

Graphical abstract
Keywords: Drug resistance, Falcipains, Missense mutations, Artemisinin, Dynamic residue network, Centrality, Residue contact maps, MD-TASK, MDM-TASK-web, Allostery, Pocket identification
Highlights
-
•
MD-TASK and MDM-TASK-web tools were applied for mutation analysis.
-
•
Cryptic allosteric pocket observed in the presence of M245IPocket 2 became highly rigid in the presence of A353T mutation.
-
•
Allosteric communication paths were identified via centrality hubs.
-
•
Centrality hubs and interactions to other residues changed in some mutants.
Abstract
Continually emerging resistant strains of malarial parasites to current drugs present challenges. Understanding the underlying resistance mechanisms, especially those linked to allostery is, thus, highly crucial for drug design. This forms the main concern of the paper through a case study of falcipain 2 (FP-2) and its mutations, some of which are linked to artemisinin (ART) drug resistance. Here, we applied a variety of in silico approaches and tools that we developed recently, together with existing computational tools. This included novel essential dynamics and dynamic residue network (DRN) analysis algorithms. We identified six pockets demonstrating dynamic differences in the presence of some mutations. We observed striking allosteric effects in two mutant proteins. In the presence of M245I, a cryptic pocket was detected via a unique mechanism in which Pocket 2 fused with Pocket 6. In the presence of the A353T mutation, which is located at Pocket 2, the pocket became the most rigid among all protein systems analyzed. Pocket 6 was also highly stable in all cases, except in the presence of M245I mutation. The effect of ART linked mutations was more subtle, and the changes were at residue level. Importantly, we identified an allosteric communication path formed by four unique averaged BC hubs going from the mutated residue to the catalytic site and passing through the interface of three identified pockets. Collectively, we established and demonstrated that we have robust tools and a pipeline that can be applicable to the analysis of mutations.
Nomenclature
Abbreviations
- 3D
Three Dimension
- Å
Angstrom
- ART
Artemisinin
- BC
Betweenness Centrality
- C
Covariance matrix
- CC
Closeness Centrality
- CHPC
Centre for High Performance Computing
- COM
Center of Mass
- DC
Degree Centrality
- DRN
Dynamic Residue Network
- EC
Eigencentrality
- FPs
Falcipains
- FP-2
Falcipain-2
- GROMACS
GROningen MAchine for Chemical Simulations
- Hb
hemoglobin
- KC
Katz Centrality
- MD
Molecular Dynamics
- ns
nanoseconds
- NPT
constant Number of particles, Pressure and Temperature
- NVT
constant Number of particles, Volume and Temperature
- PDB
Protein Data Bank
- PCA
Principal Component Analysis
- Pf
Plasmodium falciparum
- RMSD
Root Mean Square Deviation
- RMSF
Root Mean Square Fluctuation
- Rg
Radius of gyration
- VMD
Visual Molecular Dynamics
- WT
Wild-type
- ΔG
Gibbs free energy
1. Introduction
Continually emerging resistant strains of pathogens to current drugs present an immense challenge for the eradication of infectious diseases. Understanding the underlying resistance mechanisms at the molecular level using structure-based computational methods and applying this knowledge to future drug design to potentially circumvent the pathogens’ resistance tactics is highly crucial. Yet, this concept has not been well established in computational drug discovery and forms the main concern of this paper through a case study of falcipain 2 (FP-2) protein and its mutations, some of which are linked to malarial drug resistance.
The recent emergence and spread of P. falciparum (Pf) strains with reduced sensitivity against artemisinin (ART) and its partner drugs is a major public health concern threatening ongoing efforts to eradicate malaria globally [1], [2], [3], [4]. Despite being the cornerstone of the current recommended first-line antimalarial drugs, the exact biological target and underlying drug mechanisms of ART remain fairly uncertain [5]. Wang et al., suggested that ART activity is mediated by free radicals generated from the cleavage of its endoperoxide bridge, which lead to plasmodial proteopathy [6]. Besides the established role of mutations in the Kelch 13 protein propeller domain [7] in conferring reduced ART sensitivity and clearance rate by Pf parasites, recent studies have shown the possible involvement of additional target loci [8], [9]. These include a nonsense (S69stop) [10] and four missense mutations in the FP-2 protein; K255R, N257E, T343P and D345G [11].
FP-2 is a papain-like cysteine protease expressed by Pf during the blood-stage parasite development phase within host erythrocytes [12]. Synthesized as zymogens, the enzyme is activated by the lower pH change of the parasite food vacuole [13], [14], [15]. During the activation process, the prodomain promoter region is cleaved off, exposing the active site. The trench-like binding pocket residues, including the catalytic triad (Cys285, His417 and Asn447), are organized into four subsites, namely S1 (Q279, C282, G283, C323, N324), S2 (L327, I328, S392, L415, N416, A418, D477), S3 (K319, N320, Y321, G325, G326) and S1′ (V393, A394, V395, S396, A400, H417, N447, W449) [16] (Fig. 1A & 1B). The catalytic triad is essential for peptide cleavage in which the thiol group of Cys285 initiates a nucleophilic attack of the substrate peptide bonds [16]. FP-2 shares a conserved structural fold with other papain-like Clan CA family enzymes, including human cathepsins (Cat K, L, S). However, together with its homologs from other Plasmodium species, the enzyme possesses unique structural features, viz., longer prodomain (inhibitory) segments and two inserts in the catalytic domain (a ∼ 17 aa nose and a ∼ 14 aa arm) [17], [18], [19], [20].
Fig. 1.

