

Abstract
Extant fold‐switching proteins remodel their secondary structures and change their functions in response to cellular stimuli, regulating biological processes and affecting human health. Despite their biological importance, these proteins remain understudied. Predictive methods are needed to expedite the process of discovering and characterizing more of these shapeshifting proteins. Most previous approaches require a solved structure or all‐atom simulations, greatly constraining their use. Here, we propose a high‐throughput sequence‐based method for predicting extant fold switchers that transition from α‐helix in one conformation to β‐strand in the other. This method leverages two previous observations: (a) α‐helix ↔ β‐strand prediction discrepancies from JPred4 are a robust predictor of fold switching, and (b) the fold‐switching regions (FSRs) of some extant fold switchers have different secondary structure propensities when expressed by themselves (isolated FSRs) than when expressed within the context of their parent protein (contextualized FSRs). Combining these two observations, we ran JPred4 on 99‐fold‐switching proteins and found strong correspondence between predicted and experimentally observed α‐helix ↔ β‐strand discrepancies. To test the overall robustness of this finding, we randomly selected regions of proteins not expected to switch folds (single‐fold proteins) and found significantly fewer predicted α‐helix ↔ β‐strand discrepancies. Combining these discrepancies with the overall percentage of predicted secondary structure, we developed a classifier to identify extant fold switchers (Matthews correlation coefficient of .71). Although this classifier had a high false‐negative rate (7/17), its false‐positive rate was very low (2/136), suggesting that it can be used to predict a subset of extant fold switchers from a multitude of available genomic sequences.
Keywords: fold‐switching proteins, metamorphic proteins, protein folding, bioinformatics
1. INTRODUCTION
Extant fold‐switching proteins remodel their secondary structures and change their functions in response to cellular stimuli.[ 1 ] These environmentally responsive shapeshifters perform over 30 diverse functions, occur in all domains of life, and are associated with diseases such as cancer,[ 2 ] autoimmune disorders,[ 3 ] and malaria.[ 4 ] Furthermore, increasing evidence suggests that extant fold switchers regulate biological processes[ 5 ] such as cyanobacterial circadian rhythms[ 6 ] and transcription/translation of bacterial virulence genes.[ 7 ]
Compared with single‐fold proteins, which maintain stable secondary and tertiary structures and typically perform one biological function, extant fold switchers are understudied. Specifically, out of the ~160 000 proteins with solved structures available in the Protein Data Bank (PDB[ 8 ]), fewer than 100 have been shown to switch folds. Increasing evidence suggests that fold switching is likely more widespread than currently appreciated,[ 1 ] but the current shortage of experimental examples makes it difficult to determine either the physical‐chemical properties or the functional scope of fold switchers. Thus, predictive tools are needed to identify more.
Recent computational studies suggest that fold switching is predictable, a prospect that—if realized—could greatly expand the small pool of experimentally determined fold switchers currently available. For example, naturally occurring extant fold switchers were predicted blindly by searching for differences between predicted and experimentally determined protein structures.[ 1 , 9 ] Furthermore, several fold‐switching proteins have been designed computationally using the Rosetta software suite.[ 10 , 11 ] Progress has also been made in predicting mutation‐induced fold switching[ 12 , 13 ] as well as other conformational changes, such as rigid body motions.[ 14 ] Finally, a classifier for extant fold switchers was recently developed as a proof of concept that fold switching is predictable from protein sequence.[ 15 ] This classifier is based on confidences of all secondary structure predictions (helix, strand, and coil), whereas the one we developed relies on discrepancies between predicted α‐helices and β‐strands.
Here we present a sequence‐based method for predicting extant fold switchers. This method builds on our previous approach designed for evolved fold switchers, which are defined to have highly similar sequences but different folds.[ 12 ] By contrast, extant fold switchers have one sequence that can assume more than one stable secondary and tertiary structure configuration. Whereas the approach for extant fold switchers compared secondary structure predictions of two (or more) different proteins with slightly different sequences, the current method identifies extant fold switchers from the secondary structure predictions of different regions from a single amino acid sequence. The following hypothesis provides the basis for our method: the JPred4 secondary structure prediction of an isolated fold‐switching region (FSR) sequence might differ from the JPred4 prediction of the same FSR within the context of its naturally occurring sequence (hereafter called a contextualized FSR). We developed this hypothesis using the previous observation[ 1 ] that extant fold‐switching proteins generally have: (a) regions that change secondary structure between the two forms (FSRs) and (b) regions that maintain the same secondary structure (structurally constant regions, or SCRs[ 16 ]). By definition, FSRs assume multiple stable secondary structures, and several studies have suggested that at least one FSR conformation is stabilized by exogenous interactions.[ 17 , 18 ] Together, these observations indicate that the dominant secondary structure of a given FSR might differ depending on the context of its sequence. Thus, we tested our approach on 99 extant fold switchers with the aim of developing a classifier that could distinguish extant fold switchers from single‐folding proteins.
2. METHODS
2.1. Selection of extant fold switchers
We selected 93 extant fold switchers from a previous dataset.[ 1 ] We excluded 2GED/1NRJB and 3VO9B/3VPAA because they had nearly identical structures but were misclassified due to missing crystal density. We also excluded 1MBYA/2N19A because they come from different organisms, their FSRs differ by three amino acids, and their resting states appear to assume different conformations. Thus, they appear to be evolved—rather than extant—fold switchers. In addition to these 93 extant fold switchers, we included the bacterial cell‐division protein MinE,[ 19 , 20 ] SARS‐CoV‐2 ORF9b,[ 21 ] and the human apoptosis regulator BAX,[ 22 ] which have all been shown to switch folds, as well as 3 KaiB homologs presumed to switch folds since they come from cyanobacterial strains similar to Synechococcus elongatus.
