ms duration resulted in compound muscle action potentials (CMAP), measured with a recording needle electrode placed s.c. over the dorsal foot muscles. The amplitude (mA) for supramaximal stimulation was determined for each analysed animal individually. Furthermore, the grounding electrode was placed between the stimulation and recording electrode. To calculate the NCV, the distance between both electrodes was divided by the difference of the latency; 10 F-waves’ persistence and minimum latencies were evoked by stimulating the electrode in the popliteal fossa and recorded for the right hind-paw.22 Study report is based on the ‘Checklist for performance and reporting NCS in rats’ by Kohle et al.23 Proposed normative values based on meta-analyses by Kohle et al. are as follows: NCV 47.24 m/s (43.65–50.84); CMAP sciatic notch 12.16 mV (9.72–14.60); CMAP ankle 17.41 mV (11.87–22.95).
Flow cytometric analysis of mononuclear cells in blood, spleen, lymph nodes and bone marrow tissue
Blood samples were obtained immediately by cardiac puncture. After transcardial perfusion with phosphate-buffered saline (PBS), spleen and inguinal lymph nodes were extracted and stored in ice-cold RPMI-1640 medium (Gibco, Thermo Fischer). The right femur was gently removed (once for the toxic setting and three times for the therapeutic setting), cleaned from muscle tissues and stored in ice-cold ethanol. Spleen and lymph nodes single-cell suspensions were obtained using 100 and 70 µm cell strainer (Sarstedt) and washed twice. Bone marrow tissue was prepared as described before.24 Red blood cells were lysed with BD FACS lysing solution according to the manufacturer (BD Biosciences). Based on the manufacturer’s protocol, flow cytometry was used to evaluate the cell count for CD8+ T cells, CD4+ T cells, CD11b+ monocytes, CD4+CD11b+ dendritic cells (DCs), CD4+CD25+FOXP3+ regulatory T cells (Tregs), CD4+CD11b–MHCII+ plasmacytoid DCs in the toxic setting. Additionally, CD45R+ and CD19+ (B-cells), and CD138+ IgK light chainhigh+ (plasma cells) were evaluated in the therapeutic setting.25 Intracellular staining for FOXP3 and IgK light chain was performed by using the eBioscience™ FOXP3/Transcription Factor Staining Buffer Set according to the manufacturer (Thermo Fischer Scientific). Flow cytometry was performed using the BD FACSCanto™ II system and the BD FACSDiva™ Software and compensation controls with compensation particles (BD Biosciences). Used flow cytometry antibodies are listed in Supplementary Table 1.
Histopathological analyses of the sciatic nerve
Dissected sciatic nerves were divided into four parts of equal length and cryosections (8 µm) were prepared. To evaluate toxic side effects of BTZ, cross-sections were stained with antibodies against the regeneration-associated sensory axon marker SCG10 (1:1000; Novus Biologicals, NBP1-49461) and the regeneration-associated protein GAP43 (1:1000; Invitrogen) to assess axonal injury and regeneration. Labelled axons were quantified, as previously described.26,27
For the therapeutic setting, demyelination was assessed with FluoroMyelin™-Red Fluorescent Myelin Stain (1:300, Thermo Fischer Scientific, F34652). Primary antibodies against CD3 (1:1000, Thermo Fischer Scientific) and CD68 (1:1000, Hycult Biotech, HM3029) were used to determine T cell and macrophage infiltration and incubated at 4°C overnight. Secondary antibodies conjugated with Alexa Fluor 488 (1:1000, Invitrogen) were used for detection. Nuclear staining was performed with DAPI Fluoromount-G (4', 6'-diamino-2-phenylindole-2HCl, Biozol). Fluorescent signals of CD3, CD68 and FluoroMyelin™-Red were detected using an inverted fluorescence microscope (BX51; Olympus) equipped with an Olympus DP50 digital camera. The four transversal sciatic nerve sections of each rat were recorded four times (technical n = 16, ×20 magnification) and analysed by two blinded investigators using the image analysis software ImageJ (National Institutes of Health). Demyelinated lesions are reported as the percentage of the whole analysed area. CD3 and CD68 positive cell counts are reported as the number of infiltrating cells per mm2.
Histopathological analysis of DRG
For the therapeutic setting, a minimum of six and a maximum of eight DRGs per segment (lumbar, thoracic and cervical) were dissected and washed with PBS, stored in 4% paraformaldehyde (PFA) for 1 h and cryoprotected in 30% sucrose overnight. Cryosections (8 µm) were stained with primary antibodies against TRPV1 (1:500, Thermo Fisher Scientific, PA1-29421) and NeuN (1:300, Merck Millipore, MAB377), anti-rabbit Alexa 568 (1:1000, Invitrogen) to visualize TRPV1 staining and anti-mouse Alexa 488 (1:1000, Invitrogen) as a secondary antibody for NeuN. DAPI Fluoromount-G (Biozol) was used to visualize cell nuclei. The fluorescence signal of TRPV1 was detected using an inverted fluorescence microscope (BX51; Olympus) equipped with an Olympus DP50 digital camera. Three replicate pictures of each segment/rat were recorded three times (biological n = 4, technical n = 9, ×20 magnification) and analysed by two blinded investigators using the image analysis software ImageJ (National Institutes of Health, Bethesda, USA). TRPV1high+ cells were counted and reported as the percentage of all neuronal cells in DRG.
Histopathological analysis of bone marrow tissue
In the therapeutic setting, the right femur was dissected, stored in 4% PFA and further processed and stained for CD138+ plasma cells in cooperation with the department of pathology, University Hospital Bergmannsheil Bochum. Plasma cells were stained as described recently.28
CD138+ plasma cells in bone marrow tissue were counted by a blinded investigator using a light microscope (Olympus). The number of infiltrated cells per mm2 was determined.
Quantitative PCR
Dissected sciatic nerves were immediately flash-frozen in liquid nitrogen and stored at –80°C. Frozen samples were lysed and homogenized with TRIzol™ reagent (Thermo Fischer Scientific), using the Tissue Lyser II system and stainless-steel beads of 5 mm diameter (Qiagen). Total RNA was isolated using the RNeasy Plus Mini kit (Qiagen) and transcribed into cDNA using the Go Script Reverse Transcription Mix Oligo(dT) and random primers (Promega) according to the manufacturer’s protocol. GoTaq® qPCR Master Mix (Promega) and target-specific primers (Microsynth) were used to evaluate relative mRNA expression levels of Il4, Il10, Ifng, Tnfa, Trpv1, Ikba (also known as Nfkbia), Cgrp (also known as Calcb) and Nfkb with the QuantStudio 3 Real-Time-PCR system (Thermo Fischer Scientific). qPCR runs were performed at 95°C for denaturation, 60°C for annealing/extension and 40 cycles. Post-amplification melting-curve analysis was performed to check for primer-dimer artefacts and to verify reaction specificity. Detailed primer information is shown in Supplementary Table 2. The relative target expression was determined using the modified relative quantification model by Pfaffl29 with efficiency correction and were normalized to Gapdh and Actb as reference genes. Relative target gene expression is described as fold-change relative to the corresponding experimental control (CTR) group. All experiments were carried out in duplicates, and the mean Ct was used in the equation.