Structural representation of the FP-2 protein. A) and B) Subsites are illustrated as a pale green surface. The ART linked mutations are in firebrick, and the rest of the mutations are in grey spheres. The three catalytic residues (C285, H417, N447) are also labelled. C) and D) Identified pockets are shown in surface presentation. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
FP-2, with other proteases, is responsible for hydrolyzing up to 75% of the host hemoglobin (Hb) as a means of nutrient acquisition [12], [21], [22], [23], [24], [25]. As such, the enzyme is a potential antimalarial drug target [23], [24], [25], [26], [27]. Genetic manipulation via knockout and chemical inhibition of FP-2 resulted in Pf parasites with decreased sensitivity to ART, an indication that Hb digestion process is linked to ART activity [11], [28]. Additionally, the occurrence of missense and nonsense (codon 69) mutations in FP-2 also confer parasites with reduced ART sensitivity, possibly due to altered enzyme efficiency and transient reduction in Hb degradation [11], [29]. Several studies have suggested the involvement of ferrous iron (Fe2+) from Hb degradation in the activation process of ART in situ, but the exact mechanism is still controversial [30], [31], [32].
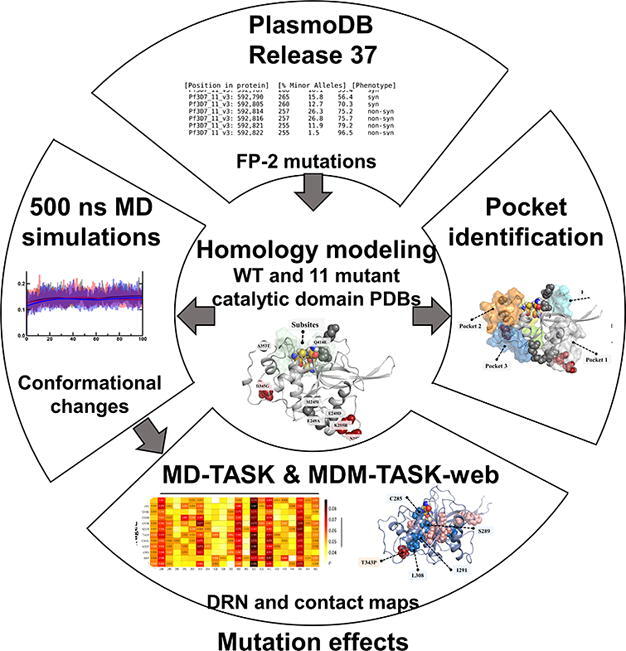
Here, we investigated 11 FP-2 missense mutations retrieved from the Pf genome resource database (PlasmoDB) [33], four of which are linked to ART resistance (Fig. 1A & 1B). The interpretation of structural and dynamic impacts of existing missense mutations in FP-2 may provide significant insight towards understanding their possible effects on the catalytic efficiency, as well as in the prediction of probable resistance mechanisms. The allosteric effect of distant mutations on enzyme function and drug resistance has previously been discussed [34], [35], [36], [37]. Since we proposed the use of dynamic residue network (DRN) in mutation analysis [38], [39] and established the relevant tools [40], [41], this approach has become an integral approach in studying mutation-induced effects in protein structure and function [34], [35], [42]. This article takes advantage of an extended version of DRN to assess the impact of these mutations.
In our recent study, we established new algorithms to investigate the relationships and effectiveness of five DRN metrics (betweenness centrality (BC), closeness centrality (CC), degree centrality (DC), eigencentrality (EC) and katz centrality (KC)) in characterizing allosteric behavior of a protein in the presence of mutations [43]. In the same recent study, we also introduced the concept of analyzing globally central nodes and developed an algorithm to pinpoint the highest centrality nodes for any given averaged centrality metric. These approaches, together with some other MDM-TASK-web tools, traditional post-MD trajectory analysis, pocket analysis, as well as an energy-based approach, formed the basis of the current study. Starting from global analysis, we gradually zoomed into residue level effects of the mutations. These steps included the analysis of mutation effects on the entire protein structure; the identification of changes in the substrate binding pocket and potential allosteric pockets; the description of functionally important persistent hubs; the observation of newly formed communication hubs in the presence of certain mutations travelling to the catalytic triad; the inspection of newly formed or lost residue interactions in the functionally important parts of the protein; and different energy distributions due to distal mutations.
Overall, our results provided novel insights on the possible allosteric mechanisms through which missense mutations may impact the Hb degradation function of FP-2, leading to the development of ART resistance. Further, our results demonstrated the importance of DRN and related approaches that we established previously and extended here to identify subtle communication changes within the protein, which may be more difficult to detect via conventional methods.
2. Methodology
2.1. Data retrieval
The 3D structure of FP-2 [PDB ID: 2OUL] [44] was retrieved from the RCSB Protein Data Bank (PDB) [45], [46]. All crystallographic waters and bound the ligand (chagasin) were removed in PyMOL [47]. Despite the structural annotation of FP-2 indicating that it lacked mutations, available UniProt data (UniProt ID: Q8I6U4) [48] revealed the existence of four missense mutations viz. K255R, N257E, T343P and D345G in the crystal structure. From PlasmoDB 9.3 [49] (release 37, accessed 21-Aug-2018), FP-2 catalytic domain missense mutations (including those linked to ART resistance) were also identified. These comprised M245I, E248D, E249A, K255R, N257E, T343P, D345G, A353T, V393I, A400P, Q414E; mutation numbering is based on the whole protein sequence.
2.2. Wild type and mutant FP-2 3D structures via homology modeling
Due to the mutations present in the retrieved FP-2 crystal structure (PDB ID: 2OUL), a wild-type (WT) catalytic domain 3D structure was predicted in MODELLER version 9.19 [50] using the retrieved structure as a template and a very slow refinement. From the 100 models generated, the structure with the lowest normalized discrete protein energy (z-DOPE) score [51] was selected. Mutant structures were predicted using the modelled WT as a template. The quality of the modeled structures was evaluated by z-DOPE score, Verify3D [52], ProSA [53] and PROHECK [54], and all models showed high quality (Table S1). The characteristic presence of four disulphide bonds involved in the maintenance of structural integrity in the modeled structures was assessed using the protein interaction calculator (PIC) web server [55]. The protein structures were protonated using the PROPKA tool from PDB2PQR (version 2.1.1) [56], [57] to a pH 5.5 to match the acidic environment in the food vacuole before further analysis.
2.3. Identification of potential allosteric pockets
A combination of structure-based approaches was utilized to identify topologically distinct pockets on the FP-2 surface. The FTMAP [58], FTSite [59], Allosite [60] and the Protein Allosteric and Regulatory sites (PARS) [61] web servers were used with default parameters to scan for potential binding pockets with a minimum volume of 100 Å3. Additionally, SiteMap [62] and AutoLigand [63] stand-alone packages from Schrödinger and AutoDock, respectively, were also used to determine the druggability potential of the identified sites based on their physicochemical (D-score) and geometric properties (size and volume).
2.4. Determination of global conformational variability in WT FP-2 and its mutants using molecular dynamics simulations
To determine the effect of the mutations on the structure and dynamics of the catalytic domains of FP-2, MD simulations of up to 500 ns (ns) were performed using the GROMACS 5.1.2 package [64]. At first, test MD runs were performed to determine the most suitable force field parameters. Subsequently, using the AMBER99SB-ILDN [65], the force field of choice, gro and top GROMACS input files for the WT and corresponding mutant FP-2 proteins were prepared. MD simulations were carried out under periodic boundary conditions (PBC) using a triclinic box, with a distance of 1.75 nm placed between the protein and the edge of the box). The simulated boxes were solvated using TIP3P water molecules, and 0.15 M NaCl was added to neutralize the system charges. Energy minimization of the system was performed using the steepest descent algorithm with an initial step of 0.01 nm without constraints until a maximum force of<1000.0 kJ mol−1 nm−1 was achieved. Once the system converged, a two-step equilibration (each 100 ps) was applied to ensure the correct temperature and pressure of the solvated system was attained. The temperature was set at 300 K (NVT – constant number of particles, volume and temperature) using the Berendsen thermostat. Subsequently, pressure equilibration at 1 atm (NPT – constant number of particles, pressure and temperature) was performed using the Parrinello-Rahman barostat [66]. The equilibrated systems were subjected to production MD for 500 ns, with an integration step of 2 fs (femtoseconds) and an average CPU time of ∼ 18,000 h. During the equilibration process and production phase, all bonds were constrained using the LINCS algorithm [67]. Long-range electrostatics were calculated using the Particle-mesh Ewald (PME) algorithm [68] with a Fourier grid spacing of 0.16 nm. A non-bonded cut-off distance of 1.4 nm was used for the Coulomb and van der Waals interactions. After the production phase, the trajectory for each system was stripped of periodic boundary conditions and fitted to the starting frame using the gmx trjconv command. Global and local conformational changes of each system were determined using gmx rms, gmx rmsf, gmx gyrate and analysed in RStudio [69] and using Python libraries including matplotlib [70], NGLview [71], Numpy [72], Pandas [73], and Seaborn [74]. To compare the conformational variability during the course of the simulation, pairwise RMSD calculations were done.
2.5. Comparative essential dynamics of the active site and predicted pockets
Comparative essential dynamics, as implemented by the MDM-TASK-web “compare_essential_dynamics.py” tool [41], was run on a local Linux cluster to assess the distribution of pocket conformational sampling across MD simulations for the WT and 11 mutant FP-2 proteins in the same eigen subspace. In essence, the comparative essential dynamics approach involves data filtering and alignment of all MD frames to a common reference (using Cα atoms) before proceeding to a single decomposition. In this case, frames for the WT and 11 mutant FP-2 protein were aligned to a single frame from the WT simulation before specifying the region of interest, which is used to compute and decompose the covariance matrix. By default, three C-terminal residues are excluded prior to structural alignment. The method was independently applied to FP-2 active site residues and to each of the predicted pockets (shown in Fig. 1). The distributions of active site or predicted pocket conformations were then represented as scatter plots from the first (PC1) and second principal component (PC2) axes. The percentages of total variance explained by each PC are also shown along the axes. Additionally, the lowest energy conformation for the whole proteins obtained from the maxima of the 2D kernel density estimates produced from the two principal components were visualized to detect any significant conformational change.
2.6. Identification of dynamic residue network changes as a result of mutations in FP-2 using centrality analysis
To determine the effect of the missense mutations on the flow of information within the protein residue network, DRN analysis was performed using the newly established MDM-TASK-web server [41]. In this approach, each residue Cβ (Cα for Gly) is treated as a node, while the inter-node proximity is used to define the edges that compose a network graph. NetworkX [75] is used to compute the following metrics at each time frame: betweenness centrality (BC), closeness centrality (CC), degree centrality (DC), eigenvector centrality (EC) and Katz centrality (KC), before averaging the values across each residue. A cut-off distance of 6.7 Å and a step size of 50 frames were used for DRN computation over the entire trajectory for each system. BC is a measure of the usage (hence importance) of a residue for protein communication and represents the number of shortest paths between all other node pairs passing through a given node of interest [40]. CC determines how fast information flows through a node by measuring how close it is to all other nodes in a network [76], [77]. DC is defined as the total number of immediate connections around a node [78]. The higher the number of edges around a node, the more central it is locally. EC measures the level of influence of a node within a network by recursively considering the importance of node neighbors. This is done by solving for the eigenvector, which explains the most variation present in the adjacency matrix, using the power iteration method [75]. Similar to EC, KC determines the relative influence of a node within a network by recursively estimating node centrality based on the centrality of neighboring nodes. The centrality is, however, controlled by two parameters (a dampening coefficient α and a constant β). Table 1 shows the mathematical formulae corresponding to the various centrality metrics used.
Table 1.
Dynamic residue network centrality metrics used to identify residues crucial for communication in FP-2 systems.
| Centrality metric | Formula | Note |
|---|---|---|
| Averaged BC | V is the complete set of nodes; m is the number of frames; is the number of shortest paths connecting nodes s and t; is the number of these paths passing through another node v; and i is the frame number. | |
| Averaged CC | d(v, u) is the shortest-path distance between v and u, and n is the number of nodes in the graph. | |
| Averaged DC | n is the number of nodes; Aijk is the jkth adjacency for the ith frame. | |
| Averaged EC | (a)(b) | (a) EC is the eigenvector, and lambda is the eigenvalue for the eigen decomposition of adjacency matrix A. In NetworkX, this is obtained by power iteration. (b) Averaged EC is computed for ith residue by computing the vector for each MD frame and averaging. |
| Averaged KC | (a) (b) | KC is a modification of EC that employs a dampening coefficient and a constant in order to influence adjacency values. |
2.7. Determination of centrality hubs
Averaged centrality values produced by MDM-TASK-web [41] for related proteins were pooled before extracting the global top 5% central nodes, separately for each metric as recently reported by Sheik Amamuddy et al. [43]. To do so, the centrality values from each protein were concatenated as a vector and sorted in descending order to determine a global cut-off value. This threshold was then used to obtain a binary matrix of the same dimension as the original dataset that contained all the samples. Finally, any residue position that contained a hub was similarly shortlisted from the other samples. The same approach was used for each DRN metric.
2.8. Residue contact map analysis
Residue contact map analysis was performed to estimate how often a residue of interest interacts with its neighboring residues throughout the entire MD trajectory, using the contact_map.py and the contact_heatmap.py scripts from MDM-TASK-web [40], [41]. A cut-off distance of 6.7 Å and a step size of 50 frames were used as parameters for the weighted contact maps, which were then aggregated into a heat map using the contact_heatmap.py. “Weighted” in this context means contact maps over the MD simulations, indicating the frequency.
2.9. Evaluation of distal effects associated with mutations via an energy-based approach
To determine the presence of distal energetic changes in FP-2 catalytic domain due to the mutations, the AlloSigma web server [79], [80], [81] was used under default settings. The server adopts a structure-based statistical mechanical model of allostery (SBSMMA) to determine the Gibbs free energy change per residue as a result of perturbation events from ligand binding and/or mutations. The type of perturbation is determined by the size of mutated residue, with changes to larger residues being considered as “Up-Mutation” whereas those leading to smaller as “Down-Mutation”. From the webserver, the allosteric free energy change (Δgresidue) upon perturbations involving either “Up-Mutation” or “Down-Mutation” was evaluated for each mutant system.
3. Results and discussion
3.1. Revisiting FP-2 structure for the identification of potential allosteric pockets and missense mutations
In the most general definition, allostery is the occurrence of changes in the active site (in the case of enzyme) or binding site (in the case of receptors) due to perturbations in a topologically distal part of the protein, which is called allosteric site; and the perturbations can be due to ligands, ions or DNA/RNA binding, residue mutations or post-translational modifications [82], [83], [84], [85]. Allosteric effects of distal mutations were reviewed [36], [86] and studied in a number of research articles [34], [35]. Further, a relationship between allosteric pockets and the mutations was reported [87].
Recent work by Siddiqui and co-workers identified four FP-2 mutations, K255R, N257E, T343P and D345G, that were linked to K13 protein mutations (F446I and P574L) known to confer parasites with reduced artemisinin sensitivity [11]. Here, as a first step to understand if these mutations, and the seven other mutations of this study, are allosteric and linked to drug resistance, we searched for potential allosteric pockets with an intention to see if any of the missense mutations are located in or around these pockets.
Apart from the trench-like active site pocket located between the left and right domains of FP-2, whose floor is formed by the central helix, no additional groove was consistently detected by the various approaches used for pocket identification. Nevertheless, an integration of results from these different tools gave a total of six potential topological pockets (named as Pocket 1–6) (Fig. 1C & 1D). Pocket 1 (Y247, E248, I251, K252, K255, G256, N257, E258, N259, F260, H262, D376, N377, K378, L379, K380, E381, A382, Y443, E465, S466, G467 and L468) is located around the nose region loop, a structural feature that is exclusive to FP-2 and related homologs from other Plasmodium parasite species. Interestingly, two of the ART resistant linked missense mutations, K255 and N257, are located in this pocket, and the other three missense mutations of this study (M245I, E248D, E249A) are near the pocket. Pocket 2 (Q311, L313, V314, D315, C316, S317, F318, K319, N320, Y321, GLY322, C323, N324, D334, V350, S351, D352, A353, P354, N355, L356, C357, N358, I359) and Pocket 4 (D398, F401, Y402, K403, E404, G405, I406, F407, D408, G409 and E410) are formed by residues located in highly dynamic loops which are adjacent to the S1 and S1′ subsites respectively. Pocket 2 contained the missense mutation A535T, while A400P was on the border of Pocket 4. Pocket 3 is located at the bottom of the L-domain and consists of residues Y298, K302, N303, K304, I306, D334, M335, I336, E337, LEU338, G339, G340, I341, T343, T363, E364, K365, Y366, G367, I368, K369, N370, L482, I483, and E484. Pocket 5 (S271, G272, V273, T274, A299, N303 and L305) and Pocket 6 (P275, V276, K277, D278, K280, T307, S309, E310, D344, D345, P348, Y349, G450 and Q451) were only identified by AutoLigand [63] as one pocket, which we separated into two. They consisted of two small grooves. The interface of Pocket 3 and 6 shared the other two ART resistance-linked mutations (T343P and D345G). From the druggability analysis using SiteMap and AutoLigand (Table S2), all the pockets, with the exception of Pocket 1, were considered to be less likely to bind small molecules (D-score < 0.6).
We further looked at the distances of mutant residues from the active site area. For that, we used three different center of mass (COM) measures: Inter-COM distances between the mutation and (1) the catalytic thiol, (2) substrate binding pocket and (3) the catalytic triad (Table S3). V393I was the nearest mutation to the binding pocket environment. All four mutations that are linked to ART resistance were located more than 20 Å away from the catalytic pocket environment. All the identified mutations are located in loop regions. Even though V393I and A400P of FP-2 were the only mutations of the active site binding pocket environment (S1′subsite), which may cause disruption of important native contacts with the Hb substrate, it is yet to be established if they confer ART resistance.
3.2. Global analysis: Mutation-induced structural changes via MD simulations
To accurately analyze the properties of biomolecular systems using MD simulations, it is critical to determine the most suitable force field parameters. Further, it is important to have long enough simulations to ensure that the systems converge to their most stable configuration. Thus, as a preliminary step, we evaluated three force fields viz. AMBER96 [88], AMBER99SB-ILDN [65] and CHARMM36 [89] in triplicate run per force field, over 500 ns, for the WT FP-2. From the Cα RMSD analysis, AMBER99SB-ILDN was the most appropriate in describing the properties of FP, which are composed of ∼ 50% loop regions (Figure S1).
Subsequently, the dynamic changes of WT FP-2 and 11 mutant proteins were assessed over 500 ns MD simulations. The cleaned MD trajectories for all systems (without PBC, solvent and ions as well as fitted to the respective first frame of each system) were visualized using VMD to observe any obvious structural conformational changes. Post-MD global analysis was done by RMSD (whole protein); Rg (whole protein, central core and the different pocket residues including those forming the active site) to determine protein stability, conformational changes in mutant proteins with respect to WT. The outcome of the calculations is discussed as follows.
3.2.1. Protein conformational changes due to mutations were analyzed via RMSD
The global conformational variability of the mutant systems was assessed using Cα RMSD, and a comparison was done against WT structure. For that, we utilized different types of RMSD analysis. At first, the most commonly used RMSD vs timeline plots were calculated to evaluate the divergence of a structure compared to the starting conformation over the course of the simulation (Figure S2). Nearly all systems displayed convergence after the first few equilibration nanoseconds. Next, to determine the conformational variability over the simulations, RMSD distribution violin plots for the whole protein, central invariant core and the binding pocket residues were calculated, excluding the first 20 ns of the equilibration time. As seen in the RMSD plots (Fig. 2A-C), majority of the protein systems displayed a unimodal distribution, indicating a single conformational equilibrium during the simulation. Exceptions to this were M245I, E248D, V393I and A400P. M245I and A400P, which demonstrated bimodal distribution for the whole protein systems, and E248D and V393I for the active site RMSDs. Even though the central core of FP-2 has a very rigid structure, A400P mutant protein showed bimodal behavior.
Fig. 2.