2.2. JPred4 predictions of extant fold switchers
All amino acid sequences from 99 extant fold switchers with solved structures were downloaded from the PDB and saved as individual FASTA files. JPred4 predictions were run remotely using a publicly downloadable scheduler available on the JPred4 website (http://compbio.dundee.ac.uk/jpred/), and jnetpred predictions were used for all calculations. Jnetpred maximizes accuracy by combining sequence profiles from HMMer[ 23 ] and PSI‐BLAST,[ 24 ] and we found previously that it identifies fold switchers more robustly than other secondary structure predictors.[ 12 ] Each residue was assigned one of three secondary structures: “H” for helix, “E” for extended β‐strand, and “C” for coil. Chain breaks were annotated “−”. PDB IDs and chains of each fold‐switched pair, as well as their FSR boundaries, are reported in Table S1. FSR boundaries were initially chosen based on the regions reported previously (bold sequences in table S2 of Ref. [1]). PimA, KaiB, and RfaH were shortened to yield secondary structure prediction discrepancies, and an additional 11 residues were also added to the N‐terminal end of PimA's FSR. Such modifications seemed reasonable since JPred4 makes predictions based on a 20‐residue window[ 25 ] that it could use to associate an isolated fragment with its contextualized secondary structure prediction. Thus, modifying short stretches of N‐ and C‐terminal sequence could decrease the association between isolated sequences and their contextualized predictions.
2.3. Observed secondary structure discrepancies
Secondary structure classifications of the 93 extant fold switchers were taken from Ref.[ 1 ], and classifications of the three KaiB variants were presumed to be the same as S. elongatus KaiB. Classifications of MinE, ORF9b, and BAX were determined using DSSP.[ 26 ] To quantify secondary structure difference, FSR sequences were aligned with their parents using Biopython[ 27 ] pairwise2.align.localxs with gap open/extension penalties of −1.0/−0.5. Secondary structure classifications in the same register as the aligned FSR sequences were extracted from both experimentally determined structures. Helix ↔ strand discrepancies between the classifications were summed residue‐by‐residue (1 for discrepancy, 0 for no discrepancy) and normalized by FSR length. Pearson correlations were calculated using the corcoef function from Numpy,[ 28 ] and linear fits were determined using Scipy[ 29 ] stats.linregress. Our benchmark set was selected by maximizing:
where TP is the number of true positives and “total” is the total number of proteins (true positives + false negatives). Since all 99 proteins switch folds, correct predictions were true positives and incorrect ones were false negatives.
2.4. Single‐fold proteins and fragments
Proteins expected not to switch folds and having fewer than 800 residues (the upper limit in JPred4), totaling 211, were taken from table S3C of Ref. [1]. One segment was selected from a random region of each protein. Segment lengths were randomly selected from a distribution of FSR lengths ranging from 20 to 41, the range of lengths in our benchmark set. Random selections were performed using the random module of Python 2.7. JPred4 was run on all 422 sequences (211 full sequences + 211 segments) using its mass‐submit scheduler (http://www.compbio.dundee.ac.uk/jpred4/api.shtml#massSubmit).
2.5. Helix ↔ strand discrepancies and distribution
Sequences of isolated FSRs were aligned with full‐length proteins using the pairwise2.align.localxs function from Biopython[ 27 ] with gap open/extension penalties of −1.0/−0.5. Secondary structure predictions were re‐registered according to the resulting alignments and compared. Helix ↔ strand discrepancies between the predictions were summed residue‐by‐residue (1 for discrepancy, 0 for no discrepancy) and normalized by FSR length. An overall view of our predictive method (Sections 2.2, 2.5) is shown in Scheme 1.
SCHEME 1.
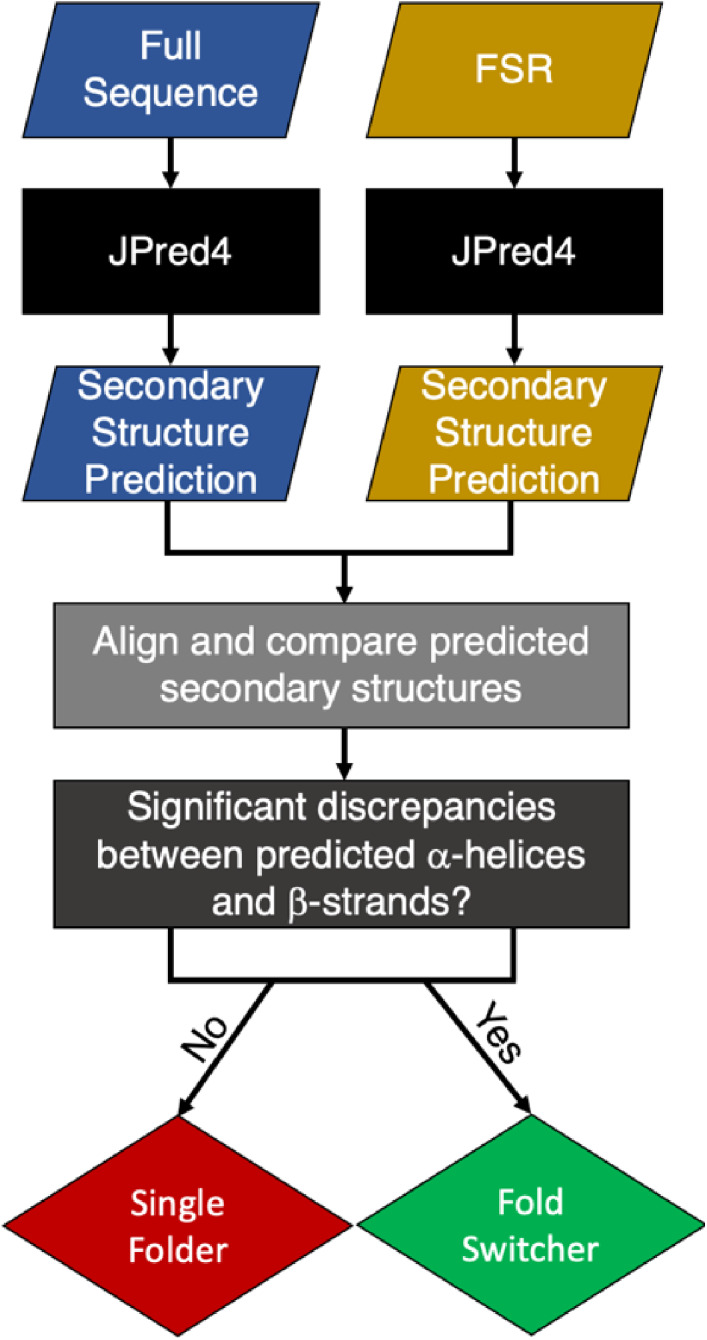
Summary of predictive approach
2.6. Distributions and statistics
The distributions in Figures 1 and 3 were generated with Matplotlib.[ 30 ] Matthews correlation coefficients[ 31 ] were calculated as follows:
where TP = number of true positives, TN = number of true negatives, FP = number of false positives, and FN = number of false negatives.