In vitro DRG outgrowth study
We evaluated sensory neuron axon growth of DRGs in a longitudinal setting. A total of eight rats were used to analyse the effect of 0.2 mg/kg BTZ on axonal growth. Rats were treated on Days 0, 4, 8 and 12. For DRG isolation, two rats were sacrificed at each time point (0, 7, 14), except Day 21, where eight rats were sacrificed. Sensory neurons were obtained from dissociated DRG, as described previously.27,30 In short, isolated DRG (T8-L6) were incubated in 0.25% trypsin/EDTA (GE Healthcare) and 0.3% collagenase Type IA (Sigma) in Dulbecco’s modified eagle media (DMEM) (Invitrogen) at 37°C and 5% CO2 for 45 min and then mechanically dissociated. Cells were re-suspended in DMEM containing 10% foetal bovine serum (GE Healthcare) and penicillin/streptomycin (500 U/ml; Merck, Millipore) and cultured at 37°C and 5% CO2 on poly-d-lysine (0.1 mg/ml; Sigma) and laminin (20 μg/ml; Sigma)-coated slides (Sarstedt). Axonal growth was determined after 48 h incubation by fixation in 4% PFA (Sigma) and immunocytochemical staining with antibodies against NeuN (1:2000; Abcam, ab177487) and βIII-tubulin (1:2000; Covance). Imaging was performed automatically with the Olympus VS210-S5 slide scanner. Total axon length and neuron numbers per well were automatically quantified with the NeuriteTracer plugin for ImageJ. The average axon length per neuron and neuron counts per experimental group were normalized to CTR groups. Data represent the mean ± SD of a technical N of 4.
Statistical analysis
Sample size was calculated using the power analysis program G*Power (α-error = 0.05, power = 0.80). Animals were randomly divided into the experimental groups and all experimental measurements were performed by two blinded investigators. Statistical analyses were performed using GraphPad Prism 9 (GraphPad Software Inc.). The normality of data was analysed before testing. If not mentioned otherwise, we used t-Test or Mann–Whitney test to compare two conditions and (multiple) comparison tests in one/two-way ANOVA analyses or Kruskal–Wallis for more than two groups.
Data availability
The data of this study are available from the corresponding author, upon reasonable request.
Results
Toxic setting
BTZ induces toxic hyperalgesia in a concentration of 0.2 mg/kg
Healthy rats were treated i.p. with BTZ on Days 0, 4, 8 and 12 with a concentration of 0.2 and 0.05 mg/kg or 5 % DMSO (n = 4/group). BTZ-induced effects on pain detection were assessed with vF test. The experiment was performed twice, and the area under the curve (AUC) was analysed. One representative experiment is shown in Fig. 1. Hyperalgesia developed immediately after BTZ treatment with 0.2 mg/kg and reached its maximum around Day 13 (mean value ± SD: 2.918 ± 0.735G, P-value = 0.0002) after four injections compared to the CTR group. After discontinuing BTZ, hyperalgesia recovered slightly and the 50% paw withdrawal threshold increased on Days 16 (3.208 ± 0.662G) and 19 (4.243 ± 0.769G). Comparing the time point with maximal hyperalgesia with Day 19, 50% paw withdrawal threshold recovered significantly (P-value = 0.0471). Overall, compared to the CTR group, there was a significant reduction of 50% paw withdrawal threshold in a high concentration of BTZ (AUC mean values: CTR = 72.99 ± 8.53, 0.2 mg/kg BTZ = 29.14 ± 8.61, 0.05 mg/kg BTZ = 63.16 ± 9.09; P-value = 0.0206). No differences between the CTR group and the low dose group (0.05 mg/kg BTZ) were detected.
Figure 1.

BTZ induced hyperalgesia in high concentrations. Von Frey hair test after i.p. treatment with 5% DMSO as control, 0.2 mg/kg BTZ and 0.05 mg/kg BTZ on Days 0, 4, 8 and 12 is shown in A (arrows). With 0.2 mg/kg BTZ, 50% paw withdrawal threshold dropped significantly on Day 4 and reached its minimum on Day 13. The homogeneity of variances was analysed with Levene test. Variances are homogeneous at Days 0, 2, 4, 6 and 19. Therefore, ordinary one-way ANOVA was performed at these days. At Days 13 (F = 9.080, df1 = 2, df2 = 8, P = 0.009) and 16 (F = 5.264, df1 = 2, df2 = 8, P = 0.035) variances are not homogeneous. In consequence, Welch ANOVA was performed followed by Dunnett’s T3 multiple comparison test (Day 13: P-value < 0.0001). Subsequently, significant recovery started (P-value = 0.0471). Overall, a significant reduction of area under the curve in the group with 0.2 mg/kg BTZ indicates hyperalgesia in rats, shown in B (Kruskal–Wallis test followed by Dunn’s multiple comparison test, P-value = 0.0206). Each group consisted of four animals (n = 4). The experiment was performed twice. Both experiments showed nearly identical P-values. The figure shows one representative experiment; therefore each data point represents one animal.
Electrophysiological measurements show no affection of large, myelinated motor fibres
Electrophysiological measurements of the right sciatic nerve were performed on Day 21. We evaluated demyelination, characterized by MNCV, axonal damage represented by mean CMAP and lumbar root involvement, depicted by prolongation of F-wave latencies. No changes in MNCV, CMAP or F-wave latencies were found (n = 4, experiment repeated twice; data not shown).
BTZ does not affect mRNA expression of Ikba and Nfkb in peripheral nerves in a dose of 0.2 mg/kg
On Day 21, we investigated relative mRNA expression of inflammatory cytokines and Ikba and Nfkb in the sciatic nerve. It is well known that IKB inhibits the transcriptional factor NFKB reducing severe immunomodulatory mechanisms.31 Since BTZ is inhibiting the immunoproteasome and the immunoproteasome inhibits IKB degradation, we expected changes of posttranscriptional Ikba and Nfkb regulation. This was examined twice with duplicates for the analysis (biological n = 4, technical n = 2). However, there was no significant posttranscriptional regulation detected for Ikba and Nfkb. No differences were detected in relative mRNA expression of Il4, Il10, Tnfa, Ifng, Cgrp (data not shown).