Cα backbone stability analysis. A) RMSD violin distribution plots of the A) whole protein, B) central invariant core, and C) binding active site residues of WT and mutant FP-2 proteins. Shown in purple are the WT ensemble plots. D) All vs all Cα RMSD of FP-2 catalytic systems. The × and y-axes denote time (ns). Color scale shows the degree of conformational variability between frames (most similar = white, distinct = dark). (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Further analysis to compare how starting conformation differed as compared to that of each frame, all vs all frame RMSD plots were calculated using pytraj [90]. As with the classical RMSD vs time plots, the starting structures in majority of the systems showed a significant variation as compared to other frames along the simulation (Fig. 2D). Again, M245I and A400P demonstrated strong bimodal behaviour. We later identified that the bimodal distribution of M245I is due to a cryptic allosteric pocket (see Section 3.3.3).
3.2.2. Mutation linked changes in the putative allosteric pockets were identified via Rg and RMSF
We investigated the effects of mutations on the compactness of the whole protein, the central invariant core, the binding pocket (active site) and the identified pockets (Pocket 1 – Pocket 6) using Rg calculations. Distribution of Rg values for the whole protein, the central invariant core, as well, as the binding pocket residues revealed insignificant changes in compactness (Figure S3). All the systems had a unimodal Rg density distribution, which signified the absence of major conformational changes associated with the selected mutations. Despite the extent of loopy regions within the protein, the globular nature and the presence of two pairs of disulphide bonds may be the reason for the perceived compactness. Mutants E248D, N257E, V393I and A400P displayed higher active site pocket Rg values compared to the WT. This might be linked to an increased loop flexibility from residue RMSF measurements (Figure S4). In both V393I and A400P, a considerable loss of stabilizing contact with the neighboring loop residue V395 was observed.
We further analysed the pockets via Rg and RMSF (Fig. 3) to understand if there are any links between mutations and changes in pockets. In general, pockets showed more variability in Rg results compared to Figure S3. In the case of Pocket 1, WT, M245I and N257E demonstrated bimodal behaviour (Fig. 3A). A353T seemed to sample a much wider range of conformations for Pocket 1, with a flatter distribution. A353T mutant protein had the highest RMSF for the residues E258-F260. Pocket 2 presented the most diverse Rg distributions among all pockets compared (Fig. 3B). M245I, E249A, N257E and V393I sampled two major conformations. Residues S351-N355 showed high flexibility for M245I, though we have not seen similar residue flexibility in the other three mutant proteins. Interestingly, in the presence of A353T mutation, which is located at Pocket 2, the pocket became the most rigid one among all protein cases according to RMSF data. In Pocket 3, most of the mutant systems displayed similar Rg distributions as that of the WT, with the exception of A353T, where a significantly higher mode was observed. This observation in A353T can be linked to the comparatively higher flexibility of loop residues K302-K304, which led to it being farther from the central core helix (Fig. 3C). In the case of Pocket 4, the majority of mutant systems sampled a single conformation as opposed to the WT and A353T mutant, and generally, the pocket residues showed similar flexibility except WT and A353T (Fig. 3D). In Pocket 5, only A353T had notable differences in conformational distribution, producing two modes distinct from all cases, one being the lowest and the other being the highest (Fig. 3E). Lastly, in the case of Pocket 6, M245I showed a distinct Rg distribution compared to all the other systems (Fig. 3F). This is linked to the observation described for Pocket 2, where a section of its residues was pulled into Pocket 6.
Fig. 3.

Rg and RMSF values for Pocket 1 – Pocket 6 (A – F) presented as violin distribution plots and heat maps respectively. In the violin plots, the purple indicates WT FP-2. Pocket residues are presented on the x-axis, and WT and mutant proteins on the y-axis of the heat maps. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
This unique mechanism is detailed in the next section on essential dynamics calculations. Interestingly, A353T seems completely opposite –a stabilizing effect on these two back to back pockets, Pockets 2 and 6, was observed. In conclusion, we observed changes in some of the pockets due to mutations, with the most distinct observations came from M245I and A353T mutations. ART linked mutations did not have obvious effects on the pocket dynamics, except in the presence of N257E mutation, Pocket 1 and 2 showed bimodal behavior.
3.2.3. Essential dynamics revealed diverse conformational changes in the active site region and the identified putative allosteric pockets
As a next step, we used one of our newly developed tools from the MDM-TASK-web, comparative essential dynamics, to investigate the behaviour of the active site and the pockets in the presence and absence of FP-2 mutations. The lowest energy equilibrium conformations for the catalytic site were very similar irrespective of mutation or lack thereof, indicating a common stable conformation in each case. Fig. 4, which shows the conformational distributions of the active site, also revealed that they were very similar, along the axis corresponding to the largest percentage of explained variance (71.09% along PC1), even though minute differences were also observed along PC2, which explained a much lower proportion of the total variance (4.06%). Despite the similarities, the most differential distribution along PC1 was observed in the case of E248D, whereby the conformation distribution slightly shifted to the left, where the pose extracted at 227,716 ps was the closest energy minimum in that region. Upon examination of the pose, we found that the major contributor of variation comprised residues from subsite S1. This correlates with the high RMSF recorded from the residues of the same site (Figure S4). This variation occurred in most of the systems as well.Fig. 5.
Fig. 4.

Comparative essential dynamic analysis of the active site pocket residues along PC1 and PC2. Colour code (dark = start, yellow = end) represents the simulation time (ns). Indicated in blue on each PCA plots is the timestamp when the structure was most stable. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Fig. 5.

Comparative essential dynamic analysis of the different putative allosteric pockets along PC1 and PC2. Colour code (dark = start, yellow = end) represents the simulation time (ns). Indicated in blue on each PCA plots is the timestamp when the structure was most stable. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
In the case of the putative allosteric pockets, the essential dynamics showed relatively more variability compared to the active site. For Pocket 1, the mutant A353T showed the highest divergence in conformational sampling, while A400P diverged the least, as observed along PC1 that accounted for 53.42% of the total variance. In the WT, the same pocket had a distribution intermediate to those of these two mutants along the same axis. While some changes were observed along PC2, the axis only accounted for 9.6% of the variance. The divergence between these mutants agreed with the observed differences in compactness inferred from Rg distributions and RMSF values (Fig. 3). Structural examination of the lowest energy conformations revealed contrasting surface topologies of the pocket in A353T and A400P. The irregularity of the pocket surfaces, however, made direct comparison difficult.
In Pocket 2, PCs 1 and 2, respectively, accounted for 38.29% and 16.56% of the total variance. Together, the axes showed a more spread out conformational distribution in the case of M245I and more compact distributions in the case of K255R, T343P and A400P. Appreciable shifts in distribution from the WT were noted for M245I, E249A and D345G. While the compact distributions point to less diverse pocket conformations, shifts in the distributions point to the favoring of other states. More importantly, examination of the pose extracted by the k-means algorithm at 448,180 ps of the M254I mutant protein MD simulation revealed a unique opening of Pocket 2. As it is not seen in the WT and the other mutants, this suggests that it is a cryptic pocket. Coincidentally, a pairwise Pearson correlation (r) of all pocket Rg values revealed a possible cross-talking mechanism between Pocket 2 and Pocket 6, with an R-value of 0.81 (Figure S5). Visualization of the trajectory revealed that some residues in Pocket 2 moved into Pocket 6, resulting in their fusion into a single groove linking the two (Movie S1).
In Pocket 3, PC1 and PC2 accounted for 75.32% and 4.19%, respectively, of the total variance. The distribution of pocket conformations was generally similar in all cases along both PC axes. However, a wider distribution along PC1 was observed in E249A and A353T when compared to the same pocket in the WT FP-2. An upper shift in the distribution of E248D (from the WT) was observed along PC2. Although M245I and D345G also showed variations from the WT along the same axis, these were supported by a much lower percentage of explained variance. Overall, from the visualization, minimal conformational changes were observed for this pocket.
In Pocket 4, both axes were comparable in terms of percentage of variance explained, with a total variance of 56.4%. We observed a general tendency of the pockets to sample a wide range of conformations. However, when compared to the WT, slightly reduced ranges were obtained in mutants M245I, E248D, K255R, N257E and A400P when measured along PC1, PC2 or along both axes. Structural visualization revealed minimal changes to this pocket, other than those resulting from side chain movements.
For Pocket 5, PC1 explained a large percentage (83.12%) of the total variance, while PC2 only explained 3.57% of the variance. Mutants N257E and A353T displayed the highest range of conformation sampling along PC1, whereas the other samples were closer to the WT. Nevertheless, examination of the maximum probability density pocket poses did not show any significant changes.
For Pocket 6, PC1 and PC2 both explain significant amounts of the total variance (74.14%) despite contributing to dissimilar amounts of variation. Mutants M245I displayed the highest range of conformation sampling along PC 2, followed by N257E, whereas E249A displayed a right shift along PC1, when compared to the WT. Visualization showed that Pocket 6 formed grooves of various depths, and agreed with the previous observation made from Pocket 2, whereby the two fused to form a wider trench in M245I.
Overall, our findings in this section agreed with the Rg and RMSF analyses of the previous section.
3.3. Identification of hubs for each of the five dynamic residue network metrics with specific attention to persistent hubs
Protein systems exist as a network of residues commonly referred to as nodes and non-covalent interactions of nodes as edges [91]. Even though most of these nodes lack a direct functional role, they are key in mediating a flexibility pattern needed for protein function [92]. The importance of a node in communication (centrality) can be determined from a given network topology. Besides altering the structure of a protein system, mutations can induce significant inter-residue contact changes depending on the biophysical characteristics of side chains [93]. The resulting perturbation not only affects the immediate community of residues but also distal residues depending on how the affected region is linked with the rest of the network. A key method in the determination of how mutations affect communication in a protein ensemble is DRN analysis [39], [40]. This method uses graph theory techniques to predict signalling effects associated with perturbations on residue backbone connectivity arising from mutations and ligand binding [38], [94], [95]. Herein, we performed network analysis using five different centrality metrics (BC, CC, DC, EC and KC) implemented in the MDM-TASK-web server [41] to identify hubs in FP-2 and the effects of mutations on their communication profiles.
In our previous study, we defined a “hub” as any node that formed part of the set of highest centrality nodes and identified these hubs with a new approach, as also explained in the methodology section here [43]. We specified these hubs as the global top 5% centrality nodes measured across all related samples for any given averaged centrality metric and analyzed the communication paths. Fig. 6 shows the heat maps of the five DRN metrics for the WT and mutant FP-2 proteins. Overall examination of the figure demonstrated that there are some residues that preserve their hub statuses. Previously, we introduced the term “persistent hub” [43]; meaning if hub remains across all systems compared, then the hub is called persistent. In this study “all systems” would be the WT and mutant FP-2 proteins. As shown previously, most of the persistent hubs are metric-specific, giving a different perspectives of the network, as each metric refers to different measures of importance within a network; hence persistent hubs may not be shared between different DRN metrics.
Fig. 6.