2.7. Chameleon sequences
All 8‐residue chameleon sequences (stringent criterion) with non‐homologous sequences from the ChSeq[ 32 ] database were tested for fold switching. Since JPred4 cannot predict the secondary structures of sequences so short, we extracted 30‐residue (mean FSR length of 28 rounded to the nearest multiple of 5) fragments from their parents centered on the chameleon sequences (or as close as possible if the sequences were near termini). JPred4 was then run on all fragments and whole sequences using the mass‐submit scheduler. Predictions of whole sequences and fragments were compared as in Section 2.5.
3. RESULTS
3.1. JPred4 predicts fold switchers that undergo α‐helix ↔ β‐strand transitions
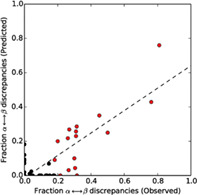
We sought to determine whether JPred4 can identify FSRs of extant fold switchers. To do this, JPred4 predictions of isolated FSR sequences and FSRs within their parent sequences (hereafter called contextualized FSRs, Methods) were compared for 99 experimentally validated fold switchers. A moderate Pearson correlation (.67) was observed between predicted and experimentally observed α‐helix ↔ β‐strand discrepancies (Figure S1), indicating that JPred4 can identify some fold switchers that undergo α‐helix ↔ β‐strand transitions. False positives with no observed α ↔ β transitions were eliminated by removing fragments with high levels of predicted coil (≥65%), improving the overall correlation substantially (.82, Figure 1). Together, these results indicate that our method can effectively identify some fold switchers that undergo α‐helix ↔ β‐strand transitions, but not fold switchers that undergo other types of secondary structure transitions, such as shifts in β‐sheet register.
FIGURE 1.
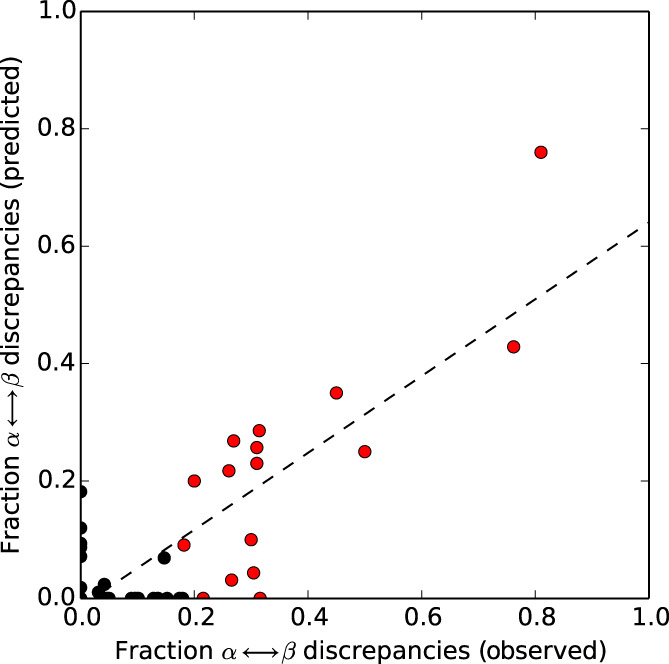
Helix ↔ strand discrepancies predicted by JPred4 correspond to experimentally observed α‐helix ↔ β‐strand differences in fold‐switching regions. Dotted line represents best linear fit of all datapoints (black and red circles; Pearson correlation: .82). Red circles correspond to benchmark set of 17‐fold switchers. Only 16 can be observed because two KaiB variants (4KSO and 1WWJ) overlap exactly at (0.31, 0.26)
3.2. Extant fold switchers with sizeable α‐helix ↔ β‐sheet transitions
We selected a benchmark set of 17 fold switchers by determining the fraction of observed α ↔ β discrepancies that maximized both the percentage and the total number of true positives (Methods, fraction = 0.17, Figure 1, Figure S2). Ten members of this set are highlighted briefly in Figure 2, and all are reported in Table S2.
FIGURE 2.
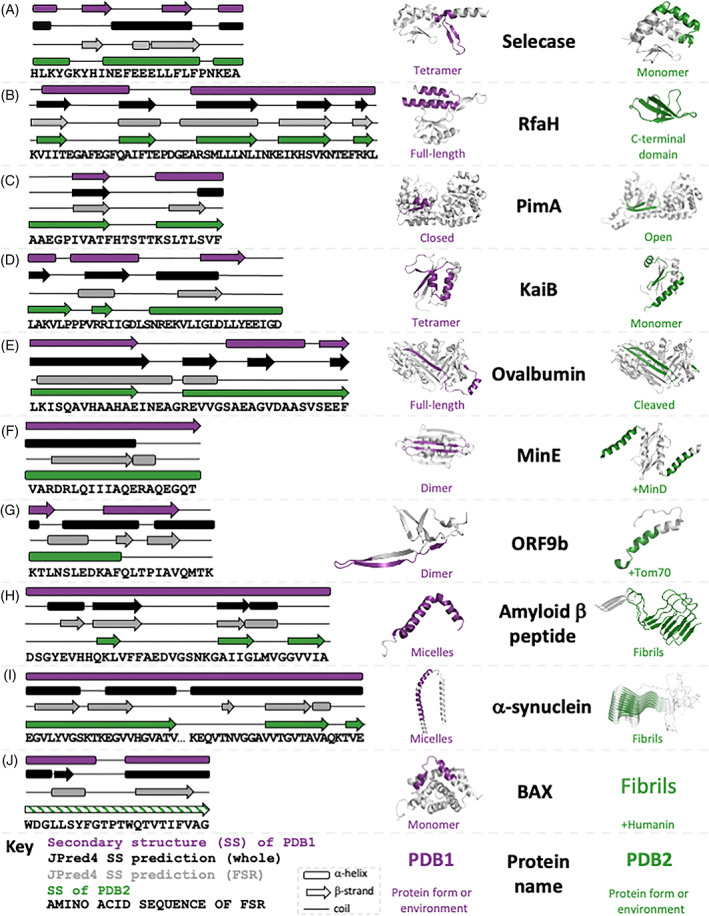
JPred4 predicts different secondary structures for isolated and contextualized FSRs of extant fold switchers with substantive transitions between α‐helix and β‐strand. Each panel shows the experimentally determined secondary structures of both conformations of the fold switcher (purple and green) along with JPred4 secondary structure predictions of the whole sequence (black) and FSR (gray). Purple and green regions of protein structure correspond to FSR sequence shown in diagram; gray corresponds to structurally constant regions (SCRs). Predicted secondary structures that were at least two contiguous residues long are shown. The KaiB variant (2QKE) represents all members of the KaiB family; the other three (1WWJ, 4SKO, 1R5P) are not shown; amylin is also not shown due to lack of space. The differential secondary structure predictions for ORF9b were reported previously.[ 33 ] The green secondary structure diagram of BAX is shaded with lines to signify that its structure has not been solved, though other experimental evidence strongly suggests that it folds into a β‐sheet. All three‐dimensional protein structures were made using PyMOL[ 34 ]
Selecase (“selective and specific caseinolytic metallopeptidase”; Figure 2A), produced by archaea and bacteria, and most studied from the archaeon Methanocaldococcus jannaschii, is an active metallopeptidase in its monomeric form. Upon forming structured higher‐order oligomers, namely dimers, tetramers, and octamers, Selecase is inactivated.[ 35 ] Its structures and activities are regulated by its concentration: mostly monomers at 0‐0.3 mg/mL; dimers at 0.3‐2 mg/mL; tetramers at 2‐6 mg/mL, and octamers at >6 mg/mL.
