

Abstract
A 41-year-old man was admitted with a chief complaint of dyspnea. Echocardiography showed diffuse severe hypokinesis in the left ventricle. Although his heart failure improved, high creatine kinase levels persisted. A muscle biopsy of the biceps brachii showed necrotic and regenerating fibers along with positive findings for major histocompatibility complex class I and membrane attack complex. He was diagnosed with antibody-negative immune-mediated necrotizing myopathy (IMNM). Steroid therapy was started, but he died due to ventricular fibrillation. Autopsy findings revealed CD68-positive macrophages in the myocardium and quadriceps. To our knowledge, this is the first case of antibody-negative IMNM with cardiac involvement.
Keywords: immune-mediated necrotizing myopathy, secondary cardiomyopathy, cardiac magnetic resonance imaging, autopsy
Introduction
Immune-mediated necrotizing myopathy (IMNM) is an unusual and rare subgroup of idiopathic inflammatory myopathies. The incidence of idiopathic inflammatory myopathies in a homogenous population is 10 to 15 per million. Although the exact prevalence is unclear, previous studies have shown that IMNM affects approximately 7 to 11 in 100,000 people per year (1-3). IMNM presents with severe proximal limb muscle weakness and elevated serum creatine kinase (CK) levels. IMNM is characterized by pathognomonic features of myofiber necrosis with minimal inflammatory cell infiltrate on a muscle biopsy. IMNM is classified into three subtypes: anti-signal recognition particle (SRP) myopathy, anti-3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR) myopathy, and antibody-negative IMNM. Cardiac involvement is relatively frequent in anti-SRP myopathy and rare in anti-HMGCR myopathy. The prevalence of antibody-negative IMNM with cardiac complications is unclear. Severe cardiac involvement can be critical; therefore, its diagnosis is particularly important.
We herein report a case diagnosed with antibody-negative IMNM with cardiac involvement.
Case Report
A 41-year-old man presented to our hospital with lower leg edema and dyspnea at rest. He had experienced exertional dyspnea with light exercise for two years but never noticed limb muscle weakness. He had no family history of heart disease or neuromuscular disease. He had no remarkable medical history, including the use of statins.
A physical examination revealed jugular venous distention and peripheral pitting edema. No evident heart murmur or rale was noted, but breath sounds were decreased bilaterally. He staggered when he walked, and a neurological examination showed proximal limb muscle weakness [manual muscle testing (MMT) score: 3/5]. His serum creatine kinase (CK) level was 4,409 U/L, and his CK-muscle/brain level was 229 U/L (normal <12 U/L). His plasma brain natriuretic peptide (BNP) level was also elevated (896.6 pg/mL; normal <18.4), as was his cardiac troponin I level (240 pg/mL; cut-off 26.2).
Chest radiography revealed cardiomegaly (cardiothoracic ratio: 64%), pulmonary congestion, and pleural effusion. An electrocardiogram showed a sinus rhythm and a heart rate of 133 beats per minute, poor R wave progression, and negative T waves in the II, III, and aVF leads. Transthoracic echocardiography showed an enlarged left ventricle (end-diastolic volume: 120 ml, end-systolic volume: 93 ml) with diffuse hypokinesis, severe systolic dysfunction (ejection fraction 20%), and no segmental wall motion abnormalities.
Tricuspid regurgitation was moderate, and mitral regurgitation was mild. He was diagnosed with acute heart failure and treated in a coronary care unit. He received intensive care, including catecholamines, tracheal intubation, and intra-aortic balloon pumping. His heart failure was improved after the therapy, and he was prescribed an angiotensin-converting enzyme inhibitor, β-blocker, and amiodarone. He was discharged after 136 days of hospitalization. However, the low ejection fraction and high serum CK, BNP, and troponin I levels persisted, and his proximal limb muscle weakness progressed despite long-term medication and rehabilitation in the outpatient clinic (Fig. 1).
Figure 1.