BTZ at a toxic concentration exerts no effects on peripheral and central immune cells in flow cytometry
Flow cytometry analyses of the spleen, blood, inguinal lymph nodes and bone marrow tissue did not show significant treatment effects with 0.2 or 0.05 mg/kg BTZ compared to the CTR group. This experiment was carried out once (n = 4/group, data not shown).
Histological signs of sciatic nerve regeneration after toxic treatment with BTZ at a dose of 0.2 mg/kg
We histologically analysed SCG10 and GAP43, a sensory and global regeneration marker, respectively, expressed only in injured and regenerating axons. Both proteins were detected in sciatic nerve axons 21 days after the high dose application of BTZ (Fig. 2, Supplementary Fig. 1), indicating previous degeneration. In contrast, the lower dose of 0.05 mg/kg showed less induction, indicating that BTZ induces axon damage, particularly at higher doses (SGC10: mean values ± SD, 0.2 mg/kg BTZ = 251.1 ± 68.46, 0.05 mg/kg BTZ = 26.30 ± 10.89; P-value < 0.0001; GAP43: mean values ± SD, 0.2 mg/kg BTZ = 416.2 ± 84.86, 0.05 mg/kg BTZ = 59.18 ± 22.76, P-value < 0.0001; n = 4/group). Yet, this degeneration seems to be transient, as vF indicates a significant regeneration on Days 16 and 18.
Figure 2.

High dose BTZ mediated degeneration in the sciatic nerve. Representative pictures of SCG10, shown in A, and GAP43, shown in C, staining in the sciatic nerve of Lewis rats. Positive cells are highlighted with arrows. SCG10+ cells represent sensory regeneration, while GAP43+ cells represent global regeneration. Treatment with 0.2 mg/kg BTZ significantly increased both regeneration markers in the sciatic nerve, indicating previous degeneration, shown in B and D (one-way ANOVA followed by the Holm–Sidak post hoc test, P-value < 0.0001), while low dose application did not have the same effect. Each data point represents one animal. Scale bar: 25 µm.
In vivo treatment with 0.2 mg/kg BTZ does not inhibit DRG outgrowth in vitro
Regeneration potential after toxic BTZ treatment was evaluated through DRG axonal outgrowth studies in vitro after treating animals with 0.2 mg/kg BTZ (Fig. 3). In agreement with the clinical findings and the histological staining, the damage after BTZ treatment is not permanent. Our examination revealed no impairment of neurite growth after toxic BTZ treatment.
Figure 3.

High dose BTZ did not affect axonal outgrowth of DRGs in vitro. Experimental setup in A shows rats were treated with 0.2 mg/kg BTZ per day. DRGs were dissected on Days 0, 7, 14 and 21. No significant difference between the baseline on Day 0 and the different time points was observed in B. Representative pictures are shown in C. Data points in C are explained in A.
Therapeutic setting
BTZ significantly ameliorates EAN signs at a concentration of 0.05 mg/kg
EAN clinical signs occurred during Days 9 and 10 with a maximum around Days 15 and 17. p.i. BTZ treatment started at Day 9 p.i. and was repeated every 4 days until Day 21 (0.05 mg/kg BTZ, 0.1 mg/kg BTZ and 5% DMSO). EAN incidence in all groups was 100%. Figure 4 shows the ameliorative effect of 0.05 mg/kg BTZ on the clinical disease course in EAN. Treatment with 0.05 mg/kg BTZ significantly reduced the AUC in comparison to the 5% DMSO CTR group (AUC mean values: CTR = 36.88 ± 7.623, 0.1 mg/kg BTZ = 30.13 ± 8.197, 0.05 mg/kg BTZ = 15.88 ± 8.38; P-value = 0.0093). We repeated this experiment three times and found AUC significantly reduced twice (AUC: P-value = 0.0093, P-value = 0.0314). However, the third experiment also showed overall significant mean differences between the CTR group and the group treated with 0.05 mg/kg BTZ (P-value < 0.0247, data not shown). No significant body weight loss or further toxic effects were detected in our experiments. Treatment with 0.1 mg/kg BTZ did not affect the EAN disease course significantly.
Figure 4.

Low dose BTZ ameliorated EAN disease course. EAN score over time after i.p. treatment with 5% DMSO as control, 0.1 mg/kg BTZ and 0.05 mg/kg BTZ on Days 9, 13, 17 and 21 p.i., shown in A (arrows). A concentration of 0.05 mg/kg BTZ ameliorates EAN disease course, shown in A, and significantly reduces area under the curve, shown in B (one-way ANOVA followed by the Dunnett’s multiple comparison test, P-value = 0.0093). Each group consisted of four animals (n = 4). The experiment was performed three times. Experiments with a condition of 0.1 mg/kg BTZ were performed twice. The figure shows one representative experiment; therefore, each data point represents one animal.
BTZ reduces significantly EAN-induced hyperalgesia in vF test
While it is known that EAN animals develop paraparesis after immunization with P2-peptide, there is no published data on nociception—small fibre involvement in EAN yet. In this concept, we could show that nociception is impaired in EAN. A reduction of 50% paw withdrawal threshold could be detected at Day 6, while motor impairment started at Day 9 (compare Figs 4A and 5A); 50% paw withdrawal threshold dropped after EAN disease onset on Day 10 to 6.924G in the CTR group, 6.715G in the 0.1 mg/kg BTZ-treated group and 10.44G with 0.05 mg/kg BTZ treatment. The lowest threshold, representing the time point with maximal hyperalgesia, was reached at Day 22 in the CTR group with a mean value of 2.765G, at Day 18 in the 0.1 mg/kg BTZ-treated group with a mean value of 3.051G, and at the same day in the group with 0.05 mg/kg BTZ with a mean value of 9.042G (Fig. 5B). Comparing those minimal threshold values, BTZ was significantly able to prevent rats from further deterioration of mechanical allodynia in EAN (P-value = 0.0285, Fig. 5B). Furthermore, the most significant ameliorative effect was observed on Day 22 between the CTR group and 0.05 mg/kg BTZ with a mean difference of 9.889G (P-value = 0.0009). We performed this experiment three times and found the 50% paw withdrawal threshold increased twice in animals with low dose BTZ treatment on Day 22. The third experiment showed same tendencies but did not reach significance (data not shown). The results correlate well with the previously presented results in EAN disease course and verify the robust therapeutic effects of BTZ at a concentration of 0.05 mg/kg.
Figure 5.