RfaH (Figure 2B) regulates the expression of virulence proteins from enterobacteria such as Escherichia coli.[ 36 ] It has two domains: an N‐terminal NGN‐binding domain (NTD) and a C‐terminal domain (CTD) that switches folds. RfaH's CTD folds into an α‐helical bundle that forms a binding interface with the NTD, masking its RNA polymerase (RNAP) binding site. Upon binding both RNAP and a specific DNA consensus sequence, called ops, the CTD dissociates from the NTD, unmasking the NTD's RNAP binding site. This binding event also triggers the CTD to reversibly refold into a β‐barrel able to bind the integral S10 unit of the ribosome and foster efficient translation.[ 37 ] When expressed in isolation, RfaH's CTD folds into a β‐barrel with no trace of α‐helical content (green structure).[ 37 ]
PimA (Figure 2C) is a membrane‐associated bacterial glycosyltransferase (phosphatidyl‐myo‐inositol mannosyltransferase) that initiates the biosynthesis of virulence factors produced by Mycobacterium tuberculosis. This enzyme has both a closed GDP‐bound form and an open form with reshuffled secondary structure. PimA's FSR is highly conserved in mycobacterial orthologs, and both crystallographic and near‐UV CD evidence indicate that its open form could play an important role in membrane interactions.[ 38 ]
KaiB (Figure 2D) is a major component of the cyanobacterial circadian clock of S. elongatus.[ 6 ] Unlike most other circadian clocks, which are driven by transcription‐translation oscillation, the cyanobacterial circadian clock is maintained through a periodic phosphorylation cycle, known as a post‐translational oscillator (PTO).[ 39 ] At night, KaiB's active monomeric form helps to regulate the dephosphorylation of the PTO, while in the morning it primarily populates an inactive tetramer with a different fold, allowing phosphorylation of the PTO.
Ovalbumin (Figure 2E) is a member of the serpin family (serine protease inhibitor; although ovalbumin is not known to have in situ inhibitory activity—it constitutes 60%‐65% of egg whites and appears to be a storage protein[ 40 ]) with a zymogenic form (i.e., an inactive precursor, as has plasmepsin). Specifically, inactive ovalbumin has a reactive center loop (RCL) that, when cleaved by a serine protease such as subtilisin, forms a β‐strand inserted between two pairs of β‐hairpins on its surface. Additionally, the α‐helix formed by ovalbumin's uncleaved RCL is regular and less flexible than the distorted helices of inhibitory serpins.[ 41 ]
MinE (Figure 2F) is part of a three‐component protein oscillator that helps to regulate bacterial cell division.[ 19 ] In its resting state, MinE forms a homodimer with six β‐strands (three from each monomer) and four α‐helices (two from each monomer). When bound to MinD, another component of the oscillator, MinE's two central β‐strands are extruded from its dimer interface and refold into helices that bind MinD,[ 20 ] stimulating MinD's ATPase activity and leading to membrane release.
ORF9b (Figure 2G) is from the genome of the Severe Acute Respiratory Syndrome Coronavirus 2 (SARS‐CoV‐2). Expressed in isolation, it forms a homodimer composed of β‐sheets. When bound to human Tom70, however, it refolds into an α‐helix with one of two possible cellular effects[ 21 ]: (a) modulating interferon and apoptosis signaling or (b) decreasing mitochondrial import efficiency, leading to mitophagy. JPred4 has been used previously to predict ORF9b's fold switching.[ 33 ]
The human amyloid‐forming proteins α‐Synuclein and amyloid β (Figure 2H,I, respectively), along with amylin (Table S2), are all believed to interact with membranes, where they form α‐helices.[ 42 , 43 , 44 ] While the cognate functions of helical α‐synuclein and amyloid β remain under investigation, amylin is an endocrine hormone (co‐secreted with insulin) that regulates glycemic metabolism.[ 43 ] All three peptides can also form fibrillar deposits associated with diseases such as Parkinson's (α‐Synuclein),[ 45 ] type 2 diabetes (amylin),[ 46 ] and Alzheimer's (amyloid β).[ 47 ]
BAX is a human protein involved in mitochondrial apoptosis. It assumes an all α‐helical fold in the cytosol, and membrane insertion of its C‐terminal helix appears to foster its apoptotic function. Several lines of experimental evidence (e.g., mass spectrometry, electron microscopy, and circular dichroism spectroscopy) indicate that BAX refolds into β‐sheet fibrils when bound to the humanin peptide.[ 22 ] Furthermore, light‐scattering experiments demonstrate that its C‐terminal helix propagates fibril formation.[ 22 ] This refolding is believed to sequester BAX, preventing it from initiating mitochondrial apoptosis.
3.3.
In all cases shown in Figure 2, along with 5/7 of the other proteins in our benchmark set (Table S2), we found that JPred4 predicted different secondary structures for isolated and contextualized FSR sequences. JPred4 secondary structure predictions tend to correspond reasonably well (<6% α‐helix ↔ β‐strand discrepancies[ 9 ]) with at least one experimentally determined protein structure for all 14 proteins. In fact, in all 17 cases, α‐helix and β‐strand secondary structure elements correspond well between one prediction and one experimentally determined conformation (correct secondary structures in all of the right positions, though not necessarily the experimentally determined lengths). However, the alternative JPred4 predictions generally do not correspond well with the alternative secondary structure prediction, except for PimA and KaiB. Nevertheless, as in previous work,[ 12 ] we use discrepancies between predictions to infer fold switching; for our purposes, the accuracies of the JPred4 predictions have no bearing on this inference.