Heat maps for the potential hub residues (x-axis) according to the global top 5% for each of the five DRN metrics, for the WT and 11 mutant FP-2 proteins (y-axis). Detected hubs are annotated by their centrality values, while their homologous residues in alternate samples are not, but are only shown for the sake of comparison. For each metric, low tohigh centrality values are colored white, through yellow, orange and red to black. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
According to Fig. 6, persistent hubs for the averaged BC were F288, I389, M420, Y441 and I445. Persistent hubs for the averaged CC consisted of residues S289, S290, G292, S293, P388, S390, I391, V419, M420 and L421. DC persistent hubs were S284, W286, S296, I328 and I389. EC persistent hubs were W286, S289, S290, S293, I328 and I389; and residues W286, S289, S293, I328 and I389 were for KC. The persistent hubs S284, W286 and S289 form the peptidase_C1 functional site (PS00139) motif, which is a characteristic of the papain-like family cysteine proteases [96].
Visual inspection of these hubs via mapping to the structure showed that all the identified persistent hubs were located in the central invariant core of FP-2, where the main binding pocket is located (Fig. 7). In our previous study [43], where these metrics were compared for the first time, the distribution and location of persistent hubs were more diverse, unlike the observation that we have here for FP-2. This difference between the current and previous studies can be attributed to the different sizes and organization of the proteins. In the previous study, Mpro was analysed, and this homodimeric protein has three domains per protomer with very well-defined secondary structure elements. On the other hand, FP-2 is a relatively small monomeric protein, and ∼50% of it contains flexible loops; hence the accumulation of hubs in the core area. In fact, from all metrics used, as in the previous study, we observed that residues within the α-helices and β-sheets had high values, indicating the presence of increased interactions with other residues in the network; and majority of the residues within the loop regions, including those in the nose region and the β-hairpin (in both the mutants and corresponding WT) exhibited the lowest centrality values. From PIC (Protein Interaction Calculator) [55], a dense bonding network between the identified hubs was observed. S289 of the central core helix formed H-bonds with C285, S293, S390 and V419 (central β-sheet), whereas S390 interacted with S293. I328, located at the α-helix at the interface of Pocket 2 and 3, formed hydrophobic interaction with W286 (central core α-helix) as well as A418 (subsite S2). This bonding network most likely functions in stabilizing the central core while maintaining the integrity of the main binding pocket.
Fig. 7.

Cartoon representation of FP-2 with the distribution of the persistent hubs (in spheres) as per five metrics of DRN. (A) Averaged BC (orange), (B) Averaged CC (skyblue), (C) Averaged DC (aquamarine), (D) Averaged EC (chartreuse), (E) Averaged KC (yellow), (F) Collective presentation of persistent hubs in purple spheres. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Overall, the heat map representation of the identified hubs according to the global top 5% for each of the five DRN metrics (Fig. 6) allowed us to identify persistent hubs, which indicate the key residues that are not affected in the presence of mutations. We believe that these residues are functionally highly important. Previously, we made a correlation between persistent hubs and cold spot residues [43]; and here, we reiterate that the persistent hubs of a system should be considered in inhibitor design studies.
3.4. Averaged DRN metric hub changes due to mutations were investigated
To gain insights on changes in hubs due to mutations, we further analysed the data presented in Fig. 6. For that, we mapped (1) the uniquely observed hubs of each mutant protein with respect to the WT hubs as well as (2) the rest of the hubs commonly shared (intersected) with the WT for each metric.
3.4.1. Betweenness centrality (BC)
BC is defined as the number of shortest paths going through a node/residue for a given residue interaction network, and provides a measure of usage frequency of each node within the network. BC analysis is a measure for identifying hubs responsible for both inter- and intra-protein communication. These sites were found to correlate closely with known functional sites, demonstrating how BC analysis may be utilized to identify functional sites on a protein [97]. In a previous study, BC was successfully used to identify key residues in FPs and how ligand binding on the orthosteric site affected the communication network around the active site, as well as distal sites [98]. Similar studies have also applied BC to identify potential allosteric sites and mutation-induced changes in a range of protein systems [38], [95], [97], [99].
With this in mind, we further analyzed the global top 5% averaged BC hubs for WT and 11 FP-2 mutant proteins (Fig. 8 and Figure S6). Hubs are colored according to their unique occurrence in only mutant proteins with respect to the WT or commonly shared with the WT protein (Figure S6). One striking observation (Fig. 6 and Fig. 8) is the appearance of the catalytic thiol (C285) as a new hub in all mutant cases except in the WT protein and in E249A. This is also seen with S289 as a mutant-specific averaged BC hub, located at the central α-helix, in all cases except in E248D and A400P (Fig. 8). To a lesser extent, we also identified I291 as a new common hub. I291 was identified in six mutant proteins (M245I, E249A, K255R, T343P, A353T and V393).
Fig. 8.

Cartoon representation of (A) WT and (B-L) mutant FP-2 with the distribution of the averaged BC hubs unique to each mutant protein with respect to WT (skyblue spheres). WT averaged BC hubs are in salmon color. Catalytic thiol (of C285) is marked with a black stick if it is identified as a new averaged BC hub; otherwise is red. The other common hub, residue S289, is also labelled. ART-linked mutations are represented as firebrick spheres and the rest of the mutations are in grey spheres. Catalytic residues are shown in orange if they are not hubs. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Interestingly, these three hubs are connected to each other and form a short communication path. Few of the mutant proteins possessed this short communication path. These are M245I, K255R, T343P, A353T and V393I, two of which are linked to ART resistance. More interestingly, we observed a full allosteric residue communication path (L308-I291-S289-C285), which was established from the T343P-specific averaged BC hubs (Fig. 8 and Fig. 9). The path originates from residue L308, which is in contact with the mutant residue (T343P) and ends at the catalytic site, including C285. Closer 3D visual inspection of the protein and these hubs showed that while T343P is located between Pockets 3 and 6, L308-I291 are positioned between Pockets 2, 3 and 6 (Fig. 7C and D). T343P is one of the mutations linked to ART resistance, hence we speculate that the resistance is probably due to this strong allosteric communication path formed by four unique averaged BC hubs travelling from the mutation to the catalytic thiol.
Fig. 9.

Cartoon representation of WT and T343P mutant proteins with their averaged BC hubs. (A) WT with the distribution of the averaged BC hubs depicted as salmon colored spheres. (B) T343P with its unique hubs (skyblue) and the ones common to the WT hubs (salmon). (C) and (D) show the allosteric communication path formed by four unique hubs originating from T343P and going to the catalytic site, passing through the interface of three pockets.
The WT hubs, L421 (located in the antiparallel β-sheet) and N447 (subsite S1′), on the other hand, were lost in the other FP-2 mutations studied.
3.4.2. Closeness centrality (CC)
CC identifies the central nodes which are closer to most of the other nodes, as it is calculated as the inverse of the average of the shortest path length from one node to every other node. Previously we showed that large CC values occur within the protein core [43]. Here, we did not observe any drastic changes between WT and mutant proteins, and all global top 5% averaged CC hubs were primarily located in the center of the protein. Fig. 7B outlines the locations of the persistent CC hubs, which constitute most of the global top 5% averaged CC hubs. Exceptions to these are hub residues A418 (only in A353T), V422 (in M245I, N257E and D345G) and I445 (exists in all except in the WT, E249A and A400P). A418 is one of the subsite 2 residues; and I445 is also identified as a persistent averaged BC hub.
3.4.3. Degree centrality (DC)
DC is defined as the number of neighboring nodes around a given node. Even though DC nodes do not inform us about how central they are in the entire network, they provide information on how well they are connected locally. Hence DC hubs can be regarded as functionally important at a local level.
Upon visual inspection, it was obvious that the number of averaged DC hubs increased in all mutant proteins (Figure S7). While the WT has seven DC hubs, those of the ART resistance-linked mutant proteins are roughly doubled: K255R (12 hubs), N257E (16 hubs), T343P (16 hubs) and D345G (15 hubs). This increase, of course, can also be extrapolated to unique hubs with respect to the WT protein (Fig. 10). The highest numbers of unique hubs are observed in the three ART resistance-linked mutant proteins: N257E contained 10 unique hubs, and this was followed by T343P (9 hubs) and D345G (8 hubs). K255R, A353T, A400P and Q414E had six unique averaged DC hubs compared to the WT. Overall, residue-residue contact rearrangements due to some of the mutations were evident. The large number of unique hubs indicate that a significant shift in the communication occurred. The significantly larger number of hubs, especially, in the presence of ART-linked mutations, also suggest that the mutant proteins are more compact locally than the WT. We have not observed the global compactness of the protein in Rg calculations (Figure S3). This shows us the sensitivity of DC calculations compared to Rg calculations.
Fig. 10.

Cartoon representation of (A) WT and (B-L) mutant FP-2, with the distribution of the averaged DC hubs unique to each mutant protein with respect to WT (skyblue spheres). WT averaged DC hubs are in salmon color. ART resistance-linked mutations are depicted as firebrick spheres and the rest of the mutations are in grey spheres. Catalytic residues are shown in orange if they are not hubs. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
We also observed that the averaged DC hub S289, which interacts with the catalytic thiol of C285, appeared in all mutant proteins, except in M245I and E248D. Interestingly, hub residue L308, which interacts with T343P again featured among the averaged DC hubs of this mutant protein. Further, nine mutant proteins (M245I, E248D, E249A, T343P, D345G, A353T, V393I, A400P and Q414E) commonly have hub residue 420, which interacts with S293 and S296. On the other hand, two WT hubs, S293 and I391, are lost in some mutant proteins. N257E and A400P lost hub S293; and mutants E249A, K255R, D345G, V393I and A400P lost hub I391.
3.4.4. Eigencentrality (EC)
Initial centrality calculation of EC is based on DC, and it builds further on a recursive allocation of centrality on the basis of nodes that draw importance from that of their successive connections. High connectivity nodes also tend to have a high EC, especially when surrounded by other high connectivity nodes, due to their dependence on the residue neighborhood. From our experience in applying these metrics to analyze protein dynamics, averaged EC has always been more stringent in assigning centrality compared to averaged DC, while KC has typically been in-between. Visual inspection of the EC hubs, again gave the same observation (Fig. 11 and Figure S8). In contrast to DC hubs, we have not seen any drastic increase in the number of unique EC hubs in mutant proteins with respect to hubs of the WT. Residue P388, which was identified as averaged a CC persistent hub, appeared in 9 mutant systems as a unique EC hub, except in the N257E and A353T mutant proteins. Residues S284 and F288 in N257E, and residue A418 in A353T were the unique hubs for these two proteins compared to the WT FP-2 hubs. The other commonly observed unique hub for many mutant proteins was L421, which was again a persistent hub of CC. Again, as observed with DC hubs, the T343P mutant protein had a high number of unique hubs; residues S296, P388, V419, L421 and V422. Interestingly, these hubs were forming another alternative allosteric communication path (S296-P388-V222-L421-V419), originating from S296, which is located within Pocket 5, and going towards the catalytic site, contacting H417 via V419 (Fig. 12). The route was almost in the opposite side of the allosteric communication path that we observed, which was formed via averaged BC hubs. The gap between L421 and V419 was filled with the existing EC hubs (I391 and M420) that are inherited from the WT. In this path though, we have not observed a direct contact to the mutant residue.
Fig. 11.