EAN induced hyperalgesia while low dose BTZ protected rats. Representative results in Von Frey hair test after i.p. treatment with 5% DMSO as control, 0.1 mg/kg BTZ and 0.05 mg/kg BTZ on Days 9, 13, 17 and 21 p.i. are shown in A (arrows). Each group consisted of four animals (n = 4). A concentration of 0.05 mg/kg BTZ hyperalgesia recovers earlier, shown in A (two-way ANOVA followed by Dunnett’s multiple comparison test, P-value = 0.0009), and the minimum of paw withdrawal threshold is significantly higher, shown in B (Kruskal–Wallis test followed by Dunn’s multiple comparison test, P-value = 0.0285). Each group consisted of four animals (n = 4). The experiment was performed three times. The figure shows one representative experiment, therefore each data point represents one animal.
BTZ significantly improves motor conduction velocity of the sciatic nerve
In the therapeutic setting, electrophysiological measurements of the right sciatic nerve were performed on Day 23, as described before. We evaluated the following measurements:
MNCV for demyelination
Mean CMAP for axonal damage
Prolongation of F-wave latencies for lumbar root involvement
Mean value of MNCV in the CTR group (22.38 m/s ± 2.783) was significantly reduced compared to 0.05 mg/kg BTZ group (31.15 m/s ± 1.87; P-value = 0.0019, Fig. 6). Therefore, the treatment reduced demyelination in the sciatic nerve. We repeated the experiment two more times and obtained the same results (P-value = 0.0426 and 0.0314). However, supramaximal stimulation of proximal large, myelinated fibres did not show any differences or prolonged F-wave latencies between the CTR and treatment groups at this late-recovery time point. Additionally, axonal damage represented by CMAP decrease was not detected (data not shown).
Figure 6.

Treatment with low dose BTZ reduced loss of MNCV in EAN. Motor nerve conduction velocity (m/s) measured with electrophysiology on Day 23 p.i. with P2-peptide (n = 4, experiments = 3,). A shows treatment with 0.05 mg/kg BTZ reduces the loss of MNCV in EAN significantly (unpaired t-Test, P-value = 0.0019). Representative measurements are shown in B. The experiment was performed three times. The figure shows one representative experiment, therefore each data point represents one animal.
BTZ reduces inflammatory infiltration and demyelination in the sciatic nerve
We investigated immune infiltration by staining of CD3+ T cells and CD68+ macrophages in the sciatic nerve on Day 23 p.i. Figure 7 shows the results and representative stainings, demonstrating the suppression of T cell (Fig. 7A) and macrophage (Fig. 7B) infiltration between the 5% DMSO CTR group and 0.05 mg/kg BTZ treatment. Cell counts and significance are shown in Fig. 7D for T cells (P-value < 0.0001) and 7E for macrophages (CTR versus 0.1: P-value < 0.0001; CTR versus 0.05: P-value = 0.0012; n = 16, technical n = 64, experiments = 3). Comparing infiltration proximal to distal along the sciatic nerve, no differences were detected (data not shown). Furthermore, the reduction of inflammatory infiltration and the electrophysiological findings of less demyelination correlate with a significant reduction of the histological assessed demyelination in our target dose of 0.05 mg/kg BTZ compared to 5% DMSO. Representative pictures of FluoroMyelin™ staining are shown in Fig. 7C and results are demonstrated in Fig. 7F (mean values ± SD: CTR = 38.54 ± 12.10%; 0.1 mg/kg BTZ = 30.77 ± 11.75%; 0.05 mg/kg BTZ = 19.62 ± 9.308 %; P-value < 0.0001; n = 16, experiments = 3).
Figure 7.

Low dose BTZ treatment reduced inflammation and demyelination in the sciatic nerve. Representative pictures of CD3, shown in A, CD68, shown in B, and FluoroMyelin™-Red, shown in C, staining of the sciatic nerve. Treatment with 0.05 mg/kg BTZ significantly reduced infiltration of CD3+ T cells, shown in D (Kruskal–Wallis test followed by Dunn’s multiple comparison test, P-value < 0.0001, statistical analysis for N 0 64 technical replicates) and demyelination of the sciatic nerve, shown in F (one-way ANOVA followed by Dunnett’s multiple comparison test, P-value < 0.0001, statistical analysis for n = 16 technical replicates). In addition, 0.05 as well as 0.1 mg/kg BTZ significantly reduced infiltration of CD68+ cells into the sciatic nerve, shown in E [Kruskal–Wallis test followed by Dunn’s multiple comparison test, P-value < 0.0001 (CTR versus 0.1) and P-value = 0.0012 (CTR versus 0.05); statistical analysis for n = 64 technical replicates]. The experiment was performed three times. D, E and F show the results of one representative experiment, therefore each data point represents a technical replication. Scale bar: 50 µm.
No quantitative change of immune cell populations in lymphoid organs
After examining infiltration into the sciatic nerve, we investigated overall immune activity after treatment with 0.05 and 0.1 mg/kg BTZ. Yet, flow cytometry analyses of the spleen, blood, inguinal lymph nodes and bone marrow tissue did not show significant effects of the treatment in comparison to the CTR group suggesting that the ameliorative effect of BTZ is not driven by systemic modulation of the following immune cell populations: CD8+ T cells, CD4+ T cells, CD11b+ monocytes, CD4+CD11b+ DCs, CD4+CD25+FOXP3+ Tregs, CD4+CD11b–MHCII+ plasmacytoid DCs and CD45R+ B-cells (data not shown).
Low dose BTZ exerts immunomodulatory effects through modulation of mRNA expression of Ikba and Nfkb
As a proteasome inhibitor, BTZ prevents IKB degradation and, therefore, impacts the NFKB pathway. Since we used a low dose of BTZ, we examined relative Ikba and Nfkb expression changes in the sciatic nerve via qPCR on Day 23, considering this to be the leading mechanism for the drug’s ameliorative effect. Representative results are shown in Fig. 8. The relative expression of Ikba shows a significant fold change increase for the group treated with our target dose of 0.05 mg/kg BTZ compared relative to the 5% DMSO CTR group (Fig. 8A, mean values ± SD in CTR = 1 ± 0.136; 0.1 mg/kg BTZ = 1.211 ± 0.493; 0.05 mg/kg BTZ = 1.383 ± 0.201; P-value = 0.0455; n = 4, technical n = 8, experiments = 2). Furthermore, the relative expression of Nfkb decreased (Fig. 8B; mean values ± SD in CTR = 1 ± 0.208; 0.1 mg/kg BTZ = 0.9785 ± 0.2645; 0.05 mg/kg BTZ = 0.6748 ± 0.11; P-value = 0.0071, n = 4, technical n = 8, repeated twice). There was no significant change in the relative mRNA levels of Il-4, Il-10, Tnfa, Ifng, Cgrp and Trpv1 detected (see Supplementary Fig. 2).
Figure 8.