3.4. JPred4 discriminates between FSRs and single folding regions
To determine the significance of JPred4's α‐helix ↔ β‐strand prediction discrepancies for isolated and contextualized FSRs, we randomly selected fragments from a set of 211 proteins expected not to switch folds (single‐fold proteins). Upon eliminating all predictions with ≥65% coil, 136 predictions remained.
Predictions of single folders and fold switchers are compared in Figure 3. We noticed that 11/17 of the fold‐switching proteins in our benchmark set had predicted helix ↔ strand discrepancies ≥20%, while only 2/136 of single folders had helix ↔ strand discrepancies at the same threshold. One of these false positives comprised residues 12‐48 from the glutathione S‐transferase (GST) Omega 3 expressed by the silkworm Bombyx mori. Residue 29 of this segment is an asparagine, which replaces a highly conserved cysteine in the other members of the Omega family.[ 48 ] This single amino acid change is partially responsible for Omega 3's loss of GST activity: mutating asparagine 29 to a cysteine while also deleting its flexible C‐terminal helix restores GST activity. Interestingly, running JPred4 on the same segment (residues 12‐48) with just an N29C mutation gives the same secondary structure prediction as that of the whole protein (Table S3).[ 48 ] Based on our previous work on sequence‐similar fold switchers,[ 12 ] this result suggests that this protein segment might switch folds and thus might not be a false positive after all. The other false positive comprised residues 93‐142 of Bd3460, a self‐protection protein from Bdellovibrio bacteriovorus that assumes an ankyrin‐like fold.[ 49 ] No obvious reason for the fold switching misclassification was identified.
FIGURE 3.
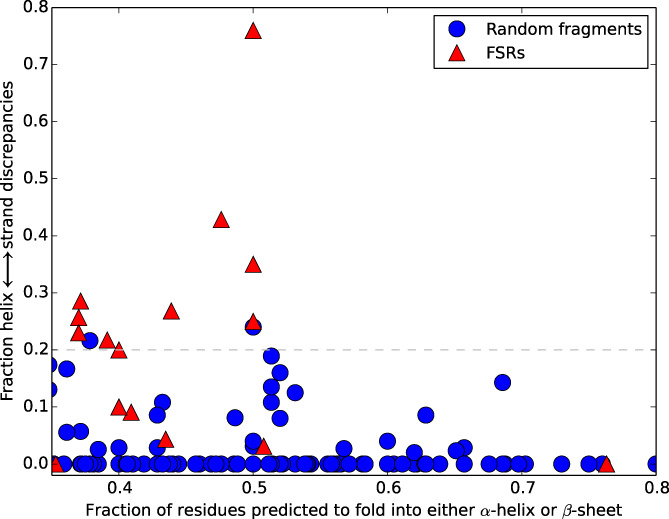
JPred4 discriminates between single folders and fold switchers. Single folders/fold switchers are blue circles/red triangles. The dashed line represents the threshold for classifying fold switchers by fraction of predicted secondary structure/fraction of α‐helix ↔ β‐strand discrepancies (0.2). Datapoints at or above this threshold are predicted to switch folds. Only 16/17‐fold switchers can be seen because two KaiB variants have identical coordinates (0.37, 0.26)
At a 20% threshold for predicted α‐helix ↔ β‐strand discrepancies, our method yielded 11 true positives, 2 false positives, 134 true negatives, and 6 false negatives, resulting in a Matthews correlation coefficient of .71 (very low false‐positive rate; moderate false‐negative rate). In 4/6 false negatives, α‐helix ↔ β‐strand discrepancies were predicted, but they were not large enough to exceed the 20% threshold for the classifier. JPred4 may have misclassified the six false negatives for two reasons. Firstly, we suspect that the sequence profiles generated for FSRs and whole proteins were similar, leading to identical JPred predictions. Secondly, database population may have played a role in the misclassification. Specifically, sequences associated with 1‐fold may have been more highly represented than sequences associated with the other.
3.5. JPred4 does not systematically classify chameleon sequences as fold switchers
To further test the robustness of our classifier, we ran our JPred4‐based method on 45 nonhomologous chameleon sequences from the ChSeq database.[ 32 ] Chameleon sequences are identical sequences that assume α‐helices in some proteins and β‐strands in others but are not associated with fold switching.[ 50 ] Of the 36 sequences with <65% coil predicted, 5 were classified as putative fold switchers (Table S4). Thus, while our method sometimes misclassifies chameleon sequences as fold switchers, it is not a systematic defect.
4. DISCUSSION
Fold switchers are exceptions to the observation that folded proteins assume one stable structure that performs one function. Nevertheless, increasing evidence suggests that these proteins may be more abundant in nature than previously thought.[ 1 ] Fold switching impacts protein function[ 5 ] and is associated with multiple diseases.[ 2 , 3 , 51 ] Thus, it would be useful to have a bioinformatic algorithm that identifies more fold switchers from their sequences. This is especially true because, up to this point, all experimentally characterized fold switchers have been stumbled upon by chance.[ 1 ]
Here we present an approach for predicting extant fold switchers from their amino acid sequences alone. This method is based on previous experimental work suggesting that the FSRs of proteins are context‐dependent: that is, their conformations are determined by their environment.[ 17 , 18 ] In light of this, we hypothesized that it might be possible to predict extant fold switchers by comparing the JPred4 secondary structure predictions of isolated FSRs with contextualized FSRs and searching for α‐helix ↔ β‐strand discrepancies. Indeed, significant discrepancies were found in 11/17‐fold switchers used in this study. We used this finding to develop a classifier for extant fold switchers that yielded a Matthews correlation coefficient of .71. We suspect that JPred4 successfully identified extant fold switchers for the same reason it identified sequence‐similar fold switchers[ 12 ]: different sequences (contextualized and isolated FSRs in this case) yielded different sequence profiles from PSI‐BLAST searches. Future work revealing how these different profiles lead to dramatically different secondary structure predictions would be useful.