Serial changes in the levels of creatine kinase, brain natriuretic peptide, and troponin I in this case. CK: creatine kinase, BNP: brain natriuretic peptide
Cardiac magnetic resonance imaging (CMRI) showed endocardial significant late gadolinium enhancement (LGE) in the anterolateral and inferior walls on LGE imaging (Fig. 2A). LGE in the inferior wall was spotty with a transmural pattern. We suspected ischemic cardiomyopathy as a cause of heart failure, but coronary angiography showed only moderate stenosis in the left anterior descending artery. Muscle MRI showed high-intensity areas in the biceps and triceps on short inversion time inversion recovery imaging (Fig. 2B).
Figure 2.

Late gadolinium enhancement (LGE) on cardiac magnetic resonance imaging (CMRI) (A) and short inversion time inversion recovery on brachial muscle MRI (B). CMRI showed endocardial LGE in the anterolateral and inferior walls and spotty transmural LGE in the inferior wall.
We suspected myopathy involving cardiomyopathy, such as muscular dystrophies (Becker muscular dystrophy or limb-girdle muscular dystrophy), endocrine myopathies (hypothyroid myopathy), lysosomal storage disease (Pompe disease or Danon disease), mitochondrial myopathy, and inflammatory myopathies (polymyositis or dermatomyositis or paraneoplastic myopathy or IMNM). None of the markers related to connective tissue disease or endocrine disease were abnormal (Table). Whole-body computed tomography showed no evidence of malignancy. There were no mutations in dystrophin genes. An endomyocardial biopsy of the right ventricular septum performed after 1,040 days of hospitalization (CK 1,638 U/L, BNP 146 pg/mL, troponin I 63 pg/mL) did not show any obvious myocardial necrosis or interstitial fibrosis. Macrophage infiltration was observed, but infiltration of lymphocytes, eosinophils, and neutrophils was minimal (Fig. 3A). Electron microscopy revealed myofibril disarrangement and proliferation of mitochondria in the myocytes but did not show any giant mitochondria consistent with mitochondrial myopathy or abnormalities in vacuoles consistent with lysosomal storage disease (Fig. 3B). A muscle biopsy of the left biceps brachii performed after 1,309 days of hospitalization (CK 4,775 U/L, BNP180 pg/mL) revealed necrosis and regeneration of muscle fibers (Fig. 4A). Macrophage infiltration was observed, but again, infiltration of lymphocytes, eosinophils, and neutrophils was minimal. Immunostaining revealed major histocompatibility complex (MHC) class I expression (Fig. 4B) and membrane attack complex (MAC) deposition on the sarcolemma (Fig. 4C). These findings were consistent with those of IMNM. Serology testing revealed that the patient had no antibodies, such as anti-SRP and anti-HMGCR. Therefore, he was diagnosed with antibody-negative IMNM with cardiac involvement.
Table.
Serologic Evaluation of Autoimmune Disease That Can Cause Myocardial Damage.
| Antibody | Results |
|---|---|
| Anti-nuclear | <40 |
| Anti-DNA | <=2.0 |
| Anti-RNP | negative |
| Anti-SSA/RO | negative |
| Anti-SSB/LA | negative |
| Ant-SCL-70 | negative |
| Anti-Centromere | <5.0 |
| Anti-JO-1 | negative |
| Anti-SRP | negative |
| Anti-HMGCR | negative |
| Anti-MitM2 | negative |
| Anti-ARS | negative |
| Anti-MDA5 | negative |
DNA: deoxyribonucleic acid, RNP: ribonucleoprotein, SRP: signal recognition particle, HMGCR: 3-hydroxy-3-methylglutaryl coenzyme A reductase, Mit: mitochondria, ARS: amino acyl-tRNA synthetases, MDA5: melanoma differentiation-associated gene 5
Figure 3.

An endomyocardial biopsy from the right ventricular septum did not show obvious myocardial necrosis or interstitial fibrosis. Macrophage infiltration was observed, but infiltration of lymphocytes, eosinophils, and neutrophils was only slight (A). Electron microscopy revealed myofibril disarrangement and the proliferation of mitochondria in the myocytes but no abnormalities in vacuoles consistent with secondary cardiomyopathy (B).
Figure 4.

A muscle biopsy of the left biceps brachii revealed necrosis and regeneration of muscle fibers (A). Macrophage infiltration was observed, but infiltration of lymphocytes, eosinophils, and neutrophils was only slight. Immunostaining revealed major histocompatibility complex (MHC) class I expression (B) and membrane attack complex (MAC) deposition on sarcolemma (C).