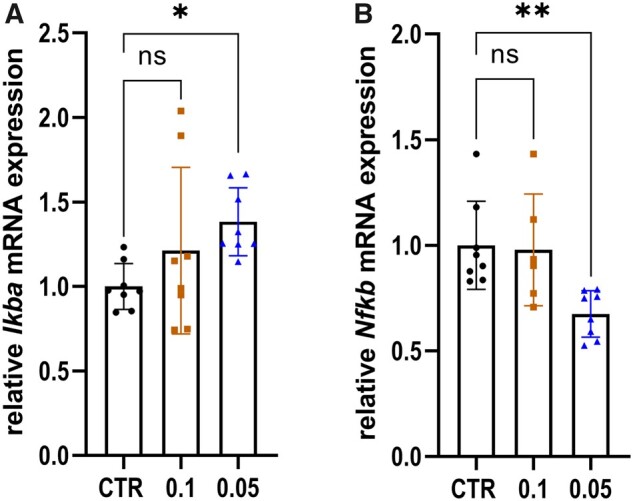
Treatment with low dose BTZ modulated relative expression of Ikba and Nfkb into an anti-inflammatory direction in the sciatic nerve. Results of qPCR of Ikba, shown in A, and Nfkb, shown in B, after treatment of EAN rats with 5% DMSO, 0.1 and 0.05 mg/kg BTZ. Data were normalized using the reference genes Actb and Gapdh and are presented relative to the control group 5% DMSO. Treatment with 0.05 mg/kg BTZ significantly increased the relative expression of Ikba (P-value = 0.0455) and reduced Nfkb (P-value = 0.0071) expression level (one-way ANOVA followed by Dunnett’s multiple comparison test). The experiment was performed twice. The figure shows the results of one representative experiment. Statistical analysis was performed with a technical N of 8, therefore each data point represents a technical replicate.
No relevant effects of BTZ on TRPV1 expression in DRGs and sciatic nerve in a dose of 0.05 mg/kg
With the vF test, we have shown that EAN induction promotes hyperalgesia in rats. We were able to demonstrate the improvement in hyperalgesia through low dose 0.05 mg/kg BTZ application. However, it remains unknown how the hyperalgesia is developing in EAN. We investigated TRPV1 channels, which are involved in hyperalgesia, as described in previous studies of BiPNP.32 Therefore, we examined the relative mRNA expression of Trpv1 in the sciatic nerve by qPCR and TRPV1 protein in DRGs by histological staining. Representative staining is shown in Supplementary Fig. 3. Neither the TRPV1 staining in DRGs nor the relative mRNA expression levels in the sciatic nerve showed any significant differences in the low BTZ concentration (data not shown).
BTZ increases CD138+ plasma cells in bone marrow tissue
Since high dose BTZ depletes plasma cells in patients with multiple myeloma, we examined CD138+ cells in the low dose context. CD138 (Syndecan1) is commonly used in immunohistochemistry to quantify plasma cells.33 Plasma cells can be found in very low numbers as circulating plasma cells in secondary lymphoid organs, and higher numbers as long-lived plasma cells in the bone marrow. Our histological analysis showed a significant increase of CD138+-cells after low dose application of 0.05 mg/kg BTZ with a mean value of 173.7 ± 80.1 cells per mm² bone marrow tissue compared to the CTR group with a mean value of 140.9 ± 113.8 plasma cells per mm² (P-value = 0.005, n = 8, pooled results of two experiments) (Supplementary Fig. 4A). The data are indicating the same effect, when BTZ is administered at a concentration of 0.1 mg/kg BTZ (mean values ± SD in CTR = 140.9 ± 113.8; 0.1 mg/kg BTZ = 194.4 ± 99.03; P-value = 0.009). Representative pictures are shown in Supplementary Fig. 4B.
Discussion
The purpose of our study was to investigate differential effects and underlying mechanisms of BTZ activity in high toxic as well as in lower-immunomodulatory concentrations. First, we performed a dose-titration study and defined the toxic threshold concentration of 0.2 mg/kg BTZ in healthy Lewis rats. Lower concentrations of BTZ (0.05 mg/kg) were not toxic. This was verified by vF test, histology and electrophysiological measurements. In contrast, this low dosage exerted significant and reproducible therapeutic efficacy in the rodent EAN model.
Our findings regarding the characteristics of the toxic effects are in accordance with clinical findings in patients with multiple myeloma and BiPNP. In these patients, high concentration of BTZ reduces clonal plasma cell expansion at the cost of toxic effects in the peripheral nerves by induction of a painful, sensory neuropathy.14–16 This toxic neuropathy is dose-dependent and is observed after 2–4 cycles of BTZ treatment for most patients.34 The toxic effects of BTZ are the main reason for drug discontinuation in multiple myeloma treatment. In accordance with clinical and preclinical data, hyperalgesia in our toxic experimental model was shown in the vF test, indicating a toxic sensory nerve fibre involvement (Fig. 1) at a concentration of 0.2 mg/kg.18,35 Electrophysiology did not show any signs of motor axon degeneration after treatment with 0.05 or 0.2 mg/kg BTZ, implying that small sensory fibres are more vulnerable in our toxic model. Surprisingly, DRG outgrowth studies have shown a crucial preserved regeneration potential of DRG after toxic BTZ treatment. This effect is of main clinical importance as immediate withdrawal of the drug after development of a toxic sensory neuropathy can reverse neuropathic symptoms in multiple myeloma patients with BiPNP.15–17 Interestingly, the high concentration of BTZ did not exert any immunomodulatory effects in the flow cytometry analyses or qPCR on immune cell populations. NFKB pathway was not modulated. Previous preclinical studies evaluated the toxicity of BTZ in a concentration of 0.2 mg/kg in rats as well. However, the results are difficult to compare. Our EAN model bases on 6–8 weeks old female Lewis rats, while Yamamoto et al.18 evaluated hyperalgesia after BTZ treatment in adult male Sprague Dawley rats. Quartu et al.32 analysed toxic effects of BTZ in Wistar rats and Duggett and Flatters35 evaluated hyperalgesia in Sprague Dawley rats through withdrawal frequencies instead of the 50% paw withdrawal threshold. Despite all these different settings, the clinical impact of high dosage BTZ on hyperalgesia was the same in each experimental design. In accordance to our data, the dose of 0.2 mg/kg initiated reversible hypersensitivity. Long-term examination of allodynia after discontinuation of treatment with BTZ revealed the recovering of hypersensitivity.35 Our DRG outgrowth study supports these findings. Indeed, modelling toxic effects of chemotherapeutic treatment on peripheral nerves represents a challenge. In particular, the examination of the underlying molecular mechanisms may be variable due to the chosen experimental design. Lehmann et al.36 described in 2019 the challenges and limitations of severe methods modelling chemotherapy induced peripheral neuropathies, underlying, for example, the impact of animal age for cell culture. However, since we aimed to analyse the therapeutic effect of BTZ in low concentrations, we decided to focus on the clinical aspects in the toxic setting. Therefore, and to guarantee the comparability of both settings, adult Lewis rats were chosen for experiments, since EAN model is well-characterized for animals of this age.