Two additional results stand out in light of our previous method,[ 12 ] which predicts evolved fold switchers with highly similar sequences. First, the method presented here predicts fold switching in all four KaiB variants tested. This positive result is an improvement over our previous method for sequence‐similar fold switchers, which failed to predict fold switching in all KaiB variants.[ 12 ] Secondly, our results strongly suggest that the fragment from Omega 3 is an FSR, even though it was in our set of proteins not expected to switch folds. Just one mutation (N29C) is sufficient to dramatically change the secondary structure predictions of this sequence, a previously identified characteristic of sequence‐similar fold switchers (proteins with highly similar—but not identical—amino acid sequences and different folds[ 12 ]). Additionally, Omega 3's GST topology[ 48 ] has been known to switch folds in other proteins, namely KaiB[ 52 ] and chloride intracellular channel 1 (CLIC1).[ 53 ] Still, further experimental work would be needed to determine whether Omega 3 switches folds.
Although we are optimistic that the approach presented here can be used to predict novel fold switchers, it has several limitations. Firstly, it can only identify fold switchers that undergo large α‐helix ↔ β‐sheet transitions. To date, these proteins are rare and comprise only 17% of known fold switchers. Biologically important fold switchers like lymphotactin,[ 54 ] which maintains β‐sheets that change their hydrogen bonding register, and most β‐pore proteins,[ 55 ] which extend already existing β‐sheet structures, will be missed. Secondly, it will not identify all fold switchers that undergo large α‐helix ↔ β‐sheet transitions, as evidenced by the fact that only 11/17 of the fold switchers tested gave a robust enough signal to be classified positively. Thirdly, because the FSRs of undiscovered fold switchers are not known a priori, our method will likely need to test many putative FSRs (different sizes and different regions) within the same protein to determine whether or not it is a fold switcher. Although this approach is much less computationally intensive than all‐atom simulations, it will still require substantial time and computational power to predict fold switching in thousands of genomic sequences. Furthermore, the more sequences probed, the more likely false positives will be hit. Additional work will be needed to more accurately distinguish between these false positives and true fold switchers. Finally, our training set was small, comprising only 17 known fold switchers suitable for the predictive method presented here. Thus, it is likely that our statistics, especially for true positives and false negatives, are noisy. As more fold switchers are discovered, we are optimistic that it will be possible to develop methods that can predict more types of fold switchers with higher accuracy.
5. CONCLUSIONS
Our results suggest that the α‐helix ↔ β‐strand transitions of some extant fold switchers can be predicted from their sequences alone using the homology‐based secondary structure predictor JPred4. Although this method will not identify all extant fold switchers whose secondary structures transition from α‐helix ↔ β‐strand, its low false positive (2/136) and moderate true positive (11/17) rates suggest that many positive predictions will likely correspond to true extant fold switchers. Thus, we are optimistic that this approach can be used to predict a subset of extant fold switchers from the broad base of available genomic sequences.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Supporting information
Figure S1. Predicted and observed α‐helix ↔ β‐strand discrepancies for all 99 extant fold switchers.
Figure S2. The number and fraction of true positives are maximized at % observed helix ↔ strand discrepancies of 0.18 (red triangle). This cutoff was used to determine our benchmark set of proteins.
Table S2. JPred4 predictions of extant fold switchers.
Table S3. JPred4 predictions of Omega 3 GST secondary structure (residues 12‐48) for native FSR, native whole protein, and FSR with N29C mutation.
Table S4. False‐positive chameleon sequences.
Table S1 FSR PDB IDs and fragment sequences.
ACKNOWLEDGMENTS
This work utilized the computational resources of the NIH HPS Biowulf cluster (http://hpc.nih.gov). This work was supported in part by the Intramural Research Program of the National Library of Medicine, National Institutes of Health.
Mishra S., Looger L. L., Porter L. L., Biopolymers 2021, 112(10), e23471. 10.1002/bip.23471
Funding information Howard Hughes Medical Institute; U.S. National Library of Medicine; National Institutes of Health
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are openly available in Github at https://github.com/porterll/extant_fold_switchers
REFERENCES
- 1. Porter L. L., Looger L. L., Proc. Natl. Acad. Sci. U. S. A. 2018, 115, 5968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Li B. P., Mao Y. T., Wang Z., Chen Y. Y., Wang Y., Zhai C. Y., Shi B., Liu S. Y., Liu J. L., Chen J. Q., Cell. Physiol. Biochem. 2018, 46, 907. [DOI] [PubMed] [Google Scholar]
- 3. Lei Y., Takahama Y., Microbes Infect. 2012, 14, 262. [DOI] [PubMed] [Google Scholar]
- 4. Jain V., Kikuchi H., Oshima Y., Sharma A., Yogavel M., J. Struct. Funct. Genomics 2014, 15, 181. [DOI] [PubMed] [Google Scholar]
- 5. Kim A. K., Porter L. L., Structure 2021, 29, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chang Y. G., Cohen S. E., Phong C., Myers W. K., Kim Y. I., Tseng R., Lin J., Zhang L., Boyd J. S., Lee Y., Kang S., Lee D., Li S., Britt R. D., Rust M. J., Golden S. S., LiWang A., Science 2015, 349, 324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kang J. Y., Mooney R. A., Nedialkov Y., Saba J., Mishanina T. V., Artsimovitch I., Landick R., Darst S. A., Cell 2018, 173, 1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Berman H. M., Battistuz T., Bhat T. N., Bluhm W. F., Bourne P. E., Burkhardt K., Feng Z., Gilliland G. L., Iype L., Jain S., Fagan P., Marvin J., Padilla D., Ravichandran V., Schneider B., Thanki N., Weissig H., Westbrook J. D., Zardecki C., Acta Crystallogr. D Biol. Crystallogr. 2002, 58, 899. [DOI] [PubMed] [Google Scholar]
- 9. Mishra S., Looger L. L., Porter L. L., Protein Sci. 2019, 28, 1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ambroggio X. I., Kuhlman B., J. Am. Chem. Soc. 2006, 128, 1154. [DOI] [PubMed] [Google Scholar]