After 1,634 days of hospitalization (CK 1,638 U/L, BNP 146 pg/mL, troponin I 63 pg/mL), he was started on steroid pulse therapy (intravenous administration of methylprednisolone 1,000 mg for one day and dexamethasone 40 mg for 2 days) followed by 8 mg dexamethasone. After the steroid therapy, his serum CK levels gradually normalized, but his muscle weakness, serum BNP levels, and left ventricular ejection fraction did not improve (Fig. 5).
Figure 5.
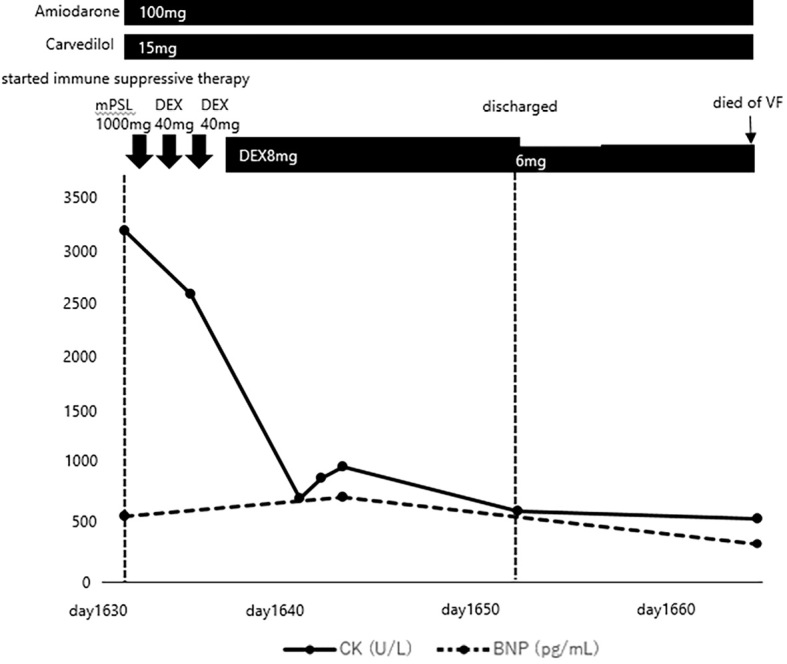
Treatment course of this case. CK: creatine kinase, BNP: brain natriuretic peptide, mPSL: methylprednisolone, DEX: dexamethasone, VF: ventricular fibrillation
Eventually, he died of ventricular fibrillation after 1,663 days of hospitalization despite treatment, and an autopsy was performed with the consent of his family.
Pathological findings
The heart was dilated and weighed 500 g (normal weight for men, 300-350 g). The cut surface revealed a broad, grayish-white tissue in the left and right ventricular subendocardium (Fig. 6A). The grayish-white tissue was mainly observed in the anterior, lateral, and inferior walls of the left ventricle and less frequently in the septum. Microscopically, cardiomyocytes in the left and right ventricles were extensively replaced by fibrotic tissues in the endocardium predominantly (Fig. 6B). Masson trichrome staining showed the replacement of collagen fibers in the myocardium (Fig. 6D).
Figure 6.

Pathological findings of the heart. The cut surface revealed broad, grayish-white tissue in the left and right ventricular subendocardium (A). Hematoxylin and Eosin staining showed that the cardiomyocytes in the ventricle had been extensively replaced by fibrotic tissues in the endocardium predominantly (B), and there was loss of myocytes, interstitial fibrosis, hypertrophic myocytes, binucleated myocytes, and variations in the sizes of the myocyte nuclei. No inflammatory cell infiltration, such as with lymphocytes and eosinophils, was detected (C). Masson-Trichrome staining showed the replacement by collagen fiber in the myocardium (D).
Hematoxylin and Eosin staining showed myocyte loss, interstitial fibrosis, hypertrophic myocytes, binucleated myocytes, and variations in the sizes of the myocyte nuclei (Fig. 6C). No inflammatory cell infiltration, such as lymphocytes and eosinophils, was found. However, immunostaining showed the infiltration of CD68-positive macrophages (Fig. 7A). Immunostaining was negative for CD3, CD4, CD8, CD20, and CD138 antibodies (Fig. 7B, C). The myocytes in the quadriceps muscle were highly atrophic and replaced by adipose tissue. Immunostaining of myocytes in the quadriceps muscle showed the infiltration of macrophages that were positive for CD-68 (Fig. 7D) and negative for CD-3, CD-4, CD-8, CD-20, and CD-138 antibodies, as well as cardiomyocytes (Fig. 7E, F).