We confirmed data from CIDP patients that lower concentrations of BTZ have a different mode of action and exert immunomodulatory effects in the therapeutic context in Lewis rat EAN. We applied the non-toxic concentration of 0.05 mg/kg BTZ, and we were able to verify the positive impact of BTZ on EAN disease course and EAN-induced hyperalgesia. The amelioration of clinical disease was confirmed by NCS. Low concentrations of BTZ prevented the reduction of MNCV significantly, and histological analyses revealed a reduction of infiltrating T cells and macrophages into the sciatic nerve. Moreover, further conclusive data were obtained through the study of demyelination of the PNS. FluoroMyelin™ staining was able to visualize a reduction of demyelination with low dose BTZ treatment. Overall, the clinical data and histological analyses show that BTZ has an immunomodulatory effect at a concentration of 0.05 mg/kg.
Previous studies already showed the efficiency of BTZ on other autoimmune diseases. The proteasome inhibitor has been already applied to patients with CIDP,12 myasthenia gravis,37 neuromyelitis optica38 and systemic lupus erythematodes.33,39,40 However, the appearance of neurotoxic side effects like BiPNP has restrained research on further modes of action of proteasome inhibitors.15–17 As reported above, this toxic polyneuropathy occurs mainly when BTZ is administered frequently (1.3 mg/m² given s.c. on Days 1, 4, 8 and 11, and every 21 days thereafter for three or four cycles) in multiple myeloma patients. Our experimental setup shows for the first time that one cycle of low dose BTZ application can ameliorate clinical, electrophysiological and histological signs of neuritis. In accord with these observations was the finding that low dose BTZ even ameliorated EAN-induced hyperalgesia.
Regarding the mechanisms of immunomodulation in the low concentration, we investigated a variety of possible cellular and humoral mechanisms. For example, the immune proteasome is involved in the NFKB pathway and antigen presentation.41,42 Further mechanisms such as terminal unfolded protein response are described, which were discussed to be responsible for the adverse drug events.43 Our study shows that only low-dose BTZ is involved in the NFKB pathway modulation in this context through the inhibition of IKB degradation, the inhibitor of NFKB.
Theoretically, systemic application of BTZ, even at this low concentration, may potentially modulate immune cell populations in the peripheral lymphoid organs and bone marrow. Interestingly, staining for CD8+ and CD4+ T cells, CD11b+ monocytes, CD4+CD11b+ DCs, CD4+CD25+FoxP3+ T-regs, CD4+CD11b–MHCII+ plasmacytoid DCs, CD19+ and CD45R+ B cells, and IgK light-chainhigh+ plasma cells did not reveal any quantitative effects on immune cell population in spleen, lymph nodes and peripheral blood.
The increase of Ikba and the reduction of Nfkb mRNA expression levels rather indicates a qualitative modulation on the immune system, a shift towards an anti-inflammatory milieu by decreasing NFKB, the main transcription factor promoting inflammation. No shift in the pro-inflammatory cytokine milieu was detected in the sciatic nerve which could imply that our investigations may have missed the changes in cytokine milieu in the recovery phase. Further experiments should focus on EAN disease maximum to clarify this question.
The aspect of plasma cell depletion in the low-dose of BTZ was further investigated, and we were able to show that plasma cell depletion in the bone marrow is not induced by low-dose BTZ. On the contrary, a probably compensatory increase of CD138+ plasma cells in the bone marrow was found. This aspect could mediate EAN’s immunomodulatory effects, long-living plasma cells in the bone marrow could also exert immunomodulatory function.44 However, further studies are required to confirm this assumption.
Commenting on the finding of hyperalgesia in the context of EAN, we must stress the point that in a subgroup of patients with immune-mediated neuropathies, disabling hyperalgesia is presenting early in the disease course. The mechanisms mediating this presentation have not been clarified yet. We show that the classic EAN model in Lewis rats is characterized by hyperalgesia. This finding opens novel options for the experimental investigation of sensory neuropathies. Previous studies showed that TRPV1 and CGRP upregulation could be involved in the BiPNP in a high dose.32 We have investigated the expression of TRPV1 channel in the low and high dose BTZ concept. We could not show that its expression mediates the described clinical effects, neither in qPCR nor in histology. However, our studies were also performed during the recovery phase and may have missed the modulation of TRPV1 directly after BTZ toxic application or at EAN disease maximum.
Concluding, novel peripheral immunomodulatory mechanisms of proteasome inhibition should also be considered when applying BTZ42,45 in the context of autoimmune diseases. In low concentrations, there seems to be an unrecognized and promising therapeutic potential for these substances. Further investigations on the use of selective immunoproteasome inhibitors may be interesting since they cause fewer adverse events.41,42,46
Our results may introduce a novel potential for proteasome inhibition in the field of autoimmune polyneuropathies. In this study and in agreement with previous clinical trials and case studies, BTZ may be a therapy option for immune-mediated neuropathies. BTZ seems to exert toxic effects in high concentration and immunomodulatory effects in low concentration. Further experiments must reveal the exact role of the NFKB pathway in this context.
Supplementary material
Supplementary material is available at Brain Communications online.
Supplementary Material
Acknowledgements
We thank Hussein Bachir, former member of our working group, for his participation in early stage of this project. Furthermore, we thank Katharina Klöster and Lisa Holtkamp for excellent technical support.
Funding
R.K. received a research scholarship and travel funding from the medical faculty (FoRUM-Program, grant number: P046-17) and the research-school (Project.International PR.INT, grant number B_2018_07_003) of Ruhr-University Bochum.
Conflict of interest
J.M. received travel grants from Biogen Idec, Novartis AG, Teva, and Eisai GmbH. His research is funded by Klaus Tschira Foundation and Ruhr-University Bochum (FoRUM-Program), not related to this manuscript; T.G. received travel reimbursement from Sanofi Genzyme and Biogen Idec, none related to this manuscript; C.S.-G. has received consulting and speaker’s honoraria from Alexion Pharmaceuticals, Amicus Therapeutics, Bayer Schering, CSL Behring, Grünenthal, Lupin Pharmaceuticals and TEVA, none related to this manuscript; A.R.-S. received honoraria, research funding and has an advisory role for Amgen, Roche, Pfizer, Sanofi, Merck Serono, Shire, Celgene, Lilly, Bristol-Myers Squibb, Servier, Baxalta, Merck Sharp & Dohme, Aurikamed, Bonita Pharmaceuticals, Iomedico, MCI (managing meetings in cancer care and education), Med Publico, Promedicis, not related to this study; A.T. received honoraria from Falk Foundation, Merck, Amgen, Pfizer, Med Update, not related to this study; M.-S.Y. has received speaker honoraria from CSL Behring and Grifols, a scientific grant from CSL Behring, not related to this study; R.G. has received consultation fees and speaker honoraria from Bayer Schering, Biogen idec, Merck Serono, Novartis, Sanofi-Aventis and TEVA. He also acknowledges grant support from Bayer Schering, Biogen idec, Merck Serono, Sanofi-Aventis and TEVA, none related to this manuscript; K.P. received travel funding and speaker honoraria from Biogen Idec, Novartis, Grifols, CSL Behring, Celgene and Bayer Schering Pharma and funding from the Ruhr-University, Bochum (FORUM-Program).
Glossary
- AUC =
area under the curve
- BiPNP =
bortezomib-induced polyneuropathy
- BTZ =
bortezomib
- CFA =
complete Freund’s adjuvant
- CIDP =
chronic inflammatory demyelinating polyneuropathy
- CMAP =
compound muscle action potentials
- CTR =
control
- DAPI =
4′, 6′-diamidin-2-phenylindol
- DC =
dendritic cell
- DMEM =
Dulbecco’s modified eagle media
- DMSO =
dimethyl sulphoxide
- DRG =
dorsal root ganglion
- EAN =
experimental autoimmune neuritis
- FBS =
foetal bovine serum
- i.p. =
intraperitoneally
- MNCV =
motor nerve conduction velocity
- NCS =
nerve conduction studies
- PBS =
phosphate-buffered saline
- PFA =
paraformaldehyde
- p.i. =
post immunization
- PNS =
peripheral nervous system
- s.c. =
subcutaneous
- SD =
standard deviation
- vF =
von Frey hair test
References
- 1. Broers MC, Bunschoten C, Nieboer D, Lingsma HF, Jacobs BC.. Incidence and prevalence of chronic inflammatory demyelinating polyradiculoneuropathy: A systematic review and meta-analysis. Neuroepidemiology. 2019;52(3-4):161–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mahdi-Rogers M, Hughes RAC.. Epidemiology of chronic inflammatory neuropathies in southeast England. Eur J Neurol. 2014;21(1):28–33. [DOI] [PubMed] [Google Scholar]
- 3. McLeod JG, Pollard JD, Macaskill P, Mohamed A, Spring P, Khurana V.. Prevalence of chronic inflammatory demyelinating polyneuropathy in New South Wales, Australia. Ann Neurol. 1999;46(6):910–913. [PubMed] [Google Scholar]
- 4. Joint Task Force of the EFNS and the PNS. European Federation of Neurological Societies/Peripheral Nerve Society Guideline on management of paraproteinemic demyelinating neuropathies. Report of a Joint Task Force of the European Federation of Neurological Societies and the Peripheral Nerve Society—First revision. J Peripher Nerv Syst. 2010;15(3):185–195. [DOI] [PubMed] [Google Scholar]
- 5. Cocito D, Paolasso I, Antonini G, et al. ; on behalf of The Italian Network for CIDP Register. A nationwide retrospective analysis on the effect of immune therapies in patients with chronic inflammatory demyelinating polyradiculoneuropathy. Eur J Neurol. 2010;17(2):289–294. [DOI] [PubMed] [Google Scholar]
- 6. Fisse AL, Motte J, Grüter T, Sgodzai M, Pitarokoili K, Gold R.. Comprehensive approaches for diagnosis, monitoring and treatment of chronic inflammatory demyelinating polyneuropathy. Neurol Res Pract. 2020;2:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yoon MS, Chan A, Gold R.. Standard and escalating treatment of chronic inflammatory demyelinating polyradiculoneuropathy. Ther Adv Neurol Disord. 2011;4(3):193–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cocito D, Grimaldi S, Paolasso I, et al. ; Italian Network for CIDP Register. Immunosuppressive treatment in refractory chronic inflammatory demyelinating polyradiculoneuropathy. A nationwide retrospective analysis. Eur J Neurol. 2011;18(12):1417–1421. [DOI] [PubMed] [Google Scholar]
- 9. Harvey GK, Gold R, Hartung HP, Toyka KV.. Non-neural-specific T lymphocytes can orchestrate inflammatory peripheral neuropathy. Brain. 1995;118(5):1263–1272. [DOI] [PubMed] [Google Scholar]
- 10. Hughes RAC, Donofrio P, Bril V, et al. ; ICE Study Group. Intravenous immune globulin (10% caprylate-chromatography purified) for the treatment of chronic inflammatory demyelinating polyradiculoneuropathy (ICE study): A randomised placebo-controlled trial. Lancet Neurol. 2008;7(2):136–144. [DOI] [PubMed] [Google Scholar]
- 11. Yan WX, Taylor J, Andrias-Kauba S, Pollard J.. Passive transfer of demyelination by serum or IgG from chronic inflammatory demyelinating polyneuropathy patients. Ann Neurol. 2000;47(6):765–775. [PubMed] [Google Scholar]
- 12. Pitarokoili K, Yoon M-S, Kröger I, Reinacher-Schick A, Gold R, Schneider-Gold C.. Severe refractory CIDP: A case series of 10 patients treated with bortezomib. J Neurol. 2017;264(9):2010–2020. [DOI] [PubMed] [Google Scholar]
- 13. Chen D, Frezza M, Schmitt S, Kanwar J, Dou Q.. Bortezomib as the first proteasome inhibitor anticancer drug: Current status and future perspectives. Curr Cancer Drug Targets. 2011;11(3):239–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Utley A, Lipchick B, Lee KP, Nikiforov MA.. Targeting multiple myeloma through the biology of long-lived plasma cells. Cancers (Basel). 2020;12(8):2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dai X, Sun X, Ni H, Zhu X.. Guillain Barré syndrome in a multiple myeloma patient after the first course of bortezomib therapy: A case report. Oncol Lett. 2015;10(5):3064–3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Elghouche A, Shokri T, Qin Y, Wargo S, Citrin D, van Waes C.. Unilateral cervical polyneuropathies following concurrent bortezomib, cetuximab, and radiotherapy for head and neck cancer. Case Rep Otolaryngol. 2016;2016:2313714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Richardson PG, Briemberg H, Jagannath S, et al. Frequency, characteristics, and reversibility of peripheral neuropathy during treatment of advanced multiple myeloma with bortezomib. J Clin Oncol. 2006;24(19):3113–3120. [DOI] [PubMed] [Google Scholar]
- 18. Yamamoto S, Kawashiri T, Higuchi H, et al. Behavioral and pharmacological characteristics of bortezomib-induced peripheral neuropathy in rats. J Pharmacol Sci. 2015;129(1):43–50. [DOI] [PubMed] [Google Scholar]
- 19. Enders U. The role of the very late antigen-4 and its counterligand vascular cell adhesion molecule-1 in the pathogenesis of experimental autoimmune neuritis of the Lewis rat. Brain. 1998;121(7):1257–1266. [DOI] [PubMed] [Google Scholar]
- 20. Pitcher GM, Ritchie J, Henry JL.. Paw withdrawal threshold in the von Frey hair test is influenced by the surface on which the rat stands. J Neurosci Methods. 1999;87(2):185–193. [DOI] [PubMed] [Google Scholar]
- 21. Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL.. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53(1):55–63. [DOI] [PubMed] [Google Scholar]
- 22. Taylor JM, Pollard JD.. Neurophysiological changes in demyelinating and axonal forms of acute experimental autoimmune neuritis in the Lewis rat. Muscle Nerve. 2003;28(3):344–352. [DOI] [PubMed] [Google Scholar]
- 23. Kohle F, Sprenger A, Klein I, Fink GR, Lehmann HC.. Nerve conductions studies in experimental models of autoimmune neuritis: A meta-analysis and guideline. J Neuroimmunol. 2021;352:577470. [DOI] [PubMed] [Google Scholar]
- 24. Saad A, Palm M, Widell S, Reiland S.. Differential analysis of rat bone marrow by flow cytometry. Comp Haematol Int. 2000;10(2):97–101. [Google Scholar]
- 25. Gomez AM, Vrolix K, Martínez-Martínez P, et al. Proteasome inhibition with bortezomib depletes plasma cells and autoantibodies in experimental autoimmune myasthenia gravis. J Immunol. 2011;186(4):2503–2513. [DOI] [PubMed] [Google Scholar]
- 26. Shin JE, Miller BR, Babetto E, et al. SCG10 is a JNK target in the axonal degeneration pathway. Proc Natl Acad Sci U S A. 2012;109(52):E3696–E3705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gobrecht P, Leibinger M, Andreadaki A, Fischer D.. Sustained GSK3 activity markedly facilitates nerve regeneration. Nat Commun. 2014;5:4561. [DOI] [PubMed] [Google Scholar]
- 28. Förster S, Tannapfel A.. Einsatz monoklonaler Antikörper in der pathologischen Diagnostik.[Use of monoclonal antibodies in pathological diagnostics]. Internist (Berl). 2019;60(10):1021–1031. [DOI] [PubMed] [Google Scholar]
- 29. Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29(9):e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gobrecht P, Andreadaki A, Diekmann H, Heskamp A, Leibinger M, Fischer D.. Promotion of functional nerve regeneration by inhibition of microtubule detyrosination. J Neurosci. 2016;36(14):3890–3902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Alé A, Bruna J, Calls A, et al. Inhibition of the neuronal NFκB pathway attenuates bortezomib-induced neuropathy in a mouse model. Neurotoxicology. 2016;55:58–64. [DOI] [PubMed] [Google Scholar]
- 32. Quartu M, Carozzi VA, Dorsey SG, et al. Bortezomib treatment produces nocifensive behavior and changes in the expression of TRPV1, CGRP, and substance P in the rat DRG, spinal cord, and sciatic nerve. Biomed Res Int. 2014;2014:180428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chilosi M, Adami F, Lestani M, et al. CD138/syndecan-1: A useful immunohistochemical marker of normal and neoplastic plasma cells on routine trephine bone marrow biopsies. Mod Pathol. 1999;12(12):1101–1106. [PubMed] [Google Scholar]
- 34. Li T, Timmins HC, King T, Kiernan MC, Goldstein D, Park SB.. Characteristics and risk factors of bortezomib induced peripheral neuropathy: A systematic review of phase III trials. Hematol Oncol. 2020;38(3):229–243. [DOI] [PubMed] [Google Scholar]
- 35. Duggett NA, Flatters SJL.. Characterization of a rat model of bortezomib-induced painful neuropathy. Br J Pharmacol. 2017;174(24):4812–4825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lehmann HC, Staff NP, Hoke A.. Modeling chemotherapy induced peripheral neuropathy (CIPN) in vitro: Prospects and limitations. Exp Neurol. 2020;326:113140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Schneider-Gold C, Reinacher-Schick A, Ellrichmann G, Gold R.. Bortezomib in severe MuSK-antibody positive myasthenia gravis: First clinical experience. Ther Adv Neurol Disord. 2017;10(10):339–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhang C, Shi F-D.. Bortezomib for neuromyelitis optica spectrum disorder: A new therapeutic option for the more severe forms?-Reply. JAMA Neurol. 2018;75(1):129–130. [DOI] [PubMed] [Google Scholar]
- 39. Neubert K, Meister S, Moser K, et al. The proteasome inhibitor bortezomib depletes plasma cells and protects mice with lupus-like disease from nephritis. Nat Med. 2008;14(7):748–755. [DOI] [PubMed] [Google Scholar]
- 40. Alexander T, Sarfert R, Klotsche J, et al. The proteasome inhibitior bortezomib depletes plasma cells and ameliorates clinical manifestations of refractory systemic lupus erythematosus. Ann Rheum Dis. 2015;74(7):1474–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Verbrugge SE, Scheper RJ, Lems WF, de Gruijl TD, Jansen G.. Proteasome inhibitors as experimental therapeutics of autoimmune diseases. Arthritis Res Ther. 2015;17:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kubiczkova L, Pour L, Sedlarikova L, Hajek R, Sevcikova S.. Proteasome inhibitors—Molecular basis and current perspectives in multiple myeloma. J Cell Mol Med. 2014;18(6):947–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Arastu-Kapur S, Anderl JL, Kraus M, et al. Nonproteasomal targets of the proteasome inhibitors bortezomib and carfilzomib: A link to clinical adverse events. Clin Cancer Res. 2011;17(9):2734–2743. [DOI] [PubMed] [Google Scholar]
- 44. Wang A, Rojas O, Lee D, Gommerman JL.. Regulation of neuroinflammation by B cells and plasma cells. Immunol Rev. 2021;299(1):45–60. [DOI] [PubMed] [Google Scholar]
- 45. Groen K, van de Donk N, Stege C, Zweegman S, Nijhof IS.. Carfilzomib for relapsed and refractory multiple myeloma. Cancer Manag Res. 2019;11:2663–2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Fricker LD. Proteasome inhibitor drugs. Annu Rev Pharmacol Toxicol. 2020;60:457–476. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data of this study are available from the corresponding author, upon reasonable request.