- 11. Wei K. Y., Moschidi D., Bick M. J., Nerli S., McShan A. C., Carter L. P., Huang P. S., Fletcher D. A., Sgourakis N. G., Boyken S. E., Baker D., Proc. Natl. Acad. Sci. U. S. A. 2020, 117, 7208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kim A. K., Looger L. L., Porter L. L., Biopolymers 2021, e23416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tian P., Best R. B., PLoS Comput. Biol. 2020, 16, e1008285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sfriso P., Duran‐Frigola M., Mosca R., Emperador A., Aloy P., Orozco M., Structure 2016, 24, 116. [DOI] [PubMed] [Google Scholar]
- 15. Chen N., Das M., LiWang A., Wang L. P., Biophys. J. 2020, 119, 1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Huang I. K., Pei J., Grishin N. V., Bioinformatics 2013, 29, 175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Minor D. L. Jr., Kim P. S., Nature 1996, 380, 730. [DOI] [PubMed] [Google Scholar]
- 18. Porter L. L., He Y., Chen Y., Orban J., Bryan P. N., Biophys. J. 2015, 108, 154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cai M., Huang Y., Shen Y., Li M., Mizuuchi M., Ghirlando R., Mizuuchi K., Clore G. M., Proc. Natl. Acad. Sci. U. S. A. 2019, 116, 25446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Park K. T., Wu W., Battaile K. P., Lovell S., Holyoak T., Lutkenhaus J., Cell 2011, 146, 396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gordon D. E., Hiatt J., Bouhaddou M., Rezelj V. V., Ulferts S., Braberg H., Jureka A. S., Obernier K., Guo J. Z., Batra J., Kaake R. M., Weckstein A. R., Owens T. W., Gupta M., Pourmal S., Titus E. W., Cakir M., Soucheray M., McGregor M., Cakir Z., Jang G., O'Meara M. J., Tummino T. A., Zhang Z., Foussard H., Rojc A., Zhou Y., Kuchenov D., Huttenhain R., Xu J., Eckhardt M., Swaney D. L., Fabius J. M., Ummadi M., Tutuncuoglu B., Rathore U., Modak M., Haas P., Haas K. M., Naing Z. Z. C., Pulido E. H., Shi Y., Barrio‐Hernandez I., Memon D., Petsalaki E., Dunham A., Marrero M. C., Burke D., Koh C., Vallet T., Silvas J. A., Azumaya C. M., Billesbolle C., Brilot A. F., Campbell M. G., Diallo A., Dickinson M. S., Diwanji D., Herrera N., Hoppe N., Kratochvil H. T., Liu Y., Merz G. E., Moritz M., Nguyen H. C., Nowotny C., Puchades C., Rizo A. N., Schulze‐Gahmen U., Smith A. M., Sun M., Young I. D., Zhao J., Asarnow D., Biel J., Bowen A., Braxton J. R., Chen J., Chio C. M., Chio U. S., Deshpande I., Doan L., Faust B., Flores S., Jin M., Kim K., Lam V. L., Li F., Li J., Li Y. L., Li Y., Liu X., Lo M., Lopez K. E., Melo A. A., F. R. Moss 3rd., Nguyen P., Paulino J., Pawar K. I., Peters J. K., Pospiech T. H. Jr., Safari M., Sangwan S., Schaefer K., Thomas P. V., Thwin A. C., Trenker R., Tse E., Tsui T. K. M., Wang F., Whitis N., Yu Z., Zhang K., Zhang Y., Zhou F., Saltzberg D., Consortium Q. S. B., Hodder A. J., Shun‐Shion A. S., Williams D. M., White K. M., Rosales R., Kehrer T., Miorin L., Moreno E., Patel A. H., Rihn S., Khalid M. M., Vallejo‐Gracia A., Fozouni P., Simoneau C. R., Roth T. L., Wu D., Karim M. A., Ghoussaini M., Dunham I., Berardi F., Weigang S., Chazal M., Park J., Logue J., McGrath M., Weston S., Haupt R., Hastie C. J., Elliott M., Brown F., Burness K. A., Reid E., Dorward M., Johnson C., Wilkinson S. G., Geyer A., Giesel D. M., Baillie C., Raggett S., Leech H., Toth R., Goodman N., Keough K. C., Lind A. L., Zoonomia C., Klesh R. J., Hemphill K. R., Carlson‐Stevermer J., Oki J., Holden K., Maures T., Pollard K. S., Sali A., Agard D. A., Cheng Y., Fraser J. S., Frost A., Jura N., Kortemme T., Manglik A., Southworth D. R., Stroud R. M., Alessi D. R., Davies P., Frieman M. B., Ideker T., Abate C., Jouvenet N., Kochs G., Shoichet B., Ott M., Palmarini M., Shokat K. M., Garcia‐Sastre A., Rassen J. A., Grosse R., Rosenberg O. S., Verba K. A., Basler C. F., Vignuzzi M., Peden A. A., Beltrao P., Krogan N. J., Science 2020, 370, eabe9403.33060197 [Google Scholar]
- 22. Morris D. L., Kastner D. W., Johnson S., Strub M. P., He Y., Bleck C. K. E., Lee D. Y., Tjandra N., J. Biol. Chem. 2019, 294, 19055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Potter S. C., Luciani A., Eddy S. R., Park Y., Lopez R., Finn R. D., Nucleic Acids Res. 2018, 46, W200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Altschul S. F., Madden T. L., Schaffer A. A., Zhang J., Zhang Z., Miller W., Lipman D. J., Nucleic Acids Res. 1997, 25, 3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Drozdetskiy A., Cole C., Procter J., Barton G. J., Nucleic Acids Res. 2015, 43, W389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kabsch W., Sander C., Biopolymers 1983, 22, 2577. [DOI] [PubMed] [Google Scholar]
- 27. Cock P. J., Antao T., Chang J. T., Chapman B. A., Cox C. J., Dalke A., Friedberg I., Hamelryck T., Kauff F., Wilczynski B., de Hoon M. J., Bioinformatics 2009, 25, 1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Harris C. R., Millman K. J., van der Walt S. J., Gommers R., Virtanen P., Cournapeau D., Wieser E., Taylor J., Berg S., Smith N. J., Kern R., Picus M., Hoyer S., van Kerkwijk M. H., Brett M., Haldane A., Del Rio J. F., Wiebe M., Peterson P., Gerard‐Marchant P., Sheppard K., Reddy T., Weckesser W., Abbasi H., Gohlke C., Oliphant T. E., Nature 2020, 585, 357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Virtanen P., Gommers R., Oliphant T. E., Haberland M., Reddy T., Cournapeau D., Burovski E., Peterson P., Weckesser W., Bright J., van der Walt S. J., Brett M., Wilson J., Millman K. J., Mayorov N., Nelson A. R. J., Jones E., Kern R., Larson E., Carey C. J., Polat I., Feng Y., Moore E. W., VanderPlas J., Laxalde D., Perktold J., Cimrman R., Henriksen I., Quintero E. A., Harris C. R., Archibald A. M., Ribeiro A. H., Pedregosa F., van Mulbregt P., SciPy C., Nat. Methods 2020, 17, 261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hunter J. D., Comput. Sci. Eng. 2007, 9, 90. [Google Scholar]
- 31. Matthews B. W., Biochim. Biophys. Acta 1975, 405, 442. [DOI] [PubMed] [Google Scholar]
- 32. Li W., Kinch L. N., Karplus P. A., Grishin N. V., Protein Sci. 2015, 24, 1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Porter L. L., Protein Sci. 2021, 30, 1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.The PyMOL Molecular Graphics System, Version 2.0 Schrödinger, LLC.