Figure 7.

Immunostaining findings in the heart (A-C) and quadriceps (D-F) at the autopsy. In the heart, immunostaining showed the infiltration of CD68-positive macrophages (A). All other immunostaining antibodies were negative (B, C). Immunostaining with myocytes in the quadriceps muscle showed infiltration of CD68-positive macrophages (D) and negative findings for other antibodies, as well as cardiomyocytes (E, F).
Discussion
IMNM presents with acute or progressive proximal limb muscle weakness, elevated serum CK levels, and infrequent extramuscular involvement. Histologically, IMNM is defined by the presence of myofiber necrosis and prominent regeneration with minimal or no inflammatory cell infiltration. In IMNM patients, MHC-I and MAC are positively stained for not only necrotic fibers or vessels but also non-necrotic ones, and the presence of CD68-positive macrophages is observed mainly in the necrotic muscle fibers and endomysial connective tissue in immunohistochemical analyses (4). IMNM is associated with anti-SRP antibody, anti-HMGCR antibody, and other antibodies, such as anti-mitochondrial M2 protein antibody and aminoacyl t-RNA synthetase antibody (5).
The frequency of these antibodies in IMNM differs among reports, but IMNM is classified into three subtypes in almost all studies: anti-SRP myopathy, anti-HMGCR myopathy, and antibody-negative IMNM. Watanabe et al. reported that in IMNM, 26% had anti-HMGCR myopathy, 39% had anti-SRP myopathy, and 35% were negative for these two autoantibodies in Japan (6). Cardiac involvement is relatively frequent in anti-SRP myopathy and rare in anti-HMGCR myopathy (2). Antibody-negative IMNM has distinctive features from antibody-positive IMNM (7) but it is less well understood, and minimal diagnostic and treatment guidance is available in the literature (8). Furthermore, limited information is available on the prevalence, diagnostic methods, and treatment of antibody-negative IMNM with cardiac involvement. Severe cardiac involvement can be critical to IMNM patients; therefore, its diagnosis and treatment are particularly important.
In our case, CMRI showed significant endocardial late gadolinium enhancement, which was consistent with the pathological findings of significant endocardial fibrosis. There are few case reports on CMRI findings of IMNM. Thiébaut et al. and Takeguchi-Kikuchi et al. reported instances of anti-SRP positive IMNM with cardiac involvement (9,10). Their CMRI findings showed transmural and subendocardial LGE, similar to our CMRI findings. These findings were different from the well-known finding of significant epicardial LGE involved with lymphocytic myocarditis and may be common findings in cases of cardiomyopathy with IMNM.
It is hypothesized that the causes of myocardial dysfunction with inflammatory myopathies are myocarditis, myocardial infarction due to accelerated atherosclerosis, and coronary vasculopathy (11,12). The autopsy of this case suggested that the myocardial dysfunction in IMNM was caused by an immunological mechanism rather than an ischemic mechanism. There was no obstruction in the coronary artery, and immunostaining showed the same changes in cardiomyocytes and muscle cells of the quadriceps muscle. This result strongly suggests that the myocardium and skeletal muscle may be damaged by the same mechanism. Furthermore, steroid treatment was insufficient for myocardial and skeletal muscle disorders in this case. It is considered that steroid therapy did not suppress macrophages completely, necessitating additional immunosuppressive therapy, such as intravenous immunoglobulin and methotrexate. In addition, immunosuppressive therapy might have been more effective had it been performed earlier.
IMNM is a rare disease and difficult to diagnose and sometimes may not be diagnosed for long periods (13). In the present case, histological scrutiny was delayed despite the cardiac contraction disorders with persistently elevated troponin and CK levels. In addition, our patient had a severely impaired cardiac function and died due to ventricular fibrillation. In cases with severe cardiac involvement, device treatment, such as an implantable cardioverter-defibrillator or cardiac resynchronization therapy defibrillator, should be performed.
Conclusion
IMNM is a rare idiopathic inflammatory myopathy, and its diagnosis is difficult, especially in antibody-negative cases. To our knowledge, this is the first report of the autopsy findings of antibody-negative IMNM with severe cardiac involvement. We should consider IMNM as a differential diagnosis when encountering patients with heart failure accompanied by proximal muscle weakness and persistently high CK levels. Imaging techniques, such as CMRI, muscle biopsies, and myocardial biopsies, are necessary for the diagnosis of IMNM. We should consider early and aggressive immunotherapy and device therapy to improve the prognosis of IMNM patients with severe cardiac involvement.
The authors state that they have no Conflict of Interest (COI).
Acknowledgement
We are grateful to Dr. Ichizo Nishino (Department of Neuromuscular Research, National Institute of Neuroscience, National Center of Neurology and Psychiatry, Japan) for his helpful discussion concerning muscle pathology and Dr. Yoshihiko Ikeda (Department of Pathology, National Cerebral and Cardiovascular Center, Japan).
References
- 1. Meyer A, Meyer N, Schaeffer M, Gottenberg JE, Geny B, Sibilia J. Incidence and prevalence of inflammatory myopathies: a systematic review. Rheumatology (Oxford) 54: 50-63, 2015. [DOI] [PubMed] [Google Scholar]
- 2. Pinal-Fernandez I, Casal-Dominguez M, Mammen AL. Immune-mediated necrotizing myopathy. Curr Rheumatol Rep 20: 21, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Schmidt J. Current classification and management of inflammatory myopathies. J Neuromuscul Dis 5: 109-129, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wang Q, Li Y, Ji S, Feng F, Bu B. Immunopathological characterization of muscle biopsy samples from immune-mediated necrotizing myopathy patients. Med Sci Monit 24: 2189-2196, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Allenbach Y, Mammen AL, Benveniste O; Immune-Mediated Necrotizing Myopathies Working Group. 224th ENMC international workshop: clinico-sero-pathological classification of immune-mediated necrotizing myopathies, Zandvoort, The Netherlands, 14-16 October 2016. Neuromuscul Disord 28: 87-99, 2018. [DOI] [PubMed] [Google Scholar]
- 6. Watanabe Y, Uruha A, Suzuki S, et al. Clinical features and prognosis in anti-SRP and anti-HMGCR necrotising myopathy. J Neurol Neurosurg Psychiatry 87: 1038-1044, 2016. [DOI] [PubMed] [Google Scholar]
- 7. DA Marotta, Zadourian A, Jabaay MJ, Kesserwani A, Kesserwani H. Autoantibody-negative immune-mediated necrotizing myopathy responds to early and aggressive treatment: a case report. Cureus 12: e7827, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lim J, Rietveld A, De Bleecker JL, et al. Seronegative patients form a distinctive subgroup of immune-mediated necrotizing myopathy. Neurol Neuroimmunol Neuroinflamm 6: e513, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Thiébaut M, Terrier B, Menacer S, et al. Antisignal recognition particle antibodies-related cardiomyopathy. Circulation 127: e434-e436, 2013. [DOI] [PubMed] [Google Scholar]
- 10. Takeguchi-Kikuchi S, Hayasaka T, Katayama T, et al. Anti-signal recognition particle antibody-positive necrotizing myopathy with secondary cardiomyopathy: the first myocardial biopsy- and multimodal imaging-proven case. Intern Med 58: 3189-3194, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Opinc AH, Makowski MA, Łukasik ZM, Makowska JS. Cardiovascular complications in patients with idiopathic inflammatory myopathies: does heart matter in idiopathic inflammatory myopathies? Heart Fail Rev 26: 111-125, 2019. [DOI] [PubMed] [Google Scholar]
- 12. Schwartz T, Diederichsen LP, Lundberg IE, Sjaastad I, Sanner H. Cardiac involvement in adult and juvenile idiopathic inflammatory myopathies. RMD Open 2: e000291, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kuru S, Suzuki S, Ogata K, et al. Screening of autoantibodies associated with necrotizing myopathy among undiagnosed chronic myopathy. Clin Neurol 57: 562-566, 2017. [DOI] [PubMed] [Google Scholar]