Cartoon representation of (A) WT and (B-L) mutant FP-2, with the distribution of the averaged EC hubs unique to each mutant protein with respect to WT (skyblue spheres). WT averaged EC hubs are in salmon color. ART resistance-linked mutations are depicted as firebrick spheres and the rest of the mutations are in grey spheres. Catalytic residues are shown in orange if they are not identified as new hubs. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Fig. 12.

Cartoon representation of T343P mutant protein with its averaged EC hubs. Unique hubs of T343P mutant protein are in skyblue, and the ones common with the WT hubs are in salmon. The allosteric communication path is formed by five unique EC hubs originating from Pocket 5 (green) and going to the catalytic site, interacting with H417. Catalytic triad residues are depicted in orange. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
3.4.5. Katz centrality
Visualization of the averaged KC metric (Figure S9) for the unique hubs specific to the mutant proteins gave us relatively similar results to EC.
3.5. Residue interaction changes due to mutations identified using contact map analysis
In the previous two sections (3.4, 3.5), we analyzed the effects of mutations on the FP-2 structure by identifying DRN hubs from five network metrics. This gave us information of centrality hubs and how they change in the different mutant protein systems. Here, we further zoom into the residue-residue interactions to determine the possible effects of mutations on the native inter-residue networks.
In recent years, the considerable interest in evaluating changes in residue networks to understand protein function has led to the development of various tools that provide a reduced matrix representation of the structure [100], [101], [102], [103]. To account for the inherent dynamic properties of proteins which was lacking in the existing approaches, a contact map tool to determine residue contact frequencies around a single residue across MD trajectories was implemented in MD-TASK [40]. Contacts are inferred from interactions such as van der Waals, hydrogen bonds and electrostatic bonds [40]. By assigning values in the range of 0 (absence) to 1 (presence), residues with loss, gain, and reduced interactions can be identified. Contact maps have previously been applied to identify important interaction changes associated with ligand binding and mutations on different proteins [38], [94], [104]. A recent improvement of the contact map functionality in MD-TASK as implemented in MDM-TASK-web allows users to determine contact frequencies for multiple residues of interest [41]. This was recently used in characterizing atovaquone drug resistance in Pf cytochrome b protein [105].
Using equilibrated trajectory regions of the WT and mutant proteins, we calculated the local contact frequencies 1) around the mutated residues, 2) on the catalytic triad, and 3) around the persistent hubs. In terms of local perturbation around the mutations, notable changes were observed in the four ART resistance-conferring mutations as well as in V393I and A400P (Fig. 13A).
Fig. 13.

Residue contact analysis heatmaps showing the interaction changes around (A) the mutations and (B) catalytic triad residues (C285, H417 and N447). Dashed boxes in panel A show mutations causing significant interaction changes between the mutated residue and its surrounding contacting residues with respect to the native network in WT. Color code represents the contact frequency where 0 = no contact and 1.0 = strong contact between target and contacting residues.
As compared to the WT, the cation-Pi interaction between K255 and F385 was reduced four-fold in K255R mutant protein. Additionally, a compensatory gain and loss of interactions between N257 and N259 with position 255 was also observed. In N257E, a non-existent interaction in the WT was formed with Y254. In T343P, a two-fold reduction of the native interaction between T343 and I306 was observed. Additional new bonds in the mutant between P343 and each of C362 and Y366 were also formed. In D345G, a four-fold strengthening of the contact between position 345 and P348 was observed. In A353T, the native hydrophobic interaction with V350 was completely lost. Additionally, the main chain hydrogen bond between A353 and N355 had a three-fold reduction in the case of the mutation. However, a new bond between T353 and C323 was formed. A three-fold reduction of the hydrophobic interaction between V393 and V395, as well as A418 was observed with mutation V393I. In A400P, a nearly complete loss of the main chain interaction with D397 was experienced, as well as that of a hydrophobic interaction with V395.
To determine the effect of mutations on the catalytic triad residue interactions, contact maps on C285, H417 and N447 were also evaluated (Fig. 13B). Nearly all the mutant proteins maintained the native residue interactions around the catalytic triad residues, except with the increased contact of H417 with A394 in both N57E and T343P. Additionally, a reduction of the contact between residues N447 and F458 in mutants M245I, E248D, N257E, A353T, A400P and Q414E was observed. The native main chain hydrogen bond contact between H417 and V393 was equally maintained in the case of V393I.
In the case of the persistent hubs, notable changes in the mutant systems were observed in comparison to the WT (Table 2). We identified newly formed interactions between a number of persistent hubs or between persistent hubs and other residues that we will call “neighbor residues”. Neighbor residues forming new interactions, included W267, M335, S351, G387, A418, V422 and K439. Among them, S351 is a Pocket 2 residue, and we identified that it shows high flexibility in the M245I protein (see Section 3.3.2). A418 is a subsite S2 residue, and it was also detected as a CC and an EC hub specific to the A353T mutant protein. V422 is one of the unique CC hubs for M245I, N257E and D345G. It is also part of the allosteric communication path formed by EC hubs in T343P (see Sections 3.5.4). Our analysis showed that in all mutant systems, a new interaction between I389 and S293 was established. I389 was identified as averaged BC, DC, EC and KC persistent hub. S293 is the persistent hub of averaged CC, EC and KC. This, most likely, further stabilized the central core. An increased contact frequency between I445 (BC persistent hub) and M420 (persistent hub of BC and CC) was observed in all the four ART resistance-linked mutations as well as A353T and V393I mutant proteins.
Table 2.
Dynamic inter-residue contact analysis around persistent hubs showed significant changes (new/enhanced or lost/reduced) in interactions with respect to the WT. Neighbor residues are indicated in bold and are underlined.
| Protein | New/Increased | Lost/Reduced |
|---|---|---|
| M245I | I389-S293, I391-M420, S293-I389, S390-A418, Y441-K439 | I389-L379, F288-G292, S289-A287, S293-Q297 |
| E248D | I389-S293, I391-M420, S293-I389, S296-G387, S390-A418, Y441-K439 | I389-L379, F288-G292, S289-A287 |
| E249A | I389-S293, I391-M420, I445-V422, S289-S448, S296-G387 | |
| K255R | I389-S293, S289-S448, I445-M420 | F288-G292, S284-C323, W286-C325 |
| N257E | I389-S293, I391-S289, S284-S351, S290-M335, I445-M420, Y441-K439 | I389-L379, M420-S293, S296-V273, S390-M420, W286-S284 |
| T343P | I389-S293, I445-M420, S289-S293, S296-W267, Y441-K439 | I389-L379, S284-W286, W286-C325 |
| D345G | I389-S293, I445-M420, S296-G387, Y441-K439 | L421-L383, P388-V273 |
| A353T | I389-S293, I445-M420, S289-S448, Y441-K439, W286-S284 | F288-G292, P388-V273 |
| V393I | I389-S293, I445-M420, S289-S448, S296-W267, Y441-K439 | L421-L383, W286-C325 |
| A400P | I389-S293, I391-M420, S289-S448, S293-S289, Y441-K439 | |
| Q414E | I389-S293, I391-M420, I445-M420, S296-W267, Y441-K439 | W286-C325, S296-G387 |
Neighbor residues with lost/reduced interactions with persistent hubs included V273, L379, A287, Q297, C323 and C325. Residue V273 is part of Pocket 5. C323 is a Subsite 1 residue, while C325 is part of Subsite 3. L379 is a Pocket 1 residue. In M245I, E248D, N257E and T343P, the hydrophobic interaction between I389 and Pocket 1 residue L379 was reduced.
Overall, from these results, it can be concluded that mutations impose changes in the existing WT residue interactions. Gained interactions are mainly to make interactions with the other identified persistent hubs and neighbor residues, while the lost/weakened interactions are mainly with neighbor pocket and subsite residues.
3.6. Distal effects of mutations were observed using energy-based approaches
We also looked at the effects of mutations using an energy-based approach and applied the SBSMMA model in AlloSigMA web server [79], [80], [81]. As shown in Fig. 14, diverse distal energy modulation effects were observed in the protein as a result of the mutations. M245I, E248D, E249A mutations, located around the nose region, and D345G caused local destabilization effects in their vicinity, while a stabilization effect was observed in the other parts of the proteins. K255R and N257E mutations from the same area, on the other hand, caused a local stabilization effect around the nose region while destabilizing the other areas of the protein. The third group of mutations, if we categorize them according to our AlloSigMA results, were T343P, A353T, V393I, A400P and Q414E. This group of mutations leads to strong stability in their vicinity and only slight destabilization in the regions far from the mutation point. One interesting observation can be made on AlloSigma results for the stability of A353T mutation, which leads to rigidity of Pocket 2, in agreement with the result reported in Section 3.3.2.
Fig. 14.

Allosteric per-residue free energy changes associated with the missense mutations occurring on the catalytic domains of FP-2. The resulting destabilization (ΔG greater than 0 kcal/mol) and stabilization (ΔG < 0 kcal/mol) effects on the proteins are shown in blue and red. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
From these results, perturbations exerted by the different mutations appear to cause subtle energy changes to distal protein sites, including the active pocket environment of these proteins. This altered energetic landscape may influence the catalytic efficiency and function of the protease.
4. Conclusion
Mutations of key plasmodial enzymes, which act as pharmacological targets, continue to present the greatest challenge towards successful global control of malaria due to drug resistance [106]. Thus, a better understanding of how mutations confer resistance, especially those involving ART based drugs, is of uttermost importance. Besides ensuring the efficacy of ART therapies, this would also guide the design of new drugs aimed at potentially circumventing the complex drug resistance mechanisms employed by Plasmodium parasites. Recently, the possible association of the observed mutations in FP-2 and reduced ART sensitivity was identified [11], hence the main concern of this study. FP-2 and other hemoglobinases play an important role in the development cycle of P. falciparum during the blood stage in host erythrocytes.
Here, we utilized a range of computational approaches that we recently developed [40], [41], [43] together with existing methods to investigate the structural and dynamic effects of a collection of 11 missense mutations of FP-2. We started with general global analysis of the mutations, which are distributed within the catalytic domain of the protease. With the exception of mutations V393I and A400P (FP-2 S1′ mutations), all other mutations, including those that confer ART resistance (K255R, N257E, T343P and D345G) in FP-2, are distally located from the main orthosteric site. We identified six putative allosteric pockets and noted that some of the mutations are in or around these pockets. Two of the ART resistant linked missense mutations, K255 and N257, were located in Pocket 1, and the other three missense mutations of this study (M245I, E248D, E249A) were near this pocket. Pocket 2 contained the missense mutation A535T, while A400P was on the border of Pocket 4. The interface of Pockets 3 and 6 shared the other two ART resistance-linked mutations (T343P and D345G).
As a next step, we applied MD simulations followed by post-MD analysis approaches. At a global level, the proteins were analyzed using RMSD, Rg and RMSF. These analyses were applied both to whole WT and mutant proteins, as well as to the binding pocket and potential allosteric pockets, with the intention of identifying mutation related differences. We also combined the pocket analysis with the outcomes of one of the new tools that we developed, comparative essential dynamics tool [41]. Traditional PCA calculations provide results with different axis scales due to different covariance matrices obtained from trajectories. Our tool aligns one trajectory to a reference trajectory before performing a single decomposition to lay out all conformations on a common set of principal axes, such that the percentage of explained variance is the one shared by both trajectories. This functionality enabled us to accurately compare the pockets in the presence of different missense mutations. Collectively, we had a number of important observations: 1) In the presence of M245I, a cryptic pocket was detected via a unique mechanism in which some residues in Pocket 2 moved into Pocket 6. 2) In the presence of A353T mutation, which is located at Pocket 2, the pocket became the most rigid among all protein systems analyzed. Pocket 6 was also highly stable, seemingly an opposite effect of M245I mutation. 3) The effect of ART linked mutations was subtler.
From there, we gradually started to zoom into residue level analysis to understand the structural differences (if any) caused by these mutations. We previously proposed DRN analysis to probe the impact of mutations and their allosteric effects [39], [40]. In this study, we applied five DRN metrics (BC, CC, DC, EC and KC) in characterizing key communication residues of FP-2 and its allosteric behavior in the presence of mutations. This concept was first introduced in our previous study [43], and here, for the second time, we showed the effectiveness of looking at these metrics in a combined manner. Further, we used the global top 5% algorithm that we introduced in our previous study to pinpoint key hub residues [43]. We defined a hub as any node that forms part of the set of highest centrality nodes for any given averaged centrality metric. Again, with this approach, we obtained a number of striking results: 1) The heat map representation of the identified hubs according to the global top 5% for each of the five DRN metrics allowed us to identify persistent hubs, which indicate the key residues that are not affected in the presence of mutations. We believe that these residues are functionally highly important. Previously, we made a correlation between persistent hubs and cold spot residues [43]; 2) Averaged BC calculations revealed an interesting observation: the appearance of the catalytic thiol (C285) as a new hub in all mutant cases except WT and E249A mutant protein. This is also seen with S289 as mutant specific averaged BC hub, located at the central α-helix, in all except E248D and A400P. 3) We identified a number of short allosteric communication paths formed by the averaged BC and DC hubs in the presence of mutations. The most compelling one was in the presence of ART resistant linked T343P mutation. The allosteric communication path originated from residue L308 which is in contact with the mutant residue (T343P) and ended at the catalytic site, including C285. While T343P mutation is located between Pockets 3 and 6; a part of the continuation of the communication path, L308-I291, is positioned between Pockets 2, 3 and 6. 4) Global top 5% analysis for averaged DC hub analysis was also highly informative, indicating a drastic increase of hub numbers (hence communication network) in the central core of FP-2 in a number of mutant systems including some of those linked to ART resistance.
After DRN analysis, we further zoomed into the residue-residue interactions to determine the possible effect of mutations on the native inter-residue networks. Collectively, weighted network analysis results indicated that mutations imposed changes in the existing WT residue interactions. Gained interactions were mainly to make interactions with the other identified hubs, while the lost/weakened interactions were mainly with pocket and subsite residues. This may be the basis of altered binding pocket dynamics and stability, which may ultimately affect the catalytic efficiency of proteases. This alteration in residue interactions and dynamics may impact the nucleophile mediated cleavage reaction of substrate peptide bonds. Based on these observations, we hypothesize the existence of subtle dynamic residue interaction changes which could affect the Hb degradation process.
Funding
This work was supported through the Grand Challenges Africa Drug Discovery programme [GCA/DD/rnd3/023]. Grand Challenges Africa is a programme of the African Academy of Sciences (AAS) implemented through the Alliance for Accelerating Excellence in Science in Africa (AESA) platform, an initiative of the AAS and the African Union Development Agency (AUDA-NEPAD). GC Africa is supported by the Bill & Melinda Gates Foundation (BMGF), Swedish International Development Cooperation Agency (SIDA), German Federal Ministry of Education and Research (BMBF), Medicines for Malaria Venture (MMV), and Drug Discovery and Development Centre of University of Cape Town (H3D). The views expressed herein are those of the author(s) and not necessarily those of the AAS and its partners. V.B. and T.M.M. are funded as a PhD student and postdoctoral fellow, respectively, by the Grand Challenges Africa programme [GCA/DD/rnd3/023]. C.J.O and O.S.A. are funded as a doctoral student and a postdoctoral fellow, respectively, by H3ABioNet, which is supported by the National Human Genome Research Institute of the National Institutes of Health under Award Number U24HG006941.
6. Data and software availability
All data reported in this article are presented in the article and the Supporting Information section. Dynamic residue network analysis metric scripts are implemented in the MDM-TASK-web platform (https://mdmtaskweb.rubi.ru.ac.za/) and are available at https://github.com/RUBi-ZA/MD-TASK/tree/mdm-task-web. MD simulations will be made available upon request.
CRediT authorship contribution statement
Chiamaka Jessica Okeke: Formal analysis, Methodology, Writing – review & editing. Thommas Mutemi Musyoka: Formal analysis, Methodology, Writing – review & editing. Olivier Sheik Amamuddy: Formal analysis, Software, Writing – review & editing. Victor Barozi: Methodology. Özlem Tastan Bishop: Conceptualization, Formal analysis, Funding acquisition, Methodology, Writing – review & editing.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgement
Authors acknowledge the use of the Centre for High Performance Computing (CHPC), Cape Town, South Africa, for the molecular dynamics simulations.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.csbj.2021.10.011.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Machin J.M., Kantsadi A.L., Vakonakis I. The complex of Plasmodium falciparum falcipain-2 protease with an (E)-chalcone-based inhibitor highlights a novel, small, molecule-binding site. Malar J. 2019;18:388. doi: 10.1186/s12936-019-3043-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dondorp A.M., Yeung S., White L., Nguon C., Day N.P.J., Socheat D. Artemisinin resistance: Current status and scenarios for containment. Nat Rev Microbiol. 2010 doi: 10.1038/nrmicro2331. [DOI] [PubMed] [Google Scholar]
- 3.Fairhurst R.M., Dondorp A.M. American Society of Microbiology; 2016. Artemisinin-Resistant Plasmodium falciparum Malaria. In Emerging infections 10; pp. 409–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Severini, C., Menegon, M. Resistance to antimalarial drugs: An endless world war against Plasmodium that we risk losing. J. Glob. Antimicrob. Resist; 2015. [DOI] [PubMed]
- 5.Heller L.E., Roepe P.D. Artemisinin-Based Antimalarial Drug Therapy: Molecular Pharmacology and Evolving Resistance. Trop Med Infect Dis. 2019;4:89. doi: 10.3390/tropicalmed4020089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang J., Zhang C.-J., Chia W.N., Loh C.C.Y., Li Z., Lee Y.M. Haem-activated promiscuous targeting of artemisinin in Plasmodium falciparum. Nat Commun. 2015;6(1) doi: 10.1038/ncomms10111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mita, T., Tachibana, S.I., Hashimoto, M., Hirai, M. Plasmodium falciparum kelch 13: A potential molecular marker for tackling artemisinin-resistant malaria parasites. Expert Rev Anti Infect Ther; 2016. [DOI] [PubMed]
- 8.Miotto O., Amato R., Ashley E.A., MacInnis B., Almagro-Garcia J., Amaratunga C. Genetic architecture of artemisinin-resistant Plasmodium falciparum. Nat Genet. 2015;47(3):226–234. doi: 10.1038/ng.3189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wamae K., Okanda D., Ndwiga L., Osoti V., Kimenyi K.M., Abdi A.I. No Evidence of Plasmodium falciparum k13 Artemisinin Resistance-Conferring Mutations over a 24-Year Analysis in Coastal Kenya but a near Complete Reversion to Chloroquine-Sensitive Parasites. Antimicrob Agents Chemother. 2019;63(12) doi: 10.1128/AAC.01067-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ariey, F., Witkowski, B., Amaratunga, C., Beghain, J., Langlois, A.C., Khim, N., Kim, S., Duru, V., Bouchier, C., Ma, L., et al. A molecular marker of artemisinin-resistant Plasmodium falciparum malaria. Nature; 2014, 505, 50–55, doi:10.1038/nature12876. [DOI] [PMC free article] [PubMed]
- 11.Siddiqui, F.A., Cabrera, M., Wang, M., Brashear, A., Kemirembe, K., Wang, Z., Miao, J., Chookajorn, T., Yang, Z., Cao, Y., et al. Plasmodium falciparum Falcipain-2a Polymorphisms in Southeast Asia and Their Association With Artemisinin Resistance. J Infect Dis; 2018, 218, 434–442, doi:10.1093/infdis/jiy188. [DOI] [PMC free article] [PubMed]
- 12.Rosenthal, P.J. Falcipains and Other Cysteine Proteases of Malaria Parasites. In Advances in Experimental Medicine and Biology; 2011; pp. 30–48 ISBN 9781441984135. [DOI] [PubMed]
- 13.Sajid M., McKerrow J.H. Cysteine proteases of parasitic organisms☆. Mol Biochem Parasitol. 2002;120(1):1–21. doi: 10.1016/S0166-6851(01)00438-8. [DOI] [PubMed] [Google Scholar]
- 14.Dahl E.L., Rosenthal P.J. Biosynthesis, localization, and processing of falcipain cysteine proteases of Plasmodium falciparum. Mol Biochem Parasitol. 2005;139(2):205–212. doi: 10.1016/j.molbiopara.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 15.Rozman J., Stojan J., Kuhelj R., Turk V., Turk B. Autocatalytic processing of recombinant human procathepsin B is a bimolecular process. FEBS Lett. 1999;459:358–362. doi: 10.1016/s0014-5793(99)01302-2. [DOI] [PubMed] [Google Scholar]
- 16.Kerr I.D., Lee J.H., Pandey K.C., Harrison A., Sajid M., Rosenthal P.J. Structures of falcipain-2 and falcipain-3 bound to small molecule inhibitors: Implications for substrate specificity. J Med Chem. 2009;52(3):852–857. doi: 10.1021/jm8013663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Na, B.-K., Shenai, B.R.,Sijwali, P.S., Choe, Y., Pandey, K.C., Singh, A., Craik, C.S., Rosenthal, P.J. Identification and biochemical characterization of vivapains, cysteine proteases of the malaria parasite Plasmodium vivax. Biochem. J. 2004, 378, 529–38, doi:10.1042/BJ20031487. [DOI] [PMC free article] [PubMed]
- 18.Prasad R., Atul, Soni A., Puri S.K., Sijwali P.S., Snounou G. Expression, characterization, and cellular localization of knowpains, papain-like cysteine proteases of the Plasmodium knowlesi malaria parasite. PLoS ONE. 2012;7(12):e51619. doi: 10.1371/journal.pone.0051619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pei Y., Miller J.L., Lindner S.E., Vaughan A.M., Torii M., Kappe S.H.I. Plasmodium yoelii inhibitor of cysteine proteases is exported to exomembrane structures and interacts with yoelipain-2 during asexual blood-stage development. Cell Microbiol. 2013;15(9):1508–1526. doi: 10.1111/cmi.12124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Caldeira R.L., Gonçalves L.M.D., Martins T.M., Silveira H., Novo C., Rosário V.d. Plasmodium chabaudi: Expression of active recombinant chabaupain-1 and localization studies in Anopheles sp. Exp Parasitol. 2009;122(2):97–105. doi: 10.1016/j.exppara.2009.03.003. [DOI] [PubMed] [Google Scholar]
- 21.Sijwali P.S., Rosenthal P.J. Gene disruption confirms a critical role for the cysteine protease falcipain-2 in hemoglobin hydrolysis by Plasmodium falciparum. Proc Natl Acad Sci USA. 2004;101(13):4384–4389. doi: 10.1073/pnas.0307720101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Subramanian S., Hardt M., Choe Y., Niles R.K., Johansen E.B., Legac J. Hemoglobin cleavage site-specificity of the Plasmodium falciparum cysteine proteases falcipain-2 and falcipain-3. PLoS ONE. 2009;4(4):e5156. doi: 10.1371/journal.pone.0005156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Deu E. Proteases as antimalarial targets: strategies for genetic, chemical, and therapeutic validation. FEBS J. 2017;284(16):2604–2628. doi: 10.1111/febs.2017.284.issue-1610.1111/febs.14130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Istvan E.S., Dharia N.V., Bopp S.E., Gluzman I., Winzeler E.A., Goldberg D.E. Validation of isoleucine utilization targets in Plasmodium falciparum. Proc Natl Acad Sci USA. 2011;108(4):1627–1632. doi: 10.1073/pnas.1011560108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu J., Istvan E.S., Gluzman I.Y., Gross J., Goldberg D.E. Plasmodium falciparum ensures its amino acid supply with multiple acquisition pathways and redundant proteolytic enzyme systems. Proc Natl Acad Sci. 2006;103(23):8840–8845. doi: 10.1073/pnas.0601876103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Qidwai T. Hemoglobin Degrading Proteases of Plasmodium falciparum as Antimalarial Drug Targets. Curr Drug Targets. 2015;16:1133–1141. doi: 10.2174/1389450116666150304104123. [DOI] [PubMed] [Google Scholar]
- 27.Teixeira C., Gomes R.B., Gomes J., Falcipains P. Plasmodium falciparum Cysteine Proteases as Key Drug Targets Against Malaria. Curr Med Chem. 2011;18:1555–1572. doi: 10.2174/092986711795328328. [DOI] [PubMed] [Google Scholar]
- 28.Klonis N., Crespo-Ortiz M.P., Bottova I., Abu-Bakar N., Kenny S., Rosenthal P.J. Artemisinin activity against Plasmodium falciparum requires hemoglobin uptake and digestion. Proc Natl Acad Sci. 2011;108(28):11405–11410. doi: 10.1073/pnas.1104063108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ariey Frédéric, Witkowski Benoit, Amaratunga Chanaki, Beghain Johann, Langlois Anne-Claire, Khim Nimol. A molecular marker of artemisinin-resistant Plasmodium falciparum malaria. Nature. 2014;505(7481):50–55. doi: 10.1038/nature12876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang Jigang, Lin Qingsong. Chemical proteomics approach reveals the direct targets and the heme-dependent activation mechanism of artemisinin in plasmodium falciparum using an artemisinin-based activity probe. Microb Cell. 2016;3(5):230–231. doi: 10.15698/mic2016.05.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tilley Leann, Straimer Judith, Gnädig Nina F., Ralph Stuart A., Fidock David A. Artemisinin Action and Resistance in Plasmodium falciparum. Trends Parasitol. 2016;32(9):682–696. doi: 10.1016/j.pt.2016.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang Shiming, Gerhard Glenn S. Heme activates artemisinin more efficiently than hemin, inorganic iron, or hemoglobin. Bioorganic Med Chem. 2008;16(16):7853–7861. doi: 10.1016/j.bmc.2008.02.034. [DOI] [PubMed] [Google Scholar]
- 33.Bahl A. PlasmoDB: the Plasmodium genome resource. A database integrating experimental and computational data. Nucleic Acids Res. 2003;31:212–215. doi: 10.1093/nar/gkg081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sheik Amamuddy O., Bishop N.T., Tastan Bishop Ö. Characterizing early drug resistance-related events using geometric ensembles from HIV protease dynamics. Sci Rep. 2018;8:17938. doi: 10.1038/s41598-018-36041-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sheik Amamuddy O., Musyoka T.M., Boateng R.A., Zabo S., Tastan Bishop Ö. Determining the unbinding events and conserved motions associated with the pyrazinamide release due to resistance mutations of Mycobacterium tuberculosis pyrazinamidase. Comput Struct Biotechnol J. 2020;18:1103–1120. doi: 10.1016/j.csbj.2020.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Guarnera Enrico, Berezovsky Igor N. Allosteric drugs and mutations: chances, challenges, and necessity. Elsevier Current Trends. 2020;62:149–157. doi: 10.1016/j.sbi.2020.01.010. [DOI] [PubMed] [Google Scholar]
- 37.Guarnera E., Berezovsky I.N. Allosteric sites: remote control in regulation of protein activity. Curr Opin Struct Biol. 2016;37:1–8. doi: 10.1016/J.SBI.2015.10.004. [DOI] [PubMed] [Google Scholar]
- 38.Brown David K., Sheik Amamuddy Olivier, Tastan Bishop Özlem. Structure-Based Analysis of Single Nucleotide Variants in the Renin-Angiotensinogen Complex. Glob. Heart. 2017;12(2):121. doi: 10.1016/j.gheart.2017.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brown David K., Tastan Bishop Özlem. Role of Structural Bioinformatics in Drug Discovery by Computational SNP Analysis. Glob Heart. 2017;12(2):151. doi: 10.1016/j.gheart.2017.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brown, D.K.; Penkler, D.L.; Sheik Amamuddy, O.; Ross, C.; Atilgan, A.R.; Atilgan, C.; Tastan Bishop, Ö. MD-TASK: A software suite for analyzing molecular dynamics trajectories. Bioinformatics 2017, doi:10.1093/bioinformatics/btx349. [DOI] [PMC free article] [PubMed]
- 41.Sheik Amamuddy O., Glenister M., Tshabalala T., Tastan Bishop Ö. MDM-TASK-web: MD-TASK and MODE-TASK web server for analyzing protein dynamics. Comput Struct Biotechnol J. 2021;19:5059–5071. doi: 10.1016/j.csbj.2021.08.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sheik Amamuddy Olivier, Verkhivker Gennady M., Tastan Bishop Özlem. Impact of Early Pandemic Stage Mutations on Molecular Dynamics of SARS-CoV-2 M pro. J Chem Inf Model. 2020;60(10):5080–5102. doi: 10.1021/acs.jcim.0c0063410.1021/acs.jcim.0c00634.s001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sheik Amamuddy, O., Baoteng, R.A., Barozi, V., Nyamai, D.W., Tastan Bishop, Ö. Novel dynamic residue network analysis approaches to study homodimeric allosteric modulation in SARS-CoV-2 Mpro and in its evolutionary mutations. 2021, doi:10.33774/CHEMRXIV-2021-7THM1. [DOI] [PMC free article] [PubMed]
- 44.Wang Stephanie X., Pandey Kailash C., Scharfstein Julio, Whisstock James, Huang Rick K., Jacobelli Jordan. The Structure of Chagasin in Complex with a Cysteine Protease Clarifies the Binding Mode and Evolution of an Inhibitor Family. Structure. 2007;15(5):535–543. doi: 10.1016/j.str.2007.03.012. [DOI] [PubMed] [Google Scholar]
- 45.Berman H.M., Westbrook J., Feng Z., Gilliland G., Bhat T.N., Weissig H. The Protein Data Bank (www.rcsb.org) Nucleic Acids Res. 2000 doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Burley, S.K., Berman, H.M., Bhikadiya, C., Bi, C., Chen, L., Di Costanzo, L., Christie, C., Dalenberg, K., Duarte, J.M., Dutta, S., et al. RCSB Protein Data Bank: Biological macromolecular structures enabling research and education in fundamental biology, biomedicine, biotechnology and energy. Nucleic Acids Res. 2019, 47, D464–D474, doi:10.1093/nar/gky1004. [DOI] [PMC free article] [PubMed]
- 47.DeLano W.L. Pymol. An open-source molecular graphics tool. CCP4 Newsl. Protein Crystallogr. 2002 [Google Scholar]
- 48.Bateman A. UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res. 2019;47:D506–D515. doi: 10.1093/nar/gky1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Aurrecoechea C., Brestelli J., Brunk B.P., Dommer J., Fischer S., Gajria B. PlasmoDB: A functional genomic database for malaria parasites. Nucleic Acids Res. 2009;37(Database):D539–D543. doi: 10.1093/nar/gkn814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Šali, a MODELLER A Program for Protein Structure Modeling Release. 2013;9:r9480. [Google Scholar]
- 51.Shen Min-yi, Sali Andrej. Statistical potential for assessment and prediction of protein structures. Protein Sci. 2006;15(11):2507–2524. doi: 10.1110/(ISSN)1469-896X10.1110/ps.062416606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Eisenberg D., Lüthy R., Bowie J.U. VERIFY3D: Assessment of protein models with three-dimensional profiles. Methods Enzymol. 1997;277:396–404. doi: 10.1016/S0076-6879(97)77022-8. [DOI] [PubMed] [Google Scholar]
- 53.Wiederstein M., Sippl M.J. ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007;35:W407–W410. doi: 10.1093/NAR/GKM290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Laskowski R.A., MacArthur M.W., Moss D.S., Thornton J.M. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr. 1993;26:283–291. doi: 10.1107/S0021889892009944. [DOI] [Google Scholar]
- 55.Tina K.G., Bhadra R., Srinivasan N.P.I.C. Protein Interactions Calculator. Nucleic Acids Res. 2007;35:W473–W476. doi: 10.1093/nar/gkm423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dolinsky T.J., Czodrowski P., Li H., Nielsen J.E., Jensen J.H., Klebe G. PDB2PQR: expanding and upgrading automated preparation of biomolecular structures for molecular simulations. Nucleic Acids Res. 2007;35(Web Server):W522–W525. doi: 10.1093/nar/gkm276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dolinsky T.J., Nielsen J.E., McCammon J.A., Baker N.A. PDB2PQR: an automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 2004;32(Web Server):W665–W667. doi: 10.1093/nar/gkh381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kozakov Dima, Grove Laurie E, Hall David R, Bohnuud Tanggis, Mottarella Scott E, Luo Lingqi. The FTMap family of web servers for determining and characterizing ligand-binding hot spots of proteins. Nat Protoc. 2015;10(5):733–755. doi: 10.1038/nprot.2015.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brenke, R., Kozakov, D., Chuang, G.-Y.Y., Beglov, D., Hall, D., Landon, M.R.R., Mattos, C., Vajda, S. Fragment-based identification of druggable “hot spots” of proteins using Fourier domain correlation techniques. Bioinformatics 2009, 25, 621–7, doi:10.1093/bioinformatics/btp036. [DOI] [PMC free article] [PubMed]
- 60.Huang, W., Lu, S., Huang, Z., Liu, X., Mou, L., Luo, Y., Zhao, Y., Liu, Y., Chen, Z., Hou T., et al. Allosite: A method for predicting allosteric sites. Bioinformatics 2013, doi:10.1093/bioinformatics/btt399. [DOI] [PubMed]
- 61.Panjkovich, A., Daura, X. PARS: a web server for the prediction of Protein Allosteric and Regulatory Sites. 2014, 30, doi:10.1093/bioinformatics/btu002. [DOI] [PubMed]
- 62.Halgren Thomas A. Identifying and characterizing binding sites and assessing druggability. J Chem Inf Model. 2009;49(2):377–389. doi: 10.1021/ci800324m. [DOI] [PubMed] [Google Scholar]
- 63.Harris Rodney, Olson Arthur J., Goodsell David S. Automated prediction of ligand-binding sites in proteins. Proteins Struct. Funct. Bioinforma. 2008;70(4):1506–1517. doi: 10.1002/prot.21645. [DOI] [PubMed] [Google Scholar]
- 64.Abraham Mark James, Murtola Teemu, Schulz Roland, Páll Szilárd, Smith Jeremy C., Hess Berk. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015;1-2:19–25. doi: 10.1016/j.softx.2015.06.001. [DOI] [Google Scholar]
- 65.Lindorff-Larsen Kresten, Piana Stefano, Palmo Kim, Maragakis Paul, Klepeis John L., Dror Ron O. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins Struct Funct Bioinforma. 2010;78(8):1950–1958. doi: 10.1002/prot.22711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Parrinello M., Rahman A. Polymorphic transitions in single crystals: A new molecular dynamics method. J Appl Phys. 1981;52(12):7182–7190. doi: 10.1063/1.328693. [DOI] [Google Scholar]
- 67.Hess B., Bekker H., Berendsen H.J.C., Fraaije J.G.E.M. LINCS: A Linear Constraint Solver for molecular simulations. J Comput Chem. 1997 doi: 10.1002/(SICI)1096-987X(199709)18:12<1463::AID-JCC4>3.0.CO;2-H. [DOI] [Google Scholar]
- 68.Petersen Henrik G. Accuracy and efficiency of the particle mesh Ewald method. J Chem Phys. 1995;103(9):3668–3679. doi: 10.1063/1.470043. [DOI] [Google Scholar]
- 69.RStudio Team RStudio: Integrated Development for R. RStudio, Inc., Boston, MA. URL http://www.rstudio.com/. RStudio, Inc.; 2015.
- 70.Hunter John D. Matplotlib: A 2D Graphics Environment. Comput Sci Eng. 2007;9(3):90–95. doi: 10.1109/MCSE.2007.55. [DOI] [Google Scholar]
- 71.Nguyen, H., Case, D.A., Rose, A.S. NGLview-interactive molecular graphics for Jupyter notebooks. Bioinformatics 2018, 34, 1241–1242, doi:10.1093/bioinformatics/btx789. [DOI] [PMC free article] [PubMed]
- 72.van der Walt Stéfan, Colbert S Chris, Varoquaux Gaël. The NumPy Array: A Structure for Efficient Numerical Computation. Comput Sci Eng. 2011;13(2):22–30. doi: 10.1109/MCSE.2011.37. [DOI] [Google Scholar]
- 73.McKinney W. Data Structures for Statistical Computing in Python. Proc. 9th Python Sci. Conf. 2010 [Google Scholar]
- 74.Waskom, M., Botvinnik, O., Ostblom, J., Gelbart, M., Lukauskas, S., Hobson, P., Gemperline, D.C., Augspurger, T., Halchenko, Y., Cole, J.B., et al. mwaskom/seaborn: v0.10.1 (April 2020). Zenodo 2020.
- 75.Hagberg, A.A., Schult, D.A., Swart, P.J. Exploring network structure, dynamics, and function using NetworkX. In Proceedings of the 7th Python in Science Conference (SciPy 2008); 2008; pp. 11–15.
- 76.Golbeck, J. Network Structure and Measures. In: Analyzing the Social Web; 2013.
- 77.Liang Z., Verkhivker G.M., Hu G. Integration of network models and evolutionary analysis into high-throughput modeling of protein dynamics and allosteric regulation: Theory, tools and applications. Brief Bioinform. 2020;21:815–835. doi: 10.1093/bib/bbz029. [DOI] [PubMed] [Google Scholar]
- 78.Sharma, D., Surolia, A. Degree Centrality. In Encyclopedia of Systems Biology. Springer New York: New York, NY, 2013; pp. 558–558.
- 79.Guarnera, E., Tan, Z.W., Zheng, Z., Berezovsky, I.N. AlloSigMA: allosteric signaling and mutation analysis server. Bioinformatics 2017, 33, 3996–3998, doi:10.1093/bioinformatics/btx430. [DOI] [PubMed]
- 80.Guarnera Enrico, Berezovsky Igor N., Liu Jin. Structure-Based Statistical Mechanical Model Accounts for the Causality and Energetics of Allosteric Communication. PLoS Comput Biol. 2016;12(3):e1004678. doi: 10.1371/journal.pcbi.1004678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tee Wei-Ven, Guarnera Enrico, Berezovsky Igor N., Papaleo Elena. Reversing allosteric communication: From detecting allosteric sites to inducing and tuning targeted allosteric response. PLoS Comput Biol. 2018;14(6):e1006228. doi: 10.1371/journal.pcbi.1006228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sheik Amamuddy, O., Veldman, W., Manyumwa, C., Khairallah, A., Agajanian, S., Oluyemi, O., Verkhivker, G.M., Tastan Bishop, Ö. Integrated Computational Approaches and Tools for Allosteric Drug Discovery. Int J Mol Sci; 2020, Vol. 21, Page 847 2020, 21, 847, doi:10.3390/IJMS21030847. [DOI] [PMC free article] [PubMed]
- 83.He X., Ni D., Lu S., Zhang J. Characteristics of Allosteric Proteins, Sites, and Modulators. Adv Exp Med Biol. 2019;1163:107–139. doi: 10.1007/978-981-13-8719-7_6. [DOI] [PubMed] [Google Scholar]
- 84.Wodak Shoshana J., Paci Emanuele, Dokholyan Nikolay V., Berezovsky Igor N., Horovitz Amnon, Li Jing. Allostery in Its Many Disguises: From Theory to Applications. Structure. 2019;27(4):566–578. doi: 10.1016/j.str.2019.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Liu Jin, Nussinov Ruth. An Overview of Its History, Concepts, Methods, and Applications. PLoS Comput Biol. 2016;12(6):e1004966. doi: 10.1371/journal.pcbi.100496610.1371/journal.pcbi.1004966.g001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tee, W.V., Guarnera, E.; Berezovsky, I.N.; WV, T.; E, G.; IN, B. On the Allosteric Effect of nsSNPs and the Emerging Importance of Allosteric Polymorphism. 2019, 431, 3933–3942. [DOI] [PubMed]
- 87.Liu, X., Lu, S., Song, K., Shen, Q., Ni, D., Li, Q., He, X., Zhang, H., Wang, Q., Chen, Y., et al. Unraveling allosteric landscapes of allosterome with ASD. Nucleic Acids Res; 2020, 48, D394–D401, doi:10.1093/NAR/GKZ958. [DOI] [PMC free article] [PubMed]
- 88.Kollman Peter A. Advances and Continuing Challenges in Achieving Realistic and Predictive Simulations of the Properties of Organic and Biological Molecules. Acc Chem Res. 1996;29(10):461–469. doi: 10.1021/ar9500675. [DOI] [Google Scholar]
- 89.Best R.B., Zhu X., Shim J., Lopes P.E.M., Mittal J., Feig M. Jr Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone φ, ψ and side-chain χ1 and χ2 dihedral angles. J Chem Theory Comput. 2012;8:3257. doi: 10.1021/CT300400X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Nguyen, H., Roe, D.R., Swails, J., Case, D.A. PYTRAJ: Interactive data analysis for molecular dynamics simulations. New Brunswick, NJ Rutgers Univ.; 2016, doi:10.5281/ZENODO.44612.
- 91.Vendruscolo M., Dokholyan N.V., Paci E., Karplus M. Small-world view of the amino acids that play a key role in protein folding. Phys Rev E. 2002;65 doi: 10.1103/PhysRevE.65.061910. [DOI] [PubMed] [Google Scholar]
- 92.Saldaño Tadeo E., Tosatto Silvio C.E., Parisi Gustavo, Fernandez-Alberti Sebastian. Network analysis of dynamically important residues in protein structures mediating ligand-binding conformational changes. Eur Biophys J. 2019;48(6):559–568. doi: 10.1007/s00249-019-01384-1. [DOI] [PubMed] [Google Scholar]
- 93.Zhang Xiuwei, Perica Tina, Teichmann Sarah A. Evolution of protein structures and interactions from the perspective of residue contact networks. Curr Opin Struct Biol. 2013;23(6):954–963. doi: 10.1016/j.sbi.2013.07.004. [DOI] [PubMed] [Google Scholar]
- 94.Sanyanga T.A., Nizami B., Tastan Bishop Ö. Mechanism of Action of Non-Synonymous Single Nucleotide Variations Associated with α-Carbonic Anhydrase II Deficiency. Molecules. 2019;24:3987. doi: 10.3390/molecules24213987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Amusengeri A., Tata R.B., Tastan Bishop Ö. Understanding the Pyrimethamine Drug Resistance Mechanism via Combined Molecular Dynamics and Dynamic Residue Network Analysis. Molecules. 2020;25:904. doi: 10.3390/molecules25040904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Musyoka Thommas Mutemi, Njuguna Joyce Njoki, Tastan Bishop Özlem. Comparing sequence and structure of falcipains and human homologs at prodomain and catalytic active site for malarial peptide based inhibitor design. Malar J. 2019;18(1) doi: 10.1186/s12936-019-2790-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Penkler David L., Atilgan Canan, Tastan Bishop Özlem. Allosteric Modulation of Human Hsp90α Conformational Dynamics. J Chem Inf Model. 2018;58(2):383–404. doi: 10.1021/acs.jcim.7b0063010.1021/acs.jcim.7b00630.s001. [DOI] [PubMed] [Google Scholar]
- 98.Musyoka T., Tastan Bishop Ö. South African Abietane Diterpenoids and Their Analogs as Potential Antimalarials: Novel Insights from Hybrid Computational Approaches. Molecules. 2019;24:4036. doi: 10.3390/molecules24224036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Penkler D.L., Tastan Bishop Ö. Modulation of Human Hsp90α Conformational Dynamics by Allosteric Ligand Interaction at the C-Terminal Domain. Sci Rep. 2019;9:1600. doi: 10.1038/s41598-018-35835-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Chung J.-L., Beaver J.E., Scheeff E.D., Bourne P.E. Con-Struct Map: a comparative contact map analysis tool. Bioinformatics. 2007;23:2491–2492. doi: 10.1093/BIOINFORMATICS/BTM356. [DOI] [PubMed] [Google Scholar]
- 101.Vehlow C., Stehr H., Winkelmann M., Duarte J.M., Petzold L., Dinse J. CMView: Interactive contact map visualization and analysis. Bioinformatics. 2011;27(11):1573–1574. doi: 10.1093/bioinformatics/btr163. [DOI] [PubMed] [Google Scholar]
- 102.Kozma D., Simon I., Tusnady G.E. CMWeb: an interactive on-line tool for analysing residue–residue contacts and contact prediction methods. Nucleic Acids Res. 2012;40(W1):W329–W333. doi: 10.1093/nar/gks488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ho H.K., Kuiper M.J., Kotagiri R. PConPy - A Python module for generating 2D protein maps. Bioinformatics. 2008;24(24):2934–2935. doi: 10.1093/bioinformatics/btn566. [DOI] [PubMed] [Google Scholar]
- 104.Nyamai D.W., Tastan Bishop Ö. Identification of Selective Novel Hits against Plasmodium falciparum Prolyl tRNA Synthetase Active Site and a Predicted Allosteric Site Using In Silico Approaches. Int J Mol Sci. 2020;21:3803. doi: 10.3390/ijms21113803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Chebon-Bore, L., Sanyanga, T.A., Manyumwa, C.V., Khairallah, A., Tastan Bishop, Ö. Decoding the Molecular Effects of Atovaquone Linked Resistant Mutations on Plasmodium falciparum Cytb-ISP Complex in the Phospholipid Bilayer Membrane. Int J Mol Sci; 2021, 22, Page 2138 2021, 22, 2138, doi:10.3390/IJMS22042138. [DOI] [PMC free article] [PubMed]
- 106.Menard Didier, Dondorp Arjen. Antimalarial Drug Resistance: A Threat to Malaria Elimination. Cold Spring Harb Perspect Med. 2017;7(7):a025619. doi: 10.1101/cshperspect.a025619. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this article are presented in the article and the Supporting Information section. Dynamic residue network analysis metric scripts are implemented in the MDM-TASK-web platform (https://mdmtaskweb.rubi.ru.ac.za/) and are available at https://github.com/RUBi-ZA/MD-TASK/tree/mdm-task-web. MD simulations will be made available upon request.