- 35. Lopez‐Pelegrin M., Cerda‐Costa N., Cintas‐Pedrola A., Herranz‐Trillo F., Bernado P., Peinado J. R., Arolas J. L., Gomis‐Ruth F. X., Angew. Chem. Int. Ed. Engl. 2014, 53, 10624. [DOI] [PubMed] [Google Scholar]
- 36. Burmann B. M., Knauer S. H., Sevostyanova A., Schweimer K., Mooney R. A., Landick R., Artsimovitch I., Rosch P., Cell 2012, 150, 291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zuber P. K., Schweimer K., Rosch P., Artsimovitch I., Knauer S. H., Nat. Commun. 2019, 10, 702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Giganti D., Albesa‐Jove D., Urresti S., Rodrigo‐Unzueta A., Martinez M. A., Comino N., Barilone N., Bellinzoni M., Chenal A., Guerin M. E., Alzari P. M., Nat. Chem. Biol. 2015, 11, 16. [DOI] [PubMed] [Google Scholar]
- 39. Partch C. L., J. Mol. Biol. 2020, 432, 3426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Stein P. E., Leslie A. G., Finch J. T., Carrell R. W., J. Mol. Biol. 1991, 221, 941. [DOI] [PubMed] [Google Scholar]
- 41. Yamasaki M., Arii Y., Mikami B., Hirose M., J. Mol. Biol. 2002, 315, 113. [DOI] [PubMed] [Google Scholar]
- 42. Crescenzi O., Tomaselli S., Guerrini R., Salvadori S., D'Ursi A. M., Temussi P. A., Picone D., Eur. J. Biochem. 2002, 269, 5642. [DOI] [PubMed] [Google Scholar]
- 43. Patil S. M., Xu S., Sheftic S. R., Alexandrescu A. T., J. Biol. Chem. 2009, 284, 11982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rao J. N., Jao C. C., Hegde B. G., Langen R., Ulmer T. S., J. Am. Chem. Soc. 2010, 132, 8657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Walti M. A., Ravotti F., Arai H., Glabe C. G., Wall J. S., Bockmann A., Guntert P., Meier B. H., Riek R., Proc. Natl. Acad. Sci. U. S. A. 2016, 113, E4976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Cao Q., Boyer D. R., Sawaya M. R., Ge P., Eisenberg D. S., Nat. Struct. Mol. Biol. 2020, 27, 653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tuttle M. D., Comellas G., Nieuwkoop A. J., Covell D. J., Berthold D. A., Kloepper K. D., Courtney J. M., Kim J. K., Barclay A. M., Kendall A., Wan W., Stubbs G., Schwieters C. D., Lee V. M., George J. M., Rienstra C. M., Nat. Struct. Mol. Biol. 2016, 23, 409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chen B. Y., Ma X. X., Guo P. C., Tan X., Li W. F., Yang J. P., Zhang N. N., Chen Y., Xia Q., Zhou C. Z., J. Mol. Biol. 2011, 412, 204. [DOI] [PubMed] [Google Scholar]
- 49. Lambert C., Cadby I. T., Till R., Bui N. K., Lerner T. R., Hughes W. S., Lee D. J., Alderwick L. J., Vollmer W., Sockett R. E., Lovering A. L., Nat. Commun. 2015, 6, 8884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Mezei M., Proteins 2021, 89, 3. [DOI] [PubMed] [Google Scholar]
- 51. Jain V., Yogavel M., Oshima Y., Kikuchi H., Touquet B., Hakimi M. A., Sharma A., Structure 2015, 23, 819. [DOI] [PubMed] [Google Scholar]
- 52. Tseng R., Goularte N. F., Chavan A., Luu J., Cohen S. E., Chang Y. G., Heisler J., Li S., Michael A. K., Tripathi S., Golden S. S., LiWang A., Partch C. L., Science 2017, 355, 1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Littler D. R., Harrop S. J., Fairlie W. D., Brown L. J., Pankhurst G. J., Pankhurst S., DeMaere M. Z., Campbell T. J., Bauskin A. R., Tonini R., Mazzanti M., Breit S. N., Curmi P. M., J. Biol. Chem. 2004, 279, 9298. [DOI] [PubMed] [Google Scholar]
- 54. Dishman A. F., Tyler R. C., Fox J. C., Kleist A. B., Prehoda K. E., Babu M. M., Peterson F. C., Volkman B. F., Science 2021, 371, 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Podobnik M., Savory P., Rojko N., Kisovec M., Wood N., Hambley R., Pugh J., Wallace E. J., McNeill L., Bruce M., Liko I., Allison T. M., Mehmood S., Yilmaz N., Kobayashi T., Gilbert R. J., Robinson C. V., Jayasinghe L., Anderluh G., Nat. Commun. 2016, 7, 11598. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Predicted and observed α‐helix ↔ β‐strand discrepancies for all 99 extant fold switchers.
Figure S2. The number and fraction of true positives are maximized at % observed helix ↔ strand discrepancies of 0.18 (red triangle). This cutoff was used to determine our benchmark set of proteins.
Table S2. JPred4 predictions of extant fold switchers.
Table S3. JPred4 predictions of Omega 3 GST secondary structure (residues 12‐48) for native FSR, native whole protein, and FSR with N29C mutation.
Table S4. False‐positive chameleon sequences.
Table S1 FSR PDB IDs and fragment sequences.
Data Availability Statement
The data that support the findings of this study are openly available in Github at https://github.com/porterll/extant_fold_